 Tamara A. Sussman1Mariano Severgnini1,2Anita Giobbie-Hurder1,3Philip Friedlander4Scott J. Swanson5Michael Jaklitsch5Thomas Clancy5Laura A. Goguen6David Lautz7Richard Swanson8Heather Daley9Jerome Ritz1,9Glenn Dranoff1F. Stephen Hodi1,10*
Tamara A. Sussman1Mariano Severgnini1,2Anita Giobbie-Hurder1,3Philip Friedlander4Scott J. Swanson5Michael Jaklitsch5Thomas Clancy5Laura A. Goguen6David Lautz7Richard Swanson8Heather Daley9Jerome Ritz1,9Glenn Dranoff1F. Stephen Hodi1,10*- 1Department of Medical Oncology, Dana-Farber Cancer Institute, Boston, MA, United States
- 2Department of Clinical Sciences, Curis, Inc., Lexington, MA, United States
- 3Division of Biostatistics, Department of Data Science, Dana-Farber Cancer Institute, Boston, MA, United States
- 4Department of Hematology and Oncology, Icahn School of Medicine at Mount Sinai, New York, NY, United States
- 5Department of Surgery, Brigham and Women’s Hospital, Boston, MA, United States
- 6Division of Otolaryngology, Brigham and Women’s Hospital, Boston, MA, United States
- 7Department of Surgery, Emerson Hospital, Concord, MA, United States
- 8Department of Surgery, UMass Chan Medical School, Worcester, MA, United States
- 9Connell and O’Reilly Families Cell Manipulation Core Facility, Dana-Farber Cancer Institute, Boston, MA, United States
- 10Department of Medical Oncology, Parker Institute for Cancer Immunotherapy, Dana-Farber Cancer Institute, Boston, MA, United States
Background: In the era of immune checkpoint blockade, the role of cancer vaccines in immune priming has provided additional potential for therapeutic improvements. Prior studies have demonstrated delayed type hypersensitivity and anti-tumor immunity with vaccines engineered to secrete granulocyte-macrophage colony-stimulating factor (GM-CSF). The safety, efficacy and anti-tumor immunity of GM-CSF secreting vaccine in patients with previously treated stage III or IV melanoma needs further investigation.
Methods: In this phase II trial, excised lymph node metastases were processed to single cells, transduced with an adenoviral vector encoding GM-CSF, irradiated, and cryopreserved. Individual vaccines were composed of 1x106, 4x106, or 1x107 tumor cells, and were injected intradermally and subcutaneously at weekly and biweekly intervals. The primary endpoints were feasibility of producing vaccine in stage III patients and determining the proportion of patients alive at two years in stage IV patients.
Results: GM-CSF vaccine was successfully developed and administered in all 61 patients. Toxicities were restricted to grade 1-2 local skin reactions. The median OS for stage III patients (n = 20) was 71.1 (95% CI, 43.7 to NR) months and 14.9 (95%CI, 12.1 to 39.7) months for stage IV patients. The median PFS in stage III patients was 50.7 (95%CI, 36.3 to NR) months and 4.1 (95% CI, 3.0-6.3) months in stage IV patients. In the overall population, the disease control rate was 39.3% (95%CI, 27.1 to 52.7%). In stage III patients, higher pre-treatment plasma cytokine levels of MMP-1, TRAIL, CXCL-11, CXCL-13 were associated with improved PFS (p<0.05 for all). An increase in post-vaccination levels of IL-15 and TRAIL for stage III patients was associated with improved PFS (p=0.03 for both). Similarly, an increase in post-vaccination IL-16 level for stage IV patients was associated with improved PFS (p=0.02) and clinical benefit.
Conclusions: Vaccination with autologous melanoma cells secreting GM-CSF augments antitumor immunity in stage III and IV patients with melanoma, is safe, and demonstrates disease control. Luminex data suggests that changes in inflammatory cytokines and immune cell infiltration promote tumor antigen presentation and subsequent tumor cell destruction. Additional investigation to administer this vaccine in combination with immune checkpoint inhibitors is needed.
Introduction
Current standard of care immunotherapies for melanoma in the adjuvant and metastatic settings include immune checkpoint inhibitors directed at programmed death 1 (PD-1), namely nivolumab and pembrolizumab, and at cytotoxic T-lymphocyte antigen 4 (CTLA-4), ipilimumab, given as monotherapy or in combination. Recently, the Food and Drug Administration approved a third immune checkpoint inhibitor, relatlimab (anti-LAG-3 antibody) in combination with nivolumab for the treatment of patients with metastatic melanoma in the first and later line settings. With the advent of immunological checkpoint inhibitors, the treatment landscape for melanoma has been revolutionized. However, research is ongoing to develop new therapies to further improve outcomes while limiting toxicities.
A recent press release for a phase IIb clinical trial with an mRNA vaccine for melanoma in combination with pembrolizumab in 157 patients with resected stage III or IV melanoma identified a 44% reduction in the risk of recurrence or death when compared to standard of care therapy with pembrolizumab alone (1). This is an exciting development in which the mRNA cancer vaccine can prime patients’ immune system to generate a response to melanoma. However, developing vaccines for melanoma has been ongoing for decades, and the potential for whole cell vaccination strategies requiring further study. Initial vaccine studies included allogeneic and autologous granulocyte-macrophage colony stimulating factor (GM-CSF)-secreting tumor vaccines for pancreas cancer and melanoma (2, 3). Phase I studies demonstrated that GM-CSF secreting vaccines induced delayed-type hypersensitivity responses to autologous tumor cells and that vaccination sites showed brisk infiltrates of dendritic cells, macrophages, eosinophils, and lymphocytes contributing to enhanced tumor antigen presentation and ultimately promoting anti-tumor immunity (3–9). We previously conducted a phase I study with irradiated, autologous melanoma cells engineered to secrete GM-CSF by adenoviral mediated gene transfer, which demonstrated safety for patients and augmentation in anti-tumor immunity (6). Here, we present data from a phase II trial of irradiated, autologous melanoma cells engineered to secrete GM-CSF by adenoviral mediated gene transfer in patients with stage III and IV melanoma.
Methods
Patients
Patients were eligible if they were 18 years of age or older and had previously treated or untreated, histologically confirmed, stage III or IV melanoma with ECOG performance status 0 or 1. Patients with stage III melanoma were eligible if gross lymphadenopathy of at least 2cm was present by physical exam or on CT in a region draining a known primary melanoma; and have refused, failed or not been appropriate candidates for adjuvant high-dose interferon. Patients who received prior systemic chemotherapy, radiotherapy, immunotherapy or glucocorticoid therapy were eligible if the last dose was received at least 4 weeks prior to trial enrollment. Patients with prior bone marrow or peripheral blood stem cell transplant were eligible if they were greater than 6 months from transplant at time of trial enrollment. Key exclusion criteria were uveal melanoma, uncontrolled active infection, pregnant or nursing mothers, and infection with HIV. Full inclusion and exclusion criteria are listed in the study protocol. The study protocol was reviewed and approved by the Dana-Farber/Harvard Cancer Center institutional review board. All patients provided written informed consent. An independent data monitoring committee provided oversight to assess efficacy and safety of lethally irradiated, autologous melanoma cells engineered by adenoviral mediated gene transfer to secrete GM-CSF (NCT #00809588).
Trial design, vaccine preparation and administration
In this phase 2 trial, patients received individual vaccine doses of lethally irradiated, autologous melanoma cells engineered by adenoviral mediated gene transfer to secrete GM-CSF. Methods of vaccine production have been previously described (5). Briefly, excised melanoma metastases were processed to single cells, transduced with a replication defective adenoviral vector encoding human GM-CSF, irradiated with 10,000 cGy, and cryopreserved in liquid nitrogen. GM-CSF secretion was determined by ELISA. A portion of tumor cells was not transduced and used in delayed-type hypersensitivity analysis. Individual vaccines were composed of 1x106, 4x106, 1x107 tumor cells, or one-sixth of total depending upon overall yield, and were injected intradermally (0.5ml) and subcutaneously (0.5ml) into limbs or abdomen on a rotating basis on days 0, 7, 14 and every two weeks thereafter until the supply of vaccine was exhausted or the patient was removed from study. Administration of non-transduced, irradiated cells (1x106) were given on day 0 and with the fifth vaccination intradermally (0.5ml) for evaluation of baseline and vaccine induced delayed-type hypersensitivity. Patients could participate in a second round of tumor procurement, vaccine production, and vaccination as long as the patient continued to meet eligibility criteria. Treatment continued until the occurrence of disease progression, unacceptable adverse effects, or withdrawal of consent. Patients underwent scans at week 10 and then at four-month intervals and peripheral blood was collected for immunologic analysis monthly. Biopsies were performed for vaccination reaction (after first vaccine dose) and delayed-type hypersensitivity reaction 2-3 days after administration and again after fifth vaccine dose for vaccination reaction, and if available, for a second delayed-type hypersensitivity reaction. Please see Supplementary Figure 1 for clinical trial schema.
End points and assessments
This study consisted of two parallel cohorts: patients with stage III melanoma and patients with stage IV melanoma. The primary endpoint in the first cohort was feasibility of preparing lethally irradiated, autologous melanoma cells engineered by adenoviral mediated gene transfer to secrete GM-CSF in patients with stage III melanoma. The primary endpoint in the second cohort was to determine the proportion of patients alive at two years. Secondary endpoints for both cohorts included progression free survival, overall survival, and rate of adverse events. Progression-free survival was assessed according to RECIST, version 1.1, by blinded independent review. Adverse events were assessed continuously throughout the trial and for at least 30 days after treatment was discontinued and were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events, version 2.0. Exploratory endpoints included analysis of cytokines by Luminex platform to analyze correlation of certain biomarkers with clinical outcomes.
The distributions of OS and PFS are presented using the method of Kaplan-Meier with 95% confidence intervals estimated using log[-log(endpoint)] methodology and log-rank testing. All statistical testing is two-sided with nominal significance levels of 0.05. There are no corrections for multiple comparisons. Analyses were performed using SAS 9.4 (SAS Institute Inc., Cary, NC, USA).
Immunologic analysis
Cytokines and chemokines were quantified in serum samples using the FLEXMAP3D Luminex platform, and the xPONENT software for standard curve extrapolation. The following analytes were analyzed: MMP-1, TRAIL, TSLP, MIF, LIF, MDC, APRIL, TWEAK, CCL-2, CCL-3, CCL-4, CCL-8, CCL-11, CCL-24, CXCL-5, CXCL-10, CXCL-11, CXCL-13, CD30, CD40L, CTLA-8, IL-2, IL-15, IL-16, IL-18, IL-20, SCF, G-CSF, HGF, SDF-1a, TNF-RII, and VEGF-A. Concentrations of analytes below LLOQ were not considered in the analysis. Serum samples were diluted by two and processed for analyses as recommended by manufacturer protocol (Bio-Techne, Minneapolis MN) (10–12).
The endpoint of interest for the Luminex analysis in the Stage III cohort was PFS; for the Stage IV cohort the endpoints were clinical benefit rate (best response of CR, PR, or SD per RECIST 1.1) and PFS. Pretreatment measurements are summarized descriptively; each pretreatment biomarker is divided at its respective median and subsequent PFS summarized stratified by pretreatment high/low using the method of Kaplan-Meier. Changes at four to eight weeks relative to pretreatment are expressed as fold-changes (post/pre) and are summarized descriptively and compared with one using Wilcoxon signed-rank tests. Fold-changes are also divided into high/low according to the respective median of the fold-change. The distributions of subsequent PFS are described using the method of Kaplan-Meier and compared using log-rank tests. Comparisons of clinical benefit rates according to high/low pre-treatment biomarker levels used Fisher’s exact tests. The STROBE cohort reporting guidelines were used (13).
Results
Patients
From November 2003 through June 2009, a total of 84 patients were enrolled in this trial to receive a vaccine with lethally irradiated, autologous melanoma cells engineered to secrete GM-CSF. Four patients cancelled registration and nineteen patients did not receive any treatment; the final cohort was comprised of 61 patients, of which 20 had stage III disease and 41 had stage IV disease. The median follow up was 74 months (95%CI, 53 to 116). Overall, the median age was 56.6 years (range 14.4-80.2) with equal distribution of female (28 patients, 45.9%) to male, and predominantly Caucasian (100%) (Table 1). Across both stages, a total of 22 patients (36.1%) received prior adjuvant interferon, 12 patients (19.7%) received prior chemotherapy, and 16 patients (26.2%) received prior immunotherapy. The most frequent site of metastases was lymph nodes (36 patients, 59.0%), followed by lung (29 patients, 47.5%) and skin (18 patients, 29.5%) or other sites (19 patients, 31.1%).
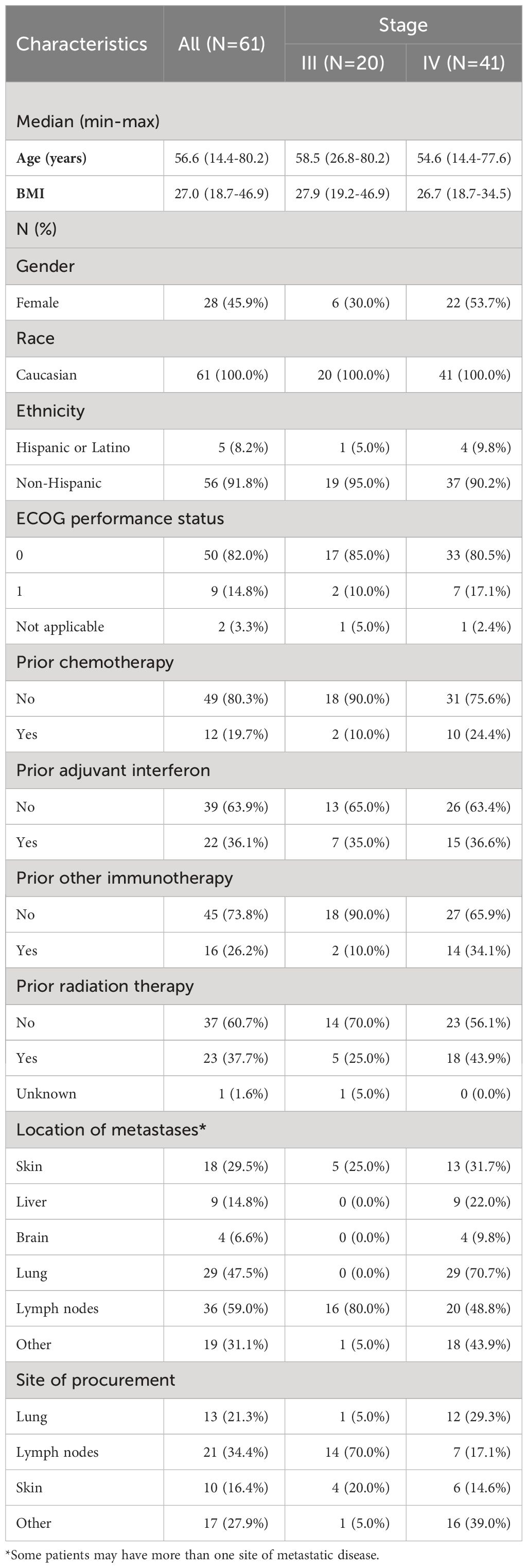
Table 1 Patient demographics and disease characteristics at baseline.
Efficacy
Among all patients, the median number of vaccinations was 7 (range 1-27). Among patients with stage IV disease, 1 patient (2.4%) had a partial response, 11 patients had stable disease (26.8%), and 29 patients (70.7%) had progressive disease (Table 2). Of patients with stage III disease, 14 (70%) had no evidence of disease and 3 (15%) had progressive disease. In the total cohort, the disease control rate (CR+PR+SD) was 39.3% (95%CI, 27.1 to 52.7%).
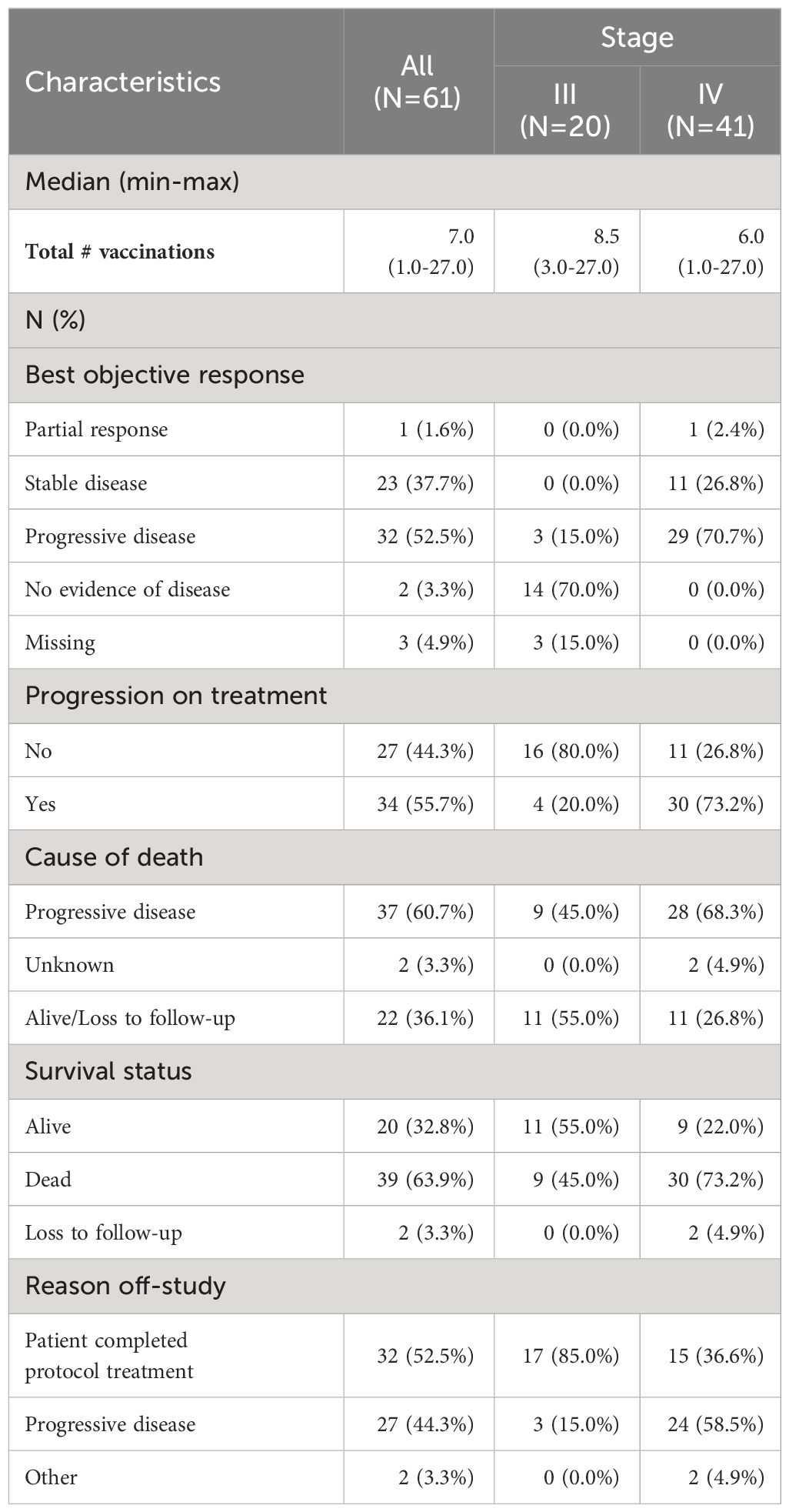
Table 2 Response rates stratified by stage.
With a median follow up of 74 months, the median progression-free survival was 7.9 (95%CI, 4.4 to 32.4) months in patients who received lethally irradiated, autologous melanoma cells engineered by adenoviral mediated gene transfer to secrete GM-CSF (Figure 1A). The median progression-free survival for patients with stage III disease was 50.7 (95%CI, 36.2 to NR) months and 4.1 (95%CI, 3.0 to 6.3) months for patients with stage IV disease (p<0.001) (Figure 1B).
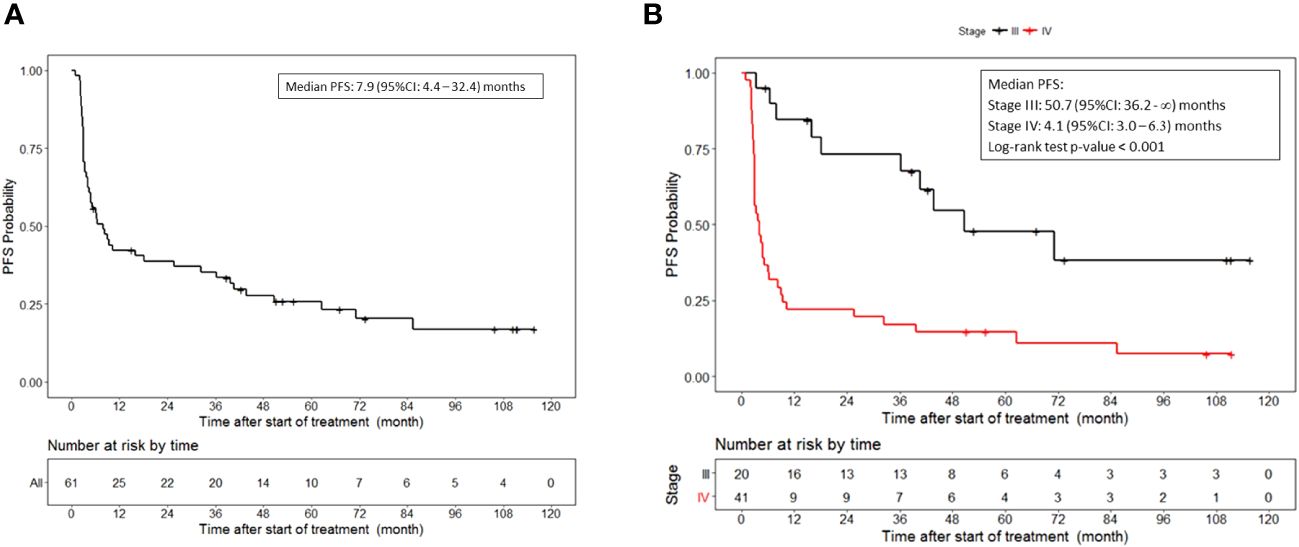
Figure 1 Progression free survival. (A) PFS for all patients who received vaccine therapy. (B) PFS stratified by stage III or IV melanoma who received vaccine therapy. Tick marks indicate censored data.
The median overall survival was 32.4 (95%CI, 16.0 to 71.1) months (Figure 2A). Additionally, the median overall survival for patients with stage III disease was 71.1 (95%CI, 43.7 to NR) months and 14.9 (95%CI, 12.1 to 39.7) months for patients with stage IV disease (p = 0.018) (Figure 2B).
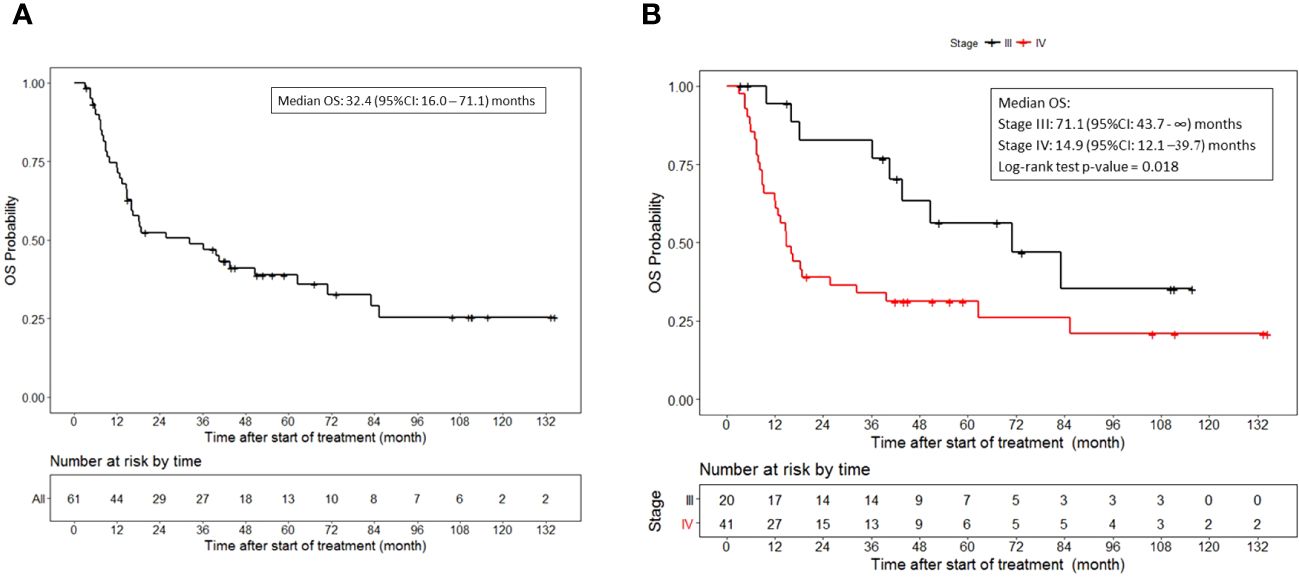
Figure 2 Overall survival. (A) OS for all patients who received vaccine therapy. (B) OS stratified by stage III or IV melanoma who received vaccine therapy. Tick marks indicate censored data.
Safety
The most frequent treatment-related adverse events are shown in Table 3. Of 61 patients, a total of 58 patients (95.1%) had any treatment-related adverse event, mostly grades 1-2. The most common grade 1 or 2 treatment-related adverse events included erythema in 46 patients (75.4%), injection site reactions in 30 patients (49.2%), and pruritis in 18 patients (29.5%). One patient had a grade 3 treatment-related adverse event of wound infection. No autoimmune reactions or adenoviral infections were observed. All other grade 3 or 4 adverse events were not related to treatment. For a full list of treatment-related adverse events and any adverse event, please see Supplementary Tables 1 and 2. Three grade 4 adverse events occurred, unlikely related to treatment. These included abdominal pain, pericarditis, and constitutional.
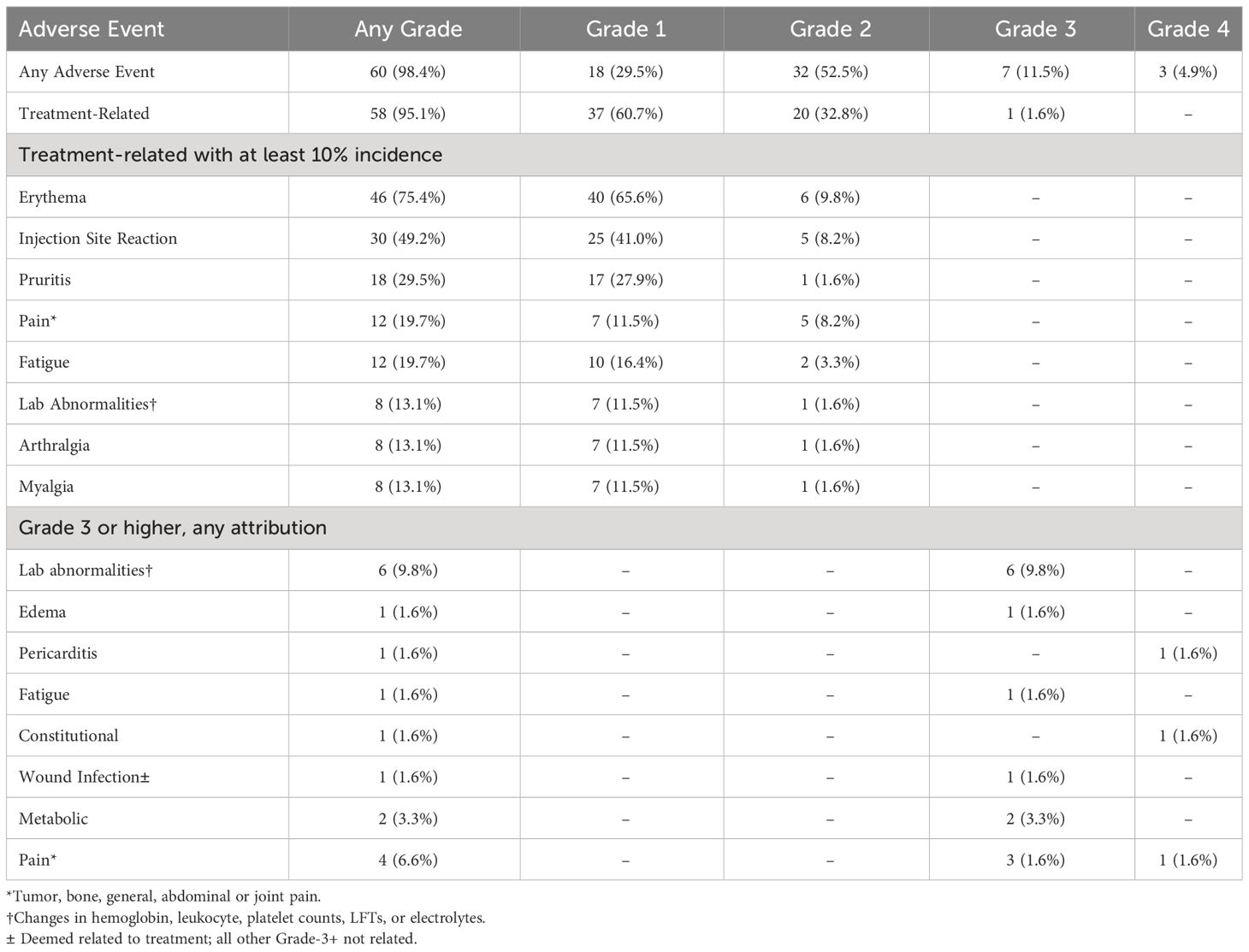
Table 3 Summary of adverse events.
Delayed-type hypersensitivity reactions
Irradiated, autologous nontransduced melanoma cells were available for delayed-type hypersensitivity testing in 60 patients (insufficient cells precluded these studies in one patient). Sixty patients received at least one injection; 88% of patients received at least two injections, for a total of 124 injections in the entire cohort. Most patients received injection at a dose of 1x106 (range: 1x106 to 4x106). About one third of injections provoked a clinical reaction at the injection site. The most common injection site reactions included erythema (33% of injections), induration (35%), redness (20%) and pruritis (9%). Supplementary Table 5 details delayed-type hypersensitivity (DTH) reactions. No association between a positive DTH reaction and PFS was observed.
Luminex
Stage III patients
A total of 32 biomarkers were analyzed by Luminex. For stage III patients, 19 had pre-treatment biomarker data and 18 also had biomarker data 4-8 weeks after the first dose of vaccine. In stage III patients, higher pre-treatment levels of MMP-1, TRAIL, CXCL-11, and CXCL-13 were statistically significantly related to improved PFS (p<0.05 for all), (Figure 3). Higher pre-treatment levels of CD40L, CTLA-8, and IL-20 trended toward improved PFS, but were not statistically significant (p=0.09, p=0.07 and p=0.08, respectively).
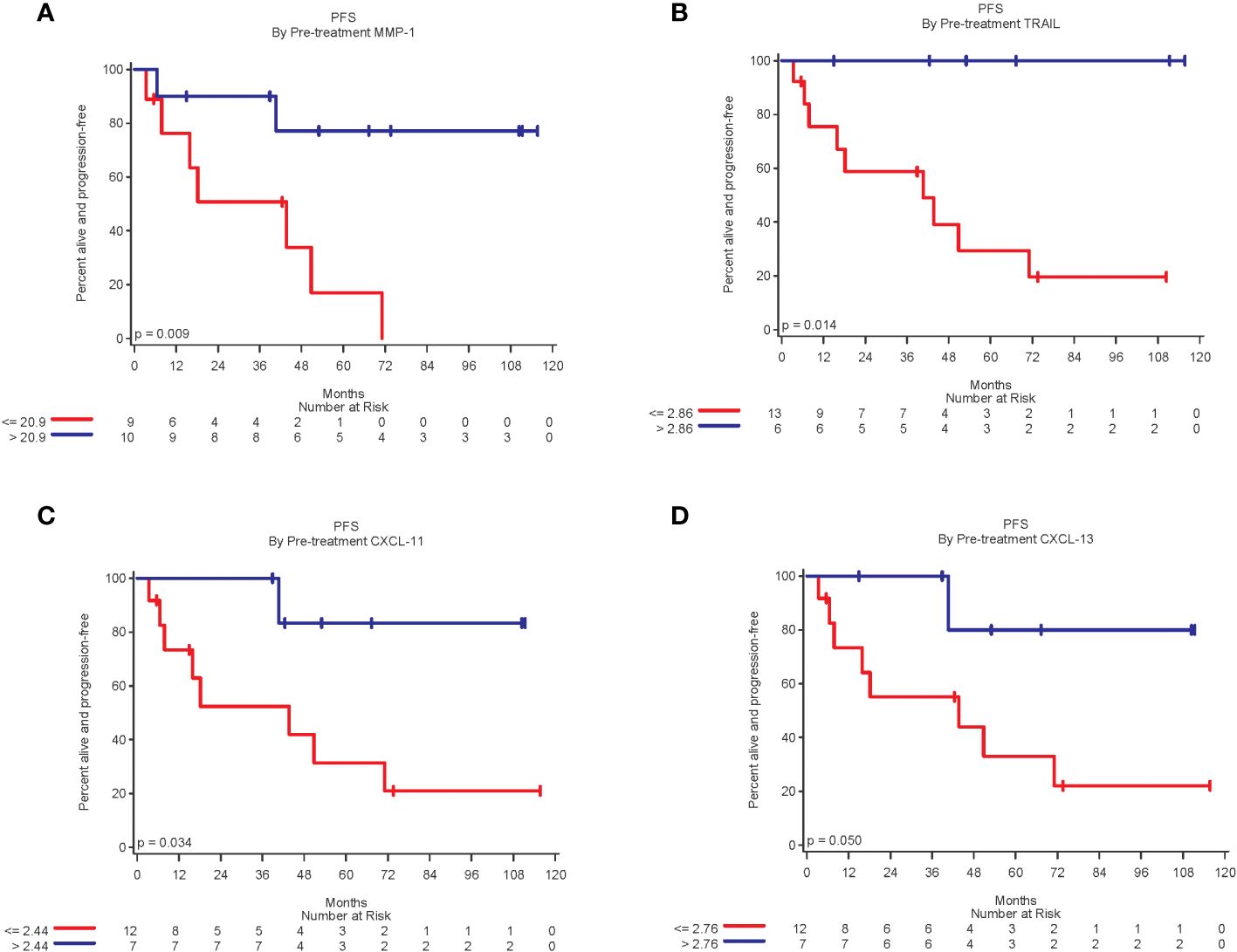
Figure 3 Kaplan-Meier plots of biomarkers related to improved PFS. (A) Patients with pre-treatment MMP-1 expression above the median of 20.9 was associated with significantly improved PFS (log-rang p=0.009); (B) Patients with TRAIL expression above the median of 2.86 was associated with significantly better PFS (p = 0.01). (C) Patients with CXCL-11 expression above median of 2.44 was associated with significantly better PFS (p=0.03); and (D) Patients with CXCL-13 expression above the median of 2.76 was associated with significantly better PFS (p = 0.05).
IL-16 and IL-20 had statistically significant increases in expression during the first 4-8 weeks after first vaccine when compared to pre-treatment levels (median increase of 76% and 58%, respectively; p<0.05 for both). For a complete list of biomarkers comparing fold-change from pre-treatment to 4-8 weeks post first vaccine, please see Supplementary Table 3.
In an exploratory analysis of 18 stage III patients, the fold-change of IL-15 and TRAIL levels when comparing pre-treatment to 8 weeks after first vaccine were significantly associated with PFS. A 5% or more decrease in IL-15 was associated with worse PFS compared to patients who had an increase in IL-15 (p=0.03) (Figure 4A). Similarly, an increase in fold-change of TRAIL was significantly associated with improved PFS (p=0.03) (Figure 4B). A decrease in fold-change in CXCL-11 demonstrated improved PFS but did not reach statistical significance (p=0.054).
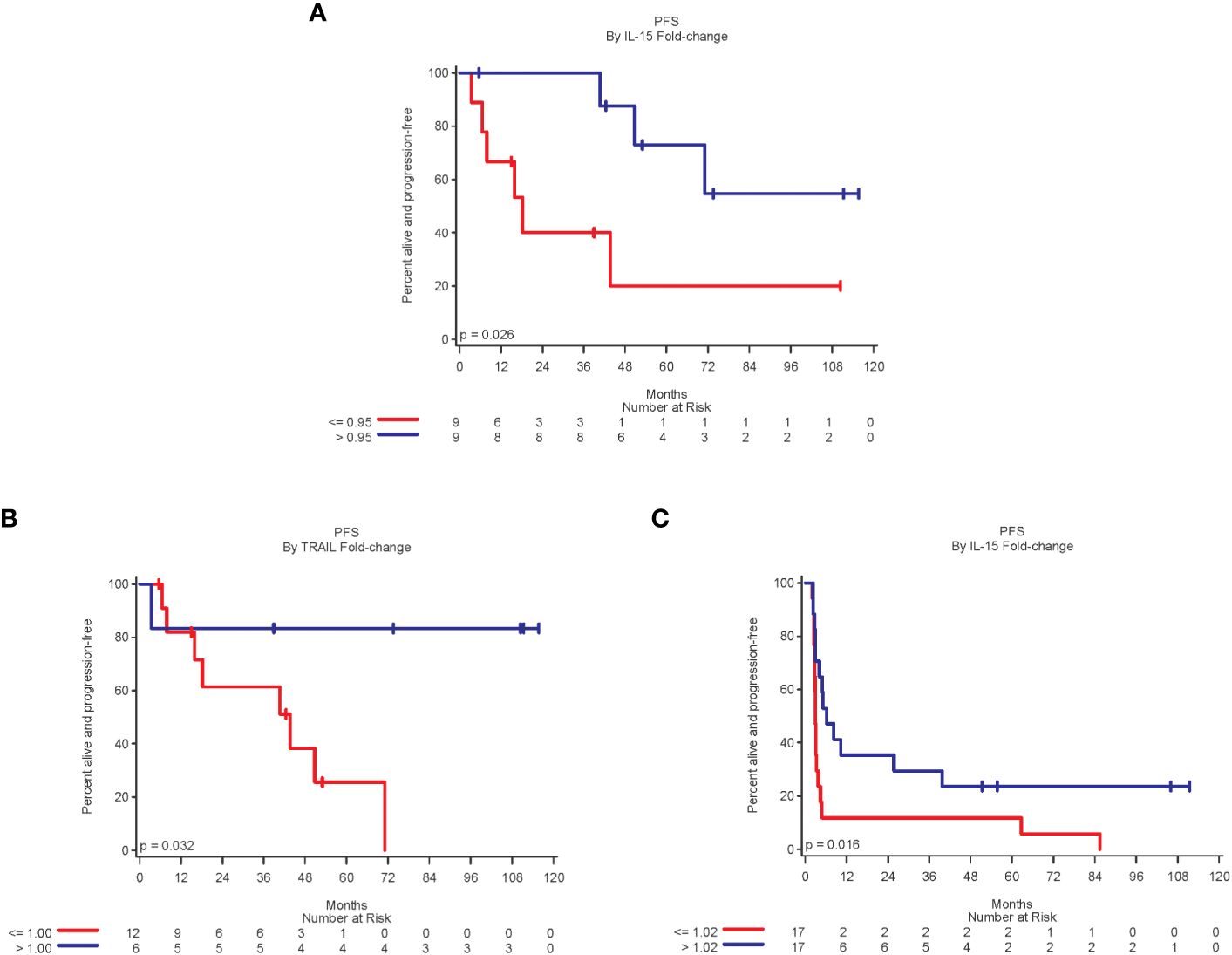
Figure 4 A fold-change above the median of 0.95 for IL-15 [(A), Stage III]; 1.00 for TRAIL [(B), Stage III]; and 1.02 for IL-16 [(C), Stage IV] was significantly associated with improved PFS.
Stage IV patients
For stage IV disease, 37 patients had pre-treatment biomarker data and 36 patients had biomarker data 4-8 weeks after the first vaccine dose. No statistically significant associations were demonstrated between pre-treatment biomarker levels and PFS. Lower pre-treatment levels of MDC and VEGF-A demonstrated better PFS, although not statistically significant (p=0.12 and p=0.14, respectively).
In comparing fold-change of cytokine expression from pre-treatment levels to 4-8 weeks after first vaccine, CTLA-8 and CXCL-5 demonstrated a statistically significant decrease (p=0.02 and p=0.01, respectively) (Supplementary Table 4). MMP-1 and IL-18 demonstrated decrease in fold-change, although not statistically significant (p=0.09 and p=0.07, respectively).
Increase in fold-change of IL-16 after treatment was significantly associated with improved PFS when compared to pre-treatment levels (p=0.02) (Figure 4C). Increases in CCL-4 and CXCL-11 during the first 4-8 weeks were associated with improved PFS, although not statistically significant (p=0.08 for both). A greater than 30% decrease in CCL-24 levels was associated with worse PFS, although not statistically significant (p=0.07). A fold-change in IL-16 was also related to clinical benefit. The clinical benefit rate (CR, PR, or SD) in patients with more than 2% increase in IL-16 was 47% (8 of 17 patients), compared to 12% in patients with a decrease or small increase in IL-16.
For stage IV patients, 11 patients experienced a DTH reaction and had pre-treatment Luminex data available and 23 did not have a DTH reaction but had Luminex data available. Pretreatment CXCL-11 levels were higher in patients who experienced a DTH reaction compared to patients who did not have a DTH reaction, although not statistically significant (p=0.10). CXCL-5 levels were lower in patients who had a DTH reaction compared to those that did not have a DTH reaction but was not statistically significant (p = 0.08). At a median of four weeks after first vaccine, 32 patients with stage IV disease had available Luminex data for analysis, of which 10 patients had a DTH reaction. CTLA-8 levels were lower in patients who had a reaction, although not statistically significant (p=0.08). Supplementary Table 6 details Luminex data for patients with and without DTH reaction.
Discussion
This phase II trial of autologous GM-CSF-secreting melanoma cell vaccines in stage III and IV patients demonstrates modest efficacy with evidence for the enhancement of anti-tumor immunity. The disease control rate in the overall cohort was 39.3% in patients who received GM-CSF secreting vaccines. Four of twenty (20%) patients with stage III disease progressed while receiving treatment, with a median PFS of 4.2 years and median OS of 5.9 years (5-year OS rate of 62%). Historically, the median recurrence free survival was about 2.2 to 2.5 years for resected high-risk stage III-IV patients receiving adjuvant interferon and median 5-year OS rate was 60% (14–17). This is comparable or inferior to our findings with adjuvant GM-CSF vaccine, which pre-dates the era of immune checkpoint inhibitor therapy. Vaccination is also associated with less toxicities compared to interferon-alpha (14–17).
Thirty of forty-one (72.2%) patients with stage IV disease progressed while receiving treatment, with a median PFS of 4.1 months and median OS of 14.9 months. These findings are comparable to historic data for ipilimumab in patients with metastatic disease, with median OS of 11.4 months in patients receiving ipilimumab for metastatic melanoma (18). Together, our data suggest that the GM-CSF secreting vaccine may be more efficacious in patients with locoregional disease compared to metastatic disease. Prior studies have demonstrated that functional tumor-specific T cells are more frequently found in melanoma patients with regional metastasis (71%), in comparison to patients with distant metastasis (23%) (19). Additionally, the greater efficacy of GM-CSF vaccination in the adjuvant setting may result in eradication of residual micro-metastasis if present, and higher frequencies of functional tumor-specific T cells may be due to lower tumor burden and ultimately less tumor-mediated immune suppression compared to metastatic disease (20).
In addition, the results of Luminex analysis demonstrated stronger correlation to changes in anti-tumor immunity for stage III patients. Higher pre-treatment levels of MMP-1, TRAIL, CXCL-11, and CXCL-13 were associated with improved PFS. Matrix metalloproteinase-1 (MMP-1) serves as an extracellular matrix degrading enzyme that facilitates tumor migration and invasion, promoting melanoma growth and metastasis (21). Higher levels of MMP-1 prior to vaccination might, however, modify immune cell trafficking or modulate dendritic cell function to facilitate an anti-tumor immune response. Similarly, TNFα-related apoptosis-inducing ligand (TRAIL) may promote extrinsic proapoptotic pathways through death receptor mediated signaling, potentially stimulating tumor cell killing in the presence of immune activation (22–25). Additionally, the presence of inflammatory (CXCL-11) and lymphoid (CXCL-13) chemokines has been associated with the recruitment of tumor infiltrating lymphocytes, namely CD4+ T cells, CD8+ T cells and mature dendritic cells, to promote melanoma cell destruction (26–28). CXCL-13 is also linked to the formation of tertiary lymphoid structures, and prior work showed that GM-CSF based vaccines stimulate coordinated T cell and antibody responses (4, 6). Together, these data raise the possibility that vaccine efficacy might be influenced, at least in part, by particular mixtures of chemokines and soluble tumor-associated factors that are present at the time of initiating therapy.
Luminex results for stage III patients also demonstrated increased expression of IL-16 and IL-20 after vaccine therapy, which suggests the initiation and/or amplification of an inflammatory response via activation of CD4+ T cells that might secrete additional pro-inflammatory cytokines including IL-1β, IL-6 and TNF-α (29–31). Moreover, increases in expression of IL-15 and TRAIL after vaccine therapy were associated with improved PFS in stage III patients. Recent studies have suggested that interleukins, like IL-15 elicit changes in natural killer cells that subsequently provide anti-tumor functionality and can remain active for weeks after first cytokine stimulation (32, 33). This further suggests that inflammatory cytokines induced by vaccination potentially contribute to tumor cell destruction and anti-tumor immunity.
Stage IV patients similarly demonstrated increases in IL-16 after vaccination that were associated with improved PFS and clinical benefit. IL-16 stimulates the production of pro-inflammatory cytokines, which would also support anti-tumor immunity (30). Levels of CTLA-8 (IL-17) and CXCL-5 decreased after vaccination, potentially indicating a reduction in tumor-promoting inflammation. Indeed, several studies have indicated that CTLA-8 (IL-17) may stimulate cancer cells to produce angiogenic factors like VEGF, thereby enhancing tumor angiogenesis and growth via STAT3 signaling (34–37). While CXCL-5 is a chemokine that recruits and activates leukocytes, it also promotes angiogenesis, tumor growth, and metastasis (38–41). High levels of CXCL-5 have similarly been associated with clinical response in a small study of patients treated with ipilimumab plus nivolumab, suggesting that the role of this chemokine in immunotherapy should be investigated in more detail (42).
The use of GM-CSF in cancer vaccines is based on the ability of the cytokine to increase antigen-specific immune responses and to function as an immune adjuvant for dendritic cells (43). Indeed, autologous dendritic cell vaccines can generate potent anti-tumor immune responses via enhanced tumor antigen presentation (44). The median OS demonstrated in our study of 14.9 months for stage IV patients is similar to prior dendritic cell vaccine trials using allogeneic and autologous tumor cells, with OS ranging from 12 to 14 months (45, 46). Moreover, Dillman et al. conducted a randomized phase II trial that compared autologous tumor cell vaccine (TC) with autologous tumor cells loaded onto dendritic cells (DCV) admixed with GM-CSF protein in 42 patients with metastatic melanoma (47–49). This trial demonstrated that the DCV arm achieved a superior 2-year survival rate of 72% versus 31% in the TC arm (p=0.007) (48). At 5-years, patients who received DCV survived longer with a median OS of 43.4 months versus 20.5 months, and showed a 70% reduction in the risk of death (HR 0.30, p=0.005) (49). Similarly, two clinical trials that administered ipilimumab to patients with metastatic melanoma or ovarian carcinoma that were previously vaccinated with autologous GM-CSF-secreting melanoma or ovarian cancer vaccines demonstrated extensive tumor necrosis or the reduction of cancer antigen-125 (CA-125) levels (50, 51). These findings suggest that the addition of CTLA-4 inhibition might intensify tumor immunity in patients who have been previously vaccinated. In this context, GM-CSF secreting melanoma vaccines may serve to prime a patient’s immune system, whereas the addition of checkpoint blockade or other immune activating mechanisms may augment the anti-tumor response and potentiate clinical efficacy.
Additional promising investigations in vaccine therapy include neoantigen vaccines in melanoma. Several studies with neoantigen vaccines using mRNA or peptides admixed with poly-ICLC (NeoVax) were recently published (52–54). Ott et al. demonstrated in six high-risk resected stage III-IV melanoma patients that personalized vaccines formulated with up to 20 predicted MHC class I restricted neoantigens induced specific CD4+ and, to a lesser extent, CD8+ T cells (54). At 25 months after vaccination, four patients showed no recurrence and the two patients who did recur subsequently achieved complete responses after anti-PD-1 therapy. mRNA vaccines similarly demonstrated the generation of antigen specific CD4+ and CD8+ T cells and a phenomenon of epitope-spreading (52–54). Neoantigen vaccines are now being broadly studied in conjunction with anti-PD-1 therapies in several disease settings.
The current study demonstrates the feasibility of an autologous whole cell vaccination strategy in stage III and stage IV metastatic melanoma patients. Limitations of this study include the small cohort size and the single-arm design. Since there was no comparator cohort, we cannot definitively demonstrate an improvement in PFS or OS that can be attributed to GM-CSF secreting melanoma vaccines. Follow up data on subsequent therapies that patients received after vaccination were not collected, so the potential impact of immune checkpoint inhibitor therapy on PFS or OS is unknown at this time. The Luminex findings are also exploratory and must be verified in a larger cohort of patients. Additionally, biopsies of metastatic sites may have provided further information on immune cell infiltration, especially in comparison to local vaccine site biopsies. Prior studies have demonstrated infiltrates composed of dendritic cells, macrophages, T and B lymphocytes, and eosinophils in vaccination sites and distant metastases (4, 6). Lastly, the efficacy of such a vaccination strategy may be of greater benefit in patients with limited disease in the adjuvant setting and with the use of immune checkpoint blockade.
In conclusion, this study demonstrates that vaccination with irradiated, autologous melanoma cells engineered to secrete GM-CSF may elicit antitumor immunity in patients with stage III and IV melanoma. The overall disease control rate of 39%, with a median follow up of 74 months, is intriguing and raises the possibility that GM-CSF vaccination might be effectively combined with immune checkpoint inhibitor therapy. The median PFS of 50.7 months and median OS of 71.1 months in high-risk resected stage III patients is comparable to or potentially superior in efficacy to interferon in the pre-ICI era and suggests that GM-CSF vaccination may be particularly efficacious when combined with immune checkpoint blockade for stage III patients. The prolonged overall survival in stage III patients suggests that the presence of minimal/locoregional disease might facilitate the priming of anti-tumor immunity with GM-CSF secreting melanoma vaccines, which might be further intensified with the addition of immune checkpoint inhibitors.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
The studies involving humans were approved by Dana-Farber/Harvard Cancer Center Institutional Review Board. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
Author contributions
TS: Writing – original draft, Writing – review & editing. MS: Data curation, Investigation, Methodology, Writing – review & editing. AG-H: Formal analysis, Methodology, Validation, Writing – review & editing. PF: Investigation, Validation, Writing – review & editing. SS: Investigation, Writing – review & editing. MJ: Investigation, Writing – review & editing. TC: Investigation, Writing – review & editing. LG: Investigation, Writing – review & editing. DL: Investigation, Writing – review & editing. RS: Investigation, Writing – review & editing. HD: Project administration, Writing – review & editing. JR: Investigation, Supervision, Validation, Writing – review & editing. GD: Investigation, Methodology, Supervision, Visualization, Writing – review & editing. FH: Investigation, Methodology, Resources, Supervision, Validation, Visualization, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
Author MS was employed by Curis, Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2024.1395978/full#supplementary-material
References
1. (2022). Available online at: https://www.cnn.com/2022/12/13/health/mrna-cancer-vaccine-study/index.html.
2. Leong SPL, Enders-Zohr P, Zhou YM, Stuntebeck S, Habib FA, Allen RE, et al. Recombinant human granulocyte macrophage-colony stimulating factor (rhGM-CSF) and autologous melanoma vaccine mediate tumor regression in patients with metastatic melanoma. J Immunother. (1999) 22:166–74. doi: 10.1097/00002371-199903000-00008
3. Jaffee EM, Hruban RH, Biedrzycki B, Laheru D, Schepers K, Sauter PR, et al. Novel allogeneic granulocyte-macrophage colony-stimulating factor-secreting tumor vaccine for pancreatic cancer: a phase I trial of safety and immune activation. J Clin Oncol. (2001) 19:145–56. doi: 10.1200/JCO.2001.19.1.145
4. Soiffer R, Lynch T, Mihm M, Jung K, Rhuda C, Schmollinger JC, et al. Vaccination with irradiated autologous melanoma cells engineered to secrete human granulocyte-macrophage colony-stimulating factor generates potent antitumor immunity in patients with metastatic melanoma. Proc Natl Acad Sci U.S.A. (1998) 95:13141–6. doi: 10.1073/pnas.95.22.13141
5. Dranoff G, Soiffer R, Lynch T, Mihm M, Jung K, Kolesar K, et al. A phase I Study of Vaccination with Autologous, Irradiated Melanoma Cells Engineered to Secrete Human Granulocyte-Macrophage Colony Stimulating Factor Vol. 8.Boston, Massachusetts: Dana-Farber Cancer Institute (2008). Available at: https://home.liebertpub.com/humhttps://www.liebertpub.com/doi/10.1089/hum.1997.8.1-111.
6. Soiffer R, Hodi FS, Haluska F, Jung K, Gillessen S, Singer S, et al. Vaccination with irradiated, autologous melanoma cells engineered to secrete granulocyte-macrophage colony-stimulating factor by adenoviral-mediated gene transfer augments antitumor immunity in patients with metastatic melanoma. J Clin Oncol. (2003) 21:3343–50. doi: 10.1200/JCO.2003.07.005
7. Eric L, Yeo CJ, Lillemoe KD, Biedrzycki B, Kobrin B, Herman J, et al. A lethally irradiated allogeneic granulocyte-macrophage colony stimulating factor-secreting tumor vaccine for pancreatic adenocarcinoma: A phase II trial of safety, efficacy, and immune activation. Ann Surg. (2011) 253:328–35. doi: 10.1097/SLA.0b013e3181fd271c
8. Slingluff CL Jr, Yamshchikov G, Neese P, Galavotti H, Eastham S, Engelhard VH, et al. Phase I trial of a melanoma vaccine with gp100(280-288) peptide and tetanus helper peptide in adjuvant: immunologic and clinical outcomes. Clin Cancer Res. (2001) 7(10):3012–24.
9. Smith JW, Walker EB, Fox BA, Haley D, Wisner KP, Doran T, et al. Adjuvant immunization of HLA-A2-positive melanoma patients with a modified gp100 peptide induces peptide-specific CD8+ T-cell responses. J Clin Oncol. (2003) 21:1562–73. doi: 10.1200/JCO.2003.09.020
10. Keenan TE, Guerriero JL, Barroso-Sousa R, Li T, O’Meara T, Giobbie-Hurder A, et al. Molecular correlates of response to eribulin and pembrolizumab in hormone receptor-positive metastatic breast cancer. Nat Commun. (2021) 12(1):5563. doi: 10.1038/s41467-021-25769-z
11. Vining KH, Marneth AE, Adu-Berchie K, Grolman JM, Tringides CM, Liu Y, et al. Mechanical checkpoint regulates monocyte differentiation in fibrotic niches. Nat Mater. (2022) 21:939–50. doi: 10.1038/s41563-022-01293-3
12. Tyan K, Baginska J, Brainard M, Giobbie-Hurder A, Severgnini M, Manos M, et al. Cytokine changes during immune-related adverse events and corticosteroid treatment in melanoma patients receiving immune checkpoint inhibitors. Cancer Immunol Immunother. (2021) 70:2209–21. doi: 10.1007/s00262-021-02855-1
13. von Elm E, Altman DG, Egger M, Pocock SJ, Gøtzsche PC, Vandenbroucke JP. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. J Clin Epidemiol. (2008) 61:344–9. doi: 10.1016/j.jclinepi.2007.11.008
14. Mocellin S, Lens MB, Pasquali S, Pilati P, Chiarion Sileni V. Interferon alpha for the adjuvant treatment of cutaneous melanoma. Cochrane Database Syst Rev. (2013) 2013(6):CD008955. doi: 10.1002/14651858
15. Eggermont AMM, Suciu S, Testori A, Santinami M, Kruit WHJ, Marsden J, et al. Long-term results of the randomized phase III trial EORTC 18991 of adjuvant therapy with pegylated interferon alfa-2b versus observation in resected stage III melanoma. J Clin Oncol. (2012) 30:3810–8. doi: 10.1200/JCO.2011.41.3799
16. Anaya DA, Xing Y, Feng L, Huang X, Camacho LH, Ross MI, et al. Adjuvant high-dose interferon for cutaneous melanoma is most beneficial for patients with early stage III disease. Cancer. (2008) 112:2030–7. doi: 10.1002/cncr.23399
17. Tarhini AA, Lee SJ, Hodi FS, Rao UNM, Cohen GI, Hamid O, et al. A phase III randomized study of adjuvant ipilimumab (3 or 10 mg/kg) versus high-dose interferon alfa-2b for resected high-risk melanoma (U.S. Intergroup E1609): Preliminary safety and efficacy of the ipilimumab arms. J Clin Oncol. (2017) 35:9500–0. doi: 10.1200/JCO.2017.35.15_suppl.9500
18. SChadendorf D, Hodi FS, Robert C, Weber JS, Margolin K, Hamid O, et al. Pooled analysis of long-term survival data from phase II and phase III trials of ipilimumab in unresectable or metastatic melanoma. J Clin Oncol. (2015) 33:1889–94. doi: 10.1200/JCO.2014.56.2736
19. Aarntzen EHJG, Bol K, Schreibelt G, Jacobs JFM, Lesterhuis WJ, Van Rossum MM, et al. Skin-test infiltrating lymphocytes early predict clinical outcome of dendritic cell-based vaccination in metastatic melanoma. Cancer Res. (2012) 72:6102–10. doi: 10.1158/0008-5472.CAN-12-2479
20. Gajewski TF. Failure at the effector phase: immune barriers at the level of the melanoma tumor microenvironment. Clin Cancer Res. (2007) 13:5256–61. doi: 10.1158/1078-0432.CCR-07-0892
21. Iida J, McCarthy JB. Expression of collagenase-1 (MMP-1) promotes melanoma growth through the generation of active transforming growth factor-beta. Melanoma Res. (2007) 17:205–13. doi: 10.1097/CMR.0b013e3282a660ad
22. Kurbanov BM, Geilen CC, Fecker LF, Orfanos CE, Eberle J. Efficient TRAIL-R1/DR4-mediated apoptosis in melanoma cells by tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). J Invest Dermatol. (2005) 125:1010–9. doi: 10.1111/j.0022-202X.2005.23900.x
23. Eberle J, Kurbanov BM, Hossini AM, Trefzer U, Fecker LF. Overcoming apoptosis deficiency of melanoma-hope for new therapeutic approaches. Drug Resist Update. (2007) 10:218–34. doi: 10.1016/j.drup.2007.09.001
24. Mace TA, Yamane N, Cheng J, Hylander BL, Repasky EA. The potential of the tumor microenvironment to influence Apo2L/TRAIL induced apoptosis. Immunol Invest. (2006) 35:279–96. doi: 10.1080/08820130600745463
25. Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, et al. Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med. (1999) 5:157–63. doi: 10.1038/5517
26. Li B, Severson E, Pignon JC, Zhao H, Li T, Novak J, et al. Comprehensive analyses of tumor immunity: implications for cancer immunotherapy. Genome Biol. (2016) 17(1):174. doi: 10.1186/s13059-016-1028-7
27. Martinet L, Le Guellec S, Filleron T, Lamant L, Meyer N, Rochaix P, et al. High endothelial venules (HEVs) in human melanoma lesions: Major gateways for tumor-infiltrating lymphocytes. Oncoimmunology. (2012) 1:829–39. doi: 10.4161/onci.20492
28. Liu H, Yang Z, Lu W, Chen Z, Chen L, Han S, et al. Chemokines and chemokine receptors: A new strategy for breast cancer therapy. Cancer Med. (2020) 9:3786–99. doi: 10.1002/cam4.3014
29. Cruikshank WW, Center DM, Nisar N, Wu M, Natke B, Theodore AC, et al. Molecular and functional analysis of a lymphocyte chemoattractant factor: association of biologic function with CD4 expression. Proc Natl Acad Sci U.S.A. (1994) 91:5109–13. doi: 10.1073/pnas.91.11.5109
30. Mathy NL, Scheuer W, Lanzendörfer M, Honold K, Ambrosius D, Norley S, et al. Interleukin-16 stimulates the expression and production of pro-inflammatory cytokines by human monocytes. Immunology. (2000) 100:63. doi: 10.1046/j.1365-2567.2000.00997.x
31. Cruikshank WW, Berman JS, Theodore AC, Bernardo J, Center DM. Lymphokine activation of T4+ T lymphocytes and monocytes. J Immunol. (1987) 138(11):3817–23.
32. Romee R, Rosario M, Berrien-Elliott MM, Wagner JA, Jewell BA, Schappe T, et al. Cytokine-induced memory-like natural killer cells exhibit enhanced responses against myeloid leukemia. Sci Transl Med. (2016) 8(357):357ra123. doi: 10.1126/scitranslmed.aaf2341
33. Gang M, Wong P, Berrien-Elliott MM, Fehniger TA. Memory-like natural killer cells for cancer immunotherapy. Semin Hematol. (2020) 57:185–93. doi: 10.1053/j.seminhematol.2020.11.003
34. Numasaki M, Watanabe M, Suzuki T, Takahashi H, Nakamura A, McAllister F, et al. IL-17 enhances the net angiogenic activity and in vivo growth of human non-small cell lung cancer in SCID mice through promoting CXCR-2-dependent angiogenesis. J Immunol. (2005) 175:6177–89. doi: 10.4049/jimmunol.175.9.6177
35. Numasaki M, Fukushi JI, Ono M, Narula SK, Zavodny PJ, Kudo T, et al. Interleukin-17 promotes angiogenesis and tumor growth. Blood. (2003) 101:2620–7. doi: 10.1182/blood-2002-05-1461
36. Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H. IL-17 can promote tumor growth through an IL-6–Stat3 signaling pathway. J Exp Med. (2009) 206:1457. doi: 10.1084/jem.20090207
37. Kirkham BW, Kavanaugh A, Reich K. Interleukin-17A: a unique pathway in immune-mediated diseases: psoriasis, psoriatic arthritis and rheumatoid arthritis. Immunology. (2014) 141:133–42. doi: 10.1111/imm.12142
38. Zhang W, Wang H, Sun M, Deng X, Wu X, Ma Y, et al. CXCL5/CXCR2 axis in tumor microenvironment as potential diagnostic biomarker and therapeutic target. Cancer Commun. (2020) 40:69. doi: 10.1002/cac2.12010
39. Duchene J, Lecomte F, Ahmed S, Cayla C, Pesquero J, Bader M, et al. A novel inflammatory pathway involved in leukocyte recruitment: role for the kinin B1 receptor and the chemokine CXCL5. J Immunol. (2007) 179:4849–56. doi: 10.4049/jimmunol.179.7.4849
40. Griffith JW, Sokol CL, Luster AD. Chemokines and chemokine receptors: positioning cells for host defense and immunity. Annu Rev Immunol. (2014) 32:659–702. doi: 10.1146/annurev-immunol-032713-120145
41. Zlotnik A, Yoshie O. The chemokine superfamily revisited. Immunity. (2012) 36:705–16. doi: 10.1016/j.immuni.2012.05.008
42. Fujimura T, Sato Y, Tanita K, Lyu C, Kambayashi Y, Amagai R, et al. Association of baseline serum levels of CXCL5 with the efficacy of nivolumab in advanced melanoma. Front Med (Lausanne). (2019) 6. doi: 10.3389/fmed.2019.00086
43. Melssen MM, Pollack KE, Meneveau MO, Smolkin ME, Pinczewski J, Koeppel AF, et al. Characterization and comparison of innate and adaptive immune responses at vaccine sites in melanoma vaccine clinical trials. Cancer Immunology Immunotherapy. (2021) 70:2151–64. doi: 10.1007/s00262-020-02844-w
44. Javed A, Sato S, Sato T. Autologous melanoma cell vaccine using monocyte-derived dendritic cells (NBS20/eltrapuldencel-T). Future Oncol. (2016) 12:751–62. doi: 10.2217/fon.16.13
45. Ribas A, Camacho LH, Lee SM, Hersh EM, Brown CK, Richards JM, et al. Multicenter phase II study of matured dendritic cells pulsed with melanoma cell line lysates in patients with advanced melanoma. J Transl Med. (2010) 8:89. doi: 10.1186/1479-5876-8-89
46. López MN, Pereda C, Segal G, Muñoz L, Aguilera R, González FE, et al. Prolonged survival of dendritic cell-vaccinated melanoma patients correlates with tumor-specific delayed type IV hypersensitivity response and reduction of tumor growth factor beta-expressing T cells. J Clin Oncol. (2009) 27:945–52. doi: 10.1200/JCO.2008.18.0794
47. Dillman RO, Selvan SR, Schiltz PM, McClay EF, Barth NM, Depriest C, et al. Phase II trial of dendritic cells loaded with antigens from self-renewing, proliferating autologous tumor cells as patient-specific antitumor vaccines in patients with metastatic melanoma: final report. Cancer Biother Radiopharm. (2009) 24:311–9. doi: 10.1089/cbr.2008.0599
48. Dillman RO, Cornforth AN, Depriest C, McClay EF, Amatruda TT, de Leon C, et al. Tumor stem cell antigens as consolidative active specific immunotherapy: a randomized phase II trial of dendritic cells versus tumor cells in patients with metastatic melanoma. J Immunother. (2012) 35:641–9. doi: 10.1097/CJI.0b013e31826f79c8
49. Dillman RO, Cornforth AN, Nistor GI, McClay EF, Amatruda TT, Depriest C. Randomized phase II trial of autologous dendritic cell vaccines versus autologous tumor cell vaccines in metastatic melanoma: 5-year follow up and additional analyses. J Immunother Cancer. (2018) 6(1):19. doi: 10.1186/s40425-018-0330-1
50. Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV, et al. Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci U.S.A. (2003) 100:4712–7. doi: 10.1073/pnas.0830997100
51. Hodi FS, Butler M, Oble DA, Seiden MV, Haluska FG, Kruse A, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U.S.A. (2008) 105:3005–10. doi: 10.1073/pnas.0712237105
52. Hu Z, Leet DE, Allesøe RL, Oliveira G, Li S, Luoma AM, et al. Personal neoantigen vaccines induce persistent memory T cell responses and epitope spreading in patients with melanoma. Nat Med. (2021) 27:515–25. doi: 10.1038/s41591-020-01206-4
53. Sahin U, Derhovanessian E, Miller M, Kloke BP, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. (2017) 547:222–6. doi: 10.1038/nature23003
Keywords: melanoma, vaccine, advanced disease, GM-CSF, stage III, Stage IV
Citation: Sussman TA, Severgnini M, Giobbie-Hurder A, Friedlander P, Swanson SJ, Jaklitsch M, Clancy T, Goguen LA, Lautz D, Swanson R, Daley H, Ritz J, Dranoff G and Hodi FS (2024) Phase II trial of vaccination with autologous, irradiated melanoma cells engineered by adenoviral mediated gene transfer to secrete granulocyte-macrophage colony stimulating factor in patients with stage III and IV melanoma. Front. Oncol. 14:1395978. doi: 10.3389/fonc.2024.1395978
Received: 04 March 2024; Accepted: 16 April 2024;
Published: 15 May 2024.
Edited by:
Suzie Chen, Rutgers, The State University of New Jersey, United StatesReviewed by:
María Marcela Barrio, Fundación Cáncer, ArgentinaEvidio Domingo-Musibay, Allina Health, United States
Copyright © 2024 Sussman, Severgnini, Giobbie-Hurder, Friedlander, Swanson, Jaklitsch, Clancy, Goguen, Lautz, Swanson, Daley, Ritz, Dranoff and Hodi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: F. Stephen Hodi, Stephen_hodi@dfci.harvard.edu