
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol., 24 April 2024
Sec. Cancer Metabolism
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1395192
This article is part of the Research TopicMetabolic Crosstalk between Cancer Cells and Immune Cells in the Tumor Microenvironment: Cellular and Molecular Insights, and their Therapeutic ImplicationsView all 12 articles
 Tie Sun
Tie Sun Xuan Xiao*
Xuan Xiao*Altered cholesterol metabolism has been identified as a critical feature of cancers. Cholesterol functions as the main component of cell membrane, cholesterol and is required for sustaining membrane integrity and mediating signaling transduction for cell survival. The intracellular level of cholesterol is dynamically regulated. Excessive cholesterol could be converted to less toxic cholesteryl esters by acyl-coenzyme A:cholesterol acyltransferases (ACATs). While ACAT2 has limited value in cancers, ACAT1 has been found to be widely participated in tumor initiation and progression. Moreover, due to the important role of cholesterol metabolism in immune function, ACAT1 is also essential for regulating anti-tumor immunity. ACAT1 inhibition may be exploited as a potential strategy to enhance the anti-tumor immunity and eliminate tumors. Herein, a comprehensive understanding of the role of ACAT1 in tumor development and anti-tumor immunity may provide new insights for anti-tumor strategies.
A hallmark of cancer is the deregulated metabolism (1). As an integral component of cell membrane, cholesterol is crucial for maintaining membrane integrity and signaling transduction for cell survival (2). Besides, cholesterol also participates in the regulation of multiple biological processes, including lipid metabolism, inflammation, apoptosis, and cell survival (3–5). Cholesterol-derived metabolites exert a wide variety of biological effects in tumor development and anti-tumor immunity responses (6). As fast-proliferating cells, tumor cells rely on cholesterol for membrane biogenesis and various biological processes (7). Therefore, targeting cholesterol metabolism may provide novel therapeutic strategies for cancer management.
Intracellular cholesterol is dynamically transported for maintaining membrane integrity (8). Excessive cholesterol is either exported by ATP-binding cassette proteins, or converted to less toxic cholesteryl esters by acyl-coenzyme A: cholesterol acyltransferases (ACATs) to store in the form of lipid droplets or lipoproteins (3). ACATs belong to membrane-bound O-acyltransferase family, are composed of two enzymes localizing in the mitochondria and cytoplasm, respectively (9). ACAT1 and ACAT2 catalyze acyl transfer from acyl-coenzyme A (CoA) to cholesterol and produce cholesterol esters that are used for storage and intercellular transport of sterol, which is important for cellular cholesterol homeostasis (10). ACAT1 is expressed in nucleated eukaryotic cells and its products are incorporated into lipid droplets (LDs) in the cytoplasm (11). ACAT2 is primarily expressed in intestinal epithelial cells and hepatocytes, and its products are incorporated into lipoproteins in the endoplasmic reticulum (12). While ACAT2 has limited value in tumors, ACAT1 has been found to be implicated in tumor occurrence and development. Recent studies introduced the complex role of ACAT1 in tumor development and anti-tumor immunity, which may provide new insights for anti-tumor strategies.
Structure analysis has identified human ACAT1 as a tetramer with two homodimers (13). Each monomer is composed of nine transmembrane segments, which enclose a cytosolic tunnel and a transmembrane tunnel that converge at the predicted catalytic site (14). ACAT1 tetramers, but not monomers, are phosphorylated and stabilized by enhanced Y407 phosphorylation observed in multiple human cancer cells. It has also been indicated that CoA could enter through the cytosolic tunnel, while cholesterol enters via the transmembrane tunnel (13). The structure of ACAT1 has been deciphered previously (13, 14). ACAT1 exerts its catalytic role in ketolysis, ketogenesis, fatty acid oxidation and isoleucine degradation (12). ACAT1 senses free cholesterol by its allosteric site. ACAT1 cannot exert its catalytic role for esterification with high efficiency under low cholesterol concentrations, whereas high amounts of cholesterol could facilitate esterification allosterically under high cholesterol concentrations (13). Herein, ACAT1 activity is determined by the level of free cholesterol to regulate cholesterol homeostasis of the endoplasmic reticulum (15). In addition to mediating cholesterol homeostasis, ACAT1 exerts its acetyltransferase activity capable of specifically acetylating various enzymes. For instance, ACAT1 regulates pyruvate dehydrogenase complex (PDC) by acetylating pyruvate dehydrogenase (PDH) and PDH phosphatase to promote glycolysis (16). ACAT1-mediated K128 acetylation of GNPAT could protect FASN from degradation and promote lipid metabolism (17). ACAT1-mediated K337 acetylation of ME1 dimerize and activate ME1 to regulate NADPH generation and lipid metabolism (18). The structure and function of ACAT1 has been illustrated in Figure 1.
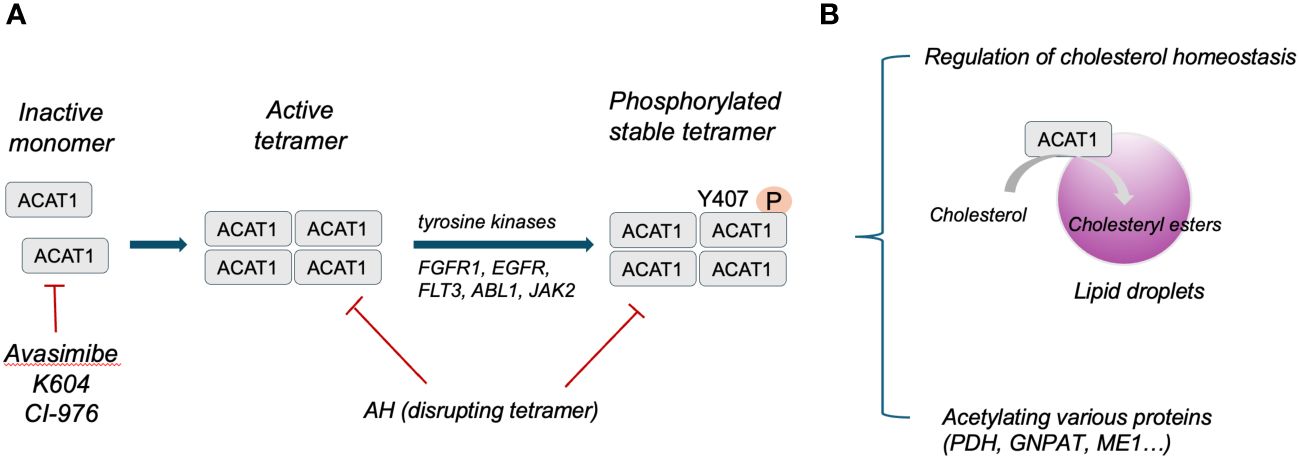
Figure 1 (A), Schematic illustration of ACAT1 structure. (B), Primary function of ACAT1 in cells.
Cholesterol metabolism has been identified to be essential for immune function (4, 19). Cholesterol biosynthesis is critical for T cell growth, activation, and anti-tumor function (4, 20, 21). It has been demonstrated that elevated levels of cholesterol in T cells could boost the anti-tumor immunity of T cells (22). CD8+ T cells play an essential role in anti-tumor immunity, but their function is always abrogated in the context of cancers (23). Therefore, remodeling the anti-tumor ability of CD8+ T cells is a key strategy for improving the efficacy of immunotherapy. ACAT1 inhibition could impair cholesterol esterification, therefore potentiating anti-tumor effect and strengthening cell proliferation of CD8+ T cells (22). Mechanistically, elevated cholesterol level of CD8+ T cells could enhance T-cell receptor clustering and signaling. ACAT1-deleted CD8+ T cells exhibited impaired tumor growth and metastasis of melanoma (22). An avasimibe-induced inhibition of cholesterol esterification has been shown to improve the antitumor response of CD8+ T cells in mice (24). Avasimibe exerted significant anti-tumor effect. Moreover, avasimibe combined with PD-1 inhibitor exhibited greater anti-tumor capabilities compared with PD-1 inhibitor alone. Avasimibe could be restrained on the T cell surface to induce rapid T cell receptor clustering and sustaine T cell activation (25). In addition, paclitaxel and immunoadjuvant αGC were co-encapsulated in liposomes modified with pH sensitive TH peptide (PTX/αGC-TH-Lip). Avasimibe could elevate the level of free cholesterol and relieve the inhibition of CD8+ T cells resulted from PTX/αGC-TH-Lip. The combination of avasimibe and PTX/αGC-TH-Lip could enhance immune responses and cytotoxic effects in xenografts of melanoma, which is a potential strategy to improve the anti-tumor effects of immune-chemotherapy (26) (Figure 2).
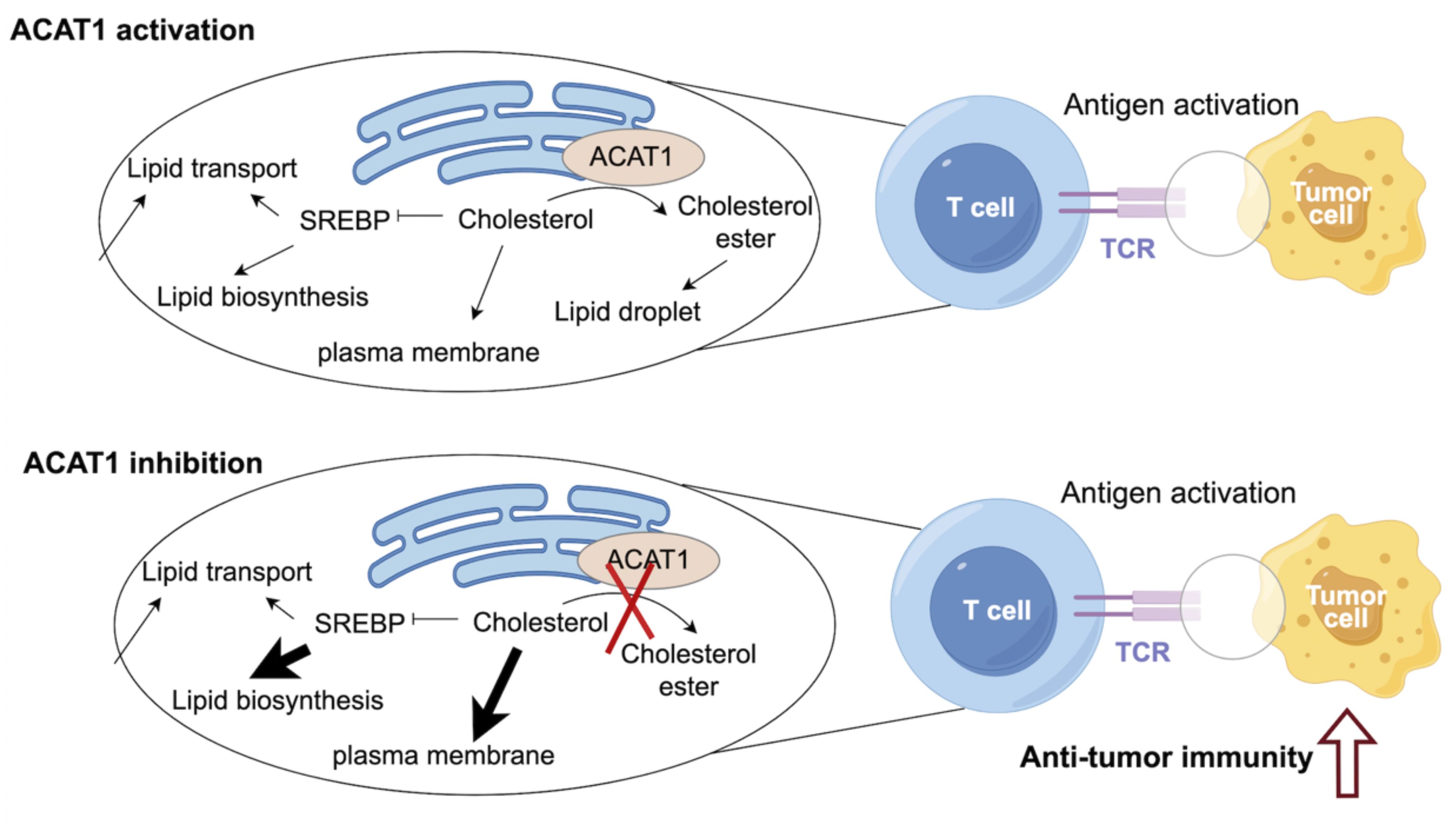
Figure 2 ACAT1 is a potential target to enhance the anti-tumor strategy.
The role of metabolic enzymes in various biological processes has been gradually discovered. It has been well-established that multiple metabolic enzymes could participate in epigenetic remodeling by providing substrates such as acetyl-CoA (27, 28). Considering the catalytic role of ACAT1 in mediating acyl transfer from CoA to cholesterol, ACAT1 may exert complex and dynamic role in tumorigenesis and progression. Mounting evidence has elucidated the significance of ACAT1 in multiple malignances. Here, we summarized the role of ACAT1 in the contexts of different cancers.
Colorectal cancer (CRC) is the third most diagnosed tumor worldwide (29). 25% of newly diagnosed CRC patients are diagnosed at the metastatic stage (30). The crosstalk between cholesterol metabolism and CRC has been under vast investigations. An elevated cholesterol level could accelerate CRC progression by activating β-catenin oncogenic signaling pathway (29). Specific liver metastases of CRC display an aberration of cholesterol biosynthesis (31). Thus, aberrant cholesterol metabolism is a hallmark of CRC, which may be exploited as potential therapeutic targets.
Numerous evidence illustrated that various molecular mechanisms are engaged in the tumorigenesis and development of CRC (32). The cytoplasmic form of malic enzymes (ME), ME1, has been identified as a primary source of NADPH for lipogenesis and glutamine biosynthesis. In CRC cells, depletion of ACAT1 could dramatically impair ME1 acetylation without influencing its protein level, whereas ACAT1 overexpression exerts the opposite effect. Moreover, ACAT1 overexpression enhances ME1 dimerization whereas deletion of ACAT1 expression impair ME1 dimerization. This ACAT1-mediated K337 acetylation positively regulates ME1 dimerization. PGAM5, a mitochondrial serine/threonine phosphatase, could dephosphorylate ME1 at S336, further promoting acetylation by ACAT1 at the adjacent K337. ACAT1-mediated ME1 K337 acetylation could enhance NADPH generation, lipogenesis, and CRC tumorigenesis (18). β-hydroxybutyrate (BHB) was previously identified as an oncogenic metabolite of CRC, which was also found to be elevated in CRC tissues. BHB has been found to promote CRC progression by ACAT1 by mediating acetylation of isocitrate dehydrogenase 1. ACAT1 abrogation could impair CRC tumorigenesis and abrogate the tumorigenic effects of BHB (33, 34). Small molecule inhibitors that target ACAT1-mediated ME1 acetylation may be a potential anti-tumor strategy for CRC patients. Besides, the development of CRC is correlated with hyperinsulinemia. Insulin-induced tumor progression of CRC is regulated by ACAT1. Insulin enhanced CRC development by upregulating ACAT1, which can be exploited as a promising therapeutic target for CRC (35). Collectively, the understanding of CRC biological features associated with ACAT1 may be beneficial for diagnosis and treatment of CRC in the clinical settings (36).
Liver cancer is the sixth most diagnosed cancer worldwide (29). Notably, it is highly refractory to most chemotherapeutic regimens. Hepatocellular carcinoma (HCC) is the most common type of liver cancer (37). In HCC cells, ACAT1 could stabilize and dimerize glyceronephosphate O-acyltransferase (GNPAT), a rate-limiting enzyme in plasmalogen synthesis and lipogenesis, by acetylation at K128. Precisely, ACAT1-mediated GNPAT acetylation could inhibit GNPAT degradation by repressing TRIM21-mediated GNPAT ubiquitination, ultimately promoting tumor growth in HCC xenografts. ACAT1 overexpression enhanced tumor growth in HCC xenografts and GNPAT deletion could attenuate ACAT1-induced HCC growth, and ACAT1 overexpression with GNPAT inhibition diminish fatty acid synthesis and lipogenesis. Combination treatment of ACAT1 inhibitor and sorafenib could significantly inhibit tumor growth in HCC xenografts, indicating that pharmaceutical inhibition of ACAT1 could be a promising target in anti-HCC strategy (17). In HEK293 cells, ACAT1 has been identified as a substrate of E3 ubiquitin ligase UBE3A/E6AP. High-fat diet could downregulate UBE3A expression, while UBE3A overexpression could lead to decreased ACAT1 protein level (38). Future studies may require combination regimens that include systemic therapies and molecularly targeted treatments, such as ACAT1.
Glioblastoma (GBM) are the most frequently diagnosed malignant primary brain tumors that originate from neuroglial progenitor cells (39). Conventional treatment could bring limited improvements in the survival of glioma patients, leading to poor survival outcomes for GBM patients (40). Herein, it is desperately required for effective molecularly targeted therapy to improve prognosis of GBM patients. In GBM patients, ACAT1 has been found to be upregulated and correlated with poor prognosis. Moreover, pharmacological inhibition of ACAT1 in GBM cells demonstrated that ACAT1 is required for GBM proliferation (41). Upon inhibition of mTORC1, ACAT1 could catalyze acetylation of glycine decarboxylase (GLDC), a critical enzyme of glycine metabolism that catalyzes the conversion of glycine into one-carbon units. The acetylation of GLDC at K514 inhibits its enzymatic activity, which promoted K33-linked polyubiquitination at K544 by NF-X1, resulting in GLDC degradation by the proteasomal pathway (42). Acetylation of GLDC at K514 could suppress glycine catabolism, pyrimidines synthesis and GBM development. K604, a potent ACAT1 inhibitor, could impair the proliferation of U251−MG cells and inactivate Akt signaling pathway in GBM cells (43). Avasimibe, another specific inhibitor of ACAT, exerts anti-tumor effect on U87, A172 and GL261 GBM cells. In GBM cell lines, avasimibe could inhibit the expression of ACAT1 and biosynthesis of cholesterol ester. Moreover, avasimibe could impair the proliferation of GBM cells resulted from caspase-8 and caspase-3 activation (44). Herein, ACAT1 functions as a novel target for HCC, providing effective assistance to the treatment of GBM.
Lung cancer is a heterogenous disease composed of multiple genetic and molecular subtypes, which is still the leading cause of cancer-related death worldwide (45). Considering that molecularly defined subtypes are potentially targetable, novel anti-tumor strategies in for lung cancer are required to be explored. Excessive intracellular cholesterol is catalyzed to cholesteryl esters via ACAT1 and exported via the cholesterol transporter ABCA1. In a cohort of patients with lung adenocarcinoma, ACAT1 has been found to be upregulated, while ABCA1 is downregulated in the lung cancer tissues. In H1299 cells, ACAT1 has been identified as the acetyltransferase of PDHA1 and PDP1. Mechanistically, PDP1 phosphorylation at Y381 recruits ACAT1 and dissociates SIRT3 to promote lysine acetylation of PDP1 and PDHA1. ACAT1 predominantly signals through inhibition of PDC by PDP1 and PDHA acetylation to enhance glycolysis and tumor growth, indicating the ACAT1-PDP1-PDHA axis a promising anti-cancer target (46). lncRNA DARS-AS1 inhibition attenuated non-small cell lung cancer development by activating miR-302a-3p to inhibit ACAT1 expression (47). It has been demonstrated that Kras-specific antigenic peptides in combination of avasimibe could promote CD8+ T cell infiltration and impair lung tumor progression (48). Collectively, ACAT1 may function as a promising therapeutic target for lung cancer.
Breast cancer is a heterogeneous malignancy with multiple molecular subtypes based on histological and genomic features (49). Gene expression profiling has proposed four intrinsic molecular subtypes: Luminal A, Luminal B, HER 2+ and basal like (50). It has been demonstrated that ER-negative breast cancer cells display accumulation of LDs, increased LDL uptake, a higher ratio of cholesteryl ester to triacylglycerol, lower cholesterol biosynthesis, increased expression of ACAT1, higher ACAT activity as compared to ER-positive breast cancer cells (51). CP-113,818, a ACAT inhibitor, could inhibit proliferation of breast tumor cells and reduce LDL-mediated proliferation of ER-negative cells. In MDA-MB-231 cells, LDL receptor (LDLR) mRNA could be markedly impaired by ACAT inhibition, indicating that high ACAT1 activity is correlated with higher LDLR expression (52). It has been found that ACAT1 upregulation in breast tumor cells could promote tumor initiation and metastasis, indicating ACAT1 as a metabolic tumor promoter (53). Nuclear receptor subfamily 2 group F member 6 (NR2F6) could transcriptionally activate ACAT1 and enhance the suppressive role of ACAT1-induced METTL3 acetylation on cell migration and invasion of breast cancer (54).
Leukemia is a heterogeneous malignancy with different genetic, morphologic and molecular feature, which is composed of multiple subtypes including acute myeloid leukemia (AML) and chronic myeloid leukemia (CML) (55). ACAT1 and SIRT3 have been identified as the upstream acetyltransferase and deacetylase of mutant isocitrate dehydrogenase 2 (mIDH2) in AML to regulate K413-acetylation of mIDH2 and inhibit mIDH2 activity (56). Spectromicroscopic analysis in multiple leukemia cell lines has revealed that aberrant accumulation of CE was found in CML (chronic myelogenous leukemia), which may be resulted from altered BCR-ABL kinase activity. Inhibiting cholesterol esterification via avasimibe could significantly suppress CML cell proliferation. Besides, combinational treatment of avasimibe and imatinib brought synergistic effects on blocking cell proliferation in K562R cells (57).
Targeting ACAT1 has been identified as a potential anti-tumor strategy (58). Avasimibe, also named as avasimin, has been developed as a potent non-specific ACAT1 inhibitor to impair cholesterol esterification in multiple cancer models. In vitro studies have elucidated that avasimibe could reduce cholesteryl-ester storage in LDs and increase levels of free cholesterol, leading to cell apoptosis and impaired proliferation (59). ACAT1 inhibitor could also enhance the cytotoxic effects of CD8+ T cells by reprogramming cholesterol metabolism. A combination of avasimin and anti-PD-1 treatment exhibited synergistic cytotoxic effects in suppressing melanoma development (22). Avasimin combined with nanoparticles of doxorubicin showed better anti-tumor efficacy in impairing breast cancer progression (60). The combination of avasimibe and immune-chemotherapy could enhance the anti-tumor effects of immune-chemotherapy. The combinational treatment could increase the level of free cholesterol and relieve the inhibition of CD8+ T cells resulted from PTX/αGC-TH-Lip. The combination of avasimibe and PTX/αGC-TH-Lip could enhance immune responses and cytotoxic effects in xenografts of melanoma, which is a potential strategy to improve the anti-tumor effects of immune-chemotherapy (26). It has been demonstrated that vaccine of Kras-specific antigenic peptides combines with avasimibe could eliminate regulatory T cells and promote CD8+ T cell infiltration (48).
Targeting tetrameric ACAT1 has been proposed as a promising anti-tumor strategy. Arecoline hydrobromide (AH) is a covalent ACAT1 inhibitor that specifically binds to and disrupts ACAT1 tetramers, thereby AH treatment leads to impaired ACAT1 activity. Due to the inhibitory effect of ACAT1 on PDC by acetylating PDH and PDH phosphatase, AH treatment could enhance PDC flux and oxidative phosphorylation to impair tumor growth, making ACAT1 a potential anti-tumor target. Combination treatment of AH with other anti-tumor strategy have shown greater anti-tumor efficacy. In HCC, AH treatment combined with sorafenib could significantly inhibit tumor growth in HCC xenografts (16). CI-976, a small molecule ACAT1 inhibitor, can bind inside the catalytic chamber and blocks the accessibility of the active site residues of ACAT1. CI-976 has been found to reduce atherosclerotic plaques and decrease plasma cholesterol levels in animals fed with high cholesterol diet (61). Another selective ACAT1 inhibitor K604 has been found to impair the proliferation of U251−MG cells and inhibit Akt signaling in glioblastoma cells (43). The current reported ACAT1 inhibitors have been illustrated in Table 1. The anti-tumor effect of ACAT1 inhibitors should be further verified in more cancer types in vitro and vivo models to explore the cancer types that can be effectively treated with ACAT1 inhibitors. Clinical trials should be accelerated to evaluate the anti-tumor effects of more ACAT1 inhibitors in different cancer types (62).
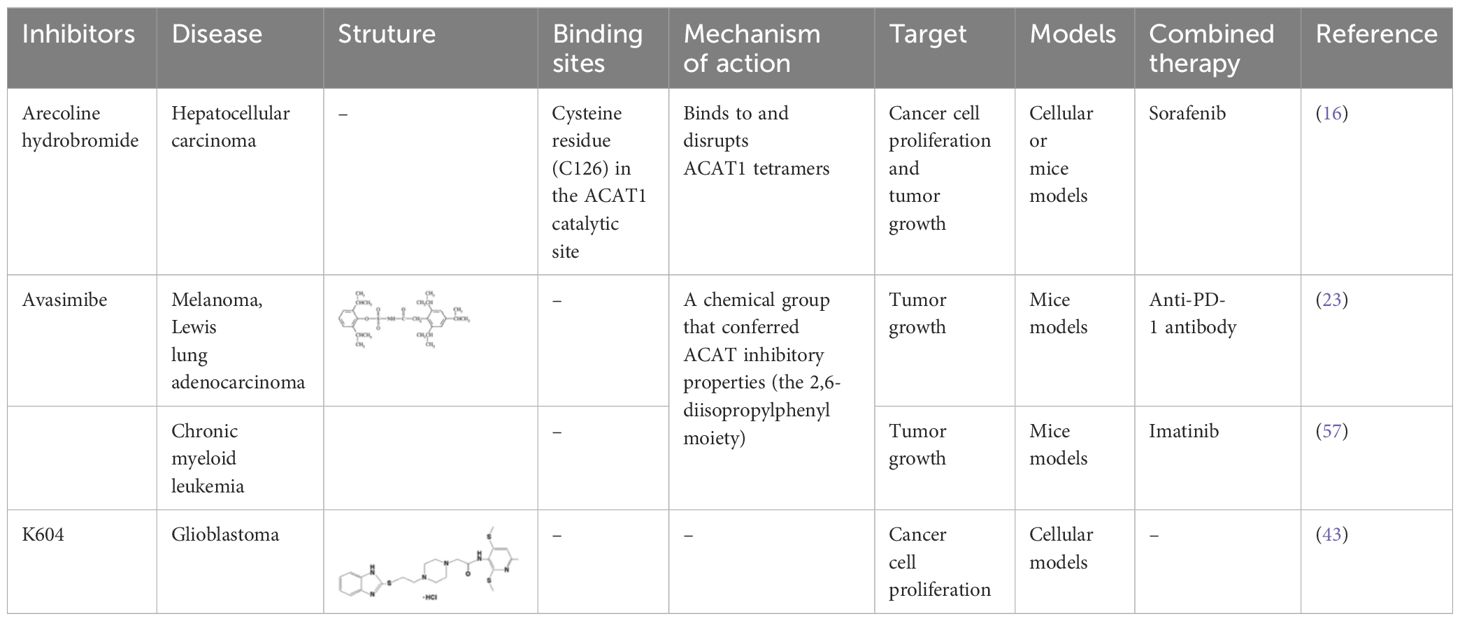
Table 1 Current ACAT1 inhibitors tested in human cancers.
TS: Writing – original draft, Writing – review & editing. XX: Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's Achilles' heel. Cancer Cell. (2008) 13:472–82. doi: 10.1016/j.ccr.2008.05.005
2. Maxfield FR, van Meer G. Cholesterol, the central lipid of mammalian cells. Curr Opin Cell Biol. (2010) 22:422–9. doi: 10.1016/j.ceb.2010.05.004
3. Luo J, Yang H, Song BL. Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol. (2020) 21:225–45. doi: 10.1038/s41580-019-0190-7
4. Tall AR, Yvan-Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. (2015) 15:104–16. doi: 10.1038/nri3793
5. Li K, Deng Y, Deng G, Chen P, Wang Y, Wu H, et al. High cholesterol induces apoptosis and autophagy through the ROS-activated AKT/FOXO1 pathway in tendon-derived stem cells. Stem Cell Res Ther. (2020) 11:131. doi: 10.1186/s13287-020-01643-5
6. Huang B, Song BL, Xu C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab. (2020) 2:132–41. doi: 10.1038/s42255-020-0174-0
7. Kuzu OF, Noory MA, Robertson GP. The role of cholesterol in cancer. Cancer Res. (2016) 76:2063–70. doi: 10.1158/0008-5472.CAN-15-2613
8. SChade DS, Shey L, Eaton RP. Cholesterol review: A metabolically important molecule. Endocr Pract. (2020) 26:1514–23. doi: 10.4158/EP-2020-0347
9. Antonenkov VD, Croes K, Waelkens E, Van Veldhoven PP, Mannaerts GP. Identification, purification and characterization of an acetoacetyl-CoA thiolase from rat liver peroxisomes. Eur J Biochem. (2000) 267:2981–90. doi: 10.1046/j.1432-1327.2000.01314.x
10. Chang TY, Li BL, Chang CC, Urano Y. Acyl-coenzyme A:cholesterol acyltransferases. Am J Physiol Endocrinol Metab. (2009) 297:E1–9. doi: 10.1152/ajpendo.90926.2008
11. Shibuya Y, Chang CC, Chang TY. ACAT1/SOAT1 as a therapeutic target for Alzheimer's disease. Future Med Chem. (2015) 7:2451–67. doi: 10.4155/fmc.15.161
12. Goudarzi A. The recent insights into the function of ACAT1: A possible anti-cancer therapeutic target. Life Sci. (2019) 232:116592. doi: 10.1016/j.lfs.2019.116592
13. Qian H, Zhao X, Yan R, Yao X, Gao S, Sun X, et al. Structural basis for catalysis and substrate specificity of human ACAT1. Nature. (2020) 581:333–8. doi: 10.1038/s41586-020-2290-0
14. Long T, Sun Y, Hassan A, Qi X, Li X. Structure of nevanimibe-bound tetrameric human ACAT1. Nature. (2020) 581:339–43. doi: 10.1038/s41586-020-2295-8
15. Lee-Rueckert M, Lappalainen J, Leinonen H, Plihtari R, Nordström T, Åkerman K, et al. Acidic extracellular pH promotes accumulation of free cholesterol in human monocyte-derived macrophages via inhibition of ACAT1 activity. Atherosclerosis. (2020) 312:1–7. doi: 10.1016/j.atherosclerosis.2020.08.011
16. Fan J, Lin R, Xia S, Chen D, Elf SE, Liu S, et al. Tetrameric acetyl-coA acetyltransferase 1 is important for tumor growth. Mol Cell. (2016) 64:859–74. doi: 10.1016/j.molcel.2016.10.014
17. Gu L, Zhu Y, Lin X, Tan X, Lu B, Li Y. Stabilization of FASN by ACAT1-mediated GNPAT acetylation promotes lipid metabolism and hepatocarcinogenesis. Oncogene. (2020) 39:2437–49. doi: 10.1038/s41388-020-1156-0
18. Zhu Y, Gu L, Lin X, Liu C, Lu B, Cui K, et al. Dynamic regulation of ME1 phosphorylation and acetylation affects lipid metabolism and colorectal tumorigenesis. Mol Cell. (2020) 77:138–149.e5. doi: 10.1016/j.molcel.2019.10.015
19. King RJ, Singh PK, Mehla K. The cholesterol pathway: impact on immunity and cancer. Trends Immunol. (2022) 43:78–92. doi: 10.1016/j.it.2021.11.007
20. Ma X, Bi E, Lu Y, Su P, Huang C, Liu L, et al. Cholesterol induces CD8+ T cell exhaustion in the tumor microenvironment. Cell Metab. (2019) 30:143–156.e5. doi: 10.1016/j.cmet.2019.04.002
21. Ma X, Xiao L, Liu L, Ye L, Su P, Bi E, et al. CD36-mediated ferroptosis dampens intratumoral CD8+ T cell effector function and impairs their antitumor ability. Cell Metab. (2021) 33:1001–1012.e5. doi: 10.1016/j.cmet.2021.02.015
22. Rashkovan M, Albero R, Gianni F, Perez-Duran P, Miller HI, Mackey AL, et al. Intracellular cholesterol pools regulate oncogenic signaling and epigenetic circuitries in early T-cell precursor acute lymphoblastic leukemia. Cancer Discovery. (2022) 12:856–71. doi: 10.1158/2159-8290.CD-21-0551
23. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature. (2016) 531:651–5. doi: 10.1038/nature17412
24. Hao M, Hou S, Li W, Li K, Xue L, Hu Q, et al. Combination of metabolic intervention and T cell therapy enhances solid tumor immunotherapy. Sci Transl Med. (2020) 12:eaaz6667. doi: 10.1126/scitranslmed.aaz6667
25. Zhao L, Li J, Liu Y, Kang L, Chen H, Jin Y, et al. Cholesterol esterification enzyme inhibition enhances antitumor effects of human chimeric antigen receptors modified T cells. J Immunother. (2018) 41:45–52. doi: 10.1097/CJI.0000000000000207
26. Li M, Yang Y, Wei J, Cun X, Lu Z, Qiu Y, et al. Enhanced chemo-immunotherapy against melanoma by inhibition of cholesterol esterification in CD8+ T cells. Nanomedicine. (2018) 14:2541–50. doi: 10.1016/j.nano.2018.08.008
27. Mews P, Donahue G, Drake AM, Luczak V, Abel T, Berger SL. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature. (2017) 546:381–6. doi: 10.1038/nature22405
28. Pietrocola F, Galluzzi L, Bravo-San Pedro JM, Madeo F, Kroemer G. Acetyl coenzyme A: a central metabolite and second messenger. Cell Metab. (2015) 21:805–21. doi: 10.1016/j.cmet.2015.05.014
29. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
30. Andrei P, Battuello P, Grasso G, Rovera E, Tesio N, Bardelli A. Integrated approaches for precision oncology in colorectal cancer: The more you know, the better. Semin Cancer Biol. (2022) 84:199–213. doi: 10.1016/j.semcancer.2021.04.007
31. Jun SY, Brown AJ, Chua NK, Yoon JY, Lee JJ, Yang JO, et al. Reduction of squalene epoxidase by cholesterol accumulation accelerates colorectal cancer progression and metastasis. Gastroenterology. (2021) 160:1194–1207.e28. doi: 10.1053/j.gastro.2020.09.009
32. Zhang KL, Zhu WW, Wang SH, Gao C, Pan JJ, Du ZG, et al. Organ-specific cholesterol metabolic aberration fuels liver metastasis of colorectal cancer. Theranostics. (2021) 11:6560–72. doi: 10.7150/thno.55609
33. Mao T, Qin F, Zhang M, Li J, Li J, Lai M. Elevated serum β-hydroxybutyrate, a circulating ketone metabolite, accelerates colorectal cancer proliferation and metastasis via ACAT1. Oncogene. (2023) 42:1889–99. doi: 10.1038/s41388-023-02700-y
34. Liu Z, Gomez CR, Espinoza I, Le TPT, Shenoy V, Zhou X. Correlation of cholesteryl ester metabolism to pathogenesis, progression and disparities in colorectal Cancer. Lipids Health Dis. (2022) 21:22. doi: 10.1186/s12944-022-01629-7
35. Chen X, Liang H, Song Q, Xu X, Cao D. Insulin promotes progression of colon cancer by upregulation of ACAT1. Lipids Health Dis. (2018) 17:122. doi: 10.1186/s12944-018-0773-x
36. Xu H, Xia H, Zhou S, Tang Q, Bi F. Cholesterol activates the Wnt/PCP-YAP signaling in SOAT1-targeted treatment of colon cancer. Cell Death Discovery. (2021) 7:38. doi: 10.1038/s41420-021-00421-3
37. Gilles H, Garbutt T, Landrum J. Hepatocellular carcinoma. Crit Care Nurs Clin North Am. (2022) 34:289–301. doi: 10.1016/j.cnc.2022.04.004
38. Peng K, Wang S, Liu R, Zhou L, Jeong GH, Jeong IH, et al. Effects of UBE3A on cell and liver metabolism through the ubiquitination of PDHA1 and ACAT1. Biochemistry. (2023) 62:1274–86. doi: 10.1021/acs.biochem.2c00624
39. Le Rhun E, Preusser M, Roth P, Reardon DA, van den Bent M, Wen P, et al. Molecular targeted therapy of glioblastoma. Cancer Treat Rev. (2019) 80:101896. doi: 10.1016/j.ctrv.2019.101896
40. Ou A, Yung WKA, Majd N. Molecular mechanisms of treatment resistance in glioblastoma. Int J Mol Sci. (2020) 22:351. doi: 10.3390/ijms22010351
41. Chi KC, Tsai WC, Wu CL, Lin TY, Hueng DY. An adult drosophila glioma model for studying pathometabolic pathways of gliomagenesis. Mol Neurobiol. (2019) 56:4589–99. doi: 10.1007/s12035-018-1392-2
42. Liu R, Zeng LW, Gong R, Yuan F, Shu HB, Li S. mTORC1 activity regulates post-translational modifications of glycine decarboxylase to modulate glycine metabolism and tumorigenesis. Nat Commun. (2021) 12:4227. doi: 10.1038/s41467-021-24321-3
43. Ohmoto T, Nishitsuji K, Yoshitani N, Mizuguchi M, Yanagisawa Y, Saito H, et al. K604, a specific acyl−CoA:cholesterol acyltransferase 1 inhibitor, suppresses proliferation of U251−MG glioblastoma cells. Mol Med Rep. (2015) 12:6037–42. doi: 10.3892/mmr.2015.4200
44. Bemlih S, Poirier MD, El Andaloussi A. Acyl-coenzyme A: cholesterol acyltransferase inhibitor Avasimibe affect survival and proliferation of glioma tumor cell lines. Cancer Biol Ther. (2010) 9:1025–32. doi: 10.4161/cbt.9.12.11875
45. Bade BC, Dela Cruz CS. Lung cancer 2020: epidemiology, etiology, and prevention. Clin Chest Med. (2020) 41:1–24. doi: 10.1016/j.ccm.2019.10.001
46. Fan J, Shan C, Kang HB, Elf S, Xie J, Tucker M, et al. Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol Cell. (2014) 53:534–48. doi: 10.1016/j.molcel.2013.12.026
47. Li J, Li Y, Sun X, Wei L, Guan J, Fu L, et al. Silencing lncRNA-DARS-AS1 suppresses nonsmall cell lung cancer progression by stimulating miR-302a-3p to inhibit ACAT1 expression. Mol Carcinog. (2024) 63(4):757–71. doi: 10.1002/mc.23686
48. Pan J, Zhang Q, Palen K, Wang L, Qiao L, Johnson B, et al. Potentiation of Kras peptide cancer vaccine by avasimibe, a cholesterol modulator. EBioMedicine. (2019) 49:72–81. doi: 10.1016/j.ebiom.2019.10.044
49. Harbeck N, Gnant M. Breast cancer. Lancet. (2017) 389:1134–50. doi: 10.1016/S0140-6736(16)31891-8
50. Waks AG, Winer EP. Breast cancer treatment: A review. JAMA. (2019) 321:288–300. doi: 10.1001/jama.2018.19323
51. Antalis CJ, Uchida A, Buhman KK, Siddiqui RA. Migration of MDA-MB-231 breast cancer cells depends on the availability of exogenous lipids and cholesterol esterification. Clin Exp Metastasis. (2011) 28:733–41. doi: 10.1007/s10585-011-9405-9
52. Antalis CJ, Arnold T, Rasool T, Lee B, Buhman KK, Siddiqui RA. High ACAT1 expression in estrogen receptor negative basal-like breast cancer cells is associated with LDL-induced proliferation. Breast Cancer Res Treat. (2010) 122:661–70. doi: 10.1007/s10549-009-0594-8
53. Ozsvari B, Sotgia F, Simmons K, Trowbridge R, Foster R, Lisanti MP. Mitoketoscins: Novel mitochondrial inhibitors for targeting ketone metabolism in cancer stem cells (CSCs). Oncotarget. (2017) 8:78340–50. doi: 10.18632/oncotarget.21259
54. Zhang G, Huang R, Zhao H, Xia Y, Huang H, Qian M, et al. ACAT1-mediated METTL3 acetylation inhibits cell migration and invasion in triple negative breast cancer. Genes Immun. (2023) 24:99–107. doi: 10.1038/s41435-023-00202-1
55. Estey E, Döhner H. Acute myeloid leukaemia. Lancet. (2006) 368:1894–907. doi: 10.1016/S0140-6736(06)69780-8
56. Chen D, Xia S, Zhang R, Li Y, Famulare CA, Fan H, et al. Lysine acetylation restricts mutant IDH2 activity to optimize transformation in AML cells. Mol Cell. (2021) 81:3833–3847.e11. doi: 10.1016/j.molcel.2021.06.027
57. Bandyopadhyay S, Li J, Traer E, Tyner JW, Zhou A, Oh ST, et al. Cholesterol esterification inhibition and imatinib treatment synergistically inhibit growth of BCR-ABL mutation-independent resistant chronic myelogenous leukemia. PloS One. (2017) 12:e0179558. doi: 10.1371/journal.pone.0179558
58. Tu T, Zhang H, Xu H. Targeting sterol-O-acyltransferase 1 to disrupt cholesterol metabolism for cancer therapy. Front Oncol. (2023) 13:1197502. doi: 10.3389/fonc.2023.1197502
59. Lee SS, Li J, Tai JN, Ratliff TL, Park K, Cheng JX. Avasimibe encapsulated in human serum albumin blocks cholesterol esterification for selective cancer treatment. ACS Nano. (2015) 9:2420–32. doi: 10.1021/nn504025a
60. Lei J, Wang H, Zhu D, Wan Y, Yin L. Combined effects of avasimibe immunotherapy, doxorubicin chemotherapy, and metal-organic frameworks nanoparticles on breast cancer. J Cell Physiol. (2020) 235:4814–23. doi: 10.1002/jcp.29358
61. Guan C, Niu Y, Chen SC, Kang Y, Wu JX, Nishi K, et al. Structural insights into the inhibition mechanism of human sterol O-acyltransferase 1 by a competitive inhibitor. Nat Commun. (2020) 11:2478. doi: 10.1038/s41467-020-16288-4
Keywords: metabolism, immunity, ACAT (acyl-CoA:cholesterol acyltransferase), cancer, cholesterol
Citation: Sun T and Xiao X (2024) Targeting ACAT1 in cancer: from threat to treatment. Front. Oncol. 14:1395192. doi: 10.3389/fonc.2024.1395192
Received: 03 March 2024; Accepted: 12 April 2024;
Published: 24 April 2024.
Edited by:
Parmanand Malvi, University of Alabama at Birmingham, United StatesReviewed by:
Chunming Cheng, University of Oklahoma, United StatesCopyright © 2024 Sun and Xiao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuan Xiao, YnJleEBzaW5hLmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.