 Jiaqi Xu
Jiaqi Xu Ji Zhang
Ji Zhang Wubing Chen3*†
Wubing Chen3*†
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 28 May 2024
Sec. Neuro-Oncology and Neurosurgical Oncology
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1388700
This article is part of the Research TopicAdvances in Brain Tumor TherapyView all 11 articles
Malignant gliomas are one of the most common and lethal brain tumors with poor prognosis. Most patients with glioblastoma (GBM) die within 2 years of diagnosis, even after receiving standard treatments including surgery combined with concomitant radiotherapy and chemotherapy. Temozolomide (TMZ) is the first-line chemotherapeutic agent for gliomas, but the frequent acquisition of chemoresistance generally leads to its treatment failure. Thus, it’s urgent to investigate the strategies for overcoming glioma chemoresistance. Currently, many studies have elucidated that cancer chemoresistance is not only associated with the high expression of drug-resistance genes in glioma cells but also can be induced by the alterations of the tumor microenvironment (TME). Numerous studies have explored the use of antifibrosis drugs to sensitize chemotherapy in solid tumors, and surprisingly, these preclinical and clinical attempts have exhibited promising efficacy in treating certain types of cancer. However, it remains unclear how tumor-associated fibrotic alterations in the glioma microenvironment (GME) mediate chemoresistance. Furthermore, the possible mechanisms behind this phenomenon are yet to be determined. In this review, we have summarized the molecular mechanisms by which tumor-associated fibrotic reactions drive glioma transformation from a chemosensitive to a chemoresistant state. Additionally, we have outlined antitumor drugs with antifibrosis functions, suggesting that antifibrosis strategies may be effective in overcoming glioma chemoresistance through TME normalization.
Gliomas are stratified into grades 1 through 4 according to the World Health Organization’s tiered grading system, and grade 4 is the most prevalent and virulent subtype, also known as glioblastoma. GBM, an unyielding primary cerebral malignancy, has a grim prognosis with a 5-year survival rate of less than 10% (1–3), despite standard therapies including the maximal tumor excision, combined with concomitant radiotherapy and temozolomide chemotherapy. So far, TMZ is the first-line chemotherapy drug for glioma. However, due to the frequent occurrence of TMZ resistance after chemotherapy, glioma is recalcitrant and refractory. To increase the prognosis of GBM patients, it’s important to summarize the potential mechanisms of glioma chemoresistance and find useful strategies to overcome TMZ resistance.
Gliomas are characterized as easily chemoresistant intracranial malignancy through demethylation of O(6)-methylguanine-DNA methyltransferase (MGMT) promoter, overexpression of cell membrane glycoprotein, and the augmentation of stemness-associated molecules (4–7). Moreover, the chemoresistance could not only be developed by the cellular alterations in cancer cells but also, in part, be modulated by the specific TME (8). Many researchers recently have focused on the chemoresistance promoted by the GME and are increasingly aware of the significance of overcoming chemoresistance by normalizing GME. The nonneoplastic immune cells and stromal components foster an immunosuppressive GME under the interaction of glioma-secreted cytokines (9, 10). The prominent nonneoplastic stromal cells in gliomas consist of endothelial cells, microglia, and tumor-associated macrophages (TAMs), etc. (2, 11–13). In solid tumor stroma, cancer-associated fibroblasts (CAFs) secrete a lot of collagen after stimulation (14), and subsequently increase the stiffness of the tumor matrix which in turn enhances the proliferation, invasiveness, and stemness as well as chemoresistance of glioma cells. Different from others’ attention on the glioma chemoresistance increased by the alterations of glioma cells themselves, in this review, we summarize the relationship between the chemoresistance and glioma-associated fibrotic reactions. Several investigators have attempted to enhance TMZ chemotherapy efficacy with reasonable combinations of some clinically approved conventional drugs (15), and among these drugs, the increased chemotherapy efficacy by some agents with antifibrosis function draws our attention. However, it is so little known why the antifibrosis medication is effective for solid tumors and how glioma-associated fibrotic reactions in GME specifically contribute to TMZ chemoresistance and poor prognosis for glioma patients. In this review, we thus explore the mechanism of the occurrence and development of tumor-associated fibrotic phenomena in GME and sum up the antifibrosis strategies for sensitizing chemotherapy, hoping to provide novel insights for glioma research and treatment.
During the malignant progression of tumor cells, changes in the tumor stroma also take place including alterations of extracellular matrix (ECM) components, stroma stiffness, excessive vascularization, hypoxia, and paracrine cytokine secretion. As a principal non-cellular component, the ECM plays a crucial role in driving tumor malignancy by providing cells with architectural and mechanical supports, regulating nutrient supply, as well as engaging in multiple cellular processes as a reservoir of diverse cytokine regulators (16–19). The ECM components would transform into a specific status that can stimulate the growth of cancer cells and tumor-associated cells. As cancer occurs and develops, malignant and stromal cells can deposit, break down, and remodel the ECM through the production of multiple ECM proteins, including collagens, fibronectins, laminins, and proteolytic enzymes, which can stimulate the growth of cancer cells and tumor-associated cells (20). In addition, alterations in the biophysical properties of the ECM, such as stiffness, density, rigidity, tension, and protein deposition, are recognized as hallmarks of tumor stromal fibrosis (21, 22). In the TME, CAFs are one of the most critical stromal cell types functioning as the architects of matrix remodeling, which provides the “soil” for tumor survival (23). CAFs could be identified with molecular markers such as αSMA, FAP-1, desmin, podoplanin, NG2 (CSPG4), and PDGFR-α/β (24). These CAFs could secrete substantial quantities of ECM components after being activated and could mediate the malignant progression of tumors through the promotion of stromal inflammation and fibrosis (24, 25). Cancer cells exhibit multiple features of cancer progression, including the recruitment of various stromal cells to form the TME (26), which encompasses different functional subtypes of stromal cells and matrix polymers (27). Among these cells, CAFs promote the formation of a dense and rigid fibrotic microenvironment by large amounts of ECM proteins and cytokines secretions (28). In TME, tumor-associated fibrosis is the result of excessive accumulation of collagen, fibronectin, laminin, tendon protein, etc. (29, 30). Among these ECM components, the most abundant one is collagen protein which constitutes the main rigid structures of tumor stroma. There are more than 28 types of collagens which are categorized into four subtypes: fibril-forming collagens (I, II, III, V, XI, XXVI, XXVII), fibril-associated collagens with interrupted triple helices (FACITs: IX, XII, XIV, XVI, XIX, XX, XXI, XXII, XXIV), network-forming collagens (IV, VIII, X), and membrane-anchored collagens (MACITs: XIII, XVII, XXIII, XXV) (31). Among these collagens, types I, III, and V collagen are predominantly secreted by CAFs, while type IV collagen is mainly produced by epithelial and endothelial cells. It is worth noting that, under certain conditions, tumor cells and TAMs can also synthesize collagen (32). Collectively, tumor-associated fibrotic reactions are induced by the interactions between tumor cells and stromal cells.
In glioma, radiotherapy and cytotoxic chemotherapy can induce epithelial-mesenchymal transition (EMT) and upregulate the transforming growth factor-β (TGF-β) signal, which is the key signaling pathway to fibrosis initiation (33). EMT is associated with increased expression of TGF-β, collagen, fibronectin, α-SMA, and S100A4, suggesting that these molecular mechanisms could be involved in inducing stromal fibrotic reactions in glioma. Some researchers have proposed a “repurposing” strategy for treating GBM by using clinically approved conventional drugs to inhibit EMT. For instance, Kast et al. summarized six clinically approved drugs including fenofibrate, quetiapine, lithium, nifedipine, itraconazole, and metformin (33). These drugs are being explored as adjunctive agents to enhance chemosensitivity in tumor therapy. Given the heterogeneity of GBM, further research is needed to determine which molecular subtypes may benefit from these non-antitumor drugs.
The ECM in the normal brain tissues predominantly consists of hyaluronic acid, proteoglycans, and laminin, but very little collagen. However, in GBM, there is a substantial presence of collagen proteins, laminin, and fibronectin, primarily distributed in the vascular basement membrane in tumor tissue (34). Recent reports highlighted the role of collagen in enhancing GBM cell stemness and promoting EMT and invasion (35, 36). Some researchers found that type-I collagen, the main component of tumor-associated fibrosis, could be used to promote the formation of an invasive, tight GBM spheroid structure when the collagen concentrations increase to some extent (37). Huijbers et al. conducted histological analyses on 90 GBM cases, revealing a significant abundance of collagen proteins within the ECM of GBM (38). They also observed that the collagen receptors Endo180 are overexpressed on the surface of GBM cells. It is worth noting that Endo180 expression is particularly pronounced in stromal-rich high-grade gliomas (39), and its regulation is linked to the TGF-β signaling pathway (40). Additionally, the interactions between Endo180 and collagen significantly potentiates GBM invasion (38). In GBM, collagen XVI induces tumor invasion by modulating the activation pattern of integrin β1, possibly impacting the interactions between glioma cells and the stroma to further enhance the invasive phenotype (41). Furthermore, in order to confirm the association between tumor-associated fibrotic reactions and glioma prognosis, we analyzed the Chinese Glioma Genome Atlas (CGGA) (http://www.cgga.org.cn/) and Gene Expression Profiling Interactive Analysis (GEPIA) (http://gepia.cancer-pku.cn/index.html) databases. According to the CGGA dataset, fibrosis-related marker genes (COL1A2, COL1A1, COL3A1, COL4A1, COL4A2, COL5A2, COL6A2, COL6A1) are highly expressed in the mesenchymal (ME) and classical (CL) GBM subtypes, which are associated with shorter overall survival (Figures 1A, B). Meanwhile, analysis of the GEPIA dataset reveals that the high expressions of collagen-related genes (COL1A1, COL3A1, COL4A1, COL5A2, and COL6A1.) are associated with poor prognosis of glioma patients (Figures 1C–G). Collectively, it suggests that heavier fibrotic reactions play a critical role in glioma progression and predict a poor prognosis for glioma patients.
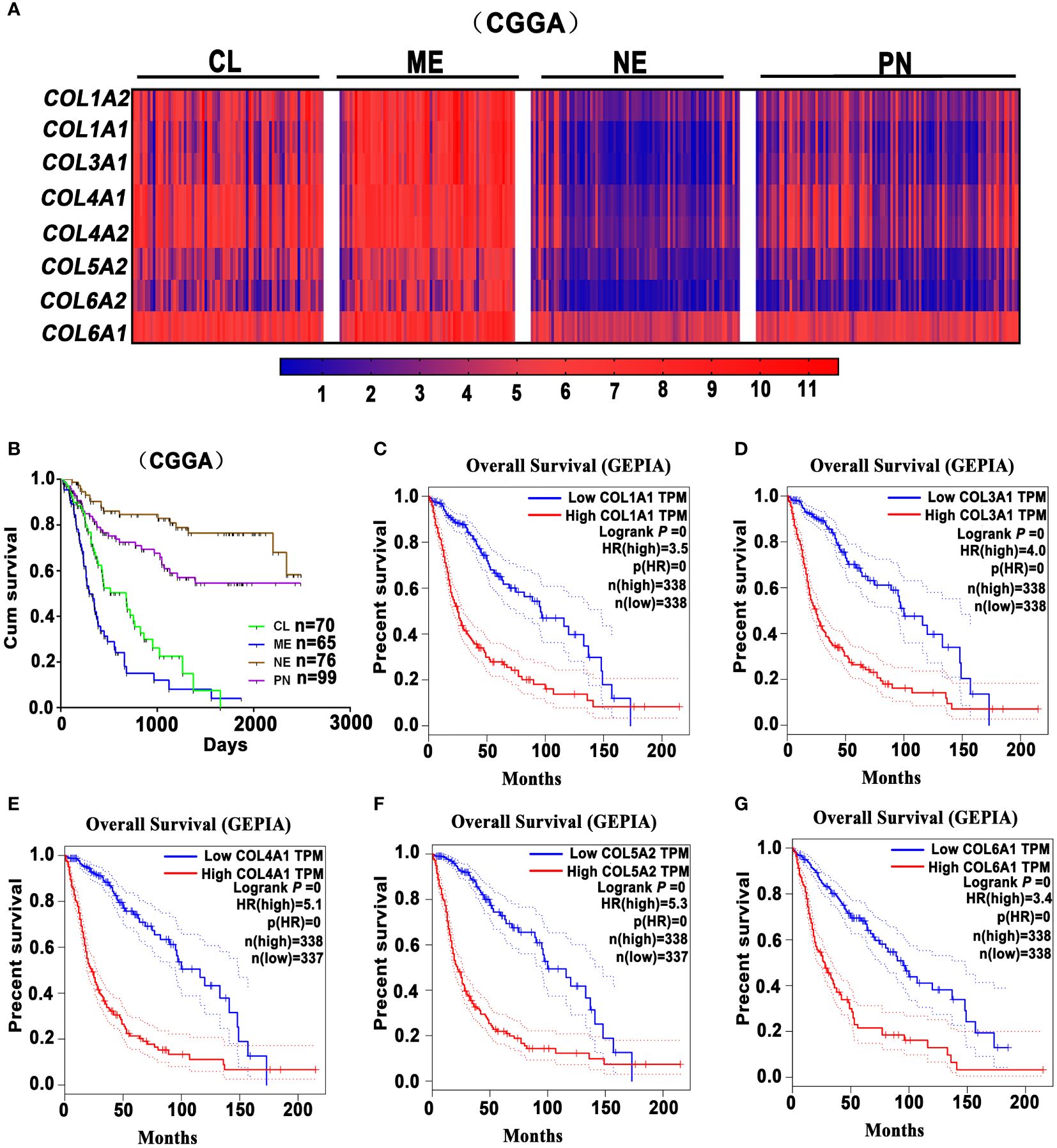
Figure 1 Glioma-associated fibrosis predicts poor prognosis of glioma patients. (A) The expression analysis of fibrosis-related marker genes COL1A2, COL1A1, COL3A1, COL4A1, COL4A2, COL5A2, COL6A2, COL6A1 in the Proneural (PN), Classical (CL), Mesenchymal (ME), and Neural (NE) GBM subtypes in the CGGA dataset. (B) Kaplan-Meier survival curves indicate that ME and CL GBM subtypes predict a poor prognosis for glioma patients in the CGGA dataset. (C–G) Kaplan-Meier survival curves indicate that the high expression of collagen-related genes (COL1A1, COL3A1, COL4A1, COL5A2, and COL6A1) correlates with a poor prognosis for glioma patients in the GEPIA dataset. (GEPIA dataset includes TCGA LGG and GBM dataset.).
Knocking out the collagen XVI gene in GBM U87MG cells resulted in a significant decrease in invasive capabilities compared to the control group (42). Experimental evidence has also shown that the GBM cell compaction promotes the expression of more collagen proteins and vascular endothelial growth factors in GBM, notably elevating the mRNA and protein levels of collagen types VI and IV, as well as the collagen crosslinker named lysyl oxidase (LOX). Notably, β-aminopropionitrile (BAPN), a collagen inhibitor, significantly inhibits collagen crosslinking in the ECM components of GBM by specifically targeting and suppressing LOX. Studies concurrently demonstrated that LOX expression controls the malignant progression of GBM. In an in situ GBM mouse model, treatment with BAPN markedly inhibited intracranial tumor growth by suppressing LOX activities (34). In conclusion, these findings underscore the pivotal role of tumor-associated fibrotic components in fostering the malignant progression of glioma, shedding light on the potential therapeutic effect of antifibrosis medications for controlling this devastating disease.
There are two aspects explaining the mechanism by which glioma-associated fibrotic reactions induce chemoresistance. Firstly, after treated with cytotoxic drugs in solid tumors, CAFs and mesenchymal stem cells (MSCs) are recruited and increased in ECM, along with the accumulation of cytokines and other secreting factors. This fortifies the tumor “stemness”, thereby leading to chemoresistance (43). Secondly, the increased stiffness of tumor tissues with heavily fibrotic ECM components hinders the delivery of chemotherapeutic agents to tumor cells. This limits the penetration of drugs into tumor cells, thereby impairing chemotherapy efficacy (29).
The increased resistance of glioma stem cells (GSCs) to chemotherapeutic agents, which contribute to glioma refractoriness and recurrence, has been extensively documented (44, 45). GSCs are known to cause TMZ resistance through the upregulation of MGMT protein levels (46, 47). In TME, Oleynikova et al. found that the CAFs form the niche for tumor stem cells and these compartments surrounding tumor cells facilitated chemotherapy resistance. In agreement, CAFs may promote the stemness of cancer cells by establishing a survival niche to sustain cancer stem cells (CSCs) and protecting them from chemotherapy-induced cell death, hence, facilitating chemotherapy resistance (25, 28, 48). While it remains unclear how glioma-associated fibrotic reactions develop and which of the molecular characteristics of gliomas is more likely to form fibrosis. It has been widely reported that fibroblast activation protein-α (FAP-α) is involved in tumor-associated fibrosis. FAP-α, typically undetectable in normal tissues, however, exhibits overexpression within glioma cells and glioma stroma (49). Its selective localization in the tissue remodeling and repairing sites enhances the invasiveness and malignant progression of solid tumors including gliomas, implicating FAP-α as a potential target for addressing tumor-associated fibrosis dysregulation (12, 50). Currently, it has been reported that PT-100 significantly reduces CAF enrichment by targeting FAP-α in the tumor stroma, which enhances chemotherapeutic efficacy and reduces drug resistance when combined with oxaliplatin for colon cancer treatment (51). Also, the bone marrow derived MSCs have been demonstrated to enhance tumor stemness after being recruited to the tumor stroma, either through direct paracrine signaling or via its transformation into CAFs (43). Jia et al. compared the gene expression profiles of glioma cells between three-dimensional culture with collagen scaffolds and the conventional two-dimensional culture, and they found that collagen scaffolds could upregulate the expression of EMT-related molecules N-cadherin and vimentin, invasion-related matrix metalloproteinases (MMPs) such as MMP1, MMP2, MMP3, and MMP7, as well as stemness-associated factors CD133, Nestin, Oct4, Sox2, c-Myc, Nanog, MSI1, MSI2 and BMI-1, etc. (35). The glioma stroma harbors a substantial population of TAMs and microglia, which secrete high levels of TGF-β. This cytokine in turn, promotes the invasiveness of CD133(+) GSCs. Moreover, the upregulated TGF-β1 levels are associated with the increased MMP9 production in GSCs (36). What’s more, high serum levels of TGF-β positively correlate with poor prognosis in GBM, hinting at its pivotal role in the maintenance of glioma stemness and malignancy (52). Currently, the strategies targeting the TGF-β signaling have exhibited promising safety and efficacy profiles for gliomas. For instance, anti-TGF-β antibodies have significantly prolonged the survival of recurrent glioma patients (53). It has been proved that after TMZ treatment, the activation of the TGF-β signaling in GBM leads to connective tissue growth factor (CTGF) overexpression, which subsequently mediates TMZ resistance by enhancing the stemness of glioma cells (54). Therefore, these findings suggest that tumor-associated fibrotic reactions play a role in promoting chemoresistance by enhancing the stemness and EMT of glioma cells.
The ECM consists of a variety of structural proteins that maintain tissue structure and regulate extracellular biochemical signals, thereby modulating cellular functions (18, 55). In the process of traveling from blood vessels to tumor cells, chemotherapeutic drugs must navigate through the ECM to reach their target cells. However, drug penetration can be hindered by low pH conditions that facilitate the binding of positively charged chemotherapy drugs to negatively charged ECM components, ultimately reducing the efficiency of drug delivery to cancer cells (29). What’s more, positively charged drugs have more difficulty in crossing the hydrophobic plasma cell membranes (56, 57). As we know, only after the chemotherapeutic drugs penetrate cell membranes and reach the nucleus can they adequately exert their cytotoxic effects (29).
It is known that tissue stiffness varies across different diseases and organs; for instance, normal liver tissue exhibits a “stiffness” at 6 kPa, while the “stiffness” of fibrotic liver tissues can reach up to 12 kPa (58). As the tumor develops, the deposition of type I and IV collagen increases in the cross-linking and tumor-associated fibrosis process, leading to an increase in the “stiffness” of the ECM (59). The increased ECM stiffness corresponds to upregulated contractile and traction forces of the cell cytoskeleton as cells attempt to balance extracellular tension (29). Intercellular mechanotransduction is the process of converting external mechanical stimuli into intracellular biochemical signals. Changes in ECM stiffness are sensed by local junctions between cells, and these junctions are protein complexes containing mechanosensitive protein molecules such as talin and integrins (60). Due to the tension between intracellular contractile forces and extracellular stiffness, talin unfolds in response to the forces, resulting in the exposure of hidden intracellular binding sites that allow effector proteins to bind (61). What’s more, focal adhesion kinase (FAK) is another element that can be activated by external rigidity, and this kinase activity can be utilized to initiate intracellular signaling pathways such as Yes-associated protein (YAP) nuclear localization (62). As mentioned before, the heightened tumor stiffness is closely associated with ECM compositions such as MMPs, hyaluronic acid, and abundant collagens and their cross-linking (18). During the progression of tumors, the accumulation of mechanical pressure can compress tumor blood vessels and lymphatic vessels, leading to reduced perfusion, hypoxia, and elevated interstitial pressure within tumor tissues, thus reducing chemotherapy efficacy (63, 64). Therefore, strategies aiming at reducing mechanical stress in glioma, such as tumor decompression therapy (65), can relieve vascular compression within tumors, enhance tissue perfusion, and improve the transport of chemotherapeutic drugs into tumor cells.
It is important to recognize that chemoresistance, in part, is modulated by collagen and hyaluronic acid in the TME (66) and the strategies specifically targeting these components may be useful tumor decompression therapies. Surprisingly, repurposing those conventionally approved drugs with antifibrosis function can indeed inhibit tumor growth by normalizing TME with the downregulated ECM synthesis. This, in turn, reduces tumor stiffness and mechanical stress, relieves vascular and lymphatic compression, and enhances drug permeability into tumor tissues (67–69). Currently, antifibrosis drugs such as tranilast, losartan, and pirfenidone, have been used to improve chemotherapy in solid tumors (63, 70, 71), while it warrants subsequent research to confirm their efficacy in glioma. As for stroma-rich tumors, researchers concentrate on developing nanomedicines targeting CAFs to reduce tumor matrix stiffness (72, 73). These nano-delivery systems have a double effect on enhancing chemotherapy. Firstly, they reverse tumor progression, immunosuppression, or drug-resistance phenotypes by inhibiting signaling between CAFs and tumor cells, thus increasing chemosensitivity. Secondly, by weakening CAFs function, nanomedicines reduce tumor solid-phase pressure, tumor tissue fluid pressure, and ECM density, leading to increased penetration depth of antitumor drugs and improved efficiency of chemotherapeutic drug delivery.
MMPs are a group of zinc-dependent endopeptidases involved in the dynamic remodeling of ECM, exhibiting proteolytic activities toward ECM components such as collagen (74). In normal circumstances, the synthesis and degradation of ECM is a homeostatic process regulated by the balanced activity of MMPs. However, in tumor tissues, this homeostasis is disrupted due to the overexpressed or hyperactivated of MMPs in gliomas, such as MMP2, MMP9 MMP3, MMP13, MMP14, MMP19, MMP26, and MMP28 (75–84). After effective treatment of U87 glioma xenografts with TMZ, MMP expression is downregulated, with the downregulation of MMP2 and MMP3 associated with the inhibitory effects of TMZ on gliomas (85). In addition, MMPs can promote tumor invasion by facilitating tumor cell degradation of the surrounding matrix or by activating paracrine signaling factors through proteolytic cleavage. For instance, MMPs can lead to the secretion of large amounts of TGF-β, which subsequently promotes CAF activation, long-term fibrosis, and MMP expression and secretion (86).
TME contains numerous signaling molecules and growth factors that, upon binding to cell surface receptors, initiate intracellular signaling in cancer cells, ultimately leading to changes in gene expression. Signaling factors through this mechanism are significantly increased in tumors, for instance, growth factors such as epidermal growth factors (EGFs), fibroblast growth factors (FGFs), platelet-derived growth factors (PDGF), and hepatocyte growth factors (HGF) are abundant in TME (87). During ECM remodeling, the secretion of MMPs promotes the release of growth factors in ECM, such as TGF-β (18, 88). TGF-β exhibits a dual regulatory role in tumor cells, promoting both apoptosis and survival. It suggests that the switch from proapoptotic to prosurvival signaling in tumor cells is influenced by the TP53 gene mutation status (89) or the stiffness of the ECM (90). After the activation of CAFs by TGF-β, they play a crucial role in mediating the maintenance of the TME through paracrine signaling pathways (91). Both glioma cells and infiltrating immune cells in TME could secrete various cytokines, including TGF-β, CTGF, IL-6, and IL-10, contributing to the formation of an immunosuppressive GME (52), many of these cytokines promote chemoresistance in gliomas. Research has shown that in GBM, when treated with TMZ, the activation of the TGF-β signaling pathway leads to the overexpression of CTGF, and subsequently, CTGF increases the expression levels of glioma stem cell markers, including ALDH1, CD44, Nestin, and Nanog (54). In conclusion, the fibrotic alterations in GME are closely related to the maintenance of glioma cell stemness and the chemoresistance of glioma.
Nowadays, numerous glioma immunotherapies have been investigated in clinical and preclinical phases. These include immune checkpoint blockade targeting IDO, CTLA-4, and PD-L1 (92), as well as inhibitors of M2 macrophages such as CSF-1R (93, 94), PI3Kγ (8), and BAPN (11), and antibodies targeting cytokines like IL-6 (95), and CCL5 (96, 97), etc. However, the efficacy of many immunotherapy strategies for GBM remains very limited due to the absence of T lymphocytes, B lymphocytes, and NK cells, as well as the presence of the blood-brain barrier (BBB). Furthermore, during the process of tumor-associated fibrotic reactions, the stiff ECM, particularly the highly crosslinked collagen, creates hypoxic conditions in and around the TME (98) and hinders the infiltration of immune cells or immunotherapeutic agents into tumor tissues (99). Therefore, tumor-associated fibrotic reactions play a role in promoting an immunosuppressive TME, which mediates the immunotherapy resistance in solid tumors. As widely known, the mesenchymal subtype of GBM is characterized by abundant immune features (100), especially the M2 macrophages and microglias (101), suggesting that targeting macrophages could be a useful strategy for treating mesenchymal subtype GBM. Interactions between CAFs and M2 macrophages play a crucial role in the formation of tumor-associated fibrotic reactions (102). As is known, macrophages, contributing to glioma progression (11, 103), can also release significant amounts of TGF-β to initiate and accelerate fibrotic reactions. Furthermore, our investigation revealed that fibrosis-related collagens expression and M2 macrophage marker CD163 expression may participate in glioma malignancy, and analysis from the GEPIA database shows that these collagens (COL1A2, COL1A1, COL3A1, COL4A1, COL4A2, COL5A2, COL6A2, COL6A1) and CD163 expression are higher in GBM compared to low-grade glioma (LGG) (Figure 2A). The mesenchymal subtype of GBM exhibits severe glioma-associated fibrotic reactions, characterized by the most prominent collagen deposition and highest macrophage infiltration in the CGGA and GEPIA datasets (Figures 1A, 2B). It also suggests that the expression level of COL1A1 positively correlates with the expression level of CD163 (the M2 macrophage marker gene) (Figures 2C, D). TAMs-secreted IL-11 and LOX factors promote glioma chemoresistance and progression, while PI3Kγ inhibition (8) and LOX inhibitors (11) could significantly improve TMZ efficacy in orthotopic GBM mouse models. Furthermore, IL-11 (104) and LOX (105), two crucial determinants of tissue fibrosis, are therapeutic targets against organ fibrosis. This suggests that antifibrosis strategies may enhance chemosensitivity in glioma. Therefore, glioma progression and chemoresistance are not only directly promoted by M2 macrophage-secreted cytokines (IL-10, TGF-β, IL-6, etc.) but also modulated by M2 macrophage-mediated fibrotic reactions. Collectively, it suggests that fibrotic reactions partly contribute to macrophage-mediated chemoresistance.
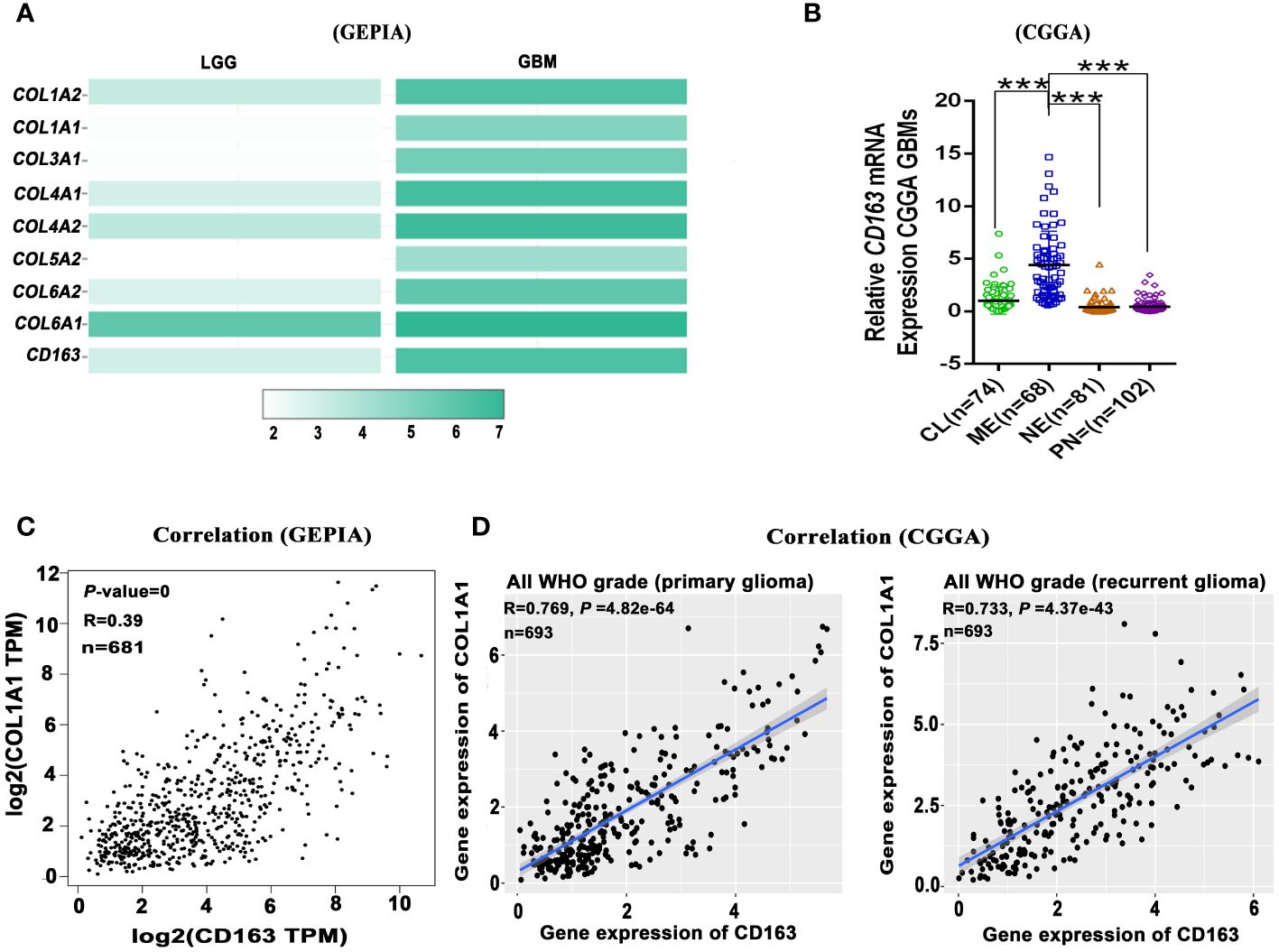
Figure 2 Glioma-associated fibrosis positively correlates with the expression of macrophage marker gene CD163. (A) The expression of fibrosis-related marker genes (COL1A2, COL1A1, COL3A1, COL4A1, COL4A2, COL5A2, COL6A2, COL6A1) and M2 macrophage marker gene (CD163) in low-grade glioma (LGG) and GBM in the GEPIA dataset. (B) Relative CD163 mRNA expression levels of four GBM subtypes in the CGGA dataset. ***p < 0.001. (C) CD163 expression positively correlates with COL1A1 expression in all grade glioma (GEPIA). (D) CD163 expression positively correlates with COL1A1 expression in primary and recurrent glioma (CGGA). (GEPIA dataset includes TCGA LGG and GBM dataset.).
Researchers have explored the antifibrosis therapies in solid tumors, showing definitive sensitization effects for chemotherapy. However, in the presence of the BBB, further research is warranted to determine the efficacy of antifibrosis drugs in sensitizing glioma chemotherapy. In this context, we have summarized the information concerning the utilization of antifibrotic therapies in glioma. According to the existing classifications of antifibrosis therapies by scholars (106), we conclude and discuss antifibrosis therapies in glioma as follows.
As previously discussed, ECM components could be transformed into pro-tumor phenotypes during tumor progression. This transformation presents numerous viable antifibrosis targets for improving chemotherapy by reducing ECM stiffness through downregulated ECM deposition and collagen-modifying enzymes.
Collagen is a prominent component of the ECM, and inhibiting collagen cross-linking has demonstrated significant efficacy in orthotopic GBM mouse models (34). LOX, a kind of collagen cross-linking enzyme, is significantly upregulated during glioma progression due to “cell compaction”. Studies indicate that BAPN, a LOX inhibitor, can effectively inhibit the growth of intracranial PTEN-null GBM mouse models (34, 107). This suggests that BAPN can be a promising strategy for inhibiting glioma growth, possibly by negatively modulating tumor-associated fibrosis. Similarly, another enzyme involved in the process of collagen cross-linking, procollagen-lysine 2-oxoglutarate 5-dioxygenase 2 (PLOD2), has also been tested in GBM treatment. Elevated PLOD2 expression is significantly associated with GBM proliferation, invasion, metastasis, and poor overall survival (108–110). PLOD2 participates in the formation of tumor-associated fibrosis through promoting EMT transition (111), possibly via FAK (108), and PI3K-Akt (111) signaling pathways. Both in vivo and in vitro studies have demonstrated that PLOD2 knockdown inhibits the proliferation, invasion, and anchorage-independent growth of GBM (108, 110, 111). Minoxidil, a confirmed PLOD2 inhibitor (112), could suppress tumor metastasis, in part, by reversing collagen cross-linking in ECM (113, 114). Moreover, studies have found that minoxidil cloud increases the antitumor drug permeability of the blood-brain tumor barrier, resulting in improved and selective delivery to brain tumors, including GBM (115, 116). In conclusion, PLOD2 could serve as a viable target against glioma, possibly by normalizing GME with its antifibrosis function, and PLOD2 inhibitors like Minoxidil may offer potential benefits for glioma patients.
The TGF-β signaling pathway is recognized as the key signal that mediates tissue fibrosis processes and contributes to cancer progression (117, 118). It has been extensively explored whether repurposing antifibrosis drugs can increase chemotherapy sensitivity by targeting TGF-β signaling.
Antifibrosis drugs such as tranilast, pirfenidone, and losartan have shown encouraging efficacy in cancer treatment. Tranilast, for instance, has been demonstrated to reduce matrix mechanical pressure, lower tissue fluid hydrostatic pressure, and enhance tumor perfusion. And, it can enhance the efficacy of chemotherapy drugs with different molecular sizes, including doxorubicin, paclitaxel, and doxorubicin liposomes, by suppressing TGF-β signaling and expression of ECM components (63). Moreover, the combination of TMZ and tranilast significantly suppresses GBM patient-derived xenografts compared to TMZ alone (119, 120). Collectively, repurposing tranilast can not only enhance the efficacy of conventional chemotherapy drugs but also improve the effectiveness of antitumor nanomedicines. Similarly, pirfenidone, another antifibrosis drug that has been clinically approved for the treatment of idiopathic pulmonary fibrosis, also exhibits the function of reducing collagen and hyaluronic acid synthesis (33). Pirfenidone is confirmed to inhibit TGF-β expression in malignant glioma cells, indicating its further application as an adjunctive drug to sensitize glioma TMZ chemotherapy (121). Losartan (LOS), an angiotensin receptor blocker, can reduce the production of collagen and hyaluronic acid by downregulating profibrotic signals such as TGF-β1, CCN2, and ET-1 (70). Therefore, LOS may enhance chemotherapy efficacy by upregulating vascular perfusion and reducing the solid-phase pressure in tumors, which improves the delivery of drugs and oxygen to tumors. Additionally, LOS could antagonize the neoangiogenetic, profibrotic, and immunosuppressive effects of angiotensin II and significantly inhibit its stimulatory effects on local estrogen production, suppressing glioma cell growth and alleviating cerebral edema (122, 123). As a cost-effective angiotensin receptor blocker with an established safety profile, LOS can be quickly repurposed as an adjuvant pharmacological tool prospectively for GBM.
Recent studies have indicated that histone deacetylase inhibitors (HDACi) exhibit antifibrotic effects in various experimental models by preventing histone deacetylation, inducing chromatin decondensation and antifibrotic genes upregulation (124, 125). Valproic acid (VPA), an HDACi agent, exerts its antifibrotic effects by upregulating Smad7 and inhibiting the TGF-β/Smad signaling pathway (126). VPA has been found to inhibit fibrosis in experimental models of various diseases, including liver (127), kidney (128), and heart diseases (129), by reducing macrophage infiltration and downregulating the TGF-β signaling pathway. Briefly, VPA exhibits a dual-purpose effect in glioma therapy, as it not only functions as antiepileptics but also sensitizes TMZ chemotherapy in brain tumor patients (130–132). Another HDACi, vorinostat, approved by the U.S. FDA for the treatment of T-cell lymphoma (133), reduces collagen formation and inhibits fibrosis (134). Studies have shown that the combination of vorinostat and TMZ significantly enhances TMZ efficacy for glioma (135, 136). However, it remains unclear whether the enhanced chemosensitivity induced by VPA and vorinostat is partly or mainly modulated by the inhibition of glioma-associated fibrosis.
Interestingly, Chinese traditional medicine with antifibrosis properties also demonstrates its antitumor efficacy. Berberine, an isoquinoline alkaloid present in many traditional Chinese medicines (137), is confirmed to reduce collagen accumulation in pulmonary fibrosis (138), diabetic nephropathy (138), and arthritis (139), the related mechanisms of which may involve inhibiting TGF-β signaling (140) and restraining EMT (141). Moreover, berberine could suppress glioma growth, migration, and invasion by inhibiting COL11A1 expression and also induce programmed cell death through ERK1/2-mediated mitochondrial damage in glioma cells (142). These studies suggest that berberine could inhibit glioma growth possibly through its antifibrosis properties. Therefore, these conventionally approved antifibrosis drugs could be used to sensitize chemotherapy in glioma through inhibition of TGF-β signaling.
Various antitumor strategies have been developed by directly targeting CAFs (74) including the depletion (73) and normalization of CAFs (143). As for certain cancers, the population of CAFs consists of a collection of multiple subsets of cells with diverse and specific phenotypes at different developmental stages. FAP, a universally acknowledged marker of CAFs, serves as a potential target in both antitumor and antifibrosis therapies. In glioma, FAP expression is detected in glioma cells, mesenchymal cells, and pericytes, etc. (144). Studies have developed an oncolytic adenovirus targeting both GBM cells and GBM-associated stromal FAP+ cells, highlighting its potential immunotherapy through depleting FAP+ CAFs (145). Additionally, FAP-targeting CAR‐T cells have demonstrated promising efficacy in a mouse xenograft model of GBM (146). Another new CAF phenotype in breast cancer, CD10+ GPR77+ CAFs, has been found to be associated with the acquisition of a chemoresistance phenotype. Targeting CD10+ GPR77+ CAFs has been demonstrated to retard tumor formation and reverse chemoresistance by destroying the survival niches for CSCs in both breast and lung cancers (25). However, apart from FAP+ CAFs, further research is needed to explore specific CAF phenotypes associated with glioma chemoresistance.
Above all, we summarized the current glioma therapies with different antifibrosis targets (Figure 3, Table 1).
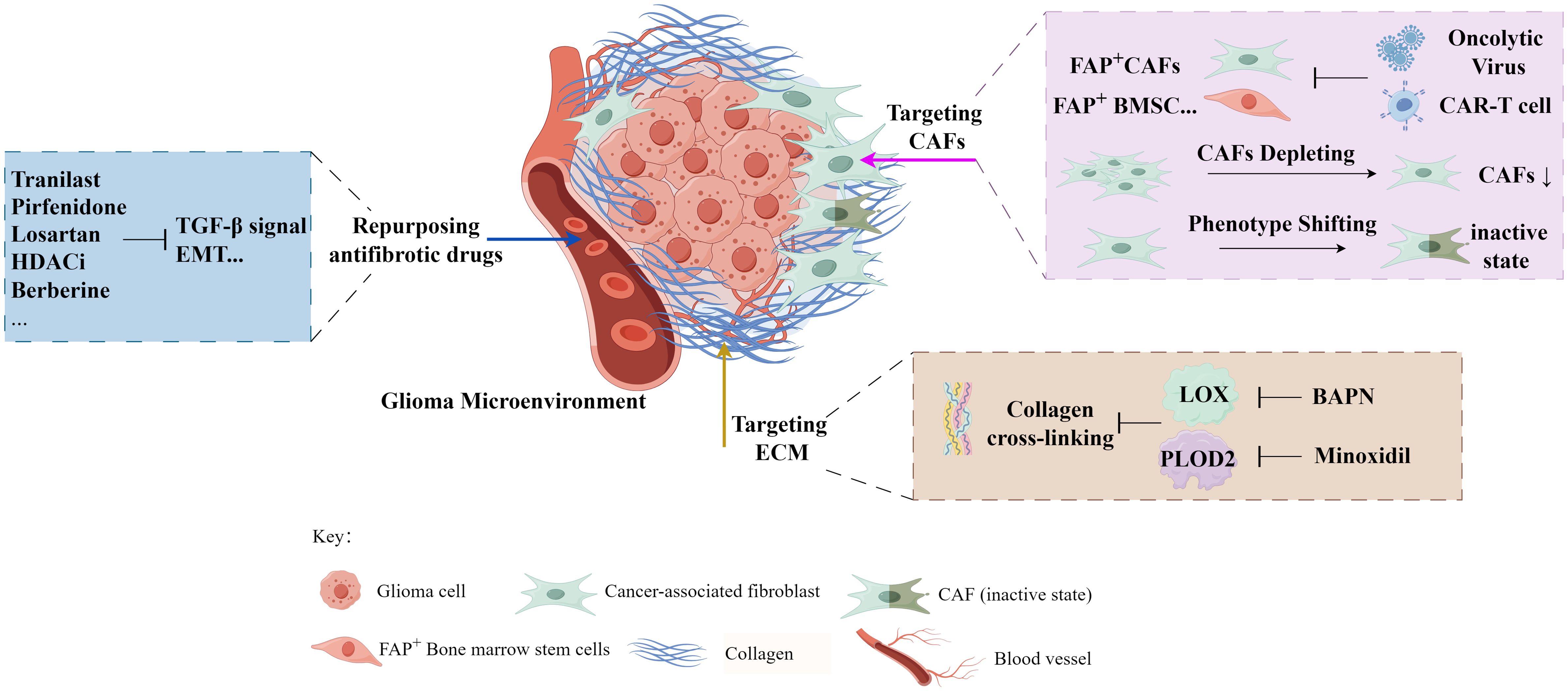
Figure 3 The exploration of antifibrosis-related strategies in glioma treatment (by Figdraw).
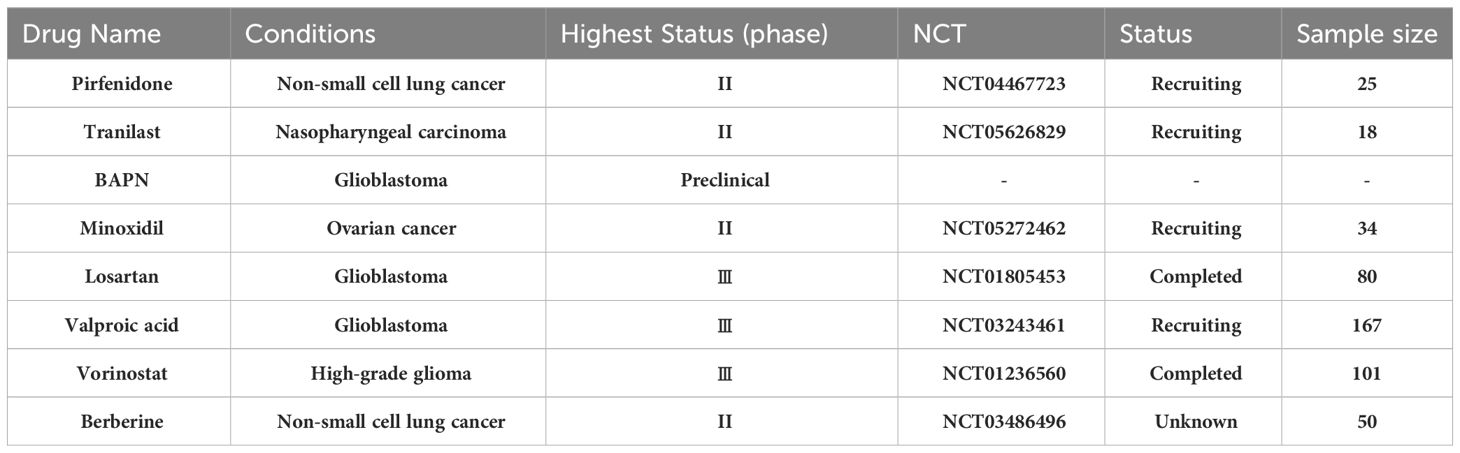
Table 1 Drugs with antifibrosis function are utilized and tested in the treatment of solid tumors including glioblastoma.
Collectively, the mechanisms associated with glioma cells chemoresistance development can be attributed to two aspects: chemoresistance-related genetic alterations within glioma cells, and the GME changes contributing to drug resistance. The latter is, in part, induced and modulated by glioma-associated fibrosis, leading to increased tumor stiffness and decreased efficiency of chemotherapeutics delivery to the cancer cell nuclei. The features of the fibrotic GME include the abnormal vascular system, heightened ECM deposition, increased tumor stiffness, upregulated growth factors, etc. We further emphasize the crucial role of glioma-associated fibrotic reactions in glioma progression, prognosis, and chemoresistance. Intense glioma-associated fibrotic reactions positively correlate with poor outcomes in glioma patients, suggesting its clinical significance as both a prognostic indicator and a promising therapeutic target for overcoming glioma chemoresistance. Additionally, we propose a theory that chemotherapy-induced activation of TGF-β signaling could lead to tumor-associated fibrotic reactions in the GME, characterized by increased ECM stiffness. This, in turn, may hinder the penetration of chemotherapeutics into glioma cells. In this review, we emphasize that tumor-associated fibrotic reactions play a role in maintaining glioma stemness, leading to the acquisition of a chemoresistant phenotype (Figure 4). A comprehensive understanding of this mechanism promises new insights into effectively reversing chemoresistance. This review underscores the urgent need to decipher the complex relationship between glioma-associated fibrosis and chemotherapy sensitivity, providing a promising strategy to develop more effective interventions for glioma.
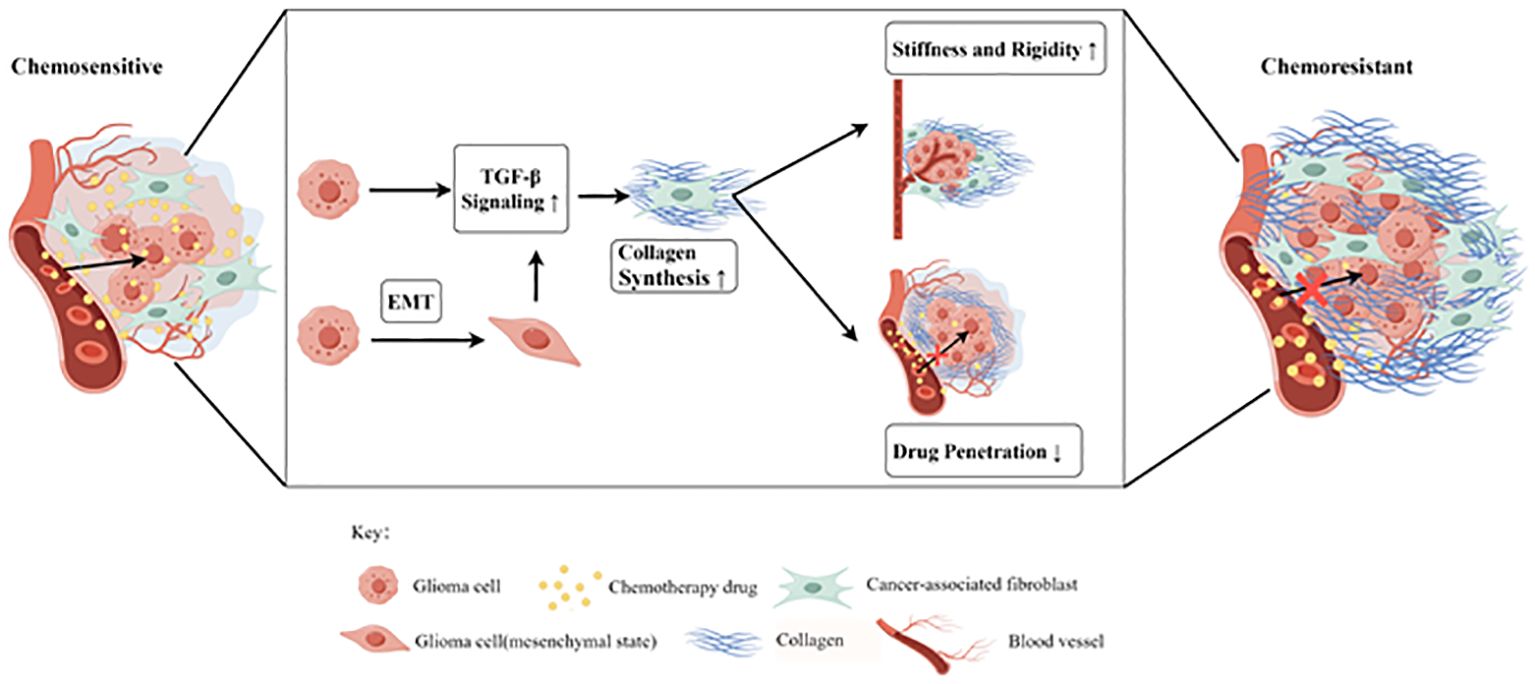
Figure 4 The tumor-associated fibrosis aggravates glioma chemoresistance by reducing the efficacy of drug delivery (by Figdraw). The TGF-β signaling pathway could be upregulated in glioma after chemotherapy and epithelial-mesenchymal transition (EMT), resulting in the increased collagen synthesis of cancer-associated fibroblasts (CAFs). In the tumor microenvironment (TME), the tumor-associated fibrosis increased the stiffness and rigidity of glioma tissues which in turn impairs the delivery of chemotherapeutic drugs to cancer cells, thus promoting chemoresistance.
Despite our extensive summarization of numerous studies on how tumor-associated fibrosis facilitates chemoresistance, the exact molecular mechanisms still remain elusive in glioma. Therefore, in the future, it’s warranted to explore which molecular characteristics of glioma are more likely to develop fibrosis, and whether the ECM stiffness promotes the expression of chemoresistance-related proteins in glioma. Such insights would contribute to a deeper understanding of the interactions among these various chemoresistance mechanisms, potentially unveiling novel strategies to overcome chemoresistance. In addition to therapeutic agents directly targeting cancer cells, several innovative drugs are under investigation for their potential to overcome chemoresistance through modulating the TME. The antifibrosis therapy for solid tumors is one of the TME normalization strategies, with some showing significant tumor inhibition effects. Numerous studies have suggested that targeting CAFs and fibrosis with conventional clinically approved agents can enhance the chemosensitivity of solid tumors. However, further in-depth research is required to determine their efficacy specifically in the context of glioma treatment.
JX: Writing – review & editing, Writing – original draft. JZ: Writing – review & editing. WC: Visualization, Writing – review & editing. XN: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was funded by President Foundation of Zhujiang Hospital, Southern Medical University (yzjj2022qn06).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. (2005) 352:987–96. doi: 10.1056/NEJMoa043330
2. Menna G, Mattogno PP, Donzelli CM, Lisi L, Olivi A, Della Pepa GM. Glioma-associated microglia characterization in the glioblastoma microenvironment through a ‘Seed-and soil’ Approach: a systematic review. Brain Sci. (2022) 12(6):718. doi: 10.3390/brainsci12060718
3. Stupp R, Hegi ME, Mason WP, van den Bent MJ, Taphoorn MJ, Janzer RC, et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. (2009) 10:459–66. doi: 10.1016/S1470-2045(09)70025-7
4. Ou A, Yung WKA, Majd N. Molecular mechanisms of treatment resistance in glioblastoma. Int J Mol Sci. (2020) 22(1):351. doi: 10.3390/ijms22010351
5. Zhang G, Tao X, Ji B, Gong J. Hypoxia-driven M2-polarized macrophages facilitate cancer aggressiveness and temozolomide resistance in glioblastoma. Oxid Med Cell Longev. (2022) 2022:1614336. doi: 10.1155/2022/1614336
6. Fidoamore A, Cristiano L, Antonosante A, d’Angelo M, Di Giacomo E, Astarita C, et al. Glioblastoma stem cells microenvironment: the paracrine roles of the niche in drug and radioresistance. Stem Cells Int. (2016) 2016:6809105. doi: 10.1155/2016/6809105
7. Kenig S, Faoro V, Bourkoula E, Podergajs N, Ius T, Vindigni M, et al. Topoisomerase IIβ mediates the resistance of glioblastoma stem cells to replication stress-inducing drugs. Cancer Cell Int. (2016) 16:58. doi: 10.1186/s12935-016-0339-9
8. Li J, Kaneda MM, Ma J, Li M, Shepard RM, Patel K, et al. PI3Kγ inhibition suppresses microglia/TAM accumulation in glioblastoma microenvironment to promote exceptional temozolomide response. Proc Natl Acad Sci U.S.A. (2021) 118(16):e2009290118. doi: 10.1073/pnas.2009290118
9. Li I, Nabet BY. Exosomes in the tumor microenvironment as mediators of cancer therapy resistance. Mol Cancer. (2019) 18:32. doi: 10.1186/s12943-019-0975-5
10. Caponnetto F, Dalla E, Mangoni D, Piazza S, Radovic S, Ius T, et al. he miRNA content of exosomes released from the glioma microenvironment can affect Malignant progression. Biomedicines. (2020) 8(12):564. doi: 10.3390/biomedicines8120564
11. Chen P, Zhao D, Li J, Liang X, Li J, Chang A, et al. Symbiotic macrophage-glioma cell interactions reveal synthetic lethality in PTEN-null glioma. Cancer Cell. (2019) 35:868–84.e6. doi: 10.1016/j.ccell.2019.05.003
12. Mentlein R, Hattermann K, Hemion C, Jungbluth AA, Held-Feindt J. Expression and role of the cell surface protease seprase/fibroblast activation protein-α (FAP-α) in astroglial tumors. Biol Chem. (2011) 392:199–207. doi: 10.1515/bc.2010.119
13. Ye XZ, Xu SL, Xin YH, Yu SC, Ping YF, Chen L, et al. Tumor-associated microglia/macrophages enhance the invasion of glioma stem-like cells via TGF-β1 signaling pathway. J Immunol. (2012) 189:444–53. doi: 10.4049/jimmunol.1103248
14. Lakins MA, Ghorani E, Munir H, Martins CP, Shields JD. Cancer-associated fibroblasts induce antigen-specific deletion of CD8 (+) T Cells to protect tumour cells. Nat Commun. (2018) 9:948. doi: 10.1038/s41467-018-03347-0
15. Xu C, Zhao Y, Wu C, Li L. Repurposing drugs for the treatment of glioma. Glioma.(2019) 2:159–64. doi: 10.4103/glioma.glioma_26_19
17. Yuan Z, Li Y, Zhang S, Wang X, Dou H, Yu X, et al. Extracellular matrix remodeling in tumor progression and immune escape: from mechanisms to treatments. Mol Cancer. (2023) 22:48. doi: 10.1186/s12943-023-01744-8
18. Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. (2014) 15:786–801. doi: 10.1038/nrm3904
19. Winkler J, Abisoye-Ogunniyan A, Metcalf KJ, Werb Z. Concepts of extracellular matrix remodelling in tumour progression and metastasis. Nat Commun. (2020) 11:5120. doi: 10.1038/s41467-020-18794-x
20. Mohan V, Das A, Sagi I. Emerging roles of ECM remodeling processes in cancer. Semin Cancer Biol. (2020) 62:192–200. doi: 10.1016/j.semcancer.2019.09.004
21. Wu JS, Sheng SR, Liang XH, Tang YL. The role of tumor microenvironment in collective tumor cell invasion. Future Oncol. (2017) 13:991–1002. doi: 10.2217/fon-2016-0501
22. Chen X, Song E. The theory of tumor ecosystem. Cancer Commun (Lond). (2022) 42:587–608. doi: 10.1002/cac2.12316
23. Chen Z, Zhuo S, He G, Tang J, Hao W, Gao WQ, et al. Prognosis and immunotherapy significances of a cancer-associated fibroblasts-related gene signature in gliomas. Front Cell Dev Biol. (2021) 9:721897. doi: 10.3389/fcell.2021.721897
24. Oleynikova NA, Danilova NV, Mikhailov IA, Semina EV, Malkov PG. [Cancer-associated fibroblasts and their significance in tumor progression]. Arkh Patol. (2020) 82:68–77. doi: 10.17116/patol20208201168
25. Su S, Chen J, Yao H, Liu J, Yu S, Lao L, et al. CD10(+)GPR77(+) cancer-associated fibroblasts promote cancer formation and chemoresistance by sustaining cancer stemness. Cell.(2018) 172(4):841–56.e16. doi: 10.1016/j.cell.2018.01.009
26. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell.(2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
27. Lambrechts D, Wauters E, Boeckx B, Aibar S, Nittner D, Burton O, et al. Phenotype molding of stromal cells in the lung tumor microenvironment. Nat Med. (2018) 24:1277–89. doi: 10.1038/s41591-018-0096-5
28. Santi A, Kugeratski FG, Zanivan S. Cancer associated fibroblasts: the architects of stroma remodeling. Proteomics. (2018) 18:e1700167. doi: 10.1002/pmic.201700167
29. Yeldag G, Rice A, Del Río Hernández A. Chemoresistance and the self-maintaining tumor microenvironment. Cancers (Basel). (2018) 10(12):471. doi: 10.3390/cancers10120471
30. Xu S, Xu H, Wang W, Li S, Li H, Li T, et al. The role of collagen in cancer: from bench to bedside. J Transl Med. (2019) 17:309. doi: 10.1186/s12967-019-2058-1
31. Ricard-Blum S. The collagen family. Cold Spring Harb Perspect Biol. (2011) 3:a004978. doi: 10.1101/cshperspect.a004978
32. Ohlund D, Lundin C, Ardnor B, Oman M, Naredi P, Sund M. Type IV collagen is a tumour stroma-derived biomarker for pancreas cancer. Br J Cancer. (2009) 101:91–7. doi: 10.1038/sj.bjc.6605107
33. Kast RE, Skuli N, Karpel-Massler G, Frosina G, Ryken T, Halatsch ME. Blocking epithelial-to-mesenchymal transition in glioblastoma with a sextet of repurposed drugs: the EIS regimen. Oncotarget.(2017) 8:60727–49. doi: 10.18632/oncotarget.v8i37
34. Mammoto T, Jiang A, Jiang E, Panigrahy D, Kieran MW, Mammoto A. Role of collagen matrix in tumor angiogenesis and glioblastoma multiforme progression. Am J Pathol. (2013) 183:1293–305. doi: 10.1016/j.ajpath.2013.06.026
35. Jia W, Jiang X, Liu W, Wang L, Zhu B, Zhu H, et al. Effects of three-dimensional collagen scaffolds on the expression profiles and biological functions of glioma cells. Int J Oncol. (2018) 52:1787–800doi: 10.3892/ijo
36. Qiu S, Deng L, Liao X, Nie L, Qi F, Jin K, et al. Tumor-associated macrophages promote bladder tumor growth through PI3K/AKT signal induced by collagen. Cancer Sci. (2019) 110:2110–8. doi: 10.1111/cas.14078
37. Calori IR, Alves SR, Bi H. Type-I collagen/collagenase modulates the 3D structure and behavior of glioblastoma spheroid. Models.(2022) 5:723–33. doi: 10.1021/acsabm.1c01138
38. Huijbers IJ, Iravani M, Popov S, Robertson D, Al-Sarraj S, Jones C, et al. A role for fibrillar collagen deposition and the collagen internalization receptor endo180 in glioma invasion. PloS One. (2010) 5:e9808. doi: 10.1371/journal.pone.0009808
39. Phillips HS, Kharbanda S, Chen R, Forrest WF, Soriano RH, Wu TD, et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell. (2006) 9:157–73. doi: 10.1016/j.ccr.2006.02.019
40. Wienke D, Davies GC, Johnson DA, Sturge J, Lambros MB, Savage K, et al. The collagen receptor Endo180 (CD280) Is expressed on basal-like breast tumor cells and promotes tumor growth in vivo. Cancer Res. (2007) 67:10230–40. doi: 10.1158/0008-5472.CAN-06-3496
41. Grässel S, Bauer RJ. Collagen XVI in health and disease. Matrix Biol. (2013) 32:64–73. doi: 10.1016/j.matbio.2012.11.001
42. Bauer R, Ratzinger S, Wales L, Bosserhoff A, Senner V, Grifka J, et al. Inhibition of collagen XVI expression reduces glioma cell invasiveness. Cell Physiol Biochem. (2011) 27:217–26. doi: 10.1159/000327947
43. Chan TS, Shaked Y, Tsai KK. Targeting the interplay between cancer fibroblasts, mesenchymal stem cells, and cancer stem cells in desmoplastic cancers. Front Oncol. (2019) 9:688doi: 10.3389/fonc.2019.00688
44. Yu Q, Xue Y, Liu J, Xi Z, Li Z, Liu Y. Fibronectin promotes the Malignancy of glioma stem-like cells via modulation of cell adhesion, differentiation, proliferation and chemoresistance. Front Mol Neurosci. (2018) 11:130. doi: 10.3389/fnmol.2018.00130
45. Staberg M, Rasmussen RD, Michaelsen SR, Pedersen H, Jensen KE, Villingshøj M, et al. Targeting glioma stem-like cell survival and chemoresistance through inhibition of lysine-specific histone demethylase KDM2B. Mol Oncol. (2018) 12:406–20. doi: 10.1002/1878-0261.12174
46. Natsume A, Ishii D, Wakabayashi T, Tsuno T, Hatano H, Mizuno M, et al. IFN-beta down-regulates the expression of DNA repair gene MGMT and sensitizes resistant glioma cells to temozolomide. Cancer Res. (2005) 65:7573–9. doi: 10.1158/0008-5472.CAN-05-0036
47. Shen D, Guo CC, Wang J, Qiu ZK, Sai K, Yang QY, et al. Interferon-α/β enhances temozolomide activity against MGMT-positive glioma stem-like cells. Oncol Rep. (2015) 34:2715–21. doi: 10.3892/or.2015.4232
48. Schiffer D, Annovazzi L, Casalone C, Corona C. Glioblastoma: microenvironment and niche concept. Cancers (Basel). (2018) 11(1):5. doi: 10.3390/cancers11010005
49. Scanlan MJ, Raj BK, Calvo B, Garin-Chesa P, Sanz-Moncasi MP, Healey JH, et al. Molecular cloning of fibroblast activation protein alpha, a member of the serine protease family selectively expressed in stromal fibroblasts of epithelial cancers. Proc Natl Acad Sci U S A. (1994) 91:5657–61. doi: 10.1073/pnas.91.12.5657
50. Juillerat-Jeanneret L, Tafelmeyer P, Golshayan D. Fibroblast activation protein-α in fibrogenic disorders and cancer: more than a prolyl-specific peptidase? Expert Opin Ther Targets. (2017) 21:977–91doi: 10.1080/14728222.2017.1370455
51. Li M, Li M, Yin T, Shi H, Wen Y, Zhang B, et al. Targeting of cancer−associated fibroblasts enhances the efficacy of cancer chemotherapy by regulating the tumor microenvironment. Mol Med Rep. (2016) 13:2476–84. doi: 10.3892/mmr.2016.4868
52. Ma Q, Long W, Xing C, Chu J, Luo M, Wang HY, et al. Cancer stem cells and immunosuppressive microenvironment in glioma. Front Immunol. (2018) 9:2924. doi: 10.3389/fimmu.2018.02924
53. Han J, Alvarez-Breckenridge CA, Wang QE, Yu J. TGF-β signaling and its targeting for glioma treatment. Am J Cancer Res. (2015) 5(3):945–55.
54. Zeng H, Yang Z, Xu N, Liu B, Fu Z, Lian C, et al. Connective tissue growth factor promotes temozolomide resistance in glioblastoma through TGF-β1-dependent activation of Smad/ERK signaling. Cell Death Dis. (2017) 8:e2885. doi: 10.1038/cddis.2017.248
55. Theocharis AD, Skandalis SS, Gialeli C, Karamanos NK. Extracellular matrix structure. Adv Drug Delivery Rev. (2016) 97:4–27. doi: 10.1016/j.addr.2015.11.001
56. Stylianopoulos T, Poh MZ, Insin N, Bawendi MG, Fukumura D, Munn LL, et al. Diffusion of particles in the extracellular matrix: the effect of repulsive electrostatic interactions. Biophys J. (2010) 99:1342–9. doi: 10.1016/j.bpj.2010.06.016
57. Tannock IF, Rotin D. Acid pH in tumors and its potential for therapeutic exploitation. Cancer Res. (1989) 49:4373–84.
58. Mueller S, Sandrin L. Liver stiffness: a novel parameter for the diagnosis of liver disease. Hepat Med. (2010) 2:49–67. doi: 10.2147/HMER
59. Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell.(2009) 139:891–906. doi: 10.1016/j.cell.2009.10.027
60. Lu P, Weaver VM, Werb Z. The extracellular matrix: a dynamic niche in cancer progression. J Cell Biol. (2012) 196:395–406. doi: 10.1083/jcb.201102147
61. Haining AWM, Rahikainen R, Cortes E, Lachowski D, Rice A, von Essen M, et al. Mechanotransduction in talin through the interaction of the R8 domain with DLC1. PloS Biol. (2018) 16:e2005599. doi: 10.1371/journal.pbio.2005599
62. Lachowski D, Cortes E, Robinson B, Rice A, Rombouts K, Del Río Hernández AE. FAK controls the mechanical activation of YAP, a transcriptional regulator required for durotaxis. FASEB J. (2018) 32:1099–107. doi: 10.1096/fj.201700721R
63. Papageorgis P, Polydorou C, Mpekris F, Voutouri C, Agathokleous E, Kapnissi-Christodoulou CP, et al. Tranilast-induced stress alleviation in solid tumors improves the efficacy of chemo- and nanotherapeutics in a size-independent manner. Sci Rep. (2017) 7:46140. doi: 10.1038/srep46140
64. Naik A, Leask A. Tumor-associated fibrosis impairs the response to immunotherapy. Matrix Biol. (2023) 119:125–40doi: 10.1016/j.matbio.2023.04.002
65. Mascheroni P, López Alfonso JC, Kalli M, Stylianopoulos T, Meyer-Hermann M, Hatzikirou H. On the impact of chemo-mechanically induced phenotypic transitions in gliomas. Cancers (Basel). (2019) 11(5):716. doi: 10.3390/cancers11050716
66. Stylianopoulos T, Martin JD, Chauhan VP, Jain SR, Diop-Frimpong B, Bardeesy N, et al. Causes, consequences, and remedies for growth-induced solid stress in murine and human tumors. Proc Natl Acad Sci U S A. (2012) 109:15101–8. doi: 10.1073/pnas.1213353109
67. Tajaldini M, Poorkhani A, Amiriani T, Amiriani A, Javid H, Aref P, et al. Strategy of targeting the tumor microenvironment via inhibition of fibroblast/fibrosis remodeling new era to cancer chemo-immunotherapy resistance. Eur J Pharmacol. (2023) 957:175991. doi: 10.1016/j.ejphar.2023.175991
68. Hauge A, Rofstad EK. Antifibrotic therapy to normalize the tumor microenvironment. J Transl Med. (2020) 18:207. doi: 10.1186/s12967-020-02376-y
69. Zhang B, Jiang T, Shen S, She X, Tuo Y, Hu Y, et al. Cyclopamine disrupts tumor extracellular matrix and improves the distribution and efficacy of nanotherapeutics in pancreatic cancer. Biomaterials.(2016) 103:12–21. doi: 10.1016/j.biomaterials.2016.06.048
70. Chauhan VP, Martin JD, Liu H, Lacorre DA, Jain SR, Kozin SV, et al. Angiotensin inhibition enhances drug delivery and potentiates chemotherapy by decompressing tumour blood vessels. Nat Commun. (2013) 4:2516. doi: 10.1038/ncomms3516
71. Polydorou C, Mpekris F, Papageorgis P, Voutouri C, Stylianopoulos T. Pirfenidone normalizes the tumor microenvironment to improve chemotherapy. Oncotarget.(2017) 8:24506–17. doi: 10.18632/oncotarget.v8i15
72. Guo J, Zeng H, Chen Y. Emerging nano drug delivery systems targeting cancer-associated fibroblasts for improved antitumor effect and tumor drug penetration. Mol Pharm. (2020) 17:1028–48. doi: 10.1021/acs.molpharmaceut.0c00014
73. Miao L, Liu Q, Lin CM, Luo C, Wang Y, Liu L, et al. Targeting tumor-associated fibroblasts for therapeutic delivery in desmoplastic tumors. Cancer Res. (2017) 77:719–31. doi: 10.1158/0008-5472.CAN-16-0866
74. Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discovery. (2019) 18:99–115. doi: 10.1038/s41573-018-0004-1
75. Moreira RK. Hepatic stellate cells and liver fibrosis. Arch Pathol Lab Med. (2007) 131:1728–34. doi: 10.5858/2007-131-1728-HSCALF
76. Zhou W, Yu X, Sun S, Zhang X, Yang W, Zhang J, et al. Increased expression of MMP-2 and MMP-9 indicates poor prognosis in glioma recurrence. BioMed Pharmacother. (2019) 118:109369. doi: 10.1016/j.biopha.2019.109369
77. Sun ZF, Wang L, Gu F, Fu L, Li WL, Ma YJ. [Expression of Notch1, MMP-2 and MMP-9 and their significance in glioma patients]. Zhonghua Zhong Liu Za Zhi. (2012) 34:26–30.
78. Sun C, Wang Q, Zhou H, Yu S, Simard AR, Kang C, et al. Antisense MMP-9 RNA inhibits Malignant glioma cell growth in vitro and in vivo. Neurosci Bull. (2013) 29:83–93. doi: 10.1007/s12264-012-1296-5
79. Lakka SS, Jasti SL, Gondi C, Boyd D, Chandrasekar N, Dinh DH, et al. Downregulation of MMP-9 in ERK-mutated stable transfectants inhibits glioma invasion in vitro. Oncogene. (2002) 21:5601–8. doi: 10.1038/sj.onc.1205646
80. Lee EJ, Kim SY, Hyun JW, Min SW, Kim DH, Kim HS. Glycitein inhibits glioma cell invasion through down-regulation of MMP-3 and MMP-9 gene expression. Chem Biol Interact. (2010) 185:18–24. doi: 10.1016/j.cbi.2010.02.037
81. Yeh WL, Lu DY, Lee MJ, Fu WM. Leptin induces migration and invasion of glioma cells through MMP-13 production. Glia. (2009) 57:454–64. doi: 10.1002/glia.20773
82. Wang L, Yuan J, Tu Y, Mao X, He S, Fu G, et al. Co-expression of MMP-14 and MMP-19 predicts poor survival in human glioma. Clin Transl Oncol. (2013) 15:139–45. doi: 10.1007/s12094-012-0900-5
83. Guo JG, Guo CC, He ZQ, Cai XY, Mou YG. High MMP-26 expression in glioma is correlated with poor clinical outcome of patients. Oncol Lett. (2018) 16:2237–42. doi: 10.3892/ol
84. Wang X, Chen X, Sun L, Bi X, He H, Chen L, et al. The function of MMP-28/TGF-β induced cell apoptosis in human glioma cells. Exp Ther Med. (2018) 16:2867–74. doi: 10.3892/etm
85. Li L, Du Y, Xiang D, Chen L, Shi Z, Tian J, et al. Prediction of the anti-glioma therapeutic effects of temozolomide through in vivo molecular imaging of MMP expression. BioMed Opt Express. (2018) 9:3193–207. doi: 10.1364/BOE.9.003193
86. Heerboth S, Housman G, Leary M, Longacre M, Byler S, Lapinska K, et al. EMT and tumor metastasis. Clin Transl Med. (2015) 4:6. doi: 10.1186/s40169-015-0048-3
87. Hanahan D, Coussens LM. Accessories to the crime: functions of cells recruited to the tumor microenvironment. Cancer Cell. (2012) 21:309–22. doi: 10.1016/j.ccr.2012.02.022
88. Costanza B, Umelo IA, Bellier J, Castronovo V, Turtoi A. Stromal modulators of TGF-β in cancer. J Clin Med. (2017) 6(1):7. doi: 10.3390/jcm6010007
89. Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell.(2009) 137:87–98. doi: 10.1016/j.cell.2009.01.039
90. Leight JL, Wozniak MA, Chen S, Lynch ML, Chen CS. Matrix rigidity regulates a switch between TGF-β1-induced apoptosis and epithelial-mesenchymal transition. Mol Biol Cell. (2012) 23:781–91. doi: 10.1091/mbc.e11-06-0537
91. Erdogan B, Webb DJ. Cancer-associated fibroblasts modulate growth factor signaling and extracellular matrix remodeling to regulate tumor metastasis. Biochem Soc Trans. (2017) 45:229–36. doi: 10.1042/BST20160387
92. Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim CK, Tobias A, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4, and PD-L1 in mice with brain tumors. Clin Cancer Res. (2014) 20:5290–301. doi: 10.1158/1078-0432.CCR-14-0514
93. Rao R, Han R, Ogurek S, Xue C, Wu LM, Zhang L, et al. Glioblastoma genetic drivers dictate the function of tumor-associated macrophages/microglia and responses to CSF1R inhibition. Neuro Oncol. (2022) 24:584–97. doi: 10.1093/neuonc/noab228
94. Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, et al. The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science. (2016) 352:aad3018doi: 10.1126/science.aad3018
95. Yang F, He Z, Duan H, Zhang D, Li J, Yang H, et al. Synergistic immunotherapy of glioblastoma by dual targeting of IL-6 and CD40. Nat Commun. (2021) 12:3424. doi: 10.1038/s41467-021-23832-3
96. Pan Y, Smithson LJ, Ma Y, Hambardzumyan D, Gutmann DH. Ccl5 establishes an autocrine high-grade glioma growth regulatory circuit critical for mesenchymal glioblastoma survival. Oncotarget. (2017) 8:32977–89. doi: 10.18632/oncotarget.v8i20
97. Zhang XN, Yang KD, Chen C, He ZC, Wang QH, Feng H, et al. Pericytes augment glioblastoma cell resistance to temozolomide through CCL5-CCR5 paracrine signaling. Cell Res. (2021) 31(10):1072–87. doi: 10.1038/s41422-021-00528-3
98. Gilkes DM, Semenza GL, Wirtz D. Hypoxia and the extracellular matrix: drivers of tumour metastasis. Nat Rev Cancer. (2014) 14:430–9. doi: 10.1038/nrc3726
99. Grout JA, Sirven P, Leader AM, Maskey S, Hector E, Puisieux I, et al. Spatial positioning and matrix programs of cancer-associated fibroblasts promote T-cell exclusion in human lung tumors. Cancer Discov. (2022) 12(11):2606–25. doi: 10.1158/2159-8290.CD-21-1714
100. Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. (2010) 17:98–110. doi: 10.1016/j.ccr.2009.12.020
101. Jia D, Li S, Li D, Xue H, Yang D, Liu Y. Mining TCGA database for genes of prognostic value in glioblastoma microenvironment. Aging (Albany NY). (2018) 10:592–605. doi: 10.18632/aging.v10i4
102. Buechler MB, Fu W, Turley SJ. Fibroblast-macrophage reciprocal interactions in health, fibrosis, and cancer. Immunity.(2021) 54:903–15. doi: 10.1016/j.immuni.2021.04.021
103. Ni X, Wu W, Sun X, Ma J, Yu J, He X, et al. Interrogating glioma-M2 macrophage interactions identifies Gal-9/Tim-3 as a viable target against PTEN-null glioblastoma. Sci Adv. (2022) 8(27):eabl5165. doi: 10.1126/sciadv.abl5165
104. Schafer S, Viswanathan S, Widjaja AA, Lim WW, Moreno-Moral A, DeLaughter DM, et al. IL-11 is a crucial determinant of cardiovascular fibrosis. Nature.(2017) 552:110–5. doi: 10.1038/nature24676
105. Tong X, Zhang S, Wang D, Zhang L, Huang J, Zhang T, et al. Azithromycin attenuates bleomycin-induced pulmonary fibrosis partly by inhibiting the expression of LOX and LOXL-2. Front Pharmacol. (2021) 12:709819. doi: 10.3389/fphar.2021.709819
106. Piersma B, Hayward MK, Weaver VM. Fibrosis and cancer: A strained relationship. Biochim Biophys Acta Rev Cancer. (2020) 1873:188356. doi: 10.1016/j.bbcan.2020.188356
107. Laczko R, Szauter KM, Jansen MK, Hollosi P, Muranyi M, Molnar J, et al. Active lysyl oxidase (LOX) correlates with focal adhesion kinase (FAK)/paxillin activation and migration in invasive astrocytes. Neuropathol Appl Neurobiol. (2007) 33:631–43. doi: 10.1111/j.1365-2990.2007.00858.x
108. Xu Y, Zhang L, Wei Y, Zhang X, Xu R, Han M, et al. Procollagen-lysine 2-oxoglutarate 5-dioxygenase 2 promotes hypoxia-induced glioma migration and invasion. Oncotarget. (2017) 8:23401–13. doi: 10.18632/oncotarget.v8i14
109. Dong S, Nutt CL, Betensky RA, Stemmer-Rachamimov AO, Denko NC, Ligon KL, et al. Histology-based expression profiling yields novel prognostic markers in human glioblastoma. J Neuropathol Exp Neurol. (2005) 64:948–55. doi: 10.1097/01.jnen.0000186940.14779.90
110. Kreße N, Schröder H, Stein KP, Wilkens L, Mawrin C, Sandalcioglu IE, et al. PLOD2 is a prognostic marker in glioblastoma that modulates the immune microenvironment and tumor progression. Int J Mol Sci. (2022) 23(11):6037. doi: 10.3390/ijms23116037
111. Song Y, Zheng S, Wang J, Long H, Fang L, Wang G, et al. Hypoxia-induced PLOD2 promotes proliferation, migration and invasion via PI3K/Akt signaling in glioma. Oncotarget. (2017) 8:41947–62. doi: 10.18632/oncotarget.v8i26
112. Lan J, Zhang S, Zheng L, Long X, Chen J, Liu X, et al. PLOD2 promotes colorectal cancer progression by stabilizing USP15 to activate the AKT/mTOR signaling pathway. Cancer Sci. (2023) 114(8):3190–202. doi: 10.1111/cas.15851
113. Eisinger-Mathason TS, Zhang M, Qiu Q, Skuli N, Nakazawa MS, Karakasheva T, et al. Hypoxia-dependent modification of collagen networks promotes sarcoma metastasis. Cancer Discovery. (2013) 3:1190–205. doi: 10.1158/2159-8290.CD-13-0118
114. Zuurmond AM, van der Slot-Verhoeven AJ, van Dura EA, De Groot J, Bank RA. Minoxidil exerts different inhibitory effects on gene expression of lysyl hydroxylase 1, 2, and 3: implications for collagen cross-linking and treatment of fibrosis. Matrix Biol. (2005) 24:261–70. doi: 10.1016/j.matbio.2005.04.002
115. Ningaraj NS, Sankpal UT, Khaitan D, Meister EA, Vats T. Activation of KATP channels increases anticancer drug delivery to brain tumors and survival. Eur J Pharmacol. (2009) 602:188–93. doi: 10.1016/j.ejphar.2008.10.056
116. Gu YT, Xue YX, Wang YF, Wang JH, Chen X, ShangGuan QR, et al. Minoxidil sulfate induced the increase in blood-brain tumor barrier permeability through ROS/RhoA/PI3K/PKB signaling pathway. Neuropharmacology.(2013) 75:407–15. doi: 10.1016/j.neuropharm.2013.08.004
117. Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. (2011) 19:257–72. doi: 10.1016/j.ccr.2011.01.020
118. Shi X, Young CD, Zhou H, Wang X. Transforming growth factor-β Signaling in fibrotic diseases and cancer-associated fibroblasts. Biomolecules. (2020) 10(12):1666. doi: 10.3390/biom10121666
119. Gao L, Huang S, Zhang H, Hua W, Xin S, Cheng L, et al. Suppression of glioblastoma by a drug cocktail reprogramming tumor cells into neuronal like cells. Sci Rep. (2019) 9(1):3462. doi: 10.1038/s41598-019-39852-5
120. Khazaei M, Pazhouhi M, Khazaei S. Temozolomide and tranilast synergistic antiproliferative effect on human glioblastoma multiforme cell line (U87MG). Med J Islam Repub Iran. (2019) 33:39. doi: 10.47176/mjiri
121. Burghardt I, Tritschler F, Opitz CA, Frank B, Weller M, Wick W. Pirfenidone inhibits TGF-beta expression in Malignant glioma cells. Biochem Biophys Res Commun. (2007) 354:542–7. doi: 10.1016/j.bbrc.2007.01.012
122. Panza S, Malivindi R, Caruso A, Russo U, Giordano F, Győrffy B, et al. Novel Insights into the Antagonistic Effects of Losartan against Angiotensin II/AGTR1 Signaling in Glioblastoma Cells. Cancers (Basel). (2021) 13(18):4555. doi: 10.3390/cancers13184555
123. Datta M, Chatterjee S, Perez EM, Gritsch S, Roberge S, Duquette M, et al. Losartan controls immune checkpoint blocker-induced edema and improves survival in glioblastoma mouse models. Proc Natl Acad Sci USA. (2023) 120(6):e2219199120. doi: 10.1073/pnas.2219199120
124. Reddy RG, Bhat UA, Chakravarty S, Kumar A. Advances in histone deacetylase inhibitors in targeting glioblastoma stem cells. Cancer Chemother Pharmacol. (2020) 86(2):165–79. doi: 10.1007/s00280-020-04109-w
125. Han W, Guan W. Valproic acid: A promising therapeutic agent in glioma treatment. Front Oncol. (2021) 11:687362. doi: 10.3389/fonc.2021.687362
126. Costalonga EC, de Freitas LJ, Aragone D, Silva FMO, Noronha IL. Anti-fibrotic effects of valproic acid in experimental peritoneal fibrosis. PloS One. (2017) 12:e0184302. doi: 10.1371/journal.pone.0184302
127. Aher JS, Khan S, Jain S, Tikoo K, Jena G. Valproate ameliorates thioacetamide-induced fibrosis by hepatic stellate cell inactivation. Hum Exp Toxicol. (2015) 34:44–55. doi: 10.1177/0960327114531992
128. Van Beneden K, Geers C, Pauwels M, Mannaerts I, Verbeelen D, van Grunsven LA, et al. Valproic acid attenuates proteinuria and kidney injury. J Am Soc Nephrol. (2011) 22:1863–75. doi: 10.1681/ASN.2010111196
129. Kee HJ, Sohn IS, Nam KI, Park JE, Qian YR, Yin Z, et al. Inhibition of histone deacetylation blocks cardiac hypertrophy induced by angiotensin II infusion and aortic banding. Circulation.(2006) 113:51–9. doi: 10.1161/CIRCULATIONAHA.105.559724
130. Nakada M, Furuta T, Hayashi Y, Minamoto T, Hamada J. The strategy for enhancing temozolomide against Malignant glioma. Front Oncol. (2012) 2:98. doi: 10.3389/fonc.2012.00098
131. Fu J, Shao CJ, Chen FR, Ng HK, Chen ZP. Autophagy induced by valproic acid is associated with oxidative stress in glioma cell lines. Neuro Oncol. (2010) 12:328–40. doi: 10.1093/neuonc/nop005
132. Kerkhof M, Dielemans JC, van Breemen MS, Zwinkels H, Walchenbach R, Taphoorn MJ, et al. Effect of valproic acid on seizure control and on survival in patients with glioblastoma multiforme. Neuro Oncol. (2013) 15:961–7. doi: 10.1093/neuonc/not057
133. Ververis K, Hiong A, Karagiannis TC, Licciardi PV. Histone deacetylase inhibitors (HDACIs): multitargeted anticancer agents. Biologics. (2013) 7:47–60. doi: 10.2147/BTT
134. Io K, Nishino T, Obata Y, Kitamura M, Koji T, Kohno S. SAHA suppresses peritoneal fibrosis in mice. Perit Dial Int. (2015) 35:246–58. doi: 10.3747/pdi.2013.00089
135. Hsu CC, Chang WC, Hsu TI, Liu JJ, Yeh SH, Wang JY, et al. Suberoylanilide hydroxamic acid represses glioma stem-like cells. J BioMed Sci. (2016) 23:81. doi: 10.1186/s12929-016-0296-6
136. Chen J, Luo B, Wen S, Pi R. Discovery of a novel rhein-SAHA hybrid as a multi-targeted anti-glioblastoma drug. Invest New Drugs. (2020) 38:755–64doi: 10.1007/s10637-019-00821-4
137. Kumar A, Ekavali, Chopra K, Mukherjee M, Pottabathini R, Dhull DK. Current knowledge and pharmacological profile of berberine: An update. Eur J Pharmacol. (2015) 761:288–97. doi: 10.1016/j.ejphar.2015.05.068
138. Ni WJ, Ding HH, Zhou H, Qiu YY, Tang LQ. Renoprotective effects of berberine through regulation of the MMPs/TIMPs system in streptozocin-induced diabetic nephropathy in rats. Eur J Pharmacol. (2015) 764:448–56. doi: 10.1016/j.ejphar.2015.07.040
139. Chitra P, Saiprasad G, Manikandan R, Sudhandiran G. Berberine inhibits Smad and non-Smad signaling cascades and enhances autophagy against pulmonary fibrosis. J Mol Med (Berl). (2015) 93:1015–31doi: 10.1007/s00109-015-1283-1
140. Zhu L, Gu P, Shen H. Protective effects of berberine hydrochloride on DSS-induced ulcerative colitis in rats. Int Immunopharmacol. (2019) 68:242–51. doi: 10.1016/j.intimp.2018.12.036
141. Du H, Gu J, Peng Q, Wang X, Liu L, Shu X, et al. Berberine suppresses EMT in liver and gastric carcinoma cells through combination with TGFβR regulating TGF-β/smad pathway. Oxid Med Cell Longev. (2021) 2021:2337818. doi: 10.1155/2021/2337818
142. Sun Y, Huang H, Zhan Z, Gao H, Zhang C, Lai J, et al. Berberine inhibits glioma cell migration and invasion by suppressing TGF-β1/COL11A1 pathway. Biochem Biophys Res Commun. (2022) 625:38–45. doi: 10.1016/j.bbrc.2022.07.101
143. Nasiraee MR, Shahrivari S, Sayad S, Mahdavi H, Saraygord-Afshari N, Bagheri Z. An agarose-alginate microfluidic device for the study of spheroid invasion, ATRA inhibits CAFs-mediated matrix remodeling. Cytotechnology.(2023) 75:309–23. doi: 10.1007/s10616-023-00578-y
144. Shi Y, Kong Z, Liu P, Hou G, Wu J, Ma W, et al. Oncogenesis, microenvironment modulation and clinical potentiality of FAP in glioblastoma: lessons learned from other solid tumors. Cells. (2021) 10(5):1142. doi: 10.3390/cells10051142
145. Li M, Li G, Kiyokawa J, Tirmizi Z, Richardson LG, Ning J, et al. Characterization and oncolytic virus targeting of FAP-expressing tumor-associated pericytes in glioblastoma. Acta Neuropathol Commun. (2020) 8(1):221. doi: 10.1186/s40478-020-01096-0
Keywords: chemoresistance, tumor-associated fibrotic reaction, glioma, antifibrosis therapy, tumor microenvironment (TME), cancer-associate fibroblasts
Citation: Xu J, Zhang J, Chen W and Ni X (2024) The tumor-associated fibrotic reactions in microenvironment aggravate glioma chemoresistance. Front. Oncol. 14:1388700. doi: 10.3389/fonc.2024.1388700
Received: 20 February 2024; Accepted: 10 May 2024;
Published: 28 May 2024.
Edited by:
Ramcharan Singh Angom, Mayo Clinic Florida, United StatesReviewed by:
Alak Manna, Mayo Clinic Florida, United StatesCopyright © 2024 Xu, Zhang, Chen and Ni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangrong Ni, bnhyMTk5MzAyMTdAc211LmVkdS5jbg==; Wubing Chen, MTM5MjExMDEwOTBAMTYzLmNvbQ==; Ji Zhang, emhhbmdqaUBzeXN1Y2Mub3JnLmNu
†ORCID: Ji Zhang, orcid.org/0000-0002-1314-5589
Wubing Chen, orcid.org/0009-0006-9268-5124
Xiangrong Ni, orcid.org/0000-0002-4513-3152
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.