
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 20 June 2024
Sec. Hematologic Malignancies
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1396435
This article is part of the Research Topic Allogenic Hematopoietic Cell Transplant in Hematological Malignancies: Controversies and Perspective View all 13 articles
 Paola Ranalli1,2
Paola Ranalli1,2 Annalisa Natale1Francesco Guardalupi2Stella Santarone1Chiara Cantò1Gaetano La Barba1
Annalisa Natale1Francesco Guardalupi2Stella Santarone1Chiara Cantò1Gaetano La Barba1 Mauro Di Ianni1,2*
Mauro Di Ianni1,2*New available drugs allow better control of systemic symptoms associated with myelofibrosis (MF) and splenomegaly but they do not modify the natural history of progressive and poor prognosis disease. Thus, hematopoietic stem cell transplantation (HSCT) is still considered the only available curative treatment for patients with MF. Despite the increasing number of procedures worldwide in recent years, HSCT for MF patients remains challenging. An increasingly complex network of the patient, disease, and transplant-related factors should be considered to understand the need for and the benefits of the procedure. Unfortunately, prospective trials are often lacking in this setting, making an evidence-based decision process particularly arduous. In the present review, we will analyze the main controversial points of allogeneic transplantation in MF, that is, the development of more sophisticated models for the identification of eligible patients; the need for tools offering a more precise definition of expected outcomes combining comorbidity assessment and factors related to the procedure; the decision-making process about the best transplantation time; the evaluation of the most appropriate platform for curative treatment; the impact of splenomegaly; and splenectomy on outcomes.
Myelofibrosis (MF) includes primary myelofibrosis (PMF) and secondary myelofibrosis (SMF). The latter includes post-essential thrombocythemia (PET) and post-polycythemia vera (PPV) myelofibrosis. PET and PPV are associated with inferior overall survival (OS) rates compared to PMF, often due to its higher risk of leukemic transformation (LT). According to WHO and International Consensus Classification of Myeloid Meoplasms and Acute Leukemias (ICC’s) current diagnostic criteria, PMF may present two different clinical pictures: prefibrotic or early myelofibrosis (pre-PMF) and overt myelofibrosis, differing from each other essentially in the bone marrow grade of fibrosis (1–3).
MF treatment options are still limited. The treatment of low-risk MF is generally related to symptom severity. The treatment of high-risk diseases includes JAK inhibitors (JAKi) and allogeneic hematopoietic stem cell transplantation (HSCT). HSCT remains the only chance of a definitive cure for patients with both primary and secondary myelofibrosis (4). After a failure of conventional treatments, in MF patients ineligible for HSCT, enrollment in clinical trials represents an alternative option, when available.
Guidelines recommend upfront allogeneic bone marrow transplantation in patients with high-risk disease, following data from a retrospective study showing patients with intermediate-2 or high-risk score in the Dynamic International Prognostic Scoring System (DIPSS) who benefited the most from transplant than conventional therapy only (5–8).
Thus, an accurate assessment of MF-related risk should be provided by clinicians to promptly identify patients with < 5 years of expected survival, potentially candidates for hematopoietic cell transplantation. Information about transplant-related morbidity and mortality, the expected post-HSCT outcome, as well as MF-related risk, should be adequately shared with patients and their families. Such an integrated evaluation may allow proper counseling about global post-transplant prognosis. Finally, the decision has to be always taken on an individual basis (9).
Moreover, novel strategies for patient and donor selection, conditioning regimens, and post-transplant care in the last years allowed the reduction of disease relapse incidence, 5-year non-relapse mortality and survival in related and unrelated donor transplants in patients with myeloproliferative neoplasms (10), thus, justifying allo-HSCT as a curative option in younger patients of all risk categories and not just in high-risk diseases (11, 12) or in carriers of high-risk non-driver mutations (EZH2, ASXL1, IDH1/2, and SRSF2), predictive of inferior OS and disease-free survival (DFS) (13).
HSCT for MF patients remains challenging, particularly in older age patients or in those with cytopenias, splenomegaly, and severe bone marrow fibrosis (14).
Certainly, the global number of HSCTs in MF rose recently, signifying the increased interest in the only available curative treatment for the disease.
In the present review, we will analyze the main controversial points concerning allogeneic bone marrow transplantation in myelofibrosis.
Older patients are often carriers of metabolic or systemic comorbidities making them more vulnerable to toxicity associated with treatment (10, 15). In studies with a long follow-up, the recipient’s age was shown to have an impact on overall survival (OS) and on the risk of treatment failure (16). In a single-center retrospective study on patients with MF who underwent a reduced intensity conditioned (RIC) HSCT, older male patients reported an increased incidence of poor graft function (P = 0.05). This phenomenon was not associated with an increased risk of relapse/progression and did not impact OS (17).
Accordingly, age has been considered the most important factor adversely impacting transplant outcomes in myeloproliferative neoplasms (MPN) (17, 18) and clinicians have been traditionally reluctant to offer the procedure to older patients.
The age of 70 represented the upper limit established in European Society for Blood and Marrow Transplantation (EBMT)/Europena Leukemia Net (ELN) recommendations to proceed with allogeneic transplantation in subjects with intermediate-2 or high-risk disease (19).
Recently, encouraging results were shown in studies involving older patients (>70) undergoing HSCT for MF (20). Engraftment, rates of graft-vs. -host disease (GvHD), progression-free survival (PFS), and OS comparable to those reported in younger patients were shown in selected patients with primary or secondary myelofibrosis aged 60 to 78 years old who received HSCT from Human Leukocyte Antigen (HLA)-identical siblings or unrelated donors (21). Thus, age should not represent an absolute contraindication to allo-HSCT, in case of absent or well-controlled comorbidities (6).
The use of RIC regimens allow more favorable survival in older patients aged > 65 years old with no or minimal comorbidities (22). EBMT/ELN recommendations suggest considering the possibility of HSCT case by case, taking into account patients’ and disease variables and also the recipient’s preferences (6).
Traditional prognostic models include the International Prognostic Scoring System or IPSS (only applicable to newly diagnosed patients) (23); the dynamic IPSS or DIPSS (24) (applicable at any time point after diagnosis); and the DIPSS plus (25). They all include clinical parameters only (age, anemia, leukocytosis, circulating blasts, and constitutional symptoms), each independently predicting inferior survival. DIPSS plus also includes thrombocytopenia (platelets <100 × 109/L), unfavorable karyotyping (traditionally established), and the need for transfusion support (26).
In the pre-ruxolitinib era, only high-risk patients seemed to gain the greatest survival advantage from transplantation. A retrospective multicenter study including 438 patients with primary or PET and PPV myelofibrosis aged less than 65 years clearly showed a significantly lower risk of death after HSCT in comparison with subjects treated with conventional therapies only in case of DIPSS intermediate-2 and high-risk patients (respectively, p: 0.005 and 0.0007 vs. conventional therapies) (5).
Scoring systems including more prognostic parameters, mainly molecular data, were later introduced (Table 1). Among driver mutations, it has become clear that CALR, particularly type I mutation, is associated with a more indolent course, thus its protective function against progressive disease has been recognized and with better post-transplant outcome [higher 4-year OS and lower 4-year non-relapse mortality (NRM) after allo-HSCT] (27–29). On the opposite, the worst prognosis is associated with the JAK2/CALR/MPL triple-negative profile (30) (31).
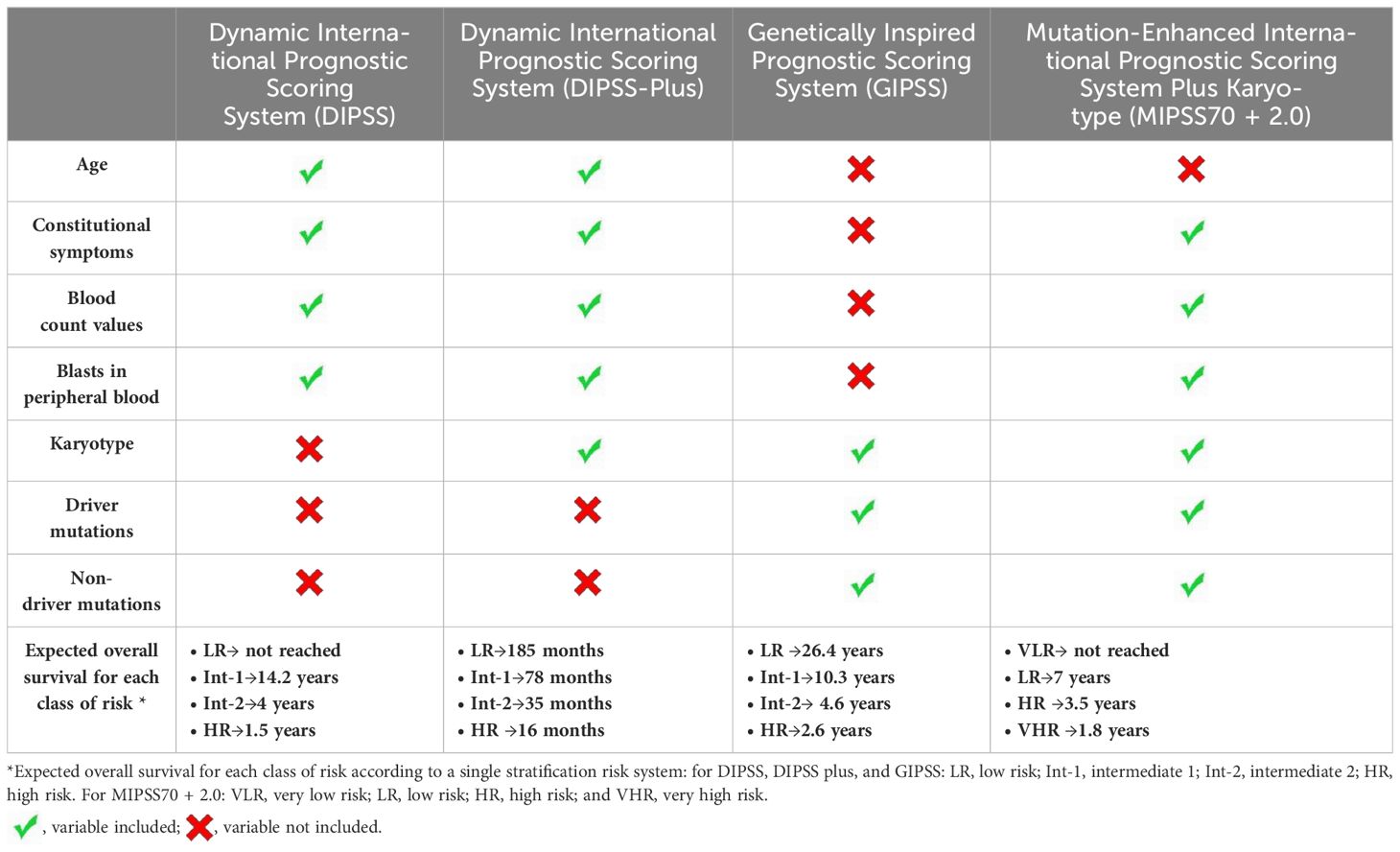
Table 1 Variables included in prognostic scores applied in myelofibrosis and identification of patients with poor OS.
Among non-driver mutations, ASXL1, EZH2, IDH1/2, and SRSF2 were identified as mutations associated with a poor prognosis, defining the group of “high molecular risk (HMR) mutations,” linked with poorer prognosis (31–33). The Mutation Enhanced International Prognostic Score Systems (MIPSS) were designed for HSCT decision-making in patients aged ≤70 years.
MIPSS70 identifies as significant risk factors for OS both clinical factors (anemia, leukocytosis, thrombocytopenia, constitutional symptoms, and circulating blasts ≥2%) and nonclinical conditions, not included in the previous traditional models, such as bone marrow fibrosis grade ≥2, absence of CALR type-1 mutation, presence of high-molecular risk mutation (ASXL1, EZH2, SRSF2, and IDH1/2), and the number of two or more high-molecular risk mutations.
The MIPSS70-plus is enriched with cytogenetic information (34), adding a more refined definition of cytogenetic risk, thus providing three risk categories. MIPSS70-plus version 2.0 represents a more complex system including more non-driver mutations (U2AF1) and also considering the number of high-risk mutations and a three-tiered cytogenetic risk classification as independent prognostic factors (34–37). New prognostic thresholds considering severity and sex-adjusted values for hemoglobin levels were also integrated (38).
Genetically inspired prognostic scoring system for primary myelofibrosis (GIPSS) focuses only on genetic and molecular factors, particularly on a limited number of non-driver lesions, without considering clinical parameters at all (39).
Patients may be identified for the HSCT path also according to the best response obtained to pharmacological therapy used as “bridge to transplant,” usually JAKi. Lower rates of responses to ruxolitinib have been shown in cytopenic vs. non-cytopenic MF, making cytopenic patients more often considered for earlier HSCT (40).
Moreover, in a recent retrospective study, a positive correlation has been found between peripheral blood CD34 cells and spleen length in both PMF and SMF, thus identifying a possible tool facilitating the assessment of spleen response more objectively than deep palpation of the abdomen (41).
Discrepancies emerged from the application of standard prognostic scores in patients with secondary MF (42). MYelofibrosis SECondary to PV and ET prognostic Model (MYSEC-PM) was validated as the only specific prognostic tool suitable for patients with MF secondary to PV and ET including both clinical and molecular data (43).
Disagreement between modern prognostic scores and traditional scores based on only clinical parameters have been observed (34, 44). GIPSS and clinical-only scores may differ quite frequently as they do not share any variable. That is why at the same risk class, an inferior OS and a worse leukemia-free survival (LFS) were shown in genomically vs. clinically established higher-risk patients (p = 0.08 and p = 0.04, respectively) (44).
To ensure the most complete evaluation of disease-associated risk, a simultaneous assessment of as many scores as possible could be facilitated by a PMF-specific calculator (45). However, recent EBMT/ELN recommendations indicate allogeneic HSCT based exclusively on DIPSS, MIPSS70, and MIPSS70 plus scores (6).
Many other disease-specific factors have an impact on the outcome as increased circulating CD34+ cells, increased bone marrow or circulating blasts (46, 47), TP53, CBL, N/KRAS mutations (48), triple negativity (31), cytopenic PMF (49). The latter is associated with both higher rates of leukemic transformation and worse survival, and generally, it pairs up with JAK2 V617F allele burden, less prominent splenomegaly, greater genomic complexity and increased risk for infections and bleeding (50).
Splenomegaly itself is not included among the relevant parameters of the prognostic scores used for myelofibrosis despite the fact that larger baseline spleen volume correlates with an increased risk of death in the COntrolled MyeloFibrosis study with ORal jak inhibitor Treatment (COMFORT) studies (51).
Studying a large cohort of patients with MPN (n = 2,035), not necessarily with MF, Grinfeld et al. identified distinct subgroups of MF patients with distinct clinical, cytogenetic, and mutational features, thus developing a personalized MPN risk calculator predicting survival and leukemic transformation, and demonstrating for the first time, the detrimental effect of mutated TP53 on survival in MF patients (52).
The role of current prognostic systems in predicting outcomes after HSCT is still uncertain (12, 53–55). DIPSS and DIPSS plus have been shown as predictive tools also for survival following HSCT despite not including the evaluation of transplant-specific variables (5, 53, 56). In SMF, MYSEC was predictive also for survival after allogeneic HSCT, as shown in a recent study (57).
A recent retrospective study published this year by Polverelli et al. on behalf of the Chronic Malignancies Working Party of EBMT confirmed the relevant and negative impact of comorbidities on HSCT outcomes for patients with MF, underlining the need to integrate such an information in the selection process (58).
The hematopoietic cell transplantation-specific comorbidity index, better known as Sorror index provides a reliable scoring of pretransplant comorbidities to more precisely define both non-relapse mortality (NRM) and survival (OS), showing a better prediction power than the Charlson Comorbidity Index (CCI). It is usually applied to all hematological diagnoses, MF included, despite having patients diagnosed with MF who were not included in the validation cohort (59).
Undoubtedly, patient- and transplant-specific risk factors like the intensity of the conditioning regimen, recipient age, cytomegalovirus serostatus, performance status or HLA matching of the donor, influence the patient’s post-HSCT outcome (19, 60).
“Myelofibrosis transplant scoring system” or MTSS, a four-level clinical-molecular score including clinical data, donor type, and mutation status for ASXL1/CALR/MPL, has been validated as a specific prognostic tool for an objective evaluation of the risk/benefit ratio of HSCT in the counseling phase, before transplantation (61).
MTSS identified independent risk factors for poor survival after transplant (pretransplantation thrombocytopenia, leukocytosis, older age, poor performance according to Karnofsky performance status, a non-CALR/MPL driver mutation genotype, ASXL1-mutation and transplantation from an HLA-mismatched unrelated donor). It should be noted that it does not include significant risk factors (from existing scoring systems) like anemia and transfusion dependency, constitutional symptoms, cytogenetic risk stratification, or the presence of two or more HMR mutations (61).
Unfortunately, the MTSS scoring system did not maintain its predictive role in other series of cases (62). Another limitation on the use of MTSS is the lack of information about comorbidities. Therefore, the application of MTSS does not disregard the need for a comorbidity index evaluation as well.
Outside the MTSS score, other variables such as spleen size, transfusion history, donor type (11), JAKV617F status, age, and constitutional symptoms are predictive of 5-year OS (55).
Recently, a detrimental effect on transplant outcome was shown in carriers of TP53 mutations of a large multicenter cohort. In particular, higher mortality was demonstrated as a consequence of higher rates of early leukemic transformation, almost a case of “multi-hit constellation” (63).
In conclusion, there is experts’ consensus on the eligibility to transplant for intermediate-2/high-risk DIPSS patients, high-risk MIPSS70 or MIPSS70-plus, high-risk or intermediate-2 MYSEC-PM who, at the same time, present a low to intermediate-risk profile according to MTSS.
Allogeneic HSCT should also be offered to DIPSS intermediate 1 risk patients and to MIPSS70 or MIPSS intermediate patients who present a low-risk profile with MTSS, taking into great consideration patients’ preferences, response to treatment, and other issues such as availability of clinical trial or additive data (6).
Traditionally, variables to consider in non-high-risk patients with MF, suggesting eligibility for transplant are represented by (1) transfusion-dependent anemia, (2) a percentage of blasts in peripheral blood > 2%, (3) adverse cytogenetics, and (4) high-risk mutations (64).
Splenomegaly is a hallmark of both primary and PET and PPV myelofibrosis, as it represents the malignant clone expanding outside the bone marrow. The real impact of spleen size and eventual splenectomy on HSCT outcomes in myelofibrosis is still debated.
Several studies have shown that splenomegaly can adversely impact transplant outcomes, as it may promote the sequestration of hematopoietic progenitors (65, 66).
In a retrospective study involving a limited number of patients with myelofibrosis, the authors considered massive splenomegaly as one of the variables adversely affecting the outcome of HSCT. This variable was included in a scoring tool used for decision-making, alongside other variables such as a transfusion history of > 20 red blood cell units before transplantation and the type of alternative donor (11).
The effect of a huge spleen on OS and relapse after allo-HSCT is not completely clear, as conflicting data emerged from other works (60, 67, 68).
Furthermore, splenomegaly was associated with a higher risk of relapse after transplantation in recent studies (69, 70).
Potentially, splenectomy before HSCT could be useful for disease debulking and also to favor a faster hematopoietic recovery (66, 71) but some other data, in contrast, did not confirm it (12, 72).
A retrospective EBMT study on 1,000 cases of MF splenectomized in comparison with non-splenectomized patients, did not report a different OS (P = 0.274) rate, but the results seemed associated with a lower rate of NRM (P = 0.018) and increased risk of relapse (P = 0.042). However, in a subanalysis considering splenectomy in different subgroups of patients, an improved outcome was reported with splenectomy in subjects with a palpable spleen length ≥ 15 cm (better OS, significant reduction in NRM, not significantly increased relapse risk, P <.001, P <.001, and P = .147 for each phenomenon) (73).
How splenectomy affects the risk of disease relapse and survival after HSCT is still unclear, thus, making it mandatory for future more prospective randomized trials.
Moreover, data available from retrospective studies on GvHD in previously splenectomized patients are quite conflicting (65, 67, 74).
The course of splenectomy can be complicated by thrombosis, bleeding, infections in up to 30% of patients, disease transformation, and death (peri-operative mortality is in the range of 5%–10%) (75). All complications eventually preclude or simply delay allo-HSCT (72, 76–78).
A multicenter retrospective study on 530 patients with a diagnosis of myelofibrosis from the French bone marrow transplantation registry (RFGM) who underwent splenectomy in the period 2008–2017 showed reassuring results, as pretransplant splenectomy did not preclude allo-HSCT; in particular, splenectomized patients had a higher rate of transplantation in the first 4 months after splenectomy [HR (95% CI) = 7.2 (5.1–10.3)] but not after this time point (79).
As spleen size in patients with MF sensitively benefit from JAKi (80–82), the need for splenectomy has to be discussed rarely nowadays.
Despite not being routinely performed or recommended, splenectomy remains useful in patients who did not benefit from therapy with JAKi, with residual massive splenomegaly.
Splenic irradiation represents a further alternative to splenectomy to reduce spleen size and alleviate splenic discomfort, although the results of such therapy are generally short-lasting and associated with the risk of severe cytopenias, eventually difficult to manage (83).In contrast, the results of a recent retrospective study on HSCT for MF preceded by splenic irradiation are encouraging, showing a reduced relapse after HSCT, without association between total irradiation dose and efficacy (84).
Therefore, it may be considered for patients not eligible for surgery or who were no longer responsive to JAKi (85, 86).
The impact of splenic radiotherapy in leukemic transformation (LT) is still unclear; conversely, similar engraftment rates and GvHD incidence have been described in patients who underwent splenic irradiation or not (74).
The option of upfront HSCT is still recommended for patients stratified as intermediate 2 and high-risk DIPSS, with an expected survival of fewer than 5 years (6), as it provides the best gain in life expectancy. This indication was reinforced by a decision analysis recently published (87).
In the case of a patient with intermediate-risk disease, the decision about HSCT requires a more tailored approach, and generally, the procedure can be delayed; usually, in this setting, more prognostic factors, even outside traditional scores, have to be considered, identifying those patients less likely to have lasting response from non-transplant therapy (6, 88).
The best timing of HSCT has become a more controversial point in the era of JAKi as these drugs produce a better action on spleen size, constitutional symptoms, and also on survival outcomes.
Studies showed better response in patients treated earlier during the disease course with both HSCT (56) and JAKi, thus, decision-making about transplant becomes even more complex. It should be underlined that JAKi are not curative (89, 90), and they do not prevent the progression to blast phase or leukemic transformation, the main determinant of death in MF (23, 91, 92).
Furthermore, despite the success with ruxolitinib in the frontline setting, discontinuation of JAKi therapy may occur because of intolerance or refractoriness, events associated with a poor OS according to retrospective studies (93, 94).
Comparative studies testing the results of upfront HSCT approaches with non-transplant therapies of the JAKi era are still lacking, thus leading to a wide variability of conducts on the use of HSCT in MF. In 2024, upfront JAKi therapy was compared with upfront HSCT strategy in MF patients not older than 70 years old in a large, multicenter and retrospective study; in patients treated with upfront HSCT, an earlier mortality was observed and in general, they do not report significative benefit (95).
Thus, one can imagine that in the “JAKi era,” HSCT is limited to cases of cytopenic myelofibrosis, not manageable with cytoreductive or JAKi therapy; could be delayed until response to JAKi is lost, and that delaying time could become even longer as more than one JAKi has become available (21). In fact, according to some recent data, fedratinib or other JAKi may improve upon the poor prognosis associated with ruxolitinib discontinuation (96, 97).
Advanced-stage disease, increasing age, or leukemic transformation, often associated with the emergence of acquired unfavorable mutations, could represent the dramatic consequences of delaying the HSCT procedure, as impactful disease-modifying therapy other than HSCT still does not exist.
For this reason, many authors underline that patients whose therapeutic goal is cure should still undergo HSCT even if responding to JAKi (98).
The possibility to rapidly obtain a spleen response with JAKi represents an attractive option for clinicians looking for a “bridge to transplant strategy,” thus eventually making engraftment time more rapid (99, 100). The use of JAKi as pre-HSCT strategy is increasing and offers encouraging results. With this approach, eligible patients should undergo HSCT at the time of the best response to JAKi (100).
In non-randomized and retrospective studies, the treatment of patients with ruxolitinib in the phase preceding HSCT is well-tolerated and associated with better post-transplant outcomes and survival (101, 102). The prospective phase-2 trial JAK ALLO study showed that a short course of ruxolitinib administered before HSCT and stopped progressively or abruptly before the conditioning regimen is safe and associated with a high probability of HSCT for those with a donor and no increased risk of disease progression (103).
The initiation of ruxolitinib is recommended ≥2 months before HSCT, careful weaning 5–7 days before conditioning, and complete withdrawal on the day before conditioning according to the European guidelines for primary MF (19). Adverse events happened in patients who stopped JAK inhibitor ≥ 6 days before conditioning therapy (104), while they were infrequent in those treated with JAK inhibitor until HSCT conditioning therapy was started (105).
Future studies will clarify the hypothesis that JAKi treatment in candidates for HSCT may reduce the incidence of poor graft function (17).
It should be noted that in MF patients pre-treated with ruxolitinib for 6 months before HSCT, different outcomes were shown according to the type of donor. In particular, poorer mortality and GvHD outcomes were associated with patients receiving HSCT from an unrelated donor compared to those with a matched sibling donor in JAK ALLO phase-2 trial. These results could be explained by many factors such as advanced disease, loss of response to ruxolitinb at the time of HSCT or insufficient period of treatment, thus not showing a direct impact of ruxolitinib on post-HSCT outcomes (103).
In a multicenter German study reporting the experience of ruxolitinib pretreatment in 159 MF patients who underwent RIC HSCT between 2000 and 2015 from different types of donors, ruxolitinib did not negatively impact HSCT outcomes, as similar outcomes were shown in non-ruxolitinib pre-treated patients. Similar OS, DFS, and GvHD were reported among ruxolitinib responders and those who failed to respond or were no longer responsive to JAKi (106).
Following JAKi failure (93, 107), HSCT should be considered in any patient (108), according to little data from retrospective studies showing improved survival with HSCT in this setting (109).
The treatment landscape has become more intricate with the availability of fedratinib and novel combination strategies involving ruxolitinib within clinical trials. Nevertheless, HSCT remains a viable option for eligible candidates. Despite the efficacy of fedratinib on splenomegaly, there is still lack of information on the use of this agent or other novel agents, as an alternative to ruxolitinib, before HSCT (6).
Thus, response to ruxolitinib should be systematically assessed 6 months after initiating therapy (6), as recently recommended by EBMT/ELN.
The model, named Response to Ruxolitinib After 6 Months (RR6), was validated as a prognostic model allowing the identification of MF patients who have already been treated with ruxolitinib for 6 months and in need of second-line treatment strategies, HSCT included. The predictive role of such a prognostic tool, evaluating three variables (drug dose, spleen response, and transfusion requirement) and thus stratifying the risk into three categories (low, intermediate, and high) overcomes conventional risk stratification in MF treated with ruxolitinib (110). According to the RR6 model, high-risk patients need a prompt evaluation for HSCT (6).
Donor type is an important predictor of outcome for MF transplanted patients, with HLA-matched sibling donors (MSD) being preferred over matched unrelated donors (MUD) and mismatched unrelated donors (mMUD). Gupta et al. reported HSCT outcomes of 233 MF patients for CIMBTR. In multivariate analysis, donor type was the sole independent factor associated with survival (5-year OS was 56%, 48%, and 34% for MSD, MUD, and mMUD, respectively) (111).
Alternative donor options in MF expand the donor pool in patients who do not have a suitable sibling or unrelated donor. Unrelated cord blood units are rarely used in MF patients, with graft failure remaining a major concern. A retrospective study from the EBMT registry evaluated 35 patients who received cord blood HSCT reporting 2 years of OS and EFS rates being 44% and 30%, respectively (112).
The haploidentical setting is still under investigation with improving results over time. Bregante et al. evaluated the outcome of 95 patients with myelofibrosis who were allografted between 2001 and 2014. The 3-year HSCT-related mortality (TRM), relapse rate, and overall survival were 16% vs. 32%, 16% vs. 40%, and 70% vs. 39%, respectively, in the 2011 to 2014 period versus the 2000 to 2010 period. Improved survival was most pronounced in alternative donors (69% vs. 21%), compared with MSD (72% vs. 45%) (113).
Kunte et al. reported the results from a multicenter retrospective study of 69 patients who underwent haploidentical HSCT with post-procedural cyclophosphamide (PTCy) with 3-year OS being 72%, 3-year relapse-free survival of 44%, and non-relapse mortality of 23% (69).
Primary graft failure and poor graft function are two difficult challenges after HSCT.
Primary graft failure is defined according to the EBMT criteria by an ANC < 0.5 × 109/L by day +28 following stem cell infusion, Hb <8.0 g/L, and platelets <20 × 109 (114).
In a retrospective EBMT study involving 2,916 MF patients who underwent allo-HSCT from an HLA-identical sibling or unrelated donor between 2000 and 2016, the 5-year survival rate in patients who developed graft failure was 14% (115).
Recognized risk factors for transplanted patients are related to donor, conditioning, cell dose, and HLA sensitization if the recipient is heavily transfused (116).
No consensus is available about therapeutic options for patients with graft failure, second allo-HSCT using either the same or alternative stem cell donor is warranted (116).
Recently, a retrospective study from the Francophone Society of Bone Marrow Transplantation and Cellular Therapy demonstrated the rescuable potential of salvage haplo-HSCT with PTCy for graft failure. The median time to neutrophil engraftment was 18 days and the cumulative incidence of neutrophil engraftment at day 30 was 79%. One-year overall survival (OS) was 56% and HSCT complications accounted for 80% of causes of death, with multiple organ failure as the leading cause (117).
According to the EBMT criteria, poor graft function (PGF) is defined by the presence of bi- or tri-lineage cytopenia lasting for more than 2 weeks, after day +28 in the presence of donor chimerism >5% (114).
Moreover, PGF is also defined by the presence of mild/moderate cytopenias in at least two hematopoietic lines (ANC < 1.5 × 109/L, platelet count < 30 × 109/L, Hb < 8.5 g/dL) lasting for more than 2 consecutive weeks following engraftment beyond day +14. This definition was recently introduced by an expert panel of the EBMT Chronic Malignancies Working Party, because it is easier to apply in clinical practice than the former one (116).
In a cohort of 100 patients with primary MF or post-ET/PV MF who received a reduced-intensity HSCT, the cumulative incidence of poor graft function was 17% and all cases occurred before day 100 after HSCT at a median of 49 days (range: 24–99 days). In univariate analysis, recipients of older age and splenomegaly at day 30 after HSCT showed an increased cumulative incidence of poor graft function (17).
An expert panel from the EBMT/ELN International Working Group recommends the use of growth factors for anemia (erythropoietin) or neutropenia (granulocyte colony-stimulating factor), whereas data on the use of thrombopoietin analogs in patients with myelofibrosis who underwent allogeneic HSCT are scarce. The most definitive treatment for poor graft function is a CD34+ stem-cell boost from the original donor, either fresh or cryopreserved, without further conditioning in patients without active GvHD (6).
In the Hamburg cohort, CD34+ selected stem cell boost infusion in patients with PGF achieved similar outcomes at 3 years when compared to patients who did not have PGF (17).
Management of persistent splenomegaly in patients with PGF after HSCT is challenging. Splenectomy was reported to be an option in selected patients (17) but it is not without risks. JAK2 inhibitors have not been tested for the indication of post-procedural poor graft function. In majority of the patients, tri-lineage hematologic recovery can be achieved, but will require several months.
Unfortunately, 10%–30% of transplanted patients experience MF relapse after a median of 7 months after HSCT with a median overall survival from the time of relapse of 2 years (116, 118).
Ataganduz et al. also described a late relapse in 14% of patients later than 5 years after HSCT at a median of 7.1 years (119) (Table 2).
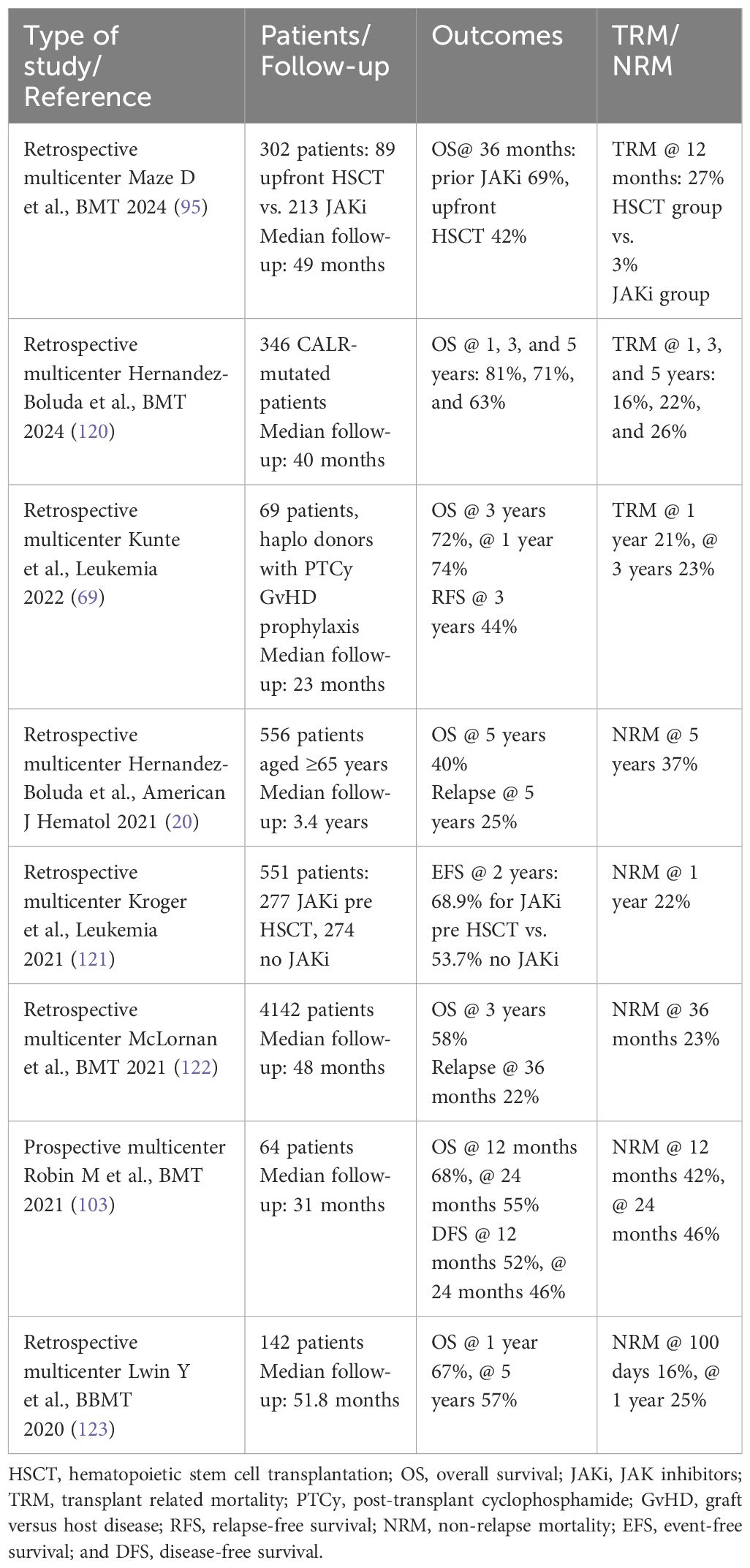
Table 2 Transplant outcomes in MF patients.
The EBMT Chronic Malignancies Working Party defined MF relapses after HSCT as molecular relapse only, cytogenetic relapse only (rarely reported), molecular and cytogenetic relapse only, and morphological/clinical relapse (116).
The expert panel from the EBMT/ELN International Working Group recommends molecular monitoring by sensitive PCR for one of the driver mutations (JAK2, CALR, or MPL) or highly sensitive chimerism for triple-negative MF after HSCT at 1 month and at 3-month intervals thereafter, for up to 1 year and annual testing thereafter (6).
In case of detection of a molecular relapse, early intervention with the aim of reduction of immunosuppressive therapy and use of adoptive immunotherapy with donor lymphocyte infusions (DLI) can achieve molecular remission avoiding progression to overt hematological relapse in responders (116, 124, 125).
Moreover, Gaglemann et al. showed higher rates of complete molecular remission after DLI for molecular relapse comparing hematological relapse (88% and 60%, respectively) (125).
Second HSCT is a valid option to rescue selected fit patients. Nabergoj et al. for the Chronic Malignancies Working Party of EBMT analyzed 216 patients undergoing a second allo-HSCT for either relapse (56%) or graft failure (31%), achieving 42% 3-year overall survival and 39% relapse-free survival (RFS) (126).
Date are insufficient to recommend the use of JAKi after HSCT as maintenance therapy to prevent relapse and in molecular relapse to prevent overt hematological relapse (6, 116). In patients experiencing hematological relapse after HSCT JAKi represent a valid option to reduce constitutional symptoms and/or splenomegaly (127).
Results of transplant outcomes in MF are showed in Table 2 (20, 69, 95, 103, 120–123).
HSCT remains a challenging and controversial procedure in MF; the assessment of the opportunity and modality of HSCT is usually carried out taking into account specific disease variables but also the recipient’s conditions and preferences, case by case.
As the number of HSCT rises rapidly, the best approach to patient and donor selection, splenomegaly management, and timing of HSCT in the era of new drugs need to be clarified. Further studies are required and will test, last but not the least, how to improve HSCT outcomes in this setting. The most appropriate transplant platforms, GvHD prophylaxis, infections management, and thrombosis prophylaxis need to be addressed undoubtedly, as soon as possible.
PR: Writing – original draft, Writing – review & editing. AN: Writing – original draft, Writing – review & editing. FG: Writing – original draft. SS: Writing – review & editing. CC: Writing – review & editing. GL: Writing – review & editing. MD: Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Barbui T, Thiele J, Vannucchi AM, Tefferi A. Rationale for revision and proposed changes of the WHO diagnostic criteria for polycythemia vera, essential thrombocythemia and primary myelofibrosis. Blood Cancer J. (2015) 5:e337. doi: 10.1038/bcj.2015.64
2. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. (2016) 127:2391–405. doi: 10.1182/blood-2016-03-643544
3. Arber DA, Orazi A, Hasserjian RP, Borowitz MJ, Calvo KR, Kvasnicka HM, et al. International Consensus Classification of Myeloid Neoplasms and Acute Leukemias: integrating morphologic, clinical, and genomic data. Blood. (2022) 140:1200–28. doi: 10.1182/blood.2022015850
4. Iurlo A, Cattaneo D, Bucelli C. Management of myelofibrosis: from diagnosis to new target therapies. Curr Treat Opt Oncol. (2020) 21:46. doi: 10.1007/s11864-020-00734-y
5. Kroger N, Giorgino T, Scott BL, Ditschkowski M, Alchalby H, Cervantes F, et al. Impact of allogeneic stem cell transplantation on survival of patients less than 65 years of age with primary myelofibrosis. Blood. (2015) 125:3347–50. doi: 10.1182/blood-2014-10-608315
6. Kroger N, Bacigalupo A, Barbui T, Ditschkowski M, Gagelmann N, Griesshammer M, et al. Indication and management of allogeneic haematopoietic stem–cell transplantation in myelofibrosis: updated recommendations by the EBMT/ELN International Working Group. Lancet Haematol. (2024) 11:e62–74. doi: 10.1016/S2352-3026(23)00305-8
7. Gerds AT, Gotlib J, Ali H, Bose P, Dumbar A, Elshoury A, et al. Myeloproliferative Neoplasms, Version 3.2022, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw. (2022) 20(9):1033–62. doi: 10.6004/jnccn.2022.0046
8. Tefferi A. Primary Myelofibrosis: 2023 update on diagnosis, risk-stratification and managment. Am J Hematol. (2021) 98:801–21. doi: 10.1002/ajh.26857
9. Passamonti F. Stem cell transplant in MF: it's time to personalize. Blood. (2019) 133:2118–20. doi: 10.1182/blood-2019-03-900860
10. Kerbauy DM, Gooley TA, Sale GE, Flowers ME, Doney KC, Georges GE, et al. Hematopoietic cell transplantation as curative therapy for idiopathic myelofibrosis, advanced polycythemia vera, and essential thrombocythemia. Biol Blood Marrow Transplant. (2007) 13:355–65. doi: 10.1016/j.bbmt.2006.11.004
11. Bacigalupo A, Soraru M, Dominietto A, Pozzi S, Geroldi S, Van Lint MT, et al. Allogeneic hemopoietic SCT for patients with primary myelofibrosis: a predictive transplant score based on transfusion requirement, spleen size and donor type. Bone Marrow Transplant. (2010) 45:458–63. doi: 10.1038/bmt.2009.188
12. Scott BL, Gooley TA, Sorror ML, Rezvani AR, Linenberger ML, Grim J, et al. The Dynamic International Prognostic Scoring System for myelofibrosis predicts outcomes after hematopoietic cell transplantation. Blood. (2012) 119:2657–64. doi: 10.1182/blood-2011-08-372904
13. Guglielmelli P, Lasho TL, Rotunno G, Score J, Mannarelli C, Pancrazzi A, et al. The number of prognostically detrimental mutations and prognosis in primary myelofibrosis: an international study of 797 patients. Leukemia. (2014) 28:1804–10. doi: 10.1038/leu.2014.76
14. Ali H, Bacigalupo A. 2021 Update on allogeneic hematopoietic stem cell transplant for myelofibrosis: A review of current data and applications on risk stratification and management. Am J Hematol. (2021) 96:1532–8. doi: 10.1002/ajh.26349
15. Barosi G, Bacigalupo A. Allogeneic hematopoietic stem cell transplantation for myelofibrosis. Curr Opin Hematol. (2006) 13:74–8. doi: 10.1097/01.moh.0000203191.99447.98
16. Guardiola P, Anderson JE, Gluckman E. Myelofibrosis with myeloid metaplasia. N Engl J Med. (2000) 343:659. doi: 10.1056/NEJM200008313430913
17. Alchalby H, Yunus DR, Zabelina T, Ayuk F, Kroger N. Incidence and risk factors of poor graft function after allogeneic stem cell transplantation for myelofibrosis. Bone Marrow Transplant. (2016) 51:1223–7. doi: 10.1038/bmt.2016.98
18. Lussana F, Rambaldi A, Finazzi MC, van Biezen A, Scholten M, Oldani E, et al. Allogeneic hematopoietic stem cell transplantation in patients with polycythemia vera or essential thrombocythemia transformed to myelofibrosis or acute myeloid leukemia: a report from the MPN Subcommittee of the Chronic Malignancies Working Party of the European Group for Blood and Marrow Transplantation. Haematologica. (2014) 99:916–21. doi: 10.3324/haematol.2013.094284
19. Kroger NM, Deeg JH, Olavarria E, Niederwieser D, Bacigalupo A, Barbui T, et al. Indication and management of allogeneic stem cell transplantation in primary myelofibrosis: a consensus process by an EBMT/ELN international working group. Leukemia. (2015) 29:2126–33. doi: 10.1038/leu.2015.233
20. Hernandez–Boluda JC, Pereira A, Kroger N, Cornelissen JJ, Finke J, Beelen D, et al. Allogeneic hematopoietic cell transplantation in older myelofibrosis patients: A study of the chronic Malignancies working party of EBMT and the Spanish Myelofibrosis Registry. Am J Hematol. (2021) 96:1186–94. doi: 10.1002/ajh.26279
21. Samuelson S, Sandmaier BM, Heslop HE, Popat U, Carrum G, Champlin RE, et al. Allogeneic haematopoietic cell transplantation for myelofibrosis in 30 patients 60–78 years of age. Br J Haematol. (2011) 153:76–82. doi: 10.1111/j.1365-2141.2011.08582.x
22. Daghia G, Zabelina T, Zeck G, von Pein UM, Christopeit M, Wolschke C, et al. Allogeneic stem cell transplantation for myelofibrosis patients aged >/=65 years. Eur J Haematol. (2019) 103:370–8. doi: 10.1111/ejh.13294
23. Cervantes F, Dupriez B, Pereira A, Passamonti F, Reilly JT, Morra E, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. (2009) 113:2895–901. doi: 10.1182/blood-2008-07-170449
24. Passamonti F, Cervantes F, Vannucchi AM, Morra E, Rumi E, Pereira A, et al. A dynamic prognostic model to predict survival in primary myelofibrosis: a study by the IWG–MRT (International Working Group for Myeloproliferative Neoplasms Research and Treatment). Blood. (2010) 115:1703–8. doi: 10.1182/blood-2009-09-245837
25. Gangat N, Caramazza D, Vaidya R, George G, Begna K, Schwager S, et al. DIPSS plus: a refined Dynamic International Prognostic Scoring System for primary myelofibrosis that incorporates prognostic information from karyotype, platelet count, and transfusion status. J Clin Oncol. (2011) 29:392–7. doi: 10.1200/JCO.2010.32.2446
26. Tefferi A, Lasho TL, Jimma T, Finke CM, Gangat N, Vaidya R, et al. One thousand patients with primary myelofibrosis: the mayo clinic experience. Mayo Clin Proc. (2012) 87:25–33. doi: 10.1016/j.mayocp.2011.11.001
27. Tefferi A, Nicolosi M, Mudireddy M, Szuber N, Finke CM, Lasho TL, et al. Driver mutations and prognosis in primary myelofibrosis: Mayo–Careggi MPN alliance study of 1,095 patients. Am J Hematol. (2018) 93:348–55. doi: 10.1002/ajh.24978
28. Panagiota V, Thol F, Markus B, Fehse B, Alchalby H, Badbaran A, et al. Prognostic effect of calreticulin mutations in patients with myelofibrosis after allogeneic hematopoietic stem cell transplantation. Leukemia. (2014) 28:1552–5. doi: 10.1038/leu.2014.66
29. Kroger N, Panagiota V, Badbaran A, Zabelina T, Triviai I, Araujo Cruz MM, et al. Impact of molecular genetics on outcome in myelofibrosis patients after allogeneic stem cell transplantation. Biol Blood Marrow Transplant. (2017) 23:1095–101. doi: 10.1016/j.bbmt.2017.03.034
30. Tefferi A, Lasho TL, Finke CM, Knudson RA, Ketterling R, Hanson CH, et al. CALR vs JAK2 vs MPL–mutated or triple–negative myelofibrosis: clinical, cytogenetic and molecular comparisons. Leukemia. (2014) 28:1472–7. doi: 10.1038/leu.2014.3
31. Rumi E, Pietra D, Pascutto C, Guglielmelli P, Martinez–Trillos A, Casetti I, et al. Clinical effect of driver mutations of JAK2, CALR, or MPL in primary myelofibrosis. Blood. (2014) 124:1062–9. doi: 10.1182/blood-2014-05-578435
32. Guglielmelli P, Rotunno G, Fanelli T, Pacilli A, Brogi G, Calabresi L, et al. Validation of the differential prognostic impact of type 1/type 1–like versus type 2/type 2–like CALR mutations in myelofibrosis. Blood Cancer J. (2015) 5:e360. doi: 10.1038/bcj.2015.90
33. Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, et al. Mutations and prognosis in primary myelofibrosis. Leukemia. (2013) 27:1861–9. doi: 10.1038/leu.2013.119
34. Guglielmelli P, Lasho TL, Rotunno G, Mudireddy M, Mannarelli C, Nicolosi M, et al. MIPSS70: mutation–enhanced international prognostic score system for transplantation–age patients with primary myelofibrosis. J Clin Oncol. (2018) 36:310–8. doi: 10.1200/JCO.2017.76.4886
35. Tefferi A, Guglielmelli P, Lasho TL, Gangat N, Ketterling RP, Pardanani A, et al. MIPSS70+ Version 2.0: mutation and karyotype–enhanced international prognostic scoring system for primary myelofibrosis. . J Clin Oncol. (2018) 36:1769–70. doi: 10.1200/JCO.2018.78.9867
36. Tefferi A, Nicolosi M, Mudireddy M, Lasho TL, Gangat N, Begna KH, et al. Revised cytogenetic risk stratification in primary myelofibrosis: analysis based on 1002 informative patients. Leukemia. (2018) 32:1189–99. doi: 10.1038/s41375-018-0018-z
37. Tefferi A, Finke CM, Lasho TL, Hanson CA, Ketterling RP, Gangat N, et al. U2AF1 mutation types in primary myelofibrosis: phenotypic and prognostic distinctions. Leukemia. (2018) 32:2274–8. doi: 10.1038/s41375-018-0078-0
38. Nicolosi M, Mudireddy M, Lasho TL, Hanson CA, Ketterling RP, Gangat N, et al. Sex and degree of severity influence the prognostic impact of anemia in primary myelofibrosis: analysis based on 1109 consecutive patients. Leukemia. (2018) 32:1254–8. doi: 10.1038/s41375-018-0028-x
39. Tefferi A, Guglielmelli P, Nicolosi M, Mannelli F, Mudireddy M, Bartalucci N, et al. GIPSS: genetically inspired prognostic scoring system for primary myelofibrosis. Leukemia. (2018) 32:1631–42. doi: 10.1038/s41375-018-0107-z
40. Palandri F, Breccia M, Mazzoni C, Auteri G, Elli EM, Trawinska MM, et al. Ruxolitinib in cytopenic myelofibrosis: Response, toxicity, drug discontinuation, and outcome. Cancer. (2023) 129:1704–13. doi: 10.1002/cncr.34722
41. Iurlo A, Galli N, Bucelli C, Artuso S, Consonni D, Cattaneo D. Trend of circulating CD34(+) cells in patients with myelofibrosis: Association with spleen response during ruxolitinib treatment. Br J Haematol. (2023) 200:315–22. doi: 10.1111/bjh.18526
42. Gowin K, Coakley M, Kosiorek H, Mesa R. Discrepancies of applying primary myelofibrosis prognostic scores for patients with post polycythemia vera/essential thrombocytosis myelofibrosis. Haematologica. (2016) 101:e405–e6. doi: 10.3324/haematol.2016.149013
43. Passamonti F, Giorgino T, Mora B, Guglielmelli P, Rumi E, Maffioli M, et al. A clinical–molecular prognostic model to predict survival in patients with post polycythemia vera and post essential thrombocythemia myelofibrosis. Leukemia. (2017) 31:2726–31. doi: 10.1038/leu.2017.169
44. Kuykendall AT, Talati C, Padron E, Sweet K, Sallman D, List AF, et al. Genetically inspired prognostic scoring system (GIPSS) outperforms dynamic international prognostic scoring system (DIPSS) in myelofibrosis patients. Am J Hematol. (2019) 94:87–92. doi: 10.1002/ajh.25335
46. Masarova L, Bose P, Pemmaraju N, Daver NG, Zhou L, Pierce S, et al. Prognostic value of blasts in peripheral blood in myelofibrosis in the ruxolitinib era. Cancer. (2020) 126:4322–31. doi: 10.1002/cncr.33094
47. Mannelli F, Bencini S, Coltro G, Loscocco GG, Peruzzi B, Rotunno G, et al. Integration of multiparameter flow cytometry score improves prognostic stratification provided by standard models in primary myelofibrosis. Am J Hematol. (2022) 97:846–55. doi: 10.1002/ajh.26548
48. Luque Paz D, Riou J, Verger E, Cassinat B, Chauveau A, Ianotto JC, et al. Genomic analysis of primary and secondary myelofibrosis redefines the prognostic impact of ASXL1 mutations: a FIM study. Blood Adv. (2021) 5:1442–51. doi: 10.1182/bloodadvances.2020003444
49. Coltro G, Mannelli F, Loscocco GG, Mannarelli C, Rotunno G, Maccari C, et al. Differential prognostic impact of cytopenic phenotype in prefibrotic vs overt primary myelofibrosis. Blood Cancer J. (2022) 12:116. doi: 10.1038/s41408-022-00713-6
50. Vachhani P, Verstovsek S, Bose P. Cytopenic myelofibrosis: prevalence, relevance, and treatment. Expert Opin Pharmacother. (2023) 24:901–12. doi: 10.1080/14656566.2023.2203318
51. Vannucchi AM, Kantarjian HM, Kiladjian JJ, Gotlib J, Cervantes F, Mesa RA, et al. A pooled analysis of overall survival in COMFORT–I and COMFORT–II, 2 randomized phase III trials of ruxolitinib for the treatment of myelofibrosis. Haematologica. (2015) 100:1139–45. doi: 10.3324/haematol.2014.119545
52. Grinfeld J, Nangalia J, Baxter EJ, Wedge DC, Angelopoulos N, Cantrill R, et al. Classification and personalized prognosis in myeloproliferative neoplasms. N Engl J Med. (2018) 379:1416–30. doi: 10.1056/NEJMoa1716614
53. Samuelson Bannow BT, Salit RB, Storer BE, Stevens EA, Wu D, Yeung C, et al. Hematopoietic cell transplantation for myelofibrosis: the dynamic international prognostic scoring system plus risk predicts post–transplant outcomes. Biol Blood Marrow Transplant. (2018) 24:386–92. doi: 10.1016/j.bbmt.2017.09.016
54. Ditschkowski M, Elmaagacli AH, Trenschel R, Gromke T, Steckel NK, Koldehoff M, et al. Dynamic International Prognostic Scoring System scores, pre–transplant therapy and chronic graft–versus–host disease determine outcome after allogeneic hematopoietic stem cell transplantation for myelofibrosis. Haematologica. (2012) 97:1574–81. doi: 10.3324/haematol.2011.061168
55. Alchalby H, Yunus DR, Zabelina T, Kobbe G, Holler E, Bornhauser M, et al. Risk models predicting survival after reduced–intensity transplantation for myelofibrosis. Br J Haematol. (2012) 157:75–85. doi: 10.1111/j.1365-2141.2011.09009.x
56. Gowin K, Ballen K, Ahn KW, Hu ZH, Ali H, Arcasoy MO, et al. Survival following allogeneic transplant in patients with myelofibrosis. Blood Adv. (2020) 4:1965–73. doi: 10.1182/bloodadvances.2019001084
57. Gagelmann N, Eikema DJ, de Wreede LC, Koster L, Wolschke C, Arnold R, et al. Comparison of dynamic international prognostic scoring system and MYelofibrosis SECondary to PV and ET prognostic model for prediction of outcome in polycythemia vera and essential thrombocythemia myelofibrosis after allogeneic stem cell transplantation. Biol Blood Marrow Transplant. (2019) 25:e204–e8. doi: 10.1016/j.bbmt.2019.03.024
58. Polverelli N, Bonneville EF, de Wreede LC, Koster L, Kroger NM, Schroeder T, et al. Impact of comorbidities and body mass index on the outcomes of allogeneic hematopoietic cell transplantation in myelofibrosis: A study on behalf of the Chronic Malignancies Working Party of EBMT. Am J Hematol. (2024) 99:993–6. doi: 10.1002/ajh.27262
59. Sorror ML, Maris MB, Storb R, Baron F, Sandmaier BM, Maloney DG, et al. Hematopoietic cell transplantation (HCT)–specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood. (2005) 106:2912–9. doi: 10.1182/blood-2005-05-2004
60. Kroger N, Holler E, Kobbe G, Bornhauser M, Schwerdtfeger R, Baurmann H, et al. Allogeneic stem cell transplantation after reduced–intensity conditioning in patients with myelofibrosis: a prospective, multicenter study of the Chronic Leukemia Working Party of the European Group for Blood and Marrow Transplantation. Blood. (2009) 114:5264–70. doi: 10.1182/blood-2009-07-234880
61. Gagelmann N, Ditschkowski M, Bogdanov R, Bredin S, Robin M, Cassinat B, et al. Comprehensive clinical–molecular transplant scoring system for myelofibrosis undergoing stem cell transplantation. Blood. (2019) 133:2233–42. doi: 10.1182/blood-2018-12-890889
62. Hernandez–Boluda JC, Pereira A, Alvarez–Larran A, Martin AA, Benzaquen A, Aguirre L, et al. Predicting survival after allogeneic hematopoietic cell transplantation in myelofibrosis: performance of the myelofibrosis transplant scoring system (MTSS) and development of a new prognostic model. Biol Blood Marrow Transplant. (2020) 26:2237–44. doi: 10.1016/j.bbmt.2020.07.022
63. Gagelmann N, Badbaran A, Salit RB, Schroeder T, Gurnari C, Pagliuca S, et al. Impact of TP53 on outcome of patients with myelofibrosis undergoing hematopoietic stem cell transplantation. Blood. (2023) 141:2901–11. doi: 10.1182/blood.2023019630
64. Barbui T, Tefferi A, Vannucchi AM, Passamonti F, Silver RT, Hoffman R, et al. Philadelphia chromosome–negative classical myeloproliferative neoplasms: revised management recommendations from European LeukemiaNet. Leukemia. (2018) 32:1057–69. doi: 10.1038/s41375-018-0077-1
65. Guardiola P, Anderson JE, Bandini G, Cervantes F, Runde V, Arcese W, et al. Allogeneic stem cell transplantation for agnogenic myeloid metaplasia: a European Group for Blood and Marrow Transplantation, Societe Francaise de Greffe de Moelle, Gruppo Italiano per il Trapianto del Midollo Osseo, and Fred Hutchinson Cancer Research Center Collaborative Study. Blood. (1999) 93:2831–8.
66. Li Z, Gooley T, Applebaum FR, Deeg HJ. Splenectomy and hemopoietic stem cell transplantation for myelofibrosis. Blood. (2001) 97:2180–1. doi: 10.1182/blood.V97.7.2180
67. Robin M, Zine M, Chevret S, Meignin V, Munoz–Bongrand N, Moatti H, et al. The impact of splenectomy in myelofibrosis patients before allogeneic hematopoietic stem cell transplantation. Biol Blood Marrow Transplant. (2017) 23:958–64. doi: 10.1016/j.bbmt.2017.03.002
68. Patriarca F, Masciulli A, Bacigalupo A, Bregante S, Pavoni C, Finazzi MC, et al. Busulfan– or thiotepa–based conditioning in myelofibrosis: A phase II multicenter randomized study from the GITMO group. Biol Blood Marrow Transplant. (2019) 25:932–40. doi: 10.1016/j.bbmt.2018.12.064
69. Kunte S, Rybicki L, Viswabandya A, Tamari R, Bashey A, Keyzner A, et al. Allogeneic blood or marrow transplantation with haploidentical donor and post–transplantation cyclophosphamide in patients with myelofibrosis: a multicenter study. Leukemia. (2022) 36:856–64. doi: 10.1038/s41375-021-01449-1
70. Jain T, Tsai HL, DeZern AE, Gondek LP, Elmariah H, Bolanos–Meade J, et al. Post–transplantation cyclophosphamide–based graft– versus–host disease prophylaxis with nonmyeloablative conditioning for blood or marrow transplantation for myelofibrosis. Transplant Cell Ther. (2022) 28:259 e1–e11. doi: 10.1016/j.jtct.2022.02.004
71. Lissandre S, Bay JO, Cahn JY, Porcher R, Cacheux V, Cabrespine A, et al. Retrospective study of allogeneic haematopoietic stem–cell transplantation for myelofibrosis. Bone Marrow Transplant. (2011) 46:557–61. doi: 10.1038/bmt.2010.276
72. Ciurea SO, Sadegi B, Wilbur A, Alagiozian–Angelova V, Gaitonde S, Dobogai LC, et al. Effects of extensive splenomegaly in patients with myelofibrosis undergoing a reduced intensity allogeneic stem cell transplantation. Br J Haematol. (2008) 141:80–3. doi: 10.1111/j.1365-2141.2008.07010.x
73. Polverelli N, Mauff K, Kroger N, Robin M, Beelen D, Beauvais D, et al. Impact of spleen size and splenectomy on outcomes of allogeneic hematopoietic cell transplantation for myelofibrosis: A retrospective analysis by the chronic Malignancies working party on behalf of European society for blood and marrow transplantation (EBMT). Am J Hematol. (2021) 96:69–79. doi: 10.1002/ajh.26020
74. Akpek G, Pasquini MC, Logan B, Agovi MA, Lazarus HM, Marks DI, et al. Effects of spleen status on early outcomes after hematopoietic cell transplantation. Bone Marrow Transplant. (2013) 48:825–31. doi: 10.1038/bmt.2012.249
75. Cervantes F. How I treat splenomegaly in myelofibrosis. Blood Cancer J. (2011) 1:e37. doi: 10.1038/bcj.2011.36
76. Santos FP, Tam CS, Kantarjian H, Cortes J, Thomas D, Pollock R, et al. Splenectomy in patients with myeloproliferative neoplasms: efficacy, complications and impact on survival and transformation. Leuk Lymph. (2014) 55:121–7. doi: 10.3109/10428194.2013.794269
77. Tefferi A, Mudireddy M, Gangat N, Hanson CA, Ketterling RP, Pardanani A, et al. Risk factors and a prognostic model for postsplenectomy survival in myelofibrosis. Am J Hematol. (2017) 92:1187–92. doi: 10.1002/ajh.24881
78. MiguelA. Sanz GS, Lorenzo I, Tilly H, Bastit D, Dauce J–P, Monconduit M, et al. Splenectomy before bone marrow transplantation in chronic granulocytic leukaemia. Lancet. (1986) 327:212–3. doi: 10.1016/S0140-6736(86)90686-0
79. Bossard JB, Beuscart JB, Robin M, Mohty M, Barraco F, Chevallier P, et al. Splenectomy before allogeneic hematopoietic cell transplantation for myelofibrosis: A French nationwide study. Am J Hematol. (2021) 96:80–8. doi: 10.1002/ajh.26034
80. Harrison C, Kiladjian JJ, Al–Ali HK, Gisslinger H, Waltzman R, Stalbovskaya V, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. (2012) 366:787–98. doi: 10.1056/NEJMoa1110556
81. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double–blind, placebo–controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. (2012) 366:799–807. doi: 10.1056/NEJMoa1110557
82. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. Efficacy, safety and survival with ruxolitinib in patients with myelofibrosis: results of a median 2–year follow–up of COMFORT–I. Haematologica. (2013) 98:1865–71. doi: 10.3324/haematol.2013.092155
83. Malato A, Rossi E, Tiribelli M, Mendicino F, Pugliese N. Splenectomy in myelofibrosis: indications, efficacy, and complications. Clin Lymph Myeloma Leuk. (2020) 20:588–95. doi: 10.1016/j.clml.2020.04.015
84. Gagelmann N, Hobbs GS, Campodonico E, Helbig G, Novak P, Schroeder T, et al. Splenic irradiation for myelofibrosis prior to hematopoietic cell transplantation: A global collaborative analysis. Am J Hematol. (2024) 99:844–53. doi: 10.1002/ajh.27252
85. Bouabdallah R, Coso D, Gonzague–Casabianca L, Alzieu C, Resbeut M, Gastaut JA. Safety and efficacy of splenic irradiation in the treatment of patients with idiopathic myelofibrosis: a report on 15 patients. Leuk Res. (2000) 24:491–5. doi: 10.1016/S0145-2126(00)00018-7
86. Wagner H Jr., McKeough PG, Desforges J, Madoc–Jones H. Splenic irradiation in the treatment of patients with chronic myelogenous leukemia or myelofibrosis with myeloid metaplasia. Results of daily and intermittent fractionation with and without concomitant hydroxyurea. Cancer. (1986) 58:1204–7. doi: 10.1002/(ISSN)1097-0142
87. Cipkar C, Kumar S, Thavorn K, Kekre N. Optimal timing of allogeneic stem cell transplantation for primary myelofibrosis. Transplant Cell Ther. (2022) 28:189–94. doi: 10.1016/j.jtct.2022.01.018
88. Devlin R, Gupta V. Myelofibrosis: to transplant or not to transplant? Hematol Am Soc Hematol Educ Program. (2016) 2016:543–51. doi: 10.1182/asheducation-2016.1.543
89. Spiegel JY, McNamara C, Kennedy JA, Panzarella T, Arruda A, Stockley T, et al. Impact of genomic alterations on outcomes in myelofibrosis patients undergoing JAK1/2 inhibitor therapy. Blood Adv. (2017) 1:1729–38. doi: 10.1182/bloodadvances.2017009530
90. Gupta V, Griesshammer M, Martino B, Foltz L, Tavares R, Al–Ali HK, et al. Analysis of predictors of response to ruxolitinib in patients with myelofibrosis in the phase 3b expanded–access JUMP study. Leuk Lymph. (2021) 62:918–26. doi: 10.1080/10428194.2020.1845334
91. Mylonas E, Yoshida K, Frick M, Hoyer K, Christen F, Kaeda J, et al. Single–cell analysis based dissection of clonality in myelofibrosis. Nat Commun. (2020) 11:73. doi: 10.1038/s41467-019-13892-x
92. Mesa RA, Li CY, Ketterling RP, Schroeder GS, Knudson RA, Tefferi A. Leukemic transformation in myelofibrosis with myeloid metaplasia: a single–institution experience with 91 cases. Blood. (2005) 105:973–7. doi: 10.1182/blood-2004-07-2864
93. Newberry KJ, Patel K, Masarova L, Luthra R, Manshouri T, Jabbour E, et al. Clonal evolution and outcomes in myelofibrosis after ruxolitinib discontinuation. Blood. (2017) 130:1125–31. doi: 10.1182/blood-2017-05-783225
94. Schain F, Vago E, Song C, He J, Liwing J, Lofgren C, et al. Survival outcomes in myelofibrosis patients treated with ruxolitinib: A population–based cohort study in Sweden and Norway. Eur J Haematol. (2019) 103:614–9. doi: 10.1111/ejh.13330
95. Maze D, Arcasoy MO, Henrie R, Cerquozzi S, Kamble R, Al–Hadidi S, et al. Upfront allogeneic transplantation versus JAK inhibitor therapy for patients with myelofibrosis: a North American collaborative study. Bone Marrow Transplant. (2024) 59:196–202. doi: 10.1038/s41409-023-02146-6
96. Claire Harrison J–JK, Verstovsek S, Vannucchi A, Mesa R, Reiter A, Zhang J, et al. MPN–164: overall survival (OS) and progression–free survival (PFS) in patients treated with fedratinib as first–line myelofibrosis (MF) therapy and after prior ruxolitinib (RUX): results from the JAKARTA and JAKARTA2 trials. Myeloproliferative Neoplasms. (2021) 21:S356. doi: 10.1016/S2152-2650(21)01822-X
97. Srdan Verstovsek MP, Egyed M, Lech–Marańda E, Sacha T, Dubruille V, Oh ST, et al. Myeloproliferative syndromes: clinical robust overall survival and sustained efficacy outcomes during long term exposure to momelotinib in JAK inhibitor naïve and previously JAK inhibitor treated intermediate/high risk myelofibrosis patients. Blood. (2020) 136:51–2. doi: 10.1182/blood-2020-135872
98. Gupta V, Hari P, Hoffman R. Allogeneic hematopoietic cell transplantation for myelofibrosis in the era of JAK inhibitors. Blood. (2012) 120:1367–79. doi: 10.1182/blood-2012-05-399048
99. Tiribelli M, Palandri F, Sant'Antonio E, Breccia M, Bonifacio M. The role of allogeneic stem–cell transplant in myelofibrosis in the era of JAK inhibitors: a case–based review. Bone Marrow Transplant. (2020) 55:708–16. doi: 10.1038/s41409-019-0683-1
100. McLornan DP, Yakoub–Agha I, Robin M, Chalandon Y, Harrison CN, Kroger N. State–of–the–art review: allogeneic stem cell transplantation for myelofibrosis in 2019. Haematologica. (2019) 104:659–68. doi: 10.3324/haematol.2018.206151
101. Gupta V, Kosiorek HE, Mead A, Klisovic RB, Galvin JP, Berenzon D, et al. Ruxolitinib therapy followed by reduced–intensity conditioning for hematopoietic cell transplantation for myelofibrosis: myeloproliferative disorders research consortium 114 study. Biol Blood Marrow Transplant. (2019) 25:256–64. doi: 10.1016/j.bbmt.2018.09.001
102. Zhang L, Yang F, Feng S. Allogeneic hematopoietic stem–cell transplantation for myelofibrosis. Ther Adv Hematol. (2020) 11:2040620720906002. doi: 10.1177/2040620720906002
103. Robin M, Porcher R, Orvain C, Bay JO, Barraco F, Huynh A, et al. Ruxolitinib before allogeneic hematopoietic transplantation in patients with myelofibrosis on behalf SFGM–TC and FIM groups. Bone Marrow Transplant. (2021) 56:1888–99. doi: 10.1038/s41409-021-01252-7
104. Shanavas M, Popat U, Michaelis LC, Fauble V, McLornan D, Klisovic R, et al. Outcomes of allogeneic hematopoietic cell transplantation in patients with myelofibrosis with prior exposure to janus kinase 1/2 inhibitors. Biol Blood Marrow Transplant. (2016) 22:432–40. doi: 10.1016/j.bbmt.2015.10.005
105. Hanif A, Hari PN, Atallah E, Carlson KS, Pasquini MC, Michaelis LC. Safety of ruxolitinib therapy prior to allogeneic hematopoietic stem–cell transplantation for myeloproliferative neoplasms. Bone Marrow Transplant. (2016) 51:617–8. doi: 10.1038/bmt.2015.295
106. Shahnaz Syed Abd Kadir S, Christopeit M, Wulf G, Wagner E, Bornhauser M, Schroeder T, et al. Impact of ruxolitinib pretreatment on outcomes after allogeneic stem cell transplantation in patients with myelofibrosis. Eur J Haematol. (2018) 101:305–17. doi: 10.1111/ejh.13099
107. Palandri F, Breccia M, Bonifacio M, Polverelli N, Elli EM, Benevolo G, et al. Life after ruxolitinib: Reasons for discontinuation, impact of disease phase, and outcomes in 218 patients with myelofibrosis. Cancer. (2020) 126:1243–52. doi: 10.1002/cncr.32664
108. England J, Gupta V. Novel therapies vs hematopoietic cell transplantation in myelofibrosis: who, when, how? Hematol Am Soc Hematol Educ Program. (2021) 2021:453–62. doi: 10.1182/hematology.2021000279
109. Kuykendall AT, Shah S, Talati C, Al Ali N, Sweet K, Padron E, et al. Between a rux and a hard place: evaluating salvage treatment and outcomes in myelofibrosis after ruxolitinib discontinuation. Ann Hematol. (2018) 97:435–41. doi: 10.1007/s00277-017-3194-4
110. Maffioli M, Mora B, Ball S, Iurlo A, Elli EM, Finazzi MC, et al. A prognostic model to predict survival after 6 months of ruxolitinib in patients with myelofibrosis. Blood Adv. (2022) 6:1855–64. doi: 10.1182/bloodadvances.2021006889
111. Gupta V, Malone AK, Hari PN, Ahn KW, Hu ZH, Gale RP, et al. Reduced–intensity hematopoietic cell transplantation for patients with primary myelofibrosis: a cohort analysis from the center for international blood and marrow transplant research. Biol Blood Marrow Transplant. (2014) 20:89–97. doi: 10.1016/j.bbmt.2013.10.018
112. Robin M, Giannotti F, Deconinck E, Mohty M, Michallet M, Sanz G, et al. Unrelated cord blood transplantation for patients with primary or secondary myelofibrosis. Biol Blood Marrow Transplant. (2014) 20:1841–6. doi: 10.1016/j.bbmt.2014.06.011
113. Bregante S, Dominietto A, Ghiso A, Raiola AM, Gualandi F, Varaldo R, et al. Improved outcome of alternative donor transplantations in patients with myelofibrosis: from unrelated to haploidentical family donors. Biol Blood Marrow Transplant. (2016) 22:324–9. doi: 10.1016/j.bbmt.2015.09.028
114. Carreras E, Dufour C, Mohty M, Kroger N eds. The EBMT Handbook: Hematopoietic Stem Cell Transplantation and Cellular Therapies. 7th ed. Cham (CH): Springer (2019). doi: 10.1007/978-3-030-02278-5
115. Hernandez–Boluda JC, Pereira A, Kroger N, Beelen D, Robin M, Bornhauser M, et al. Determinants of survival in myelofibrosis patients undergoing allogeneic hematopoietic cell transplantation. Leukemia. (2021) 35:215–24. doi: 10.1038/s41375-020-0815-z
116. McLornan DP, Hernandez–Boluda JC, Czerw T, Cross N, Joachim Deeg H, Ditschkowski M, et al. Allogeneic haematopoietic cell transplantation for myelofibrosis: proposed definitions and management strategies for graft failure, poor graft function and relapse: best practice recommendations of the EBMT Chronic Malignancies Working Party. Leukemia. (2021) 35:2445–59. doi: 10.1038/s41375-021-01294-2
117. Prata PH, Resche–Rigon M, Blaise D, Socie G, Rohrlich PS, Milpied N, et al. Outcomes of salvage haploidentical transplant with post–transplant cyclophosphamide for rescuing graft failure patients: a report on behalf of the francophone society of bone marrow transplantation and cellular therapy. Biol Blood Marrow Transplant. (2019) 25:1798–802. doi: 10.1016/j.bbmt.2019.05.013
118. McLornan DP, Szydlo R, Robin M, van Biezen A, Koster L, Blok HJP, et al. Outcome of patients with Myelofibrosis relapsing after allogeneic stem cell transplant: a retrospective study by the Chronic Malignancies Working Party of EBMT. Br J Haematol. (2018) 182:418–22. doi: 10.1111/bjh.15407
119. Atagunduz IK, Christopeit M, Ayuk F, Zeck G, Wolschke C, Kroger N. Incidence and outcome of late relapse after allogeneic stem cell transplantation for myelofibrosis. Biol Blood Marrow Transplant. (2020) 26:2279–84. doi: 10.1016/j.bbmt.2020.09.006
120. Hernandez–Boluda JC, Eikema DJ, Koster L, Kroger N, Robin M, de Witte M, et al. Allogeneic hematopoietic cell transplantation in patients with CALR–mutated myelofibrosis: a study of the Chronic Malignancies Working Party of EBMT. Bone Marrow Transplant. (2023) 58:1357–67. doi: 10.1038/s41409-023-02094-1
121. Kroger N, Sbianchi G, Sirait T, Wolschke C, Beelen D, Passweg J, et al. Impact of prior JAK–inhibitor therapy with ruxolitinib on outcome after allogeneic hematopoietic stem cell transplantation for myelofibrosis: a study of the CMWP of EBMT. Leukemia. (2021) 35:3551–60. doi: 10.1038/s41375-021-01276-4
122. McLornan D, Eikema DJ, Czerw T, Kroger N, Koster L, Reinhardt HC, et al. Trends in allogeneic haematopoietic cell transplantation for myelofibrosis in Europe between 1995 and 2018: a CMWP of EBMT retrospective analysis. Bone Marrow Transplant. (2021) 56:2160–72. doi: 10.1038/s41409-021-01305-x
123. Lwin Y, Kennedy G, Gottlieb D, Kwan J, Ritchie D, Szer J, et al. Australasian trends in allogeneic stem cell transplantation for myelofibrosis in the molecular era: A retrospective analysis from the australasian bone marrow transplant recipient registry. Biol Blood Marrow Transplant. (2020) 26:2252–61. doi: 10.1016/j.bbmt.2020.08.024
124. Wolschke C, Badbaran A, Zabelina T, Christopeit M, Ayuk F, Triviai I, et al. Impact of molecular residual disease post allografting in myelofibrosis patients. Bone Marrow Transplant. (2017) 52:1526–9. doi: 10.1038/bmt.2017.157
125. Gagelmann N, Wolschke C, Badbaran A, Janson D, Berger C, Klyuchnikov E, et al. Donor lymphocyte infusion and molecular monitoring for relapsed myelofibrosis after hematopoietic cell transplantation. Hemasphere. (2023) 7:e921. doi: 10.1097/HS9.0000000000000921
126. Nabergoj M, Mauff K, Robin M, Kroger N, Angelucci E, Poire X, et al. Outcomes following second allogeneic haematopoietic cell transplantation in patients with myelofibrosis: a retrospective study of the Chronic Malignancies Working Party of EBMT. Bone Marrow Transplant. (2021) 56:1944–52. doi: 10.1038/s41409-021-01271-4
Keywords: myelofibrosis, bone marrow transplantation, JAK inhibitors, scoring algorithm, splenomegaly
Citation: Ranalli P, Natale A, Guardalupi F, Santarone S, Cantò C, La Barba G and Di Ianni M (2024) Myelofibrosis and allogeneic transplantation: critical points and challenges. Front. Oncol. 14:1396435. doi: 10.3389/fonc.2024.1396435
Received: 05 March 2024; Accepted: 23 May 2024;
Published: 20 June 2024.
Edited by:
Salvatore Leotta, Independent researcher, Catania, ItalyReviewed by:
Andrea Duminuco, Gaspare Rodolico Ospedale, ItalyCopyright © 2024 Ranalli, Natale, Guardalupi, Santarone, Cantò, La Barba and Di Ianni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mauro Di Ianni, bWF1cm8uZGlpYW5uaUB1bmljaC5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.