
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 18 June 2024
Sec. Cancer Genetics
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1390221
 Daniele Paixão1*
Daniele Paixão1* Thalitta Hetamaro Ayala Lima1Rafaela Rogério Floriano de Souza1Juliana Emilia Prior Carnavalli1Clarissa Gondim Picanço-Albuquerque2Isabelle Joyce de Lima Silva-Fernandes2
Thalitta Hetamaro Ayala Lima1Rafaela Rogério Floriano de Souza1Juliana Emilia Prior Carnavalli1Clarissa Gondim Picanço-Albuquerque2Isabelle Joyce de Lima Silva-Fernandes2 Paulo Goberlânio de Barros Silva2
Paulo Goberlânio de Barros Silva2 Miguel Mitne-Neto3Caroline Mônaco Moreira1Wagner Antônio da Rosa Baratela1
Miguel Mitne-Neto3Caroline Mônaco Moreira1Wagner Antônio da Rosa Baratela1Introduction: Lynch syndrome (LS) is an inherited cancer predisposition syndrome characterized by a high risk of colorectal and extracolonic tumors. Germline pathogenic variants (GPV) in the PMS2 gene are associated with <15% of all cases. The PMS2CL pseudogene presents high homology with PMS2, challenging molecular diagnosis by next-generation sequencing (NGS). Due to the high methodological complexity required to distinguish variants between PMS2 and PMS2CL, most laboratories do not clearly report the origin of this molecular finding.
Objective: The aim of this study was to confirm the GPVs detected by NGS in regions of high homology segments of the PMS2 gene in a Brazilian sample.
Methods: An orthogonal and gold standard long-range PCR (LR-PCR) methodology to separate variants detected in the PMS2 gene from those detected in the pseudogene.
Results: A total of 74 samples with a PMS2 GPV detected by NGS in exons with high homology with PMS2CL pseudogene were evaluated. The most common was NM_000535.6:c.2182_2184delinsG, which was previously described as deleterious mutation in a study of African-American patients with LS and has been widely reported by laboratories as a pathogenic variant associated with the LS phenotype. Of all GPVs identified, only 6.8% were confirmed by LR-PCR. Conversely, more than 90% of GPV were not confirmed after LR-PCR, and the diagnosis of LS was ruled out by molecular mechanisms associated with PMS2.
Conclusion: In conclusion, the use of LR-PCR was demonstrated to be a reliable approach for accurate molecular analysis of PMS2 variants in segments with high homology with PMS2CL. We highlight that our laboratory is a pioneer in routine diagnostic complementation of the PMS2 gene in Brazil, directly contributing to a more assertive molecular diagnosis and adequate genetic counseling for these patients and their families.
Colorectal cancer (CRC) is the second most frequent cancer among men and women in Brazil, corresponding approximately to more than 45 thousand new cases per year (1). Approximately 5% of CRCs are associated with germline variants, and Lynch syndrome (LS) is the most prevalent cause of hereditary CRC and is an autosomal dominant disorder related to monoallelic germline pathogenic variants (GPVs) in DNA mismatch repair (MMR) genes MLH1, MSH2, MSH6, and PMS2, and deletions in the EPCAM gene (2–4). It is clinically characterized by predisposition to a broad spectrum of tumors, including early-onset CRC and extracolonic tumors, including endometrial, ovarian, gastric, ureter, renal pelvis, pancreatic, prostate, biliary tract, central nervous system, and small bowel (5, 6).
While GPV in MLH1 and MSH2 genes account for almost 70% of LS cases, mutations in PMS2 contribute to <15% (7, 8). Molecular testing of PMS2 is challenging due to high homology of PMS2 gene to its counterpart PMS2CL pseudogene, which is considered biologically inactive. Both are located on chromosome 7, and interpreting the clinical relevance of variants detected in these regions is essential for patients’ follow-up and is considered an important challenge nowadays (9).
The PMS2CL pseudogene presents high homology (>98%) with PMS2, with the greatest identity being found over exon 9 and between exons 11 and 15 (10, 11). NGS is able to identify variants along all the coding segments of this gene; however, mapping and variant calling pipelines struggle to differentiate whether a variant is present in the gene or in the pseudogene. Because of the high methodological complexity required to distinguish variants between PMS2 and PMS2CL, most laboratories do not clearly report the origin of this molecular finding.
Thus, due to the extreme importance of correctly reporting reliable variants in the PMS2 gene, the aim of this study was to confirm the GPVs detected by NGS in regions of high homology segments of the PMS2 gene in a Brazilian sample using the orthogonal and gold standard long-range PCR (LR-PCR) methodology to separate variants detected in the PMS2 gene from those detected in the pseudogene. This strategy will prompt reliable results that will directly contribute to appropriate clinical management.
We selected a total of 74 samples with PMS2 GPV detected by NGS Panels for Hereditary Cancer, performed at Fleury Genomics laboratory between December 2018 and August 2021. Samples were selected regardless of the personal or familial history of cancer. All participants provided informed consent before blood withdrawal or saliva collection. The study protocol was reviewed and approved by the Human Research Ethics Committee of Fleury Group (protocol number NP_614; Plataforma Brasil CAAE# 56961222.6.0000.5474; Fleury# 5.833.008).
Genomic DNA was extracted from peripheral blood, saliva, or swab samples using QIASymphony (QIAGEN, Inc.) with the QIASymphony DNA Mini Kit, QIAmp DNA Blood Mini Kit, and QIAamp DNA Blood Mini Kit (all from QIAGEN, Inc.), respectively. DNA fragmentation was followed by indexing, capture with custom probes, and enrichment of the regions of interest. Paired-end NGS was performed using Illumina platforms, either NovaSeq or NextSeq500 (Illumina, Inc., San Diego, CA, USA). Bioinformatics pipelines were used to perform the alignment and detection of variants based on the GRCh37 (Hg19) version of the Human Genome. The data generated by sequencing were analyzed using local customized bioinformatics processes.
All hereditary cancer predisposition panel data evaluated in this study were generated using the NGS approach. Considering the limitations of the current methodology, all the detected PMS2 GPV variants were confirmed by the orthogonal and gold standard methodology, LR-PCR, followed by nested PCR. The variants detected in the PMS2 gene were initially detected using the NGS methodology, which presents methodological limitations for evaluating regions that overlap with pseudogenes because the reads have an average size of 150 bp in the sequencing used (Illumina, Inc., San Diego, CA, USA), which is insufficient to distinguish genes from pseudogenes, considering that some of these intervals have more than 95% homology, requiring complementation using orthogonal methodology (LR_PCR).
The LR-PCR technique described by Vaughn et al. (2010) was employed. This technique involves an initial amplification of regions not anchored in regions of high homology with the pseudogene, followed by a new amplification of only the region to be evaluated. For this, we used a set of specific primers aimed at the amplification of PMS2 gene, in a similar approach as previously published in the literature.
The protocol used for LR-PCR was previously described by Vaughn et al. (2010) with some adaptations (i.e., Herculase II fusion DNA polymerase enzyme was used, from Agilent Technologies, Santa Clara). For amplification of the region under investigation, the primers listed in Table 1 were initially used for LR-PCR. For this reaction, we used a high-complexity long-range DNA polymerase enzyme (Herculase II fusion DNA polymerase—Agilent Technologies, Santa Clara). Subsequently, nested PCR was performed using the primers described in Table 2 (12–14).

Table 1 Set of primers used for long-range PCR.
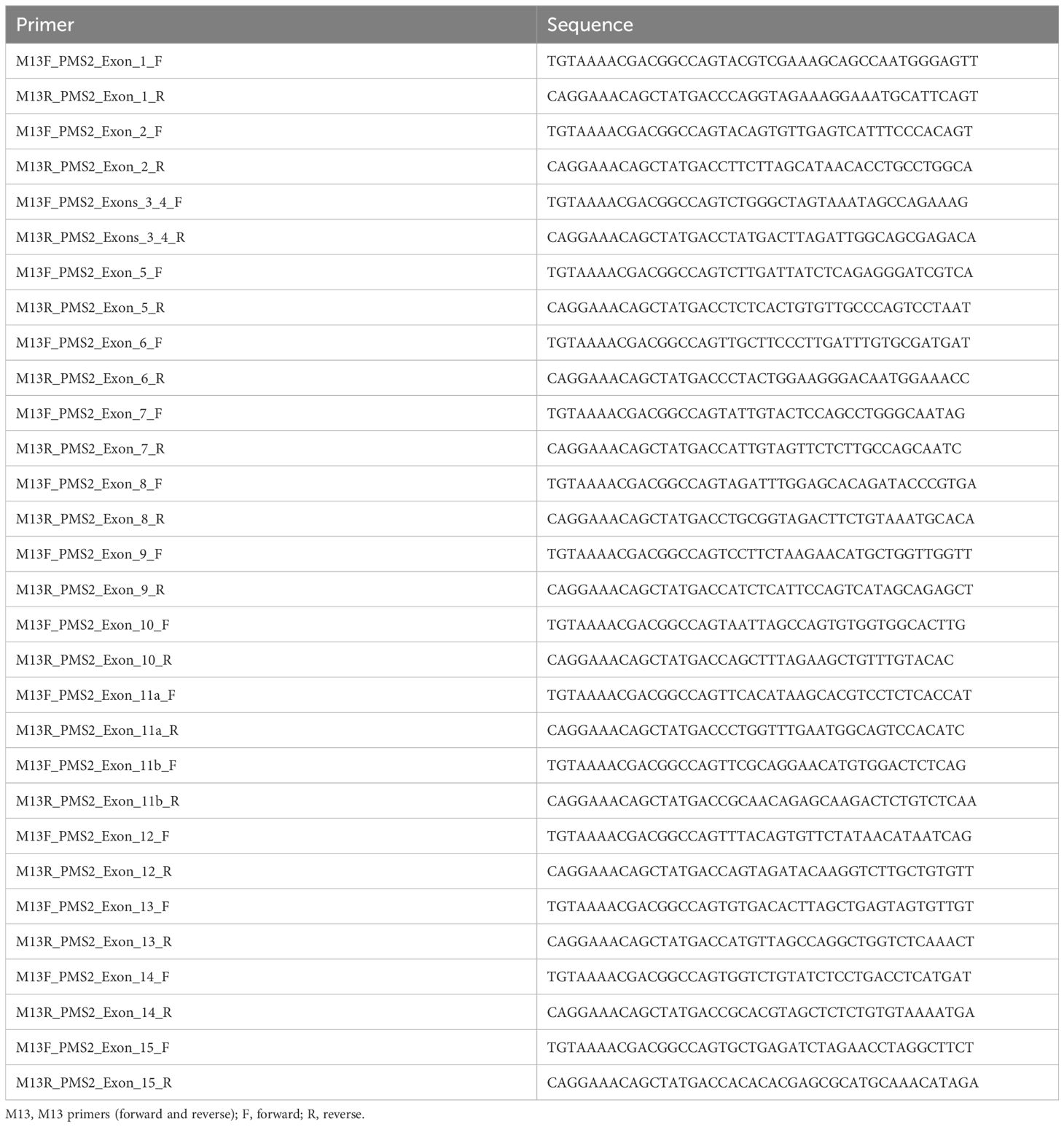
Table 2 Set of primers used for nested PCR.
We used a subset of primers described in Table 1 as amplification primers in a final volume of 50 μL, containing 150 ng of DNA, 0.5 μM each primer (Thermo Fisher Scientific Inc., Waltham, MA), 1.25 μL Herculase II fusion DNA polymerase, 1× PCR buffer (5× Herculase II reaction buffer), and 400 μM each dNTP (all from Agilent Technologies, Santa Clara). Cycling conditions were as follows: initial denaturation of 94°C for 1 min, followed by 35 cycles of 15 s at 94°C, 30 s at 65°C, and 15 min at 68°C. Final elongation entailed 10 min at 72°C. The LR-PCR was followed by nested PCR using a subset of primers described in Table 2 (Thermo Fisher Scientific Inc., Waltham, MA). The amplification primers were used in a final volume of 20 μL, containing 0.5 μM each primer, 1× AmpliTaq Gold PCR Master Mix (Thermo Fisher Scientific Inc., Waltham, MA). Cycling conditions were as follows: initial denaturation of 95°C for 15 min, followed by 30 cycles of 30 s at 95°C, 30 s at 60°C, and 45 s at 72°C. Final elongation entailed 9 min at 72°C. Amplification was evaluated on 2% agarose gel stained with GelRed (Biotium, Hayward, CA).
The amplified samples were purified using the ExoSap enzyme protocol (Thermo Fisher Scientific Inc., Waltham, MA) to perform Sanger sequencing procedures in the ABI 3130 Genetic Analyzer Applied Biosystem platform (Life Technologies). After sequencing, specific genomic coordinates were evaluated in the electropherogram using the software CLC (QIAGEN, Inc.), which allowed us to discriminate between the presence of variants detected in the PMS2 gene or its possible presence in the pseudogene (PMS2CL), demonstrating that LR-PCR can be used to amplify the PMS2 gene and avoid interference of its pseudogene counterparts through the use of anchoring primers exclusive to the PMS2 gene.
All variants were annotated according to HGVS (Sequence Variant Nomenclature) recommendations. The variants were interpreted considering the clinical features of patients and the American College of Medical Genetics (ACMG) and Association for Molecular Pathology (AMP) variant classification protocol (15). Databases such as ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), ClinGen (https://clinicalgenome.org), HGMD (Human Gene Mutation Database https://www.hgmd.cf.ac.uk/ac/index.php) , Varsome (https://varsome.com/), gnomAD (https://gnomad.broadinstitute.org/), dbSNP (https://www.ncbi.nlm.nih.gov/snp/), and Abraom—variant database from Brazilian population (http://abraom.ib.usp.br/) were consulted for clinical variant interpretation assessment.
A total of 74 samples with a PMS2 GPV detected by NGS in exons with high homology with the PMS2CL pseudogene were evaluated. Four different GPVs were identified in exons 11 and 13 (Figure 1). Of these, the most common variant detected in 68 samples was NM_000535.6: c.2182_2184delinsG (p.Thr728Alafs*7) (ClinVar ID: 231999, VCV000231999.10), located in exon 13 of the PMS2 gene, according to the NGS mapping pipeline. This variant causes a translational frameshift with a predicted stop codon and has been reported in the literature to be associated with LS (16). All detected GPVs are described in Table 3.
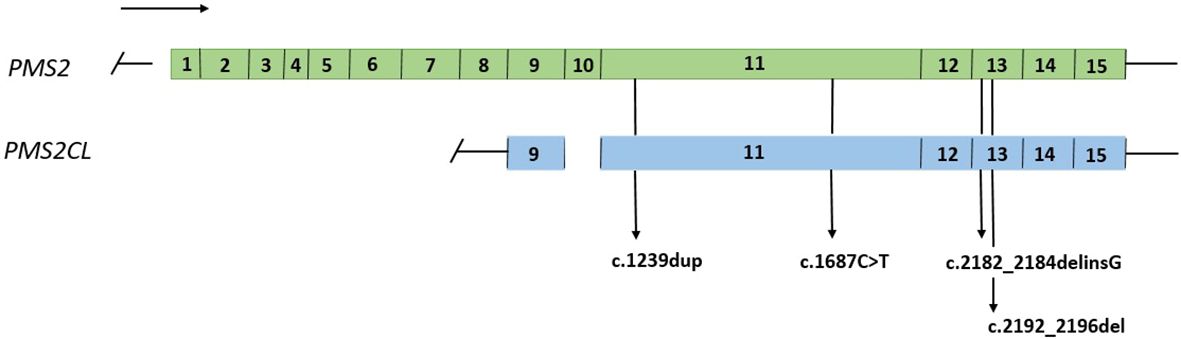
Figure 1 Representation of the PMS2 gene and the PMS2CL pseudogene, regions of high homology and variants detected in exons 11 and 13.
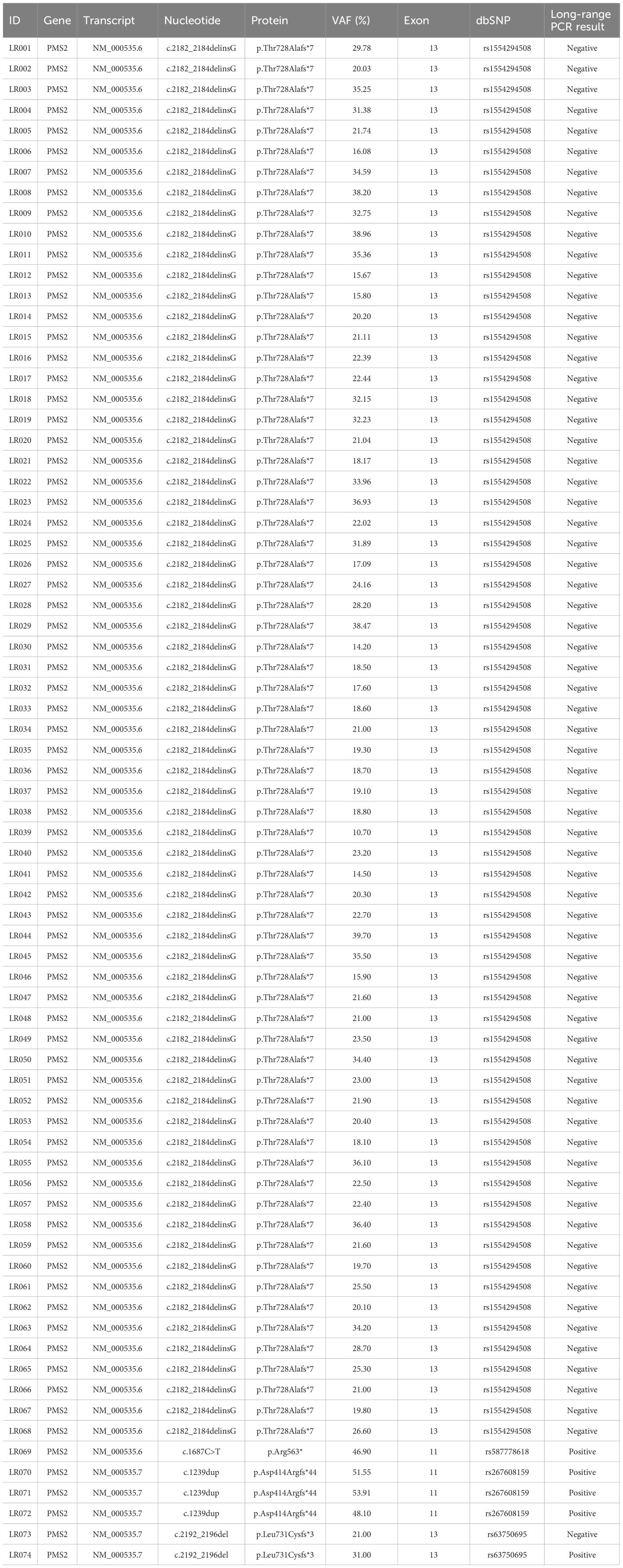
Table 3 Details of variants identified in the PMS2 gene in the region of high homology with the PMS2CL pseudogene.
It was not possible to correlate the molecular findings with the tumor MMR status (immunohistochemical) or tissue microsatellite instability analysis.
We did not have access to the correlated data or clinical information of the patients evaluated in this study. All the 68 patients harboring variant c.2182_2184delinsG, detected through NGS, have failed to confirm it by LR-PCR, indicating the absence of this variant in PMS2. The mean variant allele frequency (VAF) of these variants was 24.7% (ranging from 10.7 to 39.7%), and the median was 22.2% (Table 3).
Of the other six GPV detected, four are located in exon 11 and two in exon 13 (Table 3). Five samples evaluated by LR-PCR were confirmed and considered positive result. Three patients had the following confirmed variant NM_000535.7:c.1239dup (ClinVar ID: 216072, VCV000216072.32), located in exon 11 of PMS2. The mean VAF of these variants was 51.18% (ranging from 48.1 to 53.91%), and the median was 51.55%. The variant NM_000535.6:c.1687C>T (ClinVar ID: 135067, VCV000135067.29) was detected in one patient with VAF of 46.9% and was confirmed by LR-PCR.
The variant NM_000535.7:c.2192_2196del (ClinVar ID: 91331, VCV000091331.38) was detected in two patients and confirmed in one case. The mean VAF was 26%.
In summary, of the 74 GPV identified, five (6.8%) were confirmed by LR-PCR. Conversely, the other 69 patients (93.2%) who did not confirm the presence of the variant after LR-PCR had the diagnosis of LS ruled out by molecular mechanisms associated with the PMS2 gene, not excluding the possibility of other clinical criteria involved with this diagnosis.
Considering the five variants confirmed by LR-PCR, we found a mean VAF of 46.3% versus 24.6% of the other 69 unconfirmed variants.
NGS has some limitations, and the analysis of genes with high identity to pseudogenes is one of them. This is an important issue because the presence of the pseudogene can result in false positive or negative tests, thereby affecting clinical practice and genetic counseling. Thus, the use of different approaches is necessary to avoid interference with data interpretation, which could lead to misleading conduct.
The analysis of PMS2 variants by NGS is very complex. Even considering that the NGS approach can detect variants along all the coding segments of this gene, when a pathogenic variant is detected, the correct clinical interpretation is very challenging because of the high homology of PMS2 to its counterparts, such as the non-expressed PMS2CL pseudogene (17, 18).
The evaluation of PMS2 gene is neglected by many laboratories, due to the methodological difficulties in reporting reliable variants and not ensuring that the variant has been detected in the gene. Many commercial laboratories do not analyze the regions of these PMS2 pseudogenes, consequently generating incomplete analysis of this gene. However, some laboratories report GPV in these regions of high homology, but without confirmation using other techniques.
Although the PMS2 gene has low penetrance, a reliable identification of the presence of pathogenic variants in this gene is fundamental for the correct management of LS and genetic counseling.
In our experience, more than 90% of the pathogenic variants identified by NGS in the PMS2 gene in exons with high identity to pseudogenes were not confirmed using LR-PCR. We observed that variants confirmed by LR-PCR presented a higher VAF, near 50%. In contrast, unconfirmed variants had lower VAF, indicating that variants in the actual gene have higher VAF.
It is important to note that c.2182_2184delinsG variant identified in our cohort was not confirmed to be in the PMS2 gene in any of the patients. This variant is reported in ClinVar database as conflicting, was previously described as a deleterious mutation in a study of African-American patients with LS, and has been widely reported by laboratories as a pathogenic variant associated with the LS phenotype (16). According to Chong et al. (2020), this variant was incorrectly assigned to PMS2 in a sample of patients, suggesting reclassification and caution when interpreting these variants (19). Although we studied variants only in exons 11 and 13, this methodology was developed to confirm variants in other regions with homology to PMS2 gene.
Our findings strongly support this suggestion, and we recommend a pathogenic variant classification only if the variant is at the PMS2 gene evaluated by a LR-PCR. Thus, there are benefits for patients because the diagnosis of LS is excluded, avoiding unnecessary screening and even unequivocal indication of hysterectomy and risk-reducing salpingo-oophorectomy (20).
In conclusion, the use of LR-PCR was demonstrated to be a reliable approach for accurate molecular analysis of PMS2 gene variants in segments with high homology with the PMS2CL pseudogene. We highlight that our laboratory is a pioneer in the diagnostic complementation of the PMS2 gene in Brazil, directly contributing to more assertive molecular diagnosis. Our results indicate that using confirmation strategies such as LR-PCR in those segments is essential to avoid misdiagnosis of LS, directly impacting the genetic counseling of these patients and their families, since the correct molecular diagnosis can avoid inappropriate clinical management.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
The studies involving humans were approved by Human Research Ethics Committee (protocol number NP_614) - Fleury S.A. The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
DP: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. TL: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. RS: Methodology, Validation, Writing – review & editing. JC: Writing – review & editing. CP-A: Writing – review & editing. IS-F: Writing – review & editing. PS: Writing – review & editing. MM-N: Methodology, Validation, Writing – review & editing. CM: Project administration, Supervision, Writing – review & editing. WB: Project administration, Supervision, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The following authors are employees received salary and other bonuses of Fleury Medicina e Saude: DP, TL, RS, JC, CM, and WB.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Instituto Nacional de Câncer (Brasil). Estimativa 2023: incidência de câncer no Brasil. In: Instituto Nacional de Câncer. Rio de Janeiro: INCA (2022). Available at: https://www.inca.gov.br/publicacoes/livros/estimativa-2023-incidencia-de-cancer-no-brasil.
2. Aaltonen LA, Salovaara R, Kristo P, Canzian F, Hemminki A, Peltomäki P, et al. Incidence of hereditary nonpolyposis colorectal cancer and the feasibility of molecular screening for the disease. N Engl J Med. (1998) 338:1481–7. doi: 10.1056/NEJM199805213382101
3. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology. (2010) 138:2073–87. doi: 10.1053/j.gastro.2009.12.064
4. Kempers MJ, Kuiper RP, Ockeloen CW, Chappuis PO, Hutter P, Rahner N, et al. Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort study. Lancet Oncol. (2011) 12:49–55. doi: 10.1016/S1470–2045(10)70265–5
5. Bonadona V, Bonaïti B, Olschwang S, Grandjouan S, Huiart L, Longy M, et al. Cancer risks associated with germline mutations in MLH1, MSH2, and MSH6 genes in Lynch syndrome. JAMA. (2011) 305:2304–10. doi: 10.1001/jama.2011.743
6. Møller P, Seppälä TT, Bernstein I, Holinski-Feder E, Sala P, Gareth Evans D, et al. Cancer risk and survival in path_MMR carriers by gene and gender up to 75 years of age: a report from the Prospective Lynch Syndrome Database. Gut. (2018) 67:1306–16. doi: 10.1136/gutjnl-2017–314057
7. Palomaki GE, McClain MR, Melillo S, Hampel HL, Thibodeau SN. EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med. (2009) 11:42–65. doi: 10.1097/GIM.0b013e31818fa2db
8. Dowty JG, Win AK, Buchanan DD, Lindor NM, Macrae FA, Clendenning M, et al. Cancer risks for MLH1 and MSH2 mutation carriers. Hum Mutat. (2013) 34:490–7. doi: 10.1002/humu.22262
9. Nicolaides NC, Carter KC, Shell BK, Papadopoulos N, Vogelstein B, Kinzler KW. Genomic organization of the human PMS2 gene family. Genomics. (1995) 30:195–206. doi: 10.1006/geno.1995.9885
10. Nakagawa H, Lockman JC, Frankel WL, Hampel H, Steenblock K, Burgart LJ, et al. Mismatch repair gene PMS2: disease-causing germline mutations are frequent in patients whose tumors stain negative for PMS2 protein, but paralogous genes obscure mutation detection and interpretation. Cancer Res. (2004) 64:4721–7. doi: 10.1158/0008–5472.CAN-03–2879
11. De Vos M, Hayward BE, Picton S, Sheridan E, Bonthron DT. Novel PMS2 pseudogenes can conceal recessive mutations causing a distinctive childhood cancer syndrome. Am J Hum Genet. (2004) 74:954–64. doi: 10.1086/420796
12. Santani A, Murrell J, Funke B, Yu Z, Hegde M, Mao R, et al. Development and validation of targeted next-generation sequencing panels for detection of germline variants in inherited diseases. Arch Pathol Lab Med. (2017) 141:787–97. doi: 10.5858/arpa.2016–0517-RA
13. Hendriks YM, Jagmohan-Changur S, van der Klift HM, Morreau H, van Puijenbroek M, Tops C, et al. Heterozygous mutations in PMS2 cause hereditary nonpolyposis colorectal carcinoma (Lynch syndrome). Gastroenterology. (2006) 130:312–22. doi: 10.1053/j.gastro.2005.10.052
14. Vaughn CP, Robles J, Swensen JJ, Miller CE, Lyon E, Mao R, et al. Clinical analysis of PMS2: mutation detection and avoidance of pseudogenes. Hum Mutat. (2010) 31:588–93. doi: 10.1002/humu.21230
15. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 175:405–24. doi: 10.1038/gim.2015.30
16. Guindalini RS, Win AK, Gulden C, Lindor NM, Newcomb PA, Haile RW, et al. Mutation spectrum and risk of colorectal cancer in African American families with Lynch syndrome. Gastroenterology. (2015) 149:1446–53. doi: 10.1053/j.gastro.2015.07.052
17. Lima T, Paixão D, Bueno L, Sousa R, Carnavalli J, Bezerra MJB, et al. Pathogenic variants detected in high homology segments of the PMS2 gene in a Brazilian sample doi: 10.1016/j.gimo.2023.100094
18. Paixão D, Lima T, Bueno L, Sousa R, Carnavalli J, Picanço-Albuquerque CG, et al. Evaluation of PMS2 gene variant c.2182_2184delinsG by NGS in a Brazilian sample: How long-range PCR can solve homology. doi: 10.1016/j.gimo.2023.100094
19. Chong AS, Chong G, Foulkes WD, Saskin A. Reclassification of a frequent African-origin variant from PMS2 to the pseudogene PMS2CL. Hum Mutat. (2020) 41:749–52. doi: 10.1002/humu.23978
20. National Comprehensive Cancer Network. Genetic/Familial High-Risk Assessment: ColorectalNCCN clinical practice guidelines in oncology; version 2 (2023). Available online at: https://www.nccn.org/professionals/physician_gls/pdf/genetics_colon.pdf.
Keywords: long-range PCR, PMS2 gene, PMS2CL pseudogene, Lynch syndrome, next-generation sequencing
Citation: Paixão D, Lima THA, de Souza RRF, Carnavalli JEP, Picanço-Albuquerque CG, Silva-Fernandes IJdL, de Barros Silva PG, Mitne-Neto M, Moreira CM and Baratela WAdR (2024) Evaluation of pathogenic variants detected in high homology regions of the PMS2 gene. How effective is long-range PCR? Front. Oncol. 14:1390221. doi: 10.3389/fonc.2024.1390221
Received: 22 February 2024; Accepted: 03 June 2024;
Published: 18 June 2024.
Edited by:
Luca Ermini, Luxembourg Institute of Health, LuxembourgReviewed by:
Tiina Annikki Jokela, University of Jyväskylä, FinlandCopyright © 2024 Paixão, Lima, de Souza, Carnavalli, Picanço-Albuquerque, Silva-Fernandes, de Barros Silva, Mitne-Neto, Moreira and Baratela. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniele Paixão, ZGFuaWVsZS5wcGVyZWlyYUBncnVwb2ZsZXVyeS5jb20uYnI=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.