 Leiya Du1†Qian Zhang1†Yi Li1Ting Li1
Leiya Du1†Qian Zhang1†Yi Li1Ting Li1 Qingshan Deng2Yuming Jia1Kaijian Lei1Daohong Kan3
Qingshan Deng2Yuming Jia1Kaijian Lei1Daohong Kan3 Fang Xie1*Shenglan Huang1*
Fang Xie1*Shenglan Huang1*- 1Department of Oncology, The Second People’s Hospital of Yibin, Yibin, Sichuan, China
- 2Department of Neurosurgery, The Second People’s Hospital of Yibin, Yibin, Sichuan, China
- 3Department of Burn and Plastic Surgery, The Second People’s Hospital of Yibin, Yibin, Sichuan, China
Recent advances in immunotherapy represent a breakthrough in solid tumor treatment but the existing data indicate that immunotherapy is not effective in improving the survival time of patients with glioblastoma. The tumor microenvironment (TME) exerts a series of inhibitory effects on immune effector cells, which limits the clinical application of immunotherapy. Growing evidence shows that phosphate and tension homology deleted on chromosome ten (PTEN) plays an essential role in TME immunosuppression of glioblastoma. Emerging evidence also indicates that targeting PTEN can improve the anti-tumor immunity in TME and enhance the immunotherapy effect, highlighting the potential of PTEN as a promising therapeutic target. This review summarizes the function and specific upstream and downstream targets of PTEN-associated immune cells in glioblastoma TME, providing potential drug targets and therapeutic options for glioblastoma.
1 Introduction
Glioma is the most common primary malignant tumor of the central nervous system (1). Its pathological types and molecular characteristics are varied, and about 80% of cases manifest as glioblastoma (GBM). Primary glioblastoma is the brain tumor with the highest degree of intracranial malignancy, characterized by strong invasion and poor prognosis; the average survival time of GBM patients is only 15 months (2, 3). Currently, postoperative adjuvant chemoradiotherapy is the standard treatment for glioblastoma (GBM) but only provides limited survival benefit. Immunotherapy, represented by immune checkpoint inhibitors, has revolutionized the treatment paradigm for many solid tumors, but only a small percentage of GBM patients have shown objective efficacy (4). Compared with other tumors, GBM demonstrates stronger heterogeneity, lower tumor mutation load, and a highly immunosuppressive microenvironment. Due to the strong immunosuppressive tumor microenvironment (TME) of GBM, the application of immunotherapy in GBM remains suboptimal and requires further research (5). The most significant feature of the GBM tumor immune microenvironment is the absence of tumor-infiltrating lymphocytes (TILs) and natural killer cells (NK cells), as well as the elevated levels of tumor-associated macrophages (TAMs), myelogenic suppressor cells (MDSCs) and regulatory T cells (Tregs) (6). Enhancing the immune system’s targeting effect on GBM has emerged as a promising approach to treating tumors.
Phosphate and tension homology deleted on chromosome ten (PTEN) is the first tumor suppressor gene with protein phosphatase activity and lipid phosphatase activity discovered so far. It is located on human chromosome 10q23.3 and regulates a variety of signaling pathways through its bispecific phosphatase activity, thereby regulating the life process of various cells (7). PTEN can be involved in cell cycle regulation, inhibition of tumor cell proliferation, adhesion, metastasis, angiogenesis, and promotion of cell apoptosis, differentiation, senescence, and other physiological and pathological activities. PTEN plays a crucial role in the occurrence and development of a variety of tumors (breast, melanoma, glioblastoma, prostate, liver, lung), and even a slight decrease in PTEN enzyme activity can affect cancer susceptibility (8). Mutations in IDH, PTEN, 1p/19g, TERT, ATRX, BRAF, and H3F3A in gliomas are of great significance for patient prediction and prognosis (Table 1) (9, 10). Overall, 40% of GBM cases exhibit PTEN mutation or deficiency, which is associated with a poorer prognosis than PTEN non-deletion GBM (11). Many recent studies have shown that PTEN mediates multiple mechanisms of immunosuppression in GBM immune regulation, and targeting PTEN can enhance the immune response of GBM (12, 13). This study summarizes the direct and indirect effects of PTEN on the various pathways of immune response in GBM, the mechanisms of mutual regulation between PTEN and immune cells in the immunosuppressive microenvironment, and the latest immunotherapy strategies for glioblastoma.

Table 1. The mutations genes in GBM patients.
2 PTEN is involved in the GBM immunosuppressive pathway
In glioblastoma, PTEN deletion or mutation may affect the genomic stability, autophagy, and other aspects of the immune response, leading to immunotherapy failure (Figure 1). The P13K/Akt/mTOR signaling pathway mediates important physiological functions by regulating the cell cycle, protein synthesis, cell energy metabolism, and other pathways, and plays a central regulatory role in the process of cell proliferation, growth, and differentiation. Moreover, activation of this signal transduction pathway promotes cell survival and proliferation and participates in angiogenesis, thereby promoting tumor formation, tumor invasion, and metastasis (14). Studies (15) suggest that the P13K/Akt/mTOR signaling pathway also plays a key role in the occurrence and development of cerebral glioblastoma. The regulation of PTEN and mTOR plays an essential role in this transduction pathway. The protein encoded by the PTEN gene has phosphatase activity and can negatively regulate the P13K/Akt/mTOR signal transduction pathway by catalyzing the dephosphorylation of 3,4,5 phosphatidylinositol to 4,5 monophosphatidylinositol, thereby inducing cell apoptosis (16). As the upstream site of the P13K/Akt/mTOR signaling pathway, the PTEN gene inhibits tumor formation through negative regulation of this signaling pathway, whereas inactivating the PTEN gene reduces the negative regulation of this pathway and causes malignant changes in cells. Research (17) has shown that PTEN is involved in the tumor immune response, and PTEN deficiency activates the phosphatidylinositol 3-kinase (PI3K-AKT) pathway to form an immunosuppressive microenvironment. The combination of PI3K inhibitor and PD-1 blocker was found to have a synergistic effect in PTEN-deficient tumors and can improve patient prognosis. Furthermore, the PI3K-AKT-mTOR pathway can directly affect the immune response in PTEN-deficient glioblastoma TME (18). Increased PD-L1 cell surface expression induced by PTEN loss led to decreased T-cell proliferation and increased apoptosis. Because PTEN loss is one mechanism regulating PD-L1 expression, agents targeting the PI3K pathway may increase the antitumor adaptive immune responses (19). PIK3CA-mutated PTEN-lost tumors showed a higher prevalence of CD274-positivity than PIK3CA-wild-type PTEN-lost tumors and PTEN-expressed tumors. These findings support the role of PI3K signaling in the CD274/PDCD1 pathway (20). AKT-mediated β-catenin S552 phosphorylation and nuclear β-catenin are positively correlated with PD-L1 expression and inversely correlated with the tumor infiltration of CD8+ T cells in human glioblastoma specimens, highlighting the clinical significance of β-catenin activation in tumor immune evasion (21).
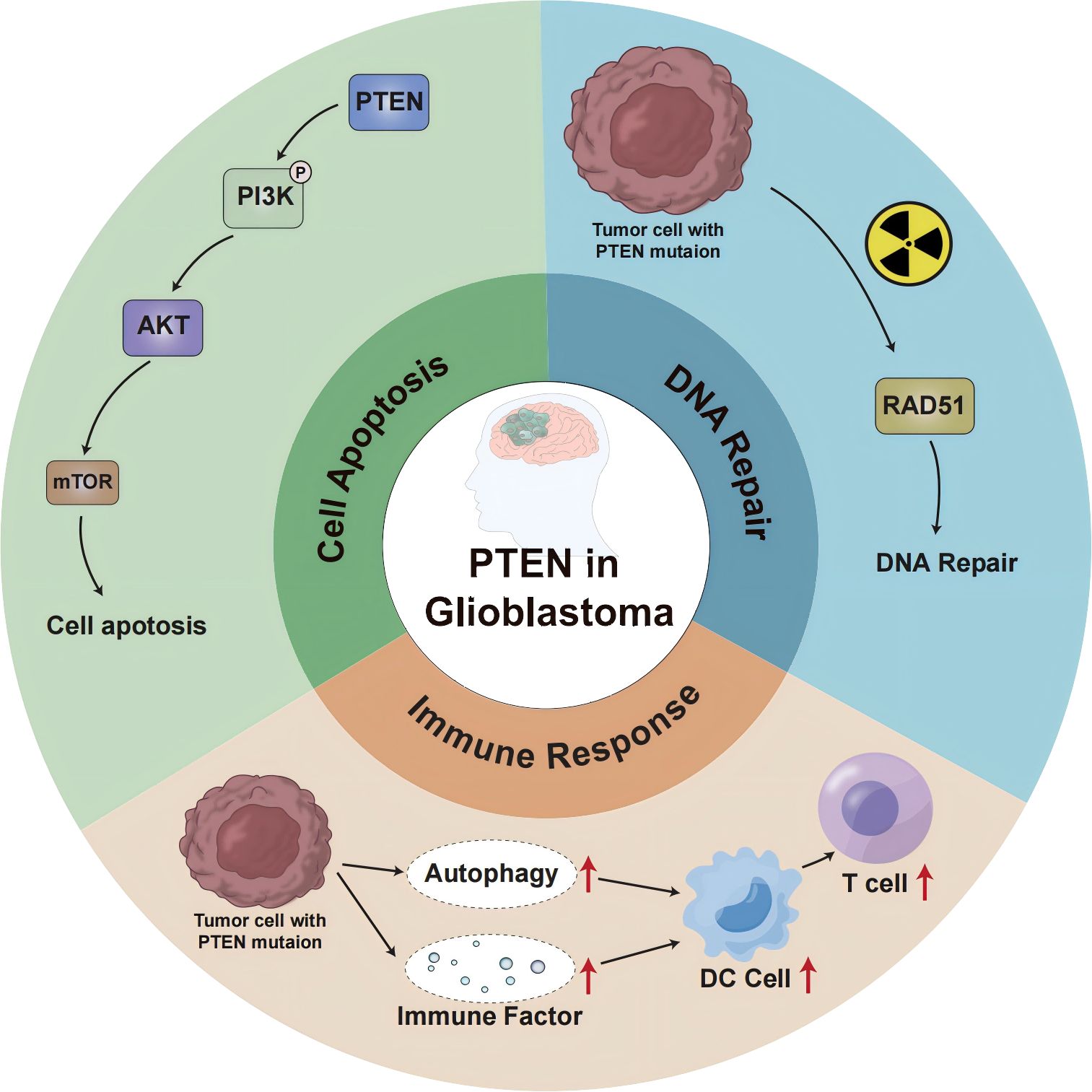
Figure 1. PTEN-mediated signaling pathway and molecular mechanism in GBM.
In addition to cytoplasmic functions that regulate cell growth and proliferation, PTEN also regulates genomic integrity and the stability of DNA repair in the nucleus. Studies (22) have shown that mice with PTEN deletion tumors exhibit increased genomic and chromosomal instability, resulting in centromeric breaks, chromosomal translocations, and spontaneous DNA double-strand breaks that occur independently of the PI3K-AKT-mTOR pathway. About 40% of GBM cases show a deficiency or mutation of the PTEN gene, which influences neurogenesis and gliogenesis, resulting in increased DNA damage repair and malignant progression of brain tumors (23). In glioblastoma (24), after cell exposure to ionizing radiation, DNA repair is weakened when nuclear PTEN is phosphorylated at position 240. Phosphorylated PTEN binds to chromatin and recruits RAD51 to facilitate DNA repair (25). Due to PTEN inactivation promoting higher genomic instability (26, 27), PTEN-deficient tumors are generally considered pro-inflammatory, exhibiting a greater mutation burden and higher immunogenicity in the TME. To counteract the effects of neoantigens, tumors with highly unstable genomes are likely to be able to suppress the host immune response against pro-inflammatory activity (28).
The expression of PTEN can induce autophagy, while the loss of PTEN function down-regulates autophagy, effectively supporting the development of tumors (29, 30). The etiology and pathogenesis of GBM remain incompletely understood, but growing evidence indicates the involvement of the ubiquitin-proteasome system (UPS) and autophagy-lysosome pathway (ALP) in the occurrence, development, and drug resistance of GBM. These effects are carried out by regulating the degradation of cancer-promoting/cancer-suppressing factors and mediating endoplasmic reticulum stress tolerance and misfolded protein reaction (31, 32). PTEN is frequently mutated in glioblastoma, and ectopic expression of functional PTEN in glioma cells induces autophagy flux and lysosomal mass. Furthermore, proteasome activity and protein ubiquitination are inhibited, restricting tumor development. Interestingly, these effects were independent of PTEN lipid phosphatase activity and the PI3K/AKT/mTOR signaling pathway (33). These findings suggest a novel mTOR-independent signaling pathway through which PTEN can act on intracellular protein degradation, regulating autophagy. In addition, studies reported that the activation of the PI3K/Akt/mTOR-mediated signaling pathway can also inhibit autophagy (34–36). Therefore, the molecular components of the proteolytic system regulated by PTEN could represent an innovative therapeutic target for cancer treatment. Moreover, proteasome inhibitors were found to induce cell death in PTEN-deficient GBM organoids and inhibit tumor growth in mice (37). Proteasome inhibitors could be used as targeted therapies for GBM. Mechanistically, PTEN-deficient GBM cells secrete high levels of galectin-9 (Gal-9) via the AKT-GSK3β-IRF1 pathway. The secreted Gal-9 drives macrophage M2 polarization by activating its receptor Tim-3 and downstream pathways in macrophages. These macrophages, in turn, secrete VEGFA to stimulate angiogenesis and support glioma growth (38). Therefore, this study suggests that blockade of Gal-9/Tim-3 signaling is effective to impair glioma progression by inhibiting macrophage M2 polarization, specifically for PTEN-null GBM. PI3Kβ inactivation in the PTEN- null setting led to reduced STAT3 signaling and increased the expression of immune stimulatory molecules, thereby promoting anti-tumor immune responses (39). These findings demonstrate a molecular mechanism linking PTEN loss and STAT3 activation in cancer and suggest that PI3Kβ controls immune escape in PTEN-mutation tumors, providing a rationale for combining PI3Kβ inhibitors with immunotherapy. NF-κB activation was necessary and sufficient for inhibition of PTEN expression. The promoter, RNA, and protein levels of PTEN are down-regulated by NF-κB. The mechanism underlying suppression of PTEN expression by NF-κB was independent of p65 DNA binding or transcription function and involved sequestration of limiting pools of transcriptional coactivators CBP/p300 by p65. Restoration of PTEN expression inhibited NF-κB transcriptional activity and augmented TNF-induced apoptosis, indicating a negative regulatory loop involving PTEN and NF-κB. PTEN is, thus, a novel target whose suppression is critical for antiapoptosis by NF-κB (40).
In the context of tumor cell death, autophagy may lead to the secretion of damage-related molecular chaperones (41, 42). In addition, dead cancer cells may also release autophagosomes containing multiple tumor antigens, which subsequently induce the maturation of dendritic cells (DCS) and cross-present to T cells, promoting tumor immunity (43, 44). PTEN inhibits autophagy, which hinders an effective anti-tumor immune response. Research (45, 46) has revealed that the biology of the immune system determines the occurrence and progression of tumors through a balance between the effects of autophagy regulation and the tolerance response. Autophagy affects the biological functions of various cell types of the immune system, including natural killer cells, dendritic cells, macrophages, and T and B lymphocytes. Autophagy also regulates the secretion of cytokines and antibodies, which in turn impact the autophagy process itself. Transforming growth factor-β, interferon-gamma-γ, and several interleukins (IL) promote autophagy, whereas IL-4, IL-10, and IL-13 are inhibitors (47). Autophagy can be stimulated by innate immune receptors such as toll-like receptors (48); in adaptive immunity, it is a determinant of antigen presentation, lymphocyte differentiation, and cytokine secretion with tumor suppressor activity (49). Therefore, the ideal treatment combination could involve the combination of existing treatment strategies and autophagy-based inducers (PTEN inducers) to trigger cancer cell death and patient response.
3 PTEN affects the GBM immune microenvironment
The glioblastoma microenvironment (TME) is composed of tumor cells, extracellular matrix (ECM), blood vessels, innate immune cells (monocytes, macrophages, mast cells, microglia, and neutrophils), T cells and neurons, astrocytes, and oligodendrocytes (Figure 2). Infiltrating immune cells in GBM are mainly composed of tumor-associated macrophages (TAMs), myelo-derived suppressor cells (MDSC), and T lymphocytes (Table 2) (59). A growing number of studies have shown that the tumor immune microenvironment (TIME) plays a crucial role in regulating the growth and metastasis of GBM. Moreover, PTEN participates in the regulation of immune cell signaling; in contrast, PTEN deficiency can lead to an immunosuppressive tumor microenvironment (60) and hinder the anti-tumor immune response. For example, previous studies revealed that the loss of PTEN is significantly associated with reduced T-cell infiltration at the tumor site and resistance to PD-1 blocking therapy (61–64). The loss of PTEN also promotes the accumulation of inhibitory immune cells, such as MDSCs and Tregs, as well as the formation of an immunosuppressive microenvironment during tumorigenation and development (65–67).
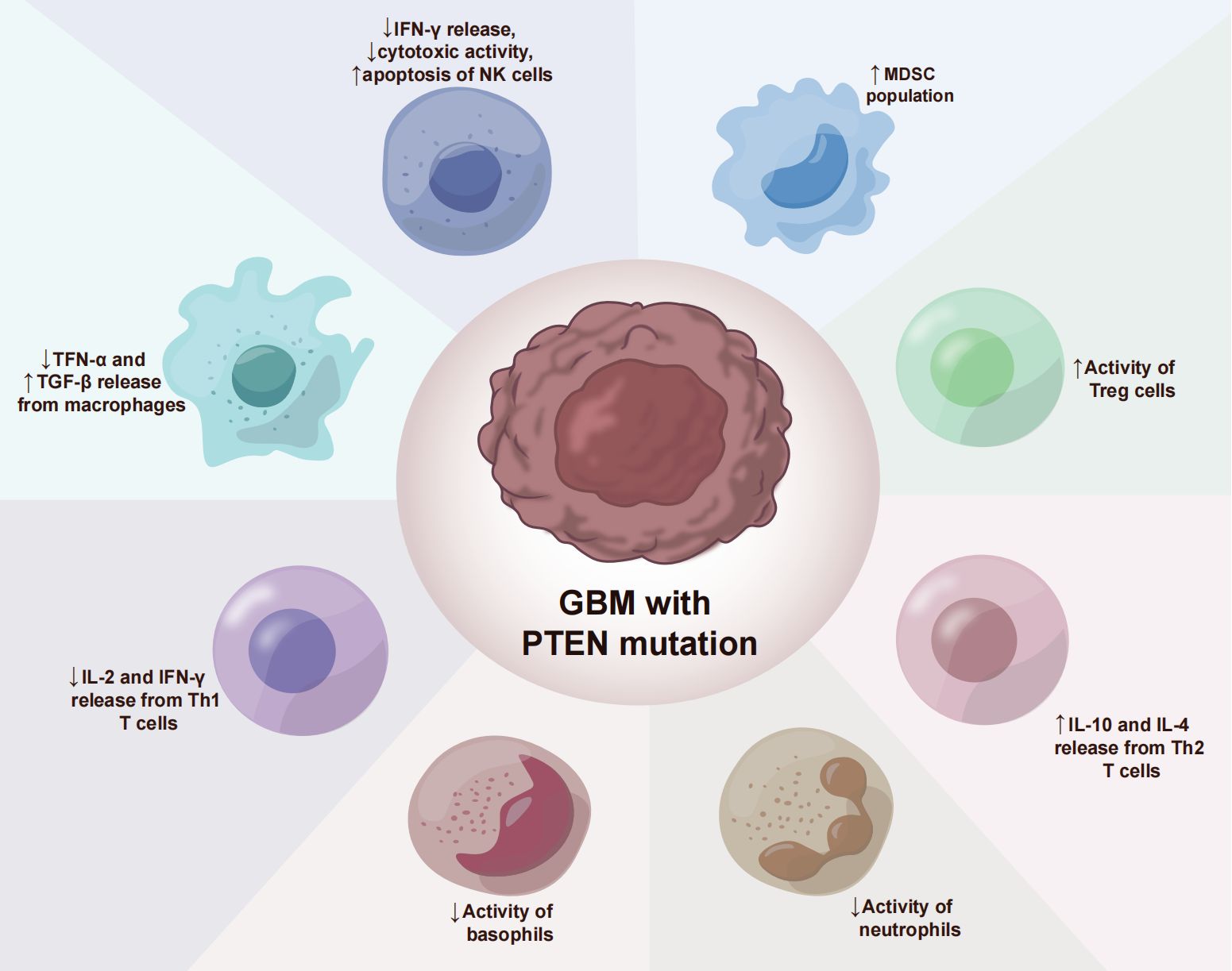
Figure 2. PTEN deficiency immunosuppressive mechanisms in GBM.
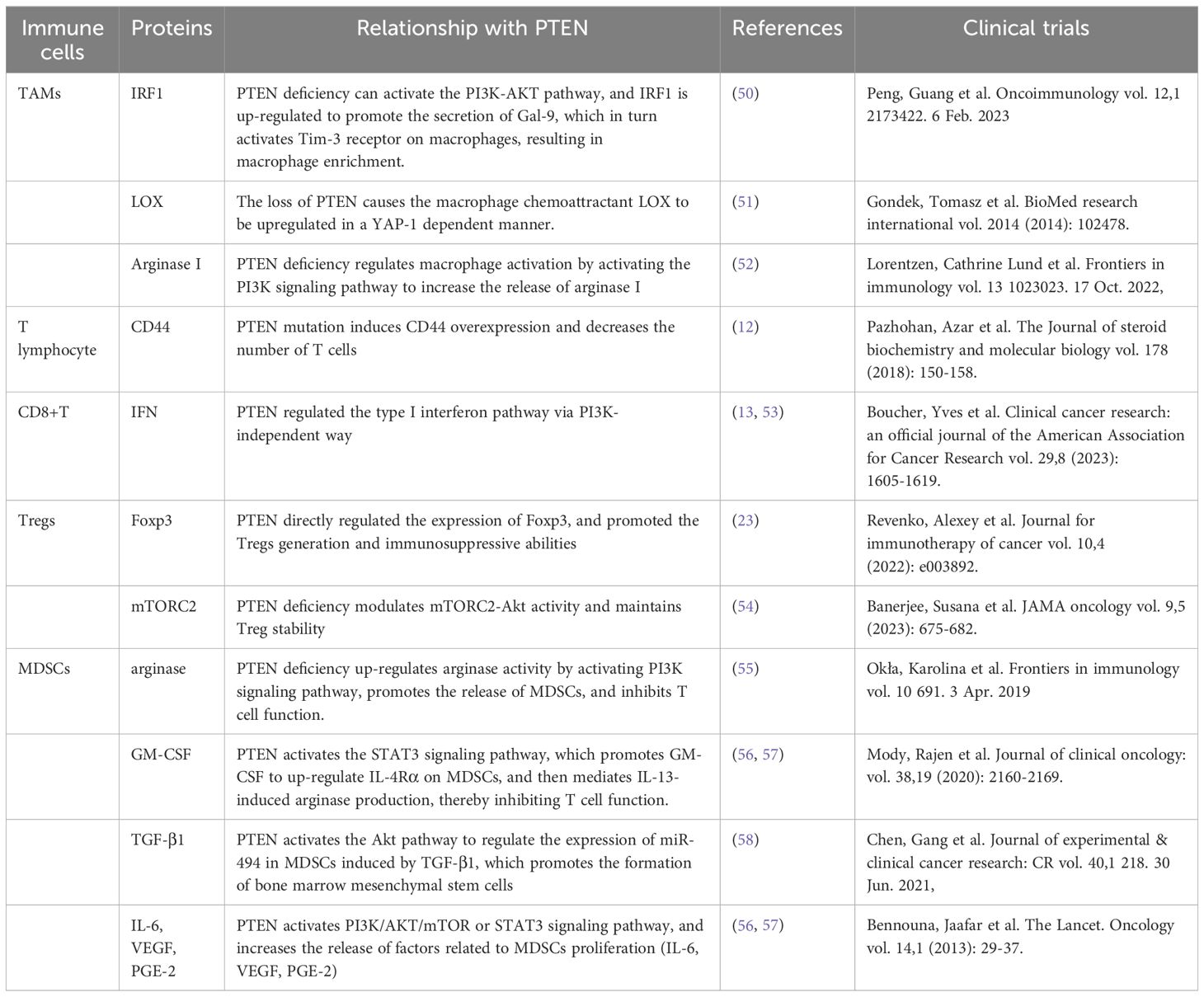
Table 2. The role of PTEN in regulating signaling proteins in immune cells.
3.1 Tumor-associated macrophages
In the glioblastoma microenvironment, tumor-associated macrophages are the most common infiltrating immune cells, accounting for 40% of the total tumor cells (68). Macrophages constitute the most prevalent non-tumor cells in GBM (23). GiomettoB also found that TAMs can be detected in 100% of GBM cases (69). Two different sources of tumor-associated macrophages have been reported in human glioma, namely from embryonic yolk sac monocytes (70) and from peripheral bone marrow-derived monocytes (50). The immunosuppressive anticancer microenvironment is maintained through the recruitment of monocytes, which are converted into macrophages in the glioma environment. TAMs can be divided into two types, M1 type and M2 type. M1-type TAMs typically express high levels of pro-inflammatory factors, promoting Th1 response and strong tumor-killing ability. In contrast, M2 TAMs promote tissue remodeling and tumor progression and secrete inhibitory inflammatory factors (51). Moreover, glioblastoma-associated macrophages have been reported to exert immunosuppressive effects (52). Previous studies have demonstrated that TAMs in the GBM microenvironment primarily adopt the M2-type polarization (53, 68), which fills the glioma microenvironment and controls tumor progression and immune escape mechanism. The M2 phenotype induces differential expression of receptors, cytokines, and chemokines, which produce IL-10, IL-1, and IL-6, thereby stimulating tumorigenesis and negatively affecting prognosis (54). M2 macrophages stimulate the proliferation and invasion of glioma cells and support the immune escape mechanism (71–73). Giotta’s study confirmed (74) the prevalence of PTEN gene mutation in GBM, which is closely associated with poor prognosis and ultra-low survival rate. A recently published report on GBM showed (75, 76) that PTEN deficiency is associated with high macrophage density. Additionally, PTEN-deficient gliomas can recruit a large number of macrophages in the glioma microenvironment. Another study by Ni et al. (38) revealed that the ability of PTEN-deficient gliomas to induce M2 polarization in macrophages was significantly stronger than that of PTEN wild-type gliomas. In PTEN-deficient glioma cells, the activated AKT pathway inactivates GSK-3β by promoting Ser9 phosphorylation, thereby reducing GSK-3β-mediated degradation of IRF1, leading to the up-regulation of the transcription factor IRF1, which enters the nucleus to promote LGALS-9 gene transcription and Gal-9 expression. The activation of the Tim-3 receptor on macrophages by the Gal-9 ligand, in turn, activates transcription factors associated with M2-type polarization and induces macrophage migration, activation, and enrichment of macrophage-associated angiogenesis pathways in PTEN-null gliomas. Gal-9/Tim-3 is a promising target for the treatment of PTEN-deficient gliomas. Blocking Gal-9/Tim-3 can inhibit the malignant progression of gliomas by inhibiting the M2 polarization of macrophages. A new study on the effect of PTEN deletion on glioblastoma demonstrated (71) increased infiltration of macrophages via the YES-associated protein 1-Lysyl oxidase b1(LOX-b1) -integrin-PYK2 axis. Furthermore, LOX expression was found to activate specific pathways in macrophages, facilitating the recruitment of macrophages to the TME. In the GBM model of PTEN deficiency (77), the loss of PTEN leads to the up-regulation of the macrophage chemotactic LOX in a YAP-1-dependent manner. In circulating monocytes, LOX-dependent up-regulation of β1 integrin receptor signaling drives its penetration into GBM tissues to obtain tumor-associated macrophage phenotype and promotes GBM survival and angiogenesis by secreting SPP1. Interfering with these interactions by inhibiting LOX signals can reduce TAM invasion and inhibit tumor growth. Other studies have found that PTEN regulates the activation of macrophages by activating the PI3K signaling pathway to increase the release of arginase I (78), resulting in a low-inflammation environment. Therefore, arginase I is also a potential therapeutic target.
3.2 T lymphocytes
GBM with PTEN mutation shows a reduced number of T cells (17). PTEN mutation can induce an immunosuppressive tumor microenvironment, which is not derived from traditional Treg cells but from tumor cells overexpressing CD44. Other studies have discovered (79) that PTEN regulates the type I interferon pathway in a PI3K-independent manner, inhibits the release of inflammatory factors, and reduces the number of CD8+T cells in GBM. Studies have shown (80) that PTEN lacks the upregulation of mTORC2-Akt activity, and loss of this activity can restore the function of Treg lacking in PTEN. From a mechanism perspective, PTEN can maintain the stability of Treg. Meanwhile, the phosphatase PTEN links Treg stability with inhibition of TH1 and follicular T-helper cell (TFH) responses. Further studies on glioblastoma (18) have revealed that anti-inflammatory cytokine release and T cell activity are significantly reduced in the absence of PTEN and dysregulation of PI3K signaling. Moreover, PTEN inducers or PI3K inhibitors may improve T cell function. Giotta’s study (74) suggested that PTEN mutations were prevalent in GBM, regulating Foxp3 expression and promoting the production of Tregs. Tregs down-regulate T cell activity and regulate innate and adaptive responses to autoantigens, allergens, and infectious agents (81–84). PTEN-deficient tumors usually exhibit a high density of Treg cells in the TME, and Tregs inhibit the function of CD4+, CD8+, and NK cells, and exert immunosuppressive effects in the TME (55, 85, 86).
In addition, T lymphocytes are down-regulated and also exhibit impaired killing function, which is related to TAMs (56). Prostaglandin E2 was found to be produced in the GBM microenvironment, further inhibiting T-cell activity by TAMs and inducing apoptosis. In addition, glioma cells can down-regulate the expression of MHC Class II molecules in microglia and induce ineffective cloning of T cells (87). However, YangI et al. reported that GBM had higher CD8+T cell infiltration compared with pilocytic astrocytoma (57). This differential expression suggests that glioblastoma has a more obvious effect on the local immune microenvironment, but the number does not necessarily represent the potency of the killer cell function. Previous studies have shown that in addition to functional downregulation, CD8+T cells in the GBM microenvironment are involved in the immune escape mechanism.
3.3 Medullary inhibitory cells MDSC
Vidotto’s study (81) reported that PTEN deficiency induces an increase in the density of tumor-infiltrating MDSC in TME. MDSCs are a heterogeneous population composed of a large number of immature bone marrow precursor cells, which are activated under pathological conditions and show strong immunosuppressive activity (88). MDSCs protect tumor cells from host immune attack by negatively regulating immune response, including the depletion of amino acids required by T cells such as arginine and cysteine, the generation of reactive oxygen species nitric oxide and peroxynitrite, direct inhibition of macrophages and natural killer cells, and promotion of tumor angiogenesis (58).
In GBM, MDSCs account for a large proportion of tumor immune cells and play an essential role in promoting tumor growth, tumor cell survival, migration, and immune suppression (89). The glioma microenvironment contributes to the immunosuppressive function of MDSCs (90, 91). MDSCs promote glioma growth, invasion, and angiogenesis as well as the proliferation of Tregs cells (92). GIELEN et al. (93) confirmed that the increase of MDSCs in GBM is related to the increase of arginase activity and that the immunosuppressive function was mediated by inhibiting T cells. Studies have found that glioma cells express many factors related to the proliferation of MDSCs (IL-6, IL-10, VEGF, PGE-2, GM-CSF, and TGF-β2); however, blocking the chemokine CCL2 signaling pathway in glioma cells effectively reduces the recruitment of MDSCs (94). Relevant research data revealed a high proportion of microglial cells/macrophages (GAMs) and MDSCs in malignant GBM, with both GAMs and MDSCs having the ability to recruit Tregs to the tumor, further inhibiting the tumor immune response (59, 95). Studies have found that multiple miRNAs in the tumor microenvironment promote the expansion and immunosuppression of MDSCs by targeting inhibiting PTEN and activating the PI3K/AKT/mTOR or STAT3 signaling pathways (96, 97). In addition, GM-CSF up-regulates IL-4Rα on MDSCs via signal transduction and the transcriptional activator STAT3, thereby mediating IL-13-induced arginase production and inhibiting T cell function.
4 Glioma immunotherapy targeting PTEN
(1) Evidence suggests that PTEN deficiency plays a crucial role in the development of immunosuppressive cancer phenotypes in glioblastoma and is involved in tumor immune responses. Furthermore, PTEN deficiency activates the phosphatidylinositol 3-kinase (PI3K-Akt) pathway to form an immunosuppressive microenvironment. Since restoring PTEN’s function is currently not feasible, suppressing PI3K signaling represents a potential approach to mitigate PTEN loss (98). Another study showed (17) that the combination of PI3K inhibitor and PD-1 blocker exerts a synergistic effect in PTEN-deficient tumors and can improve the prognosis of patients. In primary cultures of PTEN-deficient gliomas, inhibition of components of the PI3K-AKT-mTOR network resulted in reduced T cell death (99) and enhanced immune response.
(2) PTEN can regulate autophagy and affect GBM immune response through the PI3K/Akt/mTOR mediated signaling pathway and new mTOR independent signaling pathway. Therefore, the inducers of autophagy (PTEN inducers) and the molecular components of the proteolytic system associated with autophagy could be new therapeutic directions for GBM. In addition, some studies have found (37) that proteasome inhibitors specifically induce cell death in GBM organoids with PTEN defects and inhibit tumor growth in mice. Proteasome inhibitors can be used as targeted therapies for GBM.
(3) PTEN mediates immune responses independently of PI3K, so future therapies could also target other downstream pathways and signaling molecules that directly control the immune response in the microenvironment of glioblastoma. For example, PTEN-deficient glioblastomas overexpress CD44 cell-surface adhesion receptors and have a tighter tumor cell phenotype than wild-type glioblastomas (100), which can exclude angioforming and immune cells in TME, making them less responsive to immune checkpoint inhibitors (ICI) (17).
(4) From the above presentation of tumor-associated macrophages in glioblastoma with PTEN deletion or mutation, PTEN deletion or mutation was shown to lead to enhanced aggregation of macrophages into the tumor microenvironment (TME). These findings suggest that targeting M2-type TAMs may be particularly effective against gliomas with PTEN deletion. Inhibition of macrophage M2 polarization by targeting Gal-9/Tim-3 represents a potential target for precise immunotherapy for PTEN-deficient gliomas (38).
Immunotherapy is a therapeutic approach to achieve anti-tumor effects through the action of antibodies on the corresponding receptors. Currently, immunotherapy for gliomas includes vaccine therapy, immune checkpoint therapy, chimeric antigen receptor T-cell immunotherapy (CAR-T), natural killer (NK) cell therapy, and lysosomal viral therapy. However, some problems need to be solved. The main problem with immunotherapy is that normal tissues often have antigenic epitopes identical to those of tumor cells, and activation of the immune response can lead to cross-reactivity between the tumor and the body, resulting in toxicity and autoimmune disease (101). another key challenge is whether immunotherapeutic strategies can overcome the multiple mechanisms of immune evasion in gliomas and generate tumor-specific immune responses (102).In addition, the production of immunotherapeutic vaccines is often complex, with multiple methods of constructing the same vaccine, but the effects of the vaccine will vary (103), and the future of immunotherapy will not be limited to single-pharmacological treatments, but will require a combination of therapies to achieve a broad and long-lasting clinical benefit (101).
5 Conclusions and future prospects
A large number of studies have supported the role of PTEN in immune cells and illustrated the immunomodulatory effects of PTEN on glioblastoma TME. PTEN inhibits CD4+/CD8+T cells and dendritic cells while favoring M2 macrophages, Tregs, and MDSCs, participating in glioblastoma progression, metastasis, and immunity. This study outlines the function of PTEN in glioblastoma TME immune cells, as well as their cascade gene activation and clinical outcomes. Increasing evidence demonstrates that targeting PTEN can not only improve the anti-tumor immune function of TME but also enhance the immunotherapy effect, highlighting PTEN as a promising therapeutic target. Nevertheless, whether the recovery of functional PTEN can regulate TME in tumors and improve the sensitivity of tumors to ICB therapy requires further research. Investigating the effectiveness of recovering functional PTEN as a means of cancer treatment holds important clinical significance.
Author contributions
LD: Conceptualization, Funding acquisition, Supervision, Writing – original draft, Writing – review & editing. QZ: Investigation, Supervision, Validation, Writing – original draft, Writing – review & editing. YL: Resources, Validation, Writing – review & editing. TL: Funding acquisition, Investigation, Resources, Writing – review & editing. QD: Investigation, Resources, Visualization, Writing – review & editing. YJ: Investigation, Resources, Writing – review & editing. KL: Investigation, Project administration, Resources, Writing – review & editing. DK: Investigation, Resources, Validation, Writing – review & editing. FX: Data curation, Writing – original draft, Writing – review & editing. SH: Funding acquisition, Investigation, Validation, Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the 2021 hospital-level incubation Project of the Second People’s Hospital of Yibin (Grant number: 2021FY05 and 2021FY16), the Yibin Health and Wellness Medical Research Project (2022YW011, 2023YW009), and the program of Southwest Medical University Higher Education Teaching Research and Reform (JG2022270). The authors declare no conflicts of interests.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2011-2015. Neuro Oncol. (2018) 20:iv1–iv86. doi: 10.1093/neuonc/noy131
2. Fakhoury KR, Ney DE, Ormond DR, Rusthoven CG. Immunotherapy and radiation for high-grade glioma: a narrative review. Transl Cancer Res. (2021) 10:2537–70. doi: 10.21037/tcr
3. Xu S, Tang L, Li X, Fan F, Liu Z. Immunotherapy for glioma: Current management and future application. Cancer Lett. (2020) 476:1–12. doi: 10.1016/j.canlet.2020.02.002
4. Jackson CM, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol. (2019) 20:1100–9. doi: 10.1038/s41590-019-0433-y
5. Locarno CV, Simonelli M, Carenza C, Capucetti A, Stanzani E, Lorenzi E, et al. Role of myeloid cells in the immunosuppressive microenvironment in gliomas. Immunobiology. (2020) 225:151853. doi: 10.1016/j.imbio.2019.10.002
6. Majd N, De Groot J. Challenges and strategies for successful clinical development of immune checkpoint inhibitors in glioblastoma. Expert Opin Pharmacother. (2019) 20:1609–24. doi: 10.1080/14656566.2019.1621840
7. Yehia L, Keel E, Eng C. The clinical spectrum of PTEN mutations. Annu Rev Med. (2020) 71:103–16. doi: 10.1146/annurev-med-052218-125823
8. Milella M, Falcone I, Conciatori F, Cesta Incani U, Del Curatolo A, Inzerilli N, et al. PTEN: multiple functions in human Malignant tumors. Front Oncol. (2015) 5:24. doi: 10.3389/fonc.2015.00024
9. Arita H, Matsushita Y, Machida R, Yamasaki K, Hata N, Ohno M, et al. TERT promoter mutation confers favorable prognosis regardless of 1p/19q status in adult diffuse gliomas with IDH1/2 mutations. Acta neuropathologica Commun. (2020) 8:201. doi: 10.1186/s40478-020-01078-2
10. Wong QH, Li KK, Wang WW, Malta TM, Noushmehr H, Grabovska Y, et al. Molecular landscape of IDH-mutant primary astrocytoma Grade IV/glioblastomas. Modern pathology: an Off J United States Can Acad Pathology Inc. (2021) 34:1245–60. doi: 10.1038/s41379-021-00778-x
11. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. (2010) 17:98–110. doi: 10.1016/j.ccr.2009.12.020
12. Yu J, Lai M, Zhou Z, Zhou J, Hu Q, Li J, et al. The PTEN-associated immune prognostic signature reveals the landscape of the tumor microenvironment in glioblastoma. J neuroimmunology. (2023) 376:578034. doi: 10.1016/j.jneuroim.2023.578034
13. Zhou S, Wang H, Huang Y, Wu Y, Lin Z. The global change of gene expression pattern caused by PTEN mutation affects the prognosis of glioblastoma. Front Oncol. (2022) 12:952521. doi: 10.3389/fonc.2022.952521
14. Chan SM, Weng AP, Tibshirani R, Aster JC, Utz PJ. Notch signals positively regulate activity of the mTOR pathway in T-cell acute lymphoblastic leukemia. Blood. (2007) 110:278–86. doi: 10.1182/blood-2006-08-039883
15. Chakravarti A, Zhai G, Suzuki Y, Sarkesh S, Black PM, Muzikansky A, et al. The prognostic significance of phosphatidylinositol 3-kinase pathway activation in human gliomas. J Clin Oncol. (2004) 22:1926–33. doi: 10.1200/JCO.2004.07.193
16. Davidson L, Maccario H, Perera NM, Yang X, Spinelli L, Tibarewal P, et al. Suppression of cellular proliferation and invasion by the concerted lipid and protein phosphatase activities of PTEN. Oncogene. (2010) 29:687–97. doi: 10.1038/onc.2009.384
17. Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. (2019) 25:462–9. doi: 10.1038/s41591-019-0349-y
18. Cretella D, Digiacomo G, Giovannetti E, Cavazzoni A. PTEN alterations as a potential mechanism for tumor cell escape from PD-1/PD-L1 inhibition. Cancers. (2019) 11(9):1318. doi: 10.3390/cancers11091318
19. Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S, et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res. (2014) 2:361–70. doi: 10.1158/2326-6066.CIR-13-0127
20. Ugai T, Zhao M, Shimizu T, Akimoto N, Shi S, Takashima Y, et al. Association of PIK3CA mutation and PTEN loss with expression of CD274 (PD-L1) in colorectal carcinoma. Oncoimmunology. (2021) 10:1956173. doi: 10.1080/2162402X.2021.1956173
21. Du L, Lee JH, Jiang H, Wang C, Wang S, Zheng Z, et al. β-Catenin induces transcriptional expression of PD-L1 to promote glioblastoma immune evasion. J Exp Med. (2020) 217(11). doi: 10.1084/jem.20191115
22. Bassi C, Ho J, Srikumar T, Dowling RJ, Gorrini C, Miller SJ, et al. Nuclear PTEN controls DNA repair and sensitivity to genotoxic stress. Sci (New York NY). (2013) 341:395–9. doi: 10.1126/science.1236188
23. Chen Z, Hambardzumyan D. Immune microenvironment in glioblastoma subtypes. Front Immunol. (2018) 9:1004. doi: 10.3389/fimmu.2018.01004
24. Shen WH, Balajee AS, Wang J, Wu H, Eng C, Pandolfi PP, et al. Essential role for nuclear PTEN in maintaining chromosomal integrity. Cell. (2007) 128:157–70. doi: 10.1016/j.cell.2006.11.042
25. Ma J, Benitez JA, Li J, Miki S, Ponte De Albuquerque C, Galatro T, et al. Inhibition of nuclear PTEN tyrosine phosphorylation enhances glioma radiation sensitivity through attenuated DNA repair. Cancer Cell. (2019) 35:816. doi: 10.1016/j.ccell.2019.04.011
26. Turajlic S, Litchfield K, Xu H, Rosenthal R, Mcgranahan N, Reading JL, et al. Insertion-and-deletion-derived tumour-specific neoantigens and the immunogenic phenotype: a pan-cancer analysis. Lancet Oncol. (2017) 18:1009–21. doi: 10.1016/S1470-2045(17)30516-8
27. Dudley JC, Lin MT, Le DT, Eshleman JR. Microsatellite instability as a biomarker for PD-1 blockade. Clin Cancer research: an Off J Am Assoc Cancer Res. (2016) 22:813–20. doi: 10.1158/1078-0432.CCR-15-1678
28. Vidotto T, Tiezzi DG, Squire JA. Distinct subtypes of genomic PTEN deletion size influence the landscape of aneuploidy and outcome in prostate cancer. Mol cytogenetics. (2018) 11:1. doi: 10.1186/s13039-017-0348-y
29. Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem. (2001) 276:35243–6. doi: 10.1074/jbc.C100319200
30. Gozuacik D, Kimchi A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene. (2004) 23:2891–906. doi: 10.1038/sj.onc.1207521
31. Chen RH, Chen YH, Huang TY. Ubiquitin-mediated regulation of autophagy. J Biomed science. (2019) 26:80. doi: 10.1186/s12929-019-0569-y
32. Li ZY, Zhang C, Zhang Y, Chen L, Chen BD, Li QZ, et al. A novel HDAC6 inhibitor Tubastatin A: Controls HDAC6-p97/VCP-mediated ubiquitination-autophagy turnover and reverses Temozolomide-induced ER stress-tolerance in GBM cells. Cancer Lett. (2017) 391:89–99. doi: 10.1016/j.canlet.2017.01.025
33. Errafiy R, Aguado C, Ghislat G, Esteve JM, Gil A, Loutfi M, et al. PTEN increases autophagy and inhibits the ubiquitin-proteasome pathway in glioma cells independently of its lipid phosphatase activity. PLoS One. (2013) 8:e83318. doi: 10.1371/journal.pone.0083318
34. Jain MV, Paczulla AM, Klonisch T, Dimgba FN, Rao SB, Roberg K, et al. Interconnections between apoptotic, autophagic and necrotic pathways: implications for cancer therapy development. J Cell Mol Med. (2013) 17:12–29. doi: 10.1111/jcmm.12001
35. Czarny P, Pawlowska E, Bialkowska-Warzecha J, Kaarniranta K, Blasiak J. Autophagy in DNA damage response. Int J Mol Sci. (2015) 16:2641–62. doi: 10.3390/ijms16022641
36. Zhou ZW, Li XX, He ZX, Pan ST, Yang Y, Zhang X, et al. Induction of apoptosis and autophagy via sirtuin1- and PI3K/Akt/mTOR-mediated pathways by plumbagin in human prostate cancer cells. Drug design Dev Ther. (2015) 9:1511–54. doi: 10.2147/DDDT
37. Benitez JA, Finlay D, Castanza A, Parisian AD, Ma J, Longobardi C, et al. PTEN deficiency leads to proteasome addiction: a novel vulnerability in glioblastoma. Neuro Oncol. (2021) 23:1072–86. doi: 10.1093/neuonc/noab001
38. Ni X, Wu W, Sun X, Ma J, Yu Z, He X, et al. Interrogating glioma-M2 macrophage interactions identifies Gal-9/Tim-3 as a viable target against PTEN-null glioblastoma. Sci Adv. (2022) 8:eabl5165. doi: 10.14791/btrt.2022.10.Suppl
39. Bergholz JS, Wang Q, Wang Q, Ramseier M, Prakadan S, Wang W, et al. PI3Kβ controls immune evasion in PTEN-deficient breast tumours. Nature. (2023) 617:139–46. doi: 10.1038/s41586-023-05940-w
40. Vasudevan KM, Gurumurthy S, Rangnekar VM. Suppression of PTEN expression by NF-kappa B prevents apoptosis. Mol Cell Biol. (2004) 24:1007–21. doi: 10.1128/MCB.24.3.1007-1021.2004
41. Wang Y, Martins I, Ma Y, Kepp O, Galluzzi L, Kroemer G. Autophagy-dependent ATP release from dying cells via lysosomal exocytosis. Autophagy. (2013) 9:1624–5. doi: 10.4161/auto.25873
42. Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Sci (New York NY). (2011) 334:1573–7. doi: 10.1126/science.1208347
43. Su H, Luo Q, Xie H, Huang X, Ni Y, Mou Y, et al. Therapeutic antitumor efficacy of tumor-derived autophagosome (DRibble) vaccine on head and neck cancer. Int J nanomedicine. (2015) 10:1921–30. doi: 10.1016/j.bcp.2014.07.006
44. Viry E, Paggetti J, Baginska J, Mgrditchian T, Berchem G, Moussay E, et al. Autophagy: an adaptive metabolic response to stress shaping the antitumor immunity. Biochem Pharmacol. (2014) 92:31–42. doi: 10.1016/j.bcp.2014.07.006
45. Jang YJ, Kim JH, Byun S. Modulation of autophagy for controlling immunity. Cells. (2019) 25(4):214–20. doi: 10.3390/cells8020138
46. Hagerling C, Casbon AJ, Werb Z. Balancing the innate immune system in tumor development. Trends Cell Biol. (2015) 25:214–20. doi: 10.1016/j.tcb.2014.11.001
47. Monkkonen T, Debnath J. Inflammatory signaling cascades and autophagy in cancer. Autophagy. (2018) 14:190–8. doi: 10.1080/15548627.2017.1345412
48. Pan H, Chen L, Xu Y, Han W, Lou F, Fei W, et al. Autophagy-associated immune responses and cancer immunotherapy. Oncotarget. (2016) 7:21235–46. doi: 10.18632/oncotarget.v7i16
49. Shibutani ST, Saitoh T, Nowag H, Münz C, Yoshimori T. Autophagy and autophagy-related proteins in the immune system. Nat Immunol. (2015) 16:1014–24. doi: 10.1038/ni.3273
50. Franco R, Fernández-Suárez D. Alternatively activated microglia and macrophages in the central nervous system. Prog neurobiology. (2015) 131:65–86. doi: 10.1016/j.pneurobio.2015.05.003
51. Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front bioscience: J virtual library. (2008) 13:453–61. doi: 10.2741/2692
52. Grabowski MM, Sankey EW, Ryan KJ, Chongsathidkiet P, Lorrey SJ, Wilkinson DS, et al. Immune suppression in gliomas. J neuro-oncology. (2021) 151:3–12. doi: 10.1007/s11060-020-03483-y
53. Lisi L, Ciotti GM, Braun D, Kalinin S, Currò D, Dello Russo C, et al. Expression of iNOS, CD163 and ARG-1 taken as M1 and M2 markers of microglial polarization in human glioblastoma and the surrounding normal parenchyma. Neurosci letters. (2017) 645:106–12. doi: 10.1016/j.neulet.2017.02.076
54. Yunna C, Mengru H, Lei W, Weidong C. Macrophage M1/M2 polarization. Eur J Pharmacol. (2020) 877:173090. doi: 10.1016/j.ejphar.2020.173090
55. Kim JH, Kim BS, Lee SK. Regulatory T cells in tumor microenvironment and approach for anticancer immunotherapy. Immune network. (2020) 20:e4. doi: 10.4110/in.2020.20.e4
56. Khonina NA, Tsentner MI, Leplina O, Tikhonova MA, Stupak VV, Nikonov SD, et al. [Characteristics and immunologic changes in patients with Malignant brain tumors]. Voprosy onkologii. (2002) 48:196–201. doi: 10.1111/imm.12949
57. Yang I, Han SJ, Sughrue ME, Tihan T, Parsa AT. Immune cell infiltrate differences in pilocytic astrocytoma and glioblastoma: evidence of distinct immunological microenvironments that reflect tumor biology. J neurosurgery. (2011) 115:505–11. doi: 10.3171/2011.4.JNS101172
58. Vanhaver C, van der Bruggen P, Bruger AM. MDSC in mice and men: Mechanisms of immunosuppression in cancer. J Clin Med. (2021) 212(6):491–9. doi: 10.3390/jcm10132872
59. Gieryng A, Pszczolkowska D, Walentynowicz KA, Rajan WD, Kaminska B. Immune microenvironment of gliomas. Lab investigation; J Tech Methods pathology. (2017) 97:498–518. doi: 10.1038/labinvest.2017.19
60. Cao Y, Wang H, Yang L, Zhang Z, Li C, Yuan X, et al. PTEN-L promotes type I interferon responses and antiviral immunity. Cell Mol Immunol. (2018) 15:48–57. doi: 10.1038/cmi.2017.102
61. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer discovery. (2016) 6:202–16. doi: 10.1158/2159-8290.CD-15-0283
62. George S, Miao D, Demetri GD, Adeegbe D, Rodig SJ, Shukla S, et al. Loss of PTEN is associated with resistance to anti-PD-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity. (2017) 46:197–204. doi: 10.1016/j.immuni.2017.02.001
63. Barroso-Sousa R, Keenan TE, Pernas S, Exman P, Jain E, Garrido-Castro AC, et al. Tumor mutational burden and PTEN alterations as molecular correlates of response to PD-1/L1 blockade in metastatic triple-negative breast cancer. Clin Cancer research: an Off J Am Assoc Cancer Res. (2020) 26:2565–72. doi: 10.1158/1078-0432.CCR-19-3507
64. Roh W, Chen PL, Reuben A, Spencer CN, Prieto PA, Miller JP, et al. Integrated molecular analysis of tumor biopsies on sequential CTLA-4 and PD-1 blockade reveals markers of response and resistance. Sci Trans Med. (2017) 151(1):3–12. doi: 10.1126/scitranslmed.aah3560
65. Feng S, Cheng X, Zhang L, Lu X, Chaudhary S, Teng R, et al. Myeloid-derived suppressor cells inhibit T cell activation through nitrating LCK in mouse cancers. Proc Natl Acad Sci United States America. (2018) 115:10094–9. doi: 10.1073/pnas.1800695115
66. Yang R, Cai TT, Wu XJ, Liu YN, He J, Zhang XS, et al. Tumour YAP1 and PTEN expression correlates with tumour-associated myeloid suppressor cell expansion and reduced survival in colorectal cancer. Immunology. (2018) 155:263–72. doi: 10.1111/imm.12949
67. Sharma MD, Shinde R, Mcgaha TL, Huang L, Holmgaard RB, Wolchok JD, et al. The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci advances. (2015) 1:e1500845. doi: 10.1186/s12929-019-0568-z
68. Mignogna C, Signorelli F, Vismara MF, Zeppa P, Camastra C, Barni T, et al. A reappraisal of macrophage polarization in glioblastoma: Histopathological and immunohistochemical findings and review of the literature. Pathology Res practice. (2016) 212:491–9. doi: 10.1016/j.prp.2016.02.020
69. Giometto B, Bozza F, Faresin F, Alessio L, Mingrino S, Tavolato B. Immune infiltrates and cytokines in gliomas. Acta neurochirurgica. (1996) 138:50–6. doi: 10.1007/BF01411724
70. Gomez Perdiguero E, Klapproth K, Schulz C, Busch K, Azzoni E, Crozet L, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. (2015) 518:547–51. doi: 10.1038/nature13989
71. Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor-associated macrophages: an accomplice in solid tumor progression. J Biomed science. (2019) 26:78. doi: 10.1186/s12929-019-0568-z
72. Zhu C, Kros JM, Cheng C, Mustafa D. The contribution of tumor-associated macrophages in glioma neo-angiogenesis and implications for anti-angiogenic strategies. Neuro Oncol. (2017) 19:1435–46. doi: 10.1093/neuonc/nox081
73. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. (2010) 141:39–51. doi: 10.1016/j.cell.2010.03.014
74. Giotta Lucifero A, Luzzi S. Immune landscape in PTEN-related glioma microenvironment: A bioinformatic analysis. Brain Sci. (2022) 193(4):1717–27. doi: 10.4049/jimmunol.1302167
75. Fridman WH, Zitvogel L, Sautès-Fridman C, Kroemer G. The immune contexture in cancer prognosis and treatment. Nat Rev Clin Oncol. (2017) 14:717–34. doi: 10.1038/nrclinonc.2017.101
76. Chen P, Zhao D, Li J, Liang X, Li J, Chang A, et al. Symbiotic macrophage-glioma cell interactions reveal synthetic lethality in PTEN-null glioma. Cancer Cell. (2019) 35:868–84.e6. doi: 10.1016/j.ccell.2019.05.003
77. Chuntova P, Chow F, Watchmaker PB, Galvez M, Heimberger AB, Newell EW, et al. Unique challenges for glioblastoma immunotherapy-discussions across neuro-oncology and non-neuro-oncology experts in cancer immunology. Meeting Report from the 2019 SNO Immuno-Oncology Think Tank. Neuro Oncol. (2021) 23:356–75. doi: 10.1093/neuonc/noaa277
78. Sahin E, Haubenwallner S, Kuttke M, Kollmann I, Halfmann A, Dohnal AM, et al. Macrophage PTEN regulates expression and secretion of arginase I modulating innate and adaptive immune responses. J Immunol (Baltimore Md: 1950). (2014) 193:1717–27. doi: 10.4049/jimmunol.1302167
79. Xing F, Xiao J, Wu J, Liang J, Lu X, Guo L, et al. Modulating the tumor microenvironment via oncolytic virus and PI3K inhibition synergistically restores immune checkpoint therapy response in PTEN-deficient glioblastoma. Signal transduction targeted Ther. (2021) 6:275. doi: 10.1038/s41392-021-00609-0
80. Shrestha S, Yang K, Guy C, Vogel P, Neale G, Chi H. Treg cells require the phosphatase PTEN to restrain TH1 and TFH cell responses. Nat Immunol. (2015) 16:178–87. doi: 10.1038/ni.3076
81. Vidotto T, Melo CM, Castelli E, Koti M, Dos Reis RB, Squire JA. Emerging role of PTEN loss in evasion of the immune response to tumours. Br J cancer. (2020) 122:1732–43. doi: 10.1038/s41416-020-0834-6
82. Grover P, Goel PN, Greene MI. Regulatory T cells: Regulation of identity and function. Front Immunol. (2021) 12:750542. doi: 10.3389/fimmu.2021.750542
83. Boer MC, Joosten SA, Ottenhoff TH. Regulatory T-cells at the interface between human host and pathogens in infectious diseases and vaccination. Front Immunol. (2015) 6:217. doi: 10.3389/fimmu.2015.00217
84. Scheinecker C, Göschl L, Bonelli M. Treg cells in health and autoimmune diseases: New insights from single cell analysis. J autoimmunity. (2020) 110:102376. doi: 10.1016/j.jaut.2019.102376
85. Togashi Y, Shitara K, Nishikawa H. Regulatory T cells in cancer immunosuppression - implications for anticancer therapy. Nat Rev Clin Oncol. (2019) 16:356–71. doi: 10.1038/s41571-019-0175-7
86. Okeke EB, Uzonna JE. The pivotal role of regulatory T cells in the regulation of innate immune cells. Front Immunol. (2019) 10:680. doi: 10.3389/fimmu.2019.00680
87. Tran CT, Wolz P, Egensperger R, Kösel S, Imai Y, Bise K, et al. Differential expression of MHC class II molecules by microglia and neoplastic astroglia: relevance for the escape of astrocytoma cells from immune surveillance. Neuropathology Appl neurobiology. (1998) 24:293–301. doi: 10.1046/j.1365-2990.1998.00120.x
88. Tcyganov E, Mastio J, Chen E, Gabrilovich DI. Plasticity of myeloid-derived suppressor cells in cancer. Curr Opin Immunol. (2018) 51:76–82. doi: 10.1016/j.coi.2018.03.009
89. Arcuri C, Fioretti B, Bianchi R, Mecca C, Tubaro C, Beccari T, et al. Microglia-glioma cross-talk: a two way approach to new strategies against glioma. Front bioscience (Landmark edition). (2017) 22:268–309. doi: 10.2741/4486
90. Gielen PR, Schulte BM, Kers-Rebel ED, Verrijp K, Petersen-Baltussen HM, Ter Laan M, et al. Increase in both CD14-positive and CD15-positive myeloid-derived suppressor cell subpopulations in the blood of patients with glioma but predominance of CD15-positive myeloid-derived suppressor cells in glioma tissue. J neuropathology Exp neurology. (2015) 74:390–400. doi: 10.1097/NEN.0000000000000183
91. Parney IF. Basic concepts in glioma immunology. Adv Exp Med Biol. (2012) 746:42–52. doi: 10.1007/978-1-4614-3146-6_4
92. Wurdinger T, Deumelandt K, van der Vliet HJ, Wesseling P, De Gruijl TD. Mechanisms of intimate and long-distance cross-talk between glioma and myeloid cells: how to break a vicious cycle. Biochim Biophys Acta. (2014) 1846:560–75. doi: 10.1016/j.bbcan.2014.10.003
93. Gielen PR, Schulte BM, Kers-Rebel ED, Verrijp K, Bossman SA, Ter Laan M, et al. Elevated levels of polymorphonuclear myeloid-derived suppressor cells in patients with glioblastoma highly express S100A8/9 and arginase and suppress T cell function. Neuro Oncol. (2016) 18:1253–64. doi: 10.1093/neuonc/now034
94. Kohanbash G, Mckaveney K, Sakaki M, Ueda R, Mintz AH, Amankulor N, et al. GM-CSF promotes the immunosuppressive activity of glioma-infiltrating myeloid cells through interleukin-4 receptor-α. Cancer Res. (2013) 73:6413–23. doi: 10.1158/0008-5472.CAN-12-4124
95. Mei S, Xin J, Liu Y, Zhang Y, Liang X, Su X, et al. MicroRNA-200c promotes suppressive potential of myeloid-derived suppressor cells by modulating PTEN and FOG2 expression. PLoS One. (2015) 10:e0135867. doi: 10.1371/journal.pone.0135867
96. Li L, Zhang J, Diao W, Wang D, Wei Y, Zhang CY, et al. MicroRNA-155 and MicroRNA-21 promote the expansion of functional myeloid-derived suppressor cells. J Immunol (Baltimore Md: 1950). (2014) 192:1034–43. doi: 10.4049/jimmunol.1301309
97. Liu Y, Lai L, Chen Q, Song Y, Xu S, Ma F, et al. MicroRNA-494 is required for the accumulation and functions of tumor-expanded myeloid-derived suppressor cells via targeting of PTEN. J Immunol (Baltimore Md: 1950). (2012) 188:5500–10. doi: 10.4049/jimmunol.1103505
98. Mcloughlin NM, Mueller C, Grossmann TN. The therapeutic potential of PTEN modulation: Targeting strategies from gene to protein. Cell Chem Biol. (2018) 25:19–29. doi: 10.1016/j.chembiol.2017.10.009
99. Waldron JS, Yang I, Han S, Tihan T, Sughrue ME, Mills SA, et al. Implications for immunotherapy of tumor-mediated T-cell apoptosis associated with loss of the tumor suppressor PTEN in glioblastoma. J Clin neuroscience: Off J Neurosurgical Soc Australasia. (2010) 17:1543–7. doi: 10.1016/j.jocn.2010.04.021
100. Cheng F, Eng C. PTEN mutations trigger resistance to immunotherapy. Trends Mol Med. (2019) 25:461–3. doi: 10.1016/j.molmed.2019.03.003
101. Majc B, Novak M, Kopitar-Jerala N, Jewett A, Breznik B. Immunotherapy of glioblastoma: current strategies and challenges in tumor model development. Cells. (2021) 10(2). doi: 10.3390/cells10020265
102. Gilbert MR, Pugh SL, Aldape K, Sorensen AG, Mikkelsen T, Penas-Prado M, et al. NRG oncology RTOG 0625: a randomized phase II trial of bevacizumab with either irinotecan or dose-dense temozolomide in recurrent glioblastoma. J neuro-oncology. (2017) 131:193–9. doi: 10.1007/s11060-016-2288-5
103. Buchroithner J, Erhart F, Pichler J, Widhalm G, Preusser M, Stockhammer G, et al. Audencel immunotherapy based on dendritic cells has no effect on overall and progression-free survival in newly diagnosed glioblastoma: A phase II randomized trial. Cancers. (2018) 10(10). doi: 10.3390/cancers10100372
Keywords: glioblastoma, PTEN, immunity, tumor microenvironment, immunosuppressive
Citation: Du L, Zhang Q, Li Y, Li T, Deng Q, Jia Y, Lei K, Kan D, Xie F and Huang S (2024) Research progress on the role of PTEN deletion or mutation in the immune microenvironment of glioblastoma. Front. Oncol. 14:1409519. doi: 10.3389/fonc.2024.1409519
Received: 30 March 2024; Accepted: 29 July 2024;
Published: 14 August 2024.
Edited by:
Tanmay Abhay Kulkarni, Mayo Clinic, United StatesReviewed by:
Vivekanand Yadav, Children’s Mercy Kansas City, United StatesRamcharan Singh Angom, Mayo Clinic Florida, United States
Copyright © 2024 Du, Zhang, Li, Li, Deng, Jia, Lei, Kan, Xie and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shenglan Huang, NDE5MTgyMDIwQHFxLmNvbQ==; Fang Xie, cGVubnl4aWVmYW5nQGZveG1haWwuY29t
†These authors have contributed equally to this work