 Yue Wang1†Jianing Peng2†Dengyuan Yang1†Zhongjie Xing1
Yue Wang1†Jianing Peng2†Dengyuan Yang1†Zhongjie Xing1 Bo Jiang3Xu Ding1Chaoyu Jiang3Bing Ouyang1*
Bo Jiang3Xu Ding1Chaoyu Jiang3Bing Ouyang1* Lei Su3,4*
Lei Su3,4*- 1Department of Surgery, Nanjing Central Hospital, Nanjing, China
- 2Division of Biosciences, University College London, London, United Kingdom
- 3Department of General Surgery, Nanjing Drum Tower Hospital, Clinical College of Nanjing Medical University, Nanjing, China
- 4Department of General Surgery, Affiliated Drum Tower Hospital, Medical School of Nanjing University, Nanjing, China
PGC1α, a central player in mitochondrial biology, holds a complex role in the metabolic shifts seen in cancer cells. While its dysregulation is common across major cancers, its impact varies. In some cases, downregulation promotes aerobic glycolysis and progression, whereas in others, overexpression escalates respiration and aggression. PGC1α’s interactions with distinct signaling pathways and transcription factors further diversify its roles, often in a tissue-specific manner. Understanding these multifaceted functions could unlock innovative therapeutic strategies. However, challenges exist in managing the metabolic adaptability of cancer cells and refining PGC1α-targeted approaches. This review aims to collate and present the current knowledge on the expression patterns, regulators, binding partners, and roles of PGC1α in diverse cancers. We examined PGC1α’s tissue-specific functions and elucidated its dual nature as both a potential tumor suppressor and an oncogenic collaborator. In cancers where PGC1α is tumor-suppressive, reinstating its levels could halt cell proliferation and invasion, and make the cells more receptive to chemotherapy. In cancers where the opposite is true, halting PGC1α’s upregulation can be beneficial as it promotes oxidative phosphorylation, allows cancer cells to adapt to stress, and promotes a more aggressive cancer phenotype. Thus, to target PGC1α effectively, understanding its nuanced role in each cancer subtype is indispensable. This can pave the way for significant strides in the field of oncology.
Introduction
The peroxisome proliferator-activated receptor gamma coactivator 1-alpha (PGC1α) is a pivotal transcriptional coactivator with multifaceted roles in regulating cellular energy metabolism and mitochondrial biogenesis (1). Since its first identification as a binding partner of the nuclear receptor peroxisome proliferator-activated receptor gamma (PPARα) in 1998, the significance of PGC1α in orchestrating diverse metabolic pathways has become increasingly evident (2, 3). Its functions include mitochondrial biogenesis, fatty acid oxidation, gluconeogenesis, and oxidative phosphorylation (4).
The versatility of PGC1α is emphasized by its ability to interact with a multitude of transcription factors and coactivators (5, 6). This enables PGC1α to exert intricate control over the expression of genes relevant to these pathways. As the cornerstone of mitochondrial biogenesis (7), PGC1α can augment the number and activity of mitochondria. This amplification enhances cellular energy production and adaptability, ensuring a balance between energy homeostasis and the response to shifts in energy demands. Beyond its metabolic functions, PGC1α is pivotal in processes such as cell growth, differentiation, and survival (8), underscoring its role in preserving cellular integrity and function. The prominence of PGC1α in physiological processes implies its potential involvement in pathologies. Indeed, perturbations in PGC1α expression or activity have been linked to a spectrum of diseases, ranging from neurodegenerative disorders (9) and metabolic syndromes to cardiovascular diseases (10). Notably, emerging evidence suggests a role for PGC1α in cancer pathogenesis (11). Its dysregulation has been implicated in metabolic reprogramming and disease progression across a variety of malignancies, both solid and hematological (1).
Unfortunately, challenges exist in managing the metabolic adaptability of cancer cells and refining PGC1α-targeted approaches. There is a need to decode the intricate molecular ties of PGC1α’s interactions with cancer cells. This is a critical step in comprehending both PGC1α’s multifaceted involvement in cancer and the ability to use this knowledge for cancer prevention and treatment. Therefore, this review aims to collate and present the current knowledge on the expression patterns, regulators, binding partners, and roles of PGC1α in diverse cancers. We examined PGC1α’s tissue-specific functions and elucidated its dual nature as both a potential tumor suppressor and an oncogenic collaborator. A comprehensive understanding of PGC1α’s intricate relationship with cancer metabolism could pave the way for novel biomarker identification and therapeutic interventions against this pervasive global health challenge. We hope that this review will serve as a foundational guide for researchers interested in the further exploration of this domain.
Structure and functions of PGC1α
The PGC1α gene, located on chromosome 4 at the 4p15.1 position, features several functional domains crucial for its activity (3). These domains include the N-terminal transcriptional coactivatory domains, which facilitate interactions with various transcription factors; a central inhibitory domain that moderates its coactivatory functions; and an RNA recognition motif (RRM) located at the C-terminus. The N-terminal region is particularly significant for its role in engaging with nuclear receptors such as peroxisome proliferator-activated receptor gamma (PPARγ), nuclear respiratory factor 1 (NRF1), and estrogen-related receptor alpha (ERRα) (1, 12). These interactions are essential for the transcriptional regulation of genes that play a role in mitochondrial functionality and oxidative metabolism. Upon activation, PGC1α primarily functions by coactivating nuclear receptors and other transcription factors, thereby promoting the expression of genes related to energy metabolism (13). This gene is fundamental to the adaptation to changing metabolic demands during shifts in nutrient availability or energy requirements. Specifically, PGC1α enhances mitochondrial replication, respiratory capacity, and oxidative phosphorylation, which collectively increase cellular energy production (14). The regulation of its activity involves various post-translational modifications and protein interactions, allowing for a responsive adjustment to cellular energy conditions (15, 16). Through these multifaceted roles and complex regulatory mechanisms, PGC1α acts as a central regulator of metabolic processes, highlighting its importance in both normal physiology and various pathological conditions, including cancer.
Roles of PGC1α in cancer cells
PGC1α is an important gene that regulates metabolism in cancer cells, controlling pathways like glycolysis, tricarboxylic acid (TCA) cycle and fatty acid synthesis etc. But the interesting thing is its actual role differs a lot between different cancer types – sometimes it suppresses tumors, but other times it promotes cancer growth instead! This depends on the complex interplay between various intracellular signaling pathways. PGC1α closely interacts with molecules like β-catenin, AMP-activated protein kinase (AMPK), and can modulate downstream gene expression based on these upstream signals. So, it acts like a central integrator of signals that reprogram metabolism. Its expression and functions are super tissue-specific. For example, from the latest studies, PGC1α expression is upregulated in ovarian cancer (OC), colorectal cancer (CRC), gastric cancer (GC), nasopharyngeal (NPC) and cholangiocarcinoma (CCA), while downregulated in thyroid cancer (TC), liver cancer and renal cancer. However, in some types of cancers, like melanoma, prostate and breast cancer, low and high expressions of PGC1α are coexisted (Figure 1). PGC1α also reshapes the tumor microenvironment by coordinating metabolic crosstalk between cancer cells and immune cells. Targeting PGC1α could potentially help overcome therapeutic resistance, but overcoming metabolic plasticity of cancer cells remains a big challenge! Here, we elucidate the mechanisms of PGC1α from the perspective of different tumor cells in detail.
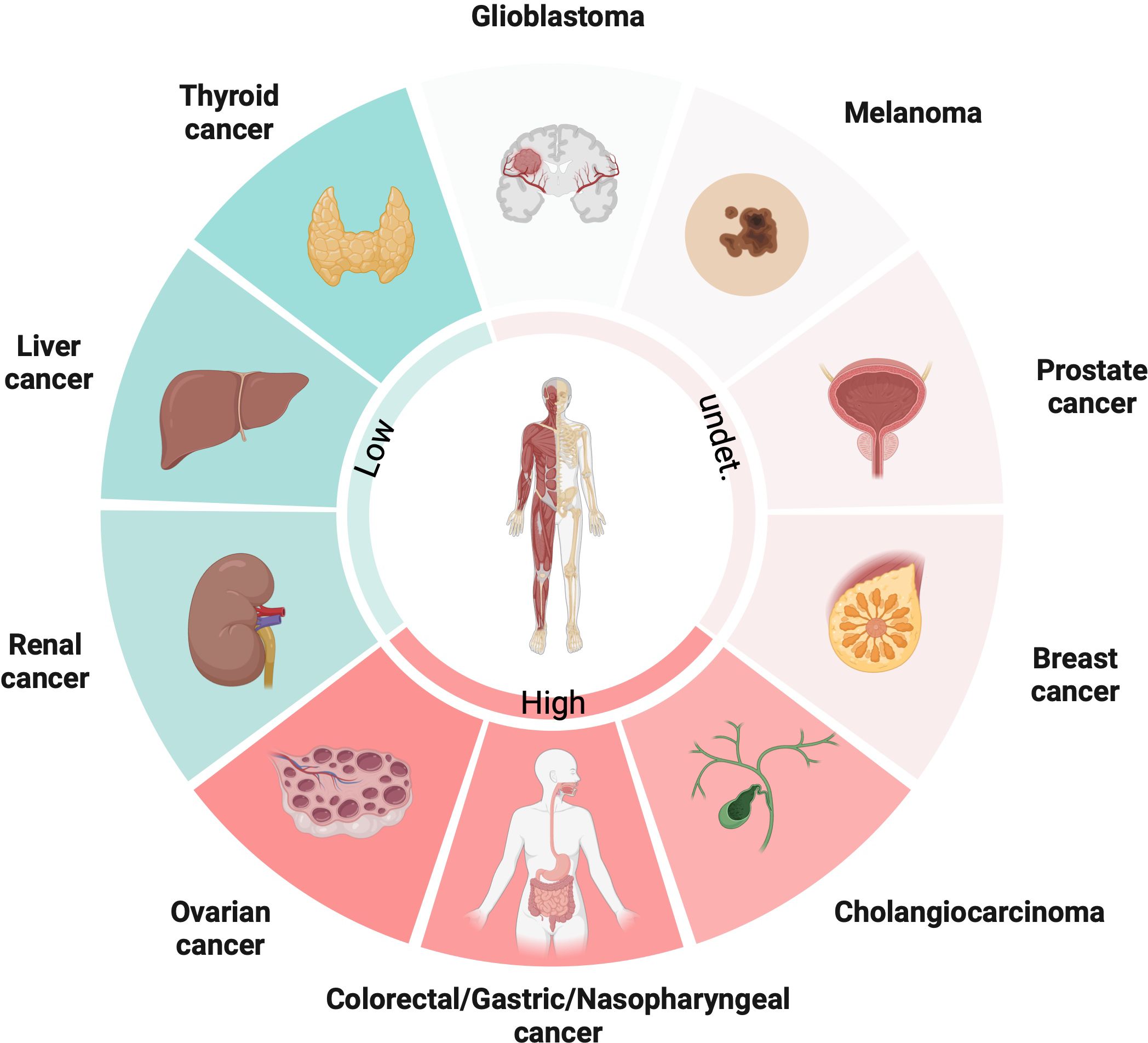
Figure 1 The expression patterns of PGC1α in diverse cancers.
PGC1α in thyroid cancer
The conventional perspective on metabolic changes observed in thyroid carcinomas is that they arise as a consequence of disease progression rather than as instigators themselves (17, 18). However, growing evidence suggests that these metabolic alterations also play regulatory roles in driving cancer progression. Studies have revealed the downregulation of PGC1α expression, particularly in the advanced stages of thyroid cancer (19) and notably in tumors harboring the BRAF V600E mutation (17), which is the most prevalent somatic oncogenic mutation in papillary thyroid carcinoma (20, 21). A comprehensive analysis on The Cancer Genome Atlas (TCGA) data from hundreds of patients with papillary thyroid carcinoma underscored the significance of PGC1α downregulation, as it correlated with a higher disease stage and an increased risk of recurrence (17). Multiple mechanisms appear to be involved in suppressing PGC1α expression in TC. The intricate AMPK signaling pathway is implicated in this regulatory process, whereby the activation of protein kinase B (AKT) leads to the suppression of PGC1α expression (17). Further, oxidative metabolism appears to inflict damage upon PGC1α mRNA, consequently dampening its expression (17). This forms a vicious feedforward loop as PGC1α loss exacerbates oxidative stress by curtailing mitochondria and antioxidant responses. Given multifaceted aspects of negative regulation, PGC1α is capable of playing important roles in TC development. Indeed, PGC1α deficiency damages mitochondrial function, elevates oxidative stress, and enhances glycolytic phenotype and disease progression (17).
PGC1α in colorectal cancer
PGC1α is frequently found to be overexpressed in CRC tissues and cell lines. It serves as a key energy mediator, and is induced by aerobic glycolysis (22) and hypoxia (23) in CRC cells. Interestingly, lactate metabolites generated from these processes contribute to the elevation of PGC1α mRNA levels in cancer cells (24, 25). Sirtuin3 (SIRT3), a principal mitochondrial deacetylase, has been implicated in modulating PGC1α levels. In metastatic CRC cell lines, the inhibition of SIRT3 using shRNAs has been found to lead to a decrease in both PGC1α mRNA and protein levels (26). Additionally, oncogenic phosphatase PRL3, a regulator of reactive oxygen species (ROS), has been found to exert its influence on upregulated PGC1α expression through the mediation of Ras-proximate-1(RAP1) (27). However, in specific scenarios where CRC cells are quiescent, the level of PGC1α has been found to be reduced. For example, linoleic acid was found to induce dormancy in CRC cells by increasing the expression of miR-494, which suppressed energy metabolism genes and maintained cell quiescence through reducing PGC1α levels (28). Interestingly, miR-494 was present at low levels in non-metastatic cases (28). In addition, in normoxic CRC cancer stem cells (CSCs), the expression of hypoxia-inducible factor 1-alpha (HIF-1α) was significantly reduced, leading to the restoration of PGC1α expression in these areas (29). This suggests that HIF1α can act as a negative regulator of PGC1α. Additionally, substances secreted from the tumor microenvironment (TME) may also impact PGC1α expression. For instance, neutrophil elastase (NE), which is released from the neutrophil extracellular traps (NETs) activated toll-like receptor 4 (TLR4) on cancer cells, has been shown to trigger the TLR4/p38/PGC1α axis, resulting in PGC1α upregulation (30). Notably, PGC1α levels have been found to not only be upregulated in cancer cells but also be heightened in adipose tissues (20), particularly in the context of obesity-related CRC (31) and in cases of cancer cachexia (32). This cachexia-related elevation of PGC1α has been shown to be achieved through the secretion of interleukin-8 (IL-8) in extracellular vesicles (EVs) from CRC cells (32). Cachexia, a late-stage complication of various cancers, is promoted by tumor growth and chemotherapy administration (33). In muscle tissues affected by wasting, abnormal PGC1α expression contributes to mitochondrial dysfunction, which in turn causes cachexia-related symptoms (34). Retrospective studies analyzing clinical tumor samples from patients with CRC have underscored the clinical relevance of PGC1α in CRC (35). Notably, PGC1α expression is positively correlated with nodal metastasis (36), and high tumor PGC1α expression is correlated with reduced overall survival (OS) (36). Hence, PGC1α might serve as a biomarker for assessing CRC invasion and progression.
Functionally, PGC1α plays a crucial role in enhancing mitochondrial biogenesis and oxidative phosphorylation, thereby reprogramming the metabolism to support cell proliferation, growth, and survival (23, 30, 37). Particularly under hypoxic conditions, PGC1α exerts an antioxidant effect to shield cancer cells from accumulation of ROS (23). Moreover, PGC1α enhances lipid biosynthesis by increasing fatty acid synthase (FASN) levels indirectly through the upregulation of Sp1 and SREBP-1c. This process provides essential building blocks for cell membranes in rapidly proliferating cells (38). Further, PGC1α activates multiple pro-tumorigenic signaling pathways in CRC. It promotes the activation of the AKT/GSK-3β pathway through physical interactions with AKT, although the exact mechanisms remain undefined (39). Additionally, PGC1α boosts leucyl-tRNA synthetase 1 (LARS1) expression, further stimulating AKT/GSK-3β signaling (37).WNT/β-catenin pathway can also be activated by PGC1α, which promotes CRC cell proliferation and inhibits apoptosis (37, 39). PGC1α activates the epithelial-mesenchymal transition (EMT) pathway by upregulating transcription factors like Snail, Slug, and Twist (37, 39), thereby facilitating cancer cell migration and invasion. Recent findings have also indicated that PGC1α can orchestrate lactate oxidation, further promoting the migration and invasion of normoxic CSCs in CRC (29). Additionally, PGC1α can upregulate oxidative phosphorylation and antioxidant genes in chemo resistant cells to adapt to metabolic stress and evade damage from chemotherapeutic agents (23, 40). In 5FU-resistant CRC cells, elevated PGC1α expression has been found to be associated with enhanced mitochondrial biogenesis (40), increased expression of BCL2 while simultaneous decreases BAX, cleaved caspase-3, and cleaved PARP-1 (23). Although SIRT1 is not found to regulate PGC1α transcriptionally, it controls deacetylation and activation of PGC1α, thus protecting CRC cells against chemotherapy (41). Suppression of PGC1α restores chemosensitivity in CRC cells (41). Consequently, monitoring changes in PGC1α expression in patient samples during and after treatment can provide insights into tumor response and the progression towards chemoresistance, enabling timely adjustments to alternative treatment regimens before the disease advances.
Above all, PGC1α integrates various oncogenic pathways in CRC, including metabolism, EMT, inflammation, and survival. Targeting PGC1α holds promise as an approach to counter metastasis and improve patient outcomes. Importantly, dietary interventions offer a potential strategy. For instance, linoleic acid (LA) has been shown to induce quiescence in CRC by suppressing PGC1α expression in mice models (28). In vitro studies have demonstrated that manuka honey (MH) reduces colon cancer cell growth in a dose-dependent manner by deactivating PGC1α (42). Clinical trials for CRC are also exploring metabolic drugs such as metformin, which indirectly inhibit PGC1α activity and reprogram metabolism (11, 43). Given their favorable safety profiles and ability to target cancer metabolism, these PGC1α-related drugs could offer new treatment avenues for patients with advanced or chemotherapy-refractory CRC.
PGC1α in gastric cancer
Mirroring its expression pattern in CRC, PGC1α has been found to be highly expressed in GC and gastric epithelial cells (44). This elevated expression appears to be influenced by oxidative stress induced by exogenous molecules. Indeed, research has demonstrated that quercetin, a potential prooxidant, increases PGC1α expression under oxidative stress conditions, thereby safeguarding gastric cells from damage (45). Notably, this effect is particularly pronounced after prolonged exposure to H2O2, whereas quercetin lacks this effect under normal circumstances (45).
Upregulated PGC1α has been found to promote GC progression through the inhibition of cell apoptosis and promotion of EMT (44). A more precise mechanism has now been revealed, wherein PGC1α orchestrates the transcription of SNAI1, subsequently affecting the levels of miR-128b (44). This regulatory cascade, which has been observed both in vitro and in vivo, enhances cell growth and metastasis in GC (44). The influence of posttranscriptional modifications on PGC1α’s function is also notable. In GC, PGC1α has been found to undergo phosphorylation by CAB39L-induced p-AMPK, culminating in the regulation of genes associated with mitochondrial respiration complexes (46). Furthermore, PGC1α possesses a pivotal role in the chemoresistance of GC, characterized by disrupted metabolism (47, 48). The HCP5/miR-3619-5p axis controls PGC1α expression, subsequently enabling its interaction with CCAAT/enhancer binding protein beta (CEBPB), thereby triggering transcription of carnitine palmitoyltransferase I (CPT1). This in turn enhances fatty acid oxidation (FAO) in GC, ultimately conferring chemoresistance to cancer cells (49). Remarkably, PGC1α suppression could sensitize GC cells to chemotherapeutic agents by inducing metabolic deficiencies and increasing oxidative stress (49).
PGC1α in liver cancer
Liver cancer, also known as hepatocellular carcinoma (HCC), is a major cause of cancer-related mortality worldwide (50). The main risk factors for HCC include chronic hepatitis B and C viral infections, alcohol abuse, nonalcoholic steatohepatitis (NASH), and aflatoxin exposure (51, 52).
Emerging evidence suggest that PGC1α is downregulated in HCC tissues and cell lines (2, 53). However, the cause of the abnormal expression remains unclear. One proposed mechanism is the accumulation of Parkin-interacting substrate (PARIS) in response to oxidative stress (54), which inhibits PGC1α expression transcriptionally. Additionally, sestrin2 (SESN2), a stress-inducible protein in HCC, has been found to mediate glutamine-dependent activation of PGC1α. SESN2 forms a complex with JNK and FOXO1, enhancing PGC1α transcription. Thus, the reduced SESN2 leads to a decreased PGC1α expression under glucose deprivation (55). The Yes-associated protein 1 (YAP1), a key effector of the Hippo signaling pathway, suppresses PGC1α expression in HCC (56). Moreover, mitochondrial transcription factor B2 (TFB2M), acts as a pivotal oncogene in HCC (57), decreases PGC1α expression at both mRNA and protein level through SIRT3/HIF-1α signaling (58). Interestingly, although hypoxia generally induces PGC1α expression in many diseases (59–61), HIF-1α has been reported to negatively regulate PGC1α expression (58, 62). However, the precise mechanism underlying this regulation remains unclear, due to a lack of Chip-Seq data and in-depth investigations.
Functional studies have demonstrated that PGC1α inhibits HCC cell proliferation and metastasis. Low PGC1α expression is associated with poor prognosis and aggressive tumor features in HCC patients (2). Mechanistically, PGC1α has been found to counter the Warburg effect, a well-known process promoting cancer progression in HCC cells (63, 64). PGC1α achieves this by promoting oxidative phosphorylation (OXPHOS) and inhibiting aerobic glycolysis, partially through PDK1 in a PPARγ-dependent manner (2). PGC1α activates PPARγ, leading to reduced β-catenin protein levels and inhibition of the WNT/β-catenin pathway and PDK1 expression (2). This results in a decrease in the Warburg effect and tumor suppression. Conversely, impaired PGC1α reverses these effects (2). Moreover, PGC1α regulates gluconeogenic genes with several coactivators, such as hepatic nuclear factor 4 alpha (HNF4α), which has been found to repress pathogenesis of HCC (65). Important targets like G6PC and PCK1, affecting the glycogen accumulation and driving HCC progression (66), are mediated by PGC1α and HNF4α. YAP reduces the ability of PGC1α to coactivate HNF4α at its promoter (56). Post-translational modifications of PGC1α also plays roles in HCC progression. Mitochondrial fission, important in promoting tumor progression in HCC (67, 68), reduces NAD+ levels and SIRT1 activity, leading to increased acetylation of PGC1α protein. Reduced PGC1α activity has been found to be associated with downregulation of CPT1A and acyl-CoA oxidase 1 (ACOX1) and inhibition of FAO in HCC cells (53), both contributing to HCC growth and metastasis (69, 70). Interestingly, general control non-depressible 5 (GCN5) has been found to inhibit PGC1α activity via acetylation (16), but the knockout of GCN5 in mouse liver has not been found to have a significant effect on cancer development (71).
PGC1α also has implications in precancerous or tumorigenic stages. High mobility group AT-hook 1 (HMGA1), a non-histone nuclear protein (72), has been found to recruit protein PGC1α to enhance HBV replication and antigen production through HBV EII/Cp promoter activation, which is associated with liver cirrhosis and HCC oncogenesis (73–75). PGC1α’s relationship with viral expression of HBV has also been observed in other studies (76, 77). Liver cirrhosis, stemming from non-alcoholic fatty liver disease (NAFLD), occasionally precedes HCC and involves Bcl-3 (78), while Bcl-3 reduces PGC1α activity, suggesting that higher PGC1α activity might protect against NAFLD-related liver cirrhosis (78, 79). Indeed, pharmacologically activating PGC1α has shown promise for NAFLD treatment (78, 80).
However, several studies have proposed an oncogenic function for PGC1α downregulation validations in HCC. For instance, SET8 inhibits Keap1 expression through PGC1α, activating the Nrf2/ARE pathway and supporting HCC progression (81, 82). Interestingly, gankyrin elevates TIGAR level, a well-known regulator of glucose metabolism, via the Nrf2/ARE pathway. This elevation promotesPGC1α nuclear importation, and drives increased glucose metabolism in HCC (83). Therefore, the synergy between Nrf2/ARE activation and nuclear localization of PGC1α could serve as a critical loop in the metabolic changes that support HCC progression. In addition, the phosphoserine aminotransferase 1 (PSAT1)’s interaction with p5372P variant in HCC cells dissociates PGC1α binding, promotes PGC1α’s nuclear translocation (84), mitochondrial transcription factor A (TFAM)-mediated OXPHOS and TCA cycle activation (15). Converse effects have been observed for wild-type p53. In HCC, CD147 promotes p53 degradation via the PI3K/AKT pathway (85). Intriguingly, when p53 is exogenously expressed, there is an upregulation of PGC1α levels (86). This suggests that CD147 might impede mitochondrial biogenesis and functionality by suppressing PGC1α/TFAM levels. In this context, PGC1α appears to play a tumor-suppressive role. Thus, targeting PGC1α upstream or downstream pathways is a promising therapeutic strategy for HCC. PPARα agonists that mimic PGC1α re-expression have shown efficacy in HCC models. For example, GW7647 diminishes hepatocarcinogenesis in-humanized mice models (87, 88).
PGC1α in renal cancer
Research has indicated a decline in PGC1α expression in clear cell renal cell carcinoma (ccRCC) tumors compared to normal tissues. This reduction in PGC1α levels aligns with higher tumor grades (89), advanced disease stage (90), worse disease progression, and worse OS (91). One possible reason for this suppression could be the activation of transforming growth factor beta (TGF-β) signaling, which is commonly observed in ccRCC. In fact, when TGF-β signaling is inhibited, PGC1α levels see an increase (92). Moreover, histone deacetylase 1 (HDAC1) and histone deacetylase 7 (HDAC7) have been identified as corepressors, playing a role in suppressing PGC1α via the TGF-β signaling pathway (92). In a related observation, retinoic acid 13 (Stra13 or Dec1) is found to transcriptionally inhibit PGC1α expression. This suggests that Stra13 could be a mediator of HIF-mediated PGC1α suppression during von Hippel-Lindau (VHL) deficiency and hypoxia in ccRCC (90). The actions of Stra13 appear to be closely related to HDAC activity (93). On another front, the epigenetic changes also seem to play a part, particularly through m6A modifications that impacts the stability of PGC1α. A decrease in FTO expression in ccRCC has been linked to a rise in methylated PGC1α mRNA, leading to reduced stability (91).
Functional experiments have uncovered PGC1α’s potential as a tumor suppressor in ccRCC. Evidence suggests that reintroducing PGC1α restores the levels of TCA cycle enzymes and mitochondrial functions, reversing the metabolic effects of TGF-β signaling in mice models (92). In addition to inducing oxidative stress, PGC1α sensitizes ccRCC cells to cytotoxic therapies (90). Moreover, PGC1α inhibits cell metastasis in vitro and in vivo by reducing collagen gene expression via miR-29a induction, including collagen type I alpha 1 chain (COL1A1) and collagen type VI alpha 2 chain (COL6A2) (94). Loss of PGC1α in metastatic RCC promotes collagen expression, discoidin domain receptor tyrosine kinase 1 (DDR1) activation, and subsequent snail family transcriptional repressor 1 (SNAIL) stabilization (89). Another tumor suppressor, mitochondrial pyruvate carrier 1 (MPC1), is also regulated by PGC1α (95, 96). PGC1α stimulates the transcription of MPC1 in conjunction with ERR-α and reduced MPC1 negates PGC1α’s effects on mitochondrial respiration and biogenesis (95).
However, PGC1α’s role seems subtype-dependent. Divergent conclusions have been drawn for the other subtypes. One instance involves the loss of MYBBP1A in 9% of renal tumors (97). MYBBP1A represses PGC1α levels, so the decline of MYBBP1A activates PGC1α directly and indirectly through c-MYB, shifting cellular metabolism from glycolysis to OXPHOS (98). This occurs primarily in the absence of c-MYB or pVHL (97, 98). Another scenario involves inactivation of SETD2 in approximately 12% of ccRCC cases (99). SETD2, a histone H3 lysine trimethyltransferase, acts as a ccRCC tumor suppressor (100, 101). Loss of SETD2 boosts PGC1α expression and mitochondrial mass in ccRCC (102), prompting a metabolic shift towards oxidative phosphorylation and lipogenesis. In both these contexts, PGC1α takes on a tumor-promoting role in ccRCC.
PGC1α in cholangiocarcinoma
The metabolic reprogramming observed in CCA plays a crucial role in driving its progression (103, 104). CCA cells exhibit increased aerobic glycolysis and glutamine anaplerosis, which allows them to produce essential biosynthetic intermediates vital for their rapid growth and survival (103). Recently, the significance of PGC1α in CCA has been emphasized. Patients with elevated levels of PGC1α expression tend to experience reduced OS and progression-free survival (PFS) and are associated with increased angioinvasion and accelerated recurrence (105). Furthermore, the upregulation of PGC1α drives CCA metastasis by elevating the expression of two critical factors: pyruvate dehydrogenase-alpha 1 (PDHA1) and mitochondrial pyruvate carrier 1 (MPC1) (96). This molecular mechanism reverses the Warburg effect, a hallmark metabolic characteristic often observes in cancer cells. Notably, PGC1α also exerts a significant influence on mitochondrial metabolism regulation and the maintenance of stem-like characteristics in CCA stem cells (105). Therefore, pharmacological interventions involving substances like metformin or SR-18292 have shown promise in inhibiting the effects associated with PGC1α upregulation, mitigating its impact on CCA progression and metastasis (105).
PGC1α in glioblastoma
Emerging evidence underscores the pivotal role of PGC1α in GBM oncogenesis, progression, and treatment resistance. Notably, data from the GBM TCGA and GBM PDX Mayo Clinic databases indicates that GBM exhibits decreased PGC1α mRNA expression compared to normal brain tissue (106). Intriguingly, protein levels of PGC1α have also been reported to be highly expressed in GBM patients, which are located not only in the perinuclear or cytoplasmic regions but also prominently within mitochondria, as proven by publicly available TMAs from US Biomax (107). However, compared to WHO grade IV gliomas, lower-grade gliomas (WHO grade II and III) show increased expression of PGC1α (108). Further survival analysis have indicated that higher PGC1α expression in patients with GBM corresponds to shorter survival times (108), implying that PGC1α loss contributes to gliomagenesis and the transition to glioblastoma. Once a GBM develops, the upregulation of PGC1α within a subset of tumors can promote aggressiveness by driving mitochondrial metabolism. Interestingly, the expression of PGC1α varies in distinct PTEN status; therefore, the protein levels of PGC1α are highest in the SF767 cells (PTEN wildtype) and lowest in the A172 cells (PTEN-deleted) (109).
Functional studies demonstrate that PGC1α seems to act as a tumor suppressor in GBM. Aurora kinase A (AURKA) has been implicated in GBM progression and is a potential therapeutic target for this aggressive brain cancer (106, 110). Research has shown that the inhibition of AURKA leads to c-Myc suppression, subsequently resulting in the upregulation of PGC1α, which in turn promotes oxidative metabolism. Furthermore, H3K27ac ChIP-seq and ATAC-seq show that chromatin accessibility at the potential c-Myc-binding region in the PGC1α promoter is increased, whereas following AURKA inhibition, the binding of c-Myc to the PGC1α promoter is reduced. Concurrently, an enhanced acetylation of the same region in PGC1α promoter has been observed following exposure to AURKA inhibition, indicating that c-Myc may act as a suppressor of PGC1α (106). Moreover, FDA-approved HDAC inhibitors, such as panobinostat, vorinostat, and romidepsin, have been shown to replicate these effects by blocking the Warburg effect in GBM cells. This interference with HDAC1/-2 reduces c-Myc levels while increasing PGC1α expression (111). Another inhibitor, crizotinib, which targets MET kinase (112, 113), induces the metabolic reprogramming of GBM cells. This reprogramming, characterized by heightened oxidative phosphorylation and fatty acid oxidation, is also mediated by upregulated PGC1α expression and facilitated by increased CREB phosphorylation after Crizotinib exposure (14, 114). The mTORC1 pathway, crucial for cell growth and proliferation in GBM (115), is often activated by epidermal growth factor receptor (EGFR). However, mTORC1 inhibition, accompanied by reduced PGC1α expression, protects GBM cells from hypoxia-induced cell death under the conditions of the TME (116, 117). Thus, preclinical experiments have shown that, rapamycin, an mTORC1 inhibitor, triggers adverse effects by promoting cell survival in GBM under hypoxic conditions (116). Concurrently, mTORC1 activation, followed by increased PGC1α expression, sensitizes GBM cells to hypoxia-induced cell death (116).
Similar to other tumors, PGC1α exhibits dual effects in GBM, displaying both anticancer and pro-cancer roles in distinct subtypes. In particular, the fusion of the FGFR3 and TACC3 genes (F3-T3), which act as potent oncogenes, has been identified in approximately 3% of GBM cases (118, 119). PGC1α has been shown to be notably overexpressed in F3-T3-positive GBM cells in the presence of PIN4. Elevated PGC1α contributes to mitochondrial biogenesis and respiration through ERRγ. Conversely, dampening PGC1α activity hinders the tumor-promoting effects of F3-T3, as demonstrated in both cellular and animal models in GBM (120).
PGC1α in melanoma
Melanoma cells exhibit two distinct transcriptional signatures, proliferative and invasive, which correspond to different cellular phenotypes (121). The metastatic spread of melanoma is thought to involve a transition in cell behavior, shifting from a proliferative program to acquiring migratory and invasive characteristics (122). The expression of PGC1α generally defines these two subsets of melanoma cells (123). In the first subset, PGC1α has been found to be expressed at high levels and plays an important role in melanoma progression and survival. Its upregulation may be triggered by the microphthalmia-associated transcription factor (MITF) via its binding to the upstream regulatory promoter (5, 123, 124), an event regulated by the Wnt/β-Catenin pathway (125) or an important lipogenic enzyme-ATP-citrate lyase (ACLY) (126). Elevated levels of PGC1α are correlated with poor survival (13, 123). In this subset, PGC1α supports melanoma through various mechanisms, with programmed cell death being key. Apoptosis, a process that triggers cell death, is regulated by PGC1α through the regulation of reactive oxygen species (ROS) levels. Thus, suppression of PGC1α leads to a decrease in the expression of genes involved in ROS detoxification, resulting in elevated ROS levels and subsequent induction of apoptosis (123). Ferroptosis, another form of cell death, is involved in melanoma progression and chemoresistance (127). Small molecules that induce ferroptosis, such as RSL3 and ML162, suppress the expression of PGC1α through the Wnt/β-Catenin-MITF pathway. Loss of PGC1α impairs mitochondrial function and antioxidant capacity, leading to excess accumulation of mitochondrial ROS and sensitizing cells to ferroptosis (125). As the activation of the Wnt/β-Catenin pathway in melanoma guides resistance to anti-PD-L1/anti-CTLA-4 treatment (128, 129), targeting the Wnt/β-Catenin signaling pathway or PGC1α may improve the effectiveness of immunotherapy by inducing ferroptosis (129). Furthermore, PGC1α tightly interacts with ERRα in melanomas, promoting mitochondrial oxidative metabolism by regulating the expression of genes involved in oxidative phosphorylation and the TCA cycle (13). Depletion or pharmacological inhibition of ERRα selectively inhibits the growth of PGC1α-positive melanomas, but not PGC1α-negative melanomas (13). BAY 1238097, a potent inhibitor of BET binding to histones, strongly represses the expression of PGC1α in melanoma cells, impairing mitochondrial function and inhibiting melanoma cell proliferation (130). These findings support the concept that PGC1α-positive melanomas depend on mitochondrial metabolism for growth.
Conversely, another subpopulation of melanoma cells exhibits lower PGC1α expression, possesses a limited number of mitochondria, and relies heavily on glycolysis to produce energy. This phenotype is often observed in invasive and metastatic melanomas (131, 132). In this subset of melanoma cells, PGC1α may be epigenetically silenced through chromatin modifications involving H3K27 trimethylation at its promoter. Pharmacological inhibition of EZH2, an enzyme involved in chromatin modifications, diminishes H3K27me3 markers (133, 134), leading to increased PGC1α level and suppression of invasion in PGC1α-silenced cells (122). Additionally, BRAF mutation (V600E) suppresses MITF and PGC1α expression in melanoma cells (135). Knocking down PGC1α in these cells promotes a pro-metastatic gene program and enhances metastasis in mice models (131). PGC1α upregulates the expression of inhibitor of DNA binding protein (ID2), which binds and inhibits a diverse array of bHLH transcription factors (136). The binding of ID2 suppresses the transcription factor TCF4, resulting in the suppression of metastasis-related genes including integrins, which are known to affect metastasis (131, 137). Moreover, ID2 suppresses the activity of TCF12, which increases the expression of WNT5A (122). As WNT5A can stabilize YAP protein levels (138, 139), inhibition of TCF12, WNT5A, or YAP blocks melanoma migration and metastasis (122). BRAF inhibitors, such as PLX4032, which have been reported to upregulate PGC1α expression in melanomas (140, 141), inhibit metastasis partly by suppressing the Wnt/β-Catenin-MITF pathway and promoting the expression of PGC1α (125). This effect is independent of their cytotoxic or growth-inhibitory properties (131). Kisspeptin-1 (KISS1) functions as a metastasis suppressor by inhibiting metastasis without affecting primary tumor growth (142). In melanoma cells, the transcriptional coactivator PGC1α plays a crucial role in mediating the effects of KISS1 on cell metabolism and metastasis suppression (143). PGC1α helps KISS1 upregulate genes that promote fatty acid oxidation, activates AMPK signaling to inhibit acetyl-CoA carboxylase (ACC), and ultimately shifts cells towards mitochondrial oxidative phosphorylation instead of glycolysis (144). The loss of PGC1α blunts these metabolic changes and abolishes KISS1’s anti-metastatic effects. The major implication of these bi-signatures is that effective melanoma therapies should target both proliferative and invasive cell types, as they coexist within tumors and can interconvert. Targeting only one phenotype may lead to the selection and outgrowth of alternative phenotypes. Indeed, suppressing of PGC1α-dependent oxidative metabolism activates glycolysis via HIF1α as a compensatory survival mechanism in melanomas. Dual inhibition of PGC1α and HIF1α causes energetic deficits, but partial rescue of melanoma cells have been observed through glutamine utilization (145). Hence, a triple targeting approach involving PGC1α, HIF1α, and glutamine metabolism is necessary to completely block melanoma growth by shutting down oxidative metabolism, glycolysis, and glutaminolysis (145), suggesting that a combination therapy targeting multiple nodes of tumor metabolism is necessary to effectively disrupt energy production and viability, However, overcoming the challenges posed by metabolic heterogeneity and redundancy remains a significant obstacle.
PGC1α in prostate cancer
The expression of PGC1α has generally been found to be reduced in PC, with a further decrease observed in metastatic tissues (146). This downregulation of PGC1α is associated with decreased disease-free survival (DFS) (147–149). The exact reasons for the downregulation of PGC1α in PC are not fully understood; however, they are believed to be a result of selective pressure during disease progression and metabolic changes. Reports suggest that miRNAs, such as miR-34a-5p, can downregulate PGC1α (150).
It has been reported that PGC1α plays a tumor-suppressor role in the development of PC, inhibiting cancer progression and metastasis (146). Interestingly, some studies have found that the protein level of PGC1α is undetectable in PC cell lines, despite comparable transcript levels to metastatic PC specimens (146, 151). The re-expression of PGC1α in vitro and in vivo has been shown to inhibit cell proliferation and cell cycle progression, supporting its antiproliferative activity (146). Moreover, PGC1α suppresses the metastatic properties of PC cells by decreasing integrin signaling, causing cytoskeletal changes (152), and downregulating MYC levels and activity (153). This effect is mediated by its interaction with the transcriptional partner estrogen-related receptor alpha (ERRα). Knockout of ERRα prevents PGC1α from inhibiting invasion, suggesting that the PGC1α/ERRα axis acts as an antagonist to the progression of PC metastasis (146, 152). Furthermore, AMPK, a metabolic regulator in PC, safeguards against cancer progression in mice models (154–156). Activation of AMPK leads to increased expression of PGC1α and its downstream targets, promoting a switch to a more oxidative and catabolic metabolism and opposing the pro-tumorigenic program of increased lipogenesis (154). However, it has been found that androgens-activated AMPK can increase the expression of PGC1α, promoting mitochondrial content and PC cell growth in cell line models (151). Intriguingly, in a mouse model of benign prostatic hyperplasia, androgen/testosterone increased prostate size but did not affect PGC1α levels (151). These findings elucidate the complex roles of the AMPK/PGC1α axis in PC development.
In a subpopulation of clinical PC samples, PGC1α level is found to be overexpressed, and PGC1α may therefore exert a tumor supporting role (151, 157). In addition to the aforementioned AMPK signaling pathway, another mechanism contributing to the abnormal expression of PGC1α is the loss or mutation of p53 (158). In PC cells with mutated or deleted p53, PGC1α has been found to be expressed at high levels. Overexpression of wild-type p53 in these cells decreases the expression of PGC1α and causes mitochondrial dysfunction (157). However, this regulation axis is highly metabolic-pattern dependence, as p53 suppresses PGC1α level and nuclear localization through redox modification (159). In these settings, the tumor-supporting role of PGC1α is found to depend on the transcription factors (TFs) it partnered with. For example, PPARG activation results in the upregulation of AKT3, which subsequently promotes the nuclear localization of PGC1α. The genes induced by PGC1α promotes mitochondrial biogenesis and energy metabolism, fueling PC progression (160).
In addition, the ETS-related gene (ERG) functions as an oncogenic transcription factor in PC (161). In such cases, PGC1α has been shown to act as a coactivator for ERG, specifically under metabolic stress conditions like glucose deprivation and serum starvation (8). This interaction and coactivation of ERG by PGC1α leads to increased expression of antioxidant genes, such as SOD1 and TXN, which can help clear ROS and benefit PC growth (8).This suggests that PGC1α allows ERG fusion-positive PC cells to adapt and survive under metabolic stress by coactivating the antioxidant transcriptional program of ERG.
PGC1α in ovarian cancer
While PGC1α activity is typically low in normal tissues, several studies have reported frequent overexpression of PGC1α in ovarian tumors compared to that in normal ovaries (162, 163). However, it is important to note that the results of the high tumor expression of PGC1α only correlates with tumor differentiation and did not exhibit significant correlations with other clinical features (164). When combined with ERRα, the overexpression of PGC1α reveals a tendency towards increased risk of metastasis and reduced OS (163). Additionally, the expression of both PGC1α and PGC1β has allowed for the classification of ovarian cancer (OC) patients into distinct subgroups. Approximately 25% of studies tumors exhibits high expression of both genes (164), indicating the presence of an overactive mitochondrial gene program. These tumors demonstrates increased mitochondrial content, oxidative metabolism, and OXPHOS (164). Mechanistic studies have shed light on how the aberrant activation of PGC1α contributes to OC progression and therapeutic resistance. Recent studies have identified PGC1α as a critical driver of OC progression, particularly in high-grade serous OC (HGSOC), which exhibits metabolic heterogeneity (165–167). In OC, the high-OXPHOS state has been linked to chronic oxidative stress (165). This stress leads to the increased aggregation of PML nuclear bodies, which subsequently activates PGC1α through deacetylation. As a result, PGC1α induces the expression of electron transport chain (ETC) components, enhancing mitochondrial respiration in high-OXPHOS cancer cells. Knockdown of PGC1α reduces both ETC gene expression and oxygen consumption rate in these cells (165). Furthermore, PGC1α plays a pivotal role in mediating the response to conventional chemotherapies. PGC1α has been found to be a key regulator of reactive ROS production (165), which are crucial determinants of the apoptotic response to cisplatin in OC cells (168). Elevated expression or activity of PGC1α is correlated with enhanced chemosensitivity by promoting mitochondrial oxidative metabolism and respiration (165). Conversely, reducing PGC1α activity and levels decreases sensitivity to chemotherapy in OC.
PGC1α in nasopharyngeal carcinoma
There is increasing evidence that metabolic reprogramming driven by PGC1α promotes NPC progression and resistance to treatment. PGC1α has been found to be upregulated in NPC and its high expression has been associated with shorter OS after radiation therapy (169). PGC1α contributes to NPC cell survival by activating FAO pathways, which provide cells with ATP and the antioxidant NADPH. These metabolic alterations allow NPC cells to adapt and thrive under challenging conditions. PGC1α works in conjunction with the transcription factor CEBPB to enhance the expression of CPT1A, a gene involved in FAO, thereby sustaining this metabolic reprogramming (169). Consequently, these changes confer radioresistance to NPC cells (169). Furthermore, TGFβ1, a signaling molecule, can upregulate PGC1α and activate FAO to facilitate EMT and invasion of NPC cells. Specifically, TGFβ1 stimulates phosphorylation and expression of AMPKα1 (170), which, in turn, phosphorylates and activates PGC1α in NPC. This activation leads to transcriptional upregulation of FAO-related genes (170). Inhibiting PGC1α expression and components of the FAO pathway have been shown to reduce EMT, invasion, and metastasis of NPC both in vitro and in vivo.
PGC1α in breast cancer
Overall, PGC1α expression has been found to be reduced in breast tumor tissues compared to that in the normal breast epithelium (171, 172). This downregulation of PGC1α potentially facilitates the Warburg effect, in which cells increase their dependence on glycolysis and glucose uptake, while decreasing mitochondrial oxidative phosphorylation, even when oxygen is available (173). Such metabolic shifts enhance the proliferation and survival of cancer cells. A key mechanism that drives this shift is the regulation of mitochondrial deacetylase SIRT3 (171, 174). Although the exact cause of PGC1α’s downregulation in BC cells is yet to be fully elucidated, certain epigenetic modifications such as negative regulation by miR-485 and miR-217 have been proposed (175, 176). Interestingly, despite its general downregulation in breast tumors, the expression of PGC1α varies according to tumor subtypes and their metastatic tendencies. Specifically, HER2+ and triple-negative breast tumors (TNBT) express high levels of PGC1α (177, 178). Moreover, elevated expression of PGC1α has been detected in BC cells that predominantly metastasize to the lungs or bone, as opposed to the liver and brain (179). Similarly, circulating tumor cells (CTCs) released from BC in mice models and patients exhibit elevated PGC1α expression (180). Indeed, PGC1α knockdown in a metastatic cell line has been found to result in reduced CTC numbers and metastasis, whereas overexpression of PGC1α has been found to increase lung metastasis in vivo (179, 180). Interestingly, BC cells with low PGC1α levels possess increased metastatic ability when overexpressing PGC1α levels (180). It is worth noting that inhibiting mitochondrial respiration with biguanides in such cells is not found to mitigate PGC1α-induced metastasis (179), suggesting that the augmented metastatic phenotype is not simply attributed to the PGC1α-induced escalation in oxidative phosphorylation. Instead, PGC1α increases overall bioenergetic capacity and flexibility to facilitate metastasis, allowing cancer cells to cope with energy disruptors (179). In these conditions, the induced PGC1α ensures the metabolic demands of aggressive breast tumors.
Early research has also highlighted PGC1α’s involvement in the initiation of BC (181). In particular, its interaction with EglN2, an enzyme involved in the regulation of the hypoxia-inducible factor (HIF) pathway, appears to be central to the modulation of mitochondrial function and has been implicated in BC tumorigenesis (182, 183). In both normoxic and hypoxia conditions, EglN2 forms a complex with both PGC1α and NRF1, leading to the induction of FDXR. This maintains mitochondrial function and contributes to breast tumorigenesis in an HIF-independent manner (182). Importantly, in the absence of PGC1α, the effects of EglN2 overexpression on BC cells are blocked.
Furthermore, PGC1α’s metabolic regulatory functions in BC often operate in collaboration with other transcription factors like ERRα or p53. For instance, the interplay between PGC1α and ERRα governs a spectrum of metabolic genes (172), driving increased mitochondrial respiration, ATP production, and other processes that culminate in heightened tumor aggression and drug resistance in BC (177, 184, 185). In ERBB2+ cancer cells, PGC1α positively regulates glutamine metabolism in conjunction with ERRα (177). This regulation contributes to increased glutamine uptake, increased flux through the citric acid cycle (CAC), and enhanced lipogenesis from glutamine, particularly under hypoxic conditions (177). The AMPK orchestrates this energy-sensor axis of PGC1α/ERRα (186). When AMPK is activated, PGC1α/ERRα represses folate cycle and one-carbon metabolism, which are vital for sustaining cell growth in cancer cells. Consequently, repression increases the sensitivity to anti-folate therapy (186). It is well established that mutant p53 confers pro-tumorigenic functions in BCs. Notably, as a key downstream of p53, its function is differentially controlled by the codon 72 variant, highlighting the importance of PGC1α as a “gain-of-function” partner of mutant p53 (187).
From a therapeutic point of view, early studies have hinted at the potential benefits of targeting PGC1α in BC treatment. For instance, interventions with vascular endothelial growth factor receptor 2 (VEGFR2) blockade or the AMPK signaling activator, 5-aminoimidazole-4-carboxamide riboside (AICAR), have shown promising shifts in mitochondrial biogenesis and cancer cell behaviors by modulating PGC1α. One study shows that VEGFR2 blockade by Ki8751 leads to increased activity of PGC1α and thereby stimulates the expression of TFAM, which is essential for mitochondrial DNA transcription and replication (188). Subsequent metabolic reprogramming contributes to increased ROS production and apoptosis in BC cells treated with Ki8751 (188). Moreover, AICAR increases PGC1α expression in triple-negative BC (TNBC) cells (189), mediating mitochondrial biogenesis and contributing to a reduced pro-tumor phenotype and increased chemosensitivity (189). Compound 11, a novel inverse agonist targeting ERRα (190), disrupts ERRα binding to its coactivator PGC1α, with promising anti-tumor activity against triple-negative BC cells and tumors (190). The use of polyethylene glycol-modified graphene oxide (PEG-GO) also results in the selective suppression of PGC1α in cancer cells (191). The reduced ATP production impairs the assembly of the F-actin cytoskeleton and formation of lamellipodia, consequently inhibiting the migration and invasion of metastatic BC cells (191). Importantly, the induction of PGC1α guides drug resistance in the course of chemotherapy of BC (5). Endocrine-resistant BC cells have shown higher PGC1α expression than the parental sensitive lines. PGC1α sensitizes BC cells to low estrogen levels during estrogen deprivation therapy (192–194). This may be an early adaptive response to endocrine therapy that potentially contributes to the development of chemoresistance over time by allowing estrogen hypersensitivity (192). Therefore, inhibiting PGC1α with SR-18292 prevents the growth of therapy resistant cell lines in a dose-dependent manner, while re-expression of PGC1α increases the viability of resistant cells when treating with certain endocrine therapies, such as tamoxifen, fulvestrant, palbociclib, or aromatase inhibitors (193).
Implications of PGC1α in the tumor microenvironment
The tumor microenvironment (TME) is a complex and dynamic landscape where cancer cells interact with, including immune cells, fibroblasts, and the extracellular matrix. The role of PGC1α in the TME is pivotal yet underexplored. Its involvement goes beyond mere energy metabolism, extending to modulating immune responses and influencing tumor progression and therapy resistance.
Significant insights have been gathered from studies on T cells. Naive T cells normally have high levels of PGC1α, which support their metabolic demands for proliferation and effector functions through mitochondrial biogenesis and oxidative metabolism. However, during T-cell activation, PGC1α expression is progressively repressed (195, 196). Notably, one study observes that, although the mRNA expression of PGC1α in memory CD8+ T cells decreases upon activation, its protein expression increases (197). This suggests that specific post-translational mechanisms may regulate the stability of PGC1α in CD8+ T cells. In melanomas, tumor-infiltrating T cells have shown a loss of PGC1α level due to the chronic AKT signal activation (195). Additionally, exhausted T cells, experiencing continuous stimulation and hypoxia increase expression of Blimp-1, which further suppress PGC1α expression (196). This impairs their adaptive metabolic responses to hypoxia via mitochondrial biogenesis. Of note, overexpressing PGC1α in these cells enhances their persistence and recall responses, particularly improving the central memory T cell formation and sustained metabolic fitness upon re-exposure to infections (197). Interestingly, the co-stimulatory molecule 4-1BB, which is abundantly expressed in exhausted T cells, promotes mitochondrial biogenesis, fusion, and respiratory capacity (198–200). Costimulation with 4-1BB elevates PGC1α levels, mediating the metabolic effects of 4-1BB signaling (199). Without PGC1α, 4-1BB agonists are less effective at enhancing mitochondrial function and improving anti-tumor responses, or enhance adoptive T cell therapy (199). Thus, restoring the PGC1α expression in functional T cells could offer a strategy to reprogram metabolism in tumor-infiltrating T cells and boost their anti-tumor activity.
Research also shows that PPARγ is essential for maturation of alternatively activated macrophages, enabling monocytes to differentiate into M2 macrophages (201, 202). Indeed, the expression of PGC1α is elevated in these macrophages (203). In breast cancer, a reduced level of miR-382 maintains PGC1α expression in tumor-associated macrophages (203), facilitating the induction of the M2 type through the PPARγ signaling pathway. Fibroblasts also respond to regulation by PGC1α. A recent study found that knocking down PGC1α in normal human lung fibroblasts reduces mitochondrial mass and function (204). This alteration increases activation of matrix synthetic fibroblasts along with secretion of soluble profibrotic factors (204). In mouse models, the loss of PGC1α in induced mouse embryonic fibroblasts (iMEFs) leads to a more aggressive and metastatic melanoma phenotype (205). Similarly, lower PGC1α expression in cancer-associated fibroblasts (CAFs) of oral squamous cell carcinoma (OSCC) enhances the proangiogenic phenotype of CAFs through the PGC1α/PFKFB3 axis (206). Moreover, PGC1α impacts mesenchymal stromal cells (MSCs) (207). In melanoma, cancer cells attract MSCs to the tumor site and induce mitochondrial biogenesis by upregulating PGC1α (207). Furthermore, PGC1α controls mitochondrial transfer from MSCs to melanoma cells, thereby supporting melanoma growth (207).
Discussion
PGC1α is rapidly establishing itself as an indispensable regulator of cancer cell metabolism across numerous malignancies. In cancers, multiple mechanisms are involved in the abnormal expression of PGC1α, particularly in the transcriptional regulation. Therefore, based on the current research progress, we have summarized the relevant findings (Figure 2).
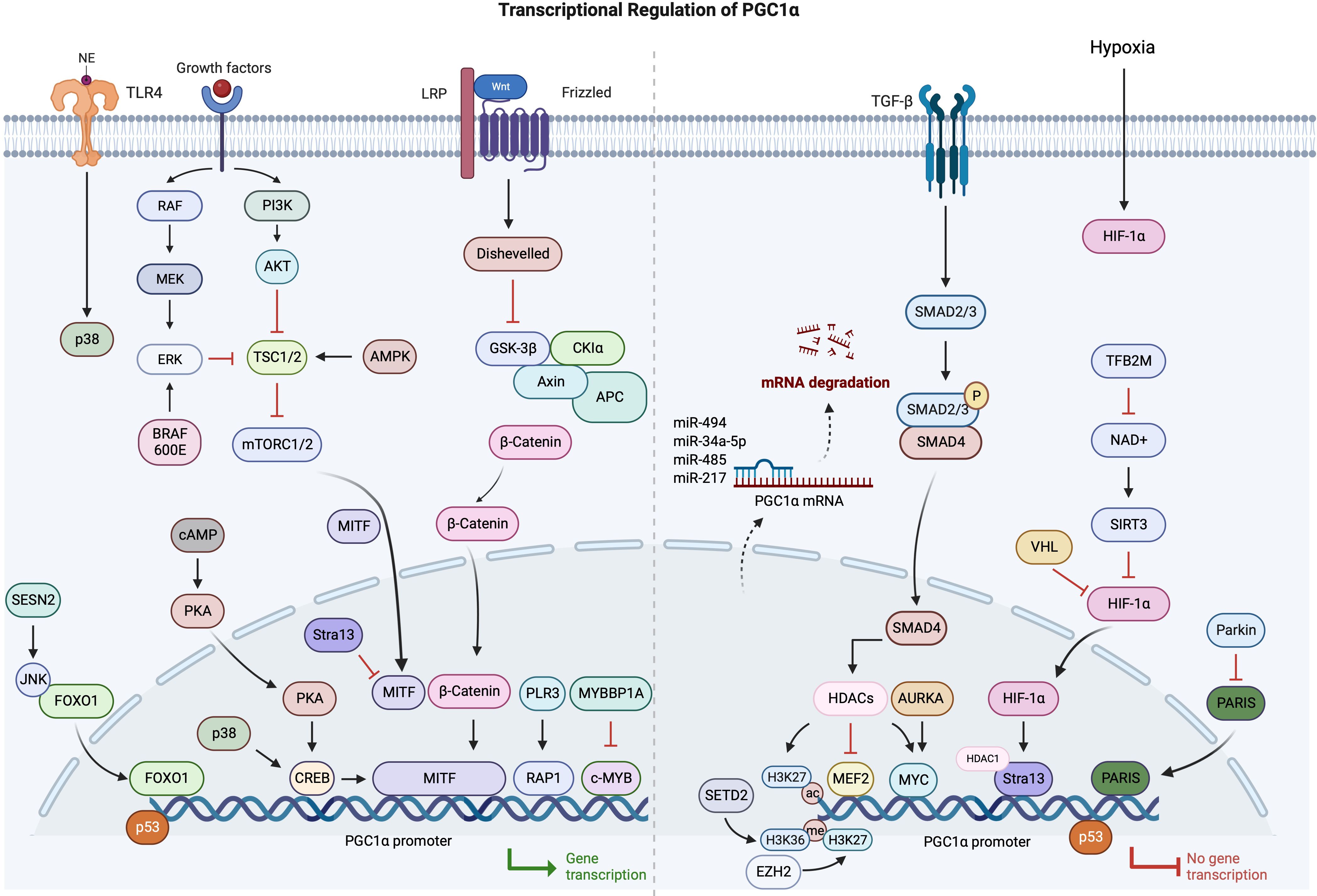
Figure 2 Schematic overview of transcriptional regulation of PGC1a in cancers.
Although the expression and functions of PGC1α are context-dependent, it primarily serves as a pivotal orchestrator of mitochondrial biogenesis, oxidative metabolism, antioxidant defenses, and other cellular processes. When PGC1α expression is downregulated, the Warburg effect is facilitated, leading to disease advancement. Subsequent metabolic aberrations can be rectified by reinstating PGC1α levels. This may halt cell proliferation and invasion, and make cells more receptive to chemotherapy. In contrast, PGC1α upregulation promotes oxidative phosphorylation, allows cancer cells to adapt to stress, and promotes a more aggressive cancer phenotype. This duality in biological behavior shows PGC1α’s adaptability in aligning with various co-regulators and executing functions tailored to its environment. Thus, to target PGC1α effectively, understanding its nuanced role in each cancer subtype is indispensable.
Central to PGC1α’s operations is its position at the crossroads of several pivotal signaling pathways involved in cancer. It processes signals from the Wnt/β-catenin, TGF-β, AMPK, AKT, and p53 pathways to regulate downstream metabolic activities. By partnering with transcription factors like ERRα, NRF1, and YAP, PGC1α can drive specific changes in gene expression. Moreover, post-translational modifications such as phosphorylation and acetylation offer another layer of control over its activity. Decoding these intricate molecular ties is a critical step in comprehending PGC1α’s multifaceted functions and how they may go awry in cancer. An intriguing development is the increasing evidence of PGC1α’s profound effect on the tumor microenvironment, particularly its interaction with immune cells.
Strategic targeting of PGC1α in cancer therapy, therefore, requires a nuanced approach that considers its dual functionality. In cases where PGC1α functions as a tumor suppressor, its upregulation or enhanced activity can shift cancer cell metabolism away from the Warburg effect. This metabolic shift involves reducing glycolysis and increasing oxidative phosphorylation, which typically slows cancer progression and may make cancer cells more amenable to interventions that induce metabolic stress. Enhancing PGC1α’s expression could be achieved through gene therapy techniques, and small molecule activators. Conversely, in cancers where PGC1α contributes to a more aggressive phenotype, its function is linked to enhanced oxidative phosphorylation, supporting cancer cell adaptation to metabolic and oxidative stress. In such cases, inhibiting PGC1α might reduce the cancer cells’ ability to sustain high energy demands and resist hostile environments, such as those imposed by chemotherapy. This can be approached through the use of small molecule inhibitors that disrupt PGC1α’s interaction with its coactivators or transcription factors it regulates. Additionally, RNA interference technologies could selectively knock down PGC1α mRNA, diminishing its protein levels and thus its functionality in cancer cells. Both strategies—enhancing or inhibiting PGC1α—must consider the cancer type, the specific metabolic profile of the tumor, and the systemic implications of altering metabolic pathways. For instance, enhancing oxidative metabolism in non-tumor cells might also affect normal cells, leading to unintended consequences like increased reactive oxygen species. Similarly, inhibiting PGC1α in aggressive tumors must be carefully managed to avoid crippling normal cells’ ability to manage oxidative stress. Effective therapeutic strategies should aim to disrupt this metabolic adaptability by targeting PGC1α along with its regulatory network to block compensatory pathways that facilitate resistance to therapy. Furthermore, PGC1α’s impact on the tumor microenvironment, particularly through its influence on the metabolic states of T cells and macrophages, is gaining attention. By modulating immune cell metabolism, PGC1α could potentially alter the immunological landscape of tumors, reducing immune suppression and enhancing the efficacy of immunotherapies. This understanding suggests that strategies which leverage PGC1α’s role in the tumor microenvironment could complement direct targeting approaches, creating a multifaceted attack on tumor growth.
In summary, PGC1α’s multifaceted roles in cancer metabolism indicate that it is a promising therapeutic target for cancer. Developing drugs that can specifically modulate PGC1α’s activity, tailored to the unique metabolic profiles of different cancer types, represents a promising approach in oncology. By doing these, we are now on the brink of translating our understanding of this metabolic mediator into its clinical benefits against cancer.
Author contributions
YW: Writing – review & editing, Writing – original draft, Investigation. JP: Resources, Methodology, Writing – review & editing, Writing – original draft. DY: Software, Writing – review & editing, Writing – original draft. ZX: Validation, Writing – review & editing, Writing – original draft. BJ: Resources, Writing – review & editing, Writing – original draft. XD: Methodology, Writing – review & editing, Writing – original draft. CJ: Investigation, Conceptualization, Writing – review & editing, Writing – original draft. BO: Visualization, Validation, Methodology, Data curation, Writing – review & editing, Writing – original draft, Funding acquisition. LS: Supervision, Software, Resources, Project administration, Investigation, Funding acquisition, Formal analysis, Conceptualization, Writing – review & editing, Writing – original draft, Visualization, Validation, Methodology, Data curation.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Nanjing Medical Science and technique Development Foundation (Grant No. QRX17105).
Acknowledgments
We would like to thank Editage (www.editage.cn) for English language editing. We also thank the bioRender website (BioRender.com) for providing us with great convenience in creating graphics.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Glossary
References
1. Tan Z, Luo X, Xiao L, Tang M, Bode AM, Dong Z, et al. The role of PGC1α in cancer metabolism and its therapeutic implications. Mol Cancer Ther. (2016) 15:774–82. doi: 10.1158/1535-7163.MCT-15-0621
2. Zuo Q, He J, Zhang S, Wang H, Jin G, Jin H, et al. PPARγ Coactivator-1α Suppresses metastasis of hepatocellular carcinoma by inhibiting warburg effect by PPARγ-dependent WNT/β-catenin/pyruvate dehydrogenase kinase isozyme 1 axis. Hepatology. (2021) 73:644–60. doi: 10.1002/hep.31280
3. Mastropasqua F, Girolimetti G, Shoshan M. PGC1α: friend or foe in cancer? Genes (Basel). (2018) 9(1):48. doi: 10.3390/genes9010048
5. Luo C, Widlund HR, Puigserver P. PGC-1 coactivators: shepherding the mitochondrial biogenesis of tumors. Trends Cancer. (2016) 2:619–31. doi: 10.1016/j.trecan.2016.09.006
6. Finck BN, Kelly DP. PGC-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. (2006) 116:615–22. doi: 10.1172/JCI27794
7. Lin J, Handschin C, Spiegelman BM. Metabolic control through the PGC-1 family of transcription coactivators. Cell Metab. (2005) 1:361–70. doi: 10.1016/j.cmet.2005.05.004
8. Dhara A, Aier I, Paladhi A, Varadwaj PK, Hira SK, Sen N. PGC1 alpha coactivates ERG fusion to drive antioxidant target genes under metabolic stress. Commun Biol. (2022) 5:416. doi: 10.1038/s42003-022-03385-x
9. Jamwal S, Blackburn JK, Elsworth JD. PPARγ/PGC1α signaling as a potential therapeutic target for mitochondrial biogenesis in neurodegenerative disorders. Pharmacol Ther. (2021) 219:107705. doi: 10.1016/j.pharmthera.2020.107705
10. Zhu X, Shen W, Yao K, Wang H, Liu B, Li T, et al. Fine-tuning of PGC1α Expression regulates cardiac function and longevity. Circ Res. (2019) 125:707–19. doi: 10.1161/CIRCRESAHA.119.315529
11. Sainero-Alcolado L, Liaño-Pons J, Ruiz-Pérez MV, Arsenian-Henriksson M. Targeting mitochondrial metabolism for precision medicine in cancer. Cell Death Differ. (2022) 29:1304–17. doi: 10.1038/s41418-022-01022-y
12. Qian L, Zhu Y, Deng C, Liang Z, Chen J, Chen Y, et al. Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) family in physiological and pathophysiological process and diseases. Signal Transduct Target Ther. (2024) 9:50. doi: 10.1038/s41392-024-01756-w
13. Luo C, Balsa E, Thomas A, Hatting M, Jedrychowski M, Gygi SP, et al. ERRα Maintains mitochondrial oxidative metabolism and constitutes an actionable target in PGC1α-elevated melanomas. Mol Cancer Res. (2017) 15:1366–75. doi: 10.1158/1541-7786.MCR-17-0143
14. Xing F, Luan Y, Cai J, Wu S, Mai J, Gu J, et al. The Anti-Warburg Effect Elicited by the cAMP-PGC1α Pathway Drives Differentiation of Glioblastoma Cells into Astrocytes. Cell Rep. (2017) 18:468–81. doi: 10.1016/j.celrep.2016.12.037
15. Qian X, Li X, Shi Z, Bai X, Xia Y, Zheng Y, et al. KDM3A senses oxygen availability to regulate PGC-1α-mediated mitochondrial biogenesis. Mol Cell. (2019) 76:885–95.e7. doi: 10.1016/j.molcel.2019.09.019
16. Dominy JE Jr., Lee Y, Jedrychowski MP, Chim H, Jurczak MJ, Camporez JP, et al. The deacetylase Sirt6 activates the acetyltransferase GCN5 and suppresses hepatic gluconeogenesis. Mol Cell. (2012) 48:900–13. doi: 10.1016/j.molcel.2012.09.030
17. Liu CL, Yang PS, Wang TY, Huang SY, Kuo YH, Cheng SP. PGC1α downregulation and glycolytic phenotype in thyroid cancer. J Cancer. (2019) 10:3819–29. doi: 10.7150/jca.30018
18. Li Y, Hei H, Zhang S, Gong W, Liu Y, Qin J. PGC-1α participates in tumor chemoresistance by regulating glucose metabolism and mitochondrial function. Mol Cell Biochem. (2023) 478:47–57. doi: 10.1007/s11010-022-04477-2
19. Cheng SP, Chen MJ, Chien MN, Lin CH, Lee JJ, Liu CL. Overexpression of teneurin transmembrane protein 1 is a potential marker of disease progression in papillary thyroid carcinoma. Clin Exp Med. (2017) 17:555–64. doi: 10.1007/s10238-016-0445-y
20. Veschi V, Turdo A, Modica C, Verona F, Di Franco S, Gaggianesi M, et al. Recapitulating thyroid cancer histotypes through engineering embryonic stem cells. Nat Commun. (2023) 14:1351. doi: 10.1038/s41467-023-36922-1
21. Chen D, Su X, Zhu L, Jia H, Han B, Chen H, et al. Papillary thyroid cancer organoids harboring BRAF(V600E) mutation reveal potentially beneficial effects of BRAF inhibitor-based combination therapies. J Transl Med. (2023) 21:9. doi: 10.1186/s12967-022-03848-z
22. Witherspoon M, Sandu D, Lu C, Wang K, Edwards R, Yeung A, et al. ETHE1 overexpression promotes SIRT1 and PGC1α mediated aerobic glycolysis, oxidative phosphorylation, mitochondrial biogenesis and colorectal cancer. Oncotarget. (2019) 10:4004–17. doi: 10.18632/oncotarget.v10i40
23. Yun CW, Lee JH, Lee SH. Hypoxia-induced PGC-1α Regulates mitochondrial function and tumorigenesis of colorectal cancer cells. Anticancer Res. (2019) 39:4865–76. doi: 10.21873/anticanres.13672
24. Lai HT, Chiang CT, Tseng WK, Chao TC, Su Y. GATA6 enhances the stemness of human colon cancer cells by creating a metabolic symbiosis through upregulating LRH-1 expression. Mol Oncol. (2020) 14:1327–47. doi: 10.1002/1878-0261.12647
25. Kitaoka Y, Takeda K, Tamura Y, Hatta H. Lactate administration increases mRNA expression of PGC-1α and UCP3 in mouse skeletal muscle. Appl Physiol Nutr Metab. (2016) 41:695–8. doi: 10.1139/apnm-2016-0016
26. Torrens-Mas M, Hernández-López R, Pons DG, Roca P, Oliver J, Sastre-Serra J. Sirtuin 3 silencing impairs mitochondrial biogenesis and metabolism in colon cancer cells. Am J Physiol Cell Physiol. (2019) 317:C398–c404. doi: 10.1152/ajpcell.00112.2019
27. Yang Y, Lian S, Meng L, Tian Z, Feng Q, Wang Y, et al. Knockdown of PRL-3 increases mitochondrial superoxide anion production through transcriptional regulation of RAP1. Cancer Manag Res. (2018) 10:5071–81. doi: 10.2147/CMAR
28. Ogata R, Mori S, Kishi S, Sasaki R, Iwata N, Ohmori H, et al. Linoleic acid upregulates microrna-494 to induce quiescence in colorectal cancer. Int J Mol Sci. (2021) 23(1):225. doi: 10.3390/ijms23010225
29. Liu S, Zhao H, Hu Y, Yan C, Mi Y, Li X, et al. Lactate promotes metastasis of normoxic colorectal cancer stem cells through PGC-1α-mediated oxidative phosphorylation. Cell Death Dis. (2022) 13:651. doi: 10.1038/s41419-022-05111-1
30. Yazdani HO, Roy E, Comerci AJ, van der Windt DJ, Zhang H, Huang H, et al. Neutrophil extracellular traps drive mitochondrial homeostasis in tumors to augment growth. Cancer Res. (2019) 79:5626–39. doi: 10.1158/0008-5472.CAN-19-0800
31. Boughanem H, Cabrera-Mulero A, Hernández-Alonso P, Bandera-Merchán B, Tinahones A, Tinahones FJ, et al. The expression/methylation profile of adipogenic and inflammatory transcription factors in adipose tissue are linked to obesity-related colorectal cancer. Cancers (Basel). (2019) 11(11):1629. doi: 10.3390/cancers11111629
32. Xiong H, Ye J, Xie K, Hu W, Xu N, Yang H. Exosomal IL-8 derived from Lung Cancer and Colon Cancer cells induced adipocyte atrophy via NF-κB signaling pathway. Lipids Health Dis. (2022) 21:147. doi: 10.1186/s12944-022-01755-2
33. Huot JR, Novinger LJ, Pin F, Bonetto A. HCT116 colorectal liver metastases exacerbate muscle wasting in a mouse model for the study of colorectal cancer cachexia. Dis Model Mech. (2020) 13(1):dmm043166. doi: 10.1242/dmm.043166
34. Barreto R, Mandili G, Witzmann FA, Novelli F, Zimmers TA, Bonetto A. Cancer and chemotherapy contribute to muscle loss by activating common signaling pathways. Front Physiol. (2016) 7:472. doi: 10.3389/fphys.2016.00472
35. Alonso-Molero J, González-Donquiles C, Fernández-Villa T, de Souza-Teixeira F, Vilorio-Marqués L, Molina AJ, et al. Alterations in PGC1α expression levels are involved in colorectal cancer risk: a qualitative systematic review. BMC Cancer. (2017) 17:731. doi: 10.1186/s12885-017-3725-3
36. Yun SH, Roh MS, Jeong JS, Park JI. Peroxisome proliferator-activated receptor γ coactivator-1α is a predictor of lymph node metastasis and poor prognosis in human colorectal cancer. Ann Diagn Pathol. (2018) 33:11–6. doi: 10.1016/j.anndiagpath.2017.11.007
37. Cho JG, Park SJ, Han SH, Park JI. PGC-1α Regulates cell proliferation, migration, and invasion by modulating leucyl-tRNA synthetase 1 expression in human colorectal cancer cells. Cancers (Basel). (2022) 15(1):159. doi: 10.3390/cancers15010159
38. Yun SH, Shin SW, Park JI. Expression of fatty acid synthase is regulated by PGC−1α and contributes to increased cell proliferation. Oncol Rep. (2017) 38:3497–506. doi: 10.3892/or
39. Yun SH, Park JI. PGC-1α Regulates cell proliferation and invasion via AKT/GSK-3β/β-catenin pathway in human colorectal cancer SW620 and SW480 cells. Anticancer Res. (2020) 40:653–64. doi: 10.21873/anticanres.13995
40. Yun CW, Han YS, Lee SH. PGC-1α Controls mitochondrial biogenesis in drug-resistant colorectal cancer cells by regulating endoplasmic reticulum stress. Int J Mol Sci. (2019) 20(7):1707. doi: 10.3390/ijms20071707
41. Vellinga TT, Borovski T, de Boer VC, van Schelven S, Trumpi K, Verheem A, et al. SIRT1/PGC1α-dependent increase in oxidative phosphorylation supports chemotherapy resistance of colon cancer. Clin Cancer Res. (2015) 21:2870–9. doi: 10.1158/1078-0432.CCR-14-2290
42. Afrin S, Giampieri F, Gasparrini M, Forbes-Hernández TY, Cianciosi D, Reboredo-Rodriguez P, et al. The inhibitory effect of Manuka honey on human colon cancer HCT-116 and LoVo cell growth. Part 2: Induction of oxidative stress, alteration of mitochondrial respiration and glycolysis, and suppression of metastatic ability. Food Funct. (2018) 9:2158–70. doi: 10.1039/C8FO00165K
43. Kamarudin MNA, Sarker MMR, Zhou JR, Parhar I. Metformin in colorectal cancer: molecular mechanism, preclinical and clinical aspects. J Exp Clin Cancer Res. (2019) 38:491. doi: 10.1186/s13046-019-1495-2
44. Wang P, Guo X, Zong W, Li Y, Liu G, Lv Y, et al. PGC-1α/SNAI1 axis regulates tumor growth and metastasis by targeting miR-128b in gastric cancer. J Cell Physiol. (2019) 234:17232–41. doi: 10.1002/jcp.28193
45. Hu XT, Ding C, Zhou N, Xu C. Quercetin protects gastric epithelial cell from oxidative damage in vitro and in vivo. Eur J Pharmacol. (2015) 754:115–24. doi: 10.1016/j.ejphar.2015.02.007
46. Li W, Wong CC, Zhang X, Kang W, Nakatsu G, Zhao Q, et al. CAB39L elicited an anti-Warburg effect via a LKB1-AMPK-PGC1α axis to inhibit gastric tumorigenesis. Oncogene. (2018) 37:6383–98. doi: 10.1038/s41388-018-0402-1
47. Pavlova NN, Thompson CB. The emerging hallmarks of cancer metabolism. Cell Metab. (2016) 23:27–47. doi: 10.1016/j.cmet.2015.12.006
48. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
49. Wu H, Liu B, Chen Z, Li G, Zhang Z. MSC-induced lncRNA HCP5 drove fatty acid oxidation through miR-3619-5p/AMPK/PGC1α/CEBPB axis to promote stemness and chemo-resistance of gastric cancer. Cell Death Dis. (2020) 11:233. doi: 10.1038/s41419-020-2426-z
50. Foda ZH, Annapragada AV, Boyapati K, Bruhm DC, Vulpescu NA, Medina JE, et al. Detecting liver cancer using cell-free DNA fragmentomes. Cancer Discovery. (2023) 13:616–31. doi: 10.1158/2159-8290.CD-22-0659
51. Toh MR, Wong EYT, Wong SH, Ng AWT, Loo LH, Chow PK, et al. Global epidemiology and genetics of hepatocellular carcinoma. Gastroenterology. (2023) 164:766–82. doi: 10.1053/j.gastro.2023.01.033
52. Åberg F, Byrne CD, Pirola CJ, Männistö V, Sookoian S. Alcohol consumption and metabolic syndrome: Clinical and epidemiological impact on liver disease. J Hepatol. (2023) 78:191–206. doi: 10.1016/j.jhep.2022.08.030
53. Wu D, Yang Y, Hou Y, Zhao Z, Liang N, Yuan P, et al. Increased mitochondrial fission drives the reprogramming of fatty acid metabolism in hepatocellular carcinoma cells through suppression of Sirtuin 1. Cancer Commun (Lond). (2022) 42:37–55. doi: 10.1002/cac2.12247
54. Kim H, Lee JY, Park SJ, Kwag E, Koo O, Shin JH. ZNF746/PARIS promotes the occurrence of hepatocellular carcinoma. Biochem Biophys Res Commun. (2021) 563:98–104. doi: 10.1016/j.bbrc.2021.05.051
55. Kumar A, Giri S, Shaha C. Sestrin2 facilitates glutamine-dependent transcription of PGC-1α and survival of liver cancer cells under glucose limitation. FEBS J. (2018) 285:1326–45. doi: 10.1111/febs.14406
56. Hu Y, Shin DJ, Pan H, Lin Z, Dreyfuss JM, Camargo FD, et al. YAP suppresses gluconeogenic gene expression through PGC1α. Hepatology. (2017) 66:2029–41. doi: 10.1002/hep.29373
57. Geng X, Geng Z, Li H, Zhang Y, Li J, Chang H. Over-expression of TFB2M facilitates cell growth and metastasis via activating ROS-Akt-NF-κB signalling in hepatocellular carcinoma. Liver Int. (2020) 40:1756–69. doi: 10.1111/liv.14440
58. Chang H, Li J, Luo Y, Wu B, Yuan C, Geng X. TFB2M activates aerobic glycolysis in hepatocellular carcinoma cells through the NAD(+) /SIRT3/HIF-1α signaling. J Gastroenterol Hepatol. (2021) 36:2978–88. doi: 10.1111/jgh.15548
59. Zhu L, Wang Q, Zhang L, Fang Z, Zhao F, Lv Z, et al. Hypoxia induces PGC-1α expression and mitochondrial biogenesis in the myocardium of TOF patients. Cell Res. (2010) 20:676–87. doi: 10.1038/cr.2010.46
60. Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. (2008) 451:1008–12. doi: 10.1038/nature06613
61. Tohme S, Yazdani HO, Liu Y, Loughran P, van der Windt DJ, Huang H, et al. Hypoxia mediates mitochondrial biogenesis in hepatocellular carcinoma to promote tumor growth through HMGB1 and TLR9 interaction. Hepatology. (2017) 66:182–97. doi: 10.1002/hep.29184
62. Yao G, Yin J, Wang Q, Dong R, Lu J. Glypican-3 enhances reprogramming of glucose metabolism in liver cancer cells. BioMed Res Int. (2019) 2019:2560650. doi: 10.1155/2019/2560650
63. Iansante V, Choy PM, Fung SW, Liu Y, Chai JG, Dyson J, et al. PARP14 promotes the Warburg effect in hepatocellular carcinoma by inhibiting JNK1-dependent PKM2 phosphorylation and activation. Nat Commun. (2015) 6:7882. doi: 10.1038/ncomms8882
64. Icard P, Simula L, Wu Z, Berzan D, Sogni P, Dohan A, et al. Why may citrate sodium significantly increase the effectiveness of transarterial chemoembolization in hepatocellular carcinoma? Drug Resist Update. (2021) 59:100790. doi: 10.1016/j.drup.2021.100790
65. Gunewardena S, Huck I, Walesky C, Robarts D, Weinman S, Apte U. Progressive loss of hepatocyte nuclear factor 4 alpha activity in chronic liver diseases in humans. Hepatology. (2022) 76:372–86. doi: 10.1002/hep.32326
66. Liu Q, Li J, Zhang W, Xiao C, Zhang S, Nian C, et al. Glycogen accumulation and phase separation drives liver tumor initiation. Cell. (2021) 184:5559–76.e19. doi: 10.1016/j.cell.2021.10.001
67. Yu Y, Peng XD, Qian XJ, Zhang KM, Huang X, Chen YH, et al. Fis1 phosphorylation by Met promotes mitochondrial fission and hepatocellular carcinoma metastasis. Signal Transduct Target Ther. (2021) 6:401. doi: 10.1038/s41392-021-00790-2
68. Li S, Han S, Zhang Q, Zhu Y, Zhang H, Wang J, et al. FUNDC2 promotes liver tumorigenesis by inhibiting MFN1-mediated mitochondrial fusion. Nat Commun. (2022) 13:3486. doi: 10.1038/s41467-022-31187-6
69. Huang D, Li T, Li X, Zhang L, Sun L, He X, et al. HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. (2014) 8:1930–42. doi: 10.1016/j.celrep.2014.08.028
70. Wu JM, Skill NJ, Maluccio MA. Evidence of aberrant lipid metabolism in hepatitis C and hepatocellular carcinoma. HPB (Oxford). (2010) 12:625–36. doi: 10.1111/j.1477-2574.2010.00207.x
71. Mutlu B, Puigserver P. GCN5 acetyltransferase in cellular energetic and metabolic processes. Biochim Biophys Acta Gene Regul Mech. (2021) 1864:194626. doi: 10.1016/j.bbagrm.2020.194626
72. Resar LM. The high mobility group A1 gene: transforming inflammatory signals into cancer? Cancer Res. (2010) 70:436–9. doi: 10.1158/0008-5472.CAN-09-1212
73. Shen Z, Wu J, Gao Z, Zhang S, Chen J, He J, et al. High mobility group AT-hook 1 (HMGA1) is an important positive regulator of hepatitis B virus (HBV) that is reciprocally upregulated by HBV X protein. Nucleic Acids Res. (2022) 50:2157–71. doi: 10.1093/nar/gkac070
74. Kar A, Samanta A, Mukherjee S, Barik S, Biswas A. The HBV web: An insight into molecular interactomes between the hepatitis B virus and its host en route to hepatocellular carcinoma. J Med Virol. (2023) 95:e28436. doi: 10.1002/jmv.28436
75. Papatheodoridis GV, Lekakis V, Voulgaris T, Lampertico P, Berg T, Chan HLY, et al. Hepatitis B virus reactivation associated with new classes of immunosuppressants and immunomodulators: A systematic review, meta-analysis, and expert opinion. J Hepatol. (2022) 77:1670–89. doi: 10.1016/j.jhep.2022.07.003
76. Melis M, Diaz G, Kleiner DE, Zamboni F, Kabat J, Lai J, et al. Viral expression and molecular profiling in liver tissue versus microdissected hepatocytes in hepatitis B virus-associated hepatocellular carcinoma. J Transl Med. (2014) 12:230. doi: 10.1186/s12967-014-0230-1
77. Deng JJ, Kong KE, Gao WW, Tang HV, Chaudhary V, Cheng Y, et al. Interplay between SIRT1 and hepatitis B virus X protein in the activation of viral transcription. Biochim Biophys Acta Gene Regul Mech. (2017) 1860:491–501. doi: 10.1016/j.bbagrm.2017.02.007
78. Gehrke N, Wörns MA, Huber Y, Hess M, Straub BK, Hövelmeyer N, et al. Hepatic B cell leukemia-3 promotes hepatic steatosis and inflammation through insulin-sensitive metabolic transcription factors. J Hepatol. (2016) 65:1188–97. doi: 10.1016/j.jhep.2016.06.026
79. Wan X, Zhu X, Wang H, Feng Y, Zhou W, Liu P, et al. PGC1α protects against hepatic steatosis and insulin resistance via enhancing IL10-mediated anti-inflammatory response. FASEB J. (2020) 34:10751–61. doi: 10.1096/fj.201902476R
80. Xu L, Huang Z, Lo TH, Lee JTH, Yang R, Yan X, et al. Hepatic PRMT1 ameliorates diet-induced hepatic steatosis via induction of PGC1α. Theranostics. (2022) 12:2502–18. doi: 10.7150/thno.63824
81. Qi J, Chen X, Wu Q, Wang J, Zhang H, Mao A, et al. Fasting induces hepatocellular carcinoma cell apoptosis by inhibiting SET8 expression. Oxid Med Cell Longev. (2020) 2020:3985089. doi: 10.1155/2020/3985089
82. Kudo Y, Sugimoto M, Arias E, Kasashima H, Cordes T, Linares JF, et al. PKCλ/ι Loss induces autophagy, oxidative phosphorylation, and NRF2 to promote liver cancer progression. Cancer Cell. (2020) 38:247–62.e11. doi: 10.1016/j.ccell.2020.05.018
83. Yang C, Cui XW, Ding ZW, Jiang TY, Feng XF, Pan YF, et al. Gankyrin and TIGAR cooperatively accelerate glucose metabolism toward the PPP and TCA cycle in hepatocellular carcinoma. Cancer Sci. (2022) 113:4151–64. doi: 10.1111/cas.15593
84. Jiang J, Chen HN, Jin P, Zhou L, Peng L, Huang Z, et al. Targeting PSAT1 to mitigate metastasis in tumors with p53-72Pro variant. Signal Transduct Target Ther. (2023) 8:65. doi: 10.1038/s41392-022-01266-7
85. Li Y, Wu J, Song F, Tang J, Wang SJ, Yu XL, et al. Extracellular membrane-proximal domain of HAb18G/CD147 binds to metal ion-dependent adhesion site (MIDAS) motif of integrin β1 to modulate Malignant properties of hepatoma cells. J Biol Chem. (2012) 287:4759–72. doi: 10.1074/jbc.M111.277699
86. Huang Q, Li J, Xing J, Li W, Li H, Ke X, et al. CD147 promotes reprogramming of glucose metabolism and cell proliferation in HCC cells by inhibiting the p53-dependent signaling pathway. J Hepatol. (2014) 61:859–66. doi: 10.1016/j.jhep.2014.04.035
87. Foreman JE, Koga T, Kosyk O, Kang BH, Zhu X, Cohen SM, et al. Diminished hepatocarcinogenesis by a potent, high-affinity human PPARα Agonist in PPARA-humanized mice. Toxicol Sci. (2021) 183:70–80. doi: 10.1093/toxsci/kfab067
88. Foreman JE, Koga T, Kosyk O, Kang BH, Zhu X, Cohen SM, et al. Species differences between mouse and human PPARα in modulating the hepatocarcinogenic effects of perinatal exposure to a high-affinity human PPARα Agonist in mice. Toxicol Sci. (2021) 183:81–92. doi: 10.1093/toxsci/kfab068
89. Nam H, Kundu A, Brinkley GJ, Chandrashekar DS, Kirkman RL, Chakravarthi B, et al. PGC1α suppresses kidney cancer progression by inhibiting collagen-induced SNAIL expression. Matrix Biol. (2020) 89:43–58. doi: 10.1016/j.matbio.2020.01.001
90. LaGory EL, Wu C, Taniguchi CM, Ding CC, Chi JT, von Eyben R, et al. Suppression of PGC-1α Is critical for reprogramming oxidative metabolism in renal cell carcinoma. Cell Rep. (2015) 12:116–27. doi: 10.1016/j.celrep.2015.06.006
91. Zhuang C, Zhuang C, Luo X, Huang X, Yao L, Li J, et al. N6-methyladenosine demethylase FTO suppresses clear cell renal cell carcinoma through a novel FTO-PGC-1α signalling axis. J Cell Mol Med. (2019) 23:2163–73. doi: 10.1111/jcmm.14128
92. Nam H, Kundu A, Karki S, Brinkley GJ, Chandrashekar DS, Kirkman RL, et al. The TGF-β/HDAC7 axis suppresses TCA cycle metabolism in renal cancer. JCI Insight. (2021) 6(22):e148438. doi: 10.1172/jci.insight.148438
93. Ivanov SV, Salnikow K, Ivanova AV, Bai L, Lerman MI. Hypoxic repression of STAT1 and its downstream genes by a pVHL/HIF-1 target DEC1/STRA13. Oncogene. (2007) 26:802–12. doi: 10.1038/sj.onc.1209842
94. Yamada Y, Sugawara S, Arai T, Kojima S, Kato M, Okato A, et al. Molecular pathogenesis of renal cell carcinoma: Impact of the anti-tumor miR-29 family on gene regulation. Int J Urol. (2018) 25:953–65. doi: 10.1111/iju.13783
95. Koh E, Kim YK, Shin D, Kim KS. MPC1 is essential for PGC-1α-induced mitochondrial respiration and biogenesis. Biochem J. (2018) 475:1687–99. doi: 10.1042/BCJ20170967
96. Dan L, Wang C, Ma P, Yu Q, Gu M, Dong L, et al. PGC1α promotes cholangiocarcinoma metastasis by upregulating PDHA1 and MPC1 expression to reverse the Warburg effect. Cell Death Dis. (2018) 9:466. doi: 10.1038/s41419-018-0494-0
97. Felipe-Abrio B, Carnero A. The tumor suppressor roles of MYBBP1A, a major contributor to metabolism plasticity and stemness. Cancers (Basel). (2020) 12(1):254. doi: 10.3390/cancers12010254
98. Felipe-Abrio B, Verdugo-Sivianes EM, Carnero A. c-MYB- and PGC1a-dependent metabolic switch induced by MYBBP1A loss in renal cancer. Mol Oncol. (2019) 13:1519–33. doi: 10.3390/cancers12010254
99. Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. (2013) 499:43–9. doi: 10.1038/nature12222
100. Xie Y, Sahin M, Sinha S, Wang Y, Nargund AM, Lyu Y, et al. SETD2 loss perturbs the kidney cancer epigenetic landscape to promote metastasis and engenders actionable dependencies on histone chaperone complexes. Nat Cancer. (2022) 3:188–202. doi: 10.1038/s43018-021-00316-3
101. Kanu N, Grönroos E, Martinez P, Burrell RA, Yi Goh X, Bartkova J, et al. SETD2 loss-of-function promotes renal cancer branched evolution through replication stress and impaired DNA repair. Oncogene. (2015) 34:5699–708. doi: 10.1038/onc.2015.24
102. Liu J, Hanavan PD, Kras K, Ruiz YW, Castle EP, Lake DF, et al. Loss of SETD2 induces a metabolic switch in renal cell carcinoma cell lines toward enhanced oxidative phosphorylation. J Proteome Res. (2019) 18:331–40. doi: 10.1021/acs.jproteome.8b00628
103. Raggi C, Taddei ML, Rae C, Braconi C, Marra F. Metabolic reprogramming in cholangiocarcinoma. J Hepatol. (2022) 77:849–64. doi: 10.1016/j.jhep.2022.04.038
104. Colyn L, Alvarez-Sola G, Latasa MU, Uriarte I, Herranz JM, Arechederra M, et al. New molecular mechanisms in cholangiocarcinoma: signals triggering interleukin-6 production in tumor cells and KRAS co-opted epigenetic mediators driving metabolic reprogramming. J Exp Clin Cancer Res. (2022) 41:183. doi: 10.1186/s13046-022-02386-2
105. Raggi C, Taddei ML, Sacco E, Navari N, Correnti M, Piombanti B, et al. Mitochondrial oxidative metabolism contributes to a cancer stem cell phenotype in cholangiocarcinoma. J Hepatol. (2021) 74:1373–85. doi: 10.1016/j.jhep.2020.12.031
106. Nguyen TTT, Shang E, Shu C, Kim S, Mela A, Humala N, et al. Aurora kinase A inhibition reverses the Warburg effect and elicits unique metabolic vulnerabilities in glioblastoma. Nat Commun. (2021) 12:5203. doi: 10.1038/s41467-021-25501-x
107. Cho SY, Kim SH, Yi MH, Zhang E, Kim E, Park J, et al. Expression of PGC1α in glioblastoma multiforme patients. Oncol Lett. (2017) 13:4055–76. doi: 10.3892/ol.2017.5972
108. Bruns I, Sauer B, Burger MC, Eriksson J, Hofmann U, Braun Y, et al. Disruption of peroxisome proliferator-activated receptor γ coactivator (PGC)-1α reverts key features of the neoplastic phenotype of glioma cells. J Biol Chem. (2019) 294:3037–50. doi: 10.1074/jbc.RA118.006993
109. Comelli M, Pretis I, Buso A, Mavelli I. Mitochondrial energy metabolism and signalling in human glioblastoma cell lines with different PTEN gene status. J Bioenerg Biomembr. (2018) 50:33–52. doi: 10.1007/s10863-017-9737-5
110. Čančer M, Drews LF, Bengtsson J, Bolin S, Rosén G, Westermark B, et al. BET and Aurora Kinase A inhibitors synergize against MYCN-positive human glioblastoma cells. Cell Death Dis. (2019) 10:881. doi: 10.1038/s41419-019-2120-1
111. Nguyen TTT, Zhang Y, Shang E, Shu C, Torrini C, Zhao J, et al. HDAC inhibitors elicit metabolic reprogramming by targeting super-enhancers in glioblastoma models. J Clin Invest. (2020) 130:3699–716. doi: 10.1172/JCI129049
112. Meng X, Zhao Y, Han B, Zha C, Zhang Y, Li Z, et al. Dual functionalized brain-targeting nanoinhibitors restrain temozolomide-resistant glioma via attenuating EGFR and MET signaling pathways. Nat Commun. (2020) 11:594. doi: 10.1038/s41467-019-14036-x
113. Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell. (2012) 22:21–35. doi: 10.1016/j.ccr.2012.05.037
114. Zhang Y, Nguyen TTT, Shang E, Mela A, Humala N, Mahajan A, et al. MET inhibition elicits PGC1α-dependent metabolic reprogramming in glioblastoma. Cancer Res. (2020) 80:30–43. doi: 10.1158/0008-5472.CAN-19-1389
115. Fan QW, Nicolaides TP, Weiss WA. Inhibiting 4EBP1 in glioblastoma. Clin Cancer Res. (2018) 24:14–21. doi: 10.1158/1078-0432.CCR-17-0042
116. Thiepold AL, Lorenz NI, Foltyn M, Engel AL, Divé I, Urban H, et al. Mammalian target of rapamycin complex 1 activation sensitizes human glioma cells to hypoxia-induced cell death. Brain. (2017) 140:2623–38. doi: 10.1093/brain/awx196
117. Ronellenfitsch MW, Brucker DP, Burger MC, Wolking S, Tritschler F, Rieger J, et al. Antagonism of the mammalian target of rapamycin selectively mediates metabolic effects of epidermal growth factor receptor inhibition and protects human Malignant glioma cells from hypoxia-induced cell death. Brain. (2009) 132:1509–22. doi: 10.1093/brain/awp093
118. Singh D, Chan JM, Zoppoli P, Niola F, Sullivan R, Castano A, et al. Transforming fusions of FGFR and TACC genes in human glioblastoma. Science. (2012) 337:1231–5. doi: 10.1126/science.1220834
119. Lasorella A, Sanson M, Iavarone A. FGFR-TACC gene fusions in human glioma. Neuro Oncol. (2017) 19:475–83. doi: 10.1093/neuonc/now240
120. Frattini V, Pagnotta SM, Tala, Fan JJ, Russo MV, Lee SB, et al. A metabolic function of FGFR3-TACC3 gene fusions in cancer. Nature. (2018) 553:222–7. doi: 10.1038/nature25171
121. Hoek KS, Eichhoff OM, Schlegel NC, Döbbeling U, Kobert N, Schaerer L, et al. In vivo switching of human melanoma cells between proliferative and invasive states. Cancer Res. (2008) 68:650–6. doi: 10.1158/0008-5472.CAN-07-2491
122. Luo C, Balsa E, Perry EA, Liang J, Tavares CD, Vazquez F, et al. H3K27me3-mediated PGC1α gene silencing promotes melanoma invasion through WNT5A and YAP. J Clin Invest. (2020) 130:853–62. doi: 10.1172/jci130038
123. Vazquez F, Lim JH, Chim H, Bhalla K, Girnun G, Pierce K, et al. PGC1α expression defines a subset of human melanoma tumors with increased mitochondrial capacity and resistance to oxidative stress. Cancer Cell. (2013) 23:287–301. doi: 10.1016/j.ccr.2012.11.020
124. Hartman ML, Czyz M. MITF in melanoma: mechanisms behind its expression and activity. Cell Mol Life Sci. (2015) 72:1249–60. doi: 10.1007/s00018-014-1791-0
125. Wang H, Zhang H, Chen Y, Wang H, Tian Y, Yi X, et al. Targeting wnt/β-catenin signaling exacerbates ferroptosis and increases the efficacy of melanoma immunotherapy via the regulation of MITF. Cells. (2022) 11(22):3580. doi: 10.3390/cells11223580
126. Guo W, Ma J, Yang Y, Guo S, Zhang W, Zhao T, et al. ATP-citrate lyase epigenetically potentiates oxidative phosphorylation to promote melanoma growth and adaptive resistance to MAPK inhibition. Clin Cancer Res. (2020) 26:2725–39. doi: 10.1158/1078-0432.CCR-19-1359
127. Talty R, Bosenberg M. The role of ferroptosis in melanoma. Pigment Cell Melanoma Res. (2022) 35:18–25. doi: 10.1111/pcmr.13009
128. DeVito NC, Sturdivant M, Thievanthiran B, Xiao C, Plebanek MP, Salama AKS, et al. Pharmacological Wnt ligand inhibition overcomes key tumor-mediated resistance pathways to anti-PD-1 immunotherapy. Cell Rep. (2021) 35:109071. doi: 10.1016/j.celrep.2021.109071
129. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature. (2015) 523:231–5. doi: 10.1038/nature14404
130. Gelato KA, Schöckel L, Klingbeil O, Rückert T, Lesche R, Toedling J, et al. Super-enhancers define a proliferative PGC-1α-expressing melanoma subgroup sensitive to BET inhibition. Oncogene. (2018) 37:512–21. doi: 10.1038/onc.2017.325
131. Luo C, Lim JH, Lee Y, Granter SR, Thomas A, Vazquez F, et al. A PGC1α-mediated transcriptional axis suppresses melanoma metastasis. Nature. (2016) 537:422–6. doi: 10.1038/nature19347
132. Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, et al. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. (2015) 527:186–91. doi: 10.1038/nature15726
133. Hoffmann F, Niebel D, Aymans P, Ferring-Schmitt S, Dietrich D, Landsberg J. H3K27me3 and EZH2 expression in melanoma: relevance for melanoma progression and response to immune checkpoint blockade. Clin Epigenetics. (2020) 12:24. doi: 10.1186/s13148-020-0818-7
134. Cai Y, Zhang Y, Loh YP, Tng JQ, Lim MC, Cao Z, et al. H3K27me3-rich genomic regions can function as silencers to repress gene expression via chromatin interactions. Nat Commun. (2021) 12:719. doi: 10.1038/s41467-021-20940-y
135. Haq R, Fisher DE, Widlund HR. Molecular pathways: BRAF induces bioenergetic adaptation by attenuating oxidative phosphorylation. Clin Cancer Res. (2014) 20:2257–63. doi: 10.1158/1078-0432.CCR-13-0898
136. Lee SB, Garofano L, Ko A, D'Angelo F, Frangaj B, Sommer D, et al. Regulated interaction of ID2 with the anaphase-promoting complex links progression through mitosis with reactivation of cell-type-specific transcription. Nat Commun. (2022) 13:2089. doi: 10.1038/s41467-022-29502-2
137. Yu Y, Wu J, Wang Y, Zhao T, Ma B, Liu Y, et al. Kindlin 2 forms a transcriptional complex with β-catenin and TCF4 to enhance Wnt signalling. EMBO Rep. (2012) 13:750–8. doi: 10.1038/embor.2012.88
138. Park HW, Kim YC, Yu B, Moroishi T, Mo JS, Plouffe SW, et al. Alternative wnt signaling activates YAP/TAZ. Cell. (2015) 162:780–94. doi: 10.1016/j.cell.2015.07.013
139. Astudillo P. An emergent Wnt5a/YAP/TAZ regulatory circuit and its possible role in cancer. Semin Cell Dev Biol. (2022) 125:45–54. doi: 10.1016/j.semcdb.2021.10.001
140. Haq R, Shoag J, Andreu-Perez P, Yokoyama S, Edelman H, Rowe GC, et al. Oncogenic BRAF regulates oxidative metabolism via PGC1α and MITF. Cancer Cell. (2013) 23:302–15. doi: 10.1016/j.ccr.2013.02.003
141. Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. (2010) 467:596–9. doi: 10.1038/nature09454
142. Corno C, Perego P. KiSS1 in regulation of metastasis and response to antitumor drugs. Drug Resist Updat. (2019) 42:12–21. doi: 10.1016/j.drup.2019.02.001
143. Liu W, Beck BH, Vaidya KS, Nash KT, Feeley KP, Ballinger SW, et al. Metastasis suppressor KISS1 seems to reverse the Warburg effect by enhancing mitochondrial biogenesis. Cancer Res. (2014) 74:954–63. doi: 10.1158/0008-5472.CAN-13-1183
144. Manley SJ, Liu W, Welch DR. The KISS1 metastasis suppressor appears to reverse the Warburg effect by shifting from glycolysis to mitochondrial beta-oxidation. J Mol Med (Berl). (2017) 95:951–63. doi: 10.1007/s00109-017-1552-2
145. Lim JH, Luo C, Vazquez F, Puigserver P. Targeting mitochondrial oxidative metabolism in melanoma causes metabolic compensation through glucose and glutamine utilization. Cancer Res. (2014) 74:3535–45. doi: 10.1158/0008-5472.CAN-13-2893-T
146. Torrano V, Valcarcel-Jimenez L, Cortazar AR, Liu X, Urosevic J, Castillo-Martin M, et al. The metabolic co-regulator PGC1α suppresses prostate cancer metastasis. Nat Cell Biol. (2016) 18:645–56. doi: 10.1038/ncb3357
147. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery. (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
148. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. (2013) 6:pl1. doi: 10.1126/scisignal.2004088
149. Siddappa M, Wani SA, Long MD, Leach DA, Mathé EA, Bevan CL, et al. Identification of transcription factor co-regulators that drive prostate cancer progression. Sci Rep. (2020) 10:20332. doi: 10.1038/s41598-020-77055-5
150. Wen Y, Huang H, Huang B, Liao X. HSA-miR-34a-5p regulates the SIRT1/TP53 axis in prostate cancer. Am J Transl Res. (2022) 14:4493–504.
151. Tennakoon JB, Shi Y, Han JJ, Tsouko E, White MA, Burns AR, et al. Androgens regulate prostate cancer cell growth via an AMPK-PGC-1α-mediated metabolic switch. Oncogene. (2014) 33:5251–61. doi: 10.1038/onc.2013.463
152. Valcarcel-Jimenez L, Macchia A, Crosas-Molist E, Schaub-Clerigué A, Camacho L, Martín-Martín N, et al. PGC1α Suppresses prostate cancer cell invasion through ERRα Transcriptional control. Cancer Res. (2019) 79:6153–65. doi: 10.1158/0008-5472.CAN-19-1231
153. Kaminski L, Torrino S, Dufies M, Djabari Z, Haider R, Roustan FR, et al. PGC1α Inhibits polyamine synthesis to suppress prostate cancer aggressiveness. Cancer Res. (2019) 79:3268–80. doi: 10.1158/0008-5472.CAN-18-2043
154. Penfold L, Woods A, Pollard AE, Arizanova J, Pascual-Navarro E, Muckett PJ, et al. AMPK activation protects against prostate cancer by inducing a catabolic cellular state. Cell Rep. (2023) 42:112396. doi: 10.1016/j.celrep.2023.112396
155. Penfold L, Woods A, Muckett P, Nikitin AY, Kent TR, Zhang S, et al. CAMKK2 promotes prostate cancer independently of AMPK via increased lipogenesis. Cancer Res. (2018) 78:6747–61. doi: 10.1158/0008-5472.CAN-18-0585
156. Yuan H, Han Y, Wang X, Li N, Liu Q, Yin Y, et al. SETD2 restricts prostate cancer metastasis by integrating EZH2 and AMPK signaling pathways. Cancer Cell. (2020) 38:350–65.e7. doi: 10.1016/j.ccell.2020.05.022
157. Li J, Li Y, Chen L, Yu B, Xue Y, Guo R, et al. p53/PGC−1α−mediated mitochondrial dysfunction promotes PC3 prostate cancer cell apoptosis. Mol Med Rep. (2020) 22:155–64. doi: 10.3892/mmr
158. McCann JJ, Vasilevskaya IA, McNair C, Gallagher P, Neupane NP, de Leeuw R, et al. Mutant p53 elicits context-dependent pro-tumorigenic phenotypes. Oncogene. (2022) 41:444–58. doi: 10.1038/s41388-021-01903-5
159. Aquilano K, Baldelli S, Pagliei B, Cannata SM, Rotilio G, Ciriolo MR. p53 orchestrates the PGC-1α-mediated antioxidant response upon mild redox and metabolic imbalance. Antioxid Redox Signal. (2013) 18:386–99. doi: 10.1089/ars.2012.4615
160. Galbraith LCA, Mui E, Nixon C, Hedley A, Strachan D, MacKay G, et al. PPAR-gamma induced AKT3 expression increases levels of mitochondrial biogenesis driving prostate cancer. Oncogene. (2021) 40:2355–66. doi: 10.1038/s41388-021-01707-7
161. Babu D, Fullwood MJ. Expanding the effects of ERG on chromatin landscapes and dysregulated transcription in prostate cancer. Nat Genet. (2017) 49:1294–5. doi: 10.1038/ng.3944
162. Signorile A, De Rasmo D, Cormio A, Musicco C, Rossi R, Fortarezza F, et al. Human ovarian cancer tissue exhibits increase of mitochondrial biogenesis and cristae remodeling. Cancers (Basel). (2019) 11(9):1350. doi: 10.3390/cancers11091350
163. Huang X, Ruan G, Liu G, Gao Y, Sun P. Immunohistochemical analysis of PGC-1α and ERRα Expression reveals their clinical significance in human ovarian cancer. Onco Targets Ther. (2020) 13:13055–62. doi: 10.2147/OTT.S288332
164. Ghilardi C, Moreira-Barbosa C, Brunelli L, Ostano P, Panini N, Lupi M, et al. PGC1α/β Expression predicts therapeutic response to oxidative phosphorylation inhibition in ovarian cancer. Cancer Res. (2022) 82:1423–34. doi: 10.1158/0008-5472.CAN-21-1223
165. Gentric G, Kieffer Y, Mieulet V, Goundiam O, Bonneau C, Nemati F, et al. PML-regulated mitochondrial metabolism enhances chemosensitivity in human ovarian cancers. Cell Metab. (2019) 29:156–73.e10. doi: 10.1016/j.cmet.2018.09.002
166. Izar B, Tirosh I, Stover EH, Wakiro I, Cuoco MS, Alter I, et al. A single-cell landscape of high-grade serous ovarian cancer. Nat Med. (2020) 26:1271–9. doi: 10.1038/s41591-020-0926-0
167. Nath A, Cosgrove PA, Mirsafian H, Christie EL, Pflieger L, Copeland B, et al. Evolution of core archetypal phenotypes in progressive high grade serous ovarian cancer. Nat Commun. (2021) 12:3039. doi: 10.1038/s41467-021-23171-3
168. Kleih M, Böpple K, Dong M, Gaißler A, Heine S, Olayioye MA, et al. Direct impact of cisplatin on mitochondria induces ROS production that dictates cell fate of ovarian cancer cells. Cell Death Dis. (2019) 10:851. doi: 10.1038/s41419-019-2081-4
169. Du Q, Tan Z, Shi F, Tang M, Xie L, Zhao L, et al. PGC1α/CEBPB/CPT1A axis promotes radiation resistance of nasopharyngeal carcinoma through activating fatty acid oxidation. Cancer Sci. (2019) 110:2050–62. doi: 10.1111/cas.14011
170. Quan J, Li N, Tan Y, Liu H, Liao W, Cao Y, et al. PGC1α-mediated fatty acid oxidation promotes TGFβ1-induced epithelial-mesenchymal transition and metastasis of nasopharyngeal carcinoma. Life Sci. (2022) 300:120558. doi: 10.1016/j.lfs.2022.120558
171. Zu Y, Chen XF, Li Q, Zhang ST, Si LN. PGC-1α activates SIRT3 to modulate cell proliferation and glycolytic metabolism in breast cancer. Neoplasma. (2021) 68:352–61. doi: 10.4149/neo_2020_200530N584
172. Deblois G, St-Pierre J, Giguère V. The PGC-1/ERR signaling axis in cancer. Oncogene. (2013) 32:3483–90. doi: 10.1038/onc.2012.529
173. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
174. Torrens-Mas M, Pons DG, Sastre-Serra J, Oliver J, Roca P. SIRT3 silencing sensitizes breast cancer cells to cytotoxic treatments through an increment in ROS production. J Cell Biochem. (2017) 118:397–406. doi: 10.1002/jcb.v118.2
175. Lou C, Xiao M, Cheng S, Lu X, Jia S, Ren Y, et al. MiR-485-3p and miR-485-5p suppress breast cancer cell metastasis by inhibiting PGC-1α expression. Cell Death Dis. (2016) 7:e2159. doi: 10.1038/cddis.2016.27
176. Zhang S, Liu X, Liu J, Guo H, Xu H, Zhang G. PGC-1 alpha interacts with microRNA-217 to functionally regulate breast cancer cell proliferation. BioMed Pharmacother. (2017) 85:541–8. doi: 10.1016/j.biopha.2016.11.062
177. McGuirk S, Gravel SP, Deblois G, Papadopoli DJ, Faubert B, Wegner A, et al. PGC-1α supports glutamine metabolism in breast cancer. Cancer Metab. (2013) 1:22. doi: 10.1186/2049-3002-1-22
178. Deblois G, Hall JA, Perry MC, Laganière J, Ghahremani M, Park M, et al. Genome-wide identification of direct target genes implicates estrogen-related receptor alpha as a determinant of breast cancer heterogeneity. Cancer Res. (2009) 69:6149–57. doi: 10.1158/0008-5472.CAN-09-1251
179. Andrzejewski S, Klimcakova E, Johnson RM, Tabariès S, Annis MG, McGuirk S, et al. PGC-1α Promotes breast cancer metastasis and confers bioenergetic flexibility against metabolic drugs. Cell Metab. (2017) 26:778–87.e5. doi: 10.1016/j.cmet.2017.09.006
180. LeBleu VS, O'Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, et al. PGC-1α mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat Cell Biol. (2014) 16:992–1003, 1-15. doi: 10.1038/ncb3039
181. Im YK, Najyb O, Gravel SP, McGuirk S, Ahn R, Avizonis DZ, et al. Interplay between shcA signaling and PGC-1α Triggers targetable metabolic vulnerabilities in breast cancer. Cancer Res. (2018) 78:4826–38. doi: 10.1158/0008-5472.CAN-17-3696
182. Zhang J, Wang C, Chen X, Takada M, Fan C, Zheng X, et al. EglN2 associates with the NRF1-PGC1α complex and controls mitochondrial function in breast cancer. EMBO J. (2015) 34:2953–70. doi: 10.15252/embj.201591437
183. Zhang J, Zheng X, Zhang Q. EglN2 positively regulates mitochondrial function in breast cancer. Mol Cell Oncol. (2016) 3:e1120845. doi: 10.1080/23723556.2015.1120845
184. Brindisi M, Fiorillo M, Frattaruolo L, Sotgia F, Lisanti MP, Cappello AR. Cholesterol and mevalonate: two metabolites involved in breast cancer progression and drug resistance through the ERRα Pathway. Cells. (2020) 9(8):1819. doi: 10.3390/cells9081819
185. Tiraby C, Hazen BC, Gantner ML, Kralli A. Estrogen-related receptor gamma promotes mesenchymal-to-epithelial transition and suppresses breast tumor growth. Cancer Res. (2011) 71:2518–28. doi: 10.1158/0008-5472.CAN-10-1315
186. Jäger S, Handschin C, St-Pierre J, Spiegelman BM. AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci USA. (2007) 104:12017–22. doi: 10.1073/pnas.0705070104
187. Basu S, Gnanapradeepan K, Barnoud T, Kung CP, Tavecchio M, Scott J, et al. Mutant p53 controls tumor metabolism and metastasis by regulating PGC-1α. Genes Dev. (2018) 32:230–43. doi: 10.1101/gad.309062.117
188. Ni H, Guo M, Zhang X, Jiang L, Tan S, Yuan J, et al. VEGFR2 inhibition hampers breast cancer cell proliferation via enhanced mitochondrial biogenesis. Cancer Biol Med. (2021) 18:139–54. doi: 10.20892/j.issn.2095-3941.2020.0151
189. Tripathi V, Jaiswal P, Assaiya A, Kumar J, Parmar HS. Anti-cancer effects of 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) on triple-negative breast cancer (TNBC) cells: mitochondrial modulation as an underlying mechanism. Curr Cancer Drug Targets. (2022) 22:245–56. doi: 10.2174/1568009622666220207101212
190. Du Y, Song L, Zhang L, Ling H, Zhang Y, Chen H, et al. The discovery of novel, potent ERR-alpha inverse agonists for the treatment of triple negative breast cancer. Eur J Med Chem. (2017) 136:457–67. doi: 10.1016/j.ejmech.2017.04.050
191. Zhou T, Zhang B, Wei P, Du Y, Zhou H, Yu M, et al. Energy metabolism analysis reveals the mechanism of inhibition of breast cancer cell metastasis by PEG-modified graphene oxide nanosheets. Biomaterials. (2014) 35:9833–43. doi: 10.1016/j.biomaterials.2014.08.033
192. Flågeng MH, Moi LL, Dixon JM, Geisler J, Lien EA, Miller WR, et al. Nuclear receptor co-activators and HER-2/neu are upregulated in breast cancer patients during neo-adjuvant treatment with aromatase inhibitors. Br J Cancer. (2009) 101:1253–60. doi: 10.1038/sj.bjc.6605324
193. Cotul EK, Zuo Q, Santaliz-Casiano A, Imir OB, Mogol AN, Tunc E, et al. Combined targeting of estrogen receptor alpha and exportin 1 in metastatic breast cancers. Cancers (Basel). (2020) 12(9):2397. doi: 10.3390/cancers12092397
194. Skildum A, Dornfeld K, Wallace K. Mitochondrial amplification selectively increases doxorubicin sensitivity in breast cancer cells with acquired antiestrogen resistance. Breast Cancer Res Treat. (2011) 129:785–97. doi: 10.1007/s10549-010-1268-2
195. Scharping NE, Menk AV, Moreci RS, Whetstone RD, Dadey RE, Watkins SC, et al. The tumor microenvironment represses T cell mitochondrial biogenesis to drive intratumoral T cell metabolic insufficiency and dysfunction. Immunity. (2016) 45:374–88. doi: 10.1016/j.immuni.2016.07.009
196. Scharping NE, Rivadeneira DB, Menk AV, Vignali PDA, Ford BR, Rittenhouse NL, et al. Mitochondrial stress induced by continuous stimulation under hypoxia rapidly drives T cell exhaustion. Nat Immunol. (2021) 22:205–15. doi: 10.1038/s41590-020-00834-9
197. Dumauthioz N, Tschumi B, Wenes M, Marti B, Wang H, Franco F, et al. Enforced PGC-1α expression promotes CD8 T cell fitness, memory formation and antitumor immunity. Cell Mol Immunol. (2021) 18:1761–71. doi: 10.1038/s41423-020-0365-3
198. Pichler AC, Carrié N, Cuisinier M, Ghazali S, Voisin A, Axisa PP, et al. TCR-independent CD137 (4-1BB) signaling promotes CD8(+)-exhausted T cell proliferation and terminal differentiation. Immunity. (2023) 56(7):1631–48.e10. doi: 10.1016/j.immuni.2023.06.007
199. Menk AV, Scharping NE, Rivadeneira DB, Calderon MJ, Watson MJ, Dunstane D, et al. 4-1BB costimulation induces T cell mitochondrial function and biogenesis enabling cancer immunotherapeutic responses. J Exp Med. (2018) 215:1091–100. doi: 10.1084/jem.20171068
200. Kawalekar OU, O'Connor RS, Fraietta JA, Guo L, McGettigan SE, Posey AD Jr., et al. Distinct signaling of coreceptors regulates specific metabolism pathways and impacts memory development in CAR T cells. Immunity. (2016) 44:380–90. doi: 10.1016/j.immuni.2016.01.021
201. Abdalla HB, Napimoga MH, Lopes AH, de Macedo Maganin AG, Cunha TM, Van Dyke TE, et al. Activation of PPAR-γ induces macrophage polarization and reduces neutrophil migration mediated by heme oxygenase 1. Int Immunopharmacol. (2020) 84:106565. doi: 10.1016/j.intimp.2020.106565
202. Chawla A. Control of macrophage activation and function by PPARs. Circ Res. (2010) 106:1559–69. doi: 10.1161/CIRCRESAHA.110.216523
203. Zhou H, Gan M, Jin X, Dai M, Wang Y, Lei Y, et al. miR−382 inhibits breast cancer progression and metastasis by affecting the M2 polarization of tumor−associated macrophages by targeting PGC−1α. Int J Oncol. (2022) 61(4):126. doi: 10.3892/ijo
204. Caporarello N, Meridew JA, Jones DL, Tan Q, Haak AJ, Choi KM, et al. PGC1α repression in IPF fibroblasts drives a pathologic metabolic, secretory and fibrogenic state. Thorax. (2019) 74:749–60. doi: 10.1136/thoraxjnl-2019-213064
205. Prieto I, Alarcón CR, García-Gómez R, Berdún R, Urgel T, Portero M, et al. Metabolic adaptations in spontaneously immortalized PGC-1α knock-out mouse embryonic fibroblasts increase their oncogenic potential. Redox Biol. (2020) 29:101396. doi: 10.1016/j.redox.2019.101396
206. Li X, Jiang E, Zhao H, Chen Y, Xu Y, Feng C, et al. Glycometabolic reprogramming-mediated proangiogenic phenotype enhancement of cancer-associated fibroblasts in oral squamous cell carcinoma: role of PGC-1α/PFKFB3 axis. Br J Cancer. (2022) 127:449–61. doi: 10.1038/s41416-022-01818-2
Keywords: PGC1α, tumor progression, cancer metabolism, signaling pathways, therapeutic target, metabolic heterogeneity
Citation: Wang Y, Peng J, Yang D, Xing Z, Jiang B, Ding X, Jiang C, Ouyang B and Su L (2024) From metabolism to malignancy: the multifaceted role of PGC1α in cancer. Front. Oncol. 14:1383809. doi: 10.3389/fonc.2024.1383809
Received: 08 February 2024; Accepted: 16 April 2024;
Published: 07 May 2024.
Edited by:
Maria Elena Pisanu, National Institute of Health (ISS), ItalyReviewed by:
Elena Rapizzi, University of Florence, ItalyMaria Letizia Taddei, University of Florence, Italy
Copyright © 2024 Wang, Peng, Yang, Xing, Jiang, Ding, Jiang, Ouyang and Su. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Su, c3VsZWluamdseXlAZmFzdG1haWwuY29t; Bing Ouyang, Ym95MjAwNDExODhAc2luYS5jb20=
†These authors have contributed equally to this work