 Jameela Lokhandwala
Jameela Lokhandwala Tracess B. Smalley
Tracess B. Smalley Timothy H. Tran
Timothy H. Tran- 1Department of Molecular Oncology, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL, United States
- 2Chemical Biology Core, H. Lee Moffitt Cancer Center and Research Institute, Tampa, FL, United States
The Kirsten rat sarcoma viral oncoprotein homolog (KRAS) is currently a primary focus of oncologists and translational scientists, driven by exciting results with KRAS-targeted therapies for non-small cell lung cancer (NSCLC) patients. While KRAS mutations continue to drive high cancer diagnosis and death, researchers have developed unique strategies to target KRAS variations. Having been investigated over the past 40 years and considered “undruggable” due to the lack of pharmacological binding pockets, recent breakthroughs and accelerated FDA approval of the first covalent inhibitors targeting KRASG12C, have largely sparked further drug development. Small molecule development has targeted the previously identified primary location alterations such as G12, G13, Q61, and expanded to address the emerging secondary mutations and acquired resistance. Of interest, the non-covalent KRASG12D targeting inhibitor MRTX-1133 has shown promising results in humanized pancreatic cancer mouse models and is seemingly making its way from bench to bedside. While this manuscript was under review a novel class of first covalent inhibitors specific for G12D was published, These so-called malolactones can crosslink both GDP and GTP bound forms of G12D. Inhibition of the latter state suppressed downstream signaling and cancer cell proliferation in vitro and in mouse xenografts. Moreover, a non-covalent pan-KRAS inhibitor, BI-2865, reduced tumor proliferation in cell lines and mouse models. Finally, the next generation of KRAS mutant-specific and pan-RAS tri-complex inhibitors have revolutionized RAS drug discovery. This review will give a structural biology perspective on the current generation of KRAS inhibitors through the lens of emerging secondary mutations and acquired resistance.
Introduction
The Kirsten rat sarcoma viral oncogene homolog (KRAS) is the most frequently mutated oncogene in cancers, and mutations in the RAS family affect approximately 30% of cancer diagnoses (1, 2). KRAS, HRAS and NRAS belong to a superfamily of small GTP-binding proteins known as GTPases, which control key biological processes such as cell division, cell differentiation, and apoptosis (3, 4). The regulation of RAS proteins is cyclic, alternating between on-GTP and off-GDP bound states, acting as a molecular switch to downstream signaling pathways (5). Common oncogenic mutations in G12, G13, and Q61 disrupt GTP hydrolysis (6, 7), ultimately locking KRAS in an active GTP-bound state that leads to the continuous activation of downstream signaling and promotion of tumor proliferation (8). Consistent within the RAS protein family, RAS activation and inactivation are controlled by RAS-guanine nucleotide exchange factors (RAS-GEFs) and RAS-GTPase activating proteins (RAS-GAPs), respectively, which in turn are controlled by receptor tyrosine kinases (RTKs) and cytokine receptors (9–12). The GTP-loaded RAS can interact with the RAS-binding domains (RBDs) in downstream effector proteins such as members of Rapidly Accelerated Fibrosarcoma (RAF) kinases ARAF, BRAF, and CRAF to activate further signaling (13). Understanding the central role that RAS plays in cancer signaling pathways has led to the creation of the NCI RAS Initiative nearly a decade ago, spearheaded by Dr. Frank McCormick and Dr. Dwight Nissley (14). This initiative is solely dedicated to the investigation of RAS cancer biology and the development of the RAS drug discovery program, highlighting the paramount importance of RAS as a direct drug target.
Discovery of KRASG12C inhibitors paving the way for drugging the undruggable
Interestingly, KRAS was first discovered in the early 1980s, yet there were no approved targeted drugs until 2021. In 2013, the Shokat laboratory achieved a breakthrough in this field using fragment screening and discovered small molecules capable of covalently binding to KRASG12C mutant at a previously unidentified allosteric switch-II pocket (15). Following this discovery, the first generation of small molecule inhibitors to KRASG12C from Amgen (Sotorasib) and Mirati (Adagrasib) therapeutics, were developed to covalently target the reactive C12 of KRASG12C (16, 17). Both inhibitors have shown some efficiency in metastatic sites. Recent reports discussing the CodeBreaK (Sotorasib) and Krystal (Adagrasib) clinical trials have illustrated effectiveness in central nervous system metastatic tumors, making them attractive candidates for later stage treatment (18). Sotorasib and Adagrasib bind to GDP-bound KRAS (inactive) and occupy the allosteric switch-II pocket located between the central β sheet, α2 (switch-II) and α3 helices of KRAS (Figure 1A, Table 1). Both compounds disrupt SOS-mediated nucleotide exchange of GDP for GTP (19, 29–31). The development of this lock and key inhibition of KRASG12C has laid the foundation for the development of KRAS-targeted therapies and has generated opportunities for future drug campaigns focusing on other mutant forms of KRAS.
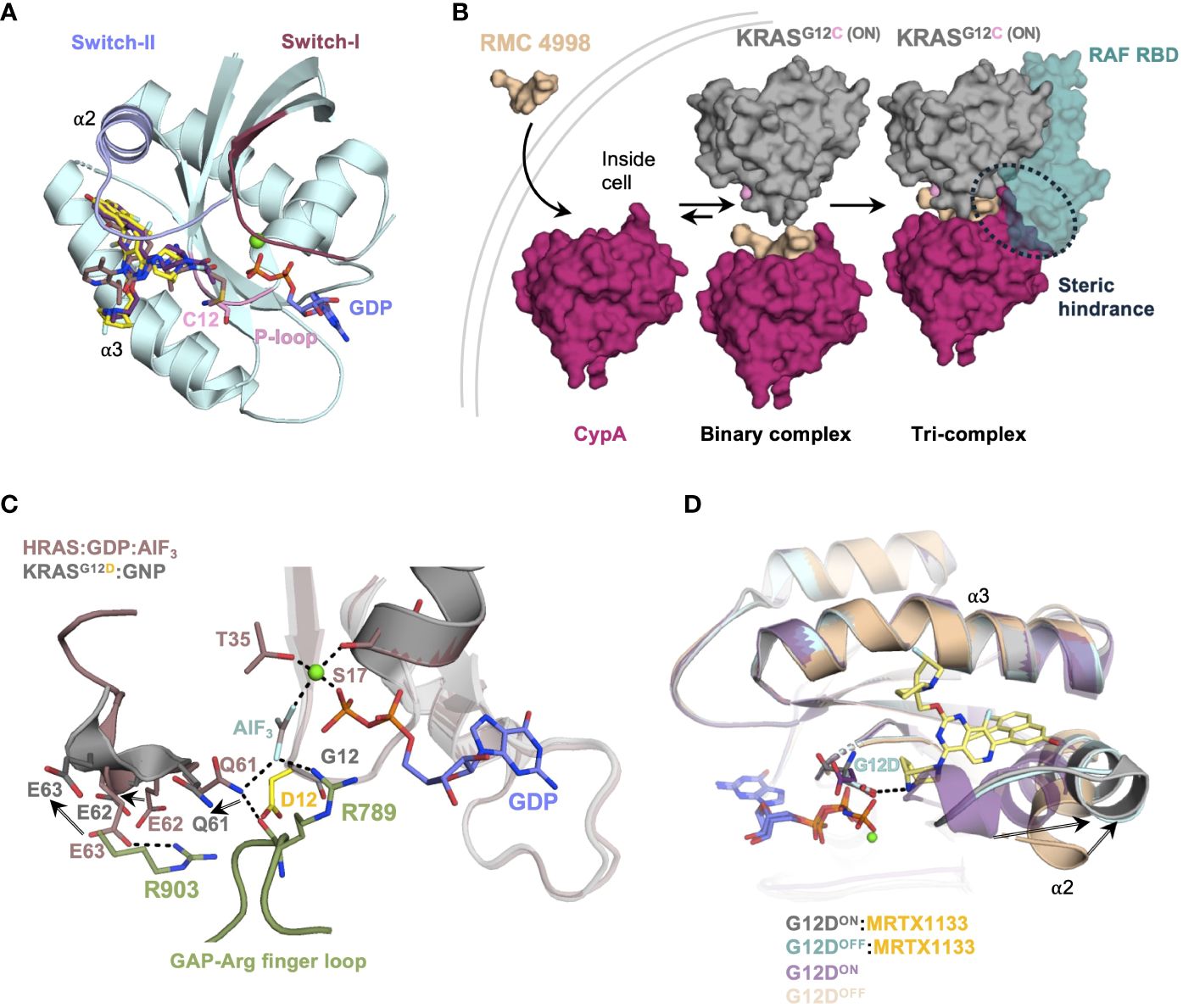
Figure 1 (A) Crystal structure (PDB: 6OIM, 7RPZ, and 6UT0) of KRASG12C/D in complex with Adagrasib (purple) and Sotorasib (salmon) and MRTX-1133 (yellow). The switch-I and switch-II regions are highlighted in pink and blue. (B) Schematic of tri-complex formation by RMC-4998 a G12CON inhibitor, the tri-complex crystal structure of KRASG12C:CypA : RMC-4998 (PDB: 8G9P) was aligned to KRAS : CRAFRBD/CRD complex structure (PDB: 6XI7). (C) Crystal structure of the complex between H-RAS and GTPase activating protein (GAP-334, green) in the active transition state (PDB: 1WQ1, gray) was aligned to the crystal structure of KRASG12D in its active state (PDB: 5USJ, cyan). (D) G12DOFF (PDB: 7RPZ, pale cyan) and G12DON (PDB: 7T47, gray) crystal structures in complex with MRTX-1133 are aligned to the G12DON (PDB: 5USJ, purple) and G12DOFF (PDB: 5US4, wheat) structures.
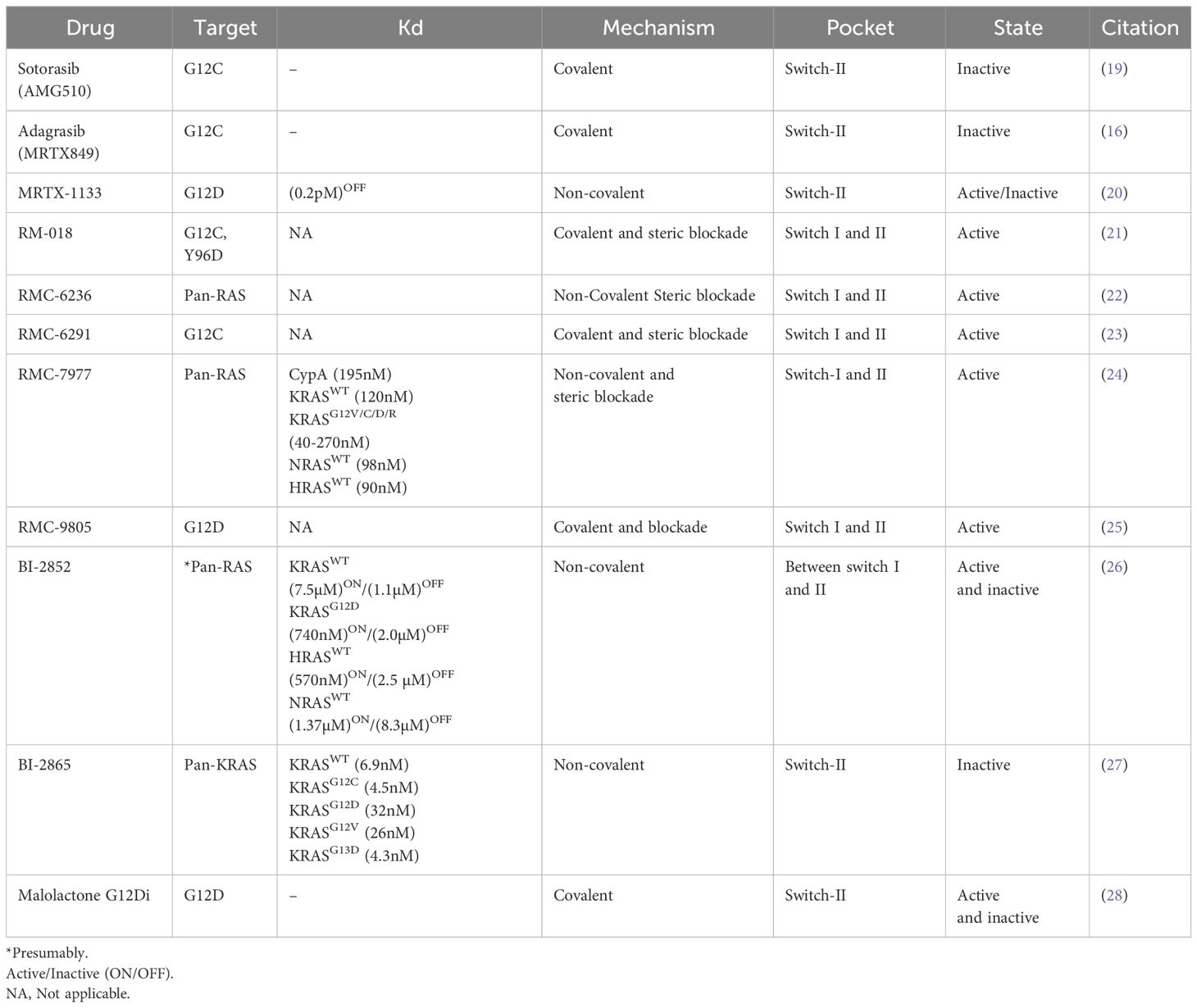
Table 1 Summary of RAS inhibitors in this review.
Targeting KRASG12C active state with molecular glue inhibitors
The early inhibitors such as Adagrasib and Sotorasib, only target the inactive GDP-bound form of KRAS, leaving a need to develop active KRAS inhibitors. Targeting the active GTP-bound KRAS remained difficult until four years ago, when Revolution Medicines came out with tri-complex covalent G12CON inhibitor, RM-018 (32, 33). This inhibitor is a unique molecular glue, which forms a trimeric complex with Cyclophilin A (CypA) and KRASG12C(ON). RM-018 is highly selective for the active form of KRASG12C(ON) with the bispecific utility of a RAS ligand and CypA ligand. The RAS ligand specific for G12C exploits a non-covalent interaction along with a warhead that forms a covalent linkage with G12C side chain as seen by several G12C inhibitors. CypA ligands consist of molecules derived from natural products, such as Sanglifehrin A, that bind strongly and reversibly to a ubiquitously expressed chaperone protein CypA (Figure 1B) (34). The innovative design of RM-018 entails remodeling the surface of CypA and has a high affinity and selectivity for the active state of KRASG12C (23). Upon cell entry, RM-018 initially forms a complex with cyclophilin A (CypA) (Figure 1B, Table 1). This binary complex then exploits non-covalent interactions with the switch-I and switch-II region of KRAS and facilitates a facile reaction with the G12C side chain to form a stable trimeric complex, which prevents effectors and regulators from binding to G12CON (21, 32, 33).
Targeting KRASG12D mutant with non-covalent and covalent inhibitors
Due to its high prevalence in various cancers- 40% in colorectal, 17% in lung adenocarcinomas, and a staggering 51% in pancreatic ductal adenocarcinoma (PDAC)- the G12D mutant has emerged as a major target of interest (35–37). This KRAS variant exhibits a low rate of both intrinsic (~2.5-fold lower than G12C variant) and GAP-mediated GTP-hydrolysis, further burdening the cell with hyperactive mutant KRAS (38, 39). The side chain of G12D, in any given rotameric conformation, prevents the key arginine finger of GAP from inserting into the active site of RAS to participate in the transition-state coordination of the GAP-mediated GTP hydrolysis reaction (6, 40–42). Additionally, the negative point charge of the G12D side chain constantly exerts a repulsive force radially outward, especially toward the negatively charged E62 and E63 of the catalytic loop. Together, E62 and E63 with the key catalytic residue Q61, are pushed away from their catalytically competent position for GTP hydrolysis (Figure 1C). These are the structural and physical bases for substantially impaired GAP-mediated and intrinsic GTP hydrolysis in KRASG12D, accounting for the highest rate of lethality among RAS-driven cancers.
The negatively charged G12D mutation has been considerably challenging to target compared to other G12 mutations. Drugs designed to specifically target G12D must be able to form a covalent bond or a hydrogen bond or an electrostatic interaction with the carboxylate group of G12D side chain, although the latter is unlikely to be a good candidate due to the plasma membrane barrier. As for the warhead design, G12D side chain, a weak nucleophile, is not as reactive compared to a cysteine residue. Hence, a drug design specific for aspartate requires a novel class of electrophiles that can execute a facile, one-step coupling reaction, similar to that of click chemistry or cysteine oxidation/S-C bond formation, with the G12D side chain. In addition, there are many other aspartate and glutamate residues on the protein surface of RAS and all other cytosolic proteins in the cell, which makes it very challenging to selectively target KRASG12D. Therefore, drug discovery targeting KRASG12D requires a novel chemistry, a highly specific pocket design, and possibly point mutations of other solvent-exposed aspartate/glutamate residues. As mentioned above, the Shokat laboratory just released the first structure of G12D in which the carboxylate of G12D side chain is alkylated with a lactone-based electrophile or malolactone (Table 1) (28). The novelty of this compound design takes advantage of the 4-membered ring strain of heterocyclic lactone in a pseudo-staggered conformation. This exposes the lowest unoccupied molecular orbital of the C-O bond and facilitates a direct strain-release SN2 ring-opening attack by G12D side chain. In addition, the authors’ judicious choice of substituents on the α-carbon of the enantioselective malolactone shielded the compound from ~55 M of competing water molecules present in the buffer. For more details, readers can examine the reaction trajectory from the reference therein (28).
Fortuitously, the G12C cancer variant has provided us with a foundation to target G12D using G12C pharmacophores. The authors of the MRTX-1133 have successfully synthesized a compound that forms a hydrogen bond with the G12D side chain via a nitrogen donor (20). This crucial hydrogen bond is critical for the specificity and anchoring of the drug, allowing the rest of the ligand to “wiggle” and adjust, taking advantage of the dynamic nature of the switch-II for induced fit. In the absence of direct ligand:G12D side chain interaction, the specificity would be significantly reduced and require additional stabilization. Since the overall design of MRTX-1133 was based on Adagrasib, other cancer drugs targeting G12C can also be modified to be reactive against G12D-driven cancers.
Interestingly, MRTX-1133 has been shown to inhibit both inactive and active forms of G12D (43). More surprisingly, superposition of both G12DON and G12DOFF structures in complex with MRTX-1133 revealed that MRTX-1133 has the same binding mode, even though GDP and GTP-bound structures have distinct conformations in the absence of MRTX-1133 (Figure 1D). This suggests that MRTX-1133 induces a novel binding mechanism. Incidentally, the critical hydrogen bond between MRTX-1133 and G12D side chain also alleviates the repulsive force of G12D point charge toward the catalytic loop as mentioned above (Figure 1C). Therefore, in addition to hindering G12D interaction with GAPs, MRTX-1133 or any drug compound that interacts with G12DON via a hydrogen bond or a covalent bond could enhance or restore its intrinsic GTP hydrolysis, allowing G12DON to return to its inactive GDP-bound form and reducing G12DON concentration in the cell.
With the advent of the novel class of G12D-specific covalent inhibitors, malolactones, a combinatorial arsenal of compounds can be designed to combat G12D-driven tumors, as the original authors have already demonstrated in cellular context as well as in mouse xenografts (Table 1). As is the case with G12CON, covalent KRASG12D specific inhibitors can be made by the tri-complex platform. This is perhaps the most remarkable breakthrough given the challenges of targeting G12D highlighted earlier. During a recent AACR conference, Revolution Medicines announced its sister version of G12CON tri-complex inhibitor, RMC-9805, targeting G12DON (Table 1) (25, 44, 45). While the structure for this version has not been published, its overall structure is most likely similar to other tri-complex inhibitors, with the exception that the G12D warhead is expected to carry a novel aspartate-reactive electrophile similar to malolactones. This would make RMC-9805, together with malolactones, the first class of covalent inhibitors specific for the carboxylate group of the G12D side chain and a very promising alternative to MRTX-1133 targeting KRASG12D driven cancers.
Pan KRAS/RAS inhibitors- targeting all Ras isoforms and mutants with one stone
Boehringer Ingelheim recently published a pan-KRAS-selective inhibitor, BI-2865, which was designed based on G12C pharmacophores targeting the inactive form of KRAS and accompanied by atomic-resolution crystal structures (27). The authors showed that BI-2865 is approximately 2-3 orders of magnitude more selective for KRAS than HRAS and NRAS, and inhibited many common G12, G13, and Q61H mutations, with the exceptions of G12R and other Q61 mutations. Based on the structure of the complex and mutagenesis studies, BI-2865 confers its selectivity around H95 (Figure 2D, Table 1), which is one of a few amino acids of the RAS G-domain that differs among RAS isoforms. However, as a pan-KRAS inhibitor, BI-2865 also inactivates WT KRAS. Presently, no clinical data demonstrating the consequences of blocking WT KRAS have been reported (46). Thus, it would be of great interest to see how cancer patients can tolerate potential toxicity of this pan-KRAS inhibitor. Nevertheless, WT KRAS activity can be potentially compensated by NRAS and HRAS isoforms.
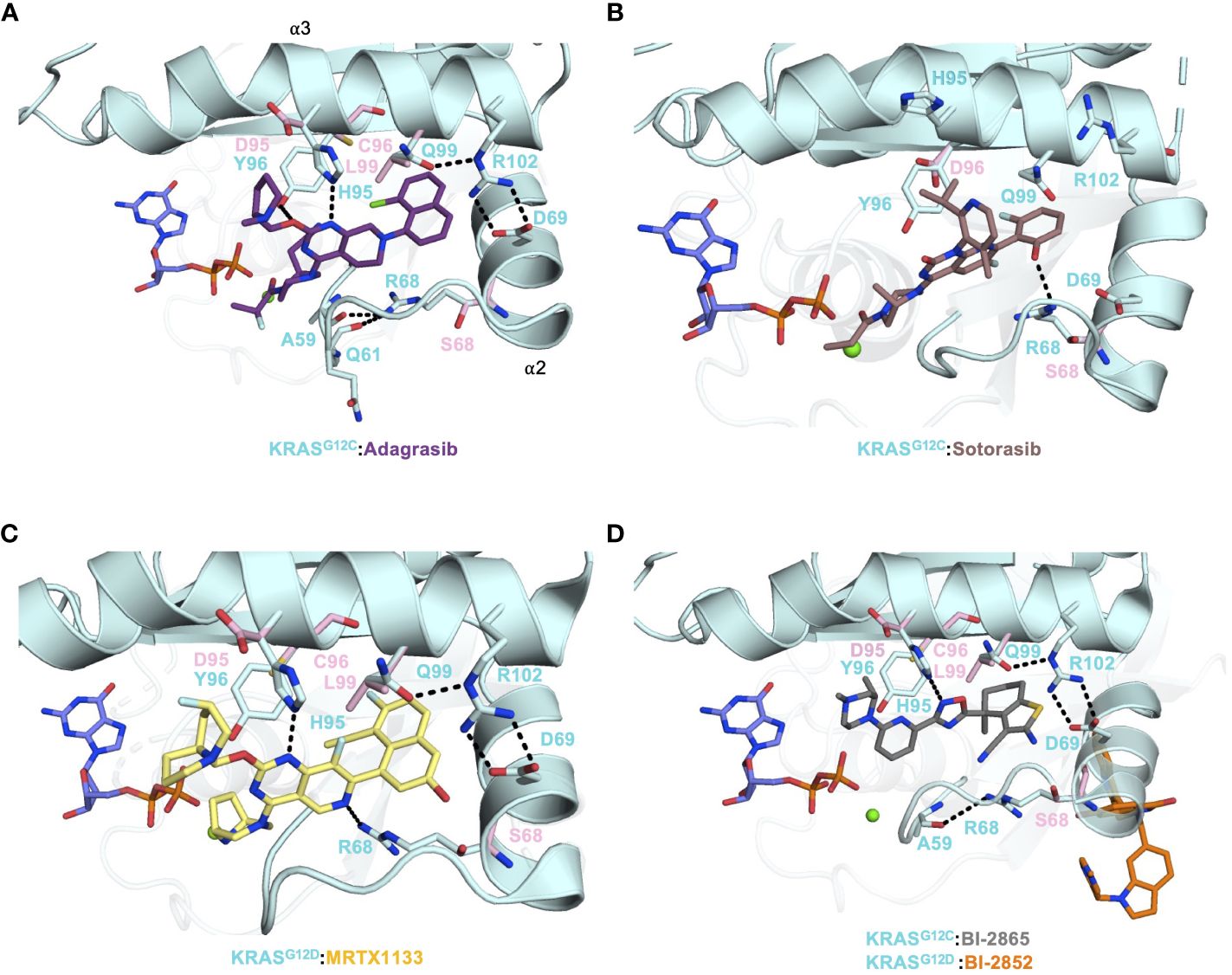
Figure 2 (A) Crystal structure of KRASG12C (PDB: 6UT0) in complex with Adagrasib (purple) and the secondary mutations were modeled on the structure and shown as pink sticks. (B) Crystal structure of KRASG12C (PDB: 6OIM) in complex with Sotorasib (salmon) and the secondary mutations were modeled on the structure and shown as pink sticks. (C) Crystal structure of KRASG12D (PDB: 7RPZ) in complex with MRTX-1133 (yellow) and the secondary mutations were modeled on the structure and shown as pink sticks. (D) The crystal structure of KRASG12C (PDB: 8AZR) in complex with pan-KRAS inhibitor BI-2865 (gray) and crystal structure KRASG12C (PDB: 6GJ8) in complex with BI-2852 (orange) were aligned and the secondary mutations were modeled on the structure and shown as pink sticks.
Several other pan-RAS inhibitors have also been reported (47–49). However, none of them have co-crystal structures. A few years earlier, Boehringer Ingelheim unveiled its first, presumably*, pan-RAS compound BI-2852 that bound and inhibited KRAS at a site distinct from the switch-II pocket, as shown in a series of KRASG12D structures (Figure 2D, Table 1) (26). BI-2852 was shown to bind stronger to KRASG12D than WT KRAS, but it also bound to NRAS and HRAS almost as tight as KRASG12D and inhibited WT KRAS, NRAS, and HRAS (Table 1). The mechanism of inhibition of BI-2852 involves dimerization of KRAS, which prevents RAF binding (50).
In addition to KRAS mutant-specific inhibitors, Revolution Medicines also reported a tri-complex pan-RAS inhibitor, RMC-6236 or RASMULTI(ON) (Table 1) (22, 51, 52). To be a pan-RAS inhibitor, the RAS ligand must interact with RAS noncovalently as seen in a series of tri-complex structures of RMC-7977 (Table 1) (24) with different WT RAS isoforms and mutants. Additionally, emerging studies show that these broad-spectrum inhibitors may have moderate tumor selectivity in NSCLC and PDAC mouse models and decreased resistance escape (53, 54). As a versatile inhibitor against the active form of RAS, RASMULTI(ON) quickly made its way into phase I clinical trials. However, despite this unprecedented breakthrough and promising results in pre-clinical studies, data from clinical trials are needed to assess its efficacy and toxicity. Nevertheless, as expected for a pan-RAS inhibitor, RASMULTI(ON) also inhibits WT HRAS and NRAS. Thus, while RASMULTI(ON) can be practically used to treat the maximum number of patients with any RAS-driven cancer, the inhibition of all WT RAS proteins may lead to higher toxicity since there are no known compensatory mechanisms. Fortunately, even if RASMULTI(ON) exhibited undesirable clinical outcomes, tri-complex platform for specific RAS mutants mentioned above for G12CON and G12DON can be used to treat KRAS mutant-specific cancers. In addition, it is also possible to design warheads for the other oncogenic variants, G13C and Q61H, which are currently in the pipeline at Revolution Medicines. Nevertheless, it is important to note that clinical results only reflect short-term outcomes. Long-term studies are needed for these drugs to assess their effectiveness against resistance by cancer cells.
Secondary mutations and acquired resistance
Adagrasib and Sotorasib have had unprecedented clinical success in NSCLC patients with KRASG12C mutation (~14% frequency). However, despite these advances in treatment options for patients in this subgroup, acquired resistance mechanisms have been reported (8, 13). There are several routes that cancer cells take to evade KRAS inhibition. Two avenues of note are secondary mutations occurring within the drug-binding pocket and KRAS gene amplification. In a study by Awad et al., 38 patients (27 non-small-cell lung cancer, 10 colorectal cancer, and 1 appendiceal cancer) treated with Adagrasib were investigated for acquired KRAS alterations. Out of 38 patients, 17 developed various resistance mechanisms to Adagrasib. The KRAS alterations included G12D/R/V/W, G13D, Q61H, R68S, H95D/Q/R, Y96C and the amplification of KRASG12C allele were observed in 7 patients (55). Covalent inhibitors, such as Adagrasib and Sotorasib, provide a strong anchor for drug design and development at the reactive cysteine of G12C. Hence, the observed secondary mutations from G12C to G12D/R/V/W would be most detrimental since that crucial anchor is severed.
Residues R68, H95, Y96 and Q99 of the switch-II pocket interact with G12C inhibitors and stabilize drug:protein interactions (Figures 2A, B). R68 side chain forms Van der Waals interactions to the piperidine and naphthyl moieties of Adagrasib (Figure 2A). Furthermore, R68 may also play a role in forming the switch-II pocket by holding A59 and Q61, via hydrogen bonds, in a favorable conformation for drug binding (55). R68S mutation would remove the favorable contact with Adagrasib and could disrupt binding. Both Adagrasib and Sotorasib bind to the H95 groove, in which H95 forms a critical hydrogen bond with the pyrimidine moiety of Adagrasib (Figure 2A) (16, 19). However, H95 does not make the equivalent hydrogen bond with Sotorasib (Figure 2B). Although H95D mutation could retain this hydrogen bond, its negative charge destabilizes the overall hydrophobic interactions between Adagrasib and KRAS (Figure 2A). In cellular studies, mutations at the H95 site were sensitive to Sotorasib whereas R68S was shown to cause resistance to Sotorasib (55). This can be explained by the fact that Sotorasib makes a critical hydrogen bond with R68 but not H95 (Figure 2B). Furthermore, resistant clones that arose after in-vitro treatment with Adagrasib and Sotorasib on Ba/F3 cells included mutations such as Q99L (56). Q99L mutation was shown to be resistant to Adagrasib but not to Sotorasib. Q99 residue is part of the α3 helix. Substituting a polar glutamine with a hydrophobic leucine resulted in a loss of a hydrogen bond that anchored the R102-D69 salt bridge connecting α2 and α3 helices, which helped confine the Adagrasib binding pocket (Figure 2A). However, this binding mode does not exist in Sotorasib and thus Q99L mutation did not affect Sotorasib (Figure 2B).
Interestingly, mutations found at the Y96 residue (to C/D/S) were reported to cause resistance to both Adagrasib and Sotorasib via in vitro experiments and in patients (55, 56). The structures of KRASG12C in complex with Adagrasib and Sotorasib indicate that although the hydroxyl group of Y96 interacts with water molecules at this solvent-accessible interface, its benzene ring forms extensive hydrophobic interactions with both compounds. Hence, mutations at Y96 to a smaller or charged residue (C/D/S) would sever the hydrophobic interactions. Moreover, molecular dynamic simulation studies have shown that Y96D mutation increases the flexibility of the switch-II and α3 helix region, causing it to become more dynamic and unstable, and subsequently weakening the Sotorasib binding pocket (57). Thus, the dynamic nature of the switch-II region also allows the drug to adjust and remain in sync with it. Gradually, this causes the drug to become less effective in altering the switch-II dynamics and allows RAS effectors/regulators to resume their normal RAS activities. Overall, these secondary mutations undoubtedly impart selective pressure on ligand stability and could result in breakage of the warhead in G12C (Adagrasib/Sotorasib), or hydrogen bonding interaction in G12D (MRTX-1133) or even direct G12 mutation as a mechanism of resistance. Therefore, secondary mutations affecting G12C inhibitors and specific to the switch-II pocket could arise during G12D-specific MRTX-1133 or pan-KRAS BI-2865 treatment. The four residues that harbored secondary mutations, R68, H95, Y96 and Q99, make crucial contacts with MRTX-1133 and BI-2865 and were shown to be mutated after Adagrasib or Sotorasib treatment. In fact, Adagrasib, MRTX-1133 and BI-2865, but not Sotorasib, have a very similar KRAS binding mode in the switch-II pocket (Figures 2A, C, D). In any event, as is the case with many cancer drugs, acquired resistance via point mutations makes KRAS a continuously moving target and must be taken into consideration in drug development.
Nonetheless, the recent breakthrough work by Revolution Medicines has demonstrated that secondary mutations can be overcome by the tri-complex platform with the parent compound of RMC-6291, RM-018, for secondary mutation KRASG12C/Y96D (21). One plausible explanation is that CypA of tri-complex inhibitors encloses the ligand and provides an extensive stabilizing interface with KRASG12C, in addition to imposing an 18 kDa physical barrier to RAS effector/regulator binding. Thus, a secondary point mutation on KRASG12C is unlikely to significantly impact the vast interaction between KRASG12C and CypA. It is not surprising that RMC-6291 is a promising and potent treatment with a pre-clinical overall response rate of 72% and a disease control rate of 92% in models with KRASG12C NSCLC. Furthermore, RMC-6291 exhibits the potential for reduced resistance during treatment and is currently recruiting for Phase 1/1b clinical trials (NCT05462717) (58, 59). Notably, that while many drug developers desire a monotherapy option for patients suffering from KRAS-driven tumors, acquired resistance continues to present difficulties in treatments. These secondary mutations could continue with newly developed inhibitors that target the switch I/II pocket. In addition to KRAS secondary mutations, resistance mechanisms can be mutations in any member of the RAS signaling pathway such as upstream tyrosine receptor kinases and downstream effectors BRAF/CRAF (60). Therefore, combination therapies offer an advantage to potentially circumvent these resistance mechanisms.
Combining immunotherapy with inhibitor treatments
The evolution of the KRAS field has yielded diverse therapeutic approaches, potentially considered for monotherapies. However, despite promising results in vitro, in vivo, and clinical studies, other strategies are needed to address the issue of acquired resistance. As KRAS has been shown to modulate the immune system, KRAS mutations have been noted to cause an immunosuppressive tumor microenvironment (TME), allowing evasion of immune detection and continued unmitigated proliferation (61–63). Several groups have expanded on novel approaches to combine immunotherapies with the current targeted approaches. These efforts are largely represented by antibody drugs, whether through bispecific T-cell engagers (BiTEs) targeting neoantigens on the cell surface or through targeting the PD-1 immunosuppressive signaling pathway (64, 65). Among these advancements, Zhang et al. leveraged the covalent bond between G12C inhibitors (ARS1620) and KRASG12C mutants to subsequently display MHC class I complexes presenting the drug-modified neoantigen on the cell surface (66). Using phage display, an antibody-fragment (P1A4) specific to this new antigen was discovered and developed into a BiTE using a CD3 T-cell targeting single-chain fragment variable (scFv – L2K-07). Zhang et al., determined that the BiTE could invoke a cytotoxic T-cell response from peripheral mononuclear blood cells in co-cultures with KRASG12C cell lines (H358, Miapaca and SW1573), which included those cells resistant to direct KRASG12C inhibition. While these studies are still in the preliminary stages of development, there is an ongoing emphasis on the importance of understanding what role different immune landscapes play in the tumor’s response to immunotherapies (67). Highlighted by this is Amgen’s early-phase clinical trial combining Sotorasib with anti-PD-1, which was inhibited by high-grade liver toxicities in many patients (68). While the cause of the toxicity is not fully understood, results still suggest that many variables need to be considered when designing combination immunotherapies. Nevertheless, novel mechanisms are emerging from preclinical research, suggesting alternative candidates to target in addition to KRAS. An example of this would be the inositol-requiring enzyme 1α branch of the unfolded protein response (IRE1α), which gets inactivated by oncogenic KRAS (69). The stabilization of IRE1α disrupts the normal programming of proteostasis mechanisms, highlighting a new druggable target. Continued efforts to synergistically combine therapeutics to KRAS inhibitors are ongoing.
Conclusions
Exciting advances in RAS cancer biology have launched new drug campaigns aimed at treating patients with KRAS mutations. By exploiting the nucleophilicity of the G12C residue in the switch-II pocket, drug expansion efforts have led to the first FDA-approved targeted therapies for KRAS mutants. However, targeting KRAS has historically posed challenges as other mutant forms of KRAS are not susceptible to G12C-specific inhibitors. Expanding beyond the inhibition of G12C is highlighted by the success of the non-covalent inhibitor to KRASG12D having shown promising results in PDAC mouse models, and covalent G12D-specific inhibitors in mouse xenografts. Furthermore, the current perspectives in the KRAS field are positive, underscored by the next generation of small molecule tools aimed at addressing KRAS in cancer. Tools such as the active form pan-RAS and KRAS mutant-specific tri-complex inhibitors have shown promising results in vivo and are moving toward the clinic. With this toolbox in hand, clinicians, researchers and drug developers can address KRAS secondary mutations that are already emerging in clinical settings. Further combination therapies specifically with immunotherapy, have shown mixed but overall promising results and should continue to be pursued to determine clinical effectiveness.
Author contributions
JL: Writing – original draft, Writing – review & editing. TS: Writing – original draft, Writing – review & editing. TT: Writing – original draft, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work has been supported in part by the Chemical Biology Core at the H. Lee Moffitt Cancer Center & Research Institute, a comprehensive cancer center designated by the National Cancer Institute and funded in part by Moffitt’s Cancer Center Support Grant (P30-CA076292).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Cox AD, SW F, AC K, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discovery. (2014) 13(11):828–51. doi: 10.1038/nrd4389
2. Tang D, Kang R. Glimmers of hope for targeting oncogenic KRAS-G12D. Cancer Gene Ther. (2022) 30(3):391–3. doi: 10.1038/s41417-022-00561-3
3. Crespo P, León J. Ras proteins in the control of the cell cycle and cell differentiation. Cell Mol Life Sci CMLS. (2000) 57(11):1613–36. doi: 10.1007/PL00000645
4. Colicelli J. Human RAS superfamily proteins and related GTPases. Science’s STKE. (2004). doi: 10.1126/stke.2502004re13
5. Simanshu DK, Nissley DV, McCormick F. RAS proteins and their regulators in human disease. Cell. (2017) 170(1):17–33. doi: 10.1016/j.cell.2017.06.009
6. Vatansever S, Erman B, Gümüş ZH. Oncogenic G12D mutation alters local conformations and dynamics of k-ras. Sci Rep. (2019) 9(1):11730. doi: 10.1038/s41598-019-48029-z
7. Malumbres M, Barbacid M. RAS oncogenes: the first 30 years. Nat Rev Cancer. (2003) 3(6):459–65. doi: 10.1038/nrc1097
8. Liu J, Kang R, Tang D. The KRAS-G12C inhibitor: activity and resistance. Cancer Gene Ther. (2023) 30(12):1715–5. doi: 10.1038/s41417-023-00692-1
9. Tcherkezian J, Lamarche-Vane N. Current knowledge of the large RhoGAP family of proteins. Biol Cell. (2007) 99(2):67–86. doi: 10.1042/bc20060086
10. Mitin N, Rossman KL, Der CJ. Signaling interplay in ras superfamily function. Curr Biol. (2005) 15(14):R563–74. doi: 10.1016/j.cub.2005.07.010
11. Bos JL, Rehmann H, Wittinghofer A. GEFs and GAPs: Critical elements in the control of small g proteins. Cell. (2007) 129(5):865–77. doi: 10.1016/j.cell.2007.05.018
12. Nimnual A, Bar-Sagi D. The two hats of SOS. Science’s STKE. (2002) 2002(145):36. doi: 10.1126/stke.2002.145.pe36
13. Punekar SR, Velcheti V, Neel BG, Wong KK. The current state of the art and future trends in RAS-targeted cancer therapies. Nat Rev Clin Oncol. (2022) 19(10):637–55. doi: 10.1038/s41571-022-00671-9
14. Nissley DV, McCormick F. RAS at 40: Update from the RAS initiative. Cancer Discovery. (2022) 12(4):895–8. doi: 10.1158/2159-8290.CD-21-1554
15. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature. (2013) 503(7477):548–51. doi: 10.1038/nature12796
16. Fell JB, Fischer JP, Baer BR, Blake JF, Bouhana K, Briere DM, et al. Identification of the clinical development candidate MRTX849, a covalent KRASG12CInhibitor for the treatment of cancer. J Medicinal Chem. (2020) 63(13):6679–93. doi: 10.1021/acs.jmedchem.9b02052
17. Lanman BA, Allen JR, Allen JG, Amegadzie AK, Ashton KS, Booker SK, et al. Discovery of a covalent inhibitor of KRASG12C (AMG 510) for the treatment of solid tumors. J Medicinal Chem. (2020) 63(1):52–65. doi: 10.1021/acs.jmedchem.9b01180
18. Dingemans AMC, Syrigos K, Livi L, Paulus A, Kim SW, Chen Y, et al. Intracranial efficacy of sotorasib versus docetaxel in pretreated KRAS G12C-mutated advanced non-small cell lung cancer (NSCLC): Practice-informing data from a global, phase 3, randomized, controlled trial (RCT). J Clin Oncol. (2023) 41(17_suppl):LBA9016–LBA9016. doi: 10.1200/JCO.2023.41.17_suppl.LBA9016
19. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature. (2019) 575(7781):217–23. doi: 10.1038/s41586-019-1694-1
20. Wang X, Allen S, Blake JF, Bowcut V, Briere DM, Calinisan A, et al. Identification of MRTX1133, a noncovalent, potent, and selective KRASG12DInhibitor. J Medicinal Chem. (2022) 65(4):3123–33. doi: 10.1021/acs.jmedchem.1c01688
21. Tanaka N, Lin JJ, Li C, Ryan MB, Zhang J, Kiedrowski LA, et al. Clinical acquired resistance to krasg12c inhibition through a novel kras switch-ii pocket mutation and polyclonal alterations converging on ras–mapk reactivation. Cancer Discovery. (2021) 11(8):1913–22. doi: 10.1158/2159-8290.CD-21-0365
22. Gustafson WC, Wildes D, Rice MA, Lee BJ, Jiang J, Wang Z, et al. Direct targeting of RAS in pancreatic ductal adenocarcinoma with RMC-6236, a first-in-class, RAS-selective, orally bioavailable, tri-complex RAS MULTI (ON) inhibitor. J Clin Oncol. (2022) 40(4_suppl):591–1. doi: 10.1200/JCO.2022.40.4_suppl.591
23. Schulze CJ, Seamon KJ, Zhao Y, Yang YC, Cregg J, Kim D, et al. Chemical remodeling of a cellular chaperone to target the active state of mutant KRAS. Science. (2023) 381(6659):794–9. doi: 10.1126/science.adg9652
24. Holderfield M, Lee BJ, Jiang J, Tomlinson A, Seamon KJ, Mira A, et al. Concurrent inhibition of oncogenic and wild-type RAS-GTP for cancer therapy. Nature. (2024). doi: 10.1038/s41586-024-07205-6
25. Jiang L, Menard M, Weller C, Wang Z, Burnett L, Aronchik I, et al. Abstract 526: RMC-9805, a first-in-class, mutant-selective, covalent and oral KRASG12D(ON) inhibitor that induces apoptosis and drives tumor regression in preclinical models of KRASG12D cancers. Cancer Res. (2023) 83(7_Supplement):526–6. doi: 10.1158/1538-7445.AM2023-526
26. Kessler D, Gmachl M, Mantoulidis A, Martin LJ, Zoephel A, Mayer M, et al. Drugging an undruggable pocket on KRAS. Proc Natl Acad Sci United States America. (2019) 116(32):15823–9. doi: 10.1073/pnas.1904529116
27. Kim D, Herdeis L, Rudolph D, Zhao Y, Böttcher J, Vides A, et al. Pan-KRAS inhibitor disables oncogenic signalling and tumour growth. Nature. (2023) 619(7968):160–6. doi: 10.1038/s41586-023-06123-3
28. Zheng Q, Zhang Z, Guiley KZ, Shokat KM. Strain-release alkylation of Asp12 enables mutant selective targeting of k-Ras-G12D. Nat Chem Biol. (2024). doi: 10.1038/s41589-024-01565-w
29. Patricelli MP, Janes MR, Li LS, Hansen R, Peters U, Kessler LV, et al. Selective inhibition of oncogenic KRAS output with small molecules targeting the inactive state. Cancer Discovery. (2016) 6(3):316–29. doi: 10.1158/2159-8290.CD-15-1105
30. Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS mutant cancers with a covalent G12C-specific inhibitor. Cell. (2018) 172(3):578–589.e17. doi: 10.1016/j.cell.2018.01.006
31. Lito P, Solomon M, Li LS, Hansen R, Rosen N. Cancer therapeutics: Allele-specific inhibitors inactivate mutant KRAS G12C by a trapping mechanism. Science. (2016) 351(6273):604–8. doi: 10.1126/science.aad6204
32. Nichols R, Schulze C, Bermingham A, Choy T, Cregg J, Kiss G, et al. A06 tri-complex inhibitors of the oncogenic, GTP-bound form of KRASG12C overcome RTK-mediated escape mechanisms and drive tumor regressions in preclinical models of NSCLC. J Thorac Oncol. (2020) 15(2):S13–4. doi: 10.1016/j.jtho.2019.12.035
33. Schulze CJ, Bermingham A, Choy TJ, Cregg JJ, Kiss G, Marquez A, et al. Abstract PR10: Tri-complex inhibitors of the oncogenic, GTP-bound form of KRASG12C overcome RTK-mediated escape mechanisms and drive tumor regressions. vivo. Mol Cancer Ther. (2019) 18(12_Supplement):PR10–0. doi: 10.1158/1535-7163.TARG-19-PR10
34. Sanglier JJ, Quesniaux V, Fehr T, Hofmann H, Mahnke M, Memmert K, et al. Sanglifehrins a, b, c and d, novel cyclophilin-binding compounds isolated from streptomyces sp. A92-308110. i. taxonomy, fermentation, isolation and biological activity. J Antibiotics. (1999) 52(5):466–73. doi: 10.7164/antibiotics.52.466
35. Waters AM, Der CJ. KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harbor Perspect Med. (2018) 8(9):a031435. doi: 10.1101/cshperspect.a031435
36. Escher TE, Satchell KJF. RAS degraders: The new frontier for RAS-driven cancers. Mol Ther. (2023) 31(7):1904–19. doi: 10.1016/j.ymthe.2023.03.017
37. Cook JH, Melloni GEM, Gulhan DC, Park PJ, Haigis KM. The origins and genetic interactions of KRAS mutations are allele- and tissue-specific. Nat Commun. (2021) 12(1). doi: 10.1038/s41467-021-22125-z
38. Moore AR, Rosenberg SC, McCormick F, Malek S. RAS-targeted therapies: is the undruggable drugged? Nat Rev Drug Discovery. (2020), 533–52. doi: 10.1038/s41573-020-0068-6
39. Hunter JC, Manandhar A, Carrasco MA, Gurbani D, Gondi S, Westover KD. Biochemical and structural analysis of common cancer-associated KRAS mutations. Mol Cancer Res. (2015) 13(9):1325–35. doi: 10.1158/1541-7786.MCR-15-0203
40. Lu S, Jang H, Nussinov R, Zhang J. The structural basis of oncogenic mutations G12, G13 and Q61 in small GTPase k-Ras4B. Sci Rep. (2016) 6. doi: 10.1038/srep21949
41. Scheffzek K, Ahmadian MR, Kabsch W, Wiesm̈ller L, Lautwein A, Schmitz F, et al. The ras-RasGAP complex: Structural basis for GTPase activation and its loss in oncogenic ras mutants. Science. (1997) 277(5324):333–9. doi: 10.1126/science.277.5324.333
42. Tong L, de Vos AM, Milburn MV, Kim SH. Crystal structures at 2.2 Å resolution of the catalytic domains of normal ras protein and an oncogenic mutant complexed with GDP. J Mol Biol. (1991) 217(3):503–16. doi: 10.1016/0022-2836(91)90753-S
43. Hallin J, Bowcut V, Calinisan A, Briere DM, Hargis L, Engstrom LD, et al. Anti-tumor efficacy of a potent and selective non-covalent KRASG12D inhibitor. Nat Med. (2022) 28(10):2171–82. doi: 10.1038/s41591-022-02007-7
44. Knox JE, Jiang J, Burnett GL, Liu Y, Weller CE, Wang Z, et al. Abstract 3596: RM-036, a first-in-class, orally-bioavailable, tri-complex covalent KRASG12D(ON) inhibitor, drives profound anti-tumor activity in KRASG12D mutant tumor models. Cancer Res. (2022) 82(12_Supplement):3596–6. doi: 10.1158/1538-7445.AM2022-3596
45. Bannoura SF, Khan HY, Azmi AS. KRAS G12D targeted therapies for pancreatic cancer: Has the fortress been conquered? Front Oncol. (2022) 12:1013902. doi: 10.3389/fonc.2022.1013902
46. Wu X, Song W, Cheng C, Liu Z, Li X, Cui Y, et al. Small molecular inhibitors for KRAS-mutant cancers. Front Immunol. (2023) 14:1223433. doi: 10.3389/fimmu.2023.1223433
47. Welsch ME, Kaplan A, Chambers JM, Stokes ME, Bos PH, Zask A, et al. Multivalent small-molecule pan-RAS inhibitors. Cell. (2017) 168(5):878–889.e29. doi: 10.1016/j.cell.2017.02.006
48. Tisi R, Spinelli M, Palmioli A, Airoldi C, Cazzaniga P, Besozzi D, et al. The multi-level mechanism of action of a pan-ras inhibitor explains its antiproliferative activity on cetuximab-resistant cancer cells. Front Mol Biosciences. (2021) 8:625979. doi: 10.3389/fmolb.2021.625979
49. Vidimar V, Beilhartz GL, Park M, Biancucci M, Kieffer MB, Gius DR, et al. An engineered chimeric toxin that cleaves activated mutant and wild-type RAS inhibits tumor growth. Proc Natl Acad Sci. (2020) 117(29):16938–48. doi: 10.1073/pnas.2000312117
50. Tran TH, Alexander P, Dharmaiah S, Agamasu C, Nissley DV, McCormick F, et al. The small molecule BI-2852 induces a nonfunctional dimer of KRAS. Proc Natl Acad Sci. (2020) 117(7):3363–4. doi: 10.1073/pnas.1918164117
51. Koltun ES, Rice MA, Gustafson WC, Wilds D, Jiang J, Lee BJ, et al. Abstract 3597: Direct targeting of KRASG12X mutant cancers with RMC-6236, a first-in-class, RAS-selective, orally bioavailable, tri-complex RASMULTI(ON) inhibitor. Cancer Res. (2022) 82(12_Supplement):3597–7. doi: 10.1158/1538-7445.AM2022-3597
52. Arbour KC, Punekar S, Garrido-Laguna I, Hong DS, Wolpin B, Pelster MS, et al. 652O preliminary clinical activity of RMC-6236, a first-in-class, RAS-selective, tri-complex RAS-MULTI(ON) inhibitor in patients with KRAS mutant pancreatic ductal adenocarcinoma (PDAC) and non-small cell lung cancer (NSCLC). Ann Oncol. (2023) 34:S458. doi: 10.1016/j.annonc.2023.09.1838
53. Wasko UN, Jiang J, Dalton TC, Curiel-Garcia A, Cole Edwards A, Wang Y, et al. Tumor-selective activity of RAS-GTP inhibition in pancreatic cancer. doi: 10.1038/s41586-024
54. Jiang J, Jiang L, Maldonato BJ, Wang Y, Holderfield M, Aronchik I, et al. Translational and therapeutic evaluation of RAS-GTP inhibition by RMC-6236 in RAS-driven cancers. Cancer Discovery. (2024), OF1–OF24. doi: 10.1158/2159-8290.CD-24-0027
55. Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired resistance to KRAS G12C inhibition in cancer. New Engl J Med. (2021) 384(25):2382–93. doi: 10.1056/nejmoa2105281
56. Koga T, Suda K, Fujino T, Ohara S, Hamada A, Nishino M, et al. KRAS secondary mutations that confer acquired resistance to KRAS G12C inhibitors, sotorasib and adagrasib, and overcoming strategies: Insights from In vitro experiments. J Thorac Oncol. (2021) 16(8):1321–32. doi: 10.1016/j.jtho.2021.04.015
57. Zhuang H, Fan J, Li M, Zhang H, Yang X, Lin L, et al. Mechanistic insights into the clinical Y96D mutation with acquired resistance to AMG510 in the KRASG12C. Front Oncol. (2022) 12:915512. doi: 10.3389/fonc.2022.915512
58. Nichols RJ, Yang YC, Cregg J, Schulze CJ, Wang Z, Dua R, et al. Abstract 3595: RMC-6291, a next-generation tri-complex KRASG12C(ON) inhibitor, outperforms KRASG12C(OFF) inhibitors in preclinical models of KRASG12C cancers. Cancer Res. (2022) 82(12_Supplement):3595–5. doi: 10.1158/1538-7445.AM2022-3595
59. Batrash F, Kutmah M, Zhang J. The current landscape of using direct inhibitors to target KRASG12C-mutated NSCLC. Exp Hematol Oncol. (2023) 12(1):93. doi: 10.1186/s40164-023-00453-8
60. Blaquier JB, Cardona AF, Recondo G. Resistance to KRASG12C inhibitors in non-small cell lung cancer. Front Oncol. (2021) 11:787585. doi: 10.3389/fonc.2021.787585
61. Liu J, Huang X, Liu H, Wei C, Ru H, Qin H, et al. Immune landscape and prognostic immune-related genes in KRAS-mutant colorectal cancer patients. J Trans Med. (2021) 19(1):27. doi: 10.1186/s12967-020-02638-9
62. Hamarsheh S, Groß O, Brummer T, Zeiser R. Immune modulatory effects of oncogenic KRAS in cancer. Nat Commun. (2020) 11(1). doi: 10.1038/s41467-020-19288-6
63. Huang L, Guo Z, Wang F, Fu L. KRAS mutation: from undruggable to druggable in cancer. Signal Transduction Targeted Ther. (2021) 6(1):386. doi: 10.1038/s41392-021-00780-4
64. Mugarza E, van Maldegem F, Boumelha J, Moore C, Rana S, Llorian Sopena M, et al. Therapeutic KRAS G12C inhibition drives effective interferon-mediated antitumor immunity in immunogenic lung cancers. Sci Advances. (2022) 8(29). doi: 10.1126/sciadv.abm8780
65. Liu C, Zheng S, Jin R, Wang X, Wang F, Zang R, et al. The superior efficacy of anti-PD-1/PD-L1 immunotherapy in KRAS-mutant non-small cell lung cancer that correlates with an inflammatory phenotype and increased immunogenicity. Cancer Letters. (2020) 470:95–105. doi: 10.1016/j.canlet.2019.10.027
66. Zhang Z, Rohweder PJ, Ongpipattanakul C, Basu K, Bohn MF, Dugan EJ, et al. A covalent inhibitor of k-Ras(G12C) induces MHC class i presentation of haptenated peptide neoepitopes targetable by immunotherapy. Cancer Cell. (2022) 40(9):1060–1069.e7. doi: 10.1016/j.ccell.2022.07.005
67. Boumelha J, de Carné Trécesson S, Law EK, Romero-Clavijo P, Coelho MA, Ng KW, et al. An immunogenic model of KRAS-mutant lung cancer enables evaluation of targeted therapy and immunotherapy combinations. Cancer Res. (2022) 82(19):3435–48. doi: 10.1158/0008-5472.CAN-22-0325
68. Chour A, Denis J, Mascaux C, Zysman M, Bigay-Game L, Swalduz A, et al. Brief report: Severe sotorasib-related hepatotoxicity and non-liver adverse events associated with sequential anti–programmed cell death (Ligand)1 and sotorasib therapy in KRASG12C-mutant lung cancer. In: Journal of thoracic oncology. Elsevier Inc (2023). p. 1408–15. doi: 10.1016/j.jtho.2023.05.013
Keywords: KRAS G12D, KRAS G12C, combination therapy, structural analysis, secondary mutation, acquired resistance, covalent inhibitor, non-covalent inhibitor
Citation: Lokhandwala J, Smalley TB and Tran TH (2024) Structural perspectives on recent breakthrough efforts toward direct drugging of RAS and acquired resistance. Front. Oncol. 14:1394702. doi: 10.3389/fonc.2024.1394702
Received: 02 March 2024; Accepted: 24 April 2024;
Published: 22 May 2024.
Edited by:
Gary Piazza, Auburn University, United StatesReviewed by:
Carmen Guerra, Spanish National Cancer Research Center, SpainCopyright © 2024 Lokhandwala, Smalley and Tran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Timothy H. Tran, dGltb3RoeS50cmFuQG1vZmZpdHQub3Jn