 Lin Feng
Lin Feng Hang Zhang
Hang Zhang Ting Liu
Ting Liu- Department of Hematology, Institute of Hematology, West China Hospital of Sichuan University, Chengdu, Sichuan, China
The IKZF1 gene encodes a transcription factor that belongs to the family of zinc-finger DNA-binding proteins associated with chromatin remodeling. The protein product, IKAROS, had been proved to regulate lymphopoiesis. Subsequent mouse model studies have further confirmed its regulating role in lymphopoiesis as well as in hematopoiesis; besides, it associates with immune function, certain immune disorders like common variable immunodeficiency and dysgammaglobulinemia have been proved to be associated with germline IKZF1 mutations. Dysfunction of IKAROS also bears paramount significance in leukemic transformation and alterations of IKZF1 gene predicts a poor prognosis in hematological malignancies. As an independent prognostic marker, IKZF1 has been incorporated in the risk stratification of BCP-ALL and stratification-guided therapy has also been generated. In this review, we provide a concise and comprehensive overview on the multifaceted roles of IKZF1 gene.
Introduction
IKZF1, namely LyF-1, had been proved to play an important role for the first time in lymphopoiesis (1). IKAROS as a founding member of a family of zinc finger transcription factors, encoded by the IKZF1 gene, associates with other zinc finger bearing transcription factors (HELIOS, AIOLOS, EOS, and PEGASUS encoded by IKZF2, IKZF3, IKZF4 and IKZF5, respectively) in the regulation of both myeloid and erythroid lineage, in addition to lymphoid lineage (2–9). Germline or somatic mutations of IKZF1 demonstrate quite variable clinical phenotypes, ranging from primary immunodeficiency (PID)/inborn errors of immunity (IEI) (10–12) to autoimmune diseases (13–16) and to even hematological malignancies such as acute lymphoblastic leukemia (17, 18). As in acute lymphoblastic leukemia, especially of high-risk B-cell-precursor ALL (BCP-ALL), in which IKZF1 gene mutation is of high frequency (19, 20), such as BCR-ABL positive ALL and BCR-ABL-like ALL, genetic alteration of IKZF1 gene is associated of poor outcome (21–23) and confers resistance in conventional chemotherapy (24, 25). In terms of the types of mutations in the IKZF1 gene, the most common are characterized by large fragment deletions mapping to IKZF1 exons, as well as by other point mutations (missense, nonsense) or frameshift mutation, which cause diversity in clinical phenotypes (Figure 1).
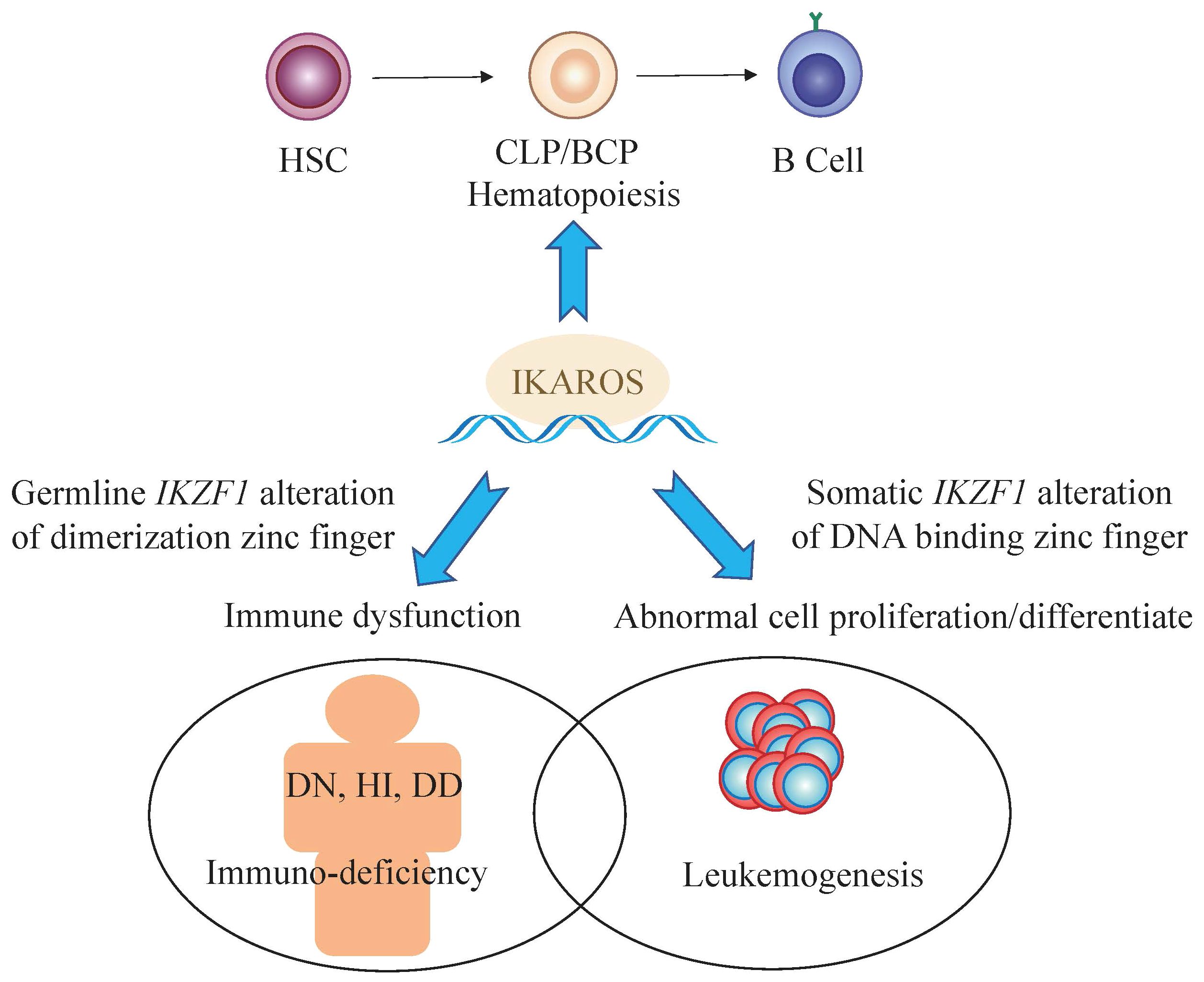
Figure 1 The multifaceted roles of IKZF1. * HSC, hematopoietic stem cell; CLP, common lymphoid precursor; BCP, B cell progenitor; DN, Dominant negative effect on IKAROS function; HI, Haploinsufficiency due to quantitative or qualitative decrease of IKAROS; DD, Dimerization defects caused by deletion of dimerization zinc fingers.
Structure and function of IKZF1 gene
From early 1990s, the studies in vitro and in vivo revealed the molecular structure of IKZF1 gene, followed by precise elaboration of the functional domains of its corresponding protein IKAROS (1, 26). The IKZF1 gene is mapped on chromosome 7 at 7p12.2 and consists of 8 exons, coding for 519 amino acids (27). It is worth noting that expression of IKZF1 gene is restricted to the fetal and adult hemo-lymphopoietic system, and IKAROS is one of the most important and founding member of a family of zinc finger transcription factors, which associates with other transcription factors, HELIOS, AIOLOS, EOS, and PEGASUS, in regulation of hemopoiesis.
IKAROS functions as a master transcription factor and is characterized by the DNA-binding ability through its zinc-finger domains to regulate target genes involved in hematopoiesis, particularly in lymphocyte differentiation, through association with transcriptional complexes or transcription factors including the nucleosome remodeling and deacetylase (NuRD) complex and epigenetic modification interferon regulatory factor 4 (IRF4) (28–30). Among these zinc-finger domains, four zinc-fingers (ZF1–4), coding by IKZF1 exons 4–6, are located at the N-terminus of the protein and bind to the targeted DNA sequences when the protein is phosphorylated and being subsequently transferred from the cell cytoplasm to nucleus to exerts its function (31, 32). Homo- or heterodimerization of wild type IKAROS with other IKAROS family members facilitate the localization of the dimers to the pericentromeric heterochromatin (PC-HC) and regulates the expression of its target genes (33, 34). IKZF1 mutations which locate in the DNA-binding domains would cause compromised DNA-binding ability of mutant IKAROS and demonstrate a bizarre staining pattern of PC-HC (35). While, the rest of the zinc-fingers (ZF5–6), being coded by exon 8 of IKZF1, are in the location of the C-terminus of the protein, required for homo- and heterodimerization between the different IKAROS proteins (26, 36). Dimerization between IKAROS zinc-finger proteins with a functional DNA binding domain facilitate their DNA-binding ability and transcriptional activity. The mutations affecting zinc-finger 5 and/or zinc-finger 6 would limit homo-and heterodimerization, in addition to a compromised DNA-binding ability and abnormal PC-HC localization (17). According to NCBI database (27), there are at least 18 isoforms of IKZF1, which are generated by alternative splicing transcript variants. The isoforms share the C-terminal dimerization zinc-fingers in common, and have variant numbers and locations of DNA binding zinc-fingers in the N-terminal (26) (Figure 2).
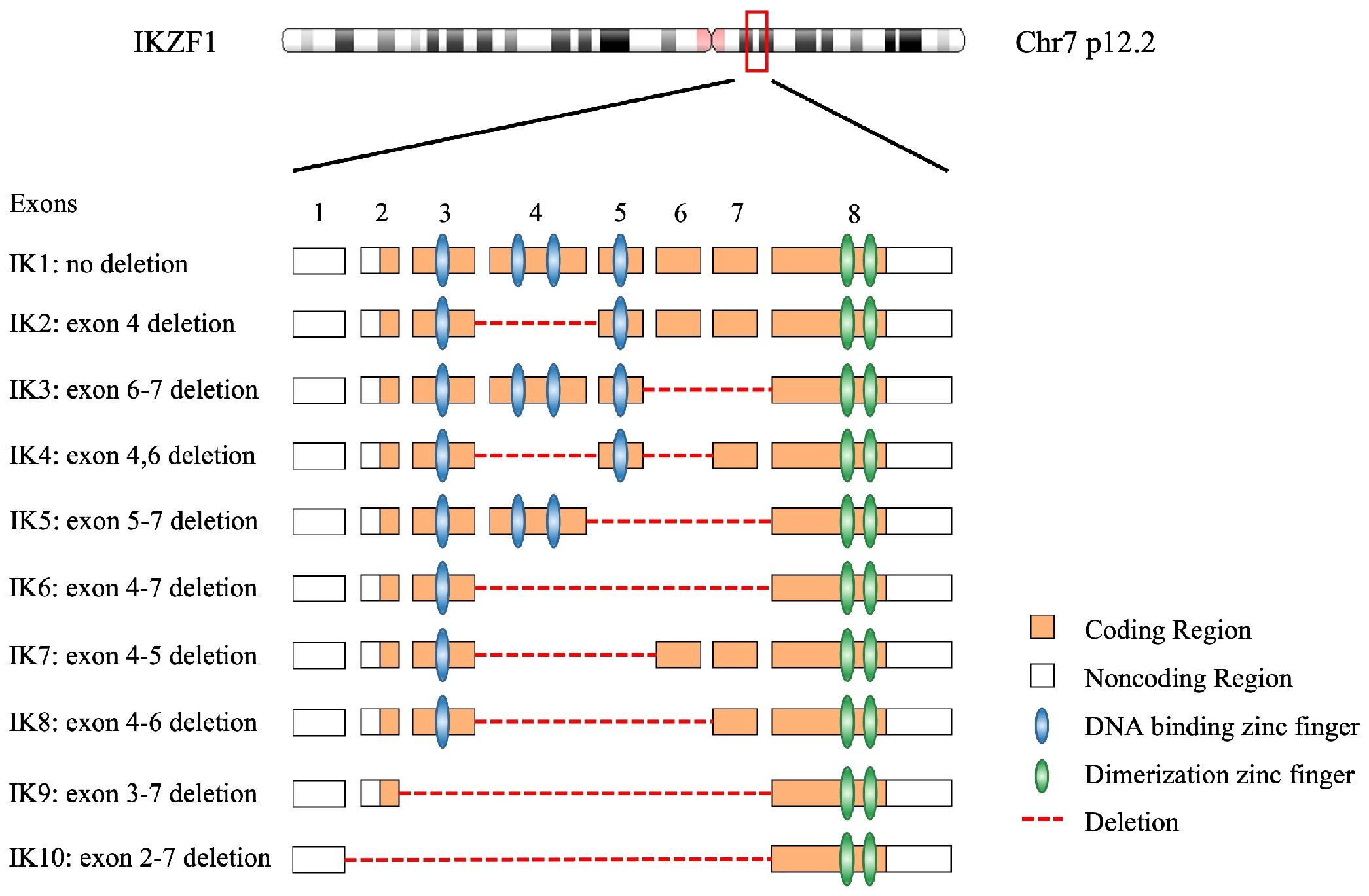
Figure 2 The genome of IKZF1 and most common 10 IKAROS isoforms with their functional domains. A total of 18 isoforms have been described on NCBI database.
Among all IKAROS isoforms, IK6 (Δ4–7) is the most common, with a proportion of up to 42%, followed by IK10 (Δ2–7) ranging from 7% to 20%, which is consistent in adult and pediatric patients (21–23). The remaining approximately one third are comprised of the so-called rare isoforms because frequency of each of these variants is no more than 10% in BCP-ALL patients, such as Δ2–3, Δ2–7, Δ2–8, etc. (Table 1). Interestingly, in terms of prognosis, there seems to be no significant difference between IK6 and IK10 isoforms (21), possibly because IK6 and IK10 share the similar structure that both isoforms lack all four N-terminal zinc-finger containing domains, thus generating a similar clinical phenotype. Nevertheless, almost all isoforms due to various IKZF1 deletions confer a poor prognosis including those rare IKAROS isoforms.
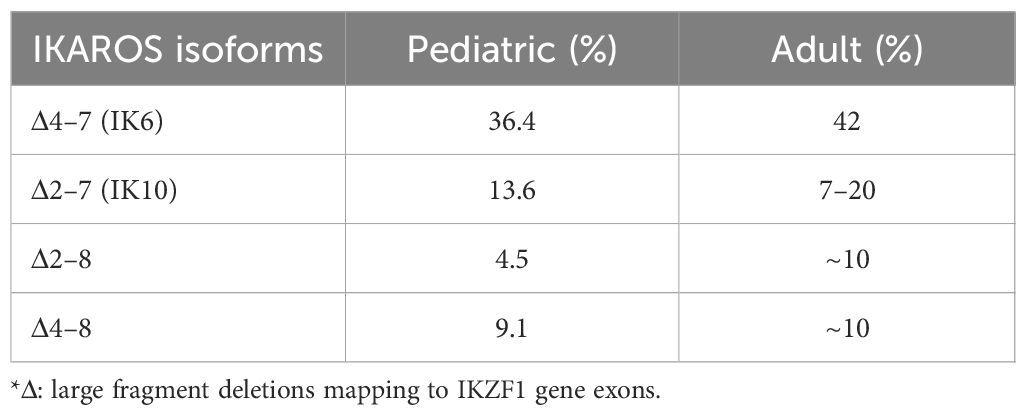
Table 1 Percentages of common IKZF1 isoforms in Pediatric and Adult BCP-ALL.
Roles of IKZF1 gene in hematopoiesis
B-cell and T-cell lineages develop from an uncommitted hematopoietic stem cell. The somatic gene rearrangements that generate the highly diverse repertoire of antigen receptors, immunoglobulin for B cells, and the T-cell receptor for T cells, occur in the early stages of the development of T cells and B cells from a common bone marrow–derived lymphoid progenitor. During the development of these lymphoid cells, a large amount of transcription factors cooperates and play vital roles in the process. The IKAROS functions as a master transcription regulator in the differentiation of lymphoid cells. Unlike its transcription factor counterparts such as EA2, FOXO1, EBF and PAX5/BASP, whose expression persists only in certain stages of B cell development in the bone marrow, IKZF1 is expressed throughout the whole process from lymphoid-myeloid primed multipotent progenitor (LMPP) to immature B cell within the bone marrow (37–39) (Figure 3). Transcription factors abnormalities could be harmful to the cell homeostasis and mediate leukemic transformation (40). There is a high frequency of transcription factors abnormalities in B-cell precursor lymphoblastic leukemia (BCP-ALL) and are recognized as the main genetic-hit events in the leukemogenesis (41). Mouse model studies by Kirstetter P et al. showed that IKAROS activity fluctuations due to various types of IKZF1 mutation had huge impact on multiple aspects of B cell lineage development (42).
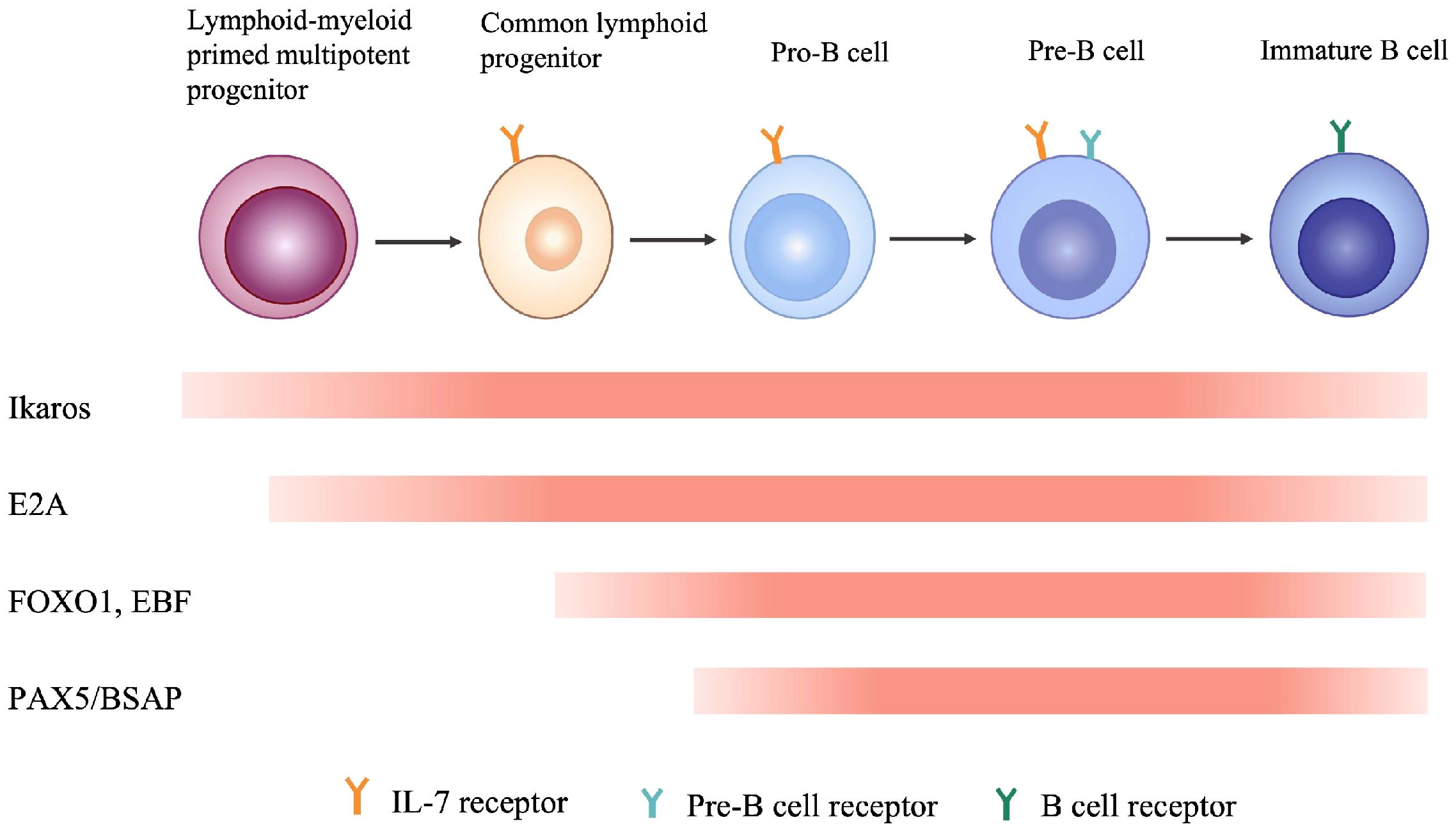
Figure 3 Multiple transcription factors are involved in different stages of B cell development in bone marrow.
Other mouse model studies by Winandy S et al. have evidenced that IKAROS is required for both early and late stage of lymphocyte differentiation in the thymus. IKAROS dysfunction will enhance the proliferation of maturing thymocytes and eventually lead to malignant transformation of such thymocytes (6, 43). Furthermore, Georgopoulos K et al. demonstrated that mice harboring homozygous germline mutation of IKZF1 were lack of not only adaptive immune lymphocytes of T and B cells, but innate immune lymphocytes of natural killer cells, in addition to deficiency of their corresponding progenitor cells, yet with preservation of normal erythroid and myeloid cells (6, 44).
However, Lopez R et al. showed in their mouse model studies a different phenotype that the IKAROS null mice had anemia and megakaryocytic abnormalities, besides lymphoid and stem cell defects (45). One possible explanation for this divergent manifestation could be the variant location of the lost zinc-fingers between the two studies. In the studies of Georgopoulos K et al., the lost zinc fingers located in the N-terminal domains for DNA-binding, which leads to the truncate IKAROS protein. Lopez R et al. deleted the entire C-terminal zinc fingers required for IKAROS protein dimerization and function, the loss of C-terminal zinc fingers probably interferes with the normal PYR complex formation on DNA (44, 46), followed by the subsequent development block of whole blood lineages. In consistent with studies by Lopez R et al., Francis O et al. and Malinge S et al. also found that IKAROS functioned as a regulator of myeloid and erythroid differentiation (7–9).
Roles of IKZF1 mutations in immune function
During the whole 1990s and early 2000s, researches on IKAROS mainly focus on the its role in regulation of hematopoiesis, especially lymphopoiesis. In the new era, more studies are carried out, striving to elaborate the correlation between IKZF1 mutation and the resultant impact on immune function. As described above in mouse models that IKZF1 null mice are prone to T-cell malignancies like T-ALL or lymphoma, nevertheless in the real world, humans harboring germline mutation of IKZF1 are more frequently linked to various types of immunodeficiency instead of malignancies (10, 11, 14–16, 47–53).
Studies from mouse models and humans suggest that there is strong correlation between genotype of the numerous variants of IKZF1 gene and phenotype of the clinical patients. The key factor defining the phenotype is the protein expression of IKAROS which is dependent on the germline alterations of IKZF1 gene, and according to these genetic and proteic variations, three clinical groups are established among those primary immunodeficiencies/inborn errors of immunity patients (12): Haploinsufficiency (HI), Dimerization defective (DD) and Dominant negative (DN). 1). HI due to quantitative or qualitive decrease of IKAROS: Large deletions of IKZF1 exons or nonsense mutation leading to impaired protein expression results in reduction of wild type IKAROS expression, or IKAROS function loss resulting from disruption of DNA-binding and PC-HC targeting, yet without influencing the wild type IKAROS protein; 2) DD due to genetic alterations (ZF5–6 involved) affecting IKAROS dimerization ability: In contrast to loss of ability to interact with IKAROS family members and generate homo- or heterodimers, DD mutants conserve the DNA-binding ability as monomers and have no impact on wild type IKAROS; 3) DN: One special IKAROS mutants that conserve the dimerization ability but are unable to bind IKAROS target sequences and disrupt interaction between PC-HC and wild type IKAROS proteins (Figure 4). In terms of IKAROS dosage effect, theoretically speaking, DN type has the most adverse impact on the normal immune function, for one thing mutants of DN type exhibit HI effect, for another DN mutants hijack the wild type IKAROS protein to worsen the normal IKAROS functional defects. Boutboul D et al. evidenced that patients with DN mutations would probably have an earlier onset of combined immunodeficiency compared to patients harboring HI or DD mutations, in addition, patients with DN mutations had a more severe loss of Pan-B cells ranging from progenitor B cells to antibody-secreting plasma cells (47). Winandy S et al. and Georgopoulos K et al. also showed in their studies that normal hemo-lymphopoiesis was more strongly influenced among DN genotype mouse who were devoid of both innate immune NK cells and adaptive immune T, B cells and the corresponding progenitor cells (6, 44). Other studies found that nearly all patients with DN mutations had low numbers of B cells in the peripheral blood in contrast to a significant lower prevalence of decreased PB B cells in patients with HI and DD mutations (17, 37, 38, 48, 51). Additionally, patients with DN mutations had more naïve T cells (CD45RA+CD62L+ or CD45RA+CD45RO–) and an abnormal ratio of Th subsets of which both CD4+ memory T cells and Treg cells were decreased, owing to impaired sensitivity to IL-2 stimulation, reduced STAT5 pathway activation (47). As one of professional antigen-presenting cells, dendritic cells (DC) play important roles not just in the initiation and activation of adaptive immune response but also in the process of T cell maturation in the thymus when early T-cell precursor cells (ETPs) differentiate from double-negative stage to single-positive naïve T cell (37). Mouse model and human studies also showed that DN genotype had lower numbers of DC (5, 44, 54). Monocytes and neutrophils are classical phagocytes responsible for defensing against invading bacteria and other microbes. Patients with DN mutations had reduced granulopoiesis of neutrophils and were incapable of full responses to stimulations to Toll-like receptors and the subsequent scant production of proinflammatory cytokines by monocytes. It is rational that DN genotype would confer a more severe clinical phenotype, in consistent with the idea, Thaventhiran J et al. and other researchers found that patients with DN mutations were susceptible to an earlier onset and broader as well as more severe invasive infections, including pneumocystis pneumonia which was infrequent in patients harboring HI or DD mutations (53, 55). Although DN genotype often implies an inferior phenotype, patients with DN mutations would barely have chances of developing autoimmune diseases like systemic lupus erythematosus (SLE), immune thrombocytopenia (ITP), rheumatoid arthritis (RA), antiphospholipid syndrome (APS) and others, of which are more frequently seen in DD patients followed by HI patients (13, 15, 16, 49–51, 56). Unlike DN genotype which is characterized by devoid of B cells, allelic variants of DD and HI are more likely to generate self-reactive B cells due to incapable of controlling B cell anergy and TLR signaling (57). As clearly described in Janeway’s immunobiology textbook, IKAROS is a rare transcription factor that expressed throughout the B cell development in the bone marrow and regulates many vital events like maintaining a suitable intensity of TLR signaling and induction of B cell anergy, which contribute to B cell central tolerance (37). Furthermore, Kuehn H et al. showed that DD mutants had an abnormal sumoylation and affected the normal interaction between IKAROS and NuRD complex, leading to aberrant epigenetic modification and the consequent B cell central intolerance which counts for much in the pathogenesis of autoimmune diseases (17).
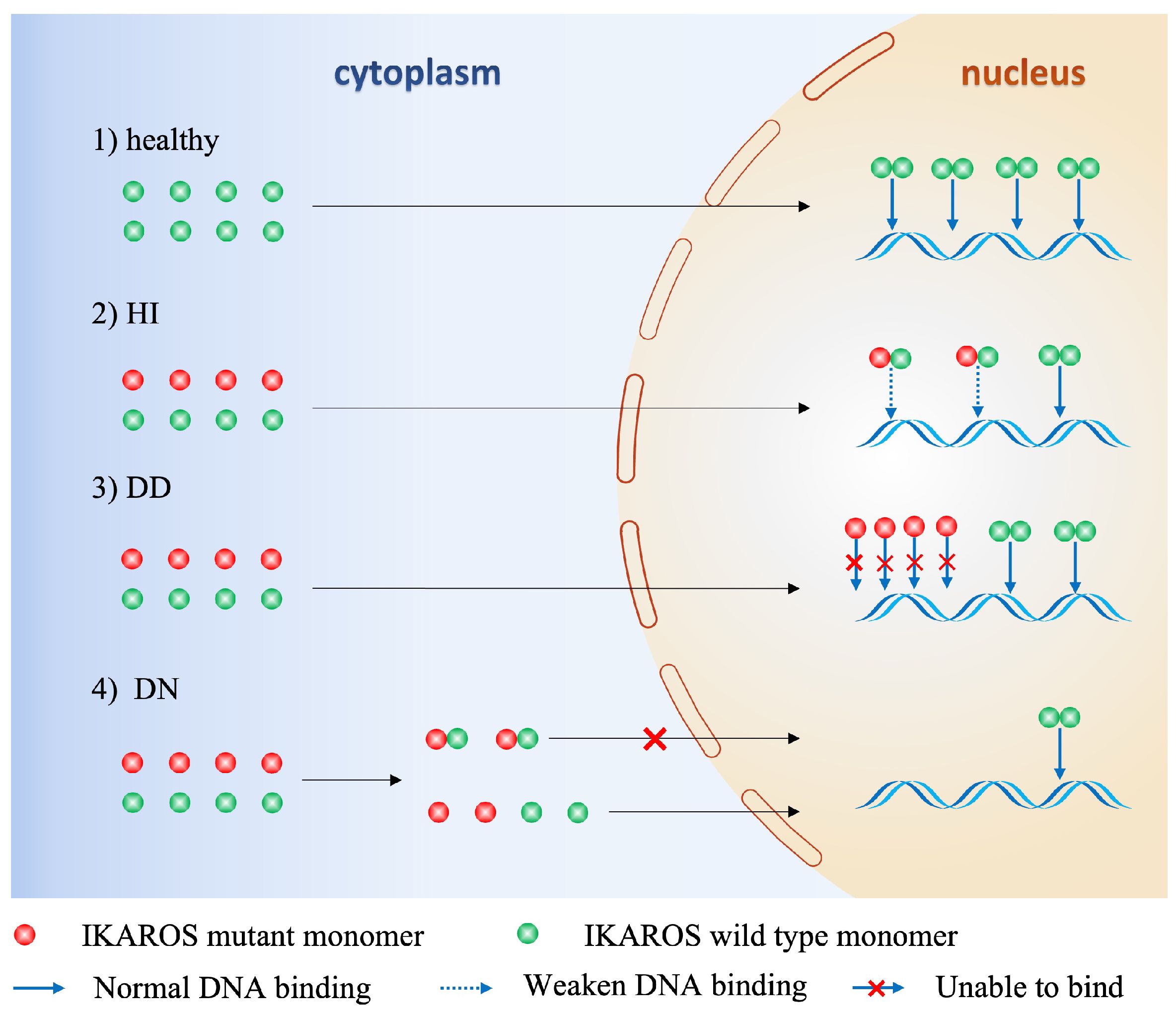
Figure 4 Mechanisms of functional defects of IKAROS caused by germline alterations of IKZF1. HI, Haploinsufficiency due to quantitative or qualitative decrease of IKAROS, without influencing the wild-type IKAROS. DD, Dimerization defects caused by deletion of dimerization zinc fingers lead to more monomers that cannot bind to DNA sequence, without influencing the wild-type IKAROS. DN, Dominant negative effect on IKAROS function. The mutant monomer can combine with wild-type monomer, but the dimer lacks DNA-binding or PC-HC targeting function, thus affecting the function of the wild-type IKAROS.
Roles of IKZF1 mutations in leukemia
Frequency of IKZF1 alterations in leukemia
Most of our knowledge of T-cell development in the thymus comes from the mouse, experiments on mice demonstrates that T-cell precursors migrate from the bone marrow to the thymus, where they commit to the T-cell lineage after Notch receptor signaling. Unlike those transcription factors (TCF1, GATA3, Bcl 11b) expressed at a later stage of T-cell development in the thymus (37), Georgopoulos K et al. found that IKZF1 gene was expressed throughout the ontogeny of the T cell lineage from as early in the hemopoietic progenitors in the fetal stage to the adult stage of T cell maturation (1). Soon afterwards, further findings from IKAROS heterozygous mice studies by Winandy S et al. showed that compromised IKAROS function leads to aberrant proliferation of clonal T cells and the consequent development of T cell leukemia and lymphoma (6). There is a strong link between IKAROS inactivation and Notch activation in murine T-ALL, so it is rationale to deduce that IKAROS defect could promote Notch activation in human T-ALL. However, IKZF1 is rarely found in human T-ALL (<5%) (23, 58–60). The patients with high Notch signaling expression appear to express normal IKAROS, except for the most aggressive T-ALL subtype, ETP-ALL, in which the frequency of IKZF1 somatic mutations is 13% (61). It is also similar in terms of acute myeloid leukemia (AML), the frequency of IKZF1 somatic mutations is only 3.83% (62). Interestingly, Grossmann V et al. found that chronic myeloid leukemia (CML) patients had a slightly higher mutation rate of IKZF1(17.9%) when progressing to blast crisis (63). Jäger R et al. also showed that transformed MPN patients had a higher incidence of heterozygous loss of IKZF1 compared to that in non-leukemic MPN patients (21% vs 0.2%) (64). In sharp contrast to the low frequency among the aforementioned hematological malignancies, IKZF1 alterations is of much higher frequency in BCP-ALL (19, 22), particularly in kinase and cytokine- receptor signaling activating leukemias including Ph+ALL and Ph-like ALL (59, 60, 65–68). Despite a high incidence of IKZF1 mutations in BCP-ALL, these alterations are mainly linked to somatic abnormalities, which are not characterized by point mutation (missense, nonsense) or frameshift mutation, but by large fragment deletions mapping to IKZF1 exons (21–23). Churchman M et al. showed that germline IKZF1 alterations is extremely low in pediatric BCP-ALL (69).
IKZF1 alterations promote leukemogenesis and induction of chemo-resistance
IKZF1 alterations promote leukemogenesis had been studied in mouse and human by Churchman M et al. and other researchers (22, 70, 71). IKZF1 alterations conferred stem cell-like properties characterized by overexpression of genes indicative of a stem cell phenotype and lead to upregulation of adhesion molecules (FAK, RHO, CTNND1, etc.) as well as integrins, altogether facilitating self-renewal or leukemic transformation. Furthermore, as a master regulator of B cell differentiation, functional impairment of IKAROS due to IKZF1 somatic alterations would affect its binding to IgH locus and disturb arrangement of the heavy chain of pre-BCR and BCR, thus signaling through the pre-BCR that mediates the transition from pre-B cell to immature B cell would be blocked (42). Besides, downstream of pre-BCR signaling is impacted where expression of c-Myc is upregulated and k-light chain is downregulated which enhance the cell cycle in pro-B and pre-B cells, and block their differentiation into immature B cells (72–74). Li JF et al. reported that IKZF1 N159Y is a rare subtype of B-ALL characterized by significant upregulation of the transcriptional coactivator YAP1, SALL1 and ARHGEF28B, and downregulation of the B-cell receptor signaling and JAK-STAT signaling pathways (75).
In addition, IKZF1 alterations also make BCP-ALL, in particular high-risk subtype such as Ph+ALL and Ph-like ALL, refractory to treatment by induction of chemo-resistance through various mechanisms. Marke R et al. demonstrated in their mouse models studies and in vitro experiments that compromised functional IKAROS due to IKZF1 alterations affected the transcription function of TSC22D3, DUSP1, IRS2, and so on, combined with the abnormal regulation of GC-activated genes in response to prednisolone, contributing to resistance to GC-induced apoptosis (23). Scheijen B et al. showed that IKZF1 haplodeficient B-cells enhanced cell survival as compared to wild-type (WT) IKAROS, when co-existing with BTG1 deficiency, these B-cells showed an even stronger GC-induced apoptosis (76). Of note, under physiological conditions, WT IKAROS controls the transition of pre-B cells from a stroma-adherent proliferative phase to a relative non-dividing but differentiating phase. As evidenced by Joshi I et al. and Churchman M et al. (77, 78), IKZF1 alterations resulted in overexpression of adhesion molecules and upregulation of focal adhesion kinase (FAK). The dysregulated FAK pathway synergizing with other upregulated intracellular signaling pathways promoted the survival of leukemic cells. Furthermore, IKZF1 deletions that co-occurred with deletions in CDKN2A, CDKN2B, PAX5, or PAR1 in the absence of ERG deletion conferred the worst outcome and, consequently, were grouped as IKZF1plus (79).
IKZF1 alterations and hereditary leukemia
Germline mutations have been known for many years that they could increase risks of various cancers, as TP53 germline mutation could increase risks of acute lymphoblastic leukemia. Until 2018, Churchman et al. demonstrated in their study that germline variants in IKZF1 predispose to ALL (69). Winer P et al. also reported that IKZF1 germline mutation contribute to acute lymphoblastic leukemia in children with Down syndrome (DS) (80). By germline whole-exome sequencing (WES) study, they discovered pathogenic variant p.Arg162Trp in IKZF1 gene to ALL predisposition, which was also reported in sporadic B-ALL patients (69). Buitenkamp T et al. showed that DS-ALL patients had a similar frequency of IKZF1 deletions to that of high-risk (HR) non-DS-ALL patients (~30%) (81). Palmi C et al. discovered that the IKZF1plus feature in DS-ALL was 3 times more frequent than in non-DS-ALL (18% versus 6%), and IKZF1plus patients had even worse outcome as well as higher relapse (82). Furthermore, they also found that the blasts of IKZF1plus DS-ALL patients were particularly sensitive to drugs which had been described to be effective in Ph-like cases, such as HDAC inhibitors. These findings could help refine stratification-directed therapy for DS-ALL patients, who are intrinskly more fragile and are of inferior outcome compared with non-DS children with ALL.
IKZF1 deletion is of high prevalence and a pivotal prognostic factor in BCP-ALL
As early as 2007, a large cohort study enrolling 1522 adult patients of ALL including Ph- and Ph+ ALL from multicenter demonstrated that karyotype could be an independent prognostic factor in adult ALL (83), however, karyotype itself has limited potency to stratify patients to the ideal risk groups and ensure that patients received the most appropriate treatment. Acute lymphoblastic leukemia patients can only benefit the most from the pretreatment evaluation of combing high quality cytogenetic data with precise molecular genetic analysis. So, we can see that during the recent two decades, extensive researches have been done trying to find out the optimal combination of the methods, and IKZF1 alterations are just one of the most important constituents to this combination. As described above, IKZF1 is a master regulator of hematopoiesis, especially lymphopoiesis. Loss of function of IKAROS due to IKZF1 alterations causes a predisposition to lymphoid malignancies. Mullighan C et al. and others reported that IKZF1 alterations is frequent in BCP-ALL, about 15% in pediatric patients and increasing to 30%-50% in adult patients, and remarkably higher in high-risk subtypes, notably, the frequency is as high as 84% and 70% in adult Ph+ALL and Ph-like ALL, respectively (19, 22, 59, 60, 65–68, 84, 85).
Either the initial study from Mullighan C (22) in which 258 ALL pediatric patients were enrolled or the following numerous large scale clinical researches (67, 86–90) which included more than 1000 pediatric patients and the relatively small-scale adult clinical researches (91, 92), all showed that IKZF1 deletion was a strong prognostic factor and confer an increased risk of relapse and poor outcome with inferior EFS as well as OS in BCP-ALL. This is in consistence with our studies (93). Furthermore, IKZF1 deletion demonstrates heterogeneous impact on the prognosis of BCP-ALL patients when coexisting with other molecular abnormalities. Ribera J et al. and others (94, 95) reported that patients with coexistence of IKZF1 and CDKN2A/B alterations had worse prognosis. Stanulla M et al. and Zaliova M et al. also pointed out that patients harboring IKZF1plus alterations had the worst prognosis compared to those with sole IKZF1 deletion (79, 96). The patients with IKZF1plus, that have co-occurring deletions in CDKN2A, CDKN2B, PAX5, or PAR1, worsen the B-ALL phenotype, as the co-aberrations enhance B cell development arrest and abnormal proliferation. It is of great interest that BCP-ALL pediatric patients with ERG deletions had a high co-occurring rate of IKZF1 deletions, and such patients confer a good outcome compared to patients with the sole-IKZF1 deletions or the sole-EGR deletions (97, 98). Ultimately, the prognostic significance of IKZF1 deletions translate into optimal therapeutic strategies to benefit patients from unnecessary treatment related toxicity and ensure sufficient treatment intensity to avoid chemo-resistance or relapse. It is noteworthy that combing MRD and IKZF1 status could refine the risk stratification and better predict the disease relapse (99, 100).
IKZF1 mutations in acute myeloid leukemia
The role and implications of IKZF1 mutations and deletions are well studied in B-ALL. However, the prevalence and impact in AML remain sparse. Eckardt J et al. reported the 2.8% of patients harboring IKZF1 alterations in 1606 adult AML patients, and the heterozygous SNVs are the most common mode of alteration. They also identified a mutational hotspot in the second N-terminal zinc finger domain at p.N159S, which was present in 19 of 45 (42.2%) IKZF1 alterations cases. AML patients with mutated IKZF1 was usually associated with aberration in RUNX1, GATA2, KRAS, KIT, SF3B1, and ETV6. IKZF1 mutation could be an independent marker of adverse risk regarding complete remission rate, event-free, relapse-free, and overall survival (101). Zhang X et al. investigated 522 newly diagnosed AML patients, 20 of whom harboring IKZF1 mutations with a significant co-occurrence of mutations in SF3B1, CSF3R, and CEBPA. The authors describe a significantly reduced CR rate for patients with IKZF1 mutations (62). Wang Y et al. found that 23 out of 475 (4.8%) AML patients bear mutated IKZF1, and delineated three clusters of IKZF-mutated as: N159S (40%), co-occurring CEBPA mutations (43%), and others (17%) (102). Jäger et al. found deletions of IKZF1 to occur in ~20% of AML cases that arose secondary to myeloproliferative neoplasms suggesting a differential role of deletions and mutations in myeloid leukemogenesis (64). Intriguingly, IKZF2, which encodes another member of IKZF family of transcription factors (HELIOS) also has a role in AML, as shown in the study by Park S et al. (103), IKZF2 has an oncogenic role in AML, and IKZF2 is required for leukemic stem cells survival and function by controlling chromatin accessibility of self-renewal and differentiation programs.
Treatment of BCP-ALL patients target to transcriptional regulation of genes by IKAROS
As reviewed above, IKZF1 alterations promote leukemogenesis, it seems quite rational to restore wild type IKZF1 expression in the treatment of BCP-ALL with IKZF1 alterations. Mullighan C et al. pioneered this work and showed that retinoid receptor agonists could induce expression of wild-type IKZF1, reversed the stem cell features of IKZF1-altered leukemic cells and increased responsiveness to dasatinib in Ph+ ALL (104). Song C et al. demonstrated that TBB and CX-4945, different kinds of casein kinase II inhibitors, restored IKAROS activity and showed an antileukemia effect by inhibiting the transcription of the genes involved in the PI3K pathway (105). Palmi C et al. studied some compounds in vitro cell culture that show one of histone deacetylase inhibitors, Givinostat, has the highest efficacy on IKZF1plus blasts in comparison to controls (82). In the clinical setting, the direct targeting of IKZF1 to restore its function have not been yet. These studies suggested that targeting IKAROS pathways could be used as a therapeutic approach for the high-risk BCP-ALL in the future.
Conclusions and perspectives
In conclusion, it has been clearly demonstrated that IKZF1 alterations have great impacts on many aspects. Germline or somatic alterations as well as heterozygous or homozygous deletions, each exhibits a heterogeneous clinical phenotype. As is evidenced by mouse model and human studies that germline IKZF1 alterations are highly associated with immune dysfunction including various types of immunodeficiency, in addition to autoimmune diseases; while for somatic deletions of IKZF1, patients are susceptible to acute B cell lymphoblastic leukemia. The roles of IKZF1 alterations in leukemogenesis and potency in prognosis prediction have been elucidating. In the future, targeting IKAROS-regulated signaling pathways could be a highly effectively therapeutic approach. Identification of IKAROS target genes would be much important, both to gain insight into IKAROS to function as a tumor suppressor, as well as to identify novel therapeutic targets for the high-risk acute lymphoblastic leukemia.
Author contributions
LF: Conceptualization, Writing – review & editing, Data curation, Formal analysis, Methodology. HZ: Data curation, Formal analysis, Writing – original draft. TL: Conceptualization, Funding acquisition, Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Grant of Tianfu Qingcheng Science and Technology Project, Sichuan, China (No. 1232 to TL).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Georgopoulos K, Moore DD, Derfler B. Ikaros, an early lymphoid-specific transcription factor and a putative mediator for T cell commitment. Science. (1992) 258:808–12. doi: 10.1126/science.1439790
2. Morgan B, Sun L, Avitahl N, Andrikopoulos K, Ikeda T, Gonzales E, et al. Aiolos, a lymphoid restricted transcription factor that interacts with Ikaros to regulate lymphocyte differentiation. EMBO J. (1997) 16:2004–13. doi: 10.1093/emboj/16.8.2004
3. Kelley CM, Ikeda T, Koipally J, Avitahl N, Wu L, Georgopoulos K, et al. Helios, a novel dimerization partner of Ikaros expressed in the earliest hematopoietic progenitors. Curr Biol. (1998) 8:508–15. doi: 10.1016/S0960-9822(98)70202-7
4. Perdomo J, Holmes M, Chong B, Crossley M. Eos and pegasus, two members of the Ikaros family of proteins with distinct DNA binding activities. J Biol Chem. (2000) 275:38347–54. doi: 10.1074/jbc.M005457200
5. Wu L, Nichogiannopoulou A, Shortman K, Georgopoulos K. Cell-autonomous defects in dendritic cell populations of Ikaros mutant mice point to a developmental relationship with the lymphoid lineage. Immunity. (1997) 7:483–92. doi: 10.1016/S1074-7613(00)80370-2
6. Winandy S, Wu P, Georgopoulos K. A dominant mutation in the Ikaros gene leads to rapid development of leukemia and lymphoma. Cell. (1995) 83:289–99. doi: 10.1016/0092-8674(95)90170-1
7. Malinge S, Thiollier C, Chlon TM, Doré LC, Diebold L, Bluteau O, et al. Ikaros inhibits megakaryopoiesis through functional interaction with GATA-1 and NOTCH signaling. Blood. (2013) 121:2440–51. doi: 10.1182/blood-2012-08-450627
8. Francis OL, Payne JL, Su RJ, Su RJ, Payne KJ. Regulator of myeloid differentiation and function: The secret life of Ikaros. World J Biol Chem. (2011) 2:119–25. doi: 10.4331/wjbc.v2.i6.119
9. Dijon M, Bardin F, Murati A, Batoz M, Chabannon C, Tonnelle C. The role of Ikaros in human erythroid differentiation. Blood. (2008) 111:1138–46. doi: 10.1182/blood-2007-07-098202
10. Eskandarian Z, Fliegauf M, Bulashevska A, Proietti M, Hague R, Smulski CR, et al. Assessing the functional relevance of variants in the IKAROS family zinc finger protein 1 (IKZF1) in a cohort of patients with primary immunodeficiency. Front Immunol. (2019) 10:568. doi: 10.3389/fimmu.2019.00568
11. Okano T, Imai K, Naruto T, Okada S, Yamashita M, Yeh TW, et al. Whole-exome sequencing-based approach for germline mutations in patients with inborn errors of immunity. J Clin Immunol. (2020) 40:729–40. doi: 10.1007/s10875-020-00798-3
12. Kuehn HS, Nunes-Santos CJ, Rosenzweig SD. Germline IKZF1 mutations and their impact on immunity: IKAROS-associated diseases and pathophysiology. Expert Rev Clin Immunol. (2021) 17:407–16. doi: 10.1080/1744666X.2021.1901582
13. Han JW, Zheng HF, Cui Y, Sun LD, Ye DQ, Hu Z, et al. Genome-wide association study in a Chinese Han population identifies nine new susceptibility loci for systemic lupus erythematosus. Nat Genet. (2009) 41:1234–7. doi: 10.1038/ng.472
14. van Nieuwenhove E, Garcia-Perez JE, Helsen C, Rodriguez PD, van Schouwenburg PA, Dooley J, et al. A kindred with mutant IKAROS and autoimmunity. J Allergy Clin Immunol. (2018) 142:699–702.e12. doi: 10.1016/j.jaci.2018.04.008
15. Sriaroon P, Chang Y, Ujhazi B, Csomos K, Joshi HR, Zhou Q, et al. Familial immune thrombocytopenia associated with a novel variant in IKZF1. Front Pediatr. (2019) 7:139. doi: 10.3389/fped.2019.00139
16. Dieudonné Y, Guffroy A, Vollmer O, Carapito R, Korganow AS. IKZF1 loss-of-function variant causes autoimmunity and severe familial antiphospholipid syndrome. J Clin Immunol. (2019) 39:353–7. doi: 10.1007/s10875-019-00643-2
17. Kuehn HS, Niemela JE, Stoddard J, Mannurita SC, Shahin T, Goel S, et al. Germline IKAROS dimerization haploinsufficiency causes hematologic cytopenias and Malignancies. Blood. (2021) 137:349–63. doi: 10.1182/blood.2020007292
18. Kastner P, Dupuis A, Gaub MP, Herbrecht R, Lutz P, Chan S. Function of Ikaros as a tumor suppressor in B cell acute lymphoblastic leukemia. Am J Blood Res. (2013) 3:1–13.
19. Tokunaga K, Yamaguchi S, Iwanaga E, Nanri T, Shimomura T, Suzushima H, et al. High frequency of IKZF1 genetic alterations in adult patients with B-cell acute lymphoblastic leukemia. Eur J Haematol. (2013) 91:201–8. doi: 10.1111/ejh.12155
20. Schwab CJ, Chilton L, Morrison H, Jones L, Al-Shehhi H, Erhorn A, et al. Genes commonly deleted in childhood B-cell precursor acute lymphoblastic leukemia: association with cytogenetics and clinical features. Haematologica. (2013) 98:1081–8. doi: 10.3324/haematol.2013.085175
21. Martinelli G, Iacobucci I, Storlazzi CT, Vignetti M, Paoloni F, Cilloni D, et al. IKZF1 (Ikaros) deletions in BCR-ABL1-positive acute lymphoblastic leukemia are associated with short disease-free survival and high rate of cumulative incidence of relapse: a GIMEMA AL WP report. J Clin Oncol. (2009) 27:5202–7. doi: 10.1200/JCO.2008.21.6408
22. Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. (2009) 360:470–80. doi: 10.1056/NEJMoa0808253
23. Dörge P, Meissner B, Zimmermann M, Möricke A, Schrauder A, Bouquin JP, et al. IKZF1 deletion is an independent predictor of outcome in pediatric acute lymphoblastic leukemia treated according to the ALL-BFM 2000 protocol. Haematologica. (2013) 98:428–32. doi: 10.3324/haematol.2011.056135
24. Marke R, Havinga J, Cloos J, Demkes M, Poelmans G, Yuniati L, et al. Tumor suppressor IKZF1 mediates glucocorticoid resistance in B-cell precursor acute lymphoblastic leukemia. Leukemia. (2016) 30:1599–603. doi: 10.1038/leu.2015.359
25. Shao C, Yang J, Kong Y, Cheng C, Lu W, Guan H, et al. Overexpression of dominant-negative Ikaros 6 isoform is associated with resistance to TKIs in patients with Philadelphia chromosome positive acute lymphoblastic leukemia. Exp Ther Med. (2017) 14:3874–9. doi: 10.3892/etm.2017.4941
26. Molnár A, Georgopoulos K. The Ikaros gene encodes a family of functionally diverse zinc finger DNA-binding proteins. Mol Cell Biol. (1994) 14:8292–303. doi: 10.1128/MCB.14.12.8292
27. National Library of Medicine. IKZF1 . Available online at: https://www.ncbi.nlm.nih.gov/gene/10320 (Accessed June 4, 2024).
28. Kim J, Sif S, Jones B, Jackson A, Koipally J, Heller E, et al. Ikaros DNA-binding proteins direct formation of chromatin remodeling complexes in lymphocytes. Immunity. (1999) 10:345–55. doi: 10.1016/S1074-7613(00)80034-5
29. Powell MD, Read KA, Sreekumar BK, Oestreich KJ. Ikaros zinc finger transcription factors: regulators of cytokine signaling pathways and CD4(+) T helper cell differentiation. Front Immunol. (2019) 10:1299. doi: 10.3389/fimmu.2019.01299
30. Ochiai K, Kondo H, Okamura Y, Shima H, Kurokochi Y, Kimura K, et al. Zinc finger-IRF composite elements bound by Ikaros/IRF4 complexes function as gene repression in plasma cell. Blood Adv. (2018) 2:883–94. doi: 10.1182/bloodadvances.2017010413
31. Uckun FM, Ma H, Zhang J, Ozer Z, Dovat S, Mao C, et al. Serine phosphorylation by SYK is critical for nuclear localization and transcription factor function of Ikaros. Proc Natl Acad Sci U S A. (2012) 109:18072–7. doi: 10.1073/pnas.1209828109
32. Ma H, Qazi S, Ozer Z, Qazi S, Ozer Z, Zhang J, et al. Regulatory phosphorylation of Ikaros by Bruton’s tyrosine kinase. PloS One. (2013) 8:e71302. doi: 10.1371/journal.pone.0071302
33. Brown KE, Guest SS, Smale ST, Hahm K, Merkenschlager M, Fisher AG. Association of transcriptionally silent genes with Ikaros complexes at centromeric heterochromatin. Cell. (1997) 91:845–54. doi: 10.1016/S0092-8674(00)80472-9
34. Koipally J, Heller EJ, Seavitt JR, Georgopoulos K. Unconventional potentiation of gene expression by Ikaros. J Biol Chem. (2002) 277:13007–15. doi: 10.1074/jbc.M111371200
35. Cobb BS, Morales-Alcelay S, Kleiger G, Brown KE, Fisher AG, Smale ST. Targeting of Ikaros to pericentromeric heterochromatin by direct DNA binding. Genes Dev. (2000) 14:2146–60. doi: 10.1101/gad.816400
36. Payne MA. Zinc finger structure-function in Ikaros. World J Biol Chem. (2011) 26:161–6. doi: 10.4331/wjbc.v2.i6.161
37. Kenneth M, Casey W, Leslie B. Janeway’s immunobiology. 10th Edition. New York: W. W. Norton & Company Ltd (2022).
38. Bogaert DJ, Kuehn HS, Bonroy C, Calvo KR, Dehoorne J, Vanlander AV, et al. A novel IKAROS haploinsufficiency kindred with unexpectedly late and variable B-cell maturation defects. J Allergy Clin Immunol. (2018) 141:432–5.e7. doi: 10.1016/j.jaci.2017.08.019
39. Ng SY, Yoshida T, Zhang J, Georgopoulos K. Genome-wide lineage-specific transcriptional networks underscore Ikaros-dependent lymphoid priming in hematopoietic stem cells. Immunity. (2009) 30:493–507. doi: 10.1016/j.immuni.2009.01.014
40. Heizmann B, Kastner P, Chan S. Ikaros is absolutely required for pre-B cell differentiation by attenuating IL-7 signals. J Exp Med. (2013) 210:2823–32. doi: 10.1084/jem.20131735
41. Yin H, Wang J, Tan Y, Jiang M, Zhang H, Meng G. Transcription factor abnormalities in B-ALL leukemogenesis and treatment. Trends Cancer. (2023) 9:855–70. doi: 10.1016/j.trecan.2023.06.004
42. Kirstetter P, Thomas M, Dierich A, Kastner P, Chan S. Ikaros is critical for B cell differentiation and function. Eur J Immunol. (2002) 32:720–30. doi: 10.1002/1521-4141(200203)32:3<720::AID-IMMU720>3.0.CO;2-P
43. Avitahl N, Winandy S, Friedrich C, Jones B, Ge Y, Georgopoulos K. Ikaros sets thresholds for T cell activation and regulates chromosome propagation. Immunity. (1999) 10:333–43. doi: 10.1016/S1074-7613(00)80033-3
44. Georgopoulos K, Bigby M, Wang JH, Molnar A, Wu P, Winandy S, et al. The Ikaros gene is required for the development of all lymphoid lineages. Cell. (1994) 79:143–56. doi: 10.1016/0092-8674(94)90407-3
45. Lopez RA, Schoetz S, DeAngelis K, O’Neill D, Bank A. Multiple hematopoietic defects and delayed globin switching in Ikaros null mice. Proc Natl Acad Sci U S A. (2002) 99:602–7. doi: 10.1073/pnas.022412699
46. O’Neill DW, Schoetz SS, Lopez RA, O’Neill DW, Schoetz SS, Lopez RA, et al. An ikaros-containing chromatin-remodeling complex in adult-type erythroid cells. Mol Cell Biol. (2000) 20:7572–82. doi: 10.1128/MCB.20.20.7572-7582.2000
47. Boutboul D, Kuehn HS, Van de Wyngaert Z, Niemela JE, Callebaut I, Stoddard J, et al. Dominant-negative IKZF1 mutations cause a T, B, and myeloid cell combined immunodeficiency. J Clin Invest. (2018) 128:3071–87. doi: 10.1172/JCI98164
48. Kuehn HS, Boisson B, Cunningham-Rundles C, Reichenbach J, Stray-Pedersen A, Gelfand EW, et al. Loss of B cells in patients with heterozygous mutations in IKAROS. N Engl J Med. (2016) 374:1032–43. doi: 10.1056/NEJMoa1512234
49. Groth DJ, Lakkaraja MM, Ferreira JO, Feuille EJ, Bassetti JA, Kaicker SM. Management of chronic immune thrombocytopenia and presumed autoimmune hepatitis in a child with IKAROS haploinsufficiency. J Clin Immunol. (2020) 40:653–7. doi: 10.1007/s10875-020-00781-y
50. Banday AZ, Jindal AK, Kaur A, Saka R, Parwaiz A, Sachdeva MUS, et al. Cutaneous IgA vasculitis-presenting manifestation of a novel mutation in the IKZF1 gene. Rheumatol (Oxford). (2021) 60:e101–3. doi: 10.1093/rheumatology/keaa492
51. Yilmaz E, Kuehn HS, Odakir E, Niemela JE, Ozcan A, Eken A, et al. Common variable immunodeficiency, autoimmune hemolytic anemia, and pancytopenia associated with a defect in IKAROS. J Pediatr Hematol Oncol. (2021) 43:e351–351e357. doi: 10.1097/MPH.0000000000001976
52. Belot A, Rice GI, Omarjee SO, Rouchon Q, Smith EMD, Moreews M, et al. Contribution of rare and predicted pathogenic gene variants to childhood-onset lupus: a large, genetic panel analysis of British and French cohorts. Lancet Rheumatol. (2020) 2:e99–e109. doi: 10.1016/S2665-9913(19)30142-0
53. Thaventhiran J, Lango Allen H, Burren OS, Rae W, Greene D, Staples E, et al. Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature. (2020) 583:90–5. doi: 10.1038/s41586-020-2265-1
54. Allman D, Dalod M, Asselin-Paturel C, Delale T, Robbins SH, Trinchieri G, et al. Ikaros is required for plasmacytoid dendritic cell differentiation. Blood. (2006) 108:4025–34. doi: 10.1182/blood-2006-03-007757
55. Kellner ES, Krupski C, Kuehn HS, Rosenzweig SD, Yoshida N, Kojima S, et al. Allogeneic hematopoietic stem cell transplant outcomes for patients with dominant negative IKZF1/IKAROS mutations. J Allergy Clin Immunol. (2019) 144:339–42. doi: 10.1016/j.jaci.2019.03.025
56. Nunes-Santos CJ, Kuehn HS, Rosenzweig SD. IKAROS family zinc finger 1-associated diseases in primary immunodeficiency patients. Immunol Allergy Clin North Am. (2020) 40:461–70. doi: 10.1016/j.iac.2020.04.004
57. Schwickert TA, Tagoh H, Schindler K, Fischer M, Jaritz M, Busslinger M. Ikaros prevents autoimmunity by controlling anergy and Toll-like receptor signaling in B cells. Nat Immunol. (2019) 20:1517–29. doi: 10.1038/s41590-019-0490-2
58. Marçais A, Jeannet R, Hernandez L, Soulier J, Sigaux F, Chan S, et al. Genetic inactivation of Ikaros is a rare event in human T-ALL. Leuk Res. (2010) 34:426–9. doi: 10.1016/j.leukres.2009.09.012
59. Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. (2007) 446:758–64. doi: 10.1038/nature05690
60. Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. (2008) 453:110–4. doi: 10.1038/nature06866
61. Zhang J, Ding L, Holmfeldt L, Wu G, Heatley SL, Payne-Turner D, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. (2012) 481:157–63. doi: 10.1038/nature10725
62. Zhang X, Huang A, Liu L, Qin J, Wang C, Yang M, et al. The clinical impact of IKZF1 mutation in acute myeloid leukemia. Exp Hematol Oncol. (2023) 12:33. doi: 10.1186/s40164-023-00398-y
63. Grossmann V, Kohlmann A, Zenger M, Schindela S, Eder C, Weissmann S, et al. A deep-sequencing study of chronic myeloid leukemia patients in blast crisis (BC-CML) detects mutations in 76. 9% cases. Leukemia. (2011) 25:557–60. doi: 10.1038/leu.2010.298
64. Jäger R, Gisslinger H, Passamonti F, Rumi E, Berg T, Gisslinger B, et al. Deletions of the transcription factor Ikaros in myeloproliferative neoplasms. Leukemia. (2010) 24:1290–8. doi: 10.1038/leu.2010.99
65. Roberts KG, Morin RD, Zhang J, Hirst M, Zhao Y, Su X, et al. Genetic alterations activating kinase and cytokine receptor signaling in high-risk acute lymphoblastic leukemia. Cancer Cell. (2012) 22:153–66. doi: 10.1016/j.ccr.2012.06.005
66. van der Veer A, Zaliova M, Mottadelli F, De Lorenzo P, Te Kronnie G, Harrison CJ, et al. IKZF1 status as a prognostic feature in BCR-ABL1-positive childhood ALL. Blood. (2014) 123:1691–8. doi: 10.1182/blood-2013-06-509794
67. Roberts KG, Li Y, Payne-Turner D, Harvey RC, Yang YL, Pei D, et al. Targetable kinase-activating lesions in Ph-like acute lymphoblastic leukemia. N Engl J Med. (2014) 371:1005–15. doi: 10.1056/NEJMoa1403088
68. Fedullo AL, Messina M, Elia L, Piciocchi A, Gianfelici V, Lauretti A, et al. Prognostic implications of additional genomic lesions in adult Philadelphia chromosome-positive acute lymphoblastic leukemia. Haematologica. (2019) 104:312–8. doi: 10.3324/haematol.2018.196055
69. Churchman ML, Qian M, Te Kronnie G, Zhang R, Yang W, Zhang H, et al. Germline genetic IKZF1 variation and predisposition to childhood acute lymphoblastic leukemia. Cancer Cell. (2018) 33:937–48.e8. doi: 10.1016/j.ccell.2018.03.021
70. Tonnelle C, Imbert MC, Sainty D, Granjeaud S, N’Guyen C, Chabannon C. Overexpression of dominant-negative Ikaros 6 protein is restricted to a subset of B common adult acute lymphoblastic leukemias that express high levels of the CD34 antigen. Hematol J. (2003) 4:104–9. doi: 10.1038/sj.thj.6200235
71. Iacobucci I, Iraci N, Messina M, Lonetti A, Chiaretti S, Valli E, et al. IKAROS deletions dictate a unique gene expression signature in patients with adult B-cell acute lymphoblastic leukemia. PloS One. (2012) 7:e40934. doi: 10.1371/journal.pone.0040934
72. Reynaud D, Demarco IA, Reddy KL, Schjerven H, Bertolino E, Chen Z, et al. Regulation of B cell fate commitment and immunoglobulin heavy-chain gene rearrangements by Ikaros. Nat Immunol. (2008) 9:927–36. doi: 10.1038/ni.1626
73. Ma S, Pathak S, Mandal M, Trinh L, Clark MR, Lu R. Ikaros and Aiolos inhibit pre-B-cell proliferation by directly suppressing c-Myc expression. Mol Cell Biol. (2010) 30:4149–58. doi: 10.1128/MCB.00224-10
74. Alkhatib A, Werner M, Hug E, Herzog S, Eschbach C, Faraidun H, et al. FoxO1 induces Ikaros splicing to promote immunoglobulin gene recombination. J Exp Med. (2012) 209:395–406. doi: 10.1084/jem.20110216
75. Li JF, Dai YT, Lilljebjorn H, Shen SH, Cui BW, Bai L, et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc Natl Acad Sci U S A. (2018) 115:E11711–E20. doi: 10.1073/pnas.1814397115
76. Scheijen B, Boer JM, Marke R, Tijchon E, van Ingen Schenau D, Waanders E, et al. Tumor suppressors BTG1 and IKZF1 cooperate during mouse leukemia development and increase relapse risk in B-cell precursor acute lymphoblastic leukemia patients. Haematologica. (2017) 102:541–51. doi: 10.3324/haematol.2016.153023
77. Joshi I, Yoshida T, Jena N, Qi X, Zhang J, Van Etten RA, et al. Loss of Ikaros DNA-binding function confers integrin-dependent survival on pre-B cells and progression to acute lymphoblastic leukemia. Nat Immunol. (2014) 15:294–304. doi: 10.1038/ni.2821
78. Churchman ML, Evans K, Richmond J, Robbins A, Jones L, Shapiro IM, et al. Synergism of FAK and tyrosine kinase inhibition in Ph(+) B-ALL. JCI Insight. (2016) 1:e86082. doi: 10.1172/jci.insight.86082
79. Stanulla M, Dagdan E, Zaliova M, Möricke A, Palmi C, Cazzaniga G, et al. IKZF1(plus) defines a new minimal residual disease-dependent very-poor prognostic profile in pediatric B-cell precursor acute lymphoblastic leukemia. J Clin Oncol. (2018) 36:1240–9. doi: 10.1200/JCO.2017.74.3617
80. Winer P, Muskens IS, Walsh KM, Vora A, Moorman AV, Wiemels JL, et al. Germline variants in predisposition genes in children with Down syndrome and acute lymphoblastic leukemia. Blood Adv. (2020) 4:672–5. doi: 10.1182/bloodadvances.2019001216
81. Buitenkamp TD, Pieters R, Gallimore NE, van der Veer A, Meijerink JP, Beverloo HB, et al. Outcome in children with Down’s syndrome and acute lymphoblastic leukemia: role of IKZF1 deletions and CRLF2 aberrations. Leukemia. (2012) 26:2204–11. doi: 10.1038/leu.2012.84
82. Palmi C, Bresolin S, Junk S, Fazio G, Silvestri D, Zaliova M, et al. Definition and prognostic value of ph-like and IKZF1plus status in children with down syndrome and B-cell precursor acute lymphoblastic leukemia. Hemasphere. (2023) 7:e892. doi: 10.1097/HS9.0000000000000892
83. Moorman AV, Harrison CJ, Buck GA, Richards SM, Secker-Walker LM, Martineau M, et al. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood. (2007) 109:3189–97. doi: 10.1182/blood-2006-10-051912
84. Kuiper RP, Schoenmakers EF, van Reijmersdal SV, Hehir-Kwa JY, van Kessel AG, van Leeuwen FN, et al. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. (2007) 21:1258–66. doi: 10.1038/sj.leu.2404691
85. Den Boer ML, van Slegtenhorst M, De Menezes RX, Cheok MH, Buijs-Gladdines JG, Peters ST, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: a genome- wide classification study. Lancet Oncol. (2009) 10:125–34. doi: 10.1016/S1470-2045(08)70339-5
86. Boer JM, van der Veer A, Rizopoulos D, Fiocco M, Sonneveld E, de Groot-Kruseman HA, et al. Prognostic value of rare IKZF1 deletion in childhood B-cell precursor acute lymphoblastic leukemia: an international collaborative study. Leukemia. (2016) 30:32–8. doi: 10.1038/leu.2015.199
87. Chen IM, Harvey RC, Mullighan CG, Gastier-Foster J, Wharton W, Kang H, et al. Outcome modeling with CRLF2, IKZF1, JAK, and minimal residual disease in pediatric acute lymphoblastic leukemia: a Children’s Oncology Group study. Blood. (2012) 119:3512–22. doi: 10.1182/blood-2011-11-394221
88. Asai D, Imamura T, Suenobu S, Saito A, Hasegawa D, Deguchi T, et al. IKZF1 deletion is associated with a poor outcome in pediatric B-cell precursor acute lymphoblastic leukemia in Japan. Cancer Med. (2013) 2:412–9. doi: 10.1002/cam4.87
89. Clappier E, Grardel N, Bakkus M, Rapion J, De Moerloose B, Kastner P, et al. IKZF1 deletion is an independent prognostic marker in childhood B-cell precursor acute lymphoblastic leukemia, and distinguishes patients benefiting from pulses during maintenance therapy: results of the EORTC Children’s Leukemia Group study 58951. Leukemia. (2015) 29:2154–61. doi: 10.1038/leu.2015.134
90. Hamadeh L, Enshaei A, Schwab C, Alonso CN, Attarbaschi A, Barbany G, et al. Validation of the United Kingdom copy-number alteration classifier in 3239 children with B-cell precursor ALL. Blood Adv. (2019) 3:148–57. doi: 10.1182/bloodadvances.2018025718
91. Ribera J, Morgades M, Zamora L, Montesinos P, Gómez-Seguí I, Pratcorona M, et al. Prognostic significance of copy number alterations in adolescent and adult patients with precursor B acute lymphoblastic leukemia enrolled in PETHEMA protocols. Cancer. (2015) 121:3809–17. doi: 10.1002/cncr.29579
92. Kobitzsch B, Gökbuget N, Schwartz S, Reinhardt R, Brüggemann M, Viardot A, et al. Loss-of-function but not dominant-negative intragenic IKZF1 deletions are associated with an adverse prognosis in adult BCR-ABL-negative acute lymphoblastic leukemia. Haematologica. (2017) 102:1739–47. doi: 10.3324/haematol.2016.161273
93. Zhang W, Kuang P, Li H, Wang F, Wang Y. Prognostic significance of IKZF1 deletion in adult B cell acute lymphoblastic leukemia: a meta-analysis. Ann Hematol. (2017) 96:215–25. doi: 10.1007/s00277-016-2869-6
94. Ribera J, Zamora L, Morgades M, Vives S, Granada I, Montesinos P, et al. Molecular profiling refines minimal residual disease-based prognostic assessment in adults with Philadelphia chromosome-negative B-cell precursor acute lymphoblastic leukemia. Genes Chromosomes Cancer. (2019) 58:815–9. doi: 10.1002/gcc.22788
95. Fang Q, Yuan T, Li Y, Feng J, Gong X, Li Q, et al. Prognostic significance of copy number alterations detected by multi-link probe amplification of multiple genes in adult acute lymphoblastic leukemia. Oncol Lett. (2018) 15:5359–67. doi: 10.3892/ol
96. Zaliova M, Stuchly J, Winkowska L, Musilova A, Fiser K, Slamova M, et al. Genomic landscape of pediatric B-other acute lymphoblastic leukemia in a consecutive European cohort. Haematologica. (2019) 104:1396–406. doi: 10.3324/haematol.2018.204974
97. Clappier E, Auclerc MF, Rapion J, Bakkus M, Caye A, Khemiri A, et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia. (2014) 28:70–7. doi: 10.1038/leu.2013.277
98. Zaliova M, Zimmermannova O, Dörge P, Eckert C, Möricke A, Zimmermann M, et al. ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia. (2015) 29:1222. doi: 10.1038/leu.2015.77
99. Li H, Zhang W, Kuang P, Ye Y, Yang J, Dai Y, et al. Combination of IKZF1 deletion and early molecular response show significant roles on prognostic stratification in Philadelphia chromosome-positive acute lymphoblastic leukemia patients. Leuk Lymphoma. (2018) 59:1890–8. doi: 10.1080/10428194.2017.1406933
100. Waanders E, van der Velden VH, van der Schoot CE, van Leeuwen FN, van Reijmersdal SV, de Haas V, et al. Integrated use of minimal residual disease classification and IKZF1 alteration status accurately predicts 79% of relapses in pediatric acute lymphoblastic leukemia. Leukemia. (2011) 25:254–8. doi: 10.1038/leu.2010.275
101. Eckardt JN, Stasik S, Röllig C, Petzold A, Sauer T, Scholl S, et al. Mutated IKZF1 is an independent marker of adverse risk in acute myeloid leukemia. Leukemia. (2023) 37:2395–403. doi: 10.1038/s41375-023-02061-1
102. Wang Y, Cheng W, Zhang Y, Zhang Y, Sun T, Zhu Y, et al. Identification of IKZF1 genetic mutations as new molecular subtypes in acute myeloid leukaemia. Clin Transl Med. (2023) 13:e1309. doi: 10.1002/ctm2.1309
103. Park SM, Cho H, Thornton AM, Barlowe TS, Chou T, Chhangawala S, et al. IKZF2 drives leukemia stem cell self-renewal and inhibits myeloid differentiation. Cell Stem Cell. (2019) 24:153–65. doi: 10.1016/j.stem.2018.10.016
104. Churchman ML, Low J, Qu C, Paietta EM, Kasper LH, Chang Y, et al. Efficacy of retinoids in IKZF1-mutated BCR-ABL1 acute lymphoblastic leukemia. Cancer Cell. (2015) 28:343–56. doi: 10.1016/j.ccell.2015.07.016
Keywords: IKZF1 gene, IKAROS, BCP-ALL, Ph+ALL, transcription factor, MRD
Citation: Feng L, Zhang H and Liu T (2024) Multifaceted roles of IKZF1 gene, perspectives from bench to bedside. Front. Oncol. 14:1383419. doi: 10.3389/fonc.2024.1383419
Received: 07 February 2024; Accepted: 10 June 2024;
Published: 24 June 2024.
Edited by:
Sebastien Malinge, University of Western Australia, AustraliaReviewed by:
Chandrika Gowda, The Pennsylvania State University, United StatesAnilkumar Gopalakrishnapillai, Alfred I. duPont Hospital for Children, United States
Sung Chiu, St John of God Midland Public Hospital, Australia
Copyright © 2024 Feng, Zhang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ting Liu, bGl1dGluZ0BzY3UuZWR1LmNu