 Oluwaseun Adebayo Bamodu1,2*
Oluwaseun Adebayo Bamodu1,2* Chen-Chih Chung3,4,5
Chen-Chih Chung3,4,5 Thomas R. Pisanic II6,7,8
Thomas R. Pisanic II6,7,8 Alexander T. H. Wu9,10,11,12
Alexander T. H. Wu9,10,11,12- 1Directorate of Postgraduate Studies, School of Clinical Medicine, Muhimbili University of Health and Allied Sciences, Dar es Salaam, Tanzania
- 2Ocean Road Cancer Institute, Dar es Salaam, Tanzania
- 3Department of Neurology, Taipei Medical University - Shuang Ho Hospital, New Taipei City, Taiwan
- 4Department of Neurology, School of Medicine, College of Medicine, Taipei Medical University, Taipei, Taiwan
- 5Taipei Neuroscience Institute, Taipei Medical University - Shuang Ho Hospital, New Taipei City, Taiwan
- 6Johns Hopkins Institute for NanoBioTechnology, Baltimore, MD, United States
- 7Sidney Kimmel Comprehensive Cancer Center, The Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 8Department of Oncology - Cancer Genetics and Epigenetics, Johns Hopkins University, Baltimore, MD, United States
- 9The Program for Translational Medicine, Graduate Institute of Biomedical Informatics, College of Medical Science and Technology, Taipei Medical University, Taipei, Taiwan
- 10TMU Research Center of Cancer Translational Medicine, Taipei Medical University, Taipei, Taiwan
- 11Clinical Research Center, Taipei Medical University Hospital, Taipei Medical University, Taipei, Taiwan
- 12Graduate Institute of Medical Sciences, National Defense Medical Center, Taipei, Taiwan
Background: Cancer stem cells (CSCs) have emerged as pivotal players in tumorigenesis, disease progression, and resistance to therapies.
Objective: This comprehensive review delves into the intricate relationship between CSCs and the cell-of-origin in diverse cancer types.
Design: Comprehensive review of thematically-relevant literature.
Methods: We explore the underlying molecular mechanisms that drive the conversion of normal cells into CSCs and the impact of the cell-of-origin on CSC properties, tumor initiation, and therapeutic responses. Moreover, we discuss potential therapeutic interventions targeting CSCs based on their distinct cell-of-origin characteristics.
Results: Accruing evidence suggest that the cell-of-origin, the cell type from which the tumor originates, plays a crucial role in determining the properties of CSCs and their contribution to tumor heterogeneity.
Conclusion: By providing critical insights into the complex interplay between CSCs and their cellular origins, this article aims to enhance our understanding of cancer biology and pave the way for more effective and personalized cancer treatments.
Introduction
Cancer is a complex and multifactorial disease characterized by uncontrolled cell proliferation and invasion (1). The concept of cancer stem cells (CSCs) has revolutionized our understanding of tumor initiation, progression, and therapeutic resistance. CSCs possess self-renewal and differentiation capabilities, contributing to tumor heterogeneity and therapeutic resistance (2, 3). Recent studies suggest the cellular origin of cancer impacts CSC properties and behavior (4, 5). This review explores the relationship between CSCs and cell-of-origin across cancer types. We analyze mechanisms driving CSC formation, examine how cell-of-origin influences CSCs and tumor progression, discuss implications for cancer therapy, and suggest future research directions to enhance understanding of this complex interplay.
The notion of CSCs challenges the traditional view of cancer as a homogeneous mass of rapidly dividing cells, and instead, emphasizes the presence of a hierarchical organization within tumors (6). According to this model, CSCs serve as the “seeds” of the tumor, while the non-tumorigenic, differentiated cancer cells constitute the “bulk” or “bulk tumor” (7). This hierarchical organization implies that the eradication of CSCs is crucial for achieving long-term tumor remission and preventing relapse, as CSCs have the capacity to regenerate and drive tumor regrowth even after initial therapy-induced tumor regression (8).
Notably, recent research has illuminated a fascinating and nuanced relationship between the cellular origin of cancer and the properties of CSCs. The cell-of-origin refers to the normal cell type that accumulates the initiating mutations and transforms into a cancerous cell (9). Evidence suggests that the cell-of-origin exerts a profound influence on CSC behavior, dictating their characteristics, tumorigenic potential, and response to therapeutic interventions (10, 11).
In conclusion, the relationship between CSCs and the cell-of-origin represents a compelling and intricate area of cancer research. Understanding the impact of the cellular origin of cancer on CSC phenotypes has the potential to unlock novel therapeutic avenues and advance precision medicine in oncology. By elucidating the molecular and cellular underpinnings of this relationship, we aim to contribute significantly to the growing body of knowledge in cancer biology and ultimately pave the way for more effective and personalized cancer treatments.
Cancer stem cells origin and properties
CSCs, a small subset of tumor cells possessing stem cell properties, including self-renewal and multipotent differentiation capacity, are the root cause of tumor initiation, therapeutic resistance, metastasis, and relapse (12, 13). Though functionally defined by their tumor-propagating ability, their cell-of-origin and relationship to normal stem cells is debated (14).
CSCs were initially thought to arise from normal tissue stem cells that accumulate mutations enabling aberrant self-renewal (15). Evidence now suggests that more committed progenitors or differentiated cells may acquire self-renewal capacity through dysregulation of embryonic stem cell programs (14). Regardless of origin, CSCs are defined by expression of stem cell markers and pathways regulating self-renewal and survival (7). Well-studied CSC markers include CD44, CD133, and aldehyde dehydrogenase (ALDH), but their specificity is context-dependent (4). Intrinsic and microenvironmental factors all contribute to generate CSC populations with heterogeneous phenotypes and plasticity (16).
CSCs’ increased tumorigenicity upon limiting dilution xenotransplantation reflects their self-renewal capacity (17). CSCs propagate tumors long-term and recapitulate intratumor heterogeneity, differentiating into non-CSC bulk tumor cells (18). Beyond initiating tumor growth, CSCs mediate metastasis and therapeutic resistance through quiescence, enhanced DNA repair, drug efflux pumps, anti-apoptotic signaling, and immunosuppressive effects (19).
However, CSCs exhibit plasticity, readily interconverting between stem and non-stem states (20). The variability in intratumoral proportion of CSCs, coupled with the non-CSCs’ capacity to dedifferentiate into CSCs, especially post-therapy, questions proposed CSC rarity (11, 16). This plasticity enables dynamic maintenance of CSC populations, challenging efforts to definitively isolate stable CSC subsets.
The presence of CSCs within tumors has critical implications for cancer therapy. Conventional cytotoxic treatments, such as chemotherapy and radiation, target rapidly dividing cells, which primarily constitute the non-CSC tumor bulk population, leading to initial shrinkage of tumor size, and often temporary tumor regression (21). However, CSCs upregulate pro-survival signaling pathways and overexpress ATP-binding cassette (ABC) drug efflux transporters, conferring resistance to chemotherapeutics, survival advantage, metastasis, and recurrence (4, 22, 23). These intrinsic resistance mechanisms allow CSCs to evade conventional treatments, survive initial therapy and eventually repopulate the tumor, leading to recurrence and metastasis (5). For instance, glioblastoma CSCs expressing Hedgehog, Notch, and angiogenic pathways were found to persist after temozolomide chemotherapy, reconstituting recurrent tumors (24). Therefore, understanding the properties and behaviors of CSCs is vital for developing tailored strategies, like combination therapies or inhibitors targeting specific resistance pathways, to eradicate these resilient cell populations and improve long-term treatment outcomes (25).
Cell-of-origin and its influence on cancer stem cells
The cell-of-origin concept proposes that stemness properties are largely shaped by the normal cell type from which CSCs arise (11). Extensive evidence now indicate that the specific cellular context in which cancer-initiating mutations occur influences downstream CSC behavior, including tumorigenicity, differentiation capacity, and therapeutic vulnerability (4, 14).
Studies across cancer types show the relationship between cell-of-origin and CSC properties. In glioblastoma, neural stem cell-derived CSCs propagated more infiltrative, aggressive tumors compared to CSCs from committed neural progenitors (24). In prostate cancer, basal cell-derived CSCs generated tumors with higher Gleason scores and metastases than luminal cell-derived CSCs (26), and in colorectal cancer, studies have demonstrated that colorectal CSCs derived from intestinal stem cells exhibit a higher tumorigenic potential and metastatic ability compared to those originating from more differentiated cell types. Specifically, Lgr5+ intestinal stem cell-derived CSCs preferentially metastasized to the liver and express liver homing chemokine receptors, such as CCR6 (27).
In pancreatic cancer, the cell-of-origin plays a crucial role in determining the characteristics of pancreatic CSCs (PCSCs). PCSCs derived from pancreatic progenitor cells display enhanced self-renewal and tumorigenic potential compared to those originating from differentiated acinar or ductal cells (28, 29). Additionally, the cell-of-origin influences the activation of specific oncogenic signaling pathways in PCSCs, with progenitor-derived PCSCs exhibiting aberrant Hedgehog pathway activation (30).
The cell-of-origin has been implicated in shaping the behavior of lung CSCs (LCSCs). LCSCs derived from basal stem cells in the airway epithelium exhibit increased invasiveness and metastatic potential compared to those originating from other cell types (31, 32). Furthermore, the cell-of-origin determines the expression of specific stem cell markers and the activation of distinct signaling pathways in LCSCs, influencing their response to targeted therapies (30, 32).
More so, mesenchymal stem cells (MSCs) have emerged as potential cells-of-origin for CSCs in various cancer types, including glioblastoma. Recent studies have suggested that MSCs may contribute to the formation and maintenance of CSCs through various mechanisms - In glioblastoma, MSCs have been implicated as potential cell-of-origin for a CSC subset exhibiting mesenchymal phenotype (33–35). These mesenchymal glioblastoma stem cells, akin to their neural stem cell-derived CSCs counterparts, are associated with increased invasiveness, resistance to therapy, and poor prognosis (35). Emerging evidence suggests that the transformation of MSCs may give rise to this aggressive glioblastoma subpopulation (34, 35).
Aside glioblastoma, MSCs have been proposed as potential cells-of-origin for CSCs in various other cancer types, including breast cancer (36, 37), prostate cancer (38), and osteosarcoma (37, 39). The capacity of MSCs to differentiate into multiple lineages and their inherent migratory and self-renewal properties may contribute to their potential role in CSC formation and tumor progression (33, 34, 36, 37). The role of MSCs as cells-of-origin for CSCs is an active area of research, and further investigation is needed to elucidate the mechanisms underlying this potential relationship and its implications for cancer development and therapeutic strategies.
Mechanistically, the cell-of-origin imprints durable epigenetic, transcriptional, and signaling programs that shape CSC behavior (40). The cell-of-origin dictates activation of distinct oncogenic pathways, as basal breast CSCs upregulate EGFR while luminal CSCs activate HER2 signaling (41). Additionally, the cell-of-origin determines CSC differentiation trajectories, with mature cells generating unipotent CSCs while early progenitors produce multipotent CSCs (10).
Importantly, the cell-of-origin also modulates therapeutic response. Breast CSCs from basal cells resist radiation but remain DNA damage-sensitive, whereas those from luminal cells upregulate ABC drug transporters (42). In melanoma, CSCs derived from blocked differentiated cells retain DNA damage response mechanisms and are readily targetable, compared to those from neural crest stem cells (43). Thus, personalized, context-specific anti-CSC therapies are possible, but challenged by intratumor heterogeneity and CSC plasticity. Integrating lineage tracing, single-cell profiling, and functional validation is critical for understanding these pivotal interactions.
Cell-of-origin based molecular mechanisms driving cancer stem cell formation
The cell-of-origin influences the molecular events that enable normal cells transform into CSCs. This cellular context determines the signaling pathways, epigenetic programs, and mutations that confer aberrant self-renewal ability. Cells-of-origin shape mutations arising during CSC formation. Different cells-of-origin possess distinct DNA repair deficiencies that allow specific mutations. For example, melanocyte stem cells have high levels of reactive oxygen species and rely heavily on nucleotide excision repair (NER) (44). Mutations in NER genes like ERCC2 in melanocyte stems cells allow DNA damage accumulation, hypermutability, and formation of melanoma CSCs (45). In contrast, mammary stem cells are deficient in homologous recombination (HR) repair due to epigenetic repression of BRCA1 (46). Hence, loss of BRCA1/2 occurs early during breast CSC formation, enabling genomic instability through HR deficiency (47).
Several key signaling pathways involved in normal stem cell biology become dysregulated in CSCs in a cell-of-origin dependent manner. For instance, Wnt pathway mutations in intestinal stem cells promote colorectal CSC formation, while Hedgehog activation drives CSC properties in Sonic Hedgehog-responsive cerebellar stem cells giving rise to medulloblastoma (48, 49). The cell-of-origin also determines which signaling pathways are leveraged for CSC formation. For example, normal hematopoietic stem cells require FGF signaling but rely on BMP signaling for differentiation (50). Mutations activating FGF signaling while inhibiting BMP signaling promote aberrant self-renewal, allowing hematopoietic stem cells to transform into leukemic stem cells (50). A similar scenario occurs in glioblastoma, where mutations activating growth factor signaling like EGFR/PDGFRα in neural stem cells drive unrestrained proliferation during CSC genesis (51).
The epigenetic and metabolic states established by the cell-of-origin play important roles in CSC reprogramming. Normal intestinal stem cells exhibit an “open” chromatin landscape at Wnt target gene loci which primes them for CSC formation when APC mutations occur, enabling aberrant TCF/β-catenin transcriptional activation of stemness signals (52, 53). Similarly, mammary stem cells possess an epigenetic landscape suppressing BRCA1 but permitting proliferation, which fosters CSC properties when BRCA1 is mutated (46). Moreover, normal intestinal stem cells rely heavily on oxidative phosphorylation, which is co-opted during colon CSC formation to sustain stemness, and mediated in part by mutations in fumarate hydratase (54, 55).
Appreciating how the cell-of-origin-dependent genomic, epigenetic, and metabolic states shape the specific mechanisms enabling CSC formation is crucial for developing novel approaches to prevent oncogenic transformation by targeting these early vulnerabilities in a cell-context-specific manner.
Microenvironmental regulation of cancer stem cell maintenance and survival
The tumor microenvironment (TME) is now recognized as a critical regulator of CSC fate and function (Figure 1). Rather than acting in isolation, CSCs engage in dynamic crosstalk with surrounding stromal, immune, endothelial, and ECM components that maintain stemness properties and confer therapeutic resilience (56). Elucidating these complex TME interactions offers exciting opportunities for new anti-CSC therapies.
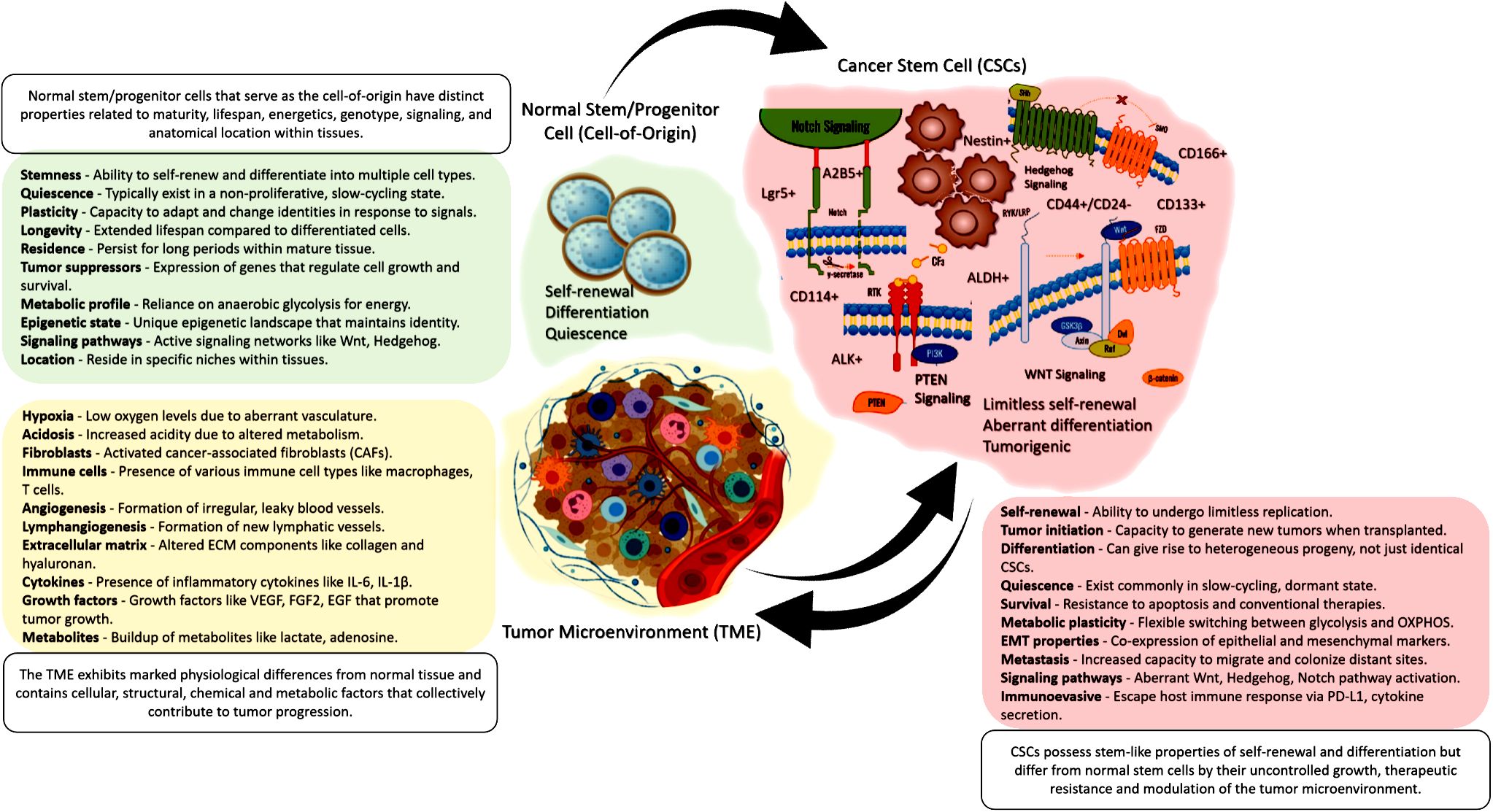
Figure 1 A visual summary showing that normal tissue stem/progenitor cells give rise to CSCs, which then interact with and reshape the surrounding tumor microenvironment. This illustrates the critical interplay between cell-of-origin, CSCs, and the TME in cancer.
Hypoxia
Hypoxia, a characteristic feature of the tumor microenvironment (TME) which is almost always coupled with chaotic vasculature and rapid cell proliferation, has emerged as a critical regulator of CSC maintenance and survival. Accruing evidence implicate hypoxia in the upregulation of multi-drug resistance transporters, maintenance of undifferentiated state, and enhancement of tumorigenic potential of CSCs (57, 58). Low oxygen tension stabilizes hypoxia-inducible factors (HIFs), which transcriptionally activate genes involved in stemness, self-renewal, and therapy resistance (59–62). Specifically, HIF-1α and HIF-2α promote the expression of pluripotency factors like Oct4, Nanog, and Sox2, as well as the upregulation of ABC drug efflux transporters, contributing to the chemoresistance of CSCs (59, 60). Additionally, hypoxia induces the expression of CSC markers, such as CD44 and CD133, and enhances the sphere-forming ability of CSCs, indicative of their self-renewal capacity (13, 61, 62). Targeting hypoxia-activated pathways thus represents a promising approach to disrupt the CSC-promoting effects of hypoxic TMEs.
Inflammation
Beyond hypoxia, the TME is often characterized by chronic inflammation, which plays a crucial role in shaping the CSC phenotype. TME-derived inflammatory signals expand CSCs through cytokine-mediated activation of NF-κB, STAT3, and other critical stemness pathways (63). Inflammatory cytokines, such as IL-6, IL-8, and TNF-α, secreted by tumor-associated immune cells and stromal components, activate pro-survival signaling pathways like NF-κB, STAT3, and Akt in CSCs (63, 64). These pathways upregulate stemness-associated transcription factors, including Oct4, Nanog, and c-Myc, promoting self-renewal and therapy resistance (64, 65). Furthermore, inflammation-induced EMT has been linked to the acquisition of stem-like properties and the generation of CSCs from non-stem tumor cells (66, 67). It is also notable that checkpoint blockade elicits T cell production of inflammatory cytokines that may inadvertently expand CSCs by stimulating these pathways (63).
Stromal cell interactions
The dynamic crosstalk between CSCs and non-malignant stromal components of the TME, such as cancer-associated fibroblasts (CAFs), endothelial cells, and mesenchymal stem cells (MSCs), plays a pivotal role in regulating CSC behavior. Paracrine signaling networks involving cytokines, growth factors, and metabolites facilitate bidirectional communication between CSCs and stromal cells (68–70). For instance, CAF-derived factors like TGF-β, CXCL12, and IL-6 induce EMT, stemness, and therapy resistance in CSCs, while CSC-secreted factors like IL-1β and IL-8 promote a pro-tumorigenic phenotype of CAFs, including EMT, drug resistance, and metastasis (71–74). Similarly, endothelial cell-derived factors stimulate CSC self-renewal and angiogenesis, while MSCs contribute to the maintenance of CSC populations through cytokine signaling and cell-cell interactions (71, 74). Targeting the homeostatic mechanisms by which non-malignant stroma supports CSC maintenance may offer new therapeutic inroads.
Extracellular matrix remodeling
The extracellular matrix (ECM) cues within the TME provides crucial biochemical and biomechanical cues that regulate CSC behavior and fate. The composition, organization, and stiffness of the ECM influence CSC self-renewal, division patterns, differentiation, plasticity, and therapy resistance (75, 76). Adhesive ECM proteins like laminin induce symmetric division that propagates the CSC pool, whereas fibrillar collagen I promotes differentiation (77). Specific ECM components, such as laminin and hyaluronic acid, have been shown to promote CSC self-renewal and stemness, while others, like collagen I, induce differentiation (75–77). Additionally, matrix metalloproteinases (MMPs) secreted by CSCs and stromal cells remodel the ECM, releasing bioactive fragments that modulate CSC signaling and promote invasion and metastasis (76, 78, 79). ECM stiffness has also been implicated in regulating CSC plasticity, with increased matrix rigidity favoring the acquisition of a mesenchymal-like CSC phenotype associated with enhanced invasiveness and therapy resistance (78, 79). Manipulating ECM signals to silence self-renewal and promote differentiation offers a promising approach to deplete CSCs.
Cell-of-origin dynamics shaping cancer stem cell-driven tumorigenesis, invasion, and metastasis
The cell-of-origin is a critical determinant of cancer pathogenesis, shaping tumor initiation, growth rate, and metastatic potential. Comparative studies exploring different cell-of-origin scenarios within specific cancers have shed light on the complexity of tumor heterogeneity and have implications for personalized cancer therapies.
Tumor initiation
Different cell types possess distinct genetic and epigenetic susceptibilities, making some more prone to oncogenic transformation (3). In glioblastoma, CSCs derived from neural stem cells generate more rapidly expanding and invasive tumors than CSCs from progenitor cells, reflecting greater self-renewal and motility (24). This indicates cells-of-origin dictate the degree of stemness and aggressiveness of resultant CSC populations.
Moreover, certain cell-of-origin contexts favor more aggressive tumor initiation. For example, basal cell-derived prostate cancer is more aggressive and associated with higher Gleason scores than luminal cell-derived cancers (80). Similarly, in breast cancer, mammary stem cells have been identified as a cell-of-origin that gives rise to tumors with a basal-like phenotype, which typically carries a poor prognosis (9). These findings underscore the significant influence of cellular context on the initial transformation events that set the stage for tumor development.
Tumor heterogeneity
The cell-of-origin controls CSC differentiation trajectories, thus, influencing downstream heterogeneity. For instance, mature luminal cells transformed into prostate CSCs produce unipotent tumors composed predominantly of luminal cells (26). However, multipotent basal stem cells yield heterogeneous prostate cancers encompassing luminal, basal, and neuroendocrine cells via their multipotent CSC derivatives (26).
Tumor growth rate
The cell-of-origin can also impact the growth rate of tumors. CSCs derived from highly proliferative and undifferentiated progenitor cells may contribute to more rapidly growing tumors (11), whereas, those originating from more differentiated cell types with limited self-renewal capacity may exhibit slower growth rates (5). The cellular background can imprint specific molecular signatures on CSCs, influencing their proliferative capacity and tumor growth dynamics (7).
Moreover, the cell-of-origin can influence the overall aggressiveness and invasive behavior of the tumor. In glioblastoma, for instance, tumors arising from neural stem cells tend to exhibit more extensive infiltration into surrounding brain tissues compared to those originating from neural progenitors (81). These distinctions in growth rate and invasiveness emphasize the importance of cell-of-origin in understanding tumor behavior and designing effective therapeutic strategies.
Metastatic potential
The cell-of-origin shapes organotropism and patterns of metastasis, by influencing migratory pathways activated in CSCs. Intestinal stem cell-derived colorectal CSCs preferentially metastasize to liver due to expression of liver homing chemokine receptors like CCR6 (27). Gastric stem cell-derived CSCs favor distant metastasis through activation of a gastric carcinoma related protein CARP-1 (82). Similarly, luminal breast cancers have been shown to preferentially metastasize to bone, while basal-like breast cancers tend to metastasize to the brain and lungs (83). In essence, the cell-of-origin has profound impacts on subsequent tumor evolution and progression kinetics. This knowledge promises to elucidate the molecular determinants of tumorigenesis and reveal actionable differences between cancers arising from distinct cells-of-origin.
Epithelial-to-mesenchymal transition
The epithelial-to-mesenchymal transition (EMT) is a fundamental process in embryonic development and tissue remodeling, wherein epithelial cells lose their polarity and cell-cell adhesion properties, acquiring a mesenchymal phenotype with increased migratory and invasive capabilities (66, 67, 72, 78). Emerging evidence suggests that EMT plays a crucial role in the generation and maintenance of CSCs, and this process is closely linked to the cell-of-origin. In epithelial cancers, such as breast and prostate cancer, EMT has been implicated in the formation of CSCs from more differentiated epithelial cells (84, 85). During EMT, epithelial tumor cells acquire stem-like properties, including self-renewal, increased expression of CSC markers (e.g., CD44, CD24, and ALDH), and enhanced resistance to therapies (66, 67, 72, 78, 84, 85). The induction of EMT in these cells is often mediated by transcription factors like Twist, Snail, Slug, and Zeb1, which suppress epithelial markers like E-cadherin and upregulate mesenchymal markers like vimentin and N-cadherin (85–87).
Importantly, the propensity for EMT and the subsequent generation of CSCs is influenced by the cell-of-origin. In breast cancer, basal-like or triple-negative breast cancer cells, which are thought to originate from more primitive mammary stem/progenitor cells, exhibit a higher propensity for EMT and CSC formation compared to luminal subtypes derived from more differentiated epithelial cells (88). Similarly, in prostate cancer, basal cells are more prone to undergoing EMT and acquiring stem-like properties compared to luminal cells (89, 90). EMT not only contributes to the formation of CSCs but also plays a critical role in metastasis, another key feature of CSCs. EMT enables CSCs to disseminate from the primary tumor site, invade surrounding tissues, and eventually colonize distant organs, establishing metastatic lesions (89, 91). The interplay between EMT and CSCs is bidirectional, as CSCs can also induce EMT in neighboring tumor cells, further promoting metastasis and therapy resistance (89, 90, 92, 93).
Metastatic cascades and organotropism
CSCs are pivotal drivers of metastatic dissemination and the establishment of secondary tumors in specific distant organs, a process known as organotropism. CSCs possess several characteristics that facilitate their ability to initiate and sustain metastatic cascades. CSCs exhibit enhanced invasive and migratory capabilities, enabling them to detach from the primary tumor, invade the surrounding stroma, and intravasate into the circulatory or lymphatic systems (89–93). This invasive behavior is mediated by the activation of EMT programs, as discussed earlier, and the expression of specific cell surface markers and proteases that facilitate extracellular matrix degradation and cell motility (72, 78, 85–87). Once in the circulation, CSCs possess intrinsic mechanisms that allow them to survive and evade immune surveillance. These include enhanced expression of anti-apoptotic proteins, increased DNA repair capacity, and the ability to enter a quiescent or dormant state (94, 95). This dormancy enables CSCs to withstand the harsh conditions of the circulatory system and later reactivate their proliferative and self-renewal capabilities upon reaching a suitable microenvironment. Upon extravasation into distant organs, CSCs demonstrate remarkable adaptability to the new microenvironment. They secrete factors that remodel the local niche, promoting angiogenesis, immune evasion, and the recruitment of supportive stromal cells (94, 95). This niche formation creates a permissive environment for CSC self-renewal, proliferation, and eventual establishment of metastatic lesions.
The preferential dissemination of CSCs to specific organs is known as organotropism, and it is influenced by the interplay between CSC-intrinsic factors and the unique microenvironmental cues of distant organs (96, 97). For example, in gallbladder tumors, CSCs expressing high levels of CXCR4 preferentially metastasize to the liver, where its ligand CXCL12 is abundantly expressed (98). Similarly, in pancreatic cancer, CSCs expressing the TEK receptor tyrosine kinase and integrin α6β4 exhibit a propensity for liver and lung metastasis, respectively (99, 100). In prostate cancer, CSCs expressing the bone-specific chemokine receptor CCR3 preferentially establish bone metastases (101). Lung metastasis in various cancers, including breast and colon cancer, has been linked to the expression of specific adhesion molecules and chemokine receptors on CSCs that facilitate their homing to the lung microenvironment (102, 103).
Understanding the intricate mechanisms underlying CSC-driven metastasis and organotropism, will help facilitate identification of potential therapeutic targets and develop strategies to disrupt these processes, ultimately improving patient outcomes.
Epigenetic mechanisms in cancer stem cells: influence of the cell-of-origin
Epigenetic modifications, including DNA methylation, histone modifications, and non-coding RNA regulation, play crucial roles in shaping the behavior and properties of CSCs (104). Importantly, the epigenetic landscape of the cell-of-origin can have a profound impact on the epigenetic patterns observed in CSCs, influencing their stemness, self-renewal, and tumorigenic potential.
DNA methylation patterns and CSC stemness
DNA methylation, the addition of methyl groups to cytosine residues in CpG dinucleotides, is a key epigenetic mechanism that regulates gene expression. Aberrant DNA methylation patterns, including hypermethylation of tumor suppressor genes and hypomethylation of oncogenes, are hallmarks of cancer and contribute to the acquisition of stem-like properties in CSCs (105–107).
Importantly, the cell-of-origin can impart specific DNA methylation patterns that shape the behavior of CSCs. For instance, in colorectal cancer, intestinal stem cells exhibit a distinct DNA methylation landscape primed for the activation of Wnt signaling, a critical pathway for stem cell self-renewal (108). When these cells acquire mutations in genes like APC, the pre-existing methylation patterns facilitate the aberrant activation of Wnt signaling, driving the formation of colorectal CSCs (53, 108, 109).
Similarly, in breast cancer, mammary stem cells possess an epigenetic landscape that represses the expression of the DNA repair gene BRCA1 (46). Mutations in BRCA1 in this context lead to genomic instability and the acquisition of stem-like properties, contributing to the formation of breast CSCs (46, 47). These examples illustrate how the cell-of-origin’s epigenetic landscape can predispose certain cell types to CSC formation upon accumulation of specific genetic alterations.
Histone modifications and gene regulation in CSCs
Histone modifications, such as acetylation, methylation, phosphorylation, and ubiquitination, regulate chromatin structure and gene expression patterns. These modifications can influence the stemness and self-renewal properties of CSCs, and their dysregulation has been implicated in various cancers (110–112).
The cell-of-origin can contribute to the histone modification patterns observed in CSCs. For example, in glioblastoma, the histone methyltransferase G9a is markedly depleted in CD133+ neural stem cells, the proposed cell-of-origin for glioblastoma CSCs (113). Upregulated G9a expression induces histone H3 lysine 9 methylation (H3K9me2), which in turn downregulates the expression of stemness-associated genes. Conversely, aberrant G9a activity in NSCLC CSCs leads to increased stemness and tumorigenicity, potentially influenced by the epigenetic landscape inherited from the lung epithelial cells-of-origin, also known as tumor-initiating cells (107). In addition, in NSCLC, histone ubiquitination patterns have been linked to the regulation of CSC properties. The histone E3 ubiquitin ligase TRIM37 is highly expressed in NSCLC CSCs and promotes stemness and metastasis through the ubiquitination of histone H2A (114), suggesting a potential link between the epigenetic landscape of the cell-of-origin and the histone modification patterns observed in CSCs.
Non-coding RNAs and CSC regulation
Non-coding RNAs, particularly microRNAs (miRNAs) and long non-coding RNAs (lncRNAs), have emerged as critical regulators of CSC properties and behavior. These non-coding RNAs can modulate gene expression by targeting mRNAs for degradation or translational repression, or by influencing epigenetic mechanisms (106, 110).
The expression and function of non-coding RNAs in CSCs are cell-of-origin-dependent. For instance, in glioblastoma, the lncRNA NEAT1 is highly expressed in neural stem cells and contributes to the maintenance of stemness and self-renewal (115). Conversely, NEAT1 has been shown to suppress stem cell self-renewal and leukemogenesis by inactivating Wnt signaling (116), consistent with its role as a tumor suppressor, and as a direct transcriptional target of the tumor suppressor gene p53 (117). Furthermore, NEAT1 has been found to mitigate multidrug resistance in leukemia by inhibiting the ABCG2 gene (118).
The long non-coding RNA MIR22HG is also upregulated in glioblastoma CSCs, where it promotes stemness and therapy resistance by regulating epigenetic changes and gene expression programs (119). However, MIR22HG plays tumor-suppressive role in various other types of cancer, including lung cancer, hepatocellular carcinoma, endometrial cancer, gastric cancer, and cholangiocarcinoma (120–122). Its low expression is associated with poor prognosis in these cancers. MIR22HG exerts its tumor-suppressive effects through various mechanisms, including the suppression of proliferation, invasion, and metastasis, and the attenuation of CSC-activating NOTCH2 signaling (120–123). This indicates that the cell-of-origin’s lncRNA expression patterns is retained in CSCs and contribute to their stemness properties.
Moreover, in breast cancer, the miRNA expression profiles of CSCs have been shown to be influenced by the cell-of-origin. Basal-like breast CSCs derived from more primitive mammary stem/progenitor cells exhibit distinct miRNA signatures compared to luminal breast CSCs derived from more differentiated cells (124, 125). These miRNA profiles can regulate key stemness-associated pathways and contribute to the differences in CSC properties observed between different breast cancer subtypes.
Increased understanding of the interplay between epigenetic mechanisms and the cell-of-origin in shaping CSC behavior will aid identification of potential therapeutic targets and develop epigenetic-based strategies to disrupt CSC maintenance and self-renewal, potentially leading to more effective cancer treatments.
Leveraging the integrative understanding of cancer stem cell biology and cell-of-origin effects in cancer therapies
Deciphering the intricate relationship between CSCs and their cell-of-origin provides unprecedented opportunities to transform cancer therapies through personalized, mechanism-based approaches. However, to effectively leverage these paradigm-shifting discoveries to conquer tumor heterogeneity, evolution, and resistance necessitates deliberate integration of this knowledge into complex therapeutic designs rooted in a deep appreciation of cancer biology.
Cell-of-origin insights can guide predictive biomarker-driven patient stratification to optimize chemotherapy regimens. For instance, gene expression profiling of breast CSCs revealed distinct chemotherapy vulnerability profiles based on the cell-of-origin-linked intrinsic subtype, which facilitates predictive selection of taxane versus anthracycline-based regimens to improve outcomes (126, 127). Furthermore, rational co-targeting of activated oncogenic drivers in CSCs through combinatorial chemotherapy regimens, as demonstrated by EGFR blockade enhancing taxane therapy in basal breast cancers, disables key survival pathways perpetuating drug resistance (128). However, conquering acquired resistance requires accounting for CSC plasticity and inevitable clonal selection. Innovative adaptive designs that dynamically adjust dosing in response to altered CSC composition show promise in this context (129).
Regarding radiotherapy, the radiosensitivity phenotype of CSCs strongly associates with cell-of-origin determinants. Compared to CSCs arising from differentiated cells that retain residual DNA repair capacity, those originating from undifferentiated stem/progenitor cells upregulate robust pro-survival signaling and display relative radioresistance (24). Targeted radiosensitization through inhibitors disabling these radioprotective CSC programs, informed by cell-of-origin insights, as with Chk1 inhibitors in laryngeal and tongue CSCs, can dramatically improve therapeutic index (130). However, the sequencing and schedule of radiotherapy combinations warrants careful optimization to avoid potential expansion of aggressive CSC subclones.
Moreover, integrative genomic and single-cell profiling can help identify cell-of-origin-associated neoantigens for personalized vaccines or CAR-T cells targeting CSCs, as demonstrated against EGFRvIII-expressing glioblastoma CSCs of neural stem origin (131, 132). However, mitigating the immunosuppression conferred by the protective CSC niche remains crucial, highlighting opportunities for immunomodulators blocking inhibitory ligands like PD-L1 in CSCs (133). Cell-of-origin insights also empower selection of targeted therapies, such as KRAS wild-type status in intestinal crypt-derived colon CSCs, predicting sensitivity to EGFR blockade (53). Nevertheless, acquired resistance is inevitable due to subclonal evolution; single-cell genomic monitoring of CSC dynamics during therapy facilitates real-time adjustment of targeted regimens to overcome resistance emergence (134).
Further, cell-of-origin-associated CSC biomarker panels can enhance early detection and therapeutic monitoring. Longitudinal tracking of circulating CSCs expressing normal mammary stem cell markers may signal occult metastases, while shifting to mesenchymal markers can indicate early treatment failure (135). Liquid biopsy assessing CSC dynamics thereby enables personalized surveillance strategies.
Therapeutic targeting of cancer stem cells based on cell-of-origin vulnerabilities
The cell-of-origin concept provides a framework for developing more precise therapies tailored to the unique vulnerabilities of CSCs based on their cellular origin.
Small molecule inhibitors
Small molecule inhibitors designed against oncogenic drivers and signaling pathways activated preferentially in CSCs based on cell-of-origin are a promising therapeutic approach. In basal breast cancers enriched in mammary stem cell-derived CSCs, EGFR tyrosine kinase inhibitors like neratinib suppress pro-survival signaling in the CSCs (136). Wnt-targeting porcupine inhibitors, including LGK974, selectively inhibit Wnt-dependent intestinal stem cell-derived colorectal CSCs (137). Sonic Hedgehog (SHH) inhibitors vismodegib and glasdegib demonstrate activity against cerebellar stem cell-derived medulloblastoma CSCs exhibiting aberrant SHH pathway activation (138). Ongoing research aim to expand the repertoire of targeted CSC inhibitors informed by cell-of-origin (see Tables 1, 2).
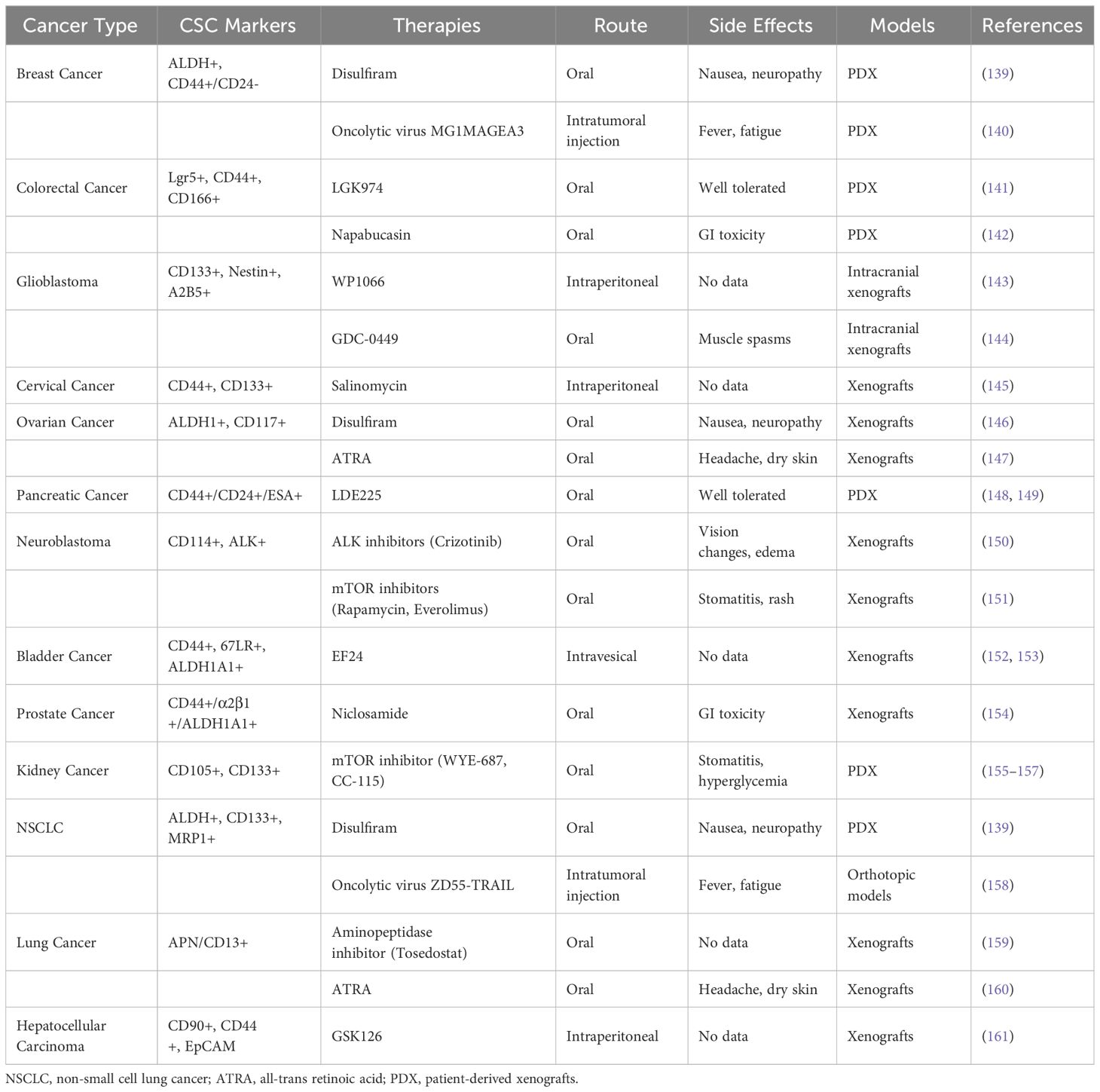
Table 1 Novel preclinical therapies targeting CSCs based on cell-of-origin to resolve chemotherapy resistance in solid tumors.
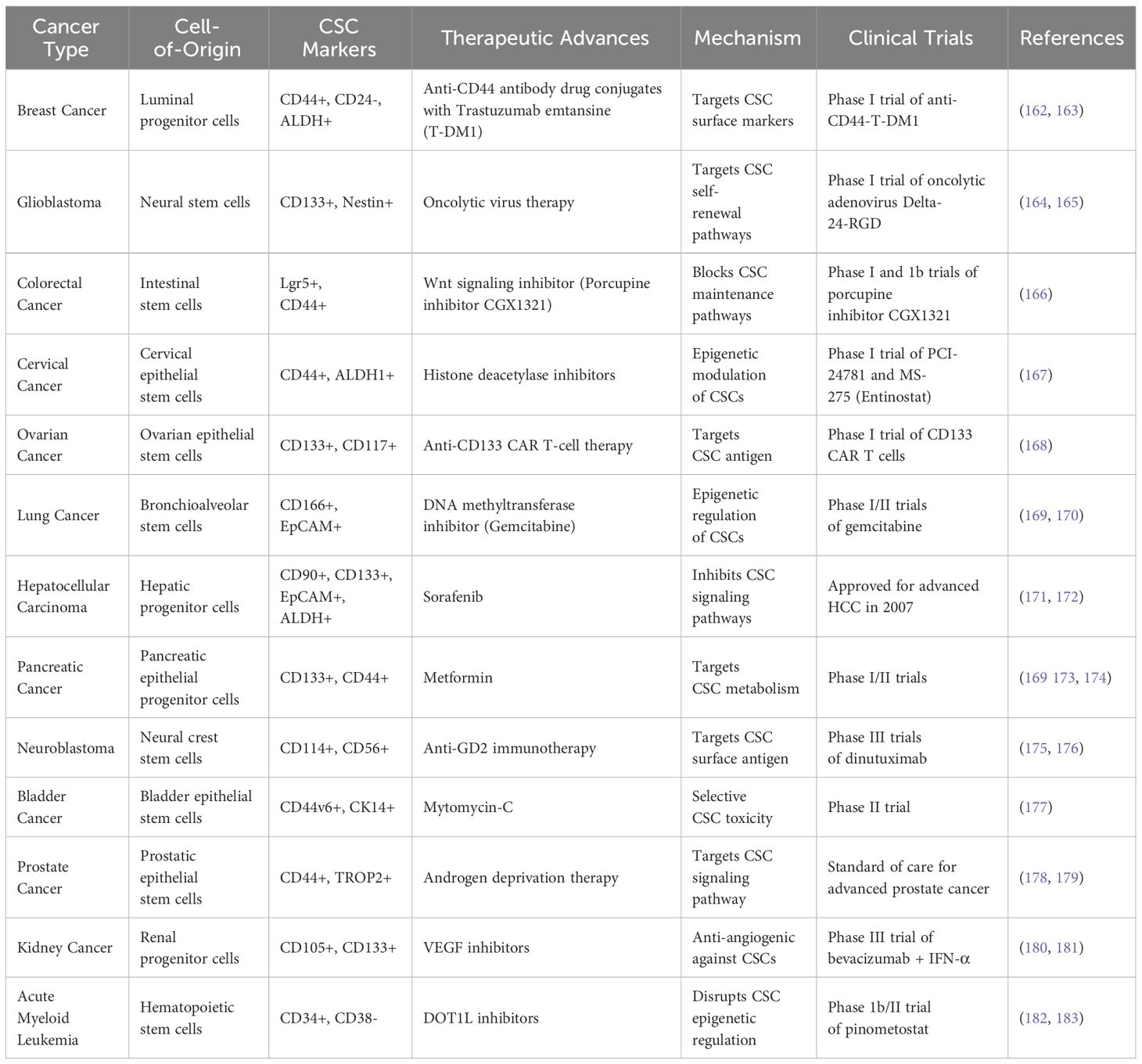
Table 2 Key clinical trial-based therapeutic advances in targeting cancer stem cells based on cell-of-origin between 2015 and 2023.
Monoclonal and bispecific antibodies
Antibodies targeting surface antigens and pathways selectively enriched in CSCs based on cell-of-origin are another promising approach. Glembatumumab vedotin targets glycoprotein NMB overexpressed on CSCs across cancer types including breast cancers of basal/myoepithelial origin (184). Additionally, antibodies against the epithelial cell adhesion molecule (EpCAM) preferentially expressed on liver CSCs derived from hepatic progenitor cells, and anti-CD47 antibodies blocking “don’t eat me” signaling in leukemia CSCs arising from hematopoietic stem cells have entered clinical testing (185, 186). Ongoing research aims to identify novel cell-origin-associated CSC antigens amenable to antibody targeting.
Gene therapies
Leveraging knowledge of genetic drivers and dependencies based on cell-of-origin offers opportunities for gene therapy. Delivering mutant KRAS-targeted CRISPR constructs preferentially suppresses acinar cell-derived pancreatic CSCs exhibiting aberrant KRAS activity (187). Additionally, suicide gene strategies using a herpes simplex virus thymidine kinase transgene and ganciclovir prodrug show promise against glioblastoma CSCs derived from neural stem cells (188). Enhancing selective targeting of gene therapies against cell-origin-defined CSC populations is warranted.
Combination strategies
Given the marked heterogeneity of CSCs, concurrently targeting bulk tumor cells and CSC subpopulations dependent on their unique cell-of-origin are under active evaluation. For example, simultaneously targeting HER2 and EGFR or PIK3CA signaling using trastuzumab and lapatinib in HER2+ progenitor-derived luminal breast CSCs shows synergistic activity (189, 190). Combined inhibition of MEK and Bcl-2 selectively suppresses intestinal crypt stem cell-derived colorectal CSCs exhibiting co-activation of MAPK and anti-apoptotic pathways (191). Moving forward, high-dimensional mapping of cell-origin CSC vulnerabilities using single-cell omics promises to inform rational combination therapies (192). Combining personalized and multi-modal approaches holds great promise for achieving long-term remissions and overcoming therapy resistance, which requires extensive research efforts focused on precision targeting of heterogeneous and adaptable CSC populations (Figure 2).
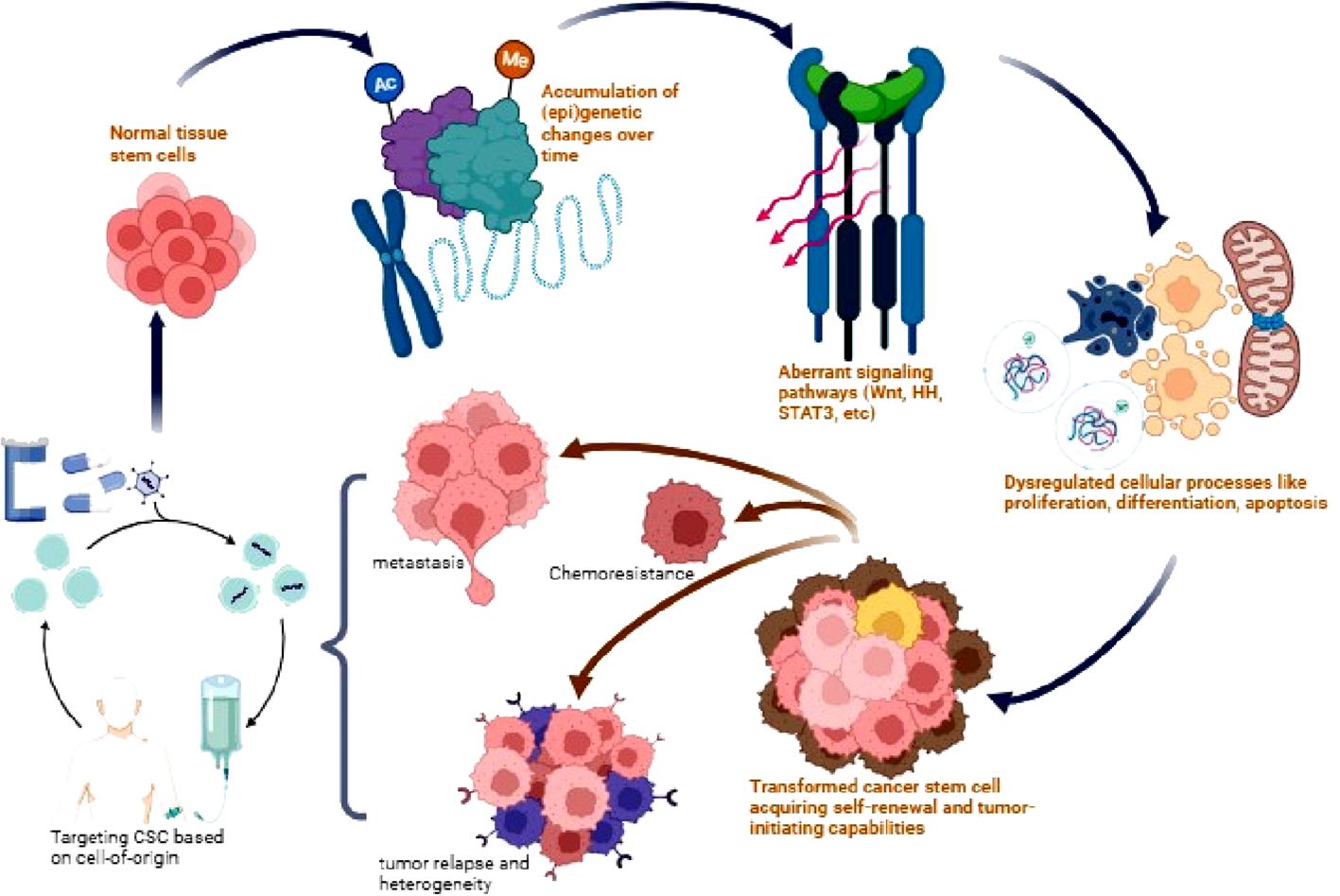
Figure 2 Origin and therapeutic targeting of CSCs. Normal tissue stem cells accumulate genetic and epigenetic alterations over time leading to dysregulation of signaling pathways such as Wnt, Hedgehog (HH), STAT3, and AKT. This results in aberrant stem cell processes and the transformation into CSCs with acquired self-renewal and tumor-initiating capabilities. CSCs drive tumor heterogeneity, metastasis, chemotherapy resistance, and relapse. Targeting dysregulated CSC signaling pathways provides a potential therapeutic approach to eliminate this tumorigenic population and improve patient outcomes.
Precision targeting of chemoresistant cancer stem cells in solid tumors based on cell-of-origin
Eradicating chemoresistant CSCs is critical to improve outcomes in solid tumors. This section discusses emerging strategies against cell-origin-defined CSCs across diverse cancer types.
Breast cancer
In triple negative breast cancer (TNBC), CSCs arise from basal/myoepithelial progenitors and are marked by ALDH+ and CD44+/CD24- (193). Overexpression of ALDH1A3 isoform in these CSCs contributes to chemoresistance. Accordingly, the ALDH inhibitor disulfiram preferentially inhibits ALDH1A3 activity, reduces CSC populations, and re-sensitizes TNBC models to taxanes (139). Additionally, oncolytic viruses like MG1MAGEA3 specifically target MAGE-A3 expressed on ALDH+ TNBC CSCs, diminishing these cells and enhancing chemotherapy efficacy (140).
Colorectal cancer
Colorectal CSCs marked by Lgr5 and CD44 arise from intestinal crypt base stem cells and drive chemoresistance via Wnt pathway activation (53). Porcupine inhibitor LGK974 suppresses Wnt signaling specifically in Lgr5+ CSCs, potentiating oxaliplatin therapy (141). Napabucasin inhibits STAT3-NANOG signaling preferentially activated in Lgr5+/CD44+ CSCs, reducing this subset and sensitizing to irinotecan (12, 142).
Glioblastoma
Glioblastoma CSCs expressing CD133 and Nestin originate from neural stem cells and utilize aberrant Hedgehog pathway signaling (144). Hedgehog inhibitor GDC-0449 selectively antagonizes SHH ligands in Nestin+ CSCs, decreasing these cells, enhancing chemoradiation response, and delaying recurrence (144). STAT3 inhibitor WP1066 suppresses stemness transcription factors like NANOG in CD133+ CSCs, improving temozolomide efficacy (143).
Cervical cancer
Cervical CSCs marked by ALDH1A1 and CD133 arise from transformed cervical epithelial progenitors and upregulate oxidative stress response pathways (194). Salinomycin selectively inhibits stress response enzymes highly expressed in CD133+/CD44+ CSCs, reducing this subset and re-sensitizing tumors to docetaxel (145).
Ovarian cancer
Ovarian CSCs expressing ALDH1 and CD117 originate from transformed fallopian tube epithelia and exhibit platinum resistance. All-trans retinoic acid (ATRA) binds retinoic acid receptors on ALDH1A1+/CD117+ CSCs, inducing differentiation and re-sensitizing tumors to platinum therapy (147). ALDH inhibitor disulfiram decreases ALDH1A1+ CSCs, abrogating taxane resistance (146).
Lung cancer
Lung CSCs marked by CD133 and ALDH derive from transformed lung epithelial cells and drive platinum resistance (195). ATRA induces differentiation of ALDH+ CSCs, reducing these cells and sensitizing tumors to cisplatin (160). Oncolytic adenovirus ZD55-TRAIL targets SOX2highNANOGhighMRP1high lung CSCs via CAR/integrin receptors, diminishing these cells and augments doxorubicin, vinblastine, cisplatin, and 5-FU efficacy (158).
Hepatocellular carcinoma
Hepatocellular carcinoma CSCs marked by CD90 originate from hepatic progenitor cells and exhibit sorafenib resistance mediated by H3K9 methylation and stemness genes (196). Histone methyltransferase inhibitor GSK126 suppresses aberrant H3K9 methylation in CD90+/EZH2+ CSCs, impairing stemness and sensitizing to sorafenib (161, 196).
Pancreatic cancer
Pancreatic CSCs expressing CD44 and ESA derive from transformed pancreatic progenitors and utilize hedgehog signaling to maintain stemness and confer gemcitabine resistance (148). Hedgehog inhibitor LDE225 blocks SMO-mediated hedgehog pathway activation specifically in CD44+/ESA+ CSCs, diminishing these cells and sensitizing tumors to gemcitabine (148, 149).
Neuroblastoma
Neuroblastoma CSCs marked by CD114 and ALK originate from neural crest progenitors and exhibit chemotherapy resistance (197). ALK and mTOR inhibitors like crizotinib and everolimus preferentially target ALK and mTOR-driven survival pathways upregulated in ALK+ and CD114+ CSCs respectively, re-sensitizing tumors to chemotherapy (150, 151).
Bladder cancer
Bladder CSCs enriched in CD44 arise from transformed basal urothelial progenitors and drive cisplatin resistance via NF-κB, Wnt and Notch signaling (152). Curcumin analog EF24 potently inhibits these pathways preferentially activated in CD44+ CSCs, reducing these cells and re-sensitizing tumors to cisplatin (153).
Prostate cancer
ALDH1A1+ castration-resistant prostate CSCs originate from transformed basal epithelial progenitors and exhibit aberrant STAT3/AKT signaling (154). Niclosamide suppresses STAT3 and AKT signaling specifically in ALDH1A1+ CSCs, augmenting tumor response to androgen deprivation (154).
Kidney cancer
CD105+ Kidney CSCs derive from nephron progenitors in clear cell renal carcinoma and confer radioresistance via mTOR and Chk1 signaling (198). The combination of mTOR and Chk1 inhibition, demonstrating cytotoxic and anti-proliferative effects, with potential translational value for treatment, has been shown to effectively target oncogenic pathways in CD105+ renal CSCs, leading to increased radiosensitivity in ccRCC (155–157). Additionally, the inhibition of PLK1, a cell-cycle-related kinase, has been identified as a potential therapeutic target in clear cell renal cell carcinoma, further supporting the potential of combined mTOR and Chk1 inhibition in this context (199).
Despite advances in CSC-targeting therapies based on cell-of-origin properties, several challenges remain. The intratumoral and intertumoral heterogeneity of CSCs presents significant challenge to identifying universal CSC-specific targets, thus, identifying robust and universal cell-origin-based CSC-specific markers remains an ongoing area of research (Tables 1, 2). The dynamic interplay between CSCs and the TME further complicates therapy responses and necessitates consideration. Preclinical models that accurately depict the cell-of-origin context and TME are essential for evaluating therapeutic interventions (Table 1). Additionally, Biomarker discovery and patient stratification based on cell-of-origin characteristics are critical for implementing personalized CSC-targeting therapies in clinical settings (Table 2). Overcoming these challenges requires integrative advanced omics technologies, preclinical models that mimic the TME, and well-designed clinical trials.
Personalized and multi-modal cancer treatments using small-molecule inhibitors, monoclonal antibodies, gene therapies, and combination approaches offer promising avenues for improving cancer treatment outcomes. Continued research on the complex interactions between CSCs and their cell-of-origin is essential for developing transformative anticancer therapeutic strategies and achieving lasting remissions (Figure 3). Overall, despite lingering challenges including tumor heterogeneity, dynamic CSC plasticity, and resistance mechanisms, unraveling cell-origin allows tailored targeting of chemoresistant CSC subsets across diverse solid tumors.
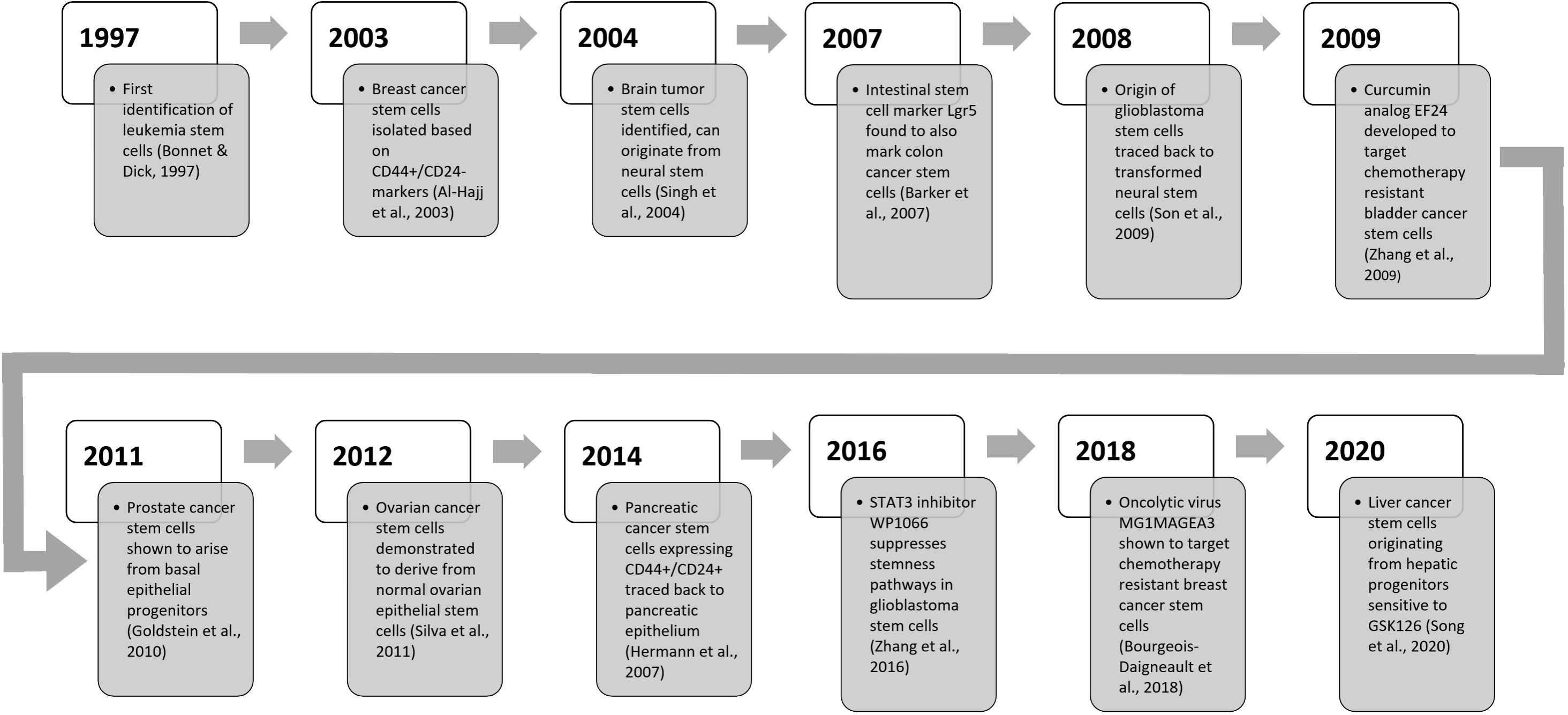
Figure 3 An updated timeline of key discoveries related to cancer stem cell research.
Critical appraisal and controversy surrounding cancer stem cells, cells-of-origin, and their clinical/therapeutic utility
The concepts of CSCs and cell-of-origin have transformed and advanced our understanding of tumor biology, but their intricacies have also sparked debate and exposed critical gaps in knowledge (14). CSCs are characterized by their ability to self-renew and recreate the full heterogeneity of the original tumor, but isolating pure CSC populations and identifying immutable CSC markers has proven extremely challenging (200). Several candidate CSC markers have been proposed, but demonstrating specificity and reproducibility has been difficult.
Against initial propositions that CSCs are exceptionally rare within tumors, accounting for <5% of cells, mounting evidence indicate non-CSC differentiated tumor cells can readily de-differentiate and acquire stem-like behavior, questioning the rarity of CSCs (201). This plasticity allows non-CSCs convert into CSCs, especially in response to therapies targeting the differentiated tumor bulk. Furthermore, CSCs exhibit inherent plasticity, dynamically transitioning between stem-like and non-stem cell states, enabling CSC flexibility and adaptability which consequently complicates therapeutic efforts to permanently eliminate them (20). In summary, CSCs demonstrate plasticity, context-dependency, and resistance to rigid definitions, creating complex challenges in isolating, studying, and targeting these shifting cells.
The cell-of-origin concept refers to the normal cell type, whether a stem cell, progenitor cell, or mature differentiated cell, that initially acquires the first cancer-causing mutations and undergoes transformation to give rise to full malignancy. However, it is difficult to definitively determine the cell-of-origin retrospectively due to limitations and challenges (9). After a tumor has developed, reconstructing the initiating events to pinpoint the cell-of-origin is difficult, if not impossible, in many cases. Moreover, different potential cells-of-origin, including progenitor or stem cells, can ultimately converge on very similar tumor phenotypes after accumulating mutations. Emerging evidence indicates that combined mutations occurring simultaneously across multiple cell types within a tissue may be required for full malignant transformation, rather than mutations within a single discrete cell (202), thus, obscuring efforts to clearly delineate the initiating cell-of-origin.
The goal of identifying the cell-of-origin is to develop therapies that specifically target malignant stem cells while sparing normal stem cells. However, developing therapies that discriminate between malignant and normal stem cells remains difficult. For example, despite preclinical data showing DOT1L inhibitor pinometostat exhibits10-fold higher toxicity against leukemia stem cells than normal hematopoietic stem cells, nearly 50% of acute myeloid leukemia patients relapsed after DOT1L inhibition, highlighting the challenges of translating cell-of-origin insights into clinical practice (182, 203).
Clinically, emerging therapies directly targeting CSCs, such as STAT3 inhibitor napabucasin and ABL inhibitor asciminib, have shown initial efficacy in early phase trials, offering proof-of-concept for CSC-directed approaches (204). However, resistance and relapse continue to pose substantial challenges. In studies of ALDH inhibitors, suppression of CSC populations was only transient before their reconstitution, highlighting the need for combination treatments (205). However, directly linking patient clinical response to the specific effects on CSCs is complicated by the realities of intratumor heterogeneity and CSC plasticity (5). CSCs can dynamically transition between cell states and repopulate the CSC pool, limiting durable responses to single agent CSC-targeting drugs.
The rarity of CSCs poses inherent difficulties for analysis and evaluation, especially in the context of tumor heterogeneity. The low CSC frequency makes obtaining sufficient material for in-depth profiling challenging. Furthermore, low initial CSC frequency can cloud clinical response assessments if the resolution is inadequate, since even small surviving CSCs can re-initiate tumor growth (206). This has stirred debate on whether rarity versus abundance is more therapeutically disadvantageous. Thus, despite promising preliminary results, realizing the full potential of CSC-targeted therapies will require overcoming hurdles related to tumor evolution, plasticity, heterogeneity, and rarity through rational combination strategies and high-resolution monitoring.
The paradigm-shifting concept of CSCs and cell-of-origin holds immense promise for transforming cancer prevention, treatment, and overcoming therapeutic resistance. However, capitalizing on their full potential requires navigating tremendous knowledge gaps and inherent complexities. A key challenge is prospectively isolating definitive purified CSC subsets for characterization because of biomarker ambiguity and interconversions between states (11, 14). Preventing resistance requires innovative, adaptive therapies blocking CSC evolution and plasticity (59). Selectively eliminating CSCs while sparing normal stem cells remains an ongoing quest requiring deeper appreciation of stemness vulnerabilities (182). Realizing clinical advances necessitates embracing CSC complexity through emerging tools like high-dimensional single-cell profiling, organoids, and computational modeling that capture dynamic stem cell ecosystem interactions (207, 208). By unraveling the intricacies of malignant stem cell biology, pioneering interdisciplinary science can transform cancer into a durable chronic condition rather than a deadly disease.
Challenges and future directions
Despite advances in our understanding of the interplay between CSCs and the cell-of-origin, several challenges persist. First, cell-origin-based identification and characterization of CSCs remains challenging due to CSC and TME heterogeneity. Improved single-cell sequencing technologies coupled with innovative lineage tracing approaches may provide deeper insights into distinct cellular origins of CSCs within heterogeneous tumors (201, 209). Second, CSC plasticity complicates their targeting because of their dynamic transitions between stem-like and differentiated states. Improved understanding of the epigenetic and signaling mechanisms regulating CSC plasticity could lead to novel strategies to maintain CSCs in a more differentiated state, susceptible to conventional therapies (210, 211). Third, TME plays a critical role in regulating CSC behavior and therapeutic responses. Interactions with immune cells, fibroblasts, and the ECM contribute to CSC maintenance and therapy resistance (5, 212). Investigating the crosstalk between CSCs and TME may unveil new therapeutic targets and combination strategies to overcome therapy resistance. Fourth, the development of reliable CSC-specific biomarkers based on cell-of-origin properties is essential for clinical translation. Biomarkers that can accurately identify and isolate CSCs from patient samples would facilitate the design of targeted therapies and monitoring of treatment responses (213). Continued efforts in biomarker discovery and validation are crucial for advancing CSC-targeted therapies.
Furthermore, integrated multi-omics approaches are required to fully characterize and map the cell-of-origin-specific molecular vulnerabilities and dependencies of CSCs. Additionally, novel ex vivo and in vivo models that more accurately recapitulate the complex native TME are required to better understand the extrinsic regulation of CSC behavior by the surrounding niche. Standardization of protocols for isolating and validating CSCs across research groups would allow for improved comparisons and reproducibility between studies. As evident in Table 2, clinical trials evaluating cell-of-origin-guided therapies and combination approaches thatspecifically target CSCs are critical for translating research findings to benefit patients (162-159). Finally, the development and validation of reliable biomarkers and assays to track and profile CSCs longitudinally in patients could enable earlier detection of minimal residual disease and facilitate more tailored, adaptive treatment monitoring approaches.
Conclusion
The intricate relationship between CSCs and their cell-of-origin is an exciting frontier in cancer research. Understanding this interaction provides insights into the molecular basis of tumor initiation, therapeutic resistance, and patient outcomes. The path forward demands interdisciplinary collaboration to unlock transformative insights that propel more effective, adaptive, and durable therapies targeting the self-renewing core of tumors; ultimately making curative outcomes feasible and fundamentally altering the prognosis of cancer patients worldwide.
Author contributions
OB: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. C-CC: Formal analysis, Funding acquisition, Methodology, Writing – original draft. TP: Formal analysis, Methodology, Writing – review & editing, Supervision. AW: Formal analysis, Methodology, Project administration, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was funded in part by grants from the National Science and Technology Council, NSTC 111-2314-B-038-132-MY3 and Taipei Medical University, TMU111-AE1-B23 to C-CC.
Acknowledgments
We acknowledge the significant contributions of many researchers in the field whose important work could not be cited in this review due to page constraints. The advancements highlighted here have only been made possible through the diligent efforts of colleagues across disciplines to unlock the immense therapeutic potential of complex interplay between cancer stem cells and their cellular origins.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
2. Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. (2005) 434:843–50. doi: 10.1038/nature03319
3. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumors: accumulating evidence and unresolved questions. Nat Rev Cancer. (2008) 8:755–68. doi: 10.1038/nrc2499
4. Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. (2013) 501:328–37. doi: 10.1038/nature12624
5. Batlle E, Clevers H. Cancer stem cells revisited. Nat Med. (2017) 23:1124–34. doi: 10.1038/nm.4409
6. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res. (2006) 66:9339–44. doi: 10.1158/0008-5472.CAN-06-3126
7. Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. (2012) 21:283–96. doi: 10.1016/j.ccr.2012.03.003
8. Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. (2009) 15:1010–2. doi: 10.1038/nm0909-1010
10. Rycaj K, Tang DG. Cell-of-origin of cancer versus cancer stem cells: assays and interpretations. Cancer Res. (2015) 75:4003–11. doi: 10.1158/0008-5472.CAN-15-0798
11. Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumor formation by single human melanoma cells. Nature. (2008) 456:593–8. doi: 10.1038/nature07567
12. Li Y, Rogoff HA, Keates S, Gao Y, Murikipudi S, Mikule K, et al. Suppression of cancer relapse and metastasis by inhibiting cancer stemness. Proc Natl Acad Sci U S A. (2015) 112:1839–44. doi: 10.1073/pnas.1424171112
13. Bamodu OA, Chang HL, Ong JR, Lee WH, Yeh CT, Tsai JT. Elevated PDK1 expression drives PI3K/AKT/MTOR signaling promotes radiation-resistant and dedifferentiated phenotype of hepatocellular carcinoma. Cells. (2020) 9:746. doi: 10.3390/cells9030746
14. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell. (2014) 14:275–91. doi: 10.1016/j.stem.2014.02.006
15. Ayob AZ, Ramasamy TS. Cancer stem cells as key drivers of tumour progression. J BioMed Sci. (2018) 25:20. doi: 10.1186/s12929-018-0426-4
16. Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clin Chem. (2013) 59:168–79. doi: 10.1373/clinchem.2012.184655
17. Qureshi-Baig K, Ullmann P, Haan S, Letellier E. Tumor-Initiating Cells: a criTICal review of isolation approaches and new challenges in targeting strategies. Mol Cancer. (2017) 16:40. doi: 10.1186/s12943-017-0602-2
18. Beck B, Blanpain C. Unravelling cancer stem cell potential. Nat Rev Cancer. (2013) 13:727–38. doi: 10.1038/nrc3597
19. Kalli M, Poskus MD, Stylianopoulos T, Zervantonakis IK. Beyond matrix stiffness: targeting force-induced cancer drug resistance. Trends Cancer. (2023) 9:937–54. doi: 10.1016/j.trecan.2023.07.006
20. Gupta PB, Fillmore CM, Jiang G, Shapira SD, Tao K, Kuperwasser C, et al. Stochastic state transitions give rise to phenotypic equilibrium in populations of cancer cells. Cell. (2011) 146:633–44. doi: 10.1016/j.cell.2011.07.026
21. Pattabiraman DR, Weinberg RA. Tackling the cancer stem cells-what challenges do they pose? Nat Rev Drug Discovery. (2019) 13(7):497–512. doi: 10.1038/nrd4253
22. Takebe N, Miele L, Harris PJ, Jeong W, Bando H, Kahn M, et al. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: clinical update. Nat Rev Clin Oncol. (2015) 12:445–64. doi: 10.1038/nrclinonc.2015.61
23. Kuroda T, Hirohashi Y, Torigoe T, Yasuda K, Takahashi A, Asanuma H, et al. ALDH1-high ovarian cancer stem-like cells can be isolated from serous and clear cell adenocarcinoma cells, and ALDH1 high expression is associated with poor prognosis. PLoS One. (2013) 8(6):e65158. doi: 10.1371/journal.pone.0065158
24. Chen J, Li Y, Yu TS, McKay RM, Burns DK, Kernie SG, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. (2012) 488:522–6. doi: 10.1038/nature11287
25. Bhat GR, Sethi I, Sadida HQ, Rah B, Mir R, Algehainy N, et al. Cancer cell plasticity: from cellular, molecular, and genetic mechanisms to tumor heterogeneity and drug resistance. Cancer Metastasis Rev. (2024) 43(1):197–228. doi: 10.1007/s10555-024-10172-z
26. Goldstein AS, Huang J, Guo C, Garraway IP, Witte ON. Identification of a cell-of-origin for human prostate cancer. Science. (2010) 329:568–71. doi: 10.1126/science.1189992
27. Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. (2009) 457:608–11. doi: 10.1038/nature07602
28. Mayerle J. Pancreatic cancer: why the cell of origin matters. Nat Rev Gastroenterol Hepatol. (2022) 19:279. doi: 10.1038/s41575-022-00595-w
29. Xu L. Cancer stem cell in the progression and therapy of pancreatic cancer. Front Biosci (Landmark Ed). (2013) 18:795–802. doi: 10.2741/4143
30. Patil K, Khan FB, Akhtar S, Ahmad A, Uddin S. The plasticity of pancreatic cancer stem cells: implications in therapeutic resistance. Cancer Metastasis Rev. (2021) 40:691–720. doi: 10.1007/s10555-021-09979-x
31. Ferone G, Song JY, Sutherland KD, Bhaskaran R, Monkhorst K, Lambooij JP, et al. SOX2 is the determining oncogenic switch in promoting lung squamous cell carcinoma from different cells of origin. Cancer Cell. (2016) 30:519–32. doi: 10.1016/j.ccell.2016.09.001
32. Wu J, Feng J, Zhang Q, He Y, Xu C, Wang C, et al. Epigenetic regulation of stem cells in lung cancer oncogenesis and therapy resistance. Front Genet. (2023) 14:1120815. doi: 10.3389/fgene.2023.1120815
33. Jiménez G, Hackenberg M, Catalina P, Boulaiz H, Griñán-Lisón C, García MÁ, et al. Mesenchymal stem cell’s secretome promotes selective enrichment of cancer stem-like cells with specific cytogenetic profile. Cancer Lett. (2018) 429:78–88. doi: 10.1016/j.canlet.2018.04.042
34. Hossain A, Gumin J, Gao F, Figueroa J, Shinojima N, Takezaki T, et al. Mesenchymal stem cells isolated from human gliomas increase proliferation and maintain stemness of glioma stem cells through the IL-6/gp130/STAT3 pathway. Stem Cells. (2015) 33:2400–15. doi: 10.1002/stem.2053
35. Huang T, Kim CK, Alvarez AA, Pangeni RP, Wan X, Song X, et al. MST4 phosphorylation of ATG4B regulates autophagic activity, tumorigenicity, and radioresistance in glioblastoma. Cancer Cell. (2017) 32:840–55. doi: 10.1016/j.ccell.2017.11.005
36. Chauhan A, Agarwal S, Masih M, Gautam PK. The multifunction role of tumor-associated mesenchymal stem cells and their interaction with immune cells in breast cancer. Immunol Invest. (2023) 52:856–78. doi: 10.1080/08820139.2023.2249025
37. Molloy AP, Martin FT, Dwyer RM, Griffin TP, Murphy M, Barry FP, et al. Mesenchymal stem cell secretion of chemokines during differentiation into osteoblasts, and their potential role in mediating interactions with breast cancer cells. Int J Cancer. (2009) 124:326–32. doi: 10.1002/ijc.23939
38. Brennen WN, Denmeade SR, Isaacs JT. Mesenchymal stem cells as a vector for the inflammatory prostate microenvironment. Endocr Relat Cancer. (2013) 20:R269–90. doi: 10.1530/ERC-13-0151
39. Tsukamoto S, Honoki K, Fujii H, Tohma Y, Kido A, Mori T, et al. Mesenchymal stem cells promote tumor engraftment and metastatic colonization in rat osteosarcoma model. Int J Oncol. (2012) 40:163–9. doi: 10.3892/ijo.2011.1220
40. Skoda AM, Simovic D, Karin V, Kardum V, Vranic S, Serman L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn J Basic Med Sci. (2018) 18:8–20. doi: 10.17305/bjbms.2018.2756
41. Meyer MJ, Fleming JM, Lin AF, Hussnain SA, Ginsburg E, Vonderhaar BK, et al. CD44posCD49fhiCD133/2hi defines xenograft-initiating cells in estrogen receptor-negative breast cancer. Cancer Res. (2010) 70:4624–33. doi: 10.1158/0008-5472.CAN-09-3619
42. Kabeche L, Compton DA. Mapping the cancer stem cell state in triple-negative breast cancer. Nat Commun. (2022) 13:1–17. doi: 10.3389/fphar.2020.599965
43. Rambow F, Rogiers A, Marin-Bejar O, Aibar S, Femel J, Dewaele M, et al. Toward minimal residual disease-directed therapy in melanoma. Cell. (2018) 174:843–855.e19. doi: 10.1016/j.cell.2018.06.025
44. Hoerter JD, Bradley P, Casillas A, Chambers D, Denholm C, Johnson K, et al. Extrafollicular dermal melanocyte stem cells and melanoma. Stem Cells Int. (2012) 2012:407079. doi: 10.1155/2012/407079
45. Scaini MC, Catoni C, Poggiana C, Pigozzo J, Piccin L, Leone K, et al. A multiparameter liquid biopsy approach allows to track melanoma dynamics and identify early treatment resistance. NPJ Precis Oncol. (2024) 8(1):78. doi: 10.1038/s41698-024-00567-0
46. Proia TA, Keller PJ, Gupta PB, Klebba I, Jones AD, Sedic M, et al. Genetic predisposition directs breast cancer phenotype by dictating progenitor cell fate. Cell Stem Cell. (2011) 8:149–63. doi: 10.1016/j.stem.2010.12.007
47. Gorodetska I, Kozeretska I, Dubrovska A. BRCA genes: the role in genome stability, cancer stemness and therapy resistance. J Cancer. (2019) 10:2109–27. doi: 10.7150/jca.30410
48. Zhao H, Ming T, Tang S, Ren S, Yang H, Liu M, et al. Wnt signaling in colorectal cancer: pathogenic role and therapeutic target. Mol Cancer. (2022) 21:144. doi: 10.1186/s12943-022-01616-7
49. Hanna A, Shevde LA. Hedgehog signaling: modulation of cancer properies and tumor mircroenvironment. Mol Cancer. (2016) 15:24. doi: 10.1186/s12943-016-0509-3
50. Pouget C, Peterkin T, Simões FC, Lee Y, Traver D, Patient R. FGF signalling restricts haematopoietic stem cell specification via modulation of the BMP pathway. Nat Commun. (2014) 5:5588. doi: 10.1038/ncomms6588
51. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. (2010) 17:98–110. doi: 10.1016/j.ccr.2009.12.020
52. Kim TH, Li F, Ferreiro-Neira I, Ho LL, Luyten A, Nalapareddy K, et al. Broadly permissive intestinal chromatin underlies lateral inhibition and cell plasticity. Nature. (2014) 506:511–5. doi: 10.1038/nature12903
53. Vermeulen L, De Sousa E Melo F, van der Heijden M, Cameron K, de Jong JH, Borovski T, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. (2010) 12:468–76. doi: 10.1038/ncb2048
54. Smith AL, Whitehall JC, Bradshaw C, Gay D, Robertson F, Blain AP, et al. Age-associated mitochondrial DNA mutations cause metabolic remodelling that contributes to accelerated intestinal tumorigenesis. Nat Cancer. (2020) 1:976–89. doi: 10.1038/s43018-020-00112-5
55. Lamb R, Ozsvari B, Lisanti CL, et al. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget. (2015) 6:4569–84. doi: 10.18632/oncotarget.3174
56. Plaks V, Kong N, Werb Z. The cancer stem cell niche: how essential is the niche in regulating stemness of tumor cells? Cell Stem Cell. (2015) 16:225–38. doi: 10.1016/j.stem.2015.02.015
57. Hirschhaeuser F, Sattler UG, Mueller-Klieser W. Lactate: a metabolic key player in cancer. Cancer Res. (2011) 71:6921–5. doi: 10.1158/0008-5472.CAN-11-1457
58. Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, et al. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. (2009) 15:501–13. doi: 10.1016/j.ccr.2009.03.018
59. Binó L, Kučera J, Štefková K, Švihálková Šindlerová L, Lánová M, Kudová J, et al. The stabilization of hypoxia inducible factor modulates differentiation status and inhibits the proliferation of mouse embryonic stem cells. Chem Biol Interact. (2016) 244:204–14. doi: 10.1016/j.cbi.2015.12.007
60. Piccoli C, D’Aprile A, Ripoli M, Scrima R, Boffoli D, Tabilio A, et al. The hypoxia-inducible factor is stabilized in circulating hematopoietic stem cells under normoxic conditions. FEBS Lett. (2007) 581:3111–9. doi: 10.1016/j.febslet.2007.05.077
61. Forristal C, Nowlan B, Barbier V, McCarthy D, Walkinshaw G, Winkler IG, et al. Oxygen-independent stabilization of the oxygen-labile transcription factor HIF-1α with dimethyloxalyl glycine or FG-4497 increases hematopoietic stem cell quiescence in vivo and mobilization in response to G-CSF. Blood. (2011) 118:2334. doi: 10.1182/blood.V118.21.2334.2334
62. Szablowska-Gadomska I, Zayat V, Buzanska L. Influence of low oxygen tensions on expression of pluripotency genes in stem cells. Acta Neurobiol Exp (Wars). (2011) 71:86–93. doi: 10.55782/ane-2011-1825
63. Wu B, Shi X, Jiang M, Liu H. Cross-talk between cancer stem cells and immune cells: potential therapeutic targets in the tumor immune microenvironment. Mol Cancer. (2023) 22:38. doi: 10.1186/s12943-023-01748-4
64. Liu Z, Ren Y, Meng L, Li L, Beatson R, Deng J, et al. Epigenetic signaling of cancer stem cells during inflammation. Front Cell Dev Biol. (2021) 9:772211. doi: 10.3389/fcell.2021.772211
65. Mitra A, Yan J, Xia X, Zhou S, Chen J, Mishra L, et al. IL6-mediated inflammatory loop reprograms normal to epithelial-mesenchymal transition+ metastatic cancer stem cells in preneoplastic liver of transforming growth factor beta-deficient β2-spectrin+/- mice. Hepatology. (2017) 65:1222–36. doi: 10.1002/hep.28951
66. Zhang L, Jiao M, Wu K, Li L, Zhu G, Wang X, et al. TNF-α induced epithelial mesenchymal transition increases stemness properties in renal cell carcinoma cells. Int J Clin Exp Med. (2014) 7(12):4951–8.
67. Suarez-Carmona M, Lesage J, Cataldo D, Gilles C. EMT and inflammation: inseparable actors of cancer progression. Mol Oncol. (2017) 11:805–23. doi: 10.1002/1878-0261.12095
68. McAndrews KM, Vazquez-Arreguin K, Kwak C, Sugimoto H, Zheng X, Li B, et al. alphaSMA(+) fibroblasts suppress lgr5(+) cancer stem cells and restrain colorectal cancer progression. Oncogene. (2021) 40:4440–52. doi: 10.1038/s41388-021-01866-7
69. Raghavan S, Snyder CS, Wang A, McLean K, Zamarin D, Buckanovich RJ, et al. Carcinoma-associated mesenchymal stem cells promote chemoresistance in ovarian cancer stem cells via PDGF signaling. Cancers (Basel). (2020) 12:2063. doi: 10.3390/cancers12082063
70. Sengupta S, Somasundaram K, Morvinski DF, Marin P, Reddy P, Mukherjee A, et al. Paracrine signaling between differentiated tumor cell-secreted fibromodulin and the stroma promotes tumor growth by enhancing angiogenesis. Cancer Res. (2022) 82:6383. doi: 10.1158/1538-7445.AM2022-6383
71. Luo H, Tu G, Liu Z, Liu M. Cancer-associated fibroblasts: a multifaceted driver of breast cancer progression. Cancer Lett. (2015) 361:155–63. doi: 10.1016/j.canlet.2015.02.018
72. Jia C, Wang G, Wang T, Fu B, Zhang Y, Huang L, et al. Cancer-associated Fibroblasts induce epithelial-mesenchymal transition via the Transglutaminase 2-dependent IL-6/IL6R/STAT3 axis in Hepatocellular Carcinoma. Int J Biol Sci. (2020) 16:2542–58. doi: 10.7150/ijbs.45446
73. Zhang F, Cui JY, Gao HF, Yu H, Gao FF, Chen JL, et al. Cancer-associated fibroblasts induce epithelial-mesenchymal transition and cisplatin resistance in ovarian cancer via CXCL12/CXCR4 axis. Future Oncol. (2020) 16:2619–33. doi: 10.2217/fon-2020-0095
74. Loh JJ, Ma S. The role of cancer-associated fibroblast as a dynamic player in mediating cancer stemness in the tumor microenvironment. Front Cell Dev Biol. (2021) 9:727640. doi: 10.3389/fcell.2021.727640
75. Kesh K, Gupta VK, Durden B, Garrido V, Mateo-Victoriano B, Lavania SP, et al. Therapy resistance, cancer stem cells and ECM in cancer: the matrix reloaded. Cancers (Basel). (2020) 12:3067. doi: 10.3390/cancers12103067
76. Li C, Qiu S, Liu X, Guo F, Zhai J, Li Z, et al. Extracellular matrix-derived mechanical force governs breast cancer cell stemness and quiescence transition through integrin-DDR signaling. Signal Transduct Target Ther. (2023) 8:247. doi: 10.1038/s41392-023-01453-0
77. Xu S, Xu H, Wang W, Li S, Li H, Li T, et al. The role of collagen in cancer: from bench to bedside. J Transl Med. (2019) 17:309. doi: 10.1186/s12967-019-2058-1
78. Fiori ME, Di Franco S, Villanova L, Bianca P, Stassi G, De Maria R. Cancer-associated fibroblasts as abettors of tumor progression at the crossroads of EMT and therapy resistance. Mol Cancer. (2019) 18:70. doi: 10.1186/s12943-019-0994-2
79. Liu Q, Guo Z, Li G, Zhang Y, Liu X, Li B, et al. Cancer stem cells and their niche in cancer progression and therapy. Cancer Cell Int. (2023) 23:305. doi: 10.1186/s12935-023-03130-2
80. Wang ZA, Mitrofanova A, Bergren SK, Abate-Shen C, Cardiff RD, Califano A, et al. Lineage analysis of basal epithelial cells reveals their unexpected plasticity and supports a cell-of-origin model for prostate cancer heterogeneity. Nat Cell Biol. (2013) 15:274–83. doi: 10.1038/ncb2697
81. Lee CC, Lai JH, Hueng DY, Ma HI, Chung Y, Sun YY, et al. Disrupting the CXCL12/CXCR4 axis disturbs the characteristics of glioblastoma stem-like cells of rat RG2 glioblastoma. Cancer Cell Int. (2013) 13:85. doi: 10.1186/1475-2867-13-85
82. Chang TS, Wei KL, Lu CK, Chen YH, Cheng YT, Tung SY, et al. Inhibition of CCAR1, a coactivator of β-catenin, suppresses the proliferation and migration of gastric cancer cells. Int J Mol Sci. (2017) 18:460. doi: 10.3390/ijms18020460
83. Bos PD, Zhang XH, Nadal C, Shu W, Gomis RR, Nguyen DX, et al. Genes that mediate breast cancer metastasis to the brain. Nature. (2009) 459:1005–9. doi: 10.1038/nature08021
84. Turner C, Kohandel M. Investigating the link between epithelial-mesenchymal transition and the cancer stem cell phenotype: A mathematical approach. J Theor Biol. (2010) 265:329–35. doi: 10.1016/j.jtbi.2010.05.024
85. Ruscetti M, Quach B, Dadashian EL, Mulholland DJ, Wu H. Tracking and functional characterization of epithelial-mesenchymal transition and mesenchymal tumor cells during prostate cancer metastasis. Cancer Res. (2015) 75:2749–59. doi: 10.1158/0008-5472.CAN-14-3476
86. Pai S, Bamodu OA, Lin YK, Lin CS, Chu PY, Chien MH, et al. CD47-SIRPα Signaling induces epithelial-mesenchymal transition and cancer stemness and links to a poor prognosis in patients with oral squamous cell carcinoma. Cells. (2019) 8:1658. doi: 10.3390/cells8121658
87. Chen JH, Huang WC, Bamodu OA, Chang PM, Chao TY, Huang TH. Monospecific antibody targeting of CDH11 inhibits epithelial-to-mesenchymal transition and represses cancer stem cell-like phenotype by up-regulating miR-335 in metastatic breast cancer, in vitro and in vivo. BMC Cancer. (2019) 19:634. doi: 10.1186/s12885-019-5811-1
88. Sarrio D, Franklin CK, Mackay A, Reis-Filho JS, Isacke CM. Epithelial and mesenchymal subpopulations within normal basal breast cell lines exhibit distinct stem cell/progenitor properties. Stem Cells. (2012) 30:292–303. doi: 10.1002/stem.791
89. Chiew SF, Looi LM, Cheah PL, Teoh KH, Chang SW, Abdul Sani SF. Epithelial-mesenchymal transition profiles in triple negative breast carcinoma may explain its aggressive nature. Malays J Pathol. (2023) 45(3):363–74.
90. Blick T, Hugo H, Widodo E, Waltham M, Pinto C, Mani SA, et al. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi/)CD24 (lo/-) stem cell phenotype in human breast cancer. J Mammary Gland Biol Neoplasia. (2010) 15:235–52. doi: 10.1007/s10911-010-9175-z
91. Fantozzi A, Gruber DC, Pisarsky L, Heck C, Kunita A, Yilmaz M, et al. VEGF-mediated angiogenesis links EMT-induced cancer stemness to tumor initiation. Cancer Res. (2014) 74:1566–75. doi: 10.1158/0008-5472.CAN-13-1641
92. Li YR, Fang Y, Lyu Z, Zhu Y, Yang L. Exploring the dynamic interplay between cancer stem cells and the tumor microenvironment: implications for novel therapeutic strategies. J Transl Med. (2023) 21:686. doi: 10.1186/s12967-023-04575-9
93. den Hollander P, Maddela JJ, Mani SA. Spatial and temporal relationship between epithelial-mesenchymal transition (EMT) and stem cells in cancer. Clin Chem. (2024) 70:190–205. doi: 10.1093/clinchem/hvad197
94. Skvortsov S, Debbage P, Lukas P, Skvortsova I. Crosstalk between DNA repair and cancer stem cell (CSC) associated intracellular pathways. Semin Cancer Biol. (2015) 31:36–42. doi: 10.1016/j.semcancer.2014.06.002
95. Nathansen J, Meyer F, Müller L, Schmitz M, Borgmann K, Dubrovska A. Beyond the double-strand breaks: the role of DNA repair proteins in cancer stem-cell regulation. Cancers (Basel). (2021) 13:4818. doi: 10.3390/cancers13194818
96. Wu Q, Tian P, He D, Jia Z, He Y, Luo W, et al. SCUBE2 mediates bone metastasis of luminal breast cancer by modulating immune-suppressive osteoblastic niches. Cell Res. (2023) 33:464–78. doi: 10.1038/s41422-023-00810-6
97. Jiang B, Zhao X, Chen W, Diao W, Ding M, Qin H, et al. Lysosomal protein transmembrane 5 promotes lung-specific metastasis by regulating BMPR1A lysosomal degradation. Nat Commun. (2022) 13:4141. doi: 10.1038/s41467-022-31783-6
98. Lee HJ, Lee K, Lee DG, Bae KH, Kim JS, Liang ZL, et al. Chemokine (C-X-C motif) ligand 12 is associated with gallbladder carcinoma progression and is a novel independent poor prognostic factor. Clin Cancer Res. (2012) 18:3270–80. doi: 10.1158/1078-0432.CCR-11-2417
99. Hoshino A, Costa-Silva B, Shen TL, Rodrigues G, Hashimoto A, Tesic Mark M, et al. Tumour exosome integrins determine organotropic metastasis. Nature. (2015) 527(7578):329–35. doi: 10.1038/nature15756
100. Costa-Silva B, Aiello NM, Ocean AJ, Singh S, Zhang H, Thakur BK, et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. (2015) 17:816–26. doi: 10.1038/ncb3169
101. Guérard A, Laurent V, Fromont G, Estève D, Gilhodes J, Bonnelye E, et al. The chemokine receptor CCR3 is potentially involved in the homing of prostate cancer cells to bone: implication of bone-marrow adipocytes. Int J Mol Sci. (2021) 22:1994. doi: 10.3390/ijms22041994
102. Wang C, Xu K, Wang R, Han X, Tang J, Guan X. Heterogeneity of BCSCs contributes to the metastatic organotropism of breast cancer. J Exp Clin Cancer Res. (2021) 40:370. doi: 10.1186/s13046-021-02164-6
103. Zhan Q, Liu B, Situ X, Luo Y, Fu T, Wang Y, et al. New insights into the correlations between circulating tumor cells and target organ metastasis. Signal Transduct Target Ther. (2023) 8:465. doi: 10.1038/s41392-023-01725-9
104. Pangeni RP, Zhang Z, Alvarez AA, Wan X, Sastry N, Lu S, et al. Genome-wide methylomic and transcriptomic analyses identify subtype-specific epigenetic signatures commonly dysregulated in glioma stem cells and glioblastoma. Epigenetics. (2018) 13:432–48. doi: 10.1080/15592294.2018.1469892
105. Sastry NG, Wan X, Huang T, Alvarez AA, Pangeni RP, Song X, et al. LY6K promotes glioblastoma tumorigenicity via CAV-1-mediated ERK1/2 signaling enhancement. Neuro Oncol. (2020) 22:1315–26. doi: 10.1093/neuonc/noaa032
106. Huang T, Alvarez AA, Pangeni RP, Horbinski CM, Lu S, Kim SH, et al. A regulatory circuit of miR-125b/miR-20b and Wnt signalling controls glioblastoma phenotypes through FZD6-modulated pathways. Nat Commun. (2016) 7:12885. doi: 10.1038/ncomms12885
107. Pangeni RP, Yang L, Zhang K, Wang J, Li W, Guo C, et al. G9a regulates tumorigenicity and stemness through genome-wide DNA methylation reprogramming in non-small cell lung cancer. Clin Epigenetics. (2020) 12:88. doi: 10.1186/s13148-020-00879-5
108. Joosten SPJ, Spaargaren M, Clevers H, Pals ST. Hepatocyte growth factor/MET and CD44 in colorectal cancer: partners in tumorigenesis and therapy resistance. Biochim Biophys Acta Rev Cancer. (2020) 1874:188437. doi: 10.1016/j.bbcan.2020.188437
109. de Sousa EM, Vermeulen L, Richel D, Medema JP. Targeting Wnt signaling in colon cancer stem cells. Clin Cancer Res. (2011) 17:647–53. doi: 10.1158/1078-0432.CCR-10-1204
110. Bamodu OA, Huang WC, Lee WH, Wu A, Wang LS, Hsiao M, et al. Aberrant KDM5B expression promotes aggressive breast cancer through MALAT1 overexpression and downregulation of hsa-miR-448. BMC Cancer. (2016) 16:160. doi: 10.1186/s12885-016-2108-5
111. Bamodu OA, Yang CK, Cheng WH, Tzeng DTW, Kuo KT, Huang CC, et al. 4-Acetyl-Antroquinonol B Suppresses SOD2-Enhanced Cancer Stem Cell-Like Phenotypes and Chemoresistance of Colorectal Cancer Cells by Inducing hsa-miR-324 re-Expression. Cancers (Basel). (2018) 10:269. doi: 10.3390/cancers10080269
112. Bamodu OA, Chao TY. Dissecting the functional pleiotropism of lysine demethylase 5B in physiology and pathology. J Cancer Res Pract. (2020) 7:49–59. doi: 10.4103/JCRP.JCRP_5_20
113. Tao H, Li H, Su Y, Feng D, Wang X, Zhang C, et al. Histone methyltransferase G9a and H3K9 dimethylation inhibit the self-renewal of glioma cancer stem cells. Mol Cell Biochem. (2014) 394:23–30. doi: 10.1007/s11010-014-2077-4
114. Bajbouj K, Al-Ali A, Ramakrishnan RK, Saber-Ayad M, Hamid Q. Histone modification in NSCLC: molecular mechanisms and therapeutic targets. Int J Mol Sci. (2021) 22:11701. doi: 10.3390/ijms222111701
115. Lulli V, Buccarelli M, Ilari R, Castellani G, De Dominicis C, Di Giamberardino A, et al. Mir-370-3p impairs glioblastoma stem-like cell Malignancy regulating a complex interplay between HMGA2/HIF1A and the oncogenic long non-coding RNA (lncRNA) NEAT1. Int J Mol Sci. (2020) 21:3610. doi: 10.3390/ijms21103610
116. Yan H, Wang Z, Sun Y, Hu L, Bu P. Cytoplasmic NEAT1 suppresses AML stem cell self-renewal and leukemogenesis through inactivation of wnt signaling. Adv Sci (Weinh). (2021) 8:e2100914. doi: 10.1002/advs.202100914
117. Idogawa M, Ohashi T, Sasaki Y, Nakase H, Tokino T. Long non-coding RNA NEAT1 is a transcriptional target of p53 and modulates p53-induced transactivation and tumor-suppressor function. Int J Cancer. (2017) 140:2785–91. doi: 10.1002/ijc.30689
118. Gao C, Zhang J, Wang Q, Ren C. Overexpression of lncRNA NEAT1 mitigates multidrug resistance by inhibiting ABCG2 in leukemia. Oncol Lett. (2016) 12:1051–7. doi: 10.3892/ol.2016.4738
119. Han M, Wang S, Fritah S, Wang X, Zhou W, Yang N, et al. Interfering with long non-coding RNA MIR22HG processing inhibits glioblastoma progression through suppression of Wnt/β-catenin signalling. Brain. (2020) 143:512–30. doi: 10.1093/brain/awz406
120. Zhang DY, Zou XJ, Cao CH, Zhang T, Lei L, Qi XL, et al. Identification and functional characterization of long non-coding RNA MIR22HG as a tumor suppressor for hepatocellular carcinoma. Theranostics. (2018) 8:3751–65. doi: 10.7150/thno.22493
121. Su W, Feng S, Chen X, Yang X, Mao R, Guo C, et al. Silencing of Long Noncoding RNA MIR22HG Triggers Cell Survival/Death Signaling via Oncogenes YBX1, MET, and p21 in Lung Cancer. Cancer Res. (2018) 78:3207–19. doi: 10.1158/0008-5472.CAN-18-0222
122. Li H, Wang Y, Long Noncoding RNA. (lncRNA) MIR22HG suppresses gastric cancer progression through attenuating NOTCH2 signaling. Med Sci Monit. (2019) 25:656–65. doi: 10.12659/MSM.912813
123. Tomé M, Tchorz J, Gassmann M, Bettler B. Constitutive activation of Notch2 signalling confers chemoresistance to neural stem cells via transactivation of fibroblast growth factor receptor-1. Stem Cell Res. (2019) 35:101390. doi: 10.1016/j.scr.2019.101390
124. Boo L, Ho WY, Mohd Ali N, Yeap SK, Ky H, Chan KG, et al. Phenotypic and microRNA transcriptomic profiling of the MDA-MB-231 spheroid-enriched CSCs with comparison of MCF-7 microRNA profiling dataset. PeerJ. (2017) 5:e3551. doi: 10.7717/peerj.3551
125. Gusterson B, Eaves CJ. Basal-like breast cancers: from pathology to biology and back again. Stem Cell Rep. (2018) 10:1676–86. doi: 10.1016/j.stemcr.2018.04.023
126. Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol. (2011) 5:5–23. doi: 10.1016/j.molonc.2010.11.003
127. Zheng Q, Zhang M, Zhou F, Zhang L, Meng X. The breast cancer stem cells traits and drug resistance. Front Pharmacol. (2021) 11:599965. doi: 10.3389/fphar.2020.599965
128. Yao S, Fan LY, Lam EW. The FOXO3-FOXM1 axis: A key cancer drug target and a modulator of cancer drug resistance. Semin Cancer Biol. (2018) 50:77–89. doi: 10.1016/j.semcancer.2017.11.018
129. Enriquez-Navas PM, Kam Y, Das T, Hassan S, Silva A, Foroutan P, et al. Exploiting evolutionary principles to prolong tumor control in preclinical models of breast cancer. Sci Transl Med. (2016) 8:327ra24. doi: 10.1126/scitranslmed.aad7842
130. Barker HE, Patel R, McLaughlin M, Schick U, Zaidi S, Nutting CM, et al. CHK1 inhibition radiosensitizes head and neck cancers to paclitaxel-based chemoradiotherapy. Mol Cancer Ther. (2016) 15:2042–54. doi: 10.1158/1535-7163.MCT-15-0998
131. Sampson JH, Archer GE, Mitchell DA, Heimberger AB, Herndon JE 2nd, Lally-Goss D, et al. An epidermal growth factor receptor variant III-targeted vaccine is safe and immunogenic in patients with glioblastoma multiforme. Mol Cancer Ther. (2009) 8:2773–9. doi: 10.1158/1535-7163.MCT-09-0124
132. Wu W, Klockow JL, Zhang M, Lafortune F, Chang E, Jin L, et al. Glioblastoma multiforme (GBM): An overview of current therapies and mechanisms of resistance. Pharmacol Res. (2021) 171:105780. doi: 10.1016/j.phrs.2021.105780
133. Bu LL, Yu GT, Wu L, Mao L, Deng WW, Liu JF, et al. STAT3 induces immunosuppression by upregulating PD-1/PD-L1 in HNSCC. J Dent Res. (2017) 96:1027–34. doi: 10.1177/0022034517712435
134. Lawson DA, Kessenbrock K, Davis RT, Pervolarakis N, Werb Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat Cell Biol. (2018) 20:1349–60. doi: 10.1038/s41556-018-0236-7
135. Baccelli I, Schneeweiss A, Riethdorf S, Stenzinger A, Schillert A, Vogel V, et al. Identification of a population of blood circulating tumor cells from breast cancer patients that initiates metastasis in a xenograft assay. Nat Biotechnol. (2013) 31:539–44. doi: 10.1038/nbt.2576
136. Kinnel B, Singh SK, Oprea-Ilies G, Singh R. Targeted therapy and mechanisms of drug resistance in breast cancer. Cancers (Basel). (2023) 15:1320. doi: 10.3390/cancers15041320
137. Madan B, Ke Z, Harmston N, Ho SY, Frois AO, Alam J, et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene. (2016) 35:2197–207. doi: 10.1038/onc.2015.280
138. Lospinoso Severini L, Ghirga F, Bufalieri F, Quaglio D, Infante P, Di Marcotullio L. The SHH/GLI signaling pathway: a therapeutic target for medulloblastoma. Expert Opin Ther Targets. (2020) 24:1159–81. doi: 10.1080/14728222.2020.1823967
139. Cole AJ, Fayomi AP, Anyaeche VI, Bai S, Buckanovich RJ. An evolving paradigm of cancer stem cell hierarchies: therapeutic implications. Theranostics. (2020) 10:3083–98. doi: 10.7150/thno.41647
140. Bourgeois-Daigneault MC, Roy DG, Aitken AS, El Sayes N, Martin NT, Varette O, et al. Neoadjuvant oncolytic virotherapy before surgery sensitizes triple-negative breast cancer to immune checkpoint therapy. Sci Transl Med. (2018) 10:eaao1641. doi: 10.1126/scitranslmed.aao1641
141. Hu YB, Yan C, Mu L, Mi YL, Zhao H, Hu H, et al. Exosomal Wnt-induced dedifferentiation of colorectal cancer cells contributes to chemotherapy resistance [published correction appears in Oncogene. 2019 Aug;38(35):6319-6321]. Oncogene. (2019) 38:1951–65. doi: 10.1038/s41388-018-0557-9
142. Shah MA, Yoshino T, Tebbutt NC, Grothey A, Tabernero J, Xu RH, et al. Napabucasin plus FOLFIRI in patients with previously treated metastatic colorectal cancer: results from the open-label, randomized phase III canStem303C study. Clin Colorectal Cancer. (2023) 22:100–10. doi: 10.1016/j.clcc.2022.11.002
143. Iwamaru A, Szymanski S, Iwado E, Aoki H, Yokoyama T, Fokt I, et al. A novel inhibitor of the STAT3 pathway induces apoptosis in Malignant glioma cells both in vitro and in vivo. Oncogene. (2007) 26:2435–44. doi: 10.1038/sj.onc.1210031
144. Bar EE, Chaudhry A, Lin A, Fan X, Schreck K, Matsui W, et al. Cyclopamine-mediated hedgehog pathway inhibition depletes stem-like cancer cells in glioblastoma. Stem Cells. (2007) 25:2524–33. doi: 10.1634/stemcells.2007-0166
145. Wang Q, Yen YT, Xie C, Liu F, Liu Q, Wei J, et al. Combined delivery of salinomycin and docetaxel by dual-targeting gelatinase nanoparticles effectively inhibits cervical cancer cells and cancer stem cells. Drug Deliv. (2021) 28:510–9. doi: 10.1080/10717544.2021.1886378
146. Guo F, Yang Z, Sehouli J, Kaufmann AM. Blockade of ALDH in cisplatin-resistant ovarian cancer stem cells in vitro synergistically enhances chemotherapy-induced cell death. Curr Oncol. (2022) 29:2808–22. doi: 10.3390/curroncol29040229
147. Takahashi A, Hong L, Chefetz I. How to win the ovarian cancer stem cell battle: destroying the roots. Cancer Drug Resist. (2020) 3:1021–33. doi: 10.20517/cdr.2020.93
148. Shankar S, Nall D, Tang SN, Meeker D, Passarini J, Sharma J, et al. Resveratrol inhibits pancreatic cancer stem cell characteristics in human and KrasG12D transgenic mice by inhibiting pluripotency maintaining factors and epithelial-mesenchymal transition. PloS One. (2011) 6:e16530. doi: 10.1371/journal.pone.0016530
149. Pijnappel EN, Wassenaar NPM, Gurney-Champion OJ, Klaassen R, van der Lee K, Pleunis-van Empel MCH, et al. Phase I/II study of LDE225 in combination with gemcitabine and nab-paclitaxel in patients with metastatic pancreatic cancer. Cancers (Basel). (2021) 13:4869. doi: 10.3390/cancers13194869
150. Cazes A, Lopez-Delisle L, Tsarovina K, Pierre-Eugène C, De Preter K, Peuchmaur M, et al. Activated Alk triggers prolonged neurogenesis and Ret upregulation providing a therapeutic target in ALK-mutated neuroblastoma. Oncotarget. (2014) 5:2688–702. doi: 10.18632/oncotarget.1883
151. Mahller YY, Williams JP, Baird WH, Mitton B, Grossheim J, Saeki Y, et al. Neuroblastoma cell lines contain pluripotent tumor initiating cells that are susceptible to a targeted oncolytic virus. PloS One. (2009) 4:e4235. doi: 10.1371/journal.pone.0004235
152. Chan KS, Espinosa I, Chao M, Wong D, Ailles L, Diehn M, et al. Identification, molecular characterization, clinical prognosis, and therapeutic targeting of human bladder tumor-initiating cells. Proc Natl Acad Sci U S A. (2009) 106:14016–21. doi: 10.1073/pnas.0906549106
153. Zhou S, Zhang S, Shen H, Chen W, Xu H, Chen X, et al. Curcumin inhibits cancer progression through regulating expression of microRNAs. Tumour Biol. (2017) 39:1010428317691680. doi: 10.1177/1010428317691680
154. Domingo-Domenech J, Vidal SJ, Rodriguez-Bravo V, Castillo-Martin M, Quinn SA, Rodriguez-Barrueco R, et al. Suppression of acquired docetaxel resistance in prostate cancer through depletion of notch- and hedgehog-dependent tumor-initiating cells. Cancer Cell. (2012) 22:373–88. doi: 10.1016/j.ccr.2012.07.016
155. Pan XD, Gu DH, Mao JH, Zhu H, Chen X, Zheng B, et al. Concurrent inhibition of mTORC1 and mTORC2 by WYE-687 inhibits renal cell carcinoma cell growth in vitro and in vivo. PloS One. (2017) 12:e0172555. doi: 10.1371/journal.pone.0172555
156. Cho DC, Mier JW. Dual inhibition of PI3-kinase and mTOR in renal cell carcinoma. Curr Cancer Drug Targets. (2013) 13:126–42. doi: 10.2174/1568009611313020003
157. Zheng B, Sun X, Chen XF, Chen Z, Zhu WL, Zhu H, et al. Dual inhibition of DNA-PKcs and mTOR by CC-115 potently inhibits human renal cell carcinoma cell growth. Aging (Albany NY). (2020) 12:20445–56. doi: 10.18632/aging.103847
158. Yang Y, Xu H, Huang W, Ding M, Xiao J, Yang D, et al. Targeting lung cancer stem-like cells with TRAIL gene armed oncolytic adenovirus. J Cell Mol Med. (2015) 19:915–23. doi: 10.1111/jcmm.12397
159. Lendeckel U, Karimi F, Al Abdulla R, Wolke C. The Role of the Ectopeptidase APN/CD13 in Cancer. Biomedicines. (2023) 11(3):724. doi: 10.3390/biomedicines11030724
160. MacDonagh L, Santiago RM, Gray SG, Breen E, Cuffe S, Finn SP, et al. Exploitation of the vitamin A/retinoic acid axis depletes ALDH1-positive cancer stem cells and re-sensitises resistant non-small cell lung cancer cells to cisplatin. Transl Oncol. (2021) 14:101025. doi: 10.1016/j.tranon.2021.101025
161. Tseng CF, Chen LT, Wang HD, Liu YH, Shiah SG. Transcriptional suppression of Dicer by HOXB-AS3/EZH2 complex dictates sorafenib resistance and cancer stemness. Cancer Sci. (2022) 113:1601–12. doi: 10.1111/cas.15319
162. Zhang Y, Yue S, Haag R, Sun H, Zhong Z. An intelligent cell-selective polymersome-DM1 nanotoxin toward triple negative breast cancer. J Control Release. (2021) 340:331–41. doi: 10.1016/j.jconrel.2021.11.014
163. Diessner J, Bruttel V, Stein RG, Horn E, Häusler SF, Dietl J, et al. Targeting of preexisting and induced breast cancer stem cells with trastuzumab and trastuzumab emtansine (T-DM1). Cell Death Dis. (2014) 5:e1149. doi: 10.1038/cddis.2014.115
164. van Putten EHP, Kleijn A, van Beusechem VW, Noske D, Lamers CHJ, de Goede AL, et al. Convection enhanced delivery of the oncolytic adenovirus delta24-RGD in patients with recurrent GBM: A phase I clinical trial including correlative studies. Clin Cancer Res. (2022) 28:1572–85. doi: 10.1158/1078-0432.CCR-21-3324
165. Lang FF, Conrad C, Gomez-Manzano C, Yung WKA, Sawaya R, Weinberg JS, et al. Phase I study of DNX-2401 (Delta-24-RGD) oncolytic adenovirus: replication and immunotherapeutic effects in recurrent Malignant glioma. J Clin Oncol. (2018) 36:1419–27. doi: 10.1200/JCO.2017.75.8219
166. Giannakis M, Le DT, Pishvaian MJ, Weinberg BA, Papadopoulos KP, Shen L, et al. Phase 1 study of WNT pathway Porcupine inhibitor CGX1321 and phase 1b study of CGX1321 + pembrolizumab (pembro) in patients (pts) with advanced gastrointestinal (GI) tumors. J Clin Oncol. (2023) 41:3514. doi: 10.1200/JCO.2023.41.16_suppl.3514
167. Takai N, Kira N, Ishii T, Nishida M, Nasu K, Narahara H. Novel chemotherapy using histone deacetylase inhibitors in cervical cancer. Asian Pac J Cancer Prev. (2011) 12(3):575–80.
168. Klapdor R, Wang S, Hacker U, Büning H, Morgan M, Dörk T, et al. Improved killing of ovarian cancer stem cells by combining a novel chimeric antigen receptor-based immunotherapy and chemotherapy. Hum Gene Ther. (2017) 28:886–96. doi: 10.1089/hum.2017.168
169. Liao ZJ, Guo YH, Zhao Z, Yao JT, Xu R, Nan KJ. Gemcitabine inhibits the micrometastasis of non-small cell lung cancer by targeting the EpCAM-positive circulating tumor cells via the HGF/cMET pathway. Int J Oncol. (2014) 45:651–8. doi: 10.3892/ijo.2014.2464
170. Teng JP, Yang ZY, Zhu YM, Ni D, Zhu ZJ, Li XQ. Gemcitabine and cisplatin for treatment of lung cancer in vitro and vivo. Eur Rev Med Pharmacol Sci. (2018) 22:3819–25. doi: 10.26355/eurrev_201806_15266
171. Zhang G, Wang Y, Fuchs BC, Guo W, Drum DL, Erstad DJ, et al. Improving the therapeutic efficacy of sorafenib for hepatocellular carcinoma by repurposing disulfiram. Front Oncol. (2022) 12:913736. doi: 10.3389/fonc.2022.913736
172. Kim BH, Park JW, Kim JS, Lee SK, Hong EK. Stem cell markers predict the response to sorafenib in patients with hepatocellular carcinoma. Gut Liver. (2019) 13:342–8. doi: 10.5009/gnl18345
173. Li X, Li T, Liu Z, Gou S, Wang C. The effect of metformin on survival of patients with pancreatic cancer: a meta-analysis. Sci Rep. (2017) 7:5825. doi: 10.1038/s41598-017-06207-x
174. Gou S, Cui P, Li X, Shi P, Liu T, Wang C. Low concentrations of metformin selectively inhibit CD133+ cell proliferation in pancreatic cancer and have anticancer action. PloS One. (2013) 8:e63969. doi: 10.1371/journal.pone.0063969
175. Yu AL, Gilman AL, Ozkaynak MF, Naranjo A, Diccianni MB, Gan J, et al. Long-term follow-up of a phase III study of ch14.18 (Dinutuximab) + Cytokine immunotherapy in children with high-risk neuroblastoma: COG study ANBL0032. Clin Cancer Res. (2021) 27:2179–89. doi: 10.1158/1078-0432.CCR-20-3909
176. Ladenstein R, Pötschger U, Valteau-Couanet D, Luksch R, Castel V, Yaniv I, et al. Interleukin 2 with anti-GD2 antibody ch14.18/CHO (dinutuximab beta) in patients with high-risk neuroblastoma (HR-NBL1/SIOPEN): a multicentre, randomised, phase 3 trial. Lancet Oncol. (2018) 19:1617–29. doi: 10.1016/S1470-2045(18)30578-3
177. Peng CK, Sung WW, Chen SL. Clinical efficacy of neoadjuvant intravesical mitomycin-C therapy immediately before transurethral resection of bladder tumor in patients with nonmuscle-invasive bladder cancer: preliminary results of a prospective, randomized phase II study. Letter. J Urol. (2023) 209:845–6. doi: 10.1097/JU.0000000000003407
178. Fizazi K, Faivre L, Lesaunier F, Delva R, Gravis G, Rolland F, et al. Androgen deprivation therapy plus docetaxel and estramustine versus androgen deprivation therapy alone for high-risk localised prostate cancer (GETUG 12): a phase 3 randomised controlled trial. Lancet Oncol. (2015) 16:787–94. doi: 10.1016/S1470-2045(15)00011-X
179. Crowell PD, Fox JJ, Hashimoto T, Diaz JA, Navarro HI, Henry GH, et al. Expansion of luminal progenitor cells in the aging mouse and human prostate. Cell Rep. (2019) 28:1499–1510.e6. doi: 10.1016/j.celrep.2019.07.007
180. Melichar B, Bracarda S, Matveev V, Alekseev B, Ivanov S, Zyryanov A, et al. A multinational phase II trial of bevacizumab with low-dose interferon-α2a as first-line treatment of metastatic renal cell carcinoma: BEVLiN. Ann Oncol. (2013) 24:2396–402. doi: 10.1093/annonc/mdt228
181. Escudier B, Bellmunt J, Négrier S, Bajetta E, Melichar B, Bracarda S, et al. Phase III trial of bevacizumab plus interferon alfa-2a in patients with metastatic renal cell carcinoma (AVOREN): final analysis of overall survival. J Clin Oncol. (2010) 28:2144–50. doi: 10.1200/JCO.2009.26.7849
182. Deshpande AJ, Deshpande A, Sinha AU, Chen L, Chang J, Cihan A, et al. AF10 regulates progressive H3K79 methylation and HOX gene expression in diverse AML subtypes. Cancer Cell. (2014) 26:896–908. doi: 10.1016/j.ccell.2014.10.009
183. Menghrajani K, Cai SF, Devlin SM, Armstrong SA, Piekarz R, Rudek M, et al. A phase ib/II study of the histone methyltransferase inhibitor pinometostat in combination with azacitidine in patients with 11q23-rearranged acute myeloid leukemia. Blood. (2019) 134:2655. doi: 10.1182/blood-2019-121926
184. Yardley DA, Weaver R, Melisko ME, Saleh MN, Arena FP, Forero A, et al. EMERGE: A randomized phase II study of the antibody-drug conjugate glembatumumab vedotin in advanced glycoprotein NMB-expressing breast cancer. J Clin Oncol. (2015) 33:1609–19. doi: 10.1200/JCO.2014.56.2959
185. Park DJ, Sung PS, Kim JH, Lee GW, Jang JW, Jung ES, et al. EpCAM-high liver cancer stem cells resist natural killer cell-mediated cytotoxicity by upregulating CEACAM1. J Immunother Cancer. (2020) 8:e000301. doi: 10.1136/jitc-2019-000301
186. Russ A, Hua AB, Montfort WR, Rahman B, Riaz IB, Khalid MU, et al. Blocking “don’t eat me” signal of CD47-SIRPα in hematological Malignancies, an in-depth review. Blood Rev. (2018) 32:480–9. doi: 10.1016/j.blre.2018.04.005
187. Jakubison BL, Schweickert PG, Moser SE, Yang Y, Gao H, Scully K, et al. Induced PTF1a expression in pancreatic ductal adenocarcinoma cells activates acinar gene networks, reduces tumorigenic properties, and sensitizes cells to gemcitabine treatment. Mol Oncol. (2018) 12:1104–24. doi: 10.1002/1878-0261.12314
188. Martinez-Quintanilla J, Bhere D, Heidari P, He D, Mahmood U, Shah K. Therapeutic efficacy and fate of bimodal engineered stem cells in Malignant brain tumors. Stem Cells. (2013) 31:1706–14. doi: 10.1002/stem.1355
189. Hanker AB, Pfefferle AD, Balko JM, Kuba MG, Young CD, Sánchez V, et al. Mutant PIK3CA accelerates HER2-driven transgenic mammary tumors and induces resistance to combinations of anti-HER2 therapies. Proc Natl Acad Sci U S A. (2013) 110:14372–7. doi: 10.1073/pnas.1303204110
190. Baselga J, Bradbury I, Eidtmann H, Di Cosimo S, de Azambuja E, Aura C, et al. Lapatinib with trastuzumab for HER2-positive early breast cancer (NeoALTTO): a randomised, open-label, multicentre, phase 3 trial. Lancet. (2012) 379:633–40. doi: 10.1016/S0140-6736(11)61847-3
191. Verissimo CS, Overmeer RM, Ponsioen B, Drost J, Mertens S, Verlaan-Klink I, et al. Targeting mutant RAS in patient-derived colorectal cancer organoids by combinatorial drug screening. Elife. (2016) 5:e18489. doi: 10.7554/eLife.18489
192. Huang D, Ma N, Li X, Gou Y, Duan Y, Liu B, et al. Advances in single-cell RNA sequencing and its applications in cancer research. J Hematol Oncol. (2023) 16:98. doi: 10.1186/s13045-023-01494-6
193. Honeth G, Schiavinotto T, Vaggi F, Marlow R, Kanno T, Shinomiya I, et al. Models of breast morphogenesis based on localization of stem cells in the developing mammary lobule. Stem Cell Rep. (2015) 4:699–711. doi: 10.1016/j.stemcr.2015.02.013
194. Muralikrishnan V, Hurley TD, Nephew KP. Targeting aldehyde dehydrogenases to eliminate cancer stem cells in gynecologic Malignancies. Cancers (Basel). (2020) 12:961. doi: 10.3390/cancers12040961
195. Liu YP, Yang CJ, Huang MS, Yeh CT, Wu AT, Lee YC, et al. Cisplatin selects for multidrug-resistant CD133+ cells in lung adenocarcinoma by activating Notch signaling. Cancer Res. (2013) 73:406–16. doi: 10.1158/0008-5472.CAN-12-1733
196. Chen J, Xu Z, Huang H, Tang Y, Shan H, Xiao F. SETD1A drives stemness by reprogramming the epigenetic landscape in hepatocellular carcinoma stem cells. JCI Insight. (2023) 8:e168375. doi: 10.1172/jci.insight.168375
197. Tomolonis JA, Agarwal S, Shohet JM. Neuroblastoma pathogenesis: deregulation of embryonic neural crest development. Cell Tissue Res. (2018) 372:245–62. doi: 10.1007/s00441-017-2747-0
198. Lasorsa F, Rutigliano M, Milella M, Ferro M, Pandolfo SD, Crocetto F, et al. Cancer stem cells in renal cell carcinoma: origins and biomarkers. Int J Mol Sci. (2023) 24:13179. doi: 10.3390/ijms241713179
199. Ding Y, Huang D, Zhang Z, Smith J, Petillo D, Looyenga BD, et al. Combined gene expression profiling and RNAi screening in clear cell renal cell carcinoma identify PLK1 and other therapeutic kinase targets. Cancer Res. (2011) 71:5225–34. doi: 10.1158/0008-5472.CAN-11-0076
200. Valent P, Bonnet D, De Maria R, Lapidot T, Copland M, Melo JV, et al. Cancer stem cell definitions and terminology: the devil is in the details. Nat Rev Cancer. (2012) 12:767–75. doi: 10.1038/nrc3368
201. Tang DG. Understanding cancer stem cell heterogeneity and plasticity. Cell Res. (2012) 22:457–72. doi: 10.1038/cr.2012.13
202. Eun K, Ham SW, Kim H. Cancer stem cell heterogeneity: origin and new perspectives on CSC targeting. BMB Rep. (2017) 50:117–25. doi: 10.5483/bmbrep.2017.50.3.222
203. Stein EM, Fathi AT, DiNardo CD, Pollyea DA, Roboz GJ, Collins R, et al. Enasidenib in patients with mutant IDH2 myelodysplastic syndromes: a phase 1 subgroup analysis of the multicentre, AG221-C-001 trial. Lancet Haematol. (2020) 7:e309–19. doi: 10.1016/S2352-3026(19)30284-4
204. Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. (2018) 15:81–94. doi: 10.1038/nrclinonc.2017.166
205. Awad O, Yustein JT, Shah P, Gul N, Katuri V, O'Neill A, et al. High ALDH activity identifies chemotherapy-resistant Ewing’s sarcoma stem cells that retain sensitivity to EWS-FLI1 inhibition. PloS One. (2010) 5:e13943. doi: 10.1371/journal.pone.0013943
206. Shibue T, Weinberg RA. EMT, CSCs, and drug resistance: the mechanistic link and clinical implications. Nat Rev Clin Oncol. (2017) 14:611–29. doi: 10.1038/nrclinonc.2017.44
207. Kozar S, Morrissey E, Nicholson AM, van der Heijden M, Zecchini HI, Kemp R, et al. Continuous clonal labeling reveals small numbers of functional stem cells in intestinal crypts and adenomas. Cell Stem Cell. (2013) 13:626–33. doi: 10.1016/j.stem.2013.08.001
208. Matsumoto R, Yamamoto T, Takahashi Y. Complex organ construction from human pluripotent stem cells for biological research and disease modeling with new emerging techniques. Int J Mol Sci. (2021) 22:10184. doi: 10.3390/ijms221910184
209. Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. (2014) 344:1396–401. doi: 10.1126/science.1254257
210. Keyvani-Ghamsari S, Khorsandi K, Rasul A, Zaman MK. Current understanding of epigenetics mechanism as a novel target in reducing cancer stem cells resistance. Clin Epigenetics. (2021) 13:120. doi: 10.1186/s13148-021-01107-4
211. Huang T, Song X, Xu D, Tiek D, Goenka A, Wu B, et al. Stem cell programs in cancer initiation, progression, and therapy resistance. Theranostics. (2020) 10:8721–43. doi: 10.7150/thno.41648
212. Bissell MJ, Hines WC. Why don’t we get more cancer? A proposed role of the microenvironment in restraining cancer progression. Nat Med. (2011) 17:320–9. doi: 10.1038/nm.2328
Keywords: cancer stem cells, cell-of-origin, tumorigenesis, tumor heterogeneity, cancer therapeutics, therapy resistance, personalized medicine, tumor microenvironment
Citation: Bamodu OA, Chung C-C, Pisanic TR II and Wu ATH (2024) The intricate interplay between cancer stem cells and cell-of-origin of cancer: implications for therapeutic strategies. Front. Oncol. 14:1404628. doi: 10.3389/fonc.2024.1404628
Received: 21 March 2024; Accepted: 25 April 2024;
Published: 10 May 2024.
Edited by:
Keqiang Zhang, City of Hope National Medical Center, United StatesReviewed by:
Rajendra Pangeni, Nova Southeastern University, United StatesJinghan Wang, Tongji University, China
Song-Bai Liu, Suzhou Vocational Health College, China
Copyright © 2024 Bamodu, Chung, Pisanic and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Oluwaseun Adebayo Bamodu, ZHJfYmFtb2R1QHlhaG9vLmNvbQ==