 Wei Yan1,2,3
Wei Yan1,2,3 Dean Rao
Dean Rao Huifang Liang
Huifang Liang Zunyi Zhang
Zunyi Zhang Hanhua Dong
Hanhua Dong
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 19 June 2024
Sec. Cancer Genetics
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1407434
This article is part of the Research Topic Exploring Epigenetic Mechanisms in Cancer View all 9 articles
Hepatitis B infection is substantially associated with the development of liver cancer globally, with the prevalence of hepatocellular carcinoma (HCC) cases exceeding 50%. Hepatitis B virus (HBV) encodes the Hepatitis B virus X (HBx) protein, a pleiotropic regulatory protein necessary for the transcription of the HBV covalently closed circular DNA (cccDNA) microchromosome. In previous studies, HBV-associated HCC was revealed to be affected by HBx in multiple signaling pathways, resulting in genetic mutations and epigenetic modifications in proto-oncogenes and tumor suppressor genes. In addition, transforming growth factor-β (TGF-β) has dichotomous potentials at various phases of malignancy as it is a crucial signaling pathway that regulates multiple cellular and physiological processes. In early HCC, TGF-β has a significant antitumor effect, whereas in advanced HCC, it promotes malignant progression. TGF-β interacts with the HBx protein in HCC, regulating the pathogenesis of HCC. This review summarizes the respective and combined functions of HBx and TGB-β in HCC occurrence and development.
Liver cancer is a substantial health problem globally, and it is estimated to be the 6th most frequent tumor and the 3rd primary reason for cancer mortalities (1). In 2020, approximately 905,700 and 830,200 people were diagnosed and died, respectively, due to this tumor worldwide. Between 2020 and 2040, a 55.0% annual increase in new liver cancer diagnoses is anticipated, with 1.4 million new cases expected (2). Various factors contribute to liver cancer development, including alcohol, metabolic syndrome, type 2 diabetes, obesity, non-alcoholic fatty liver disease (NAFLD), aflatoxin B1, and tobacco (3, 4). However, HBV is still the most significant cause as it leads to 56% of the cases, according to GLOBOCAN (5). The most common type of liver cancer, HCC, accounts for nearly 75% of all cases (6)(pp1978–2012).
HBV, a non-cytopathic DNA virus from the Hepadnaviridae family, causes diseases of liver when transmitted through infected blood or body fluids (7). The viral genomes contain relaxed circular DNA (rcDNA), which encodes numerous proteins and can transform into covalently closed circular DNA (cccDNA) in the nucleus (8–10) (Figure 1).
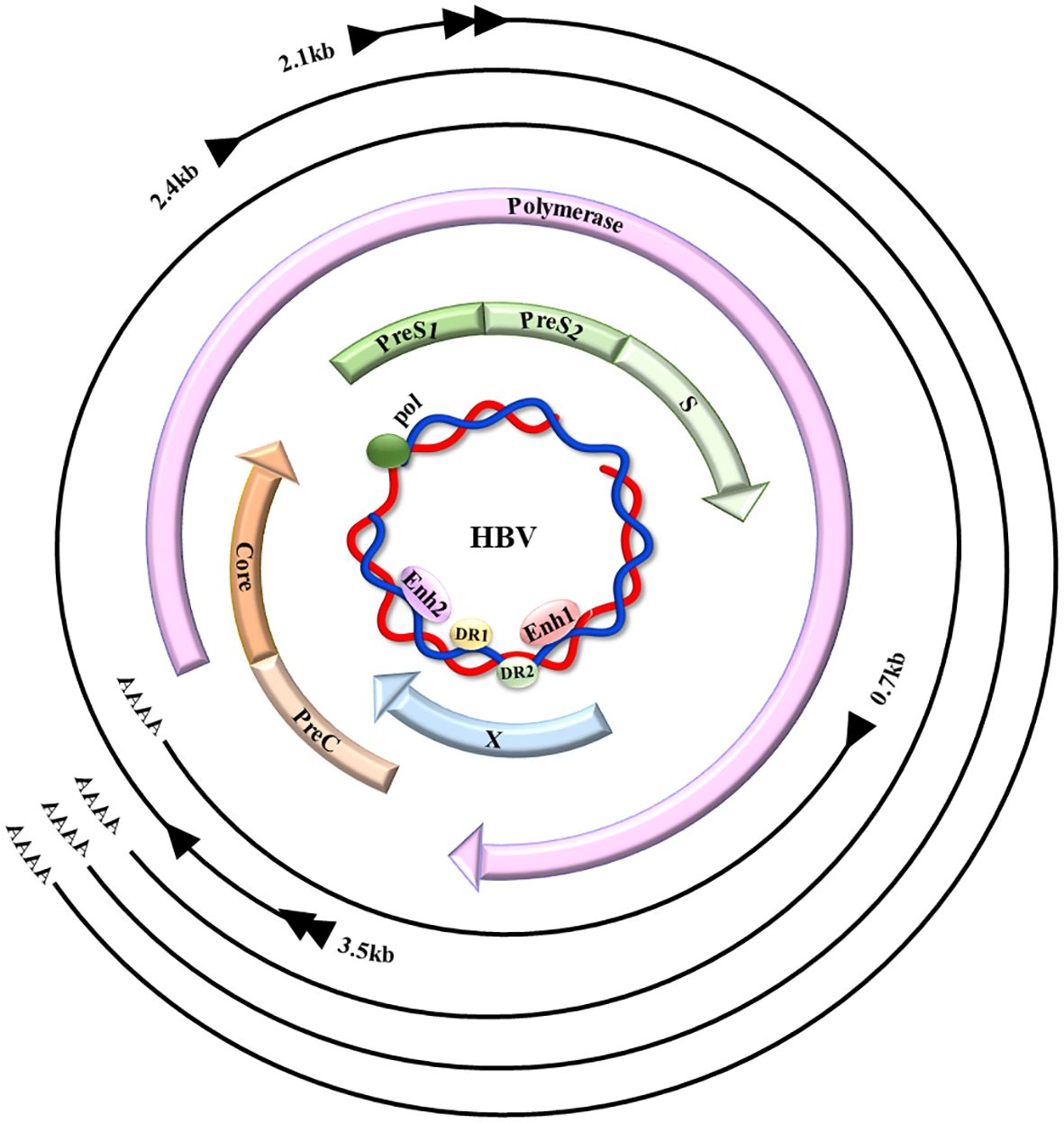
Figure 1 HBV genome map. HBV comprises a small, partially ~ dsDNA genome (the inner blue circle), which contains four promoters and two enhancer regions (Enh1/2), in addition to two direct repeats (DR1/2). The four HBV-coded overlapping ORFs: preS/S, precore/C, polymerase, and X, are indicated by the colored arrows. When the virus is replicating, the rcDNA enters the nucleus, it undergoes conversion into cccDNA through the action of host DNA polymerase and repair enzymes, that act as the viral transcription template, showing the primary HBV transcripts (outer black lines), their 5′ initiation sites (black arrowheads), aside with 3′ poly-A tails (AAAA).(This figure is modified from Figure 1 of the article PMID: 35648301) (11).
The HBx protein, comprising 154 amino acids and a 17 kDa molecular weight, and the variation in amino acid sequence of HBx protein is more pronounced between different HBV genotypes than within the same HBV genotype (12). It is named after its encoding gene due to unknown homologous proteins (13). HBx is recruited to cccDNA microchromosomes and enhances their transcription as HBV replicates; alongside being necessary for HBV cccDNA transcription/viral replication (14, 15), it is assumed to be involved in hepatocarcinogenesis. HBx is primarily found in the cytoplasm of hepatocytes, with some residing in the nucleus. HBx activates transcription by interacting with nuclear transcription factors (TFs) while indirectly bound to DNA. Additionally, HBx functions as an adaptor or kinase activator that influences signal transduction pathways (16, 17).
Transforming growth factor-β (TGF-β) is a pleiotropic cytokine involved in numerous physiological and pathological processes, including tumorigenesis (18–21). It performs contradictory roles in early and advanced liver cancer, inhibiting cell proliferation and inducing apoptosis in early-stage lesions while promoting cancer progression in advanced-stage HCC via fibrosis, invasion, and epithelial-mesenchymal transition (EMT) (22, 23). TGF-β signaling involves Samd-dependent and -independent pathways (21, 24).
As stated, TGF-β and HBx are expressed abnormally in HCC and contribute to its development and progression. HBx expression was positively correlated to TGF-β activity in hepatocytes of cirrhotic/cancerous/chronic hepatitis patients. The results suggested that HBx expression might induce TGF-β1 expression in early-stage HBV infection (25). Additionally, HBx regulates the transition of tumor-suppressive pSmad3C signaling to oncogenic pSmad3L signaling (26), deeming necessary evidence that HBx is intensely contributing to TGF-β carcinogenic effect in HCC.
In the following section, we will examine the specific mechanisms of action of HBx and TGF-β in the progression of HBV- associated HCC and their complex functional cross-talk and interactions.
HBx contributes to HCC development and progression by regulating multiple pathways, epigenetic changes, gene expression, and transcription. (Table 1, Figure 2).
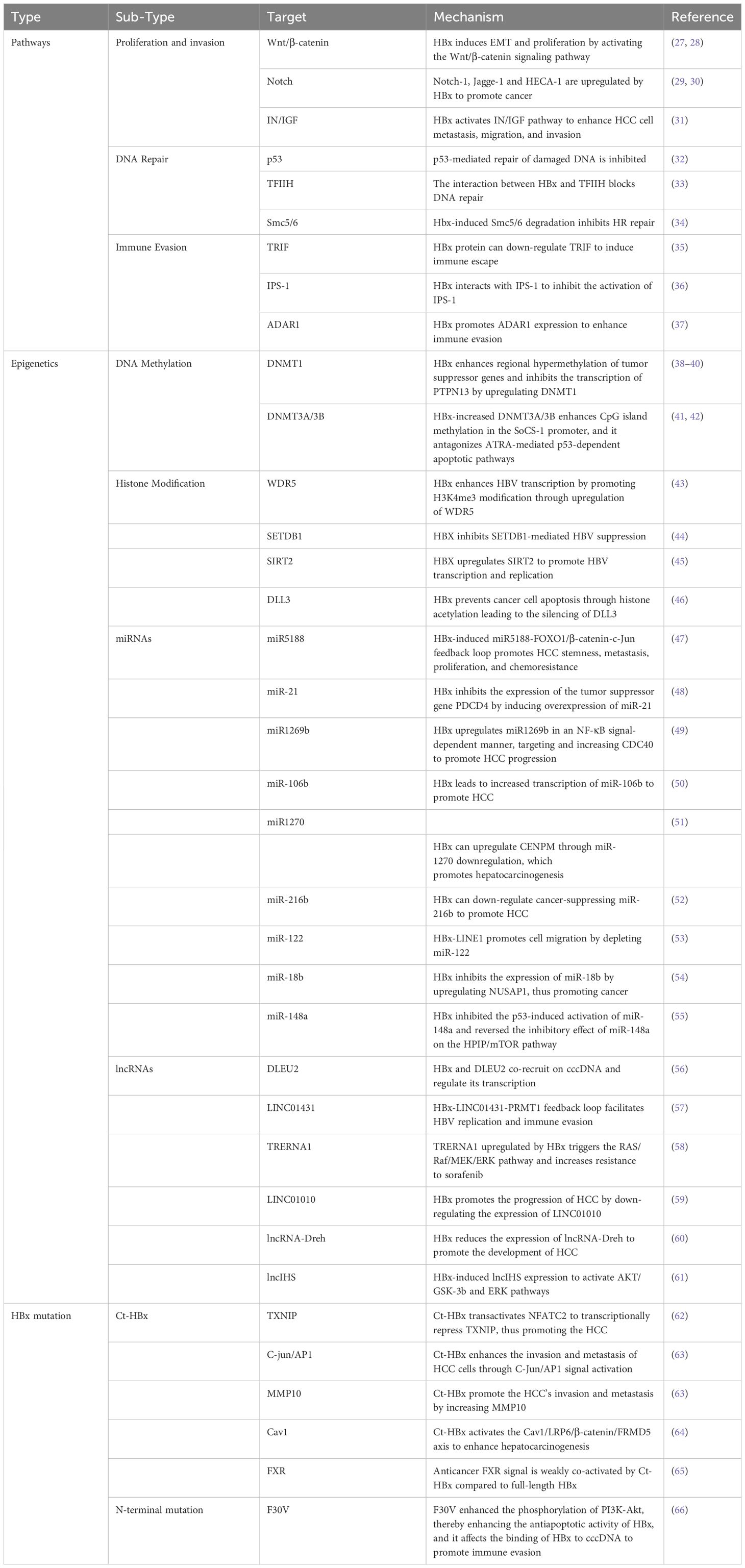
Table 1 A summary of the mechanisms by which HBx leads to HCC.
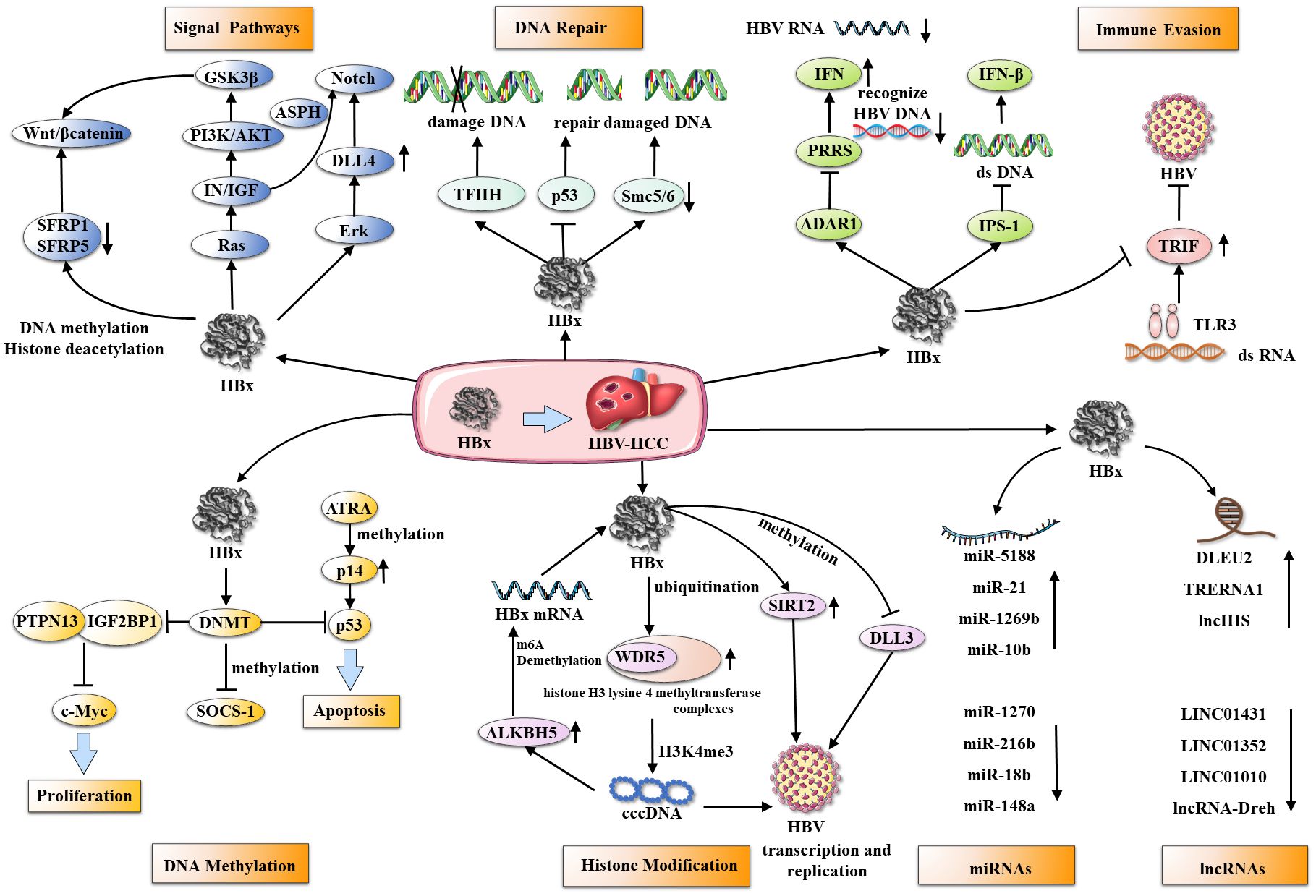
Figure 2 HBx and its multifunctional roles in hepatocarcinogenesis. HBx interacts with several cellular targets through various mechanisms, including affecting multiple signaling pathways, damaged DNA repair, immune evasion, and epigenetic changes (DNA methylation, histone acetylation, ncRNAs) to accelerate HBV transcription, replication, and malignant progression of HCC.
HBx is a key player in the etiology of HCC and manipulates various biochemical pathways within host cells, which are vital for physiological and biochemical functions. HBx redirects these pathways to promote cell proliferation and invasion, impair DNA repair, and evade immune responses, collectively contributing to tumorigenesis.
In particular, the Wnt/β-catenin, Notch signaling pathways, and the insulin-like growth factor (IN/IGF) pathway have been associated with the organization of HBx’s carcinogenic influence. The Wnt/β-catenin pathway is upregulated in over 90% of HCC cases (67), it is activated via HBx ectopic expression combined with Wnt-1, nevertheless, this activation is crucial for stabilizing the effect of HBx on β-catenin (27). In turn, the inhibition of Wnt/β-catenin pathway antagonists, SFRP1 and SFRP5, by HBx increases HCC cell proliferation and epithelial-mesenchymal transition (EMT) (28). Concurrently, HBx also leads to prolonged activation of the oncogenic pathway Notch by upregulating its receptors and ligands, particularly Notch-1 and Jagged-1 (29, 30). This Notch pathway activation is further amplified by HBx’s effect on the Erk (MEK1/2) and PI3K/AKT pathways (68). Furthermore, HBx alters the IN/IGF pathway, which acts in tandem with the Wnt/β-catenin pathway. IN a double-transgene mouse model of HBx and IRS1 that mimic human hepatocellular carcinoma, persistent activation and cross-talk of IN/IGF1, WNT/β-catenin, and Notch enhance HCC cell metastasis and invasiveness, and up-regulation of aspartate β-hydroxylase (ASPH) is central to these signaling cascades (31). The above mechanism exploration is primarily based on mouse models and cells; however, further clinical experiments are required to validate its potential as a therapeutic target for liver cancer patients.
HBx also compromises DNA repair, a critical tumorigenic facet of HBV-induced HCC. The HBx protein can impede the repair of damaged DNA mediated by the anti-oncogene p53 (32), inhibition of transcription factor IIH (TFIIH) (33), and degradation of Smc5/6 complex (34). This disruption of DNA repair processes leads to the accumulation of DNA damage, subsequently instigating the onset of hepatocarcinogenesis.
On the immune evasion front, HBx uses several strategies to avoid the innate immune response, particularly suppression of interferons. It suppresses Toll-like receptor 3 (TLR3) and its adaptor protein, TIR-domain containing adaptor protein inducing interferon-β (TRIF), thereby reducing the anti-HBV immune responsee (35). Furthermore, HBx inhibits interferon β (IFN-β) signaling by interacting with IPS-1, suppressing interferon induction during HBV infection (36). Moreover, HBx facilitates adenosine deaminases acting on RNA 1 (ADAR1)-mediated viral RNA editing, which prevents the recognition of HBV RNA by pattern recognition receptors, thereby inhibiting interferon production (37). Patients with HBV immune tolerance experience a higher incidence of HCC compared to those who receive immune activation therapy (69). Type I interferons are approved as a first-line treatment for chronic HBV. However, it is speculated that the effectiveness of IFN therapy may be limited due to inhibition by HBx, and its significant side effects limit its broader clinical application. Thus, there is a need to develop more effective immunotherapeutic drugs to curb the progression of HBV-associated HCC.
In the context of HBV or HCV infection, HCC development and progression are supported by epigenetic dynamics. This is associated with DNA methylation, histone modifications, microRNAs (miRNAs), and long noncoding RNAs (lncRNAs).
In liver tissue affected by chronic hepatitis or cirrhosis due to HBV or HCV infection, DNA methylation can be detected, which is considered a precancerous state of HCC (70). Significantly elevated HBx expression is associated with methylation abnormalities in HBV-infected HCC patients (38). DNMT1, responsible for maintaining DNA methylation during replication, and DNMT3A/3B, also known as de novo methyltransferases, are involved in methylation modifications (11). HBx upregulates DNMT1/3A1/3A2, resulting in the hypermethylation of tumor suppressor-related genes (38, 39), such as increasing CpG island methylation of the cytokine signaling-1 (SOCS-1) promoter (41) and hypermethylation of p14’s promoter (42). HBx also inhibits Tyrosine-protein phosphatase nonreceptor type 13 (PTPN13) transcription by upregulating DNMT3A and promoting DNA methylation, thereby facilitating the progression of HCC progression (40).
Epigenetic modification of histones contributes to multiple malignant tumor pathogeneses and metastases, including HCC (71–73). HBx inhibits H3 lysine 4-methyltransferase complex core subunit WDR5 ubiquitination, resulting in increased HBV transcription via H3K4me3 modification of cccDNA (43). A positive feedback loop is formed when the HBx-WDR5-H3K4me3 axis increases ALKBH5, a demethylase enzyme that catalyzes m6A demethylation of HBx mRNA (74). However, other reports indicate that H3K4me3 is generally diminished in HBV infections. HBx can alleviate the reduction in H3 acetylation and H3K4me3, as well as the methylation of histone 3 lysine 9 (H3K9me), following HBV infection. This action helps to mitigate the transcriptional silencing of cccDNA (45). HBx inhibits cell apoptosis by inducing DLL3 silencing via histone acetylation in HBV-associated HCC (46).
MicroRNAs (miRNAs) and long noncoding RNAs (lncRNAs) are two important classes of noncoding RNAs that substantially impact cellular processes, including differentiation, proliferation, and survival (75, 76).Notably, irregular expressions of these RNA types are characteristic of liver disorders, including HCC (77, 78). HBx is a key mediator in hepatocarcinogenesis, influencing the levels of both miRNAs and lncRNAs to promote or inhibit HCC development (79, 80). For example, HBx can induce the expression of miR-5188 through Wnt signaling, thereby establishing a feedback loop that promotes HCC stemness, metastasis, and chemoresistance (47). Additionally, it can modulate the expression of miR-21, miR-1269b, and miR-106b, thereby affecting tumor suppression and the progression of HCC (48–50, 81)(p12). In contrast, downregulated miRNAs affected by HBx, such as miR-1270, miR-216b, miR-122, miR-18b, and miR-148a, are involved in HCC carcinogenesis via numerous pathways (51–55). HBx influences transcription, translation, and epigenetic regulation of lncRNAs, with implications for HBV replication, immune evasion, drug resistance, and the activation of numerous signaling pathways (80). Noteworthy examples include DLEU2, LINC01431, TRERNA1, LINC01010, lncRNA-Dreh, and lncIHS (56, 57) (p1), (58–61). Consequently, these interactions between HBx and noncoding RNAs emphasize their significance in the molecular mechanisms underlying the development and progression of HBV-associated HCC.
HBV DNA integration into the host genome induces genomic instability and direct insertion mutations in cancer-related genes in early-stage clonal HCC expansion (82). Variation in the HBx sequence, specifically at the 3’-terminus, substantially affects the HCC development. Chimeric transcripts of HBx and human genes, often including 3’-terminal deletions, are frequently expressed and can encode alternative HBx versions implicated in transcriptional regulation (83, 84).
The carboxyl terminus of HBx plays multiple roles in protein-protein interactions, transcriptional transactivation, DNA repair, cellular signaling, and HCC pathogenesis (85). COOH-terminal mutations are associated with HCC and may lead to reactive oxygen species (ROS) production, which damages mitochondrial DNA (86, 87). The clustering pattern of HBx 3’ end generates a truncated X protein (Ct-HBx) (62) that promotes HCC progression by mediating glucose metabolism reprogramming, increasing matrix metalloproteinase 10 (MMP10) transcription (62, 63), and enhancing Wnt/β-Catenin signaling (64). HBX-C30 (30 aa deletion from HBx C terminus) co-activates anticancer FXR signaling less effectively than full-length HBx (65). Additionally, mutations in the N-terminal domain, such as F30V, increase HBx’s antiapoptotic activity and diminish HBV’s replication efficacy. These HBx mutations facilitate the development of HCC and immune evasion (66).
Currently, there is a lack of drugs and therapeutic methods targeting HBx, which awaits further development by researchers. During HBV infection, the host SMC complex, SMC5/6, suppresses viral transcription. HBx promotes ubiquitination and degradation of SMC5/6, enhancing cccDNA transcription and increasing viral replication. This destabilization of SMC5/6 aids HBV in evading host immune surveillance and facilitates viral persistence and propagation (15, 88, 89). Nitazoxanide, an FDA-approved thiazolide antimicrobial for protozoal enteritis, and Pevonedistat, an NAE inhibitor approved for myelodysplastic syndromes, have demonstrated in vitro efficacy in inhibiting the HBx/SMC5/6 axis. This inhibition restores SMC5/6 protein levels, effectively suppressing HBV transcription and translation (90–92). Dicoumarol, the precursor to warfarin and a competitive NQO1 inhibitor, reduces HBx level and cccDNA transcription in both HBV-infected hepatocytes and humanized mouse models (93).
The TGF-β branch, classified as the TGF-β family, is activated by three ligands: TGF-β1-3, with TGF-β1 being the most abundant and typical isomer as it is secreted by almost all cells (94). The TGF-β ligand synthesis is in a longer precursor protein that is cleaved by furin protease; additionally, TGF-β disulfide-bonded dimers and the latency-associated peptide (LAP) are joined by non-covalent bonds to form a small latent complex, which is cross-linked with the latent TGF-β binding protein (LTBP) forming a large latent complex. This inactive complex is then secreted and associated with the extracellular matrix (ECM) (95). Activation occurs when particular signals initiate the release of active TGF-β from the complex, involving elements such as extreme pH, proteases, and integrins (96). Upon TGF-β ligand binding, TβRII dimerizes and recruits TβRI to produce a heterotetrameric TβRI-TβRII complex (97, 98).
TβRI phosphorylates R-Smads (Smad2/3) at their extreme C-terminal Ser-X-Ser motifs in the canonical Smad signaling, resulting in oligomerization with Co-Smad (Smad4) and nuclear translocation (99). The activated Smad4-R-SMAD complex regulates the transcription of target genes through interactions with DNA-binding transcription factors (20). The Smad linker can also be phosphorylated to activate numerous signaling pathways involved in pathological or pathophysiological gene expressions (100). Inhibitory Smad6/7 (I-Smads) modulate canonical Smad signaling in a feedback manner via various mechanisms (101, 102). Besides canonical Smad signaling, TGF-β functions by activating non-canonical Smad pathways, including the mitogen-activated protein kinase (MAPK), extracellular signal-regulated kinases1/2 (Erk1/2), Rho-like, phosphatidylinositol-3-kinase (PI3K)/AKT, c-Jun amino-terminal kinase (JNK), p38/MAPK (103), and the Src tyrosine kinase signaling pathways (104, 105) (Figure 3).
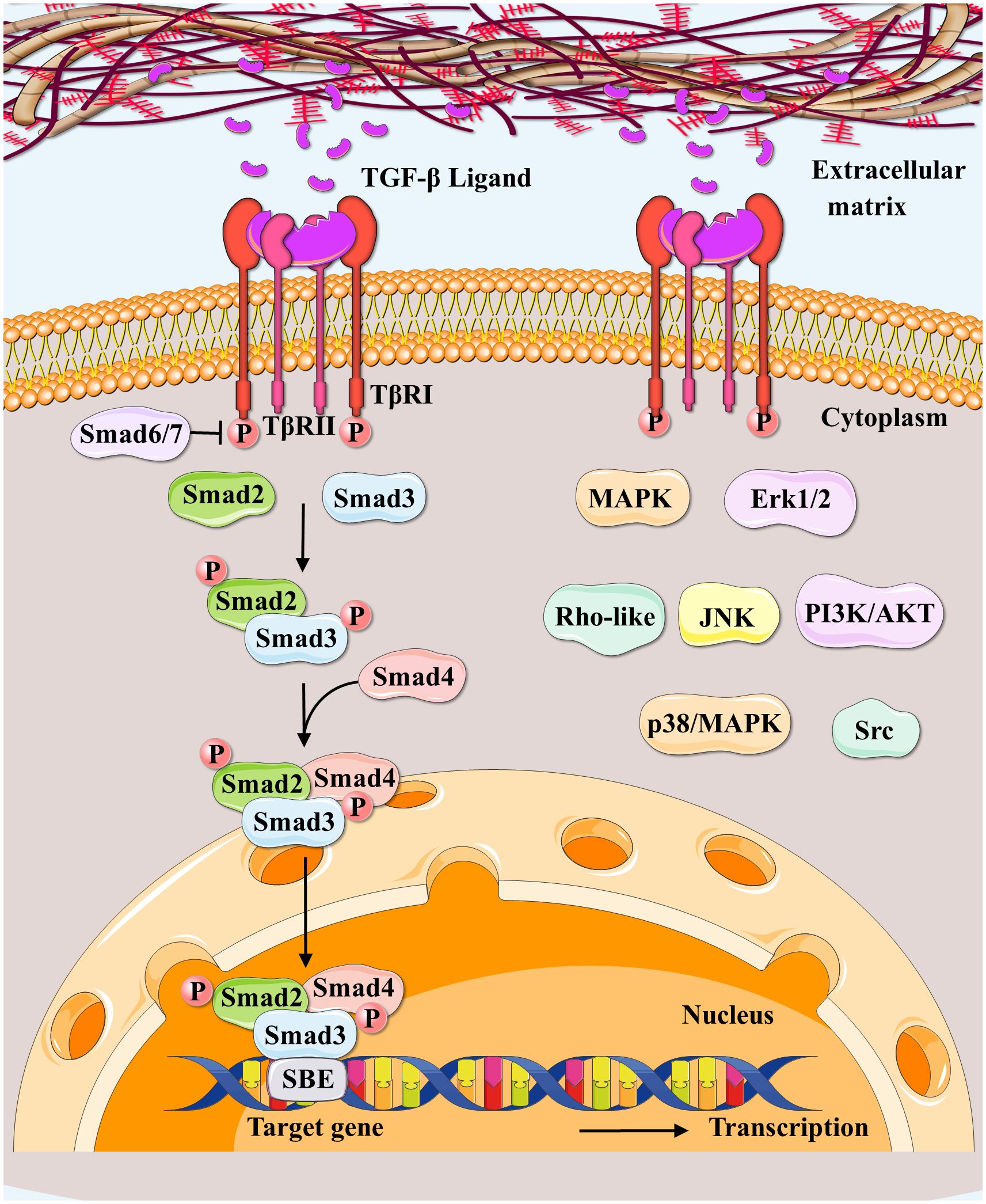
Figure 3 Canonical and non-canonical TGF-β signaling pathways. Regarding canonical TGF-β signaling pathways (SMAD‐dependent signaling pathways), the TGF-β ligand secreted by extracellular matrix binds to TGFβRII to initiate this pathway; and TGFβRII upon activation, forms a complex with TGFβRI and phosphorylates TGFβRI. Then Smad2/3/4 forms transcription complexes, entering the nucleus besides binding to DNA to regulate the target gene expressions. Smad6/7 are canonical TGF-β pathway inhibitors. Non-canonical Smad pathways (SMAD‐independent signaling pathways) include MAPK, Erk1/2, Rho-like, PI3K/AKT, JNK, p38/MAPK, and Src tyrosine kinase pathways.
TGF-β signaling is essential at every stage of liver disease progression, from initial inflammation and damage to fibrosis, cirrhosis, and ultimately liver cancer. Early on, it inhibits tumor growth by inducing senescence and apoptosis. It promotes tumor growth, EMT, and metastasis in advanced stages. Nevertheless, Smad-dependent and independent pathways are involved in the complex signaling (106). (Figure 4).
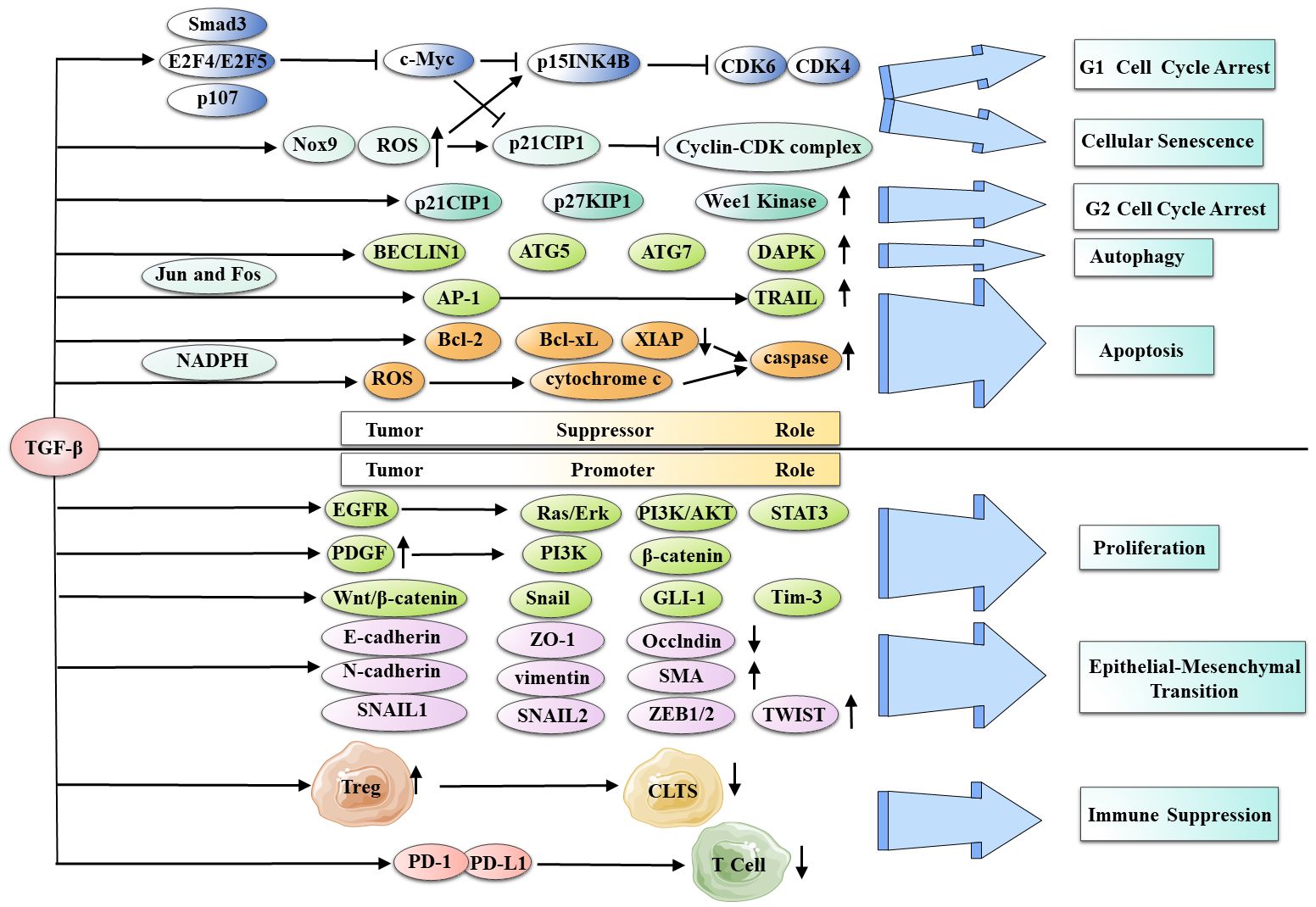
Figure 4 TGF-β dichotomous role in HCC development and progression. TGF-β has various tumor-suppressing functions, including G1 and G2 cell cycle arrest, cellular senescence, autophagy, and apoptosis. Conversely, it has the ability to serve as a tumor promoter, inducing cancer cell proliferation, EMT, and immune suppression in HCC.
TGF-β induces HCC cell cycle arrest by suppressing transcriptional factor expression, including the pro-growth TF c-Myc, which is mediated through nuclear translocation of a complex consisting of Smad3, E2F4, or E2F5, and RB-related factor p107 (107, 108). It also regulates cyclin-dependent kinases (CDKs) and their inhibitors, leading to G1 arrest (109). CDK2 binds to cyclin E to drive the cell cycle, while CDK4 or CDK6 binds to cyclin D (110). Furthermore, TGF-β reduces the inhibitory effects of c-Myc on CDK inhibitors p21CIP1 and p15INK4B, leading to cell cycle arrest (111–113)(p1), (114). The accumulation of NADPH oxidase-4 (Nox4) and ROS in well-differentiated HCC cell lines further enhances the expression of TGF-β-induced p21CIP1 and p15INK4B (115). In addition to G1 phase arrest, TGF-β causes HCC-cell G2 phase arrest by inducing CDK inhibitors p21CIP1 and p27KIP1, along with Wee1 kinase (116)(p1).
Cellular senescence is a persistent cell cycle arrest state inhibiting tumor progression (117–119). There is a significant relationship between tumor inhibition and cell senescence in HCC. Telomerase and telomeres are mainly involved in malignant cell senescence and hepatocyte aging (120), with telomerase reactivation promoting uncontrolled proliferation and malignant transformation in HCC (121). In malignant tumors, TGF-β/Smad signaling inhibits human telomerase reverse transcriptase (hTERT), demonstrating a regulatory role for TGF-β in HCC cell senescence (122–124).
Autophagy is responsible for the degradation of proteins and cells (125, 126). Through Smad and non-Smad pathways, TGF-β induces autophagy in HCC cells by upregulating the expression of autophagy-related genes, including BECLIN1, ATG5, ATG7, and DAPK. Autophagy is closely related to TGF-β-mediated growth inhibition of HCC cells. Autophagy gene knockdown reduces TGF-β-mediated growth suppression and decreases pro-apoptotic gene expression. HCC cells are more resistant to TGF-β-induced autophagy than breast cancer cells, indicating its significance in growth suppression (127, 128). However, recent studies have found that tumor cells in HCC rely on autophagy for survival. This process can promote tumor development by inducing autophagic cell death in liver (129, 130). Therefore, the role of TGF-β-induced autophagy in liver cancer requires further research to clarify.
In liver cells, TGF-β induces apoptosis and inhibits cell proliferation (131). It initiates apoptosis through the death receptor pathway by mediating the activation of the TNF-related apoptosis-inducing ligand (TRAIL) promoter’s AP-1 site via Jun, Fos, and Smad proteins (132)(p1), (133). TGF-β can induce apoptosis in liver cancer cells via the mitochondrial and death receptor pathways. TGF-β can also induce apoptosis via the mitochondrial pathway by reducing the expression of antiapoptotic B-cell lymphoma 2 (Bcl-2) family proteins and activating caspases (134). ROS generation via NADPH oxidase is essential for TGF-β-induced HCC cell apoptosis (135, 136). The apoptotic response can be suppressed by the epidermal growth factor receptor (EGFR) pathway (137, 138).
Most malignant tumors exhibit an increase in cell proliferation. TGF- promotes HCC proliferation via several pathways. Through the EGFR, TGF-β activates Ras/Erk, PI3K/AKT, and STAT3 signaling, resulting in hepatocyte proliferation. In addition, it upregulates the expression of PDGF to activate the PI3K and β-catenin pathways. TGF-β also activates other pathways, such as Wnt/β-catenin, Snail, and GLI-1, contributing to cancer cell proliferation (139–143). Furthermore, TGF-β/SMAD2 signaling induces c-KIT receptor ligand (stem cell factor) expression, activating c-KIT/JAK1/STAT3 signaling and establishing a positive feedback loop for HCC proliferation (139). The p38 and PI3K/AKT pathways mediate the PRL-3-induced TGF-β1 by activating FAK and developing another positive feedback loop (144).Thus, TGF-β interacts with multiple carcinogenic pathways in tumors, and blocking TGF-β activation could serve as a vital approach in the treatment of liver cancer.
HCC development involves genetic, epigenetic, and transcriptomic mechanisms. Approximately 40% of HCC cases contain mutations in TGF-β pathway genes. Overexpression of TGF-β pathway genes is associated with inflammation and fibrosis, whereas their down-regulation inhibits tumor growth (145). Several TGF-β target genes implicated in HCC are overexpressed, including VEGFA, COL4A1, SNAI2, DAPK2/3, CDKN1A, and CDKN1. Genomic instability in HCC is caused by mutations in the TGF-β pathway and its non-canonical targets, including the JNK/MAPK/IKK, ERK/MAPK, RHO-ROCK, and PI3K/AKT pathways (146).
Epigenetic modifications increase the carcinogenic influence of TGF-β on HCC. In early HCC, demethylation of the Smad4’s promoter suppresses tumor growth, whereas hypomethylation of Smad7 and SNAI1 in the promoter region facilitates EMT recurrence and metastasis, it has been reported that the use of decitabine may drive liver cancer progression towards a pro-carcinogenic direction through this pathway (147). Methylation of the TTP promoter eliminates the post-transcriptional regulatory function of c-Myc, shifting TGF-β signaling from proliferation inhibition to promotion (148, 149). In HCC, high methylation levels frequently lead to the inactivation of the tumor suppressors RUNX3 and Smad Interacting Protein-1 (SIP1), which interact with Smads (150, 151). In conjunction with HDACs and G9 methyltransferase, the TGF-β-induced overexpression of SNAIL2 suppresses E-cadherin and enhances the invasiveness and metastasis of HCC cells (152).
EMT triggers epithelial cells to acquire mesenchymal properties, leading to migration, invasion, stemness, and resistance to apoptosis and immune responses (153, 154). TGF-β plays a fundamental role in triggering EMT in HCC (154, 155)(p1). It downregulates epithelial markers (E-cadherin, ZO-1, and Occludin) and upregulates mesenchymal markers (N-cadherin, vimentin, and SMA) via Smad and non-Smad pathways (Rhogtase, MAPK, and PI3K/AKT/mTOR) (156, 157). TGF-β induces EMT-TFs, including SNAIL1/2, ZEB1/2, and TWIST. Smads interact with Notch, Hedgehog, Wnt, and Hippo signaling pathways to reprogram EMT-related genes. Additionally, miRNAs (158), such as the SNAIL-miR-34 and ZEB1-miR-200 feedback loops, regulate EMT during TGF-β stimulation (159). Long noncoding RNA also activated by TGF-β (lncRNA ATB) promotes ZEB1/2 overexpression via competitive binding with the miR-200 family, thereby inducing EMT and invasion (160).
TGF-β, by modulating immune cells involved in immune homeostasis and tolerance, acts as a critical inhibitor of both adaptive and innate immunity. This creates an immunosuppressive tumor microenvironment that facilitates tumor progression (161). Induction and differentiation of liver Treg cells by TGF-β contribute to immunosuppression (162) via numerous cell types, such as LSECs, HSCs, CAFs, TAMs, and MDSCs. Treg cells suppress immune responses, enhance cell proliferation, and deplete cytotoxic T lymphocytes (CTLs).TGF-β upregulates PD-1 and PD-L1, inhibiting TCR signaling and T cell proliferation and leading to T cell depletion in HCC (146). In addition to inhibition of T cells, TGF-β increases alternative macrophage activation by enhancing Tim-3 transcription in tumor-associated macrophages (TAMs), leading to the growth of HCC (163).
The TGF-β pathway plays a crucial role in the progression of HBV-associated HCC. Many drugs targeting the TGF-β pathway have shown promising results in treating HCC (164). However, the clinical applicability and efficacy of these treatments require further validation through extensive clinical trials. Studies have shown that various chemotherapeutic agents, including Fluorofenidone (AKF-PD) (165), Sanguinarine (San) (166), Aspirin (167), Praziquantel (PZQ) (168), and Ursodeoxycholic acid (UDCA) (169), along with small molecule inhibitors such as Galunisertib (LY2157299) (170–172), LY2109761 (173), SKLB023 (174), and LY3200882 (175), are effective in treating HBV-HCC. The therapeutic vaccine Belagenpumatucel-L (Lucanix) (176) also exhibits efficacy in this setting. Studies show significant activation of the TGF-β pathway in immunotherapy-resistant tumors, with TGF-β often implicated in establishing suppressive tumor microenvironments (177, 178). The TGF-β and PD-1 pathways operate through independent, yet complementary, immunosuppressive mechanisms, enhancing cancer immune evasion (179, 180). Consequently, combining TGF-β inhibitors with PD-1 monoclonal antibodies presents a promising treatment approach for HBV- HCC.
As described previously, HBx and TGF-β play distinct functions in HBV-HCC progression. In fact, HBx was shown to induce TGF-β expression early in HBV infection (25). Meanwhile, TGF-β can increase signaling pathway proteins in HBx pathogenesis (181), suggesting that TGF-β and HBx co-regulate specific signaling pathways that promote HCC. Additionally, regulating multiple pathways and epigenetic and genetic events by HBx mediates TGF-β’s participation in distinct ways in HBV-HCC progress pathogenetic mechanism.
As mentioned, the TGF-β pathway is involved in tumor suppression during early-stage tumorigenesis and tumor promotion in advanced cancers (94, 182–184). This dichotomous effect is determined by the phosphorylation status of the Smad3 protein, particularly its c-terminus or linker region (185). The linker region of Smad2/3 contains numerous conserved motifs subject to regulatory factors and post-translational modifications, such as phosphorylation (186, 187). The linker domain is phosphorylated at specific serine/threonine residues by cytoplasmic MAPKs and nuclear CDKs (20, 35–38). The phosphorylation of Smad2/3 in the linker region generates three types of phospho-isoforms: C-terminally phosphorylated Smad2/3 (pSmad2C/3C), linker-phosphorylated Smad2/3 (pSmad2L/3L), and dually phosphorylated Smad2/3 (pSmad2L/C and 3L/C) (188).
In normal epithelial homeostasis, TGF-β mediates pSmad3C signaling, inhibiting cell proliferation by interfering with cell cycle progression (189–191). This is accomplished via activating CDK inhibitors, such as p15INK4B and p21CIP1, and inhibiting c-Myc gene expression and cell cycle-related molecules (192–194). The pSmad3C pathway protects against cancer development, leading to transient Ras activation followed by growth inhibition and apoptosis. Furthermore, pSmad3C can regulate apoptosis-related protein expressions, including Bcl2 (195)(p2).
Cytoplasmic Ras-related kinase activation during carcinogenesis, including MAPKs, transforms Smad3 signaling from an antitumor pSmad3C state to the oncogenic pSmad3L and pSmad2L/C pathways. JNK, a serine/threonine kinase activated by Ras, plays a crucial role in this conversion by defeating TβRI/pSmad3C-mediated growth arrest (196, 197). Smad3 is phosphorylated at Ser-213 upon activation of JNK (198), a site where RTK pro-inflammatory cytokines, growth factors, and, to a lesser extent, TGF-β can increase phosphorylation levels. It has been demonstrated that c-Myc overexpression can inhibit the Smad3-dependent transcription of p15INK4B and p21WAF1 proteins, thereby opposing cell cycle arrest (199). The JNK/c-Myc mitotic pathway inhibits the TRI/pSmad3C/P21WAF1-mediated growth arrest (200). Ser-213 phosphorylation of Smad3L via TβRI perturbs COOH-tail phosphorylation (26, 198, 201), promoting nuclear translocation and accelerating cell proliferation signals mediated by pSmadL (198). JNK-activated pSmad3L-mediated cell proliferation signal and TβRI-activated pSmad3C-mediated cell cycle arrest signal are mutually antagonistic. Mutations in essential pathway components can cause persistent Smad3 linker phosphorylation, so highly phosphorylated Smad3L likely reduces pSmad3C’s sensitivity to growth inhibition in tumor cells (26, 202–204). The overexpression of receptors by cancer cells modifies Smad3 phosphorylation (205). The Ras/JNK pathway controls both pSmad3C and pSmad3L. Strong Smad2L/C and Smad3L/C phosphorylation is observed in colorectal cancer EMT-related tumors (206). CDK4 converts TGF-β signal-mediated pSmad2/3C to malignant pSmad2L/C and 3L/C pathways (207). The interaction between pSmad2L/C and pSmad3L induces fibrogenic signals and liver fibrosis via PAI-1 (208). Increased PAI-1 transcription and ECM synthesis positively modulate liver fibrosis in hepatocytes (209).
It has been demonstrated that HBx overexpression induces the development of hepatic tumors by stimulating DNA synthesis (210). Studies have revealed that HBx shifts TGF-β signaling from the TβRI-dependent pSmad3C tumor-suppressive pathway to the JNK-dependent pSmad3L oncogenic pathway during carcinogenesis, as observed in biopsy samples from chronically HBV-infected patients and HBx transgenic mice with liver lesions (26). The proto-oncogene c-Myc, a target of TGF-β/SMAD signaling (211, 212), is involved in HCC malignant progression (213). pSmad3L/Smad4 triggers c-Myc transcription, whereas pSmad3C/Smad4 inhibits it. The two complexes antagonize each other and govern c-Myc expression. HBx’s presence may cause the signal to upregulate c-Myc and promote cancer cell growth (214) (Figure 5).
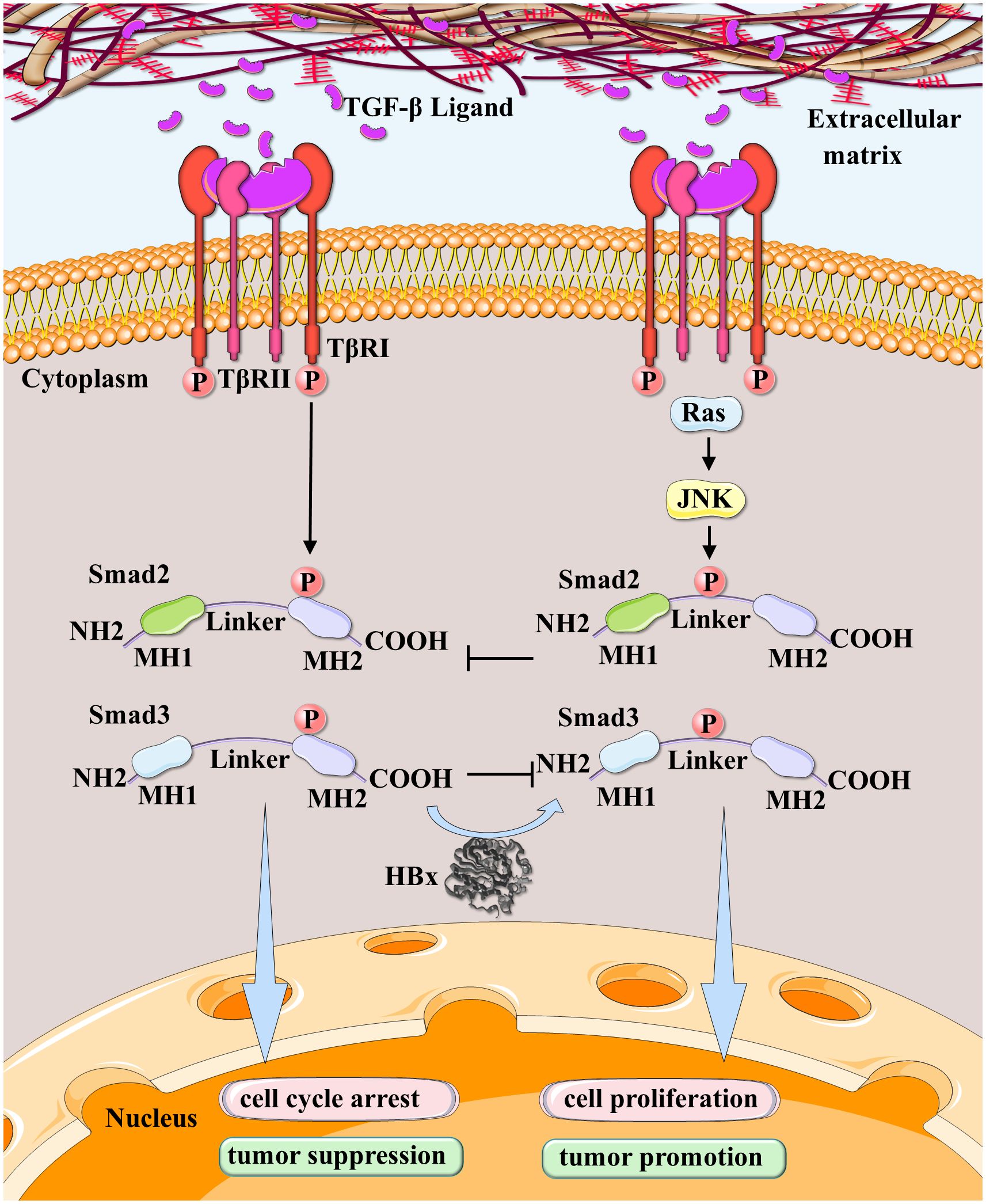
Figure 5 HBx participated in the conversion of dichotomous effects on HCC in the TGF-β pathway. JNK-activated pSmad2/3L-mediated cell proliferation signal and TβRI-activated pSmad2/3C-mediated cell cycle arrest signal are mutually antagonistic. HBx leads TGF-β signaling in hepatocytes to shift from the TβRI-dependent pSmad3C tumor-suppressive pathway to the JNK-dependent pSmad3L oncogenic pathway during carcinogenic stages.
In conclusion, the interaction between HBx and TGF-β plays an essential role in hepatocarcinogenesis at various stages of HBV infection. TGF- β signaling has a dual function, promoting tumor suppression in the early stages of tumorigenesis while promoting tumor growth in advanced malignancies. Smad3’s function is determined by its phosphorylation status, with C-terminally phosphorylated Smad3 (pSmad3C) functioning as a tumor suppressor and linker-phosphorylated Smad3 (pSmad3L) contributing to oncogenesis. HBx modifies the TGF-β signaling pathway, redirecting it to the oncogenic pSmad3L pathway. This perturbation promotes the progression of the cell cycle, inhibits apoptosis, and accelerates the development of hepatocellular carcinoma.
In prior studies, researchers revealed a correlation between HBx and TGF-β expression in HBV-infected cells. TGF-β1 level correlates positively with HBx protein expression in early-stage HBV infection, suggesting that HBx may directly or indirectly promote TGF-β1 expression. HBx forms a complex with Egr-1 protein and transactivates the TGF-β1 promoter via the Egr-1 binding site (25). HBx overexpression during HBV infection correlates with the release of secreted factors, particularly TGF-β, from HBx-transfected HCC cells or adjacent endothelial cells through increased CD133 expression, which induces invasion of these cells by EMT (210).
TGF-β1 inhibited cell proliferation and invasion in a study involving trophoblast cells (HTR-8/SVneo). Nevertheless, HBx activated the Smad pathway in HBx-transfected cells, resulting in the downregulation of E-cadherin and the upregulation of vimentin and N-cadherin, which reduced the apoptotic capacity and increased the invasive capacity of HTR-8/SVneo cells. The mechanism by which HBx shifts TGF-β signaling in HTR-8/SVneo cells needs additional investigation (211). Additionally, HBx inhibits the expression of the E-cadherin gene (CDH1) by activating TGF-β, which may be an additional mechanism for its downregulation of E-cadherin and promotion of tumor metastasis (212). The HTR-8/SVneo cell line is derived from cells that were grown from early human placental chorionic villi explants and transfected with a gene encoding the Simian Virus 40 large T antigen. Since it is not sourced from a liver cancer cell line, further foundational experiments are necessary to determine whether it can represent the general mechanisms within HBV-infected and liver cancer patients. This includes using human-derived liver cancer cell lines and replicating the studies in mouse models that simulate liver cancer to validate the results.
Epigenetic and genetic events affect the interaction between HBx and TGF-β. HBx can increase TGF-β expression via autophagy induction, and increased TGF-β upregulates lncRNA-ATB, thereby enhancing liver cancer cell migration and invasion (215). Both HBx and TGF-β1 stimulation induces significant overexpression of miR-199a-3p in hepatic progenitor cells (HPCs), thereby promoting HPCs oncogenic transformation via a JNK/c-Jun/miR-199a-3p-dependent pathway (216). C-terminal truncated mutants (ctHBx) commonly found in HCC tissue samples (217) decrease bone activin membrane-bound inhibitor (BAMBI) more than HBx alone. ctHBx significantly inhibits BAMBI promoter activity in the absence of the Wnt/β-catenin pathway, thereby reducing the inhibition of TGF-β1 and β-catenin and promoting malignancy (218).
In an HBx transgenic mouse model undergoing partial hepatectomy, TGF-β, Smad2, and phosphorylated Smad3/4 (ser423/425) were significantly overexpressed in the HBx transgenic mice liver compared to non-transgenic mice, indicating the impact of HBx on the TGF-β/Smad pathway, which promotes the progression of HCC (213). Furthermore, HBx disrupts the negative feedback loop between TGF-β and protein phosphatase magnesium-dependent 1A (PPM1A) by enhancing PPM1A ubiquitination and degradation, resulting in TGF-β pathway overactivation, HCC migration, and invasion (219). HBx also stabilizes the binding of the Smad complex to the transcriptional machinery and facilitates the nuclear transposition of Smads, thereby amplifying TGF-β signaling (220).
Contrary to previous findings, some studies suggest a negative correlation between HBx and TGF-β activation. For example, HBx inhibits TGF-β-induced apoptosis by linking Src to PI3K, thereby activating the PI3K/Akt signaling pathway (221, 222). And another study showed that cells transfected with HBx exhibited reduced expression of the TGF-β type II receptor, resulting in a weaker TGF-β1 response and reduced growth inhibition in response to TGF-β1 (214). Furthermore, research also shows that HBx plays different roles at hepatocyte cell line. In normal liver cells, HBx induces cell cycle arrest by elevating TGF-β and p27 levels, leading to G1 or G2 phase blocks that facilitate HBV replication. In HBV-infected HCC cells, however, HBx may accelerate carcinogenesis by promoting cell cycle progression through the downregulation of TGF-β and the p53/27/21 pathway (223). Despite these findings, the overall trend indicating tumor progression via the HBx/TGF-β axis aligns with earlier results. This may be attributed to the paradoxical roles of TGF-β at different stages of tumor progression or variations in experimental conditions.
It is evident that the TGF-β signaling pathway is primarily influenced and regulated by HBx, particularly within HBV-infected cells. It is worth noting that HBx has been shown to lead to malignant transformation of HCC by influencing dysregulation of TGFB and thereby activating multiple cancer-promoting mechanisms. including EMT, anti-apoptosis, proliferation, inflammatory responses, metastasis, invasion, and fibrosis. Therefore, researchers need to pay more attention to the interaction mechanism of these HBx-TGF-β axis, and develop anti-tumor drugs that can target the common pathway of this axis, bringing new hope to the drug treatment of hepatocellular carcinoma. (Figure 6).
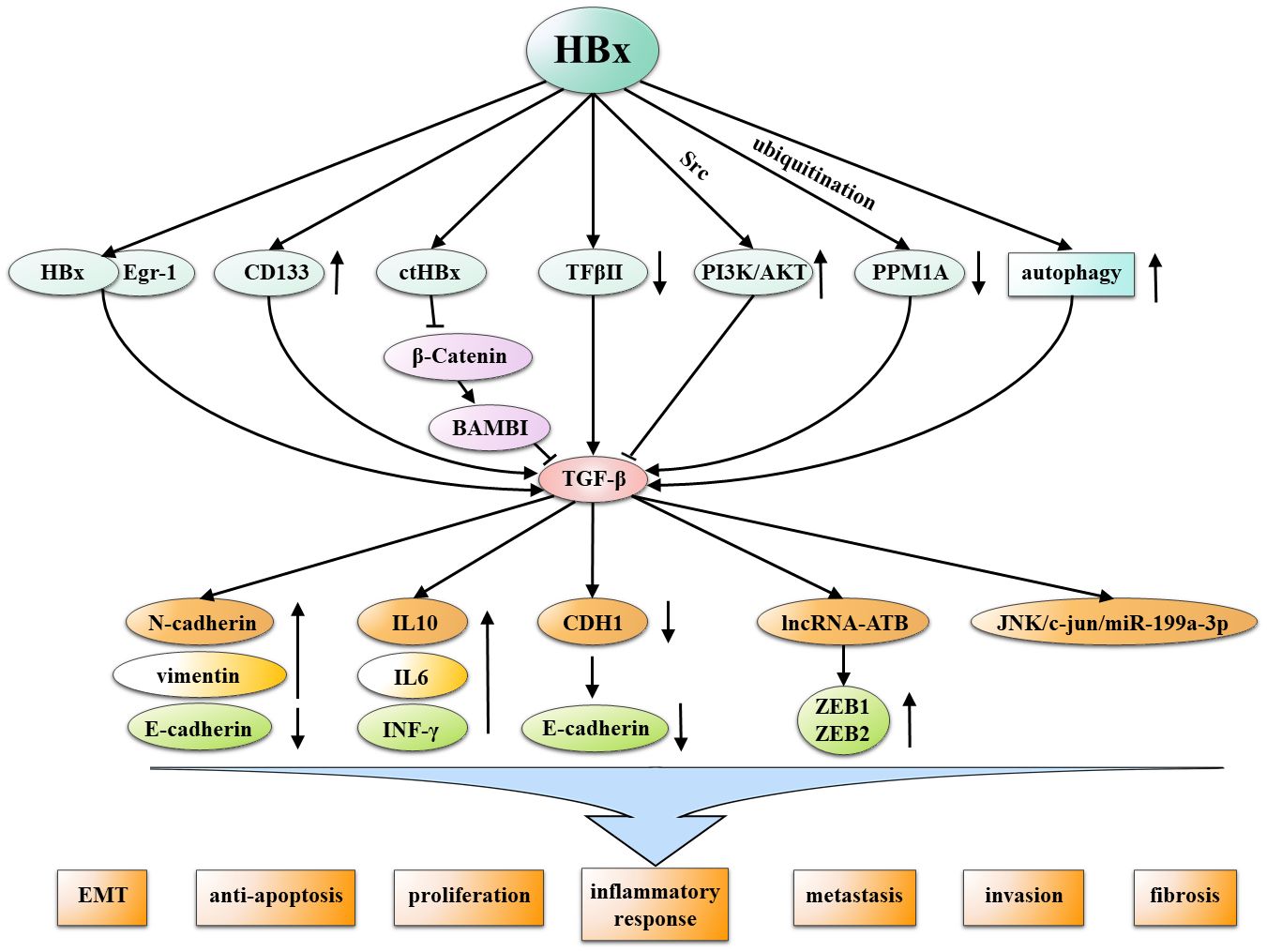
Figure 6 Multiple mechanisms by which HBx interacts with TGF-β in the non-SAMD pathways to promote HCC. HBx and TGF-β mutually interact through various mechanisms, inducing malignant characteristics in development of HCC, including EMT, anti-apoptosis, proliferation, inflammatory responses, metastasis, invasion, and fibrosis.
The incidence and mortality rates of liver cancer have garnered widespread attention within the academic community. With the advancement of liver cancer research, there has been a deeper understanding and significant progress in elucidating carcinogenic molecular mechanisms. Various mechanisms, such as DNA damage, immune evasion, epigenetic alterations, and genomic mutations, have been emphasized in studies and are considered crucial in promoting HCC through HBx. Notably, as a key signaling pathway, TGF-β plays dual roles in HCC, transitioning from an early anticancer effect to a late pro-cancer effect. HCC can acquire invasive tumor characteristics such as EMT and aberrant proliferation through this pathway. In numerous recent studies, it has been demonstrated that HBx and TGF-β signals interfere and interact with one another in HCC and collectively regulate HCC progression. With future technological advances, it is believed that more in-depth studies will be necessary to reveal the HBx oncogenic mechanism, a star protein, and TGF-β, a potential HCC therapeutic target, and to design effective clinical management strategies for HBV-correlated HCC patients. In the near future, these efforts will provide HCC patients with more effective and sensible options for receiving targeted therapies.
WY: Writing – review & editing, Writing – original draft. DR: Writing – review & editing, Writing – original draft. FF: Writing – review & editing, Writing – original draft. HL: Writing – review & editing, Writing – original draft. ZZ: Writing – review & editing, Writing – original draft. HD: Writing – review & editing, Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This research was supported by the National Natural Science Foundation of China No. 81402410 (HD), No. 81802767 (ZZ) and the Natural Science Foundation of Hubei Province 2022CFB280 (HD).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA A Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
2. Rumgay H, Arnold M, Ferlay J, Lesi O, Cabasag CJ, Vignat J, et al. Global burden of primary liver cancer in 2020 and predictions to 2040. J Hepatol. (2022) 77(6):S0168827822030227. doi: 10.1016/j.jhep.2022.08.021
3. McGlynn KA, Petrick JL, El-Serag HB. Epidemiology of hepatocellular carcinoma. Hepatology (2021) 15:4-13. doi: 10.1002/hep.31288
4. Chuang SC, Vecchia CL, Boffetta P. Liver cancer: Descriptive epidemiology and risk factors other than HBV and HCV infection. Cancer Lett. (2009) 6:9–14. doi: 10.1016/j.canlet.2008.10.040
5. Maucort-Boulch D, de Martel C, Franceschi S, Plummer M. Fraction and incidence of liver cancer attributable to hepatitis B and C viruses worldwide. Int J Cancer. (2018) 21:2471–7. doi: 10.1002/ijc.31280
6. Petrick JL, Florio AA, Znaor A, Ruggieri D, Laversanne M, Alvarez CS, et al. International trends in hepatocellular carcinoma incidence, 1978–2012. Int J Cancer. (2020) 147:317–30. doi: 10.1002/ijc.32723
7. Guidotti LG, Chisari FV. IMMUNOBIOLOGY AND PATHOGENESIS OF VIRAL HEPATITIS. Annu Rev Pathol Mech Dis. (2006) 1:23–61. doi: 10.1146/annurev.pathol.1.110304.100230
8. Xia Y, Guo H. Hepatitis B virus cccDNA: formation, regulation and therapeutic potential. Antiviral Res. (2020) 180:104824. doi: 10.1016/j.antiviral.2020.104824
9. Iannacone M, Guidotti LG. Immunobiology and pathogenesis of hepatitis B virus infection. Nat Rev Immunol. (2022) 22:19–32. doi: 10.1038/s41577-021-00549-4
10. Tsukuda S, Watashi K. Hepatitis B virus biology and life cycle. Antiviral Res. (2020) 182:104925. doi: 10.1016/j.antiviral.2020.104925
11. Yang L, Zou T, Chen Y, Zhao Y, Wu X, Li M, et al. Hepatitis B virus X protein mediated epigenetic alterations in the pathogenesis of hepatocellular carcinoma. Hepatol Int. (2022) 16:741–54. doi: 10.1007/s12072-022-10351-6
12. Wang GQ, Niu JX. The HBx protein mutation among different HBV genotypes. J Clin Hepatol. (2010) 26:392–4.
13. Miller RH, Robinson WS. Common evolutionary origin of hepatitis B virus and retroviruses. Proc Natl Acad Sci USA. (1986) 83:2531–5. doi: 10.1073/pnas.83.8.2531
14. Belloni L, Pollicino T, De Nicola F, Guerrieri F, Raffa G, Fanciulli M, et al. Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc Natl Acad Sci USA. (2009) 106:19975–9. doi: 10.1073/pnas.0908365106
15. Lucifora J, Arzberger S, Durantel D, Belloni L, Strubin M, Levrero M, et al. Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J Hepatol. (2011) 55:996–1003. doi: 10.1016/j.jhep.2011.02.015
16. Feitelson MA, Lee J. Hepatitis B virus integration, fragile sites, and hepatocarcinogenesis. Cancer Lett. (2007) 252:157–70. doi: 10.1016/j.canlet.2006.11.010
17. Diao J, Garces R. Richardson CD. X protein of hepatitis B virus modulates cytokine and growth factor related signal transduction pathways during the course of viral infections and hepatocarcinogenesis. Cytokine Growth Factor Rev. (2001) 12:189–205. doi: 10.1016/S1359-6101(00)00034-4
18. Sporn MB, Roberts AB. Transforming growth factor-beta: recent progress and new challenges. J Cell Biol. (1992) 119:1017–21. doi: 10.1083/jcb.119.5.1017
19. Heldin CH, Landström M, Moustakas A. Mechanism of TGF-β signaling to growth arrest, apoptosis, and epithelial–mesenchymal transition. Curr Opin Cell Biol. (2009) 21:166–76. doi: 10.1016/j.ceb.2009.01.021
20. Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol. (2012) 13:616–30. doi: 10.1038/nrm3434
21. Aashaq S, Batool A, Mir SA, Beigh MA, Andrabi KI, Shah ZA. TGF-β signaling: A recap of SMAD-independent and SMAD-dependent pathways. J Cell Physiol. (2022) 237:59–85. doi: 10.1002/jcp.30529
22. Dituri F, Mancarella S, Cigliano A, Chieti A, Giannelli G. TGF-β as multifaceted orchestrator in HCC progression: signaling, EMT, immune microenvironment, and novel therapeutic perspectives. Semin Liver Dis. (2019) 39:053–69. doi: 10.1055/s-0038-1676121
23. Giannelli G, Koudelkova P, Dituri F, Mikulits W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. J Hepatol. (2016) 65:798–808. doi: 10.1016/j.jhep.2016.05.007
24. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. (2003) 425:577–84. doi: 10.1038/nature02006
25. Yoo YD, Ueda H, Park K, Flanders KC, Lee YI, Jay G, et al. Regulation of transforming growth factor-1 expression by the hepatitis B virus (HBV) X transactivator. J Clin. (1996) 8:388–95.
26. Murata M, Matsuzaki K, Yoshida K, Sekimoto G, Tahashi Y, Mori S, et al. Hepatitis B virus X protein shifts human hepatic transforming growth factor (TGF)-β signaling from tumor suppression to oncogenesis in early chronic hepatitis B. Hepatology. (2009) 49:1203–17. doi: 10.1002/hep.22765
27. Cha MY, Kim CM, Park YM, Ryu WS. Hepatitis B virus X protein is essential for the activation of Wnt/?-catenin signaling in hepatoma cells. Hepatology. (2004) 39:1683–93. doi: 10.1002/hep.20245
28. Xie Q, Chen L, Shan X, Shan X, Tang J, Zhou F, et al. Epigenetic silencing of SFRP1 and SFRP5 by hepatitis B virus X protein enhances hepatoma cell tumorigenicity through Wnt signaling pathway: Epigenetic Silencing of SFRP1 and SFRP5 by HBx. Int J Cancer. (2014) 135:635–46. doi: 10.1002/ijc.28697
29. Gao J, Chen C, Hong L, Wang J, Du Y, Song J, et al. Expression of Jagged1 and its association with hepatitis B virus X protein in hepatocellular carcinoma. Biochem Biophys Res Commun. (2007) 356:341–7. doi: 10.1016/j.bbrc.2007.02.130
30. Wang F, Zhou H, Yang Y, Xia X, Sun Q, Luo J, et al. Hepatitis B virus X protein promotes the growth of hepatocellular carcinoma by modulation of the Notch signaling pathway. Oncol Rep. (2012) 27:1170–6. doi: 10.3892/or.2012.1620
31. Chung W, Kim M, de la Monte S, Longato L, Carlson R, Slagle BL, et al. Activation of signal transduction pathways during hepatic oncogenesis. Cancer Lett. (2016) 370:1–9. doi: 10.1016/j.canlet.2015.09.016
32. Jia L, Wei Wang X, Harris CC. Hepatitis B virus X protein inhibits nucleotide excision repair. Int J Cancer. (1999) 80:875–9. doi: 10.1002/(SICI)1097-0215(19990315)80:6<875::AID-IJC13>3.0.CO;2-Z
33. Qadri I, Fatima K, AbdeL-Hafiz H. Hepatitis B virus X protein impedes the DNA repair via its association with transcription factor, TFIIH. BMC Microbiol. (2011) 11:48. doi: 10.1186/1471-2180-11-48
34. Sekiba K, Otsuka M, Funato K, Miyakawa Y, Tanaka E, Seimiya T, et al. HBx-induced degradation of Smc5/6 complex impairs homologous recombination-mediated repair of damaged DNA. J Hepatol. (2022) 76:53–62. doi: 10.1016/j.jhep.2021.08.010
35. Hong Y, Zhou L, Xie H, Zheng S. Innate immune evasion by hepatitis B virus-mediated downregulation of TRIF. Biochem Biophys Res Commun. (2015) 463:719–25. doi: 10.1016/j.bbrc.2015.05.130
36. Kumar M, Jung SY, Hodgson AJ, Madden CR, Qin J, Slagle BL. Hepatitis B virus regulatory HBx protein binds to adaptor protein IPS-1 and inhibits the activation of beta interferon. J Virol. (2011) 85:987–95. doi: 10.1128/JVI.01825-10
37. Wang L, Sun Y, Song X, Wang Z, Zhang Y, Zhao Y, et al. Hepatitis B virus evades immune recognition via RNA adenosine deaminase ADAR1-mediated viral RNA editing in hepatocytes. Cell Mol Immunol. (2021) 18:1871–82. doi: 10.1038/s41423-021-00729-1
38. Park IY, Sohn BH, Yu E, Suh DJ, Chung Y, Lee J, et al. Aberrant epigenetic modifications in hepatocarcinogenesis induced by hepatitis B virus X protein. Gastroenterology. (2007) 132:1476–94. doi: 10.1053/j.gastro.2007.01.034
39. Zheng DL, Zhang L, Cheng N, Xu X, Deng Q, Teng XM, et al. Epigenetic modification induced by hepatitis B virus X protein via interaction with de novo DNA methyltransferase DNMT3A. J Hepatol. (2009) 50:377–87. doi: 10.1016/j.jhep.2008.10.019
40. Yan Y, Huang P, Mao K, He C, Xu Q, Zhang M, et al. Anti-oncogene PTPN13 inactivation by hepatitis B virus X protein counteracts IGF2BP1 to promote hepatocellular carcinoma progression. Oncogene. (2021) 40:28–45. doi: 10.1038/s41388-020-01498-3
41. Fu X, Song X, Li Y, Tan D, Liu G. Hepatitis B virus X protein upregulates DNA methyltransferase 3A/3B and enhances SOCS-1CpG island methylation. Mol Med Rep. (2016) 13:301–8. doi: 10.3892/mmr.2015.4545
42. Choi JH, Jeong H, Jang KL. Hepatitis B virus X protein suppresses all-trans retinoic acid-induced apoptosis in human hepatocytes by repressing p14 expression via DNA methylation. J Gen Virol. (2017) 98:2786–98. doi: 10.1099/jgv.0.000958
43. Gao W, Jia Z, Tian Y, Yang P, Sun H, Wang C, et al. HBx protein contributes to liver carcinogenesis by H3K4me3 modification through stabilizing WD repeat domain 5 protein. Hepatology. (2020) 71:1678–95. doi: 10.1002/hep.30947
44. Rivière L, Gerossier L, Ducroux A, Dion S, Deng Q, Michel ML, et al. HBx relieves chromatin-mediated transcriptional repression of hepatitis B viral cccDNA involving SETDB1 histone methyltransferase. J Hepatol. (2015) 63:1093–102. doi: 10.1016/j.jhep.2015.06.023
45. Cheng ST, Ren JH, Cai XF, Jiang H, Chen J. HBx-elevated SIRT2 promotes HBV replication and hepatocarcinogenesis. Biochem Biophys Res Commun. (2018) 496:904–10. doi: 10.1016/j.bbrc.2018.01.127
46. Hamamoto H, Maemura K, Matsuo K, Taniguchi K, Tanaka Y, Futaki S, et al. Delta-like 3 is silenced by HBx via histone acetylation in HBV-associated HCCs. Sci Rep. (2018) 8:4842. doi: 10.1038/s41598-018-23318-1
47. Lin X, Zuo S, Luo R, Li Y, Yu G, Zou Y, et al. HBX-induced miR-5188 impairs FOXO1 to stimulate β-catenin nuclear translocation and promotes tumor stemness in hepatocellular carcinoma. Theranostics. (2019) 9:7583–98. doi: 10.7150/thno.37717
48. Qiu X, Dong S, Qiao F, Lu S, Song Y, Lao Y, et al. HBx-mediated miR-21 upregulation represses tumor-suppressor function of PDCD4 in hepatocellular carcinoma. Oncogene. (2013) 32:3296–305. doi: 10.1038/onc.2013.150
49. xiao KX, ru LY, ping SL, Nong X, Zhang G, Zhang Y, et al. HBx-induced MiR-1269b in NF-κB dependent manner upregulates cell division cycle 40 homolog (CDC40) to promote proliferation and migration in hepatoma cells. J Transl Med. (2016) 14:189. doi: 10.1186/s12967-016-0949-y
50. Yen CS, Su ZR, Lee YP, Liu IT, Yen CJ. miR-106b promotes cancer progression in hepatitis B virus-associated hepatocellular carcinoma. World J Gastroenterol. (2016) 22:5183–92. doi: 10.3748/wjg.v22.i22.5183
51. Xiao Y, Najeeb RM, Ma D, Yang K, Zhong Q, Liu Q. Upregulation of CENPM promotes hepatocarcinogenesis through mutiple mechanisms. J Exp Clin Cancer Res. (2019) 38:458. doi: 10.1186/s13046-019-1444-0
52. Liu Fy, Zhou Sj, Deng Yl, Zhang Z, Zhang E, Wu Z, et al. MiR-216b is involved in pathogenesis and progression of hepatocellular carcinoma through HBx-miR-216b-IGF2BP2 signaling pathway. Cell Death Dis. (2015) 6:e1670–0. doi: 10.1038/cddis.2015.46
53. Liang HW, Wang N, Wang Y, Wang F, Fu Z, Yan X, et al. Hepatitis B virus-human chimeric transcript HBx-LINE1 promotes hepatic injury via sequestering cellular microRNA-122. J Hepatol. (2016) 64:278–91. doi: 10.1016/j.jhep.2015.09.013
54. Zhe Y, Jiong L, Guoxing F, Yuan W, Guang Y, Yunxia L, et al. Hepatitis B virus X protein enhances hepatocarcinogenesis by depressing the targeting of NUSAP1 mRNA by miR - 18b. Cancer Biol Med. (2019) 16:276. doi: 10.20892/j.issn.2095-3941.2018.0283
55. Xu X, Fan Z, Kang L, Han J, Jiang C, Zheng X, et al. Hepatitis B virus X protein represses miRNA-148a to enhance tumorigenesis. J Clin Invest. (2013) 123(2):JCI64265. doi: 10.1172/JCI64265
56. Salerno D, Chiodo L, Alfano V, Floriot O, Cottone G, Paturel A, et al. Hepatitis B protein HBx binds the DLEU2 lncRNA to sustain cccDNA and host cancer-related gene transcription. Gut. (2020) 69:2016–24. doi: 10.1136/gutjnl-2019-319637
57. Sun Y, Teng Y, Wang L, Zhang Z, Chen C, Wang Y, et al. LINC01431 promotes histone H4R3 methylation to impede HBV covalently closed circular DNA transcription by stabilizing PRMT1. Advanced Sci. (2022) 9:2103135. doi: 10.1002/advs.202103135
58. Song W, Zheng C, Liu M, Xu Y, Qian Y, Zhang Z, et al. TRERNA1 upregulation mediated by HBx promotes sorafenib resistance and cell proliferation in HCC via targeting NRAS by sponging miR-22-3p. Mol Ther. (2021) 29:2601–16. doi: 10.1016/j.ymthe.2021.04.011
59. Gan L, Shangguan Q, Zhang F, Tong X, Qi D, Zhao Y, et al. HBV HBx-downregulated lncRNA LINC01010 attenuates cell proliferation by interacting with vimentin. IJMS. (2021) 22:12497. doi: 10.3390/ijms222212497
60. Huang J, Guo Y, Zhao C, Yuan S, Wang Y, Tang G, et al. Hepatitis B virus X protein (HBx)-related long noncoding RNA (lncRNA) down-regulated expression by HBx (Dreh) inhibits hepatocellular carcinoma metastasis by targeting the intermediate filament protein vimentin. Hepatology. (2013) 57:1882–92. doi: 10.1002/hep.26195
61. Chen Z, Yu W, Zhou Q, Zhang J, Jiang H, Hao D, et al. A novel lncRNA IHS promotes tumor proliferation and metastasis in HCC by regulating the ERK- and AKT/GSK-3β-signaling pathways. Mol Ther - Nucleic Acids. (2019) 16:707–20. doi: 10.1016/j.omtn.2019.04.021
62. Zhang Y, Yan Q, Gong L, Xu H, Liu B, Fang X, et al. C-terminal truncated HBx initiates hepatocarcinogenesis by downregulating TXNIP and reprogramming glucose metabolism. Oncogene. (2021) 40:1147–61. doi: 10.1038/s41388-020-01593-5
63. Sze KMF, Chu GKY, Lee JMF, IOL Ng. C-terminal truncated hepatitis B virus x protein is associated with metastasis and enhances invasiveness by c-jun/matrix metalloproteinase protein 10 activation in hepatocellular carcinoma. Hepatology. (2013) 57:131–9. doi: 10.1002/hep.25979
64. Mao X, Tey SK, Ko FCF, Kwong EML, Gao Y, Ng IOL, et al. C-terminal truncated HBx protein activates caveolin-1/LRP6/β-catenin/FRMD5 axis in promoting hepatocarcinogenesis. Cancer Lett. (2019) 444:60–9. doi: 10.1016/j.canlet.2018.12.003
65. Niu Y, Chen L, Wu M, Huang W, Wu X, Huang D, et al. Partial abrogation of FXR-KNG1 signaling by carboxyl-terminal truncated HBx-C30 in hepatitis B virus-associated hepatocellular carcinoma. Virus Res. (2021) 293:198264. doi: 10.1016/j.virusres.2020.198264
66. Salpini R, Surdo M, Cortese MF, Palumbo GA, Carioti L, Cappiello G, et al. The novel HBx mutation F30V correlates with hepatocellular carcinoma in vivo, reduces hepatitis B virus replicative efficiency and enhances anti-apoptotic activity of HBx N terminus in vitro. Clin Microbiol Infection. (2019) 25:906.e1–7. doi: 10.1016/j.cmi.2018.11.017
67. Wands JR, Kim M. WNT/β-catenin signaling and hepatocellular carcinoma: WANDS AND KIM. Hepatology. (2014) 60:452–4. doi: 10.1002/hep.27081
68. Kongkavitoon P, Tangkijvanich P, Hirankarn N, Palaga T. Hepatitis B virus HBx activates notch signaling via delta-like 4/notch1 in hepatocellular carcinoma. PloS One. (2016) 11:e0146696. doi: 10.1371/journal.pone.0146696
69. Kim GA, Lim YS, Han S, Choi J, Shim JH, Kim KM, et al. High risk of hepatocellular carcinoma and death in patients with immune-tolerant-phase chronic hepatitis B. Gut. (2018) 67:945–52. doi: 10.1136/gutjnl-2017-314904
70. Kuramoto J, Arai E, Tian Y, Funahashi N, Hiramoto M, Nammo T, et al. Genome-wide DNA methylation analysis during non-alcoholic steatohepatitis-related multistage hepatocarcinogenesis: comparison with hepatitis virus-related carcinogenesis. Carcinogenesis. (2017) 38:261–70. doi: 10.1093/carcin/bgx005
71. Neganova ME, Klochkov SG, Aleksandrova YR, Aliev G. Histone modifications in epigenetic regulation of cancer: Perspectives and achieved progress. Semin Cancer Biol. (2022) 83:452–71. doi: 10.1016/j.semcancer.2020.07.015
72. Markouli M, Strepkos D, Basdra EK, Papavassiliou AG, Piperi C. Prominent role of histone modifications in the regulation of tumor metastasis. IJMS. (2021) 22:2778. doi: 10.3390/ijms22052778
73. Rajan PK, Udoh UA, Sanabria JD, Banerjee M, Smith G, Schade MS, et al. The role of histone acetylation-/methylation-mediated apoptotic gene regulation in hepatocellular carcinoma. IJMS. (2020) 21:8894. doi: 10.3390/ijms21238894
74. Qu S, Jin L, Huang H, Lin J, Gao W, Zeng Z. A positive-feedback loop between HBx and ALKBH5 promotes hepatocellular carcinogenesis. BMC Cancer. (2021) 21:686. doi: 10.1186/s12885-021-08449-5
75. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discovery. (2017) 16:203–22. doi: 10.1038/nrd.2016.246
76. He Y, Meng XM, Huang C, Wu BM, Zhang L, Lv XW, et al. Long noncoding RNAs: Novel insights into hepatocelluar carcinoma. Cancer Lett. (2014) 344:20–7. doi: 10.1016/j.canlet.2013.10.021
77. Roderburg C, Trautwein C. Cell-specific functions of miRNA in the liver. J Hepatol. (2017) 66:655–6. doi: 10.1016/j.jhep.2016.09.015
78. Zhang H, Chen X, Zhang J, Wang X, Chen H, Liu L, et al. Long non−coding RNAs in HBV−related hepatocellular carcinoma (Review). Int J Oncol. (2019) 56:18–32. doi: 10.3892/ijo.2019.4909
79. Oura K, Morishita A, Masaki T. Molecular and functional roles of microRNAs in the progression of hepatocellular carcinoma—A review. IJMS. (2020) 21:8362. doi: 10.3390/ijms21218362
80. Zhang B, Han S, Feng B, Chu X, Chen L, Wang R. Hepatitis B virus X protein-mediated non-coding RNA aberrations in the development of human hepatocellular carcinoma. Exp Mol Med. (2017) 49:e293–3. doi: 10.1038/emm.2016.177
81. Yin D, Wang Y, Sai W, Zhang L, Miao Y, Cao L, et al. HBx-induced miR-21 suppresses cell apoptosis in hepatocellular carcinoma by targeting interleukin-12. Oncol Rep. (2016) 36:2305–12. doi: 10.3892/or.2016.5026
82. Levrero M, Zucman-Rossi J. Mechanisms of HBV-induced hepatocellular carcinoma. J Hepatol. (2016) 64:S84–S101. doi: 10.1016/j.jhep.2016.02.021
83. García-García S, Caballero-Garralda A, Tabernero D, Cortese MF, Gregori J, Rodriguez-Algarra F, et al. Hepatitis B virus variants with multiple insertions and/or deletions in the X open reading frame 3′ End: common members of viral quasispecies in chronic hepatitis B patients. Biomedicines. (2022) 10:1194. doi: 10.3390/biomedicines10051194
84. Toh ST, Jin Y, Liu L, Wang J, Babrzadeh F, Gharizadeh B, et al. Deep sequencing of the hepatitis B virus in hepatocellular carcinoma patients reveals enriched integration events, structural alterations and sequence variations. Carcinogenesis. (2013) 34:787–98. doi: 10.1093/carcin/bgs406
85. Ali A. Hepatitis B virus, HBx mutants and their role in hepatocellular carcinoma. WJG. (2014) 20:10238. doi: 10.3748/wjg.v20.i30.10238
86. Ng SA, Lee C. Hepatitis B virus X gene and hepatocarcinogenesis. J Gastroenterol. (2011) 46:974–90. doi: 10.1007/s00535-011-0415-9
87. Jung SY, Kim YJ. C-terminal region of HBx is crucial for mitochondrial DNA damage. Cancer Lett. (2013) 331:76–83. doi: 10.1016/j.canlet.2012.12.004
88. Decorsière A, Mueller H, van Breugel PC, Abdul F, Gerossier L, Beran RK, et al. Hepatitis B virus X protein identifies the Smc5/6 complex as a host restriction factor. Nature. (2016) 531:386–9. doi: 10.1038/nature17170
89. Murphy CM, Xu Y, Li F, Nio K, Reszka-Blanco N, Li X, et al. Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell Rep. (2016) 16:2846–54. doi: 10.1016/j.celrep.2016.08.026
90. Mak LY, Cheung KS, Fung J, Seto WK, Yuen MF. New strategies for the treatment of chronic hepatitis B. Trends Mol Med. (2022) 28:742–57. doi: 10.1016/j.molmed.2022.06.002
91. Sekiba K, Otsuka M, Ohno M, Yamagami M, Kishikawa T, Suzuki T, et al. Inhibition of HBV transcription from cccDNA with nitazoxanide by targeting the HBx–DDB1 interaction. Cell Mol Gastroenterol Hepatol. (2019) 7:297–312. doi: 10.1016/j.jcmgh.2018.10.010
92. Sekiba K, Otsuka M, Ohno M, Yamagami M, Kishikawa T, Seimiya T, et al. Pevonedistat, a neuronal precursor cell-expressed developmentally down-regulated protein 8–activating enzyme inhibitor, is a potent inhibitor of hepatitis B virus. Hepatology. (2019) 69:1903–15. doi: 10.1002/hep.30491
93. Cheng ST, Hu JL, Ren JH, Yu HB, Zhong S, Wai Wong VK, et al. Dicoumarol, an NQO1 inhibitor, blocks cccDNA transcription by promoting degradation of HBx. J Hepatol. (2021) 74:522–34. doi: 10.1016/j.jhep.2020.09.019
94. Gungor MZ, Uysal M, Senturk S. The bright and the dark side of TGF-β Signaling in hepatocellular carcinoma: mechanisms, dysregulation, and therapeutic implications. Cancers. (2022) 14:940. doi: 10.3390/cancers14040940
95. Derynck R, Budi EH. Specificity, versatility, and control of TGF-b family signaling. Sci Signaling. (2019) 25. doi: 10.1126/scisignal.aav5183
96. Robertson IB, Rifkin DB. Regulation of the bioavailability of TGF-β and TGF-β-related proteins. Cold Spring Harb Perspect Biol. (2016) 8:a021907. doi: 10.1101/cshperspect.a021907
97. Tu S, Huang W, Huang C, Luo Z, Yan X. Contextual regulation of TGF-β Signaling in liver cancer. Cells. (2019) 8:1235. doi: 10.3390/cells8101235
98. Hata A, Chen YG. TGF-β Signaling from receptors to smads. Cold Spring Harb Perspect Biol. (2016) 8:a022061. doi: 10.1101/cshperspect.a022061
99. Wrighton KH, Lin X, Feng XH. Phospho-control of TGF-β superfamily signaling. Cell Res. (2009) 19:8–20. doi: 10.1038/cr.2008.327
100. Kamato D, Do BH, Osman N, Ross BP, Mohamed R, Xu S, et al. Smad linker region phosphorylation is a signalling pathway in its own right and not only a modulator of canonical TGF-β signalling. Cell Mol Life Sci. (2020) 77:243–51. doi: 10.1007/s00018-019-03266-3
101. Imamura T, Takase M, Nishihara A, Oeda E, Hanai J, Kawabata M, et al. Smad6 inhibits signalling by the TGF- superfamily. Nature. (1997) 389:5. doi: 10.1038/39355
102. Nakao A, Afrakhte M, Nakayama T, Christian JL, Heuchel R, Itoh S, et al. Identification of Smad7, a TGF-inducible antagonist of TGF- signalling. Nature. (1997) 389:5. doi: 10.1038/39369
103. Peng D, Fu M, Wang M, Wei Y, Wei X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol Cancer. (2022) 21:104. doi: 10.1186/s12943-022-01569-x
104. Heldin CH, Moustakas A. Signaling receptors for TGF-β Family members. Cold Spring Harb Perspect Biol. (2016) 8. doi: 10.1101/cshperspect.a022053
105. Guo X, Wang XF. Signaling cross-talk between TGF-β/BMP and other pathways. Cell Res. (2009) 19:71–88. doi: 10.1038/cr.2008.302
106. Yu Y, Feng XH. TGF-β signaling in cell fate control and cancer. Curr Opin Cell Biol. (2019) 61:56–63. doi: 10.1016/j.ceb.2019.07.007
108. Chen CR, Kang Y, Siegel PM, Massague J. E2F4/5 and p107 as Smad Cofactors Linking the TGF Receptor to c-myc Repression. Cells. (2002) 14:19–32. doi: 10.1016/s0092-8674(02)00801-2
109. Saltis’ J. TGF-/3: receptors and cell cycle arrest. Mol Cell Endocrinol. (1996) 6:227–32.doi: 10.1016/0303-7207(95)03721-7
110. Goel S, DeCristo MJ, McAllister SS, Zhao JJ. CDK4/6 inhibition in cancer: beyond cell cycle arrest. Trends Cell Biol. (2018) 28:911–25. doi: 10.1016/j.tcb.2018.07.002
111. Claassen GF, Hann SR. A role for transcriptional repression of p21CIP1 by c-Myc in overcoming transforming growth factor. Cell Biol. (2000) 6:9498–503. doi: 10.1073/pnas.150006697
112. Feng XH, Liang YY, Liang M, Zhai W, Lin X. Direct Interaction of c-Myc with Smad2 and Smad3 to Inhibit TGF--Mediated Induction of the CDK Inhibitor p15Ink4B. Mol Cell. (2016) 11:1089. doi: 10.1016/j.molcel.2016.03.026
113. Staller P, Peukert K, Kiermaier A, Seoane J, Lukas J, Karsunky H, et al. Repression of p15INK4b expression by Myc through association with Miz-1. Nat Cell Biol. (2001) 3:392–9. doi: 10.1038/35070076
114. Zhang Y, Alexander PB, Wang XF. TGF-β Family signaling in the control of cell proliferation and survival. Cold Spring Harb Perspect Biol. (2017) 9:a022145. doi: 10.1101/cshperspect.a022145
115. Senturk S, Mumcuoglu M, Gursoy-Yuzugullu O, Cingoz B, Akcali KC, Ozturk M. Transforming growth factor-beta induces senescence in hepatocellular carcinoma cells and inhibits tumor growth. Hepatology. (2010) 52:966–74. doi: 10.1002/hep.23769
116. Hashimoto O, Ueno T, Kimura R, Ohtsubo M, Nakamura T, Koga H, et al. Inhibition of proteasome-dependent degradation of Wee1 in G2-arrested Hep3B cells by TGFβ1. Mol Carcinog. (2003) 36:171–82. doi: 10.1002/mc.10111
117. Calcinotto A, Kohli J, Zagato E, Pellegrini L, Demaria M, Alimonti A. Cellular senescence: aging, cancer, and injury. Physiol Rev. (2019) 99:1047–78. doi: 10.1152/physrev.00020.2018
118. Piskorz WM, Cechowska-Pasko M. Senescence of tumor cells in anticancer therapy—Beneficial and detrimental effects. IJMS. (2022) 23:11082. doi: 10.3390/ijms231911082
119. Demirci D, Dayanc B, Mazi FA, Senturk S. The jekyll and hyde of cellular senescence in cancer. Cells. (2021) 10:208. doi: 10.3390/cells10020208
120. Satyanarayana A, Manns MP, Rudolph KL. Telomeres and telomerase: A dual role in hepatocarcinogenesis. Hepatology. (2004) 40:276–83. doi: 10.1002/hep.20308
121. Nault JC, Ningarhari M, Rebouissou S, Zucman-Rossi J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat Rev Gastroenterol Hepatol. (2019) 16:544–58. doi: 10.1038/s41575-019-0165-3
122. Lin SY, Elledge SJ. Multiple tumor suppressor pathways negatively regulate telomerase. Cell. (2003) 113:881–9. doi: 10.1016/S0092-8674(03)00430-6
123. Li H, Xu D, Toh BH, Liu JP. TGF-β and cancer: Is Smad3 a repressor of hTERT gene? Cell Res. (2006) 16:169–73. doi: 10.1038/sj.cr.7310023
124. Lacerte A, Korah J, Roy M, Yang XJ, Lemay S, Lebrun JJ. Transforming growth factor-β inhibits telomerase through SMAD3 and E2F transcription factors. Cell Signalling. (2008) 20:50–9. doi: 10.1016/j.cellsig.2007.08.012
125. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. (2008) 451:1069–75. doi: 10.1038/nature06639
126. Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science. (2000) 290:1717–21. doi: 10.1126/science.290.5497.1717
127. Kiyono K, Suzuki HI, Matsuyama H, Morishita Y, Komuro A, Kano MR, et al. Autophagy is activated by TGF-β and potentiates TGF-β–mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. (2009) 69:8844–52. doi: 10.1158/0008-5472.CAN-08-4401
128. Suzuki HI, Kiyono K, Miyazono K. Regulation of autophagy by transforming growth factor-β (TGF-β) signaling. Autophagy. (2010) 6:645–7. doi: 10.4161/auto.6.5.12046
129. Lee Y, Jang B. The role of autophagy in hepatocellular carcinoma. IJMS. (2015) 16:26629–43. doi: 10.3390/ijms161125984
130. Schneider JL. Liver autophagy: much more than just taking out the trash. Nature Reviews Gastroenterology & Hepatology. (2014) 11:187–200. doi: 10.1038/nrgastro.2013.211
131. Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology. (2014) 147:765–783.e4. doi: 10.1053/j.gastro.2014.07.018
132. Herzer K, Grosse-Wilde A, Krammer PH, Galle PR, Kanzler S. Transforming growth factor-β–mediated tumor necrosis factor-related apoptosis-inducing ligand expression and apoptosis in hepatoma cells requires functional cooperation between smad proteins and activator protein-1. Mol Cancer Res. (2008) 6:1169–77. doi: 10.1158/1541-7786.MCR-08-0073
133. Herzer K, Ganten TM, Schulze-Bergkamen H, Grosse-Wilde A, Koschny R, Krammer PH, et al. Transforming growth factor β can mediate apoptosis via the expression of TRAIL in human hepatoma cells. Hepatology. (2005) 42:183–92. doi: 10.1002/hep.20757
134. Shima Y, Nakao K, Nakashima T, Kawakami A, Nakata K, Hamasaki K, et al. Activation of caspase-8 in transforming growth factor-?-induced apoptosis of human hepatoma cells. Hepatology. (1999) 30:1215–22. doi: 10.1002/hep.510300503
135. Carmona-Cuenca I, Roncero C, Sancho P, Caja L, Fausto N, Fernández M, et al. Upregulation of the NADPH oxidase NOX4 by TGF-beta in hepatocytes is required for its pro-apoptotic activity. J Hepatol. (2008) 49:965–76. doi: 10.1016/j.jhep.2008.07.021
136. Herrera B, Álvarez Alberto M, Sánchez A, Fernández M, Roncero C, Benito M, et al. Reactive oxygen species (ROS) mediates the mitochondrial-dependent apoptosis induced by transforming growth factor ß in fetal hepatocytes. FASEB J. (2001) 15:741–51. doi: 10.1096/fj.00-0267com
137. Caja L, Sancho P, Bertran E, Fabregat I. Dissecting the effect of targeting the epidermal growth factor receptor on TGF-β-induced-apoptosis in human hepatocellular carcinoma cells. J Hepatol. (2011) 55:351–8. doi: 10.1016/j.jhep.2010.10.041
138. Moreno-Càceres J, Caballero-Díaz D, Nwosu ZC, Meyer C, López-Luque J, Malfettone A, et al. The level of caveolin-1 expression determines response to TGF-β as a tumour suppressor in hepatocellular carcinoma cells. Cell Death Dis. (2017) 8:e3098–8. doi: 10.1038/cddis.2017.469
139. Zhang K, Zhang M, Luo Z, Wen Z, Yan X. The dichotomous role of TGF-β in controlling liver cancer cell survival and proliferation. J Genet Genomics. (2020) 47:497–512. doi: 10.1016/j.jgg.2020.09.005
140. Marquardt JU, Andersen JB, Thorgeirsson SS. Functional and genetic deconstruction of the cellular origin in liver cancer. Nat Rev Cancer. (2015) 15:653–67. doi: 10.1038/nrc4017
141. Moon H, Ju HL, Chung SI, Cho KJ, Eun JW, Nam SW, et al. Transforming growth factor-β Promotes liver tumorigenesis in mice via up-regulation of snail. Gastroenterology. (2017) 153:1378–1391.e6. doi: 10.1053/j.gastro.2017.07.014
142. Sun S-L, Wang X-Y. TGF-β1 promotes proliferation and invasion of HCC cell line HepG2 by activating GLI-1 signaling. Euro Rev Med Pharmacol Sci. (2018) 8:7688–95. doi: 10.26355/eurrev_201811_16389
143. Zucman-Rossi J, Villanueva A, Nault JC, Llovet JM. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology. (2015) 149:1226–1239.e4. doi: 10.1053/j.gastro.2015.05.061
144. Zhou Q, Zhou Q, Liu Q, He Z, Yan Y, Lin J, et al. PRL-3 facilitates Hepatocellular Carcinoma progression by co-amplifying with and activating FAK. Theranostics. (2020) 10:10345–59. doi: 10.7150/thno.42069
145. Chen J, Zaidi S, Rao S, Chen JS, Phan L, Farci P, et al. Analysis of genomes and transcriptomes of hepatocellular carcinomas identifies mutations and gene expression changes in the transforming growth factor-b pathway. Gastroenterology. (2018) 154(1):16. doi: 10.1053/j.gastro.2017.09.007
146. Chen J, Gingold JA, Su X. Immunomodulatory TGF-β Signaling in hepatocellular carcinoma. Trends Mol Med. (2019) 14:1010–23. doi: 10.1016/j.molmed.2019.06.007
147. Bévant K, Desoteux M, Abdel Wahab AHA, Abdel Wahab SA, Metwally AM, Coulouarn C. DNA methylation of TGFβ Target genes: epigenetic control of TGFβ Functional duality in liver cancer. Cells. (2021) 10:2207. doi: 10.3390/cells10092207
148. Zhu H, Wu K, Yan W, Hu L, Yuan J, Dong Y, et al. Epigenetic silencing of DACH1 induces loss of transforming growth factor-b1 antiproliferative response in human hepatocellular carcinoma. Hepatology. (2013) 58(6):11. doi: 10.1002/hep.26587
149. Sohn BH, Park IY, Lee JJ, Yang J, Jang YJ, Park KC, et al. Functional switching of TGF-1 signaling in liver cancer via epigenetic modulation of a single cpG site in TTP promoter. Gastroenterology. (2010) 138(5):23. doi: 10.1053/j.gastro.2009.12.044
150. Acun T, Oztas E, Yagci T, Yakicier MC. SIP1 is downregulated in hepatocellular carcinoma by promoter hypermethylation. BMC Cancer (2011) 1. doi: 10.1186/1471-2407-11-223
151. Mori T, Nomoto S, Koshikawa K, Fujii T, Sakai M, Nishikawa Y, et al. Decreased expression and frequent allelic inactivation of the RUNX3 gene at 1p36 in human hepatocellular carcinoma. Liver Int. (2005) 25:380–8. doi: 10.1111/j.1478-3231.2005.1059.x
152. Hu Y, Zheng Y, Dai M, Wang X, Wu J, Yu B, et al. G9a and histone deacetylases are crucial for Snail2-mediated E-cadherin repression and metastasis in hepatocellular carcinoma. Cancer Sci. (2019) 11:3442–52. doi: 10.1111/cas.14173
153. Pastushenko I, Blanpain C. EMT transition states during tumor progression and metastasis. Trends Cell Biol. (2019) 29:212–26. doi: 10.1016/j.tcb.2018.12.001
154. Thiery JP, Acloque H, Huang RYJ, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. (2009) 139:871–90. doi: 10.1016/j.cell.2009.11.007
155. Su J, Morgani SM, David CJ, Wang Q, Er EE, Huang YH, et al. TGF-β orchestrates fibrogenic and developmental EMTs via the RAS effector RREB1. Nature. (2020) 577:566–71. doi: 10.1038/s41586-019-1897-5
156. Hao Y, Baker D, ten Dijke P. TGF-β-mediated epithelial-mesenchymal transition and cancer metastasis. IJMS. (2019) 20:2767. doi: 10.3390/ijms20112767
157. Derynck R, Muthusamy BP, Saeteurn KY. Signaling pathway cooperation in TGF-β-induced epithelial–mesenchymal transition. Curr Opin Cell Biol. (2014) 31:56–66. doi: 10.1016/j.ceb.2014.09.001
158. Pan G, Liu Y, Shang L, Zhou F, Yang S. EMT-associated microRNAs and their roles in cancer stemness and drug resistance. Cancer Commun. (2021) 41:199–217. doi: 10.1002/cac2.12138
160. hang YJ, Yang F, Wang F, Ma J, Guo Y, Tao Q, et al. A long noncoding RNA activated by TGF-β Promotes the invasion-metastasis cascade in hepatocellular carcinoma. Cancer Cell. (2014) 25:666–81. doi: 10.1016/j.ccr.2014.03.010
161. Batlle E, Massagué J. Transforming growth factor-β Signaling in immunity and cancer. Immunity. (2019) 50:924–40. doi: 10.1016/j.immuni.2019.03.024
162. Zhang L, Xu J, Zhou S, Yao F, Zhang R, You W, et al. Endothelial DGKG promotes tumor angiogenesis and immune evasion in hepatocellular carcinoma. J Hepatol. (2024) 80:82–98. doi: 10.1016/j.jhep.2023.10.006
163. Yan W, Liu X, Ma H, Zhang H, Song X, Gao L, et al. Tim-3 fosters HCC development by enhancing TGF-β-mediated alternative activation of macrophages. Gut. (2015) 64:1593–604. doi: 10.1136/gutjnl-2014-307671
164. Xin X, Cheng X, Zeng F, Xu Q, Hou L. The role of TGF-β/SMAD signaling in hepatocellular carcinoma: from mechanism to therapy and prognosis. Int J Biol Sci. (2024) 20:1436–51. doi: 10.7150/ijbs.89568
165. Peng X, Yang H, Tao L, Xiao J, Zeng Y, Shen Y, et al. Fluorofenidone alleviates liver fibrosis by inhibiting hepatic stellate cell autophagy via the TGF-β1/Smad pathway: implications for liver cancer. PeerJ. (2023) 11:e16060. doi: 10.7717/peerj.16060
166. Su Q, Fan M, Wang J, Ullah A, Ghauri MA, Dai B, et al. Sanguinarine inhibits epithelial–mesenchymal transition via targeting HIF-1α/TGF-β feed-forward loop in hepatocellular carcinoma. Cell Death Dis. (2019) 10:939. doi: 10.1038/s41419-019-2173-1
167. Sun Y, Liu B, Xie J, Jiang X, Xiao B, Hu X, et al. Aspirin attenuates liver fibrosis by suppressing TGF−β1/Smad signaling. Mol Med Rep. (2022) 25:181. doi: 10.3892/mmr.2022.12697
168. Liu J, Kong D, Qiu J, Xie Y, Lu Z, Zhou C, et al. Praziquantel ameliorates CCl 4 -induced liver fibrosis in mice by inhibiting TGF-β/Smad signalling via up-regulating Smad7 in hepatic stellate cells. Br J Pharmacol. (2019) 176:4666–80. doi: 10.1111/bph.14831
169. Shen Y, Lu C, Song Z, Qiao C, Wang J, Chen J, et al. Ursodeoxycholic acid reduces antitumor immunosuppression by inducing CHIP-mediated TGF-β degradation. Nat Commun. (2022) 13:3419. doi: 10.1038/s41467-022-31141-6
170. Rani B, Malfettone A, Dituri F, Soukupova J, Lupo L, Mancarella S, et al. Galunisertib suppresses the staminal phenotype in hepatocellular carcinoma by modulating CD44 expression. Cell Death Dis. (2018) 9:373. doi: 10.1038/s41419-018-0384-5
171. Hammad S, Cavalcanti E, Werle J, Caruso ML, Dropmann A, Ignazzi A, et al. Galunisertib modifies the liver fibrotic composition in the Abcb4Ko mouse model. Arch Toxicol. (2018) 92:2297–309. doi: 10.1007/s00204-018-2231-y
172. Luangmonkong T, Suriguga S, Bigaeva E, Boersema M, Oosterhuis D, De Jong KP, et al. Evaluating the antifibrotic potency of galunisertib in a human ex vivo model of liver fibrosis. Br J Pharmacol. (2017) 174:3107–17. doi: 10.1111/bph.13945
173. Fransvea E, Mazzocca A, Santamato A, Azzariti A, Antonaci S, Giannelli G. Kinase activation profile associated with TGF-β-dependent migration of HCC cells: a preclinical study. Cancer Chemother Pharmacol. (2011) 68:79–86. doi: 10.1007/s00280-010-1459-x
174. Zhang J, Li Y, Liu Q, Li R, Pu S, Yang L, et al. SKLB023 as an iNOS inhibitor alleviated liver fibrosis by inhibiting the TGF-beta/Smad signaling pathway. RSC Adv. (2018) 8:30919–24. doi: 10.1039/C8RA04955F
175. Yap TA, Vieito M, Baldini C, Sepúlveda-Sánchez JM, Kondo S, Simonelli M, et al. First-in-human phase I study of a next-generation, oral, TGFβ Receptor 1 inhibitor, LY3200882, in patients with advanced cancer. Clin Cancer Res. (2021) 27:6666–76. doi: 10.1158/1078-0432.CCR-21-1504
176. Zhao H, Wei J, Sun J. Roles of TGF-β signaling pathway in tumor microenvirionment and cancer therapy. Int Immunopharmacol. (2020) 89:107101. doi: 10.1016/j.intimp.2020.107101
177. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. (2018) 554:544–8. doi: 10.1038/nature25501
178. Tauriello DVF, Palomo-Ponce S, Stork D, Berenguer-Llergo A, Badia-Ramentol J, Iglesias M, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature. (2018) 554:538–43. doi: 10.1038/nature25492
179. Li T, Wang X, Niu M, Wang M, Zhou J, Wu K, et al. Bispecific antibody targeting TGF-β and PD-L1 for synergistic cancer immunotherapy. Front Immunol. (2023) 14:1196970. doi: 10.3389/fimmu.2023.1196970
180. Yi M, Li T, Niu M, Wu Y, Zhao Z, Wu K. TGF-β: A novel predictor and target for anti-PD-1/PD-L1 therapy. Front Immunol. (2022) 13:1061394. doi: 10.3389/fimmu.2022.1061394
181. Yang MH, Chen M, Mo HH, Tsai WC, Chang YC, Chang CC, et al. Utilizing experimental mouse model to identify effectors of hepatocellular carcinoma induced by HBx antigen. Cancers. (2020) 12:409. doi: 10.3390/cancers12020409
182. Fabregat I, Moreno-Càceres J, Sánchez A, Dooley S, Dewidar B, Giannelli G, et al. TGF-β signalling and liver disease. FEBS J. (2016) 283:2219–32. doi: 10.1111/febs.13665
183. Gough NR, Xiang X, Mishra L. TGF-β Signaling in liver, pancreas, and gastrointestinal diseases and cancer. Gastroenterology. (2021) 161:434–452.e15. doi: 10.1053/j.gastro.2021.04.064
184. Liu S, Ren J, ten Dijke P. Targeting TGFβ signal transduction for cancer therapy. Sig Transduct Target Ther. (2021) 6:8. doi: 10.1038/s41392-020-00436-9
185. Ooshima A, Park J, Kim S. Phosphorylation status at Smad3 linker region modulates transforming growth factor-β-induced epithelial-mesenchymal transition and cancer progression. Cancer Sci. (2019) 110:481–8. doi: 10.1111/cas.13922
186. Massagué J. TGF-β signal transduction. J Biol Chem. (1998) 41:32066–70. doi: 10.1074/jbc.M006496200
187. Xu P, Lin X, Feng XH. Posttranslational regulation of smads. Cold Spring Harb Perspect Biol. (2016) 8:a022087. doi: 10.1101/cshperspect.a022087
188. Matsuzaki K. Smad phosphoisoform signaling specificity: the right place at the right time. Carcinogenesis. (2011) 32:1578–88. doi: 10.1093/carcin/bgr172
189. Liu F. Smad3 phosphorylation by cyclin-dependent kinases. Cytokine Growth Factor Rev. (2006) 17:9–17. doi: 10.1016/j.cytogfr.2005.09.010
190. Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGFbeta / Smad signaling by oncogenic Ras. Genes Dev. (1999) 13:804–16. doi: 10.1101/gad.13.7.804
191. Millet C, Yamashita M, Heller M, Yu LR, Veenstra TD, Zhang YE. A negative feedback control of transforming growth factor-β Signaling by glycogen synthase kinase 3-mediated smad3 linker phosphorylation at ser-204. J Biol Chem. (2009) 284:19808–16. doi: 10.1074/jbc.M109.016667
192. Feng XH, Lin X, Derynck R. Smad2, Smad3 and Smad4 cooperate with Sp1 to induce p15Ink4B transcription in response to TGF-β. EMBO J. (2000) 19:5178–93. doi: 10.1093/emboj/19.19.5178
193. Pardali K, Kurisaki A, Morén A, ten Dijke P, Kardassis D, Moustakas A. Role of smad proteins and transcription factor sp1 in p21Waf1/cip1 regulation by transforming growth factor-β. J Biol Chem. (2000) 275:29244–56. doi: 10.1074/jbc.M909467199
194. Frederick JP, Liberati NT, Waddell DS, Shi Y, Wang XF. Transforming Growth Factor β-Mediated Transcriptional Repression of c- myc Is Dependent on Direct Binding of Smad3 to a Novel Repressive Smad Binding Element. Mol Cell Biol. (2004) 24:2546–59. doi: 10.1128/MCB.24.6.2546-2559.2004
195. Yang YA, Zhang GM, Feigenbaum L, Zhang YE. Smad3 reduces susceptibility to hepatocarcinoma by sensitizing hepatocytes to apoptosis through downregulation of Bcl-2. Cancer Cell. (2006) 9:445–57. doi: 10.1016/j.ccr.2006.04.025
196. Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. (2000) 103:239–52. doi: 10.1016/S0092-8674(00)00116-1
197. Wagner EF, Nebreda ÁR. Signal integration by JNK and p38 MAPK pathways in cancer development. Nat Rev Cancer. (2009) 9:537–49. doi: 10.1038/nrc2694
198. Sekimoto G, Matsuzaki K, Yoshida K, Zhang YE. Reversible smad-dependent signaling between tumor suppression and oncogenesis. Cancer Res. (2007) 67:5090–6. doi: 10.1158/0008-5472.CAN-06-4629
199. Moustakas A, Pardali K, Gaal A, Heldin CH. Mechanisms of TGF-β signaling in regulation of cell growth and differentiation. Immunol Lett. (2002) 82:85–91. doi: 10.1016/S0165-2478(02)00023-8
200. Hui L, Zatloukal K, Scheuch H, Stepniak E, Wagner EF. Proliferation of human HCC cells and chemically induced mouse liver cancers requires JNK1-dependent p21 downregulation. J Clin Invest. (2008) 118:3943–53. doi: 10.1172/JCI37156
201. Matsuzaki K. Smad phosphoisoform signals in acute and chronic liver injury: similarities and differences between epithelial and mesenchymal cells. Cell Tissue Res. (2012) 347:225–43. doi: 10.1007/s00441-011-1178-6
202. Matsuzaki K, Murata M, Yoshida K, Sekimoto G, Uemura Y, Sakaida N. Chronic inflammation associated with hepatitis C virus infection perturbs hepatic transforming growth factor β signaling, promoting cirrhosis and hepatocellular carcinoma. Hepatology. (2007) 46:48–57. doi: 10.1002/hep.21672
203. Nagata H, Hatano E, Tada M, Murata M, Kitamura K, Asechi H, et al. Inhibition of c-Jun NH2-terminal kinase switches Smad3 signaling from oncogenesis to tumor- suppression in rat hepatocellular carcinoma. Hepatology. (2009) 49:1944–53. doi: 10.1002/hep.22860
204. Kawamata S, Matsuzaki K, Murata M, Seki T, Matsuoka K, Iwao Y, et al. Oncogenic Smad3 signaling induced by chronic inflammation is an early event in ulcerative colitis-associated carcinogenesis. Inflammatory Bowel Dis. (2011) 17:683–95. doi: 10.1002/ibd.21395
205. Jones PA, Baylin SB. The epigenomics of cancer. Cell. (2007) 128:683–92. doi: 10.1016/j.cell.2007.01.029
206. Brown JD, DiChiara MR, Anderson KR, Gimbrone MA, Topper JN. MEKK-1, a component of the stress (Stress-activated protein kinase/c-jun N-terminal kinase) pathway, can selectively activate smad2-mediated transcriptional activation in endothelial cells. J Biol Chem. (1999) 274:8797–805. doi: 10.1074/jbc.274.13.8797
207. Matsuzaki K, Kitano C, Murata M, Sekimoto G, Yoshida K, Uemura Y, et al. Smad2 and smad3 phosphorylated at both linker and COOH-terminal regions transmit Malignant TGF-β Signal in later stages of human colorectal cancer. Cancer Res. (2009) 69:5321–30. doi: 10.1158/0008-5472.CAN-08-4203
208. Ghosh AK, Vaughan DE. PAI-1 in tissue fibrosis. J Cell Physiol. (2012) 227:493–507. doi: 10.1002/jcp.22783
209. Yoshida K, Matsuzaki K, Murata M, Yamaguchi T, Suwa K, Okazaki K. Clinico-pathological importance of TGF-β/phospho-smad signaling during human hepatic fibrocarcinogenesis. Cancers. (2018) 10:183. doi: 10.3390/cancers10060183
210. Rawal P, Siddiqui H, Hassan M, Choudhary MC, Tripathi DM, Nain V, et al. Endothelial cell-derived TGF-β Promotes epithelial-mesenchymal transition via CD133 in HBx-infected hepatoma cells. Front Oncol. (2019) 9:308. doi: 10.3389/fonc.2019.00308
211. Cui H, Li QL, Chen J, Na Q, Liu CX. Hepatitis B virus X protein modifies invasion, proliferation and the inflammatory response in an HTR-8/SVneo cell model. Oncol Rep. (2015) 34:2090–8. doi: 10.3892/or.2015.4172
212. Arzumanyan A, Friedman T, Kotei E, Ng IOL, Lian Z, Feitelson MA. Epigenetic repression of E-cadherin expression by hepatitis B virus x antigen in liver cancer. Oncogene. (2012) 31:563–72. doi: 10.1038/onc.2011.255
213. Teng C, Chang H, Tsai H, Hsieh W, Kuo Y, Su I, et al. Liver regeneration accelerates hepatitis B virus-related tumorigenesis of hepatocellular carcinoma. Mol Oncol. (2018) 12:1175–87. doi: 10.1002/1878-0261.12318
214. Oshikawa O, Tamura S, Kawata S, Ito N, Tsushima H, Kiso S, et al. The effect of hepatitis B virus X gene expression on response to growth inhibition by transforming growth factor-. Biochem AND Biophys Res Commun. (1996) 222:4. doi: 10.1006/bbrc.1996.0819
215. Zhang Y, Li J, Wang S, Yang F, Zhou Y, Liu Y, et al. HBx−associated long non−coding RNA activated by TGF−β promotes cell invasion and migration by inducing autophagy in primary liver cancer. Int J Oncol. (2019) 56:337–47. doi: 10.3892/ijo.2019.4908
216. shuai DK, Chen Y, Yang G, Liao Z, Zhang H, Liang H, et al. TGF-β1 accelerates the hepatitis B virus X-induced Malignant transformation of hepatic progenitor cells by upregulating miR-199a-3p. Oncogene. (2020) 39:1807–20. doi: 10.1038/s41388-019-1107-9
217. Siddiqui ZI, Azam SA, Khan WH, Afroz M, Farooqui SR, Amir F, et al. An in vitro study on the role of hepatitis B virus X protein C-terminal truncation in liver disease development. Front Genet. (2021) 12:633341. doi: 10.3389/fgene.2021.633341
218. Lee S. C-terminal-truncated HBV X promotes hepato-oncogenesis through inhibition of tumor-suppressive β-catenin/BAMBI signaling. Mol Med. (2016) 10. doi: 10.1038/emm.2016.107
219. Liu Y, Xu Y, Ma H, Wang B, Xu L, Zhang H, et al. Hepatitis B virus X protein amplifies TGF-β promotion on HCC motility through down-regulating PPM1a. Oncotarget. (2016) 7:33125–35. doi: 10.18632/oncotarget.8884
220. Lee DK, Park SH, Yi Y, Choi SG, Lee C, Parks WT, et al. The hepatitis B virus encoded oncoprotein pX amplifies TGF- family signaling through direct interaction with Smad4: potential mechanism of hepatitis B virus-induced liver fibrosis. Genes & Develop. (2001) 12:455–66. doi: 10.1101/gad.856201
221. Shih WL, Kuo ML, Chuang SE, Cheng AL, Doong SL. Hepatitis B virus X protein inhibits transforming growth factor-β-induced apoptosis through the activation of phosphatidylinositol 3-kinase pathway. J Biol Chem. (2000) 275:25858–64. doi: 10.1074/jbc.M003578200
222. Shih WL, Kuo ML, Chuang SE, Cheng AL, Doong SL. Hepatitis B virus X protein activates a survival signaling by linking src to phosphatidylinositol 3-kinase. J Biol Chem. (2003) 278:31807–13. doi: 10.1074/jbc.M302580200
Keywords: HBx protein, hepatitis B virus, TGF-β signaling, hepatocellular carcinoma, tumor suppressor, pro-tumorigenic
Citation: Yan W, Rao D, Fan F, Liang H, Zhang Z and Dong H (2024) Hepatitis B virus X protein and TGF-β: partners in the carcinogenic journey of hepatocellular carcinoma. Front. Oncol. 14:1407434. doi: 10.3389/fonc.2024.1407434
Received: 26 March 2024; Accepted: 21 May 2024;
Published: 19 June 2024.
Edited by:
Zaki A. Sherif, Howard University, United StatesReviewed by:
Masahiko Ito, Hamamatsu University School of Medicine, JapanCopyright © 2024 Yan, Rao, Fan, Liang, Zhang and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huifang Liang, bGlhbmdodWlmYW5nMTk5N0AxMjYuY29t; Zunyi Zhang, enVueWl6aGFuZ3RqbXVAMTYzLmNvbQ==; Hanhua Dong, aGFuaHVhX2RvbmdAaG90bWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.