 Arnaldo Marin
Arnaldo Marin Fernanda Morales
Fernanda Morales Benjamín Walbaum
Benjamín Walbaum
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 08 July 2024
Sec. Cancer Cell Signaling
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1406951
This article is part of the Research Topic FGFR signaling in breast cancer: state of art and future challenges View all 4 articles
Fibroblast Growth Factor Receptors (FGFRs) play a significant role in Estrogen Receptor-positive (ER+) breast cancer by contributing to tumorigenesis and endocrine resistance. This review explores the structure, signaling pathways, and implications of FGFRs, particularly FGFR1, FGFR2, FGFR3, and FGFR4, in ER+ breast cancer. FGFR1 is frequently amplified, especially in aggressive Luminal B-like tumors, and its amplification is associated with poor prognosis and treatment resistance. The co-amplification of FGFR1 with oncogenes like EIF4EBP1 and NSD3 complicates its role as a standalone oncogenic driver. FGFR2 amplification, though less common, is critical in hormone receptor regulation, driving proliferation and treatment resistance. FGFR3 and FGFR4 also contribute to endocrine resistance through various mechanisms, including the activation of alternate signaling pathways like PI3K/AKT/mTOR and RAS/RAF/MEK/ERK. Endocrine resistance remains a major clinical challenge, with around 70% of breast cancers initially hormone receptor positive. Despite the success of CDK 4/6 inhibitors in combination with endocrine therapy (ET), resistance often develops, necessitating new treatment strategies. FGFR inhibitors have shown potential in preclinical studies, but clinical trials have yielded limited success due to off-target toxicities and lack of predictive biomarkers. Current clinical trials, including those evaluating FGFR inhibitors like erdafitinib, lucitanib, and dovitinib, have demonstrated mixed outcomes, underscoring the complexity of FGFR signaling in breast cancer. The interplay between FGFR and other signaling pathways highlights the need for comprehensive molecular profiling and personalized treatment approaches. Future research should focus on identifying robust biomarkers and developing combination therapies to enhance the efficacy of FGFR-targeted treatments. In conclusion, targeting FGFR signaling in ER+ breast cancer presents both challenges and opportunities. A deeper understanding of the molecular mechanisms and resistance pathways is crucial for the successful integration of FGFR inhibitors into clinical practice, aiming to improve outcomes for patients with endocrine-resistant breast cancer.
The human fibroblast growth factor receptor (FGFR) family comprises FGFR1, FGFR2, FGFR3, and FGFR4. These highly conserved receptor tyrosine kinases (RTKs) are in the cell membrane (1, 2). Additionally, FGFR5 (also known as fibroblast growth factor receptor-like 1; FGFRL1), which lacks the intracellular kinase domain, is classified as the fifth member of the FGFR family (3). FGFRs are activated through the interaction of fibroblast growth factor (FGF) ligands. The mammalian FGF family comprises 22 members that are classified into 6 subfamilies by their mechanisms of action. In paracrine signaling (canonical FGFs) there are 5 subfamilies: FGF1 (FGF1 and FGF2), FGF4 (FGF4, FGF5, and FGF6), FGF7 (FGF3, FGF7, FGF10, and FGF22), FGF8 (FGF8, FGF17, and FGF18), and FGF9 (FGF9, FGF16, and FGF20). Furthermore, the endocrine subfamily consists of FGF19 (FGF19, FGF21, and FGF23) (4–6). FGF15 corresponds to the mouse ortholog of human FGF19 (FGF15/FGF19) (7, 8). In paracrine cellular communication, FGFs participate in various cellular processes that include cell proliferation, migration, differentiation and cell survival (9, 10), while endocrine secreted FGFs are released into the blood and can reach different tissues of the body, participating in the regulation of the bile acids, carbohydrates, lipids and phosphate/vitamin D metabolism (11–19).
The structure of FGFR1 – FGFR4 from N- to C-terminus contains an extracellular region comprising three immunoglobulin-like loop domains (Ig domains; DI, DII and DII) (5, 20). Between DI and DII domains is the acid box, a conserved motif rich in aspartic acid residues (21). This is followed by a transmembrane region formed by a single α-helix, a juxtamembrane domain, and two intracellular tyrosine kinase domains (1, 5, 20, 22) FGFR5 has three extracellular domains similar to immunoglobulins, a transmembrane domain and a short intracellular tail without a tyrosine kinase domain that presents a histidine-rich region (23, 24) (Figure 1).
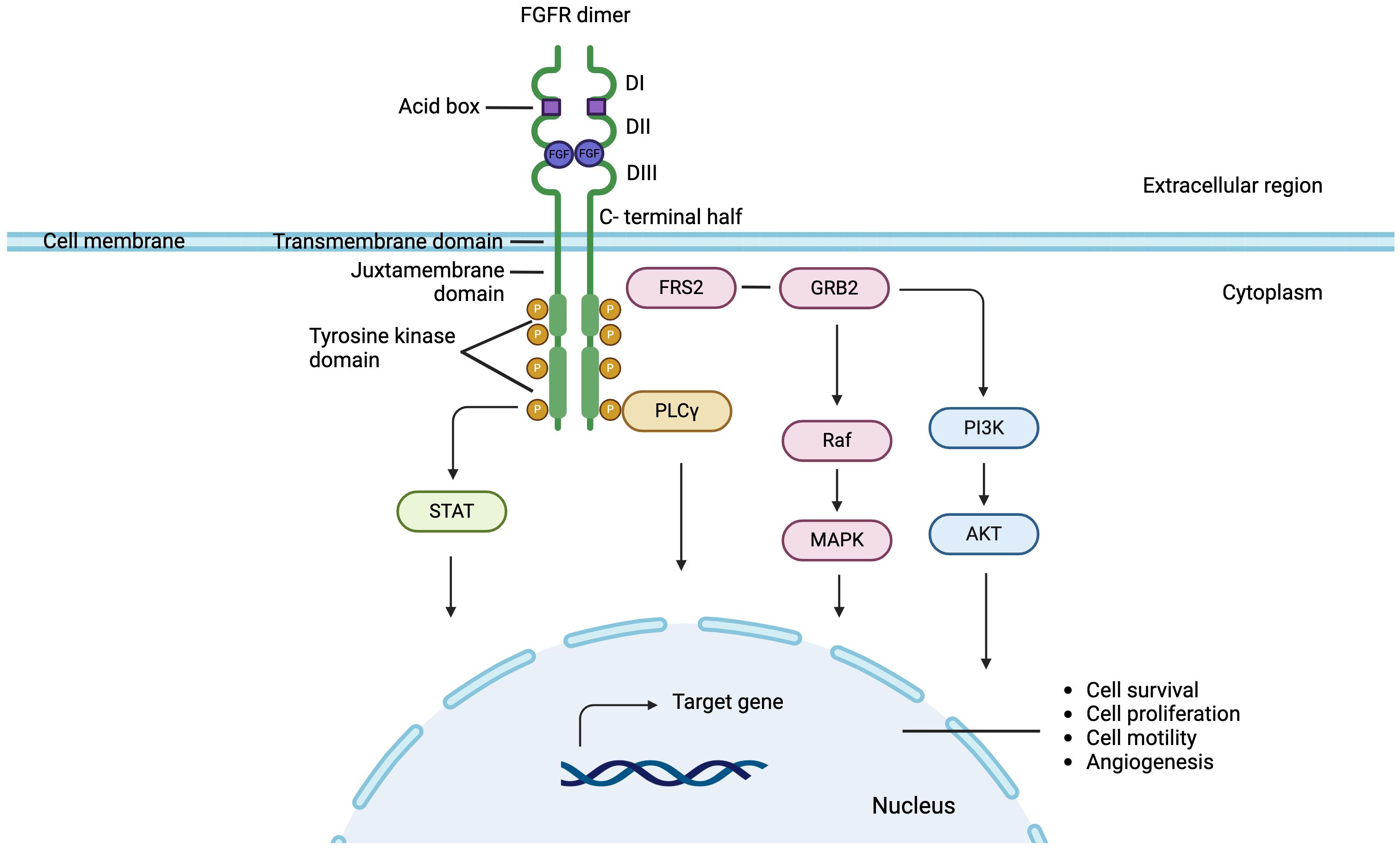
Figure 1 FGFR signaling pathway in breast cancer: FGF binds to the extracellular domain of FGFR leading to receptor dimerization and through phosphorylation inducing downstream activation of canonical pathways. Signaling pathways dependent of FGF-FGFR axis, include Fibroblast Growth Factor Receptor Substrate 2 (FRS2) Ras-mitogen-activated protein kinase (MAPK), phosphatidylinositol-3 kinase (PI3K), protein kinase B (AKT), phospholipase Cγ1 (PLCγ1) and signal transducer and activator of transcription (STAT).
FGFs are molecules that bind to FGFR monomers in their extracellular region between domains II and III (20, 25). Ligand binding induces conformational changes and dimerization of FGFR, which trigger transphosphorylation within the dimerized receptor pair, and therefore the activation of cytosolic kinase domains (20, 26–30). The autophosphorylated domains of FGFRs serve as specific binding sites for different substrates, which in turn are phosphorylated by active receptors (23, 31). Through this mechanism, various downstream effector molecules are activated and participate in the signaling pathways of the FGF-FGFR axis, including Fibroblast Growth Factor Receptor Substrate 2 (FRS2) Ras-mitogen-activated protein kinase (MAPK), phosphatidylinositol-3 kinase (PI3K), protein kinase B (AKT), phospholipase Cγ1 (PLCγ1) and signal transducer and activator of transcription (STAT). FRS2, a protein located in the plasma membrane, is the main substrate of FGFR. Following activation by phosphorylation, FRS2 binds and recruits the adapter protein Growth factor receptor-bound protein 2 (GRB2) triggering the activation of the Ras-MAPK signaling pathway, which regulates the mitogenic cellular response, cell cycle regulation and cell survival (32–37). Also activated FRS2 results in the phosphorylation and recruitment of PI3K, activating the PI3K-AKT signaling cascade involved in cell survival and angiogénesis (38–44). Furthermore, tyrosine residues of FGFR are also recognized by PLCγ1 and STATs, proteins related to cell motility and cell transformation, respectively (40, 45–48) (Figure 1).
Estrogen receptor-positive (ER+) tumors represent the most frequent subtype in breast cancer (BC) across all ages (49) yet unveiling its biology, beyond the estrogenic receptor pathway, has been challenging. Several genomic alterations in both FGFs and FGFRs genes have been reported in BC patients, which vary in their presentation according to BC subtypes and stage (50–52). Among ER+ BC, FGFR signaling pathway has emerged as a relevant factor in tumorigenesis and treatment resistance (53, 54). FGFR1 gene alterations are the most frequently detected, mainly characterized by the amplification of 8p11–12 locus, present in 10 -15% patients and reaching over 16% to 27% in aggressive tumors (Luminal B-like) (55). FGFR1 signaling appears to play a central role in proliferation, migration, and survival of mammary cells and its amplification is a relevant prognostic factor as it has shown to strongly predict poorer overall survival among BC patients (56). However, FGFR1 amplification generally occurs alongside the co-amplification of several neighboring genes, of which EIF4EBP1 and NSD3 have been identified as oncogenic, the former with a proven oncogenic role in luminal BCs (57, 58). Thus, FGFR1 is not the only oncogene present in the 8p11.23 amplicon, which challenges its role as an oncogenic driver and the real dependency or addiction to FGFR1 signaling (59). Moreover, there is a low concordance between FGFR1 amplification and final protein expression (60). While, simultaneously, FGFR1 overexpression can occur independently of gene amplification, as proven in lung cancer studies (61).
Other FGFR alterations have been implicated in ER+ breast cancer. Though infrequent (around 3%) and mainly found in ER-negative BC, FGFR2 amplification plays a predominant role as a driver oncogene. The FGFR2 amplicon is shorter, compared to FGFR1, and includes fewer coamplified genes, thus being more centered on FGFR2, better predicting its oncogenic role. FGFR2 regulates FGFR3 expression, leading to high levels of FGFR3 in FGFR2-activated tumors. Additionally, the origin of FGF2 and its interaction with FGFR2, may determine the cell line context. Either autocrine, with high FGF2 expression seen in basal-like breast cancer cell lines, or paracrine, where it is stromal FGF2 that activates hormone receptors and induces proliferation of luminal cancer cells. Consequently, preclinical studies have shown that FGFR2-amplified cell lines are more sensitive to inhibition compared to those with FGFR1 amplification (62–64).
Furthermore, FGFR4 gene amplification is detected in around 10% of patients and its overexpression tends to be found more frequently in HER2 enriched tumors (65). As opposed to other histologies, point mutations, fusions, nuclear FGFR expression and FGF ligand amplification are initially infrequent in BC, however their clonal acquisition after treatment does play a role in ER+ BC, especially after treatment exposure with endocrine therapy (ET) and Cyclin–dependent kinase 4/6 inhibitors (CDK 4/6 inhibitors) (66, 67).
Around 70% of BC are initially classified as hormone receptor-positive (68), meaning that the estrogen receptor signaling pathway is the main driver of cancer cell growth and tumor survival. Whereby endocrine therapy is the mainstay of its treatment both in early BC and in advanced BC (ABC) (69). Among patients with distant recurrence, however, around 14% of patients are primarily resistant to estrogen deprivation therapy, and the vast majority acquire resistance at some point of their treatment (70, 71). This has led to the development of targeted therapies such as CDK 4/6 inhibitors that in combination with ET have dramatically changed the natural history of this disease, with median progression free survival for first line treatment of ET plus CDK 4/6 inhibitors reaching over 24 months (72). Still, most patients eventually progress due to drug resistance, prompting for changes in the treatment strategy. Yet, further treatment lines are less effective, without one defined subsequent therapy (73). Resistant mechanisms have been extensively studied, and different treatments have been developed to overcome resistance. Estrogen receptor 1 (ESR1) mutation detection, identified as a strong predictor of standard ET resistance, has led to the approval of elacestrant, an oral selective estrogen receptor degrader (SERD) in patients progressing after ET in combination with CDK 4/6 inhibitors (74). Whereas PIK3CA gene mutations, truncal mutations present at initial stages in over 30% of patients, have shown to predict worst response to ET (75), and led to the development of PI3K inhibitors such as alpelisib (76) and the recently presented inavolisib (77). The latter showing interesting results among patients defined as endocrine resistant, due to early relapse con ET. Moreover, inhibitors of the AKT pathway have demonstrated to play a relevant role in ET sequence strategy when either PIK3CA, AKT1 or PTEN mutations are detected (78). This led to the Food and Drug Administration (FDA) approval of capivasertib for ER+/HER2- advanced BC patients following progression on at least one endocrine-based regimen (79). Loss of ER and other alterations, such as mutations in RAS/RAF/MEK pathway, have also been described as relevant resistance mechanisms favoring non estrogen dependent pathways, however drug development targeting this pathway has been elusive in BC (80).
FGFR1 gene amplification has an often-unaccounted role in endocrine resistance. Several resistance mechanisms have been described, including the activation of PI3K/AKT/mTOR pathway, persistent RAS/RAF/MEK/ERK pathway activation and cyclin D1 overexpression (81, 82). Upon inhibition of CDK4/6, AKT signaling is upregulated, which contribute to the accumulation of cyclin D1. This accumulation allows cyclin D1 to associate with CDK2, bypassing CDK4/6 inhibition, and promoting S phase progression. Notably, up to one-third of FGFR1-amplified tumors also harbor amplification of CCND1 gene, which encodes for cyclin D1 protein. Still, FGFR1 signaling promotes cell proliferation by directly upregulating the CCND1 gene, with higher levels of cyclin D1 mRNA observed even among FGFR1-amplified tumors without CCND1 amplification (83). Additionally, FGFR1 induces ET plus CDK 4/6-inhibition resistance by activating cancer cell stemness [(84) 21098263] and by upregulating Wnt/β-catenin signaling (85). ctDNA samples collected after progression on CDK 4/6-inhibitors showed an enrichment of FGFR pathway alterations, while FGFR1 amplifications detected in baseline ctDNA samples of patients included in Monaleesa -2 trial was associated with worse survival outcomes (83).
This has led to a growing body of research exploring the use of FGFR inhibitors, with preclinical data suggesting that FGFR1 amplified BC could maintain ERα pathway activated even with estrogen deprivation therapy (86). Amplification of the chromosomal region 8p11-12, the genomic location of FGFR1, is the most frequent FGFR alteration in BC, furthermore, Formisano et al. (86), measured FGFR1 prior and post preoperative ET and confirmed an increase in FGFR1 levels in post-treatment samples. Suggesting that FGFR1 amplification represents not only an intrinsic resistant mechanism, but also an adaptive mechanism of escape to antiestrogen treatment. Preclinical data have also shown that tumor cells that harbor FGFR1 amplification when treated with ET in combinations with CDK 4/6 inhibitors reach significantly lower levels of cell cycle arrest state (83, 87), with reduced cell senescence (measured by senescence-associated (SA) β-galactosidase-positive cells) (88). Ever more relevant considering the role of senescent cells as elimination route of tumor cells when treated with cytostatics therapies (89). The addition of anti FGFR1 drugs could reverse this condition and has led to an important number of clinical trials evaluating its effectiveness and safety. However, as stated one third of these tumors present other co-occurring pathway altered functions and in up to over 20% of patients there is discordance between FGFR1 amplification and overexpression (81), underscoring the need to better define and quantify FGFR amplification.
Regarding FGFR2, several reports have shown a tight interaction between progesterone receptor (PR), FGFR2 and STAT5 proteins. Wherein stromal FGF2 and progesterone exert overlapping functions by activating common signaling pathways including ERK, AKT and STAT5. Thereby enabling the formation of a nuclear complex between PR, FGFR2 and STAT5. These complexes lead to the expression of PR/STAT-5 regulated proteins, favoring mammary gland differentiation and hormone-mediated growth. This way, among patients under endocrine suppression therapy, FGFR2-altered tumors may become independent of hormonal stimulation by switching to FGF2-mediated STAT5-dependent mechanisms for continuous cell proliferation (90). However, whole exome sequencing analysis of BC patients confirmed that both FGFR2 overexpression and mutations are rarely truncal mutations but acquired in metastatic biopsy after treatment progression (91).
Other FGFR alterations have been shown to promote endocrine resistance, including co-amplification of FGF 3/4/19 and CCND1, nuclear FGFR1 expression, FGFR3 overexpression, FGFR4 overexpression and FGFR4 mutations. However, their clinical relevance has yet to be determined (66, 92–94).
Various agents ranging from pan-FGFR inhibitors and specific FGFR 1, 2 and 3 inhibitors have been developed. With several FGFR inhibitors currently under development. So far for the treatment of solid tumor patients, erdafitinib for urothelial carcinoma (FGFR 2/3 alterations) (95), and futibatinib, infigratinib and pemigatinib for cholangiocarcinoma (FGFR2 fusions or rearrangements) have been approved (96, 97). While in BC, ongoing clinical trials and preclinical data have long been evaluating possible treatment strategies to block FGFR pathway by using small molecules tyrosine kinase inhibitors (TKIs). Notwithstanding, limited benefit has so far been the norm. Several molecules have been tested in different settings. Soria et al. showed promising results in 12 patients with advanced BC with FGFR1 amplified tumor treated with first generation multikinase inhibitor lucitanib (98). Later, in the phase II FINESSE study an exploratory biomarker analysis suggested a higher overall response rate in patients with FGFR1 amplifications defined by IHC H-score ≥50. However, treatment resulted in significant toxicities limiting optimal inhibitory dose (99). More recently, a phase Ib trial including 23 patients with ER+/HER2- FGFR1 amplified metastatic BC previously exposed to CDK 4/6-inhibitors, combined erdafitinib with fulvestrant and palbociclib and showed a median PFS of only 3 months and a clinical benefit rate (CBR: complete response, partial response, or stable disease) of 28% at 6 months (100). While the phase II study EAY131 led by Gong and colleagues, which enrolled 18 patients with tumor FGFR 1-4 amplification to receive erdafitinib and included 9 patients with BC did not meet its primary endpoint, with only one patient with FGFR1-amplified BC showing prolonged survival (101). Additionally, the phase IIA RADICAL trial (102) which included 52 patients, compared the combination of anastrozole or letrozole to fexagratinib (AZD4547), a strong FGFR inhibitor (FGFR-1, 2 and 3), in endocrine resistant BC. Overall, objective response was only 10% (5/50) with a clinical benefit rate (CBR) after 28 weeks of therapy that reached 25%, with almost 27% (14) of patients discontinuing treatment due to adverse events, mainly ocular complications such as corneals ulcers, mucositis, elevated liver enzymes, anemia and fatigue. Two phase II trials have studied the benefit of the Dovitinib, a small molecule multikinase inhibitor of FGFR1, FGFR2, FGFR3 and other receptor tyrosine kinases. Preclinical data showed activity in FGFR positive and amplified BC xenograft models, reversing aromatase inhibitor resistance. Initially, single-agent Dovitinib showed activity in ER+/HER2- FGFR1 amplified BC patients, particularly among patients with higher levels of amplification. However, CBR reached a meager 25% (103). Following these results, the combination with fulvestrant was tested in a double blind, phase II trial, however the study had to be stopped early due to slow accrual. Still, the final analysis, once again showed limited benefit, with no progression free survival difference in the intention to treat analysis, but with a significant difference (according to definition of study protocol) among patients from the predefined subgroup with FGF pathway amplification; reaching a median PFS of 10.9 months vs 5.5 months with fulvestrant as monotherapy and a response rate (ORR: Complete response or partial response) of 27.7% (104). Ongoing trials including a phase II trial evaluating futibatinib plus fulvestrant (NCT04024436) and the phase I dose escalation trial (ROGABREAST) with rogaratinib in combination with palbociclib and fulvestrant (NCT04483505), are yet to show any benefit. Table 1 summarizes completed phase II or III randomized control trials.
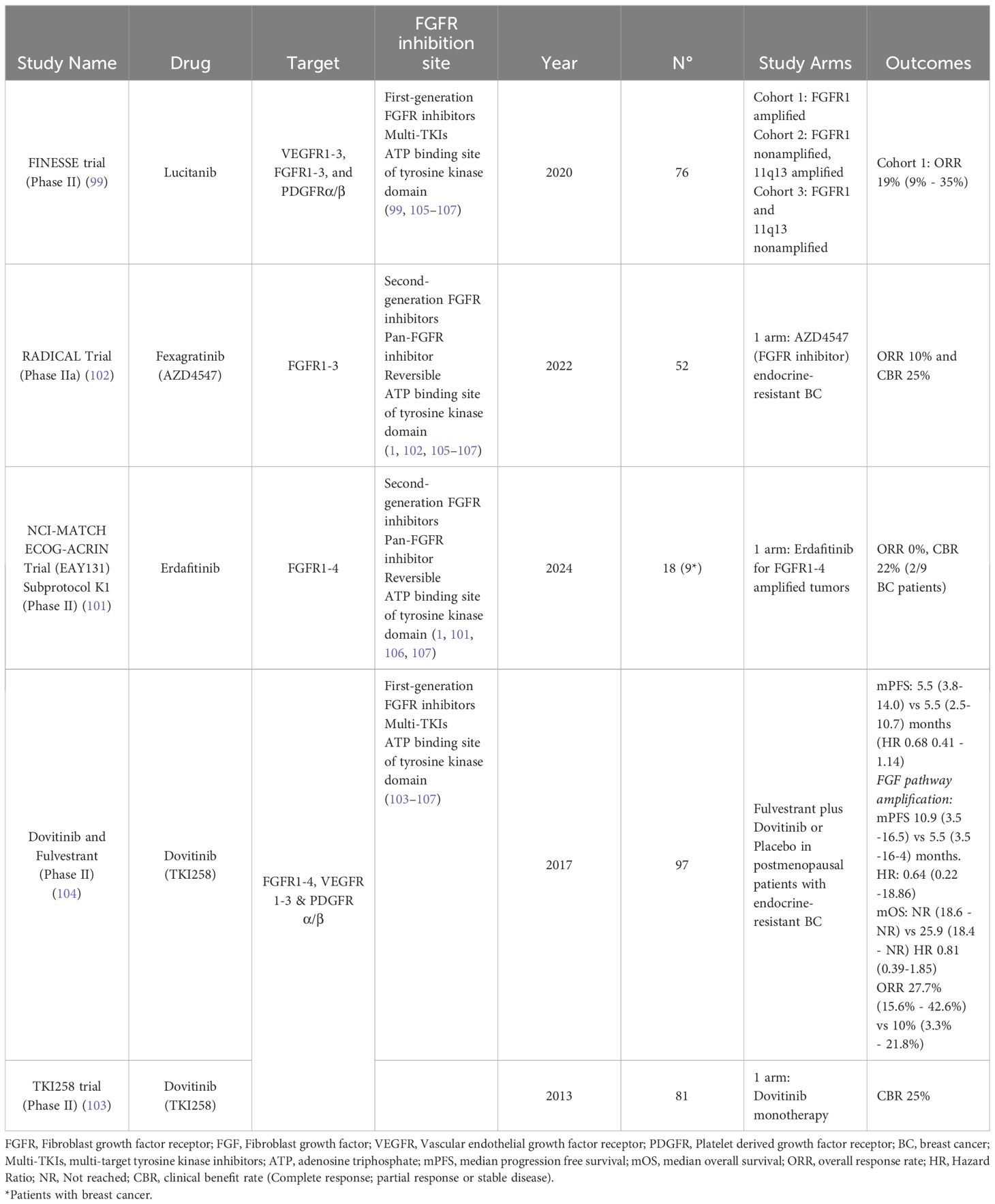
Table 1 Phase II/III trials evaluating anti FGFR treatments in breast cancer patients.
Finally, new strategies are being evaluated to better tailor therapy with FGFR inhibitors by molecularly selecting BC patients that will benefit from treatment. As such, translational research evaluating FGFR1-4 mRNA levels has shown to be a better predictive biomarker (108). Future prospective trials including molecular determinants will help better profile patients with actionable FGFR alterations that will benefit from target therapy.
The FGFR pathway represents an elusive target in the landscape of BC therapy. Particularly in ER+/HER2- BC patients, where resistance to therapy remains a significant clinical challenge. Genomic alterations and dysregulation within the FGFR signaling axis, including FGFR amplifications, fusions and point mutations, have been associated as strong prognostic factors in BC, mainly as intrinsic or acquired resistant mechanisms to endocrine therapies, favoring disease progression and poorer survival outcomes for patients. Despite promising preclinical data evaluating FGFR as a relevant resistant pathway, ongoing clinical trials testing FGFR inhibitors have not been able to translate these findings into meaningful clinical benefits. Challenges such as lack of predictive biomarkers, due to the intricacies of signaling pathways in ER+/HER2- BC, and significant adverse events due to off-target toxicities, have hindered the success of FGFR-targeted therapies in BC (109). Tumor heterogeneity mandates a deeper understanding of tumor biology including genomics analysis and new molecular technologies ranging from transcriptomics to RNA sequencing. Which may help better understand the complex interplay between FGFR signaling and other pathways involved in endocrine resistance (110). Furthermore, exploring innovative drug combinations could enable innovative approaches to overcome resistance mechanisms. Research efforts should focus on elucidating mechanisms of resistance, refining identification of predictive biomarkers, and exploring rational combination therapies to maximize efficacy while minimizing toxicity. Ultimately, the successful integration of FGFR-targeted agents into the clinical management of ER+/HER2- BC should be focused on further improving patient outcomes by overcoming the limitations posed by endocrine resistance.
AM: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. FM: Resources, Visualization, Writing – original draft. BW: Conceptualization, Investigation, Resources, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Dai S, Zhou Z, Chen Z, Xu G, Chen Y. Fibroblast growth factor receptors (FGFRs): structures and small molecule inhibitors. Cells. (2019) 8. doi: 10.3390/cells8060614
2. Santolla MF, Maggiolini M. The FGF/FGFR system in breast cancer: oncogenic features and therapeutic perspectives. Cancers (Basel). (2020) 12(10):3029. doi: 10.3390/cancers12103029
3. Sleeman M, Fraser J, McDonald M, Yuan S, White D, Grandison P, et al. Identification of a new fibroblast growth factor receptor, FGFR5. Gene. (2001) 271:171–82. doi: 10.1016/S0378-1119(01)00518-2
4. Hui Q, Jin Z, Li X, Liu C, Wang X. FGF family: from drug development to clinical application. Int J Mol Sci. (2018) 19. doi: 10.3390/ijms19071875
5. Itoh N, Ornitz DM. Evolution of the Fgf and Fgfr gene families. Trends Genet. (2004) 20:563–9. doi: 10.1016/j.tig.2004.08.007
6. Itoh N, Ornitz DM. Fibroblast growth factors: from molecular evolution to roles in development, metabolism and disease. J Biochem. (2011) 149:121–30. doi: 10.1093/jb/mvq121
7. Hansen AMK, Vienberg SG, Lykkegaard K, Zhao X, Tingqing G, Han D, et al. Differential receptor selectivity of the FGF15/FGF19 orthologues determines distinct metabolic activities in db/db mice. Biochem J. (2018) 475:2985–96. doi: 10.1042/BCJ20180555
8. Farooq M, Khan AW, Kim MS, Choi S. The role of fibroblast growth factor (FGF) signaling in tissue repair and regeneration. Cells. (2021) 10. doi: 10.3390/cells10113242
9. Boilly B, Vercoutter-Edouart AS, Hondermarck H, Nurcombe V, Bourhis X. FGF signals for cell proliferation and migration through different pathways. Cytokine Growth Factor Rev. (2000) 11:295–302. doi: 10.1016/S1359-6101(00)00014-9
10. Laestander C, Engstrom W. Role of fibroblast growth factors in elicitation of cell responses. Cell Prolif. (2014) 47:3–11. doi: 10.1111/cpr.12084
11. Potthoff MJ, Kliewer SA, Mangelsdorf DJ. Endocrine fibroblast growth factors 15/19 and 21: from feast to famine. Genes Dev. (2012) 26:312–24. doi: 10.1101/gad.184788.111
12. White KE, Carn G, Lorenz-Depiereux B, Benet-Pages A, Strom TM, Econs MJ. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. (2001) 60:2079–86. doi: 10.1046/j.1523-1755.2001.00064.x
13. Hu MC, Shiizaki K, Kuro-o M, Moe OW. Fibroblast growth factor 23 and Klotho: physiology and pathophysiology of an endocrine network of mineral metabolism. Annu Rev Physiol. (2013) 75:503–33. doi: 10.1146/annurev-physiol-030212-183727
14. Angelin B, Larsson TE, Rudling M. Circulating fibroblast growth factors as metabolic regulators–a critical appraisal. Cell Metab. (2012) 16:693–705. doi: 10.1016/j.cmet.2012.11.001
15. Beenken A, Mohammadi M. The FGF family: biology, pathophysiology and therapy. Nat Rev Drug Discovery. (2009) 8:235–53. doi: 10.1038/nrd2792
16. Kurosu H, Kuro OM. Endocrine fibroblast growth factors as regulators of metabolic homeostasis. Biofactors. (2009) 35:52–60. doi: 10.1002/biof.12
17. Lin X, Liu YB, Hu H. Metabolic role of fibroblast growth factor 21 in liver, adipose and nervous system tissues. BioMed Rep. (2017) 6:495–502. doi: 10.3892/br.2017.890
18. Wolf M. Update on fibroblast growth factor 23 in chronic kidney disease. Kidney Int. (2012) 82:737–47. doi: 10.1038/ki.2012.176
19. Kir S, Kliewer SA, Mangelsdorf DJ. Roles of FGF19 in liver metabolism. Cold Spring Harb Symp Quant Biol. (2011) 76:139–44. doi: 10.1101/sqb.2011.76.010710
20. Farrell B, Breeze AL. Structure, activation and dysregulation of fibroblast growth factor receptor kinases: perspectives for clinical targeting. Biochem Soc Trans. (2018) 46:1753–70. doi: 10.1042/BST20180004
21. Sanchez-Heras E, Howell FV, Williams G, Doherty P. The fibroblast growth factor receptor acid box is essential for interactions with N-cadherin and all of the major isoforms of neural cell adhesion molecule. J Biol Chem. (2006) 281:35208–16. doi: 10.1074/jbc.M608655200
22. Tucker JA, Klein T, Breed J, Breeze AL, Overman R, Phillips C, et al. Structural insights into FGFR kinase isoform selectivity: diverse binding modes of AZD4547 and ponatinib in complex with FGFR1 and FGFR4. Structure. (2014) 22:1764–74. doi: 10.1016/j.str.2014.09.019
23. Ornitz DM, Itoh N. The Fibroblast Growth Factor signaling pathway. Wiley Interdiscip Rev Dev Biol. (2015) 4:215–66. doi: 10.1002/wdev.176
24. Regeenes R, Silva PN, Chang HH, Arany EJ, Shukalyuk AI, Audet J, et al. Fibroblast growth factor receptor 5 (FGFR5) is a co-receptor for FGFR1 that is up-regulated in beta-cells by cytokine-induced inflammation. J Biol Chem. (2018) 293:17218–28. doi: 10.1074/jbc.RA118.003036
25. Wang F, Kan M, Xu J, Yan G, McKeehan WL. Ligand-specific structural domains in the fibroblast growth factor receptor. J Biol Chem. (1995) 270:10222–30. doi: 10.1074/jbc.270.17.10222
26. Schlessinger J, Plotnikov AN, Ibrahimi OA, Eliseenkova AV, Yeh BK, Yayon A, et al. Crystal structure of a ternary FGF-FGFR-heparin complex reveals a dual role for heparin in FGFR binding and dimerization. Mol Cell. (2000) 6:743–50. doi: 10.1016/S1097-2765(00)00073-3
27. Furdui CM, Lew ED, Schlessinger J, Anderson KS. Autophosphorylation of FGFR1 kinase is mediated by a sequential and precisely ordered reaction. Mol Cell. (2006) 21:711–7. doi: 10.1016/j.molcel.2006.01.022
28. Chen H, Xu CF, Ma J, Eliseenkova AV, Li W, Pollock PM, et al. A crystallographic snapshot of tyrosine trans-phosphorylation in action. Proc Natl Acad Sci U.S.A. (2008) 105:19660–5.
29. Bae JH, Boggon TJ, Tome F, Mandiyan V, Lax I, Schlessinger J. Asymmetric receptor contact is required for tyrosine autophosphorylation of fibroblast growth factor receptor in living cells. Proc Natl Acad Sci U.S.A. (2010) 107:2866–71.
30. Bae JH, Schlessinger J. Asymmetric tyrosine kinase arrangements in activation or autophosphorylation of receptor tyrosine kinases. Mol Cells. (2010) 29:443–8. doi: 10.1007/s10059-010-0080-5
31. Xie Y, Su N, Yang J, Tan Q, Huang S, Jin M, et al. FGF/FGFR signaling in health and disease. Signal Transduct Target Ther. (2020) 5:181. doi: 10.1038/s41392-020-00222-7
32. Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, et al. A lipid-anchored Grb2-binding protein that links FGF-receptor activation to the Ras/MAPK signaling pathway. Cell. (1997) 89:693–702. doi: 10.1016/S0092-8674(00)80252-4
33. Hadari YR, Gotoh N, Kouhara H, Lax I, Schlessinger J. Critical role for the docking-protein FRS2 alpha in FGF receptor-mediated signal transduction pathways. Proc Natl Acad Sci U.S.A. (2001) 98:8578–83.
34. Molina JR, Adjei AA. The ras/raf/MAPK pathway. J Thorac Oncol. (2006) 1:7–9. doi: 10.1016/S1556-0864(15)31506-9
35. Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. (1999) 286:1358–62. doi: 10.1126/science.286.5443.1358
36. Bahar ME, Kim HJ, Kim DR. Targeting the RAS/RAF/MAPK pathway for cancer therapy: from mechanism to clinical studies. Signal Transduct Target Ther. (2023) 8:455. doi: 10.1038/s41392-023-01705-z
37. Dailey L, Ambrosetti D, Mansukhani A, Basilico C. Mechanisms underlying differential responses to FGF signaling. Cytokine Growth Factor Rev. (2005) 16:233–47. doi: 10.1016/j.cytogfr.2005.01.007
38. Ong SH, Hadari YR, Gotoh N, Guy GR, Schlessinger J, Lax I. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc Natl Acad Sci U.S.A. (2001) 98:6074–9.
39. Okada T, Enkhjargal B, Travis ZD, Ocak U, Tang J, Suzuki H, et al. FGF-2 attenuates neuronal apoptosis via FGFR3/PI3k/akt signaling pathway after subarachnoid hemorrhage. Mol Neurobiol. (2019) 56:8203–19. doi: 10.1007/s12035-019-01668-9
40. Goetz R, Mohammadi M. Exploring mechanisms of FGF signaling through the lens of structural biology. Nat Rev Mol Cell Biol. (2013) 14:166–80. doi: 10.1038/nrm3528
41. Turner N, Grose R. Fibroblast growth factor signaling: from development to cancer. Nat Rev Cancer. (2010) 10:116–29. doi: 10.1038/nrc2780
42. Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. (2005) 24:7455–64. doi: 10.1038/sj.onc.1209085
43. Forough R, Weylie B, Patel C, Ambrus S, Singh US, Zhu J. Role of AKT/PKB signaling in fibroblast growth factor-1 (FGF-1)-induced angiogenesis in the chicken chorioallantoic membrane (CAM). J Cell Biochem. (2005) 94:109–16. doi: 10.1002/jcb.20274
44. Weylie B, Zhu J, Singh U, Ambrus S, Forough R. Phosphatidylinositide 3-kinase is important in late-stage fibroblast growth factor-1-mediated angiogenesis in vivo. J Vasc Res. (2006) 43:61–9. doi: 10.1159/000089492
45. Peters KG, Marie J, Wilson E, Ives HE, Escobedo J, Del Rosario M, et al. Point mutation of an FGF receptor abolishes phosphatidylinositol turnover and Ca2+ flux but not mitogenesis. Nature. (1992) 358:678–81. doi: 10.1038/358678a0
46. Mohammadi M, Dionne CA, Li W, Li N, Spivak T, Honegger AM, et al. Point mutation in FGF receptor eliminates phosphatidylinositol hydrolysis without affecting mitogenesis. Nature. (1992) 358:681–4. doi: 10.1038/358681a0
47. Bottcher RT, Niehrs C. Fibroblast growth factor signaling during early vertebrate development. Endocr Rev. (2005) 26:63–77. doi: 10.1210/er.2003-0040
48. Hart KC, Robertson SC, Kanemitsu MY, Meyer AN, Tynan JA, Donoghue DJ. Transformation and Stat activation by derivatives of FGFR1, FGFR3, and FGFR4. Oncogene. (2000) 19:3309–20. doi: 10.1038/sj.onc.1203650
49. Loibl S, Poortmans P, Morrow M, Denkert C, Curigliano G. Breast cancer. Lancet. (2021) 397:1750–69. doi: 10.1016/S0140-6736(20)32381-3
50. Adnane J, Gaudray P, Dionne CA, Crumley G, Jaye M, Schlessinger J, et al. two receptors to members of the FGF family, are amplified in subsets of human breast cancers. Oncogene. (1991) 6:659–63.
51. Jacquemier J, Adelaide J, Parc P, Penault-Llorca F, Planche J, deLapeyriere O, et al. Expression of the FGFR1 gene in human breast-carcinoma cells. Int J Cancer. (1994) 59:373–8. doi: 10.1002/ijc.2910590314
52. Elbauomy Elsheikh S, Green AR, Lambros MB, Turner NC, Grainge MJ, Powe D, et al. FGFR1 amplification in breast carcinomas: a chromogenic in situ hybridization analysis. Breast Cancer Res. (2007) 9:R23.
53. Karlsson E, Waltersson MA, Bostner J, Perez-Tenorio G, Olsson B, Hallbeck AL, et al. High-resolution genomic analysis of the 11q13 amplicon in breast cancers identifies synergy with 8p12 amplification, involving the mTOR targets S6K2 and 4EBP1. Genes Chromosomes Cancer. (2011) 50:775–87. doi: 10.1002/gcc.20900
54. Katoh M. Genetic alterations of FGF receptors: an emerging field in clinical cancer diagnostics and therapeutics. Expert Rev Anticancer Ther. (2010) 10:1375–9. doi: 10.1586/era.10.128
55. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery. (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
56. Drago JZ, Formisano L, Juric D, Niemierko A, Servetto A, Wander SA, et al. FGFR1 amplification mediates endocrine resistance but retains TORC sensitivity in metastatic hormone receptor-positive (HR(+)) breast cancer. Clin Cancer Res. (2019) 25:6443–51. doi: 10.1158/1078-0432.CCR-19-0138
57. Rutkovsky AC, Yeh ES, Guest ST, Findlay VJ, Muise-Helmericks RC, Armeson K, et al. Eukaryotic initiation factor 4E-binding protein as an oncogene in breast cancer. BMC Cancer. (2019) 19:491. doi: 10.1186/s12885-019-5667-4
58. Yuan G, Flores NM, Hausmann S, Lofgren SM, Kharchenko V, Angulo-Ibanez M, et al. Elevated NSD3 histone methylation activity drives squamous cell lung cancer. Nature. (2021) 590:504–8. doi: 10.1038/s41586-020-03170-y
59. Condorelli R, Mosele F, Verret B, Bachelot T, Bedard PL, Cortes J, et al. Genomic alterations in breast cancer: level of evidence for actionability according to ESMO Scale for Clinical Actionability of molecular Targets (ESCAT). Ann Oncol. (2019) 30:365–73. doi: 10.1093/annonc/mdz036
60. Shiang CY, Qi Y, Wang B, Lazar V, Wang J, Fraser Symmans W, et al. Amplification of fibroblast growth factor receptor-1 in breast cancer and the effects of brivanib alaninate. Breast Cancer Res Treat. (2010) 123:747–55. doi: 10.1007/s10549-009-0677-6
61. Ng TL, Yu H, Smith DE, Boyle TA, York ER, Leedy S, et al. Preselection of lung cancer cases using FGFR1 mRNA and gene copy number for treatment with ponatinib. Clin Lung Cancer. (2019) 20:e39–51. doi: 10.1016/j.cllc.2018.09.001
62. Cerliani JP, Vanzulli SI, Pinero CP, Bottino MC, Sahores A, Nunez M, et al. Associated expressions of FGFR-2 and FGFR-3: from mouse mammary gland physiology to human breast cancer. Breast Cancer Res Treat. (2012) 133:997–1008. doi: 10.1007/s10549-011-1883-6
63. Babina IS, Turner NC. Advances and challenges in targeting FGFR signaling in cancer. Nat Rev Cancer. (2017) 17:318–32. doi: 10.1038/nrc.2017.8
64. Turner N, Lambros MB, Horlings HM, Pearson A, Sharpe R, Natrajan R, et al. Integrative molecular profiling of triple negative breast cancers identifies amplicon drivers and potential therapeutic targets. Oncogene. (2010) 29:2013–23. doi: 10.1038/onc.2009.489
65. Penault-Llorca F, Bertucci F, Adelaide J, Parc P, Coulier F, Jacquemier J, et al. Expression of FGF and FGF receptor genes in human breast cancer. Int J Cancer. (1995) 61:170–6. doi: 10.1002/ijc.2910610205
66. Tomlinson DC, Knowles MA, Speirs V. Mechanisms of FGFR3 actions in endocrine resistant breast cancer. Int J Cancer. (2012) 130:2857–66. doi: 10.1002/ijc.26304
67. Rani A, Stebbing J, Giamas G, Murphy J. Endocrine resistance in hormone receptor positive breast cancer-from mechanism to therapy. Front Endocrinol (Lausanne). (2019) 10:245. doi: 10.3389/fendo.2019.00245
68. Giaquinto AN, Sung H, Miller KD, Kramer JL, Newman LA, Minihan A, et al. Breast cancer statistics, 2022. CA Cancer J Clin. (2022) 72:524–41. doi: 10.3322/caac.21754
69. Harbeck N, Penault-Llorca F, Cortes J, Gnant M, Houssami N, Poortmans P, et al. Breast cancer. Nat Rev Dis Primers. (2019) 5:66. doi: 10.1038/s41572-019-0111-2
70. Lambertini M, Blondeaux E, Bisagni G, Mura S, De Placido S, De Laurentiis M, et al. Prognostic and clinical impact of the endocrine resistance/sensitivity classification according to international consensus guidelines for advanced breast cancer: an individual patient-level analysis from the Mammella InterGruppo (MIG) and Gruppo Italiano Mammella (GIM) studies. EClinicalMedicine. (2023) 59:101931. doi: 10.1016/j.eclinm.2023.101931
71. Miller TW. Endocrine resistance: what do we know? Am Soc Clin Oncol Educ Book. (2013). doi: 10.1200/EdBook_AM.2013.33.e37
72. Schettini F, Giudici F, Giuliano M, Cristofanilli M, Arpino G, Mastro LD, et al. Overall survival of CDK4/6-inhibitor-based treatments in clinically relevant subgroups of metastatic breast cancer: systematic review and meta-analysis. J Natl Cancer Inst. (2020) 112:1089–97. doi: 10.1093/jnci/djaa071
73. Jackson EB, Chia SKL. Sequencing of endocrine and targeted therapies in hormone-sensitive, human epidermal growth factor receptor 2-negative advanced breast cancer. J Clin Oncol. (2023) 41:3976–83. doi: 10.1200/JCO.23.00759
74. Bidard FC, Kaklamani VG, Neven P, Streich G, Montero AJ, Forget F, et al. Elacestrant (oral selective estrogen receptor degrader) Versus Standard Endocrine Therapy for Estrogen Receptor-Positive, Human Epidermal Growth Factor Receptor 2-Negative Advanced Breast Cancer: Results From the Randomized Phase III EMERALD Trial. J Clin Oncol. (2022) 40:3246–56. doi: 10.1200/JCO.22.00338
75. Mosele F, Stefanovska B, Lusque A, Tran Dien A, Garberis I, Droin N, et al. Outcome and molecular landscape of patients with PIK3CA-mutated metastatic breast cancer. Ann Oncol. (2020) 31:377–86. doi: 10.1016/j.annonc.2019.11.006
76. Andre F, Ciruelos EM, Juric D, Loibl S, Campone M, Mayer IA, et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: final overall survival results from SOLAR-1. Ann Oncol. (2021) 32:208–17.
77. Juric D, Kalinsky K, Im S-A, Ciruelos EM, Bianchini G, Barrios CH, et al. INAVO121: Phase III study of inavolisib (INAVO) + fulvestrant (FUL) vs. alpelisib (ALP) + FUL in patients (pts) with hormone receptor-positive, HER2-negative (HR+, HER2–) PIK3CA-mutated (mut) locally advanced or metastatic breast cancer (LA/mBC). J Clin Oncol. (2023) 41:TPS1123–TPS1123.
78. Turner NC, Oliveira M, Howell SJ, Dalenc F, Cortes J, Gomez Moreno HL, et al. Capivasertib in hormone receptor-positive advanced breast cancer. N Engl J Med. (2023) 388:2058–70. doi: 10.1056/NEJMoa2214131
79. Nierengarten MB. FDA approves capivasertib with fulvestrant for breast cancer. Cancer. (2024) 130:835–6. doi: 10.1002/cncr.35238
80. Rocca A, Braga L, Volpe MC, Maiocchi S, Generali D. The predictive and prognostic role of RAS-RAF-MEK-ERK pathway alterations in breast cancer: revision of the literature and comparison with the analysis of cancer genomic datasets. Cancers (Basel). (2022) 14(21):5306. doi: 10.3390/cancers14215306
81. Mouron S, Manso L, Caleiras E, Rodriguez-Peralto JL, Rueda OM, Caldas C, et al. FGFR1 amplification or overexpression and hormonal resistance in luminal breast cancer: rationale for a triple blockade of ER, CDK4/6, and FGFR1. Breast Cancer Res. (2021) 23:21. doi: 10.1186/s13058-021-01398-8
82. Servetto A, Formisano L, Arteaga CL. FGFR signaling and endocrine resistance in breast cancer: Challenges for the clinical development of FGFR inhibitors. Biochim Biophys Acta Rev Cancer. (2021) 1876:188595. doi: 10.1016/j.bbcan.2021.188595
83. Formisano L, Lu Y, Servetto A, Hanker AB, Jansen VM, Bauer JA, et al. Aberrant FGFR signaling mediates resistance to CDK4/6 inhibitors in ER+ breast cancer. Nat Commun. (2019) 10:1373. doi: 10.1038/s41467-019-09068-2
84. Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci U.S.A. (2010) 107:21737–42.
85. Cheng Q, Ma Z, Shi Y, Parris AB, Kong L, Yang X. FGFR1 overexpression induces cancer cell stemness and enhanced akt/erk-ER signaling to promote palbociclib resistance in luminal A breast cancer cells. Cells. (2021) 10. doi: 10.3390/cells10113008
86. Formisano L, Stauffer KM, Young CD, Bhola NE, Guerrero-Zotano AL, Jansen VM, et al. Association of FGFR1 with ERalpha maintains ligand-independent ER transcription and mediates resistance to estrogen deprivation in ER(+) breast cancer. Clin Cancer Res. (2017) 23:6138–50. doi: 10.1158/1078-0432.CCR-17-1232
87. Rampioni Vinciguerra GL, Sonego M, Segatto I, Dall’Acqua A, Vecchione A, Baldassarre G, et al. CDK4/6 inhibitors in combination therapies: better in company than alone: A mini review. Front Oncol. (2022) 12:891580. doi: 10.3389/fonc.2022.891580
88. Valieva Y, Ivanova E, Fayzullin A, Kurkov A, Igrunkova A. Senescence-associated beta-galactosidase detection in pathology. Diagnostics (Basel). (2022) 12.
89. Piskorz WM, Cechowska-Pasko M. Senescence of tumor cells in anticancer therapy-beneficial and detrimental effects. Int J Mol Sci. (2022) 23. doi: 10.3390/ijms231911082
90. Cerliani JP, Guillardoy T, Giulianelli S, Vaque JP, Gutkind JS, Vanzulli SI, et al. Interaction between FGFR-2, STAT5, and progesterone receptors in breast cancer. Cancer Res. (2011) 71:3720–31. doi: 10.1158/0008-5472.CAN-10-3074
91. Mao P, Cohen O, Kowalski KJ, Kusiel JG, Buendia-Buendia JE, Cuoco MS, et al. Acquired FGFR and FGF alterations confer resistance to estrogen receptor (ER) targeted therapy in ER(+) metastatic breast cancer. Clin Cancer Res. (2020) 26:5974–89. doi: 10.1158/1078-0432.CCR-19-3958
92. Servetto A, Kollipara R, Formisano L, Lin CC, Lee KM, Sudhan DR, et al. Nuclear FGFR1 regulates gene transcription and promotes antiestrogen resistance in ER(+) breast cancer. Clin Cancer Res. (2021) 27:4379–96. doi: 10.1158/1078-0432.CCR-20-3905
93. Du S, Zhang Y, Xu J. Current progress in cancer treatment by targeting FGFR signaling. Cancer Biol Med. (2023) 20:490–9. doi: 10.20892/j.issn.2095-3941.2023.0137
94. Meijer D, Sieuwerts AM, Look MP, van Agthoven T, Foekens JA, Dorssers LC. Fibroblast growth factor receptor 4 predicts failure on tamoxifen therapy in patients with recurrent breast cancer. Endocr Relat Cancer. (2008) 15:101–11. doi: 10.1677/ERC-07-0080
95. Loriot Y, Matsubara N, Park SH, Huddart RA, Burgess EF, Houede N, et al. Erdafitinib or chemotherapy in advanced or metastatic urothelial carcinoma. N Engl J Med. (2023) 389:1961–71. doi: 10.1056/NEJMoa2308849
96. Goyal L, Meric-Bernstam F, Hollebecque A, Valle JW, Morizane C, Karasic TB, et al. Futibatinib for FGFR2-rearranged intrahepatic cholangiocarcinoma. N Engl J Med. (2023) 388:228–39. doi: 10.1056/NEJMoa2206834
97. Abou-Alfa GK, Sahai V, Hollebecque A, Vaccaro G, Melisi D, Al-Rajabi R, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicenter, open-label, phase 2 study. Lancet Oncol. (2020) 21:671–84. doi: 10.1016/S1470-2045(20)30109-1
98. Soria JC, DeBraud F, Bahleda R, Adamo B, Andre F, Dientsmann R, et al. Phase I/IIa study evaluating the safety, efficacy, pharmacokinetics, and pharmacodynamics of lucitanib in advanced solid tumors. Ann Oncol. (2014) 25:2244–51. doi: 10.1093/annonc/mdu390
99. Hui R, Pearson A, Cortes J, Campbell C, Poirot C, Azim HA Jr., et al. Lucitanib for the treatment of HR(+)/HER2(-) metastatic breast cancer: results from the multicohort phase II FINESSE study. Clin Cancer Res. (2020) 26:354–63. doi: 10.1158/1078-0432.CCR-19-1164
100. Mayer IA, Haley BB, Abramson VG, Brufsky A, Rexer B, Stringer-Reasor E, et al. Abstract PD1-03: A phase Ib trial of fulvestrant + CDK4/6 inhibitor (CDK4/6i) palbociclib + pan-FGFR tyrosine kinase inhibitor (TKI) erdafitinib in FGFR-amplified/ER+/HER2-negative metastatic breast cancer (MBC). Cancer Res. (2021) 81:PD1–03-PD1-03. doi: 10.1158/1538-7445.SABCS20-PD1-03
101. Gong J, Mita AC, Wei Z, Cheng HH, Mitchell EP, Wright JJ, et al. Phase II study of erdafitinib in patients with tumors with FGFR amplifications: results from the NCI-MATCH ECOG-ACRIN trial (EAY131) subprotocol K1. JCO Precis Oncol. (2024) 8:e2300406. doi: 10.1200/PO.23.00406
102. Coombes RC, Badman PD, Lozano-Kuehne JP, Liu X, Macpherson IR, Zubairi I, et al. Results of the phase IIa RADICAL trial of the FGFR inhibitor AZD4547 in endocrine resistant breast cancer. Nat Commun. (2022) 13:3246. doi: 10.1038/s41467-022-30666-0
103. Andre F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, et al. Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res. (2013) 19:3693–702. doi: 10.1158/1078-0432.CCR-13-0190
104. Musolino A, Campone M, Neven P, Denduluri N, Barrios CH, Cortes J, et al. Phase II, randomized, placebo-controlled study of dovitinib in combination with fulvestrant in postmenopausal patients with HR(+), HER2(-) breast cancer that had progressed during or after prior endocrine therapy. Breast Cancer Res. (2017) 19:18. doi: 10.1186/s13058-017-0807-8
105. Zheng J, Zhang W, Li L, He Y, Wei Y, Dang Y, et al. Signaling pathway and small-molecule drug discovery of FGFR: A comprehensive review. Front Chem. (2022) 10:860985. doi: 10.3389/fchem.2022.860985
106. Shan KS, Dalal S, Thaw Dar NN, McLish O, Salzberg M, Pico BA. Molecular targeting of the fibroblast growth factor receptor pathway across various cancers. Int J Mol Sci. (2024) 25(2):849. doi: 10.3390/ijms25020849
107. Chen L, Zhang Y, Yin L, Cai B, Huang P, Li X, et al. Fibroblast growth factor receptor fusions in cancer: opportunities and challenges. J Exp Clin Cancer Res. (2021) 40:345. doi: 10.1186/s13046-021-02156-6
108. Sanchez-Guixe M, Hierro C, Jimenez J, Viaplana C, Villacampa G, Monelli E, et al. High FGFR1-4 mRNA expression levels correlate with response to selective FGFR inhibitors in breast cancer. Clin Cancer Res. (2022) 28:137–49.
109. Katoh M, Loriot Y, Brandi G, Tavolari S, Wainberg ZA, Katoh M. FGFR-targeted therapeutics: clinical activity, mechanisms of resistance and new directions. Nat Rev Clin Oncol. (2024) 21:312–29. doi: 10.1038/s41571-024-00869-z
Keywords: endocrine resistance, FGFR signaling, breast cancer, ER positive, TKI - tyrosine kinase inhibitor
Citation: Marin A, Morales F and Walbaum B (2024) Fibroblast growth factor receptor signaling in estrogen receptor-positive breast cancer: mechanisms and role in endocrine resistance. Front. Oncol. 14:1406951. doi: 10.3389/fonc.2024.1406951
Received: 25 March 2024; Accepted: 25 June 2024;
Published: 08 July 2024.
Edited by:
Fabiana Napolitano, University of Texas Southwestern Medical Center, United StatesReviewed by:
Mohammad Zahid Kamran, St. Jude Children’s Research Hospital, United StatesCopyright © 2024 Marin, Morales and Walbaum. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arnaldo Marin, YXJuYWxkby5tYXJpbkB1Y2hpbGUuY2w=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.