 Lin Long
Lin Long Xiangyu Fei1†
Xiangyu Fei1†- 1School of Pharmaceutical Science, Hengyang Medical School, University of South China, Hengyang, China
- 2The First Affiliated Hospital, Hengyang Medical School, University of South China, Hengyang, China
- 3Department of Pharmacy, Central Hospital of Hengyang, Hengyang, China
Triple-negative breast cancer (TNBC) poses a significant clinical challenge due to its propensity for metastasis and poor prognosis. TNBC evades the body’s immune system recognition and attack through various mechanisms, including the Janus Kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3) signaling pathway. This pathway, characterized by heightened activity in numerous solid tumors, exhibits pronounced activation in specific TNBC subtypes. Consequently, targeting the JAK2/STAT3 signaling pathway emerges as a promising and precise therapeutic strategy for TNBC. The signal transduction cascade of the JAK2/STAT3 pathway predominantly involves receptor tyrosine kinases, the tyrosine kinase JAK2, and the transcription factor STAT3. Ongoing preclinical studies and clinical research are actively investigating this pathway as a potential therapeutic target for TNBC treatment. This article comprehensively reviews preclinical and clinical investigations into TNBC treatment by targeting the JAK2/STAT3 signaling pathway using small molecule compounds. The review explores the role of the JAK2/STAT3 pathway in TNBC therapeutics, evaluating the benefits and limitations of active inhibitors and proteolysis-targeting chimeras in TNBC treatment. The aim is to facilitate the development of novel small-molecule compounds that target TNBC effectively. Ultimately, this work seeks to contribute to enhancing therapeutic efficacy for patients with TNBC.
1 Introduction
Globally, breast cancer stands as the most prevalent malignant tumor (1). Among its subtypes, triple-negative breast cancer (TNBC), characterized by the absence of estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER-2) expression, constitutes approximately 10%-15% of all breast cancer cases (2). The treatment of TNBC presents significant challenges, notably its propensity for early metastasis (3) and a comparatively poorer prognosis than other breast cancer subtypes (4). Current clinical strategies for TNBC primarily employ taxanes and anthracycline-based cytotoxic drugs. However, these approaches frequently encounter obstacles in the form of chemotherapy resistance, and the absence of effective small-molecule targeted therapies, constituting primary impediments in TNBC clinical management (5).
Reassessing classic drug targets is crucial for advancing new precision medicine strategies. The JAK2/STAT3 pathway, clinically validated as a therapeutic target for inflammation-related conditions, has shown promise through its inhibitors in treating inflammatory and autoimmune diseases. This success paves the way for novel clinical therapy developments (6, 7). Extensive research has established a strong association between aberrations in the JAK2/STAT3 signaling pathway and key oncogenic processes such as proliferation, invasion, and metastasis in various malignancies, including TNBC. Notably, activation of this pathway has been observed in multiple solid tumors, TNBC included (8–12). Targeted inhibition of the JAK2/STAT3 signaling has demonstrated efficacy in curtailing TNBC cell proliferation, invasion, and migration (13), knockdown of JAK2 or STAT3 in triple-negative breast cancer cells significantly reduced cell proliferation, invasion and migration (14–21), tumor volume and distant metastasis were significantly inhibited in a mouse model of triple-negative breast cancer with conditional knockout of JAK2 or STAT3 (22–25). Moreover, the downregulation of this pathway has been shown to counteract paclitaxel (PTX) resistance (26). Thus, targeting JAK2/STAT3 emerges as a promising therapeutic strategy for treating TNBC and overcoming challenges associated with PTX resistance.
The signaling cascade of the JAK2/STAT3 pathway is predominantly mediated through receptor tyrosine kinases (RTKs), JAK2, and the transcription factor STAT3. RTKs are single-pass transmembrane proteins ubiquitously expressed across various cell types, including those within the tumor microenvironment. Characteristically, all RTKs possess a conserved structural composition: an extracellular ligand-binding domain, a transmembrane domain, and an intracellular tyrosine kinase domain (27). Upon ligand binding, RTKs undergo dimerization on the cell membrane and phosphorylate tyrosine residues on the receptors, facilitating their recognition and binding by downstream proteins with SH2 domains, such as JAK2. RTK dimerization brings the associated JAK2 kinase into proximity, enabling their activation through reciprocal tyrosine phosphorylation. Activated JAK2 then stimulates the RTKs to generate binding sites for STAT3. STAT3 binds to RTKs through its SH2 domain and undergoes phosphorylation under the influence of JAK2. The phosphorylated STAT3 forms homodimers and enters the nucleus to induce downstream signal transduction, effectuating various physiological or pathological roles (28). Given the characteristics of this signaling pathway, targeting its components to treat TNBC represents an effective strategy for precision therapy (Figure 1).
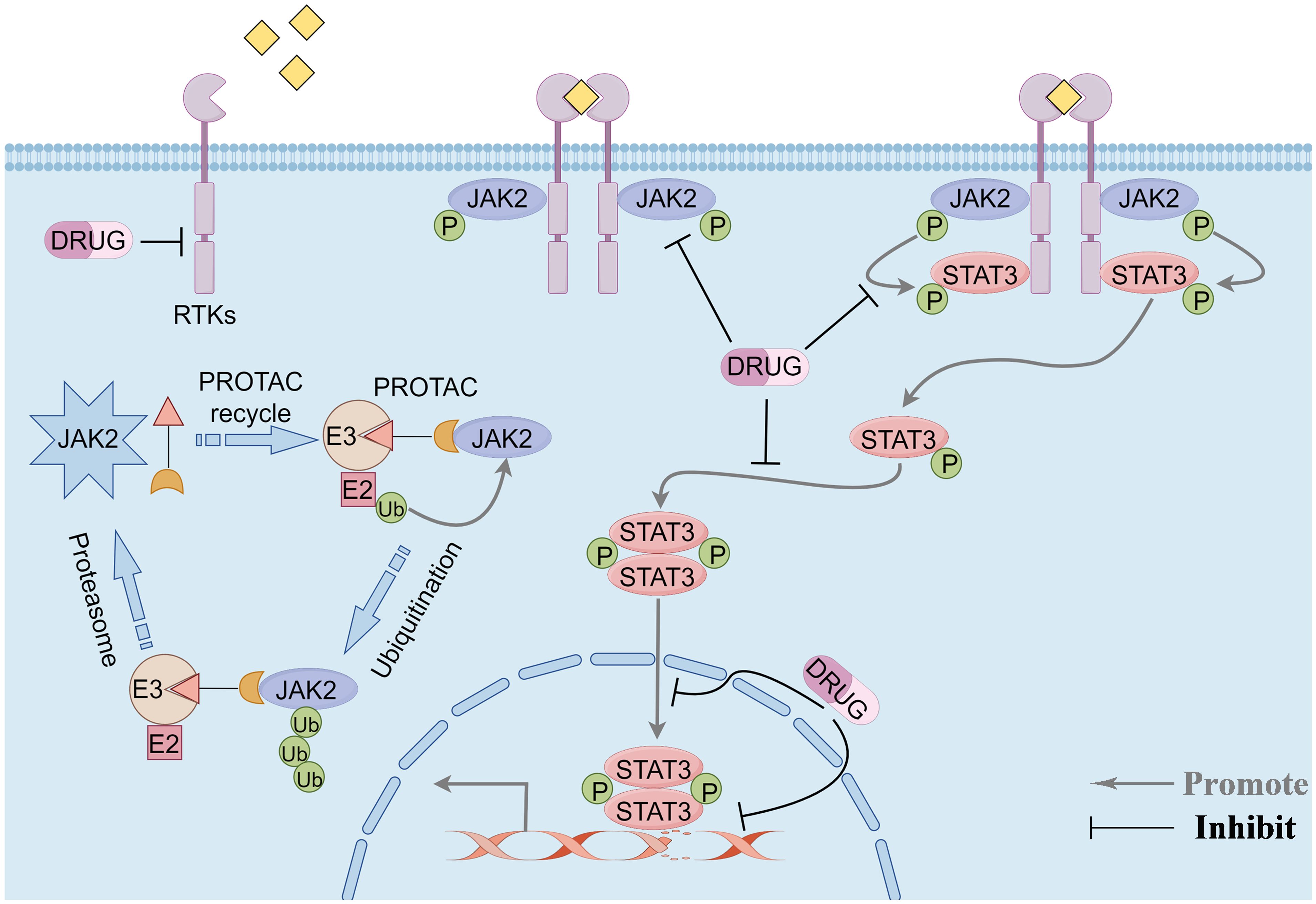
Figure 1 Overview of the JAK2/STAT3 pathway signaling modality and potential therapeutic targets of the pathway. Phosphorylation signaling blockade and protein-targeted degradation pathways. By Figdraw.
Consequently, this review systematically summarizes the roles of the JAK/STAT3 pathway in the pathogenesis of TNBC and the current advances in research on small-molecule compounds targeting the JAK/STAT3 signaling pathway as a therapeutic approach for TNBC.
2 Receptor tyrosine kinases in TNBC
Receptor tyrosine kinases represent a diverse class of enzyme-linked cell surface receptors with a high affinity for growth factors, cytokines, and hormones. These receptors not only bind specific ligands but also function as protein kinases, phosphorylating tyrosine residues on target proteins. RTKs are categorized into 20 distinct families based on the types of ligands they bind (29). TNBC expresses various RTKs, including epidermal growth factor receptors (EGFRs) (30), vascular endothelial growth factor receptors (VEGFRs) (28), insulin-like growth factor receptors (IGFRs) (31), platelet-derived growth factor receptors (PDGFRs) (32), fibroblast growth factor receptors (FGFRs) (33), leukemia inhibitory factor receptor (LIFR) (34), interleukin-6 cell factor receptor (IL-6R) (35), interleukin-13 cell factor receptor (IL-13R) (36), and glycoprotein 130 receptor (GP130) (37). Before ligand binding, RTKs exist on the cell surface as inactive monomers. Homologous ligand binding induces receptor dimerization, activating their intrinsic kinase activity (38).
2.1 Epidermal growth factor receptors: regulator of progression, metastasis, and cancer stem cells in TNBC
Epidermal growth factor receptors, the receptor for epidermal growth factor (EGF), is a key member of the HER family, which also includes Her-2, Her-3, and Her-4 (39). EGF binding to EGFR induces receptor dimerization, a critical step leading to the autophosphorylation of tyrosine residues on the activated receptor. This activation allows the receptor to recruit various signal sequence proteins, transmitting biological signals from the extracellular milieu to the intracellular domain. These signaling cascades culminate in gene transcription, modulating key cellular processes such as proliferation, differentiation, and apoptosis. In cancer, EGFR contributes to tumor progression by promoting invasion and metastasis and stimulating tumor angiogenesis (40). EGFR activates complex signal transduction pathways with primary pathways including mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) (41), JAK2/STAT3 (42), and phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) (43). Dysregulation of these pathways is intricately linked to tumor development, invasion, and metastasis. EGFR is overexpressed in several malignant tumors, including lung, colon, liver, and breast cancers (44–47). In the context of cancer prognosis, EGFR overexpression is associated with shorter recurrence times, increased recurrence rates, and reduced survival durations (48). In TNBC, the positive expression rate of EGFR is notably higher than in non-TNBC, with over 40% of patients with TNBC exhibiting EGFR overexpression, a factor closely correlated with TNBC prognosis (49, 50). Targeted inhibition of EGFR expression has demonstrated anti-cancer effects in TNBC (51). EGFR expression is also implicated in CD44+ cell aggregation, with its inhibition disrupting cancer stem cell assembly in TNBC. These lines of evidence suggest a link between EGFR expression and the progression of CD44+-mediated cancer stem cells (52).
2.2 Vascular endothelial growth factor receptors: regulator of angiogenesis and cancer stem cells in TNBC
Vascular endothelial growth factor receptors, the receptor for vascular endothelial growth factor (VEGF), comprises three primary types: VEGFR1, VEGFR2, and VEGFR3. VEGF induces angiogenesis by binding to VEGFR-2, enhancing the survival, proliferation, migration, and adhesion of endothelial cells (53). However, in pathological contexts, particularly in cancer, VEGFR expression is linked to the promotion of tumor angiogenesis and metastasis (54, 55). Numerous studies have documented the overexpression of VEGFR in a range of malignant tumors, including lung, colon, breast, liver, and ovarian cancers (56–60). In TNBC, elevated VEGF levels correlate with increased metastasis, poor treatment response, and decreased survival rates (61). Upregulation of VEGFR in TNBC is linked to heightened cell proliferation, while its downregulation inhibits this proliferation (62). A notable randomized cohort study has indicated a strong association between high VEGFR expression and 5-year and 10-year breast cancer-specific survival rates in patients (63). These findings underscore the potential of targeting the VEGF/VEGFR axis as a promising approach in the targeted therapy of TNBC. Research employing primary breast cancer mouse models and models of spontaneous breast cancer metastasis has revealed elevated VEGFR expression levels in metastatic breast cancer compared to non-metastatic forms (64). Additionally, VEGFR expression correlates with cancer stem cell characteristics. By activating the VEGFR2/STAT3 pathway, VEGF induces the upregulation of Myc and Sox2 expression, thereby promoting the self-renewal of breast cancer stem cells. The autocrine action of VEGF can establish a positive feedback loop, diminishing the efficacy of anti-angiogenic drugs and enhancing cancer stem cell renewal (28). Consequently, targeting VEGFR expression emerges as a potentially effective therapeutic strategy for the regulation of breast cancer stem cells.
2.3 Platelet-derived growth factor receptors: regulator of endothelial cell differentiation and cancer stem cells in TNBC
Tumor blood vessel development is crucial to tumor growth, making angiogenesis a potential target in cancer therapy (65). Platelet-derived growth factor receptors, which binds to platelet-derived growth factor (PDGF), exists in two forms: PDGFRα and PDGFRβ. PDGFR activation, contingent upon PDGF interaction, initiates various intracellular signaling pathways. While PDGFR contributes to vascular repair after tissue damage (66), it also promotes cell proliferation within tumor tissues (67). Studies involving mouse models with differential PDGF gene expression have yielded insightful observations. Specifically, tumors in mice with PDGF gene deficiency exhibit reduced pericyte recruitment, whereas tumors in mice with PDGF overexpression demonstrate increased pericyte recruitment. These findings suggest that tumors recruit pericytes through paracrine PDGF secretion, interacting with PDGFR, facilitating blood vessel maturation, and synergizing with VEGF-mediated angiogenesis, contributing to tumor vascularization (68). Extensive research indicates that PDGFR is overexpressed in various malignant tumors, including lung, colon, breast, and ovarian (69–72). In TNBC, PDGFRβ plays a notable role in mediating endothelial cell differentiation and vasculogenic mimicry in tumor cells (32). Further studies have identified a link between PDGFRβ expression in TNBC, and cancer stem cells, where FOXC2 induces cancer stem cell characteristics and metastasis by upregulating PDGFRβ expression (73). These findings position PDGFR as a promising therapeutic target for TNBC.
2.4 Fibroblast growth factor receptors: regulator of cell proliferation and cancer stem cells in TNBC
Fibroblast growth factor receptors, the receptor for fibroblast growth factor (FGF), comprises four subtypes: FGFR1, FGFR2, FGFR3, and FGFR4, collectively forming the FGFR family. Upon binding with FGF, FGFR is activated and modulates multiple intracellular signaling pathways crucial to various biological processes, including angiogenesis and lymphangiogenesis (74). Studies have highlighted the association of FGFR expression in various solid tumors with tumor cell proliferation (75–79). High-throughput sequencing has identified FGFR gene mutations in approximately 7.1% of malignant tumors, with breast cancer exhibiting the second-highest frequency after urothelial carcinoma (80). In TNBC, FGFR3 expression is observed, and inhibition of the FGFR3 signaling pathway reduces TNBC cell invasion and migration (81). Targeting and blocking the FGFR pathway can significantly enhance T cell infiltration and suppress tumor growth in TNBC (33). Some studies have revealed that estrogen can stimulate breast cancer stem cell proliferation via the paracrine FGF/FGFR/T-box transcription factor 3 (TBx3) signaling pathway, and inhibiting this pathway curtails cancer stem cell expansion in TNBC (82). These findings highlight the potential of targeting the FGFR pathway as a therapeutic approach in TNBC.
3 Activation of the JAK2/STAT3 signaling pathway in TNBC
Janus Kinase 2, a member of the JAK family of non-receptor tyrosine kinases, includes JAK1, JAK3, and tyrosine kinase 2 (TYK2). JAK3 is predominantly expressed in hematopoietic cells, while JAK1, JAK2, and TYK2 exhibit broader expression across various tissues (83). JAKs mediate a range of disease processes, including immune system disorders (84), hematologic conditions (85), and various malignancies (86, 87). Signal transducer and activator of transcription (STAT) protein family comprises members such as STAT1, STAT2, STAT3, STAT4, STAT5A, STAT5B, and STAT6. Notably, STAT3 is implicated in the promotion of tumor growth and the induction of immunosuppression (88–90). The JAK2/STAT3 signaling pathway is ubiquitously expressed in cells and vital in physiological functions (91), such as cell proliferation, differentiation, apoptosis, and immune regulation (92). Beyond its physiological roles, this pathway is significantly implicated in various pathologies, notably in cancer and autoimmune disorders. In breast cancer, particularly TNBC, JAK2/STAT3 signaling is known for its excessive activation (93). Advanced research has facilitated a more precise molecular classification of TNBC, identifying the mesenchymal subtype characterized by heightened JAK2/STAT3 activity (8). This insight offers a new direction and foundation for the personalized clinical treatment of TNBC, focusing on targeting the JAK2/STAT3 signaling pathway. Research has elucidated that the JAK2/STAT3/Cyclin D2 signaling pathway is pivotal in promoting cancer stem cell proliferation (94). Specifically, in TNBC, studies have demonstrated that downregulating the JAK2/STAT3 pathway can significantly inhibit cancer stem cell proliferation (95). Furthermore, additional research has indicated that suppressing this pathway may reduce TNBC cell proliferation and migration (13).
4 The role of the JAK2/STAT3 signaling pathway in multidrug resistance in TNBC
The lack of effective targeted therapies, necessitating reliance on taxanes and anthracycline cytotoxic drugs significantly hinders the treatment of TNBC. However, the emergence of multidrug resistance during treatment poses a formidable challenge to this approach (5). For instance, the activation of the JAK2/STAT3 signaling pathway in nasopharyngeal carcinoma has been demonstrated to induce forkhead box M1 transcription, thereby enhancing resistance to PTX (96). Subsequent studies have revealed the contribution of the JAK2/STAT3 pathway to the development of PTX resistance by upregulating anti-apoptotic gene expression. Targeting this pathway has proven effective in reversing PTX resistance in ovarian cancer (97). In a model of PTX-resistant cells, researchers observed differential expression of the JAK2 gene, suggesting its potential role as a candidate gene linked to PTX resistance in ovarian cancer cell lines (98). Further studies indicate that the downregulation of JAK2/STAT3 signaling pathway can counteract PTX resistance in TNBC (26). Furthermore, a separate research effort found that JAK2 inhibitors can directly bind to the drug efflux protein P-gp in resistant cell lines, thus impeding P-gp-mediated drug efflux (99). Collectively, these studies underscore the significance of the JAK2/STAT3 signaling pathway in the development of multidrug resistance. Consequently, targeting this pathway, either as a standalone therapy or in combination with PTX, presents a promising strategy for the treatment of PTX-resistant TNBC.
5 Current therapeutic applications of the JAK2/STAT3 signaling pathway in TNBC
Recent research has elucidated the pivotal role of the JAK2/STAT3 signaling pathway in driving the proliferation, invasion, and migration of TNBC. These findings position the JAK2/STAT3 pathway as a promising therapeutic target for TNBC management. In response to these insights, numerous preclinical and clinical studies are actively exploring the development of inhibitors targeting RTKs, JAK2, and STAT3. These inhibitors are categorized based on their mode of action into traditional small molecule inhibitors in the occupation-driven mode and proteolysis targeting chimera (PROTAC) molecules based on ubiquitin-mediated protein degradation in an event-driven mode. Traditional small molecule inhibitors in the occupation-driven mode function by occupying the active site or binding site of the target protein with small molecule compounds. This action blocks its interaction with downstream signaling molecules, inhibiting its function. On the other hand, PROTAC molecules employ a ligand linker to bind the target protein with an E3 ubiquitin ligase, leveraging the ubiquitin-proteasome system to drive the degradation of the target protein (Figure 2).
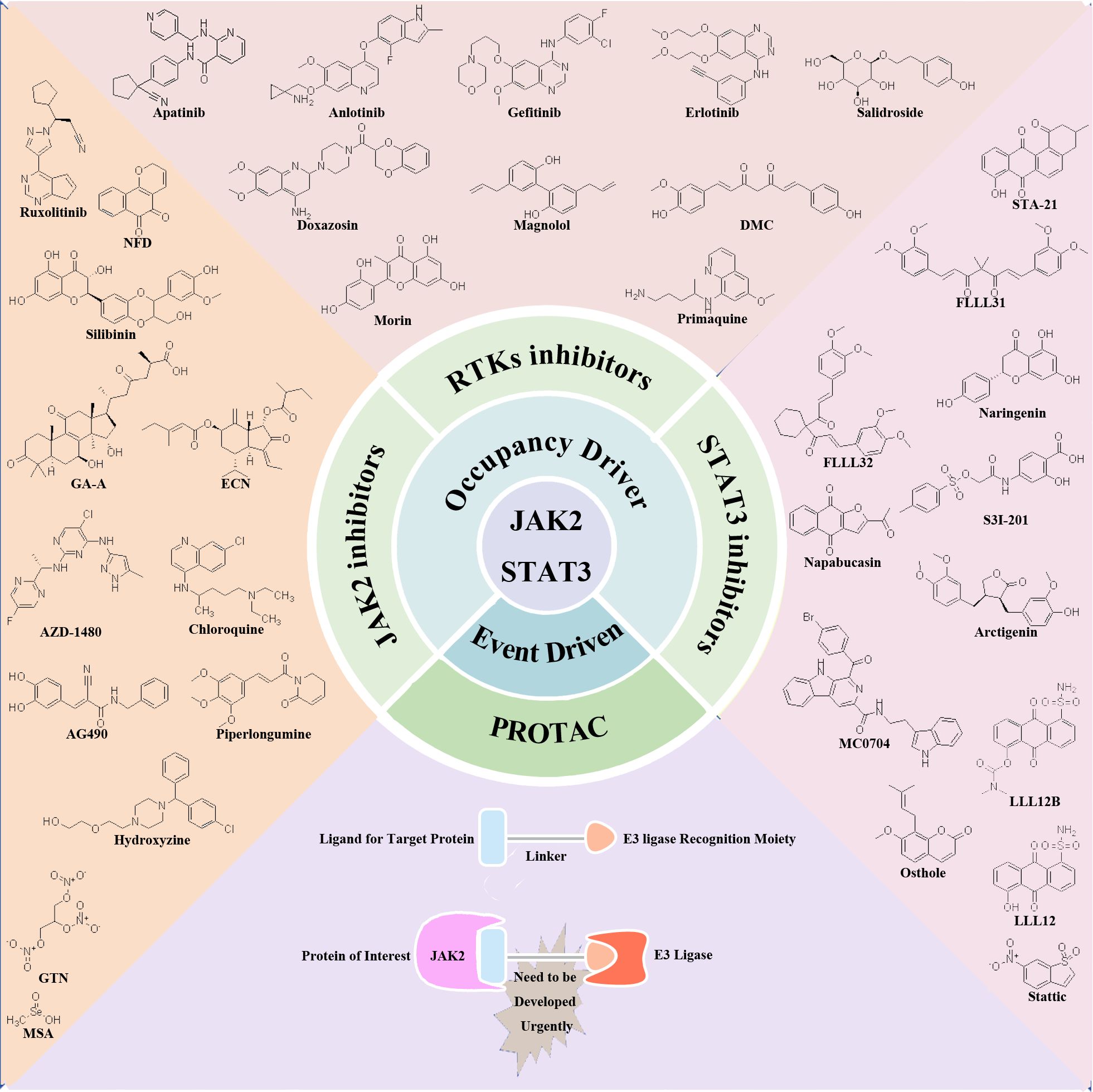
Figure 2 Potential therapeutic targets and inhibitors targeting JAK2/STAT3 signaling pathway for TNBC treatment. Occupation-driven mode: RTKs, JAK2 and STAT3-targeted inhibitors; event driven mode: small molecule compounds targeting JAK2 protein for ubiquitination degradation.
5.1 Occupation driven mode: application of small molecule inhibitors in TNBC
5.1.1 RTKs inhibitors
Currently, the U.S. Food and Drug Administration (FDA) has not approved RTK inhibitors for the treatment of TNBC. Both monoclonal antibodies and small molecule inhibitors are progressing through preclinical and clinical research stages. Preclinical studies have shown that cetuximab can effectively reduce cancer stem cells in TNBC and inhibit tumor growth (100). However, clinical trials reveal a more complex picture. For instance, a study investigating the combination of cetuximab and cisplatin in metastatic TNBC reported benefits in fewer than 20% of patients. Genomic analyses revealed limited efficacy due to cetuximab-induced activation of alternative bypass pathways. Combining cetuximab with inhibitors targeting downstream elements of the EGFR pathway is proposed for enhanced benefits in patients with TNBC (101). Preclinical research has demonstrated that bevacizumab, a VEGFR inhibitor, effectively suppresses TNBC growth in vivo (102). However, the adjunctive use of bevacizumab with chemotherapy did not improve overall survival rates in early-stage patients with TNBC compared to chemotherapy alone. Similarly, tocilizumab, an interleukin-6 receptor (IL-6R) inhibitor, exhibits potential anti-TNBC properties in preclinical studies (103), but its clinical efficacy in TNBC treatment remains unreported and warrants further investigation.
Several RTK small molecule inhibitors have been reported in clinical studies for the treatment of TNBC (Table 1). Apatinib, a highly selective VEGFR inhibitor, has exhibited promising efficacy in a Phase II clinical trial for patients with TNBC combined with chemotherapy. The results highlighted not only its effectiveness but also a manageable safety profile (104). Furthermore, combining Apatinib with a programmed death-ligand 1 (PD-L1) inhibitor in another Phase II trial resulted in favorable outcomes with a controllable safety profile (105). Integration of Apatinib with a PD-L1 inhibitor and Eribulin in a multicenter Phase II trial demonstrated significant therapeutic benefits in treating advanced TNBC, notably extending its efficacy to PD-L1–negative patients (106). Anlotinib, identified as a small molecule inhibitor targeting the VEGFR, displayed promising results in advanced TNBC treatment. Specifically, a Phase Ib clinical trial revealed that Anlotinib, when employed in a chemotherapy-free regimen alongside a PD-L1 inhibitor, effectively treated previously advanced patients with TNBC. This combination not only demonstrated favorable efficacy but also maintained a manageable safety profile (107). Another Phase II clinical trial combining Anlotinib with standard chemotherapy for metastatic TNBC demonstrated therapeutic benefits with manageable safety (108).
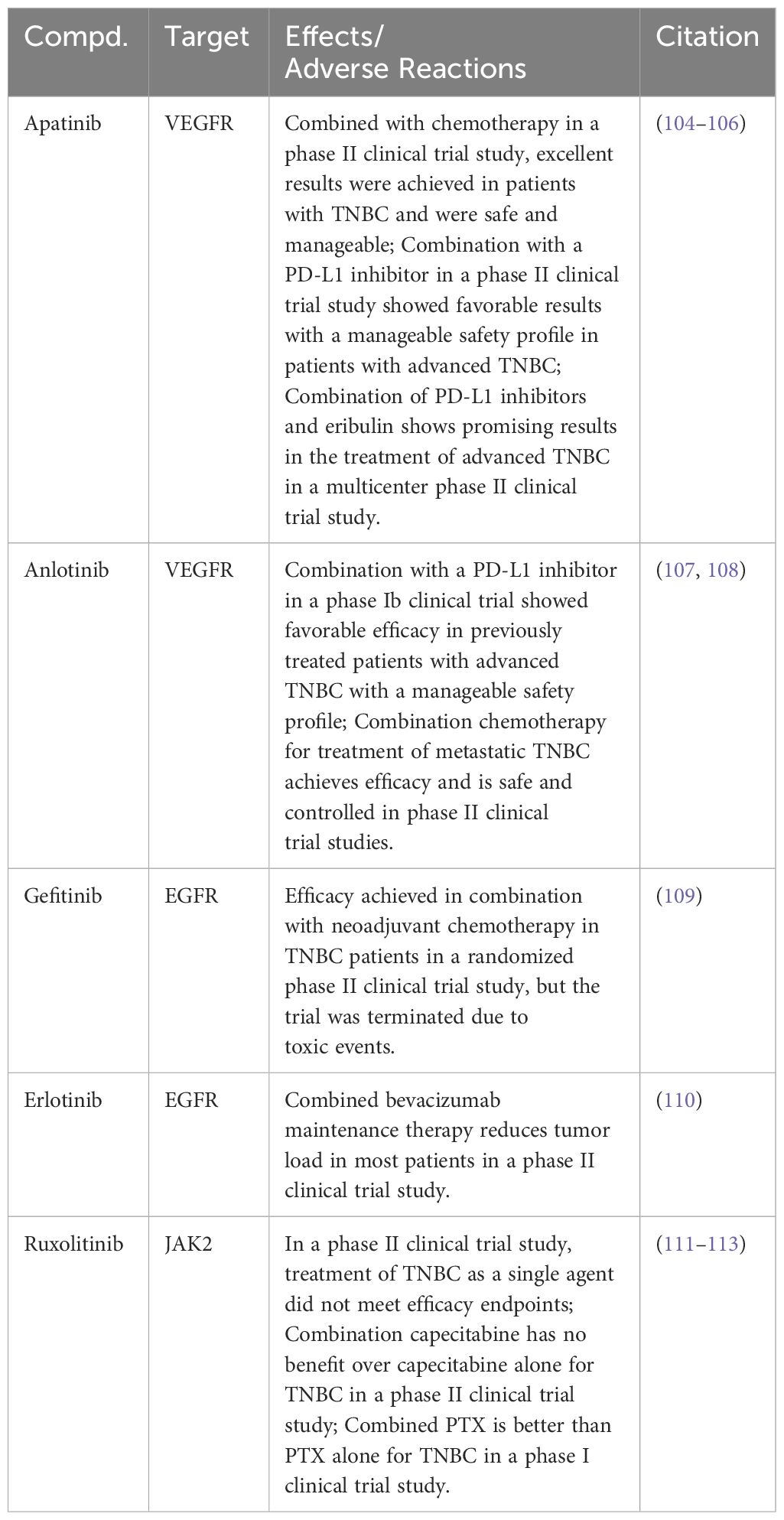
Table 1 Targeting JAK2/STAT3 signaling pathway for TNBC in preclinical studies.
Gefitinib, a small molecule EGFR inhibitor, along with neoadjuvant chemotherapy, showed a higher pathological complete response rate in a Phase II clinical trial for patients with TNBC, especially in the chemotherapy and Gefitinib combination group. However, it is critical to note that patients receiving Gefitinib exhibited a higher incidence of toxic reactions, consequently leading to the discontinuation of the trial for those patients (109). In another Phase II clinical trial, Erlotinib, a small molecule EGFR inhibitor, was evaluated for its efficacy in treating metastatic TNBC. Patients in this trial initially received treatment with albumin-bound PTX combined with bevacizumab, followed by a maintenance regimen comprising both bevacizumab and Erlotinib. Notably, a significant proportion of participants in this trial exhibited partial tumor responses (110).
Research on RTK inhibitors in TNBC is expanding to include natural products such as Salidroside extracted from Rhodiola (Table 2). Preclinical studies have shown that Salidroside inhibits phosphorylation signaling pathways of EGFR/JAK2/STAT3, thereby impacting TNBC cell viability by binding to EGFR. Salidroside’s therapeutic potential is highlighted by its selective efficacy, demonstrating minimal toxicity in normal breast epithelial cells (114). Doxazosin, primarily known as a vasodilator, has a dual-target mechanism, binding to cellular mesenchymal-epithelial transition factor (c-MET) and EGFR. This binding results in the inhibition of JAK2/STAT3 phosphorylation signaling. Research has demonstrated that doxazosin significantly affects TNBC cell proliferation, invasion, and migration, supported by in vitro and in vivo evidence. The efficacy of doxazosin in curbing TNBC lung metastasis was further substantiated through a mouse lung metastasis model (115).
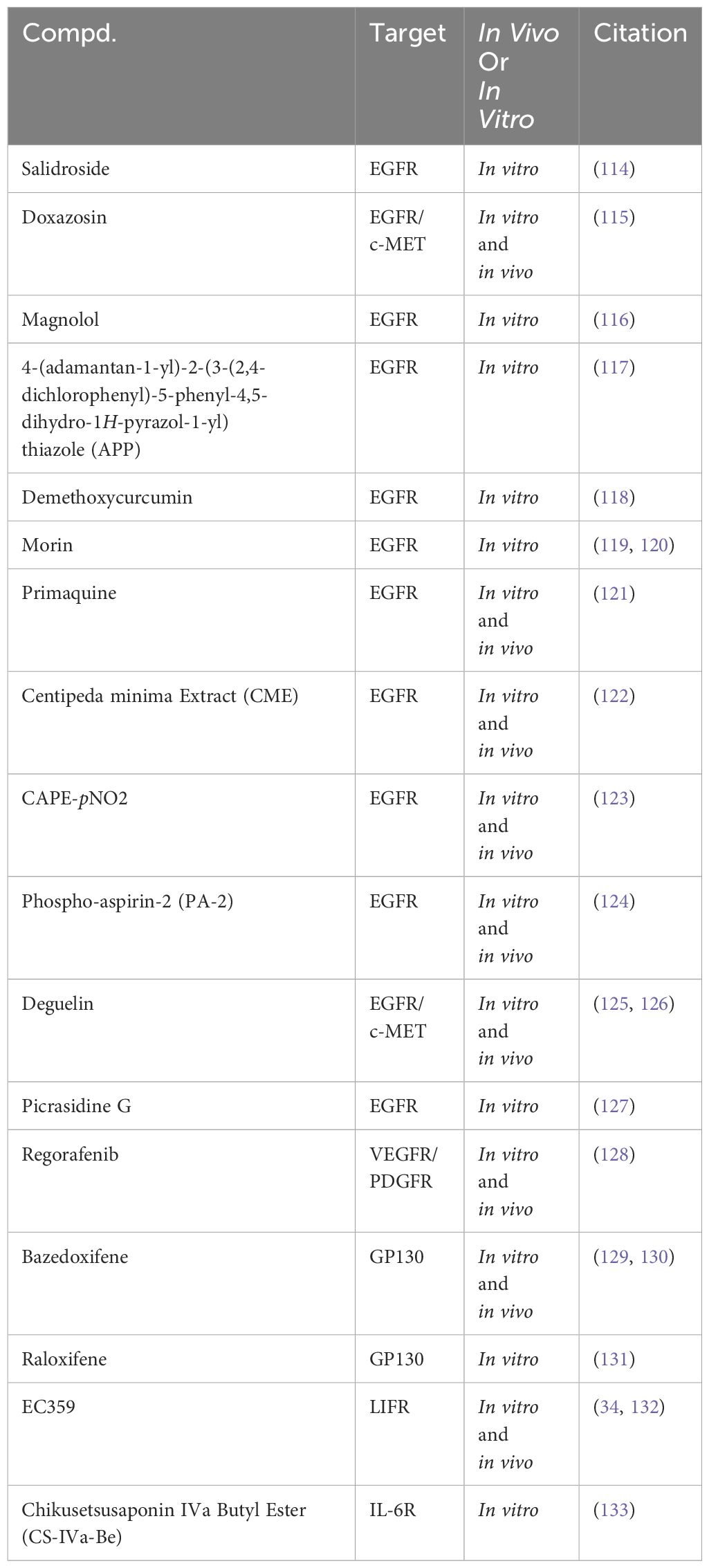
Table 2 Targeting RTKs to modulate the JAK2/STAT3 signaling pathway in preclinical studies for the treatment of TNBC.
Magnolol, a multifunctional lignan compound derived from the traditional Chinese herb Houpo, has demonstrated notable anti-cancer properties against TNBC. In vitro studies reveal that Magnolol effectively reduces the viability of TNBC cells. This inhibitory effect is primarily attributed to the suppression of phosphorylation signaling in the EGFR/JAK2/STAT3 pathway (116). The lead compound APP has also shown promising results in TNBC treatment. In vitro analyses indicate that APP induces apoptosis in TNBC cells. This apoptotic effect is mediated through the inhibition of EGFR/JAK2/STAT3 phosphorylation signaling, coupled with the regulation of apoptotic proteins (117). Demethoxycurcumin (DMC), a principal variant of curcumin predominantly found in the rhizomes of turmeric, has garnered attention in the context of TNBC research. In vitro studies have illuminated its potential in modulating TNBC cell viability. DMC achieves this by inhibiting EGFR protein expression levels. Furthermore, its mechanism involves the inhibition of specific phosphatases, thereby sustaining EGFR activation. This suggests that DMC’s influence on TNBC cells might result from its regulation of multiple signaling pathways (118). Morin, a flavonoid compound derived from plants, has shown potential in the treatment of TNBC. Studies indicate that Morin, particularly when used in conjunction with doxorubicin, promotes apoptosis in TNBC cells. This synergistic effect is attributed to the inhibition of EGFR/STAT3 phosphorylation signaling (119, 120).
Primaquine, an antimalarial drug, has been observed to inhibit TNBC cell viability and migration in vitro. This inhibition is linked to the suppression of EGFR/STAT3 phosphorylation signaling. However, the specific mechanisms by which Primaquine impedes TNBC growth in vivo remain to be explored further (121). Additionally, Centipeda minima Extract (CME), an extract from the Centipeda minima, has demonstrated efficacy in regulating TNBC cell behavior by modulating the phosphorylation signaling of multiple pathways, notably the STAT3 pathway. This modulation occurs through the inhibition of EGFR expression, thereby promoting apoptosis in TNBC cells (122). CAPE-pNO2 has also been identified as a potent inhibitor of proliferation and migration in TNBC by suppressing EGFR phosphorylation and the regulation of STAT3 and AKT phosphorylation signaling. This dual effect has been observed in both in vitro and in vivo studies (123).
Similarly, PA-2, another compound under investigation, has demonstrated its ability to promote apoptosis in TNBC cells. It achieves this through the inhibition of EGFR phosphorylation and by modulating the phosphorylation signaling of the PI3K/AKT and STAT3 pathways (124). Deguelin also contributes to this growing field of TNBC therapeutics. It influences the expression of both EGFR and c-Met, leading to the downregulation of phosphorylation signaling across several pathways, including STAT3, AKT, ERK, and NFκB. This comprehensive action results in a marked impact on the viability of TNBC cells (125, 126). Picrasidine G, a naturally derived dimeric alkaloid, has shown efficacy in inhibiting the vitality of TNBC cells in vitro by suppressing the EGFR/STAT3 phosphorylation signaling pathway (127). Regorafenib, another compound under study, exerts its anti-cancer effects by inhibiting key receptors such as VEGFR and PDGFR. This inhibition impacts the STAT3 phosphorylation signaling (128).
Bazedoxifene presents a different angle in TNBC treatment. By targeting GP130, it influences the STAT3 phosphorylation signaling pathway. This strategic inhibition inhibits TNBC both in vitro and in vivo (129, 130). Raloxifene, a compound known for its influence on the GP130 receptor, has been shown to inhibit the vitality of TNBC cells in vitro. This effect is achieved through the modulation of the STAT3 phosphorylation signaling pathway (131). EC359, another promising agent, binds to the leukemia inhibitory factor receptor (LIFR), inhibiting the LIFR/STAT3 phosphorylation signaling, thereby curbing TNBC proliferation both in vitro and in vivo (34, 132). Similarly, CS-IVa-Be targets cancer cells through the inhibition of IL-6R, impacting the JAK2/STAT3 phosphorylation signaling pathway. This specific action has been observed to inhibit TNBC in vitro (133).
In summary, RTK inhibitors can inhibit the signaling of the JAK2/STAT3 pathway. However, due to the activation of bypass pathways, clinical trial results suggest that combining these inhibitors with chemotherapy may be more beneficial for patients with TNBC.
5.1.2 JAK2 inhibitors
Directly targeting JAK2 emerges as a strategic approach for modulating the JAK2/STAT3 signaling pathway in TNBC treatment. However, to date, JAK2 inhibitors have not received FDA approval for use in TNBC therapy. Ruxolitinib, a JAK1/JAK2 inhibitor, is being evaluated for its clinical efficacy in treating TNBC in several clinical trials. In a Phase II clinical trial, Ruxolitinib monotherapy did not meet its efficacy endpoint for TNBC treatment (111). Further research explored the potential of Ruxolitinib in combination with chemotherapy drugs. While combining Ruxolitinib with Capecitabine did not enhance overall survival, another trial pairing it with PTX showed improved clinical efficacy, outperforming PTX monotherapy (112, 113) (Table 1).
In preclinical TNBC studies, several small molecules exhibit promise as JAK2 inhibitors (Table 3). For instance, Glyceryl Trinitrate (GTN), a vasodilator, inhibits STAT3 activation by blocking JAK2 phosphorylation, suppressing TNBC cell viability (134). Additionally, a range of compounds, including Withaferin A (WA) (135), Naphtho[1,2-b]furan-4,5-dione (NFD) (136), Ganoderic acid A (GA-A) (137), Methylseleninic Acid (MSA) (138), and AZD1480 (139), have been identified to inhibit the phosphorylation and signal transduction of the JAK2/STAT3 pathway, reducing the viability of TNBC cells. Recent research findings highlight the efficacy of JAK2 small molecule inhibitors in TNBC. For instance, the JAK2 inhibitor AG490 has been shown to reduce TNBC cell viability by modulating the phosphorylation and signal transduction of STAT3 and AKT (140, 141). Additionally, 3-Deoxy-2β,16-dihydroxynagilactone E (B6) interacts with the FERM-SH2 domain of JAK2, inhibiting downstream STAT3 phosphorylation and reducing TNBC cell viability (142). Another study highlights the effectiveness of ECN in suppressing TNBC cell viability by targeting the JAK2/STAT3 signaling pathway. ECN has also demonstrated the ability to inhibit TNBC tumor growth in vivo (143).
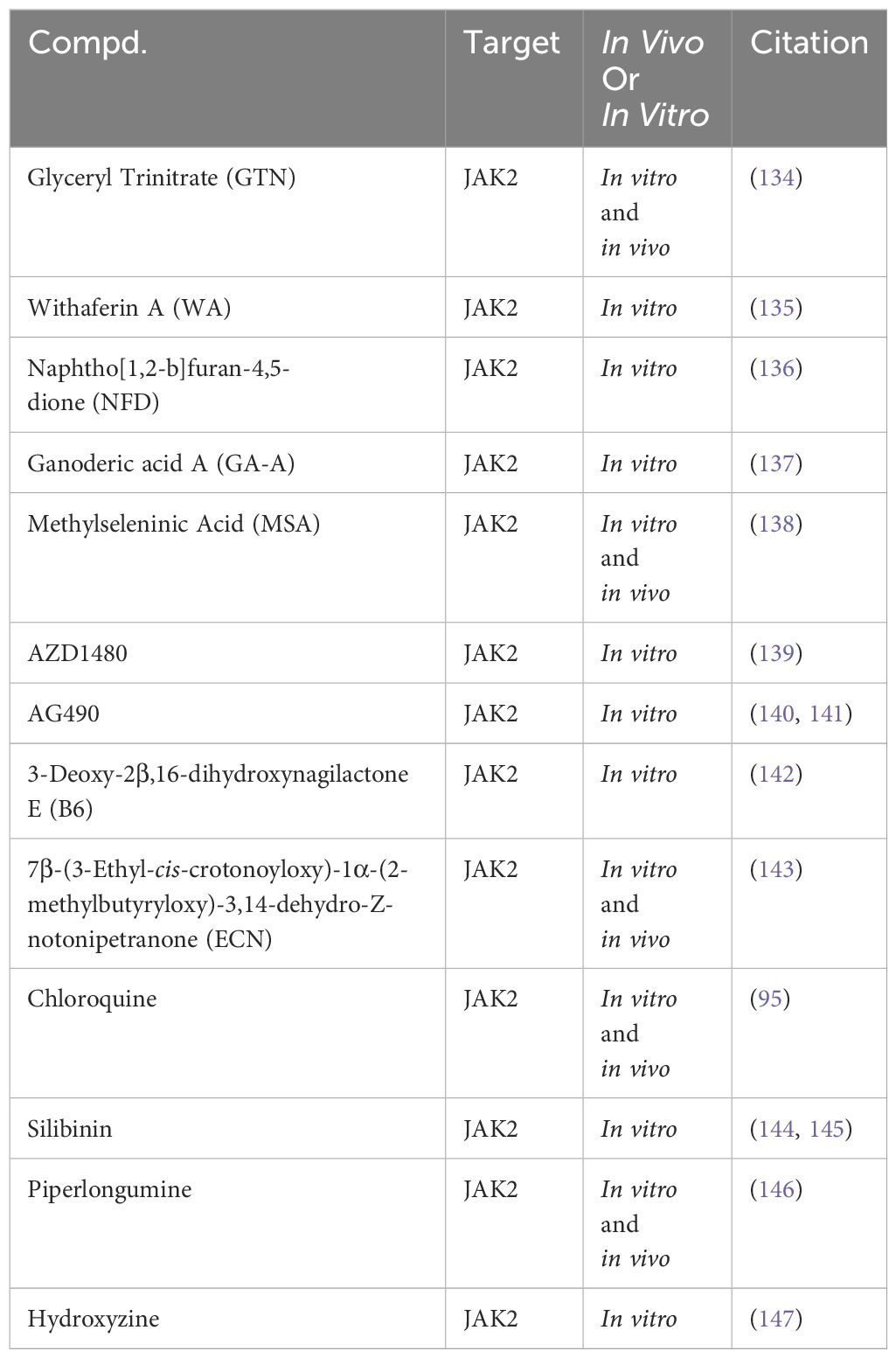
Table 3 Targeting the JAK2 for TNBC in preclinical studies.
Chloroquine enhances PTX therapeutic efficacy in TNBC by inhibiting JAK2/STAT3 pathway phosphorylation, impacting autophagy processes (95). Additionally, Silibinin suppresses TNBC cell invasive and migratory capabilities in vitro by downregulating JAK2/STAT3 pathway phosphorylation (144, 145). Piperlongumine, a bioactive alkaloid known for its antioxidant and anti-tumor properties, has been found to inhibit TNBC cell proliferation and migration by inhibiting JAK2/STAT3 pathway phosphorylation (146). Similarly, Hydroxyzine, primarily recognized as a histamine H1 receptor antagonist, has demonstrated the capability to induce apoptosis in TNBC cells through the inhibition of JAK2/STAT3 phosphorylation (147).
JAK2 inhibitors have shown significant efficacy in inhibiting TNBC in vitro. Clinical trial data in vivo also suggest that a combination of these inhibitors with the chemotherapy drug paclitaxel could be a promising therapeutic approach for TNBC. However, concerns about the safety of JAK2 inhibitors, as evidenced by FDA warnings, underscores the necessity for alternative therapeutic strategies targeting the JAK2/STAT3 pathway.
5.1.3 STAT3 inhibitor
Several STAT3 small molecule inhibitors have been reported in preclinical studies for the treatment of TNBC (Table 4). The therapeutic strategy to inhibit STAT3 involves targeting multiple stages of its functional cycle, including phosphorylation, dimerization, nuclear translocation, and DNA binding activities. This approach leverages the nuclear translocation signal of STAT3.
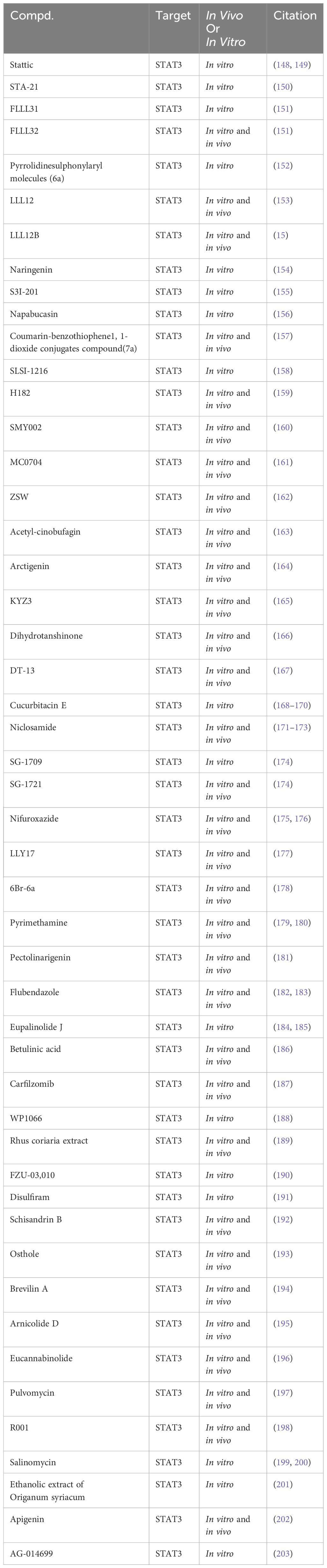
Table 4 Targeting STAT3 for TNBC in preclinical studies.
Stattic, a non-peptidic small molecule, has demonstrated notable anti-TNBC effects by selectively targeting STAT3, inhibiting its activation, dimerization, and nuclear translocation. This inhibition is facilitated through Stattic’s binding to the SH2 functional domain of STAT3 (148, 149). Similarly, STA-21, another small molecule inhibitor, induces apoptosis in TNBC cells by inhibiting DNA binding activity and dimerization of STAT3 (150). FLLL31 and FLLL32, derivatives of curcumin, have been identified as selective inhibitors of STAT3. They achieve this by binding to the SH2 functional domain of STAT3, thereby inhibiting its phosphorylation and DNA binding activities. Notably, these compounds have shown potential in synergistically inhibiting TNBC cell proliferation when combined with doxorubicin. In vivo studies further indicate that FLLL32 can effectively suppress TNBC growth by downregulating STAT3 phosphorylation levels (151). Pyrrolidine sulfonamide derivative 6a selectively inhibits STAT3 activation at phosphorylation and transcription levels, reducing TNBC cell viability in response to IL-6 stimulation (152). LLL12, a non-peptidic, cell-permeable small molecule, selectively targets STAT3 by inhibiting its DNA binding activity and phosphorylation through SH2 domain binding. It induces apoptosis in TNBC cells and suppresses TNBC growth in vivo by downregulating STAT3 phosphorylation levels (153). LLL12B, a prodrug of LLL12, is activated in the tumor microenvironment by tumor-associated plasmin, which cleaves its aminoformate bond to release active LLL12. LLL12B exhibits improved pharmacokinetic properties compared to its parent compound, LLL12. However, additional research is required to fully elucidate the comparative in vivo and in vitro pharmacology of these compounds, particularly their respective abilities to bind to STAT3 (15).
Naringenin, a naturally occurring compound, reduces TNBC cell viability by binding to the SH2 domain of STAT3, suppressing STAT3 phosphorylation. In combination with cyclophosphamide, naringenin has demonstrated enhanced efficacy in inducing apoptosis in TNBC cells (154). S3I-201, a selective STAT3 inhibitor probe, targets the SH2 functional domain of STAT3, inhibiting its DNA binding activity and dimerization. In vitro studies have revealed that S3I-201 significantly diminishes the TNBC cell viability and inhibits tumor growth by reducing STAT3 phosphorylation (155). Napabucasin, a targeted therapeutic agent, selectively inhibits the DNA binding activity and phosphorylation of STAT3 by binding to its SH2 functional domain. In vitro studies have demonstrated Napabucasin’s capability to reduce TNBC cell viability (156). Additionally, a series of compounds, such as 7a (157), SLSI-1216 (158), H182 (159), SMY002 (160), MC0704 (161), ZSW (162), and Acetyl-cinobufagin (163), have been identified to selectively inhibit STAT3 phosphorylation by binding to its SH2 domain and suppressing TNBC cell viability in vitro. Arctigenin, a bioactive lignan isolated from the seeds of Arctium lappa, inhibits STAT3 in TNBC cells by binding to its SH2 domain, thereby disrupting hydrogen bond connections between DNA and STAT3. This disruption prevents STAT3’s binding to genomic DNA, effectively reducing TNBC cell viability (164). Similarly, KYZ3, a derivative of cryptotanshinone, binds to the SH2 domain of STAT3, inhibiting its DNA binding activity and phosphorylation, leading to decreased TNBC cell viability in vitro (165). Research has identified a wide array of small molecules that exhibit the potential to inhibit TNBC cell viability. This includes Dihydrotanshinone (166), DT-13 (167), Cucurbitacin E (168–170), Niclosamide (171–173), SG-1709 (174), SG-1721 (174), Nifuroxazide (175, 176), LLY17 (177), 6Br-6a (178), Pyrimethamine (179, 180), Pectolinarigenin (181), Flubendazole (182, 183), Eupalinolide J (184, 185), Betulinic acid (186), Carfilzomib (187), WP1066 (188), Rhus coriaria extract (189), FZU-03,010 (190), Disulfiram (191), Schisandrin B (192), Osthole (193), Brevilin A (194), Arnicolide D (195), Eucannabinolide (196), Pulvomycin (197), R001 (198), Salinomycin (199, 200), the ethanolic extract of origanum syriacum (201), Apigenin (202), and AG-014699 (203). This inhibition is attributed to their ability to suppress STAT3 phosphorylation. However, direct evidence demonstrating their binding to STAT3 is currently lacking.
While preclinical studies have identified various small molecule compounds as potential STAT3 inhibitors, TTI-101 stands out as the sole compound advancing into Phase I clinical trials. Various groups of researchers are investigating the efficacy and safety of TTI-101 in patients with advanced breast cancer and those with inoperable solid tumors.
5.1.4 Adverse effects of JAK2/STAT3 pathway inhibition
Because the biological processes of normal cells also depend on the JAK2/STAT3 pathway, the long-term use of JAK2/STAT3 pathway inhibitors has certain toxic side effects. JAK2 inhibitors inevitably inhibit the normal hematopoietic function of the body, which can cause anemia, thrombocytopenia, and other adverse effects (including dizziness, headache, abdominal pain, diarrhea, and the secondary tumor).
Studies have shown that anemia and thrombocytopenia are the most common hematologic adverse effects when JAK2 inhibitors are used to treat myelofibrosis (204–210); in another study of Ruxolitinib as a drug treatment for true erythrocytosis, headache and diarrhea were the most common non-hematologic adverse effects (211); the immunosuppressive effects of JAK2 inhibitors are important in inducing infections, and in the JUMP study, the most common infection was pneumonia, followed by urinary tract infections and nasopharyngitis (205), and another study showed that 30 of 31 patients treated with Ruxolitinib developed infections, including several opportunistic infections (212); although JAK inhibitors can be used to treat hematologic cancers and inflammatory diseases, during treatment with these drugs, studies have found that some patients suffer from lymphomas and other malignancies, with a statistically significant 16-fold increase in the risk of B-cell malignancies in patients with myeloproliferative neoplasms treated with JAK1/2 inhibitors, and skin cancers being the most common secondary tumor (213); in addition to these symptoms, Ruxolitinib can also cause other adverse reactions such as abdominal pain, drowsiness, acute renal failure (211), and even some studies have reported that patients have died from cardiac arrest (204).
Since the JAK family mediates signaling of multiple cytokines and different receptors are associated with different JAKs, and comprehensive inhibition of the JAK family can result in a variety of side effects, the design and development of new targeted JAK2 inhibitors could provide a solution to these adverse effects.
5.2 Event-driven mode: application of PROTAC molecules based on ubiquitin-mediated protein degradation in TNBC
The regulation of the JAK2/STAT3 signaling pathway can be strategically achieved through the targeted degradation of the JAK2 protein employing PROTACs. These molecular constructs consist of a linker connecting two ligands, with one ligand binding to the target JAK2 protein and the other engaging with the ubiquitin E3 ligase. This dual binding facilitates the formation of a ternary complex, bringing the JAK2 protein and E3 ligase into close proximity. Subsequently, the target JAK2 protein undergoes ubiquitination, marking it for recognition by the proteasome system. This leads to the proteasomal degradation of JAK2 into peptide fragments, effectively nullifying its protein activity (214). Recent studies underscore the promising role of PROTAC molecules in TNBC treatment. MZ1, a small molecule PROTAC, targets BRD4 protein for degradation. Compared to JQ1, a conventional inhibitor targeting the protein domain, MZ1 exhibits superior anti-TNBC activity both in vitro and in vivo, which is attributed to its specific action in targeting BRD4 protein degradation (215). Another notable PROTAC molecule, NN3, is designed to target PARP1 protein degradation. Experimental findings indicate that NN3 demonstrates effective anti-TNBC activity in vitro and in vivo. Remarkably, NN3 retains its efficiency in degrading PARP1 protein even in the presence of point mutations, further underscoring its potential as an anti-tumor agent (216). Emerging research sheds light on the efficacy of PROTAC molecule 6n, designed to target the degradation of the AXL protein. Demonstrating a significant advantage over traditional AXL kinase inhibitors, 6n has shown superior anti-TNBC activity in vitro and in vivo (217). Similarly, YX-02-030, a PROTAC molecule targeting the MDM2 protein degradation, exhibits enhanced anti-tumor activity compared to specific MDM2 inhibitors. Notably, YX-02-030 achieves this therapeutic efficacy without causing harm to normal cells (218). TEP, a PROTAC molecule engineered to target c-Myc protein degradation, effectively inhibits the proliferation of TNBC cells by facilitating the specific degradation of the endogenous c-Myc/Max complex. Additionally, TEP enhances the sensitivity of TNBC cells to palbociclib, a cyclin-dependent kinase inhibitor (219). Another PROTAC molecule, CT-4, designed to target HDAC8 protein degradation, promotes apoptosis in TNBC cells through the targeted degradation of HDAC8 protein (220). The small molecule compound A4, a PROTAC developed based on DCAF16, specifically targets CDK4/6 protein degradation. Research demonstrates that A4 exhibits potent inhibitory activity against CDK4/6, offering a favorable safety profile in normal cells, which is considered superior to the established CDK4/6 inhibitor, palbociclib (221). The small molecule compounds 7f and PP-C8, which also function as PROTAC molecules, also target CDK12/13 degradation. Studies have indicated that these compounds effectively reduce TNBC cell viability by inhibiting the expression of CDK12/13 (222, 223). PROTAC molecules MS8815 and U3i, designed to target EZH2 protein degradation, induce ubiquitination and subsequent proteasome-dependent degradation of EZH2, effectively inhibiting TNBC cell growth (224, 225). Similarly, androgen receptor (AR)-PROTAC has shown efficacy in targeting AR-positive TNBC cells by mediating the ubiquitination and degradation of the AR, thereby inhibiting cell growth (226). Furthermore, C8, a PROTAC molecule developed based on the PARP1/2 inhibitor Olaparib, exhibits promising therapeutic potential against TNBC. It promotes PARP2 protein degradation, demonstrating effectiveness in vitro and in vivo (227).
Currently, PROTAC small molecules targeting the degradation of JAK2 protein have been reported. However, research indicates that the E3 ligase CUL5 mediates JAK2 protein degradation (228). Developing PROTAC small molecules that facilitate the binding of JAK2 protein to the E3 ligase CUL5 could be a feasible strategy for treating TNBC.
6 Conclusion
The JAK2/STAT3 signaling pathway, activated by cytokines, is central in governing fundamental cellular processes, including growth, differentiation, apoptosis, and immune responses. In TNBC, excessive activation of this pathway contributes to immune evasion by TNBC cells. This aberrant activation promotes tumor growth, facilitates metastasis, and develops drug resistance in TNBC. Therefore, the strategic therapeutic targeting of the JAK2/STAT3 signaling pathway emerges as a promising strategy for the effective treatment of TNBC. The JAK2/STAT3 signaling pathway presents multiple targets for therapeutic intervention in TNBC. Inhibition strategies focusing on RTKs, JAK2, and STAT3 effectively suppresses TNBC cell growth. Despite these promising results, clinical trials of inhibitors that bind to the active sites of these proteins have encountered challenges. These limitations can be attributed to several factors, including the activation of compensatory bypass pathways, overexpression of target proteins, emergence of point mutations within these targets, and the heightened expression of competitive ligands. Unlike conventional inhibitors, PROTAC molecules do not rely on sustained binding to the target protein to exert their inhibitory effect. This unique characteristic enables them to remain effective even in the presence of mutations in the protein’s active binding site. A key advantage of PROTACs lies in their catalytic mechanism; following the facilitation of ubiquitination and degradation of the target protein, PROTAC molecules can be recycled. This recycling ability potentially allows for lower drug dosages, enhancing both the safety profile and therapeutic potential of these molecules. Consequently, employing PROTACs to target the JAK2/STAT3 pathway emerges as an exceptionally promising strategy for TNBC treatment. By developing PROTACs that specifically target JAK2 protein degradation, not only is TNBC growth inhibited through the downregulation of the JAK2/STAT3 pathway, but the typical toxic side effects associated with traditional JAK2 inhibitors are also likely to be mitigated.
Author’s note
During the preparation of this work authors did not used any AI tool/service, and takes full responsibility for the content of the publication.
Author contributions
LL: Conceptualization, Resources, Writing – original draft. XF: Conceptualization, Resources, Writing – original draft. LC: Formal Analysis, Project administration, Writing – original draft. LY: Conceptualization, Supervision, Writing – review & editing. XL: Conceptualization, Supervision, Writing – review & editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
TNBC, Triple-negative breast cancer; JAK2, Janus Kinase 2; STAT3, Signal transducer and activator of transcription 3; ER, Estrogen receptor; PR, Progesterone receptor; HER-2, Human epidermal growth factor receptor 2; PTX, paclitaxel; RTKs, Receptor tyrosine kinases; EGFRs, Epidermal growth factor receptors; VEGFRs, Vascular endothelial growth factor receptors; IGFRs, Insulin-like growth factor receptors; PDGFRs, Platelet-derived growth factor receptors; FGFRs, Fibroblast growth factor receptors; LIFR, Leukemia inhibitory factor receptor; IL-6R, Interleukin-6 cell factor receptor; IL-13R, Interleukin-13 cell factor receptor; GP130, Glycoprotein 130 receptor; EGF, Epidermal growth factor; MEK, Mitogen-activated protein kinase; ERK, Extracellular signal-regulated kinase; PI3K, Phosphoinositide 3-kinase; AKT, Protein kinase B; VEGF, Vascular endothelial growth factor; PDGF, Platelet-derived growth factor; FGF, Fibroblast growth factor; TBx3, T-box transcription factor 3; TYK2, Tyrosine kinase 2; PROTAC, Proteolysis targeting chimera; PD-L1, Programmed death-ligand 1; c-MET, Cellular mesenchymal-epithelial transition; PARP, Poly ADP-ribose polymerase; AR, Androgen receptor; EZH2, Enhancer of zeste homolog 2; CDK, Cyclin-dependent kinases.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
2. Abdelwahab YA. Male breast cancer: epidemiology and risk factors. Semin Oncol. (2017) 44:267–72. doi: 10.1053/j.seminoncol.2017.11.002
3. Geyer FC, Pareja F, Weigelt B, Rakha E, Ellis IO, Schnitt SJ, et al. The spectrum of triple-negative breast disease: high- and low-grade lesions. Am J Pathol. (2017) 187:2139–51. doi: 10.1016/j.ajpath.2017.03.016
4. Ellington TD, Miller JW, Henley SJ, Wilson RJ, Wu M, Richardson LC. Trends in breast cancer incidence, by race, ethnicity, and age among women aged >/=20 years - United States, 1999-2018. Mmwr Morb Mortal Wkly Rep. (2022) 71:43–7. doi: 10.15585/mmwr.mm7102a2
5. Burguin A, Diorio C, Durocher F. Breast cancer treatments: updates and new challenges. J Pers Med. (2021) 11:808. doi: 10.3390/jpm11080808
6. Zarrin AA, Bao K, Lupardus P, Vucic D. Kinase inhibition in autoimmunity and inflammation. Nat Rev Drug Discovery. (2021) 20:39–63. doi: 10.1038/s41573-020-0082-8
7. Banerjee S, Biehl A, Gadina M, Hasni S, Schwartz DM. JAK-STAT signaling as a target for inflammatory and autoimmune diseases: current and future prospects. Drugs. (2017) 77:521–46. doi: 10.1007/s40265-017-0701-9
8. Jiang YZ, Ma D, Suo C, Shi J, Xue M, Hu X, et al. Genomic and transcriptomic landscape of triple-negative breast cancers: subtypes and treatment strategies. Cancer Cell. (2019) 35:428–40. doi: 10.1016/j.ccell.2019.02.001
9. Zheng Q, Han L, Dong Y, Tian J, Huang W, Liu Z, et al. JAK2/STAT3 targeted therapy suppresses tumor invasion via disruption of the EGFRvIII/JAK2/STAT3 axis and associated focal adhesion in EGFRvIII-expressing glioblastoma. Neuro Oncol. (2014) 16:1229–43. doi: 10.1093/neuonc/nou046
10. Xu J, Zhang L, Li N, Dai J, Zhang R, Yao F, et al. Etomidate elicits anti-tumor capacity by disrupting the JAK2/STAT3 signaling pathway in hepatocellular carcinoma. Cancer Lett. (2023) 552:215970. doi: 10.1016/j.canlet.2022.215970
11. Liang Q, Gong M, Zou JH, Luo MY, Jiang LL, Wang C, et al. A phosphoglycerate mutase 1 allosteric inhibitor overcomes drug resistance to EGFR-targeted therapy via disrupting IL-6/JAK2/STAT3 signaling pathway in lung adenocarcinoma. Drug Resist Update. (2023) 68:100957. doi: 10.1016/j.drup.2023.100957
12. Zhang L, Yang Z, Ma A, Qu Y, Xia S, Xu D, et al. Growth arrest and DNA damage 45G down-regulation contributes to Janus kinase/signal transducer and activator of transcription 3 activation and cellular senescence evasion in hepatocellular carcinoma. Hepatology. (2014) 59:178–89. doi: 10.1002/hep.26628
13. Chang R, Song L, Xu Y, Wu Y, Dai C, Wang X, et al. Loss of Wwox drives metastasis in triple-negative breast cancer by JAK2/STAT3 axis. Nat Commun. (2018) 9:3486. doi: 10.1038/s41467-018-05852-8
14. Chen M, Pockaj B, Andreozzi M, Barrett MT, Krishna S, Eaton S, et al. JAK2 and PD-L1 amplification enhance the dynamic expression of PD-L1 in triple-negative breast cancer. Clin Breast Cancer. (2018) 18:e1205–15. doi: 10.1016/j.clbc.2018.05.006
15. Pan L, Chen X, Rassool FV, Li C, Lin J. LLL12B, a novel small-molecule STAT3 inhibitor, induces apoptosis and suppresses cell migration and tumor growth in triple-negative breast cancer cells. Biomedicines. (2022) 10:2003. doi: 10.3390/biomedicines10082003
16. Wang Y, Cheng Z, Xu J, Lai M, Liu L, Zuo M, et al. Fat mass and obesity-associated protein (FTO) mediates signal transducer and activator of transcription 3 (STAT3)-drived resistance of breast cancer to doxorubicin. Bioengineered. (2021) 12:1874–89. doi: 10.1080/21655979.2021.1924544
17. Jiao K, Zhen J, Wu M, Teng M, Yang K, Zhou Q, et al. 27-Hydroxycholesterol-induced EndMT acts via STAT3 signaling to promote breast cancer cell migration by altering the tumor microenvironment. Cancer Biol Med. (2020) 17:88–100. doi: 10.20892/j.issn.2095-3941.2019.0262
18. Lim JH. Inhibition of the interleukin-11-STAT3 axis attenuates hypoxia-induced migration and invasion in MDA-MB-231 breast cancer cells. Korean J Physiol Pharmacol. (2014) 18:391–96. doi: 10.4196/kjpp.2014.18.5.391
19. Falamarzian A, Aliabadi HM, Molavi O, Seubert JM, Lai R, Uludag H, et al. Effective down-regulation of signal transducer and activator of transcription 3 (STAT3) by polyplexes of siRNA and lipid-substituted polyethyleneimine for sensitization of breast tumor cells to conventional chemotherapy. J BioMed Mater Res A. (2014) 102:3216–28. doi: 10.1002/jbm.a.34992
20. Pawlus MR, Wang L, Hu CJ. STAT3 and HIF1alpha cooperatively activate HIF1 target genes in MDA-MB-231 and RCC4 cells. Oncogene. (2014) 33:1670–79. doi: 10.1038/onc.2013.115
21. Deng XS, Wang S, Deng A, Liu B, Edgerton SM, Lind SE, et al. Metformin targets Stat3 to inhibit cell growth and induce apoptosis in triple-negative breast cancers. Cell Cycle. (2012) 11:367–76. doi: 10.4161/cc.11.2.18813
22. Tripolt S, Neubauer HA, Knab VM, Elmer DP, Aberger F, Moriggl R, et al. Opioids drive breast cancer metastasis through the delta-opioid receptor and oncogenic STAT3. Neoplasia. (2021) 23:270–79. doi: 10.1016/j.neo.2020.12.011
23. Zammarchi F, de StanChina E, Bournazou E, Supakorndej T, Martires K, Riedel E, et al. Antitumorigenic potential of STAT3 alternative splicing modulation. Proc Natl Acad Sci U.S.A. (2011) 108:17779–84. doi: 10.1073/pnas.1108482108
24. Ling X, Arlinghaus RB. Knockdown of STAT3 expression by RNA interference inhibits the induction of breast tumors in immunocompetent mice. Cancer Res. (2005) 65:2532–36. doi: 10.1158/0008-5472.CAN-04-2425
25. Wang S, Liang K, Hu Q, Li P, Song J, Yang Y, et al. JAK2-binding long noncoding RNA promotes breast cancer brain metastasis. J Clin Invest. (2017) 127:4498–515. doi: 10.1172/JCI91553
26. Han J, Yun J, Quan M, Kang W, Jung JG, Heo W, et al. JAK2 regulates paclitaxel resistance in triple negative breast cancers. J Mol Med (Berl). (2021) 99:1783–95. doi: 10.1007/s00109-021-02138-3
27. Hubbard SR, Till JH. Protein tyrosine kinase structure and function. Annu Rev Biochem. (2000) 69:373–98. doi: 10.1146/annurev.biochem.69.1.373
28. Zhao D, Pan C, Sun J, Gilbert C, Drews-Elger K, Azzam DJ, et al. VEGF drives cancer-initiating stem cells through VEGFR-2/Stat3 signaling to upregulate Myc and Sox2. Oncogene. (2015) 34:3107–19. doi: 10.1038/onc.2014.257
29. Wintheiser GA, Silberstein P. Physiology, tyrosine kinase receptors. In StatPearls. StatPearls Publishing (2024).
30. Zhang M, Zhang X, Zhao S, Wang Y, Di W, Zhao G, et al. Prognostic value of survivin and EGFR protein expression in triple-negative breast cancer (TNBC) patients. Target Oncol. (2014) 9:349–57. doi: 10.1007/s11523-013-0300-y
31. Bustamante-Marin X, Devlin KL, McDonell SB, Dave O, Merlino JL, Grindstaff EJ, et al. Regulation of IGF1R by microRNA-15b contributes to the anticancer effects of calorie restriction in a murine C3-TAg model of triple-negative breast cancer. Cancers (Basel). (2023) 15:4320. doi: 10.3390/cancers15174320
32. D’Ippolito E, Plantamura I, Bongiovanni L, Casalini P, Baroni S, Piovan C, et al. miR-9 and miR-200 regulate PDGFRbeta-mediated endothelial differentiation of tumor cells in triple-negative breast cancer. Cancer Res. (2016) 76:5562–72. doi: 10.1158/0008-5472.CAN-16-0140
33. Wu Y, Yi Z, Li J, Wei Y, Feng R, Liu J, et al. FGFR blockade boosts T cell infiltration into triple-negative breast cancer by regulating cancer-associated fibroblasts. Theranostics. (2022) 12:4564–80. doi: 10.7150/thno.68972
34. Li M, Viswanadhapalli S, Santhamma B, Pratap UP, Luo Y, Liu J, et al. LIFR inhibition enhances the therapeutic efficacy of HDAC inhibitors in triple negative breast cancer. Commun Biol. (2021) 4:1235. doi: 10.1038/s42003-021-02741-7
35. Weng YS, Tseng HY, Chen YA, Shen PC, Al HA, Chen LM, et al. MCT-1/miR-34a/IL-6/IL-6R signaling axis promotes EMT progression, cancer stemness and M2 macrophage polarization in triple-negative breast cancer. Mol Cancer. (2019) 18:42. doi: 10.1186/s12943-019-0988-0
36. Marquez-Ortiz RA, Contreras-Zarate MJ, Tesic V, Alvarez-Eraso K, Kwak G, Littrell Z, et al. IL13Ralpha2 promotes proliferation and outgrowth of breast cancer brain metastases. Clin Cancer Res. (2021) 27:6209–21. doi: 10.1158/1078-0432.CCR-21-0361
37. La Manna S, Lee E, Ouzounova M, Di Natale C, Novellino E, Merlino A, et al. Mimetics of suppressor of cytokine signaling 3: Novel potential therapeutics in triple breast cancer. Int J Cancer. (2018) 143:2177–86. doi: 10.1002/ijc.31594
38. Du Z, Lovly CM. Mechanisms of receptor tyrosine kinase activation in cancer. Mol Cancer. (2018) 17:58. doi: 10.1186/s12943-018-0782-4
39. Tebbutt N, Pedersen MW, Johns TG. Targeting the ERBB family in cancer: couples therapy. Nat Rev Cancer. (2013) 13:663–73. doi: 10.1038/nrc3559
40. Wheeler DL, Dunn EF, Harari PM. Understanding resistance to EGFR inhibitors-impact on future treatment strategies. Nat Rev Clin Oncol. (2010) 7:493–507. doi: 10.1038/nrclinonc.2010.97
41. Wise R, Zolkiewska A. Metalloprotease-dependent activation of EGFR modulates CD44(+)/CD24(-) populations in triple negative breast cancer cells through the MEK/ERK pathway. Breast Cancer Res Treat. (2017) 166:421–33. doi: 10.1007/s10549-017-4440-0
42. Bi J, Wu Z, Zhang X, Zeng T, Dai W, Qiu N, et al. TMEM25 inhibits monomeric EGFR-mediated STAT3 activation in basal state to suppress triple-negative breast cancer progression. Nat Commun. (2023) 14:2342. doi: 10.1038/s41467-023-38115-2
43. Tao JJ, Castel P, Radosevic-Robin N, Elkabets M, Auricchio N, Aceto N, et al. Antagonism of EGFR and HER3 enhances the response to inhibitors of the PI3K-Akt pathway in triple-negative breast cancer. Sci Signal. (2014) 7:ra29. doi: 10.1126/scisignal.2005125
44. Harrison PT, Vyse S, Huang PH. Rare epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer. Semin Cancer Biol. (2020) 61:167–79. doi: 10.1016/j.semcancer.2019.09.015
45. Jin H, Shi Y, Lv Y, Yuan S, Ramirez C, Lieftink C, et al. EGFR activation limits the response of liver cancer to lenvatinib. Nature. (2021) 595:730–34. doi: 10.1038/s41586-021-03741-7
46. Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature. (2012) 483:100–03. doi: 10.1038/nature10868
47. Nieto Y, Nawaz F, Jones RB, Shpall EJ, Nawaz S. Prognostic significance of overexpression and phosphorylation of epidermal growth factor receptor (EGFR) and the presence of truncated EGFRvIII in locoregionally advanced breast cancer. J Clin Oncol. (2007) 25:4405–13. doi: 10.1200/JCO.2006.09.8822
48. Coley HM, Shotton CF, Ajose-Adeogun A, Modjtahedi H, Thomas H. Receptor tyrosine kinase (RTK) inhibition is effective in chemosensitising EGFR-expressing drug resistant human ovarian cancer cell lines when used in combination with cytotoxic agents. Biochem Pharmacol. (2006) 72:941–48. doi: 10.1016/j.bcp.2006.07.022
49. Nielsen TO, Hsu FD, Jensen K, Cheang M, Karaca G, Hu Z, et al. Immunohistochemical and clinical characterization of the basal-like subtype of invasive breast carcinoma. Clin Cancer Res. (2004) 10:5367–74. doi: 10.1158/1078-0432.CCR-04-0220
50. Rakha EA, El-Sayed ME, Green AR, Lee AH, Robertson JF, Ellis IO. Prognostic markers in triple-negative breast cancer. Cancer. (2007) 109:25–32. doi: 10.1002/cncr.22381
51. Liu H, Paddock MN, Wang H, Murphy CJ, Geck RC, Navarro AJ, et al. The INPP4B tumor suppressor modulates EGFR trafficking and promotes triple-negative breast cancer. Cancer Discovery. (2020) 10:1226–39. doi: 10.1158/2159-8290.CD-19-1262
52. Liu X, Adorno-Cruz V, Chang YF, Jia Y, Kawaguchi M, Dashzeveg NK, et al. EGFR inhibition blocks cancer stem cell clustering and lung metastasis of triple negative breast cancer. Theranostics. (2021) 11:6632–43. doi: 10.7150/thno.57706
53. Vaupel P. Hypoxia and aggressive tumor phenotype: implications for therapy and prognosis. Oncologist. (2008) 13 Suppl 3:21–6. doi: 10.1634/theoncologist.13-S3-21
54. Shibuya M. Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: A crucial target for anti- and pro-angiogenic therapies. Genes Cancer. (2011) 2:1097–105. doi: 10.1177/1947601911423031
55. LeCouter J, Moritz DR, Li B, Phillips GL, Liang XH, Gerber HP, et al. Angiogenesis-independent endothelial protection of liver: role of VEGFR-1. Science. (2003) 299:890–93. doi: 10.1126/science.1079562
56. Takahashi O, Komaki R, Smith PD, Jurgensmeier JM, Ryan A, Bekele BN, et al. Combined MEK and VEGFR inhibition in orthotopic human lung cancer models results in enhanced inhibition of tumor angiogenesis, growth, and metastasis. Clin Cancer Res. (2012) 18:1641–54. doi: 10.1158/1078-0432.CCR-11-2324
57. Jayasinghe C, Simiantonaki N, Habedank S, Kirkpatrick CJ. The relevance of cell type- and tumor zone-specific VEGFR-2 activation in locally advanced colon cancer. J Exp Clin Cancer Res. (2015) 34:42. doi: 10.1186/s13046-015-0162-5
58. Sarkar S, Rajput S, Tripathi AK, Mandal M. Targeted therapy against EGFR and VEGFR using ZD6474 enhances the therapeutic potential of UV-B phototherapy in breast cancer cells. Mol Cancer. (2013) 12:122. doi: 10.1186/1476-4598-12-122
59. Bai J, Wu J, Tang R, Sun C, Ji J, Yin Z, et al. Emodin, a natural anthraquinone, suppresses liver cancer in vitro and in vivo by regulating VEGFR(2) and miR-34a. Invest New Drugs. (2020) 38:229–45. doi: 10.1007/s10637-019-00777-5
60. Spannuth WA, Nick AM, Jennings NB, Armaiz-Pena GN, Mangala LS, Danes CG, et al. Functional significance of VEGFR-2 on ovarian cancer cells. Int J Cancer. (2009) 124:1045–53. doi: 10.1002/ijc.24028
61. Bahhnassy A, Mohanad M, Shaarawy S, Ismail MF, El-Bastawisy A, Ashmawy AM, et al. Transforming growth factor-beta, insulin-like growth factor I/insulin-like growth factor I receptor and vascular endothelial growth factor-A: prognostic and predictive markers in triple-negative and non-triple-negative breast cancer. Mol Med Rep. (2015) 12:851–64. doi: 10.3892/mmr.2015.3560
62. Dong F, Ruan S, Wang J, Xia Y, Le K, Xiao X, et al. M2 macrophage-induced lncRNA PCAT6 facilitates tumorigenesis and angiogenesis of triple-negative breast cancer through modulation of VEGFR2. Cell Death Dis. (2020) 11:728. doi: 10.1038/s41419-020-02926-8
63. Ryden L, Jirstrom K, Haglund M, Stal O, Ferno M. Epidermal growth factor receptor and vascular endothelial growth factor receptor 2 are specific biomarkers in triple-negative breast cancer. Results from a controlled randomized trial with long-term follow-up. Breast Cancer Res Treat. (2010) 120:491–98. doi: 10.1007/s10549-010-0758-6
64. Roberti MP, Arriaga JM, Bianchini M, Quinta HR, Bravo AI, Levy EM, et al. Protein expression changes during human triple negative breast cancer cell line progression to lymph node metastasis in a xenografted model in nude mice. Cancer Biol Ther. (2012) 13:1123–40. doi: 10.4161/cbt.21187
65. Eichhorn ME, Strieth S, Dellian M. Anti-vascular tumor therapy: recent advances, pitfalls and clinical perspectives. Drug Resist Update. (2004) 7:125–38. doi: 10.1016/j.drup.2004.03.001
66. Noiseux N, Boucher CH, Cartier R, Sirois MG. Bolus endovascular PDGFR-beta antisense treatment suppressed intimal hyperplasia in a rat carotid injury model. Circulation. (2000) 102:1330–36. doi: 10.1161/01.cir.102.11.1330
67. Cristofanilli M, Morandi P, Krishnamurthy S, Reuben JM, Lee BN, Francis D, et al. Imatinib mesylate (Gleevec) in advanced breast cancer-expressing C-Kit or PDGFR-beta: clinical activity and biological correlations. Ann Oncol. (2008) 19:1713–19. doi: 10.1093/annonc/mdn352
68. Abramsson A, Lindblom P, Betsholtz C. Endothelial and nonendothelial sources of PDGF-B regulate pericyte recruitment and influence vascular pattern formation in tumors. J Clin Invest. (2003) 112:1142–51. doi: 10.1172/JCI18549
69. Riess JW, Neal JW. Targeting FGFR, ephrins, Mer, MET, and PDGFR-alpha in non-small cell lung cancer. J Thorac Oncol. (2011) 6:S1797–98. doi: 10.1097/01.JTO.0000407562.07029.52
70. Kuwai T, Nakamura T, Sasaki T, Kitadai Y, Kim JS, Langley RR, et al. Targeting the EGFR, VEGFR, and PDGFR on colon cancer cells and stromal cells is required for therapy. Clin Exp Metastasis. (2008) 25:477–89. doi: 10.1007/s10585-008-9153-7
71. Primac I, Maquoi E, Blacher S, Heljasvaara R, Van Deun J, Smeland HY, et al. Stromal integrin alpha11 regulates PDGFR-beta signaling and promotes breast cancer progression. J Clin Invest. (2019) 129:4609–28. doi: 10.1172/JCI125890
72. Werner TL, Wade ML, Agarwal N, Boucher K, Patel J, Luebke A, et al. A pilot study of JI-101, an inhibitor of VEGFR-2, PDGFR-beta, and EphB4 receptors, in combination with everolimus and as a single agent in an ovarian cancer expansion cohort. Invest New Drugs. (2015) 33:1217–24. doi: 10.1007/s10637-015-0288-5
73. Hollier BG, Tinnirello AA, Werden SJ, Evans KW, Taube JH, Sarkar TR, et al. FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast cancer. Cancer Res. (2013) 73:1981–92. doi: 10.1158/0008-5472.CAN-12-2962
74. Itoh N, Ornitz DM. Functional evolutionary history of the mouse Fgf gene family. Dev Dyn. (2008) 237:18–27. doi: 10.1002/dvdy.21388
75. Wu YM, Su F, Kalyana-Sundaram S, Khazanov N, Ateeq B, Cao X, et al. Identification of targetable FGFR gene fusions in diverse cancers. Cancer Discovery. (2013) 3:636–47. doi: 10.1158/2159-8290.CD-13-0050
76. Lin L, Chamberlain L, Pak ML, Nagarajan A, Gupta R, Zhu LJ, et al. A large-scale RNAi-based mouse tumorigenesis screen identifies new lung cancer tumor suppressors that repress FGFR signaling. Cancer Discovery. (2014) 4:1168–81. doi: 10.1158/2159-8290.CD-13-0747
77. Andre F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, et al. Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res. (2013) 19:3693–702. doi: 10.1158/1078-0432.CCR-13-0190
78. Zecchini S, Bombardelli L, Decio A, Bianchi M, Mazzarol G, Sanguineti F, et al. The adhesion molecule NCAM promotes ovarian cancer progression via FGFR signalling. EMBO Mol Med. (2011) 3:480–94. doi: 10.1002/emmm.201100152
79. Ye F, Dewanjee S, Li Y, Jha NK, Chen ZS, Kumar A, et al. Advancements in clinical aspects of targeted therapy and immunotherapy in breast cancer. Mol Cancer. (2023) 22:105. doi: 10.1186/s12943-023-01805-y
80. Helsten T, Elkin S, Arthur E, Tomson BN, Carter J, Kurzrock R. The FGFR landscape in cancer: analysis of 4,853 tumors by next-generation sequencing. Clin Cancer Res. (2016) 22:259–67. doi: 10.1158/1078-0432.CCR-14-3212
81. Jung SY, Yi JY, Kim MH, Song KH, Kang SM, Ahn J, et al. IM-412 inhibits the invasion of human breast carcinoma cells by blocking FGFR-mediated signaling. Oncol Rep. (2015) 34:2731–37. doi: 10.3892/or.2015.4249
82. Fillmore CM, Gupta PB, Rudnick JA, Caballero S, Keller PJ, Lander ES, et al. Estrogen expands breast cancer stem-like cells through paracrine FGF/Tbx3 signaling. Proc Natl Acad Sci U.S.A. (2010) 107:21737–42. doi: 10.1073/pnas.1007863107
83. Brooks AJ, Dai W, O’Mara ML, Abankwa D, Chhabra Y, Pelekanos RA, et al. Mechanism of activation of protein kinase JAK2 by the growth hormone receptor. Science. (2014) 344:1249783. doi: 10.1126/science.1249783
84. Lacronique V, Boureux A, Valle VD, Poirel H, Quang CT, Mauchauffe M, et al. A TEL-JAK2 fusion protein with constitutive kinase activity in human leukemia. Science. (1997) 278:1309–12. doi: 10.1126/science.278.5341.1309
85. Nagai H, Kim YS, Lee KT, Chu MY, Konishi N, Fujimoto J, et al. Inactivation of SSI-1, a JAK/STAT inhibitor, in human hepatocellular carcinomas, as revealed by two-dimensional electrophoresis. J Hepatol. (2001) 34:416–21. doi: 10.1016/s0168-8278(00)00038-6
86. Huang L, Ma B, Ma J, Wang F. Fractalkine/CX3CR1 axis modulated the development of pancreatic ductal adenocarcinoma via JAK/STAT signaling pathway. Biochem Biophys Res Commun. (2017) 493:1510–17. doi: 10.1016/j.bbrc.2017.10.006
87. Balko JM, Schwarz LJ, Luo N, Estrada MV, Giltnane JM, Davila-Gonzalez D, et al. Triple-negative breast cancers with amplification of JAK2 at the 9p24 locus demonstrate JAK2-specific dependence. Sci Transl Med. (2016) 8:334ra53. doi: 10.1126/scitranslmed.aad3001
88. Bromberg J. Stat proteins and oncogenesis. J Clin Invest. (2002) 109:1139–42. doi: 10.1172/JCI15617
89. Cimica V, Chen HC, Iyer JK, Reich NC. Dynamics of the STAT3 transcription factor: nuclear import dependent on Ran and importin-beta1. PloS One. (2011) 6:e20188. doi: 10.1371/journal.pone.0020188
90. Wu S, Lu J, Zhu H, Wu F, Mo Y, Xie L, et al. A novel axis of circKIF4A-miR-637-STAT3 promotes brain metastasis in triple-negative breast cancer. Cancer Lett. (2024) 581:216508. doi: 10.1016/j.canlet.2023.216508
91. Aaronson DS, Horvath CM. A road map for those who don’t know JAK-STAT. Science. (2002) 296:1653–55. doi: 10.1126/science.1071545
92. Darnell JJ, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science. (1994) 264:1415–21. doi: 10.1126/science.8197455
93. Yates LR, Knappskog S, Wedge D, Farmery J, Gonzalez S, Martincorena I, et al. Genomic evolution of breast cancer metastasis and relapse. Cancer Cell. (2017) 32:169–84. doi: 10.1016/j.ccell.2017.07.005
94. Park SY, Lee CJ, Choi JH, Kim JH, Kim JW, Kim JY, et al. The JAK2/STAT3/CCND2 Axis promotes colorectal Cancer stem cell persistence and radioresistance. J Exp Clin Cancer Res. (2019) 38:399. doi: 10.1186/s13046-019-1405-7
95. Choi DS, Blanco E, Kim YS, Rodriguez AA, Zhao H, Huang TH, et al. Chloroquine eliminates cancer stem cells through deregulation of Jak2 and DNMT1. Stem Cells. (2014) 32:2309–23. doi: 10.1002/stem.1746
96. Zhao C, Chen HY, Zhao F, Feng HJ, Su JP. Acylglycerol kinase promotes paclitaxel resistance in nasopharyngeal carcinoma cells by regulating FOXM1 via the JAK2/STAT3 pathway. Cytokine. (2021) 148:155595. doi: 10.1016/j.cyto.2021.155595
97. Xu Y, Zhang J, Wu J, Zhong S, Li H. Inhibition of JAK2 reverses paclitaxel resistance in human ovarian cancer cells. Int J Gynecol Cancer. (2015) 25:1557–64. doi: 10.1097/IGC.0000000000000550
98. Zhang J, Zhao J, Zhang W, Liu G, Yin D, Li J, et al. Establishment of paclitaxel-resistant cell line and the underlying mechanism on drug resistance. Int J Gynecol Cancer. (2012) 22:1450–56. doi: 10.1097/IGC.0b013e31826e2382
99. Cheon JH, Kim KS, Yadav DK, Kim M, Kim HS, Yoon S. The JAK2 inhibitors CEP-33779 and NVP-BSK805 have high P-gp inhibitory activity and sensitize drug-resistant cancer cells to vincristine. Biochem Biophys Res Commun. (2017) 490:1176–82. doi: 10.1016/j.bbrc.2017.06.178
100. Tanei T, Choi DS, Rodriguez AA, Liang DH, Dobrolecki L, Ghosh M, et al. Antitumor activity of Cetuximab in combination with Ixabepilone on triple negative breast cancer stem cells. Breast Cancer Res. (2016) 18:6. doi: 10.1186/s13058-015-0662-4
101. Carey LA, Rugo HS, Marcom PK, Mayer EL, Esteva FJ, Ma CX, et al. TBCRC 001: randomized phase II study of cetuximab in combination with carboplatin in stage IV triple-negative breast cancer. J Clin Oncol. (2012) 30:2615–23. doi: 10.1200/JCO.2010.34.5579
102. Xie W, Zhao H, Wang F, Wang Y, He Y, Wang T, et al. A novel humanized Frizzled-7-targeting antibody enhances antitumor effects of Bevacizumab against triple-negative breast cancer via blocking Wnt/beta-catenin signaling pathway. J Exp Clin Cancer Res. (2021) 40:30. doi: 10.1186/s13046-020-01800-x
103. Alraouji NN, Al-Mohanna FH, Ghebeh H, Arafah M, Almeer R, Al-Tweigeri T, et al. Tocilizumab potentiates cisplatin cytotoxicity and targets cancer stem cells in triple-negative breast cancer. Mol Carcinog. (2020) 59:1041–51. doi: 10.1002/mc.23234
104. Yang C, Zhang J, Zhang Y, Ji F, Chen Y, Zhu T, et al. Low-dose apatinib combined with neoadjuvant chemotherapy in the treatment of early-stage triple-negative breast cancer (LANCET): a single-center, single-arm, phase II trial. Ther Adv Med Oncol. (2022) 14:7437211. doi: 10.1177/17588359221118053
105. Liu J, Liu Q, Li Y, Li Q, Su F, Yao H, et al. Efficacy and safety of camrelizumab combined with apatinib in advanced triple-negative breast cancer: an open-label phase II trial. J Immunother Cancer. (2020) 8:e000696. doi: 10.1136/jitc-2020-000696
106. Liu J, Wang Y, Tian Z, Lin Y, Li H, Zhu Z, et al. Multicenter phase II trial of Camrelizumab combined with Apatinib and Eribulin in heavily pretreated patients with advanced triple-negative breast cancer. Nat Commun. (2022) 13:3011. doi: 10.1038/s41467-022-30569-0
107. Wang J, Sun T, Ouyang Q, Han Y, Xu B. A phase Ib study of TQB2450 plus anlotinib in patients with advanced triple-negative breast cancer. Iscience. (2023) 26:106876. doi: 10.1016/j.isci.2023.106876
108. Huang JY, Xie XF, Chen XL, Zhang QY, Chen LP, Bai X, et al. A single-arm phase II clinical trial of anlotinib combined with chemotherapy for the treatment of metastatic triple-negative breast cancer. Front Oncol. (2023) 13:1122294. doi: 10.3389/fonc.2023.1122294
109. Bernsdorf M, Ingvar C, Jorgensen L, Tuxen MK, Jakobsen EH, Saetersdal A, et al. Effect of adding gefitinib to neoadjuvant chemotherapy in estrogen receptor negative early breast cancer in a randomized phase II trial. Breast Cancer Res Treat. (2011) 126:463–70. doi: 10.1007/s10549-011-1352-2
110. Symonds L, Linden H, Gadi V, Korde L, Rodler E, Gralow J, et al. Combined targeted therapies for first-line treatment of metastatic triple negative breast cancer-A phase II trial of weekly nab-paclitaxel and bevacizumab followed by maintenance targeted therapy with bevacizumab and erlotinib. Clin Breast Cancer. (2019) 19:e283–96. doi: 10.1016/j.clbc.2018.12.008
111. Stover DG, Gil DAC, Brock J, Guo H, Overmoyer B, Balko J, et al. Phase II study of ruxolitinib, a selective JAK1/2 inhibitor, in patients with metastatic triple-negative breast cancer. NPJ Breast Cancer. (2018) 4:10. doi: 10.1038/s41523-018-0060-z
112. O’Shaughnessy J, DeMichele A, Ma CX, Richards P, Yardley DA, Wright GS, et al. A randomized, double-blind, phase 2 study of ruxolitinib or placebo in combination with capecitabine in patients with advanced HER2-negative breast cancer and elevated C-reactive protein, a marker of systemic inflammation. Breast Cancer Res Treat. (2018) 170:547–57. doi: 10.1007/s10549-018-4770-6
113. Lynce F, Williams JT, Regan MM, Bunnell CA, Freedman RA, Tolaney SM, et al. Phase I study of JAK1/2 inhibitor ruxolitinib with weekly paclitaxel for the treatment of HER2-negative metastatic breast cancer. Cancer Chemother Pharmacol. (2021) 87:673–79. doi: 10.1007/s00280-021-04245-x
114. Kang DY, Sp N, Kim DH, Joung YH, Lee HG, Park YM, et al. Salidroside inhibits migration, invasion and angiogenesis of MDA−MB 231 TNBC cells by regulating EGFR/Jak2/STAT3 signaling via MMP2. Int J Oncol. (2018) 53:877–85. doi: 10.3892/ijo.2018.4430
115. Kim S, Park JM, Park S, Jung E, Ko D, Park M, et al. Suppression of TNBC metastasis by doxazosin, a novel dual inhibitor of c-MET/EGFR. J Exp Clin Cancer Res. (2023) 42:292. doi: 10.1186/s13046-023-02866-z
116. Li YC, Wong CN, Hsu FT, Chen JH, Yang CC, Liu HH, et al. Accessing apoptosis induction and metastasis inhibition effect of magnolol on triple negative breast cancer in vitro. In Vivo. (2023) 37:1028–36. doi: 10.21873/invivo.13177
117. Sebastian A, Pandey V, Mohan CD, Chia YT, Rangappa S, Mathai J, et al. Novel adamantanyl-based thiadiazolyl pyrazoles targeting EGFR in triple-negative breast cancer. ACS Omega. (2016) 1:1412–24. doi: 10.1021/acsomega.6b00251
118. Shieh JM, Chen YC, Lin YC, Lin JN, Chen WC, Chen YY, et al. Demethoxycurcumin inhibits energy metabolic and oncogenic signaling pathways through AMPK activation in triple-negative breast cancer cells. J Agric Food Chem. (2013) 61:6366–75. doi: 10.1021/jf4012455
119. Maharjan S, Lee MG, Kim SY, Lee KS, Nam KS. Morin sensitizes MDA-MB-231 triple-negative breast cancer cells to doxorubicin cytotoxicity by suppressing FOXM1 and attenuating EGFR/STAT3 signaling pathways. Pharm (Basel). (2023) 16:672. doi: 10.3390/ph16050672
120. Garay JP, Karakas B, Abukhdeir AM, Cosgrove DP, Gustin JP, Higgins MJ, et al. The growth response to androgen receptor signaling in ERalpha-negative human breast cells is dependent on p21 and mediated by MAPK activation. Breast Cancer Res. (2012) 14:R27. doi: 10.1186/bcr3112
121. Kim JH, Choi HS, Lee DS. Primaquine inhibits the endosomal trafficking and nuclear localization of EGFR and induces the apoptosis of breast cancer cells by nuclear EGFR/stat3-mediated c-myc downregulation. Int J Mol Sci. (2021) 22:12961. doi: 10.3390/ijms222312961
122. Lee MM, Chan BD, Wong WY, Qu Z, Chan MS, Leung TW, et al. Anti-cancer Activity of Centipeda minima Extract in Triple Negative Breast Cancer via Inhibition of AKT, NF-kappaB, and STAT3 Signaling Pathways. Front Oncol. (2020) 10:491. doi: 10.3389/fonc.2020.00491
123. Huang Q, Li S, Zhang L, Qiao X, Zhang Y, Zhao X, et al. CAPE-pNO(2) inhibited the growth and metastasis of triple-negative breast cancer via the EGFR/STAT3/akt/E-cadherin signaling pathway. Front Oncol. (2019) 9:461. doi: 10.3389/fonc.2019.00461
124. Huang L, Wong CC, Mackenzie GG, Sun Y, Cheng KW, Vrankova K, et al. Phospho-aspirin (MDC-22) inhibits breast cancer in preclinical animal models: an effect mediated by EGFR inhibition, p53 acetylation and oxidative stress. BMC Cancer. (2014) 14:141. doi: 10.1186/1471-2407-14-141
125. Mehta R, Katta H, Alimirah F, Patel R, Murillo G, Peng X, et al. Deguelin action involves c-Met and EGFR signaling pathways in triple negative breast cancer cells. PloS One. (2013) 8:e65113. doi: 10.1371/journal.pone.0065113
126. Suh YA, Kim JH, Sung MA, Boo HJ, Yun HJ, Lee SH, et al. A novel antitumor activity of deguelin targeting the insulin-like growth factor (IGF) receptor pathway via up-regulation of IGF-binding protein-3 expression in breast cancer. Cancer Lett. (2013) 332:102–09. doi: 10.1016/j.canlet.2013.01.022
127. Yamashita N, Kondo M, Zhao S, Li W, Koike K, Nemoto K, et al. Picrasidine G decreases viability of MDA-MB 468 EGFR-overexpressing triple-negative breast cancer cells through inhibition of EGFR/STAT3 signaling pathway. Bioorg Med Chem Lett. (2017) 27:2608–12. doi: 10.1016/j.bmcl.2017.03.061
128. Su JC, Mar AC, Wu SH, Tai WT, Chu PY, Wu CY, et al. Disrupting VEGF-A paracrine and autocrine loops by targeting SHP-1 suppresses triple negative breast cancer metastasis. Sci Rep. (2016) 6:28888. doi: 10.1038/srep28888
129. Tian J, Chen X, Fu S, Zhang R, Pan L, Cao Y, et al. Bazedoxifene is a novel IL-6/GP130 inhibitor for treating triple-negative breast cancer. Breast Cancer Res Treat. (2019) 175:553–66. doi: 10.1007/s10549-019-05183-2
130. Li H, Xiao H, Lin L, Jou D, Kumari V, Lin J, et al. Drug design targeting protein-protein interactions (PPIs) using multiple ligand simultaneous docking (MLSD) and drug repositioning: discovery of raloxifene and bazedoxifene as novel inhibitors of IL-6/GP130 interface. J Med Chem. (2014) 57:632–41. doi: 10.1021/jm401144z
131. Shi W, Yan D, Zhao C, Xiao M, Wang Y, Ma H, et al. Inhibition of IL-6/STAT3 signaling in human cancer cells using Evista. Biochem Biophys Res Commun. (2017) 491:159–65. doi: 10.1016/j.bbrc.2017.07.067
132. Viswanadhapalli S, Luo Y, Sareddy GR, Santhamma B, Zhou M, Li M, et al. EC359: A first-in-class small-molecule inhibitor for targeting oncogenic LIFR signaling in triple-negative breast cancer. Mol Cancer Ther. (2019) 18:1341–54. doi: 10.1158/1535-7163.MCT-18-1258
133. Yang J, Qian S, Cai X, Lu W, Hu C, Sun X, et al. Chikusetsusaponin IVa butyl ester (CS-IVa-be), a novel IL6R antagonist, inhibits IL6/STAT3 signaling pathway and induces cancer cell apoptosis. Mol Cancer Ther. (2016) 15:1190–200. doi: 10.1158/1535-7163.MCT-15-0551
134. Bouaouiche S, Ghione S, Sghaier R, Burgy O, Racoeur C, Derangere V, et al. Nitric oxide-releasing drug glyceryl trinitrate targets JAK2/STAT3 signaling, migration and invasion of triple-negative breast cancer cells. Int J Mol Sci. (2021) 22:8449. doi: 10.3390/ijms22168449
135. Lee J, Hahm ER, Singh SV. Withaferin A inhibits activation of signal transducer and activator of transcription 3 in human breast cancer cells. Carcinogenesis. (2010) 31:1991–98. doi: 10.1093/carcin/bgq175
136. Lin KL, Su JC, Chien CM, Tseng CH, Chen YL, Chang LS, et al. Naphtho[1,2-b]furan-4,5-dione disrupts Janus kinase-2 and induces apoptosis in breast cancer MDA-MB-231 cells. Toxicol Vitro. (2010) 24:1158–67. doi: 10.1016/j.tiv.2010.02.019
137. Yang Y, Zhou H, Liu W, Wu J, Yue X, Wang J, et al. Ganoderic acid A exerts antitumor activity against MDA-MB-231 human breast cancer cells by inhibiting the Janus kinase 2/signal transducer and activator of transcription 3 signaling pathway. Oncol Lett. (2018) 16:6515–21. doi: 10.3892/ol.2018.9475
138. Qiu C, Zhang T, Zhu X, Qiu J, Jiang K, Zhao G, et al. Methylseleninic acid suppresses breast cancer growth via the JAK2/STAT3 pathway. Reprod Sci. (2019) 26:829–38. doi: 10.1177/1933719118815582
139. Kalimutho M, Sinha D, Mittal D, Srihari S, Nanayakkara D, Shafique S, et al. Blockade of PDGFRbeta circumvents resistance to MEK-JAK inhibition via intratumoral CD8(+) T-cells infiltration in triple-negative breast cancer. J Exp Clin Cancer Res. (2019) 38:85. doi: 10.1186/s13046-019-1075-5
140. Khanna P, Lee JS, Sereemaspun A, Lee H, Baeg GH. GRAMD1B regulates cell migration in breast cancer cells through JAK/STAT and Akt signaling. Sci Rep. (2018) 8:9511. doi: 10.1038/s41598-018-27864-6
141. Jeon M, You D, Bae SY, Kim SW, Nam SJ, Kim HH, et al. Dimerization of EGFR and HER2 induces breast cancer cell motility through STAT1-dependent ACTA2 induction. Oncotarget. (2017) 8:50570–81. doi: 10.18632/oncotarget.10843
142. Shan H, Yao S, Ye Y, Yu Q. 3-Deoxy-2beta,16-dihydroxynagilactone E, a natural compound from Podocarpus nagi, preferentially inhibits JAK2/STAT3 signaling by allosterically interacting with the regulatory domain of JAK2 and induces apoptosis of cancer cells. Acta Pharmacol Sin. (2019) 40:1578–86. doi: 10.1038/s41401-019-0254-4
143. Jang H, Ko H, Song K, Kim YS. A sesquiterpenoid from farfarae flos induces apoptosis of MDA-MB-231 human breast cancer cells through inhibition of JAK-STAT3 signaling. Biomolecules. (2019) 9:278. doi: 10.3390/biom9070278
144. Byun HJ, Darvin P, Kang DY, Sp N, Joung YH, Park JH, et al. Silibinin downregulates MMP2 expression via Jak2/STAT3 pathway and inhibits the migration and invasive potential in MDA-MB-231 cells. Oncol Rep. (2017) 37:3270–78. doi: 10.3892/or.2017.5588
145. Binienda A, Ziolkowska S, Pluciennik E. The anticancer properties of silibinin: its molecular mechanism and therapeutic effect in breast cancer. Anticancer Agents Med Chem. (2020) 20:1787–96. doi: 10.2174/1871520620666191220142741
146. Chen D, Ma Y, Li P, Liu M, Fang Y, Zhang J, et al. Piperlongumine induces apoptosis and synergizes with doxorubicin by inhibiting the JAK2-STAT3 pathway in triple-negative breast cancer. Molecules. (2019) 24:2338. doi: 10.3390/molecules24122338
147. Shakya R, Park GH, Joo SH, Shim JH, Choi JS. Hydroxyzine induces cell death in triple-negative breast cancer cells via mitochondrial superoxide and modulation of jak2/STAT3 signaling. Biomol Ther (Seoul). (2022) 30:585–92. doi: 10.4062/biomolther.2022.121
148. Schust J, Sperl B, Hollis A, Mayer TU, Berg T. Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol. (2006) 13:1235–42. doi: 10.1016/j.chembiol.2006.09.018
149. Poria DK, Sheshadri N, Balamurugan K, Sharan S, Sterneck E. The STAT3 inhibitor Stattic acts independently of STAT3 to decrease histone acetylation and modulate gene expression. J Biol Chem. (2021) 296:100220. doi: 10.1074/jbc.RA120.016645
150. Song H, Wang R, Wang S, Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci U.S.A. (2005) 102:4700–05. doi: 10.1073/pnas.0409894102
151. Lin L, Hutzen B, Zuo M, Ball S, Deangelis S, Foust E, et al. Novel STAT3 phosphorylation inhibitors exhibit potent growth-suppressive activity in pancreatic and breast cancer cells. Cancer Res. (2010) 70:2445–54. doi: 10.1158/0008-5472.CAN-09-2468
152. Zinzalla G, Haque MR, Basu BP, Anderson J, Kaye SL, Haider S, et al. A novel small-molecule inhibitor of IL-6 signalling. Bioorg Med Chem Lett. (2010) 20:7029–32. doi: 10.1016/j.bmcl.2010.09.117
153. Lin L, Hutzen B, Li PK, Ball S, Zuo M, DeAngelis S, et al. A novel small molecule, LLL12, inhibits STAT3 phosphorylation and activities and exhibits potent growth-suppressive activity in human cancer cells. Neoplasia. (2010) 12:39–50. doi: 10.1593/neo.91196
154. Noori S, Rezaei TM, Deravi N, Mahboobi RM, Zarghi A. Naringenin enhances the anti-cancer effect of cyclophosphamide against MDA-MB-231 breast cancer cells via targeting the STAT3 signaling pathway. Iran J Pharm Res. (2020) 19:122–33. doi: 10.22037/ijpr.2020.113103.14112
155. Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, et al. Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U.S.A. (2007) 104:7391–96. doi: 10.1073/pnas.0609757104
156. Locken H, Clamor C, Muller K. Napabucasin and related heterocycle-fused naphthoquinones as STAT3 inhibitors with antiproliferative activity against cancer cells. J Nat Prod. (2018) 81:1636–44. doi: 10.1021/acs.jnatprod.8b00247
157. Cai G, Yu W, Song D, Zhang W, Guo J, Zhu J, et al. Discovery of fluorescent coumarin-benzothiophene 1, 1-dioxide conjugates as mitochondria-targeting antitumor STAT3 inhibitors. Eur J Med Chem. (2019) 174:236–51. doi: 10.1016/j.ejmech.2019.04.024
158. Park SK, Byun WS, Lee S, Han YT, Jeong YS, Jang K, et al. A novel small molecule STAT3 inhibitor SLSI-1216 suppresses proliferation and tumor growth of triple-negative breast cancer cells through apoptotic induction. Biochem Pharmacol. (2020) 178:114053. doi: 10.1016/j.bcp.2020.114053
159. Yue P, Zhu Y, Brotherton-Pleiss C, Fu W, Verma N, Chen J, et al. Novel potent azetidine-based compounds irreversibly inhibit Stat3 activation and induce antitumor response against human breast tumor growth in vivo. Cancer Lett. (2022) 534:215613. doi: 10.1016/j.canlet.2022.215613
160. Yang Z, Xu H, Yang Y, Duan C, Zhang P, Wang Y, et al. Synthesis and evaluation of naphthalene derivatives as potent STAT3 inhibitors and agents against triple-negative breast cancer growth and metastasis. Breast Cancer Res Treat. (2023) 197:255–67. doi: 10.1007/s10549-022-06790-2
161. Byun WS, Lim H, Hong J, Bae ES, Lee SB, Kim Y, et al. Design, synthesis, and biological activity of marinacarboline analogues as STAT3 pathway inhibitors for docetaxel-resistant triple-negative breast cancer. J Med Chem. (2023) 66:3106–33. doi: 10.1021/acs.jmedchem.2c01115
162. Xie Y, Zhu S, Chen L, Liu H, Peng T, Ming Z, et al. An isoxazoloquinone derivative inhibits tumor growth by targeting STAT3 and triggering its ubiquitin-dependent degradation. Cancers (Basel). (2023) 15:2424. doi: 10.3390/cancers15092424
163. Qi Y, Wu H, Zhu T, Liu Z, Liu C, Yan C, et al. Acetyl-cinobufagin suppresses triple-negative breast cancer progression by inhibiting the STAT3 pathway. Aging (Albany Ny). (2023) 15:8258–74. doi: 10.18632/aging.204967
164. Feng T, Cao W, Shen W, Zhang L, Gu X, Guo Y, et al. Arctigenin inhibits STAT3 and exhibits anticancer potential in human triple-negative breast cancer therapy. Oncotarget. (2017) 8:329–44. doi: 10.18632/oncotarget.13393
165. Zhang W, Yu W, Cai G, Zhu J, Zhang C, Li S, et al. A new synthetic derivative of cryptotanshinone KYZ3 as STAT3 inhibitor for triple-negative breast cancer therapy. Cell Death Dis. (2018) 9:1098. doi: 10.1038/s41419-018-1139-z
166. Kim SL, Choi HS, Kim JH, Jeong DK, Kim KS, Lee DS. Dihydrotanshinone-induced NOX5 activation inhibits breast cancer stem cell through the ROS/stat3 signaling pathway. Oxid Med Cell Longev. (2019) 2019:9296439. doi: 10.1155/2019/9296439
167. He J, Wei X, Li S, Quan X, Li R, Du H, et al. DT-13 suppresses breast cancer metastasis by modulating PLOD2 in the adipocytes microenvironment. Phytomedicine. (2019) 59:152778. doi: 10.1016/j.phymed.2018.12.001
168. Lan T, Wang L, Xu Q, Liu W, Jin H, Mao W, et al. Growth inhibitory effect of Cucurbitacin E on breast cancer cells. Int J Clin Exp Pathol. (2013) 6:1799–805.
169. Nguyen T, Pham T, Nguyen C, Truong TN, Bishop C, Doan N, et al. Phytochemistry and cytotoxic activity of aquilaria crassna pericarp on MDA-MB-468 cell lines. ACS Omega. (2023) 8:42356–66. doi: 10.1021/acsomega.3c04656
170. Kong Y, Chen J, Zhou Z, Xia H, Qiu MH, Chen C. Cucurbitacin E induces cell cycle G2/M phase arrest and apoptosis in triple negative breast cancer. PloS One. (2014) 9:e103760. doi: 10.1371/journal.pone.0103760
171. Gyamfi J, Lee YH, Min BS, Choi J. Niclosamide reverses adipocyte induced epithelial-mesenchymal transition in breast cancer cells via suppression of the interleukin-6/STAT3 signalling axis. Sci Rep. (2019) 9:11336. doi: 10.1038/s41598-019-47707-2
172. Ss PS, Krishnamurthy PT, Ghanta VR, Chintamaneni PK. Phenyl boronic acid-modified lipid nanocarriers of niclosamide for targeting triple-negative breast cancer. Nanomedicine (Lond). (2020) 15:1551–65. doi: 10.2217/nnm-2020-0003
173. Pindiprolu S, Chintamaneni PK, Krishnamurthy PT, Ratna SGK. Formulation-optimization of solid lipid nanocarrier system of STAT3 inhibitor to improve its activity in triple negative breast cancer cells. Drug Dev Ind Pharm. (2019) 45:304–13. doi: 10.1080/03639045.2018.1539496
174. Ko H, Lee JH, Kim HS, Kim T, Han YT, Suh YG, et al. Novel galiellalactone analogues can target STAT3 phosphorylation and cause apoptosis in triple-negative breast cancer. Biomolecules. (2019) 9:170. doi: 10.3390/biom9050170
175. Yang F, Hu M, Lei Q, Xia Y, Zhu Y, Song X, et al. Nifuroxazide induces apoptosis and impairs pulmonary metastasis in breast cancer model. Cell Death Dis. (2015) 6:e1701. doi: 10.1038/cddis.2015.63
176. Wang X, Shi W, Wang X, Lu JJ, He P, Zhang H, et al. Nifuroxazide boosts the anticancer efficacy of palbociclib-induced senescence by dual inhibition of STAT3 and CDK2 in triple-negative breast cancer. Cell Death Discovery. (2023) 9:355. doi: 10.1038/s41420-023-01658-w
177. Pan L, Chen X, Fu S, Yu W, Li C, Wang T, et al. LLY17, a novel small molecule STAT3 inhibitor induces apoptosis and suppresses cell migration and tumor growth in triple-negative breast cancer. Breast Cancer Res Treat. (2020) 181:31–41. doi: 10.1007/s10549-020-05613-6
178. Liu Z, Ge X, Gu Y, Huang Y, Liu H, Yu M, et al. Small molecule STAT3 inhibitor, 6Br-6a suppresses breast cancer growth in vitro and in vivo. BioMed Pharmacother. (2020) 121:109502. doi: 10.1016/j.biopha.2019.109502
179. Khan MW, Saadalla A, Ewida AH, Al-Katranji K, Al-Saoudi G, Giaccone ZT, et al. The STAT3 inhibitor pyrimethamine displays anti-cancer and immune stimulatory effects in murine models of breast cancer. Cancer Immunol Immunother. (2018) 67:13–23. doi: 10.1007/s00262-017-2057-0
180. Brown JI, Wang P, Wong A, Petrova B, Persaud R, Soukhtehzari S, et al. Cycloguanil and analogues potently target DHFR in cancer cells to elicit anti-cancer activity. Metabolites. (2023) 13:151. doi: 10.3390/metabo13020151
181. Li Y, Gan C, Zhang Y, Yu Y, Fan C, Deng Y, et al. Inhibition of stat3 signaling pathway by natural product pectolinarigenin attenuates breast cancer metastasis. Front Pharmacol. (2019) 10:1195. doi: 10.3389/fphar.2019.01195
182. Oh E, Kim YJ, An H, Sung D, Cho TM, Farrand L, et al. Flubendazole elicits anti-metastatic effects in triple-negative breast cancer via STAT3 inhibition. Int J Cancer. (2018) 143:1978–93. doi: 10.1002/ijc.31585
183. Dinakar YH, Kumar H, Mudavath SL, Jain R, Ajmeer R, Jain V. Role of STAT3 in the initiation, progression, proliferation and metastasis of breast cancer and strategies to deliver JAK and STAT3 inhibitors. Life Sci. (2022) 309:120996. doi: 10.1016/j.lfs.2022.120996
184. Yang B, Shen JW, Zhou DH, Zhao YP, Wang WQ, Zhu Y, et al. Precise discovery of a STAT3 inhibitor from Eupatorium lindleyanum and evaluation of its activity of anti-triple-negative breast cancer. Nat Prod Res. (2019) 33:477–85. doi: 10.1080/14786419.2017.1396596
185. Lou C, Chen Y, Zhang J, Yang B, Zhao H. Eupalinolide J suppresses the growth of triple-negative breast cancer cells via targeting STAT3 signaling pathway. Front Pharmacol. (2019) 10:1071. doi: 10.3389/fphar.2019.01071
186. Zeng AQ, Yu Y, Yao YQ, Yang FF, Liao M, Song LJ, et al. Betulinic acid impairs metastasis and reduces immunosuppressive cells in breast cancer models. Oncotarget. (2018) 9:3794–804. doi: 10.18632/oncotarget.23376
187. Vyas D, Lopez-Hisijos N, Shah P, Deshpande KS, Basson MD, Vyas A, et al. A second-generation proteasome inhibitor and doxorubicin modulates IL-6, pSTAT-3 and NF-kB activity in MDA-MB-231 breast cancer cells. J Nanosci Nanotechnol. (2017) 17:175–85. doi: 10.1166/jnn.2017.12427
188. Cheng CC, Shi LH, Wang XJ, Wang SX, Wan XQ, Liu SR, et al. Stat3/Oct-4/c-Myc signal circuit for regulating stemness-mediated doxorubicin resistance of triple-negative breast cancer cells and inhibitory effects of WP1066. Int J Oncol. (2018) 53:339–48. doi: 10.3892/ijo.2018.4399
189. El HH, Saleh A, Al SH, Athamneh K, Attoub S, Arafat K, et al. Rhus coriaria suppresses angiogenesis, metastasis and tumor growth of breast cancer through inhibition of STAT3, NFkappaB and nitric oxide pathways. Sci Rep. (2016) 6:21144. doi: 10.1038/srep21144
190. Li W, Zhang H, Nie M, Tian Y, Chen X, Chen C, et al. Ursolic acid derivative FZU-03,010 inhibits STAT3 and induces cell cycle arrest and apoptosis in renal and breast cancer cells. Acta Biochim Biophys Sin (Shanghai). (2017) 49:367–73. doi: 10.1093/abbs/gmx012
191. Kim YJ, Kim JY, Lee N, Oh E, Sung D, Cho TM, et al. Disulfiram suppresses cancer stem-like properties and STAT3 signaling in triple-negative breast cancer cells. Biochem Biophys Res Commun. (2017) 486:1069–76. doi: 10.1016/j.bbrc.2017.03.164
192. Dai X, Yin C, Guo G, Zhang Y, Zhao C, Qian J, et al. Schisandrin B exhibits potent anticancer activity in triple negative breast cancer by inhibiting STAT3. Toxicol Appl Pharmacol. (2018) 358:110–19. doi: 10.1016/j.taap.2018.09.005
193. Dai X, Yin C, Zhang Y, Guo G, Zhao C, Wang O, et al. Osthole inhibits triple negative breast cancer cells by suppressing STAT3. J Exp Clin Cancer Res. (2018) 37:322. doi: 10.1186/s13046-018-0992-z
194. Qu Z, Lin Y, Mok DK, Bian Q, Tai WC, Chen S. Brevilin A, a natural sesquiterpene lactone inhibited the growth of triple-negative breast cancer cells via akt/mTOR and STAT3 signaling pathways. Onco Targets Ther. (2020) 13:5363–73. doi: 10.2147/OTT.S256833
195. Qu Z, Lin Y, Mok DK, Bian Q, Tai WC, Chen S. Arnicolide D inhibits triple negative breast cancer cell proliferation by suppression of akt/mTOR and STAT3 signaling pathways. Int J Med Sci. (2020) 17:1482–90. doi: 10.7150/ijms.46925
196. Zhu Z, Yuan J, Xu X, Wei Y, Yang B, Zhao H. Eucannabinolide, a novel sesquiterpene lactone, suppresses the growth, metastasis and BCSCS-like traits of TNBC via inactivation of STAT3. Neoplasia. (2021) 23:36–48. doi: 10.1016/j.neo.2020.10.012
197. Byun WS, Bae ES, Cui J, Park HJ, Oh DC, Lee SK. Antitumor activity of pulvomycin via targeting activated-STAT3 signaling in docetaxel-resistant triple-negative breast cancer cells. Biomedicines. (2021) 9:436. doi: 10.3390/biomedicines9040436
198. Zhu Y, Yue P, Dickinson CF, Yang JK, Datanagan K, Zhai N, et al. Natural product preferentially targets redox and metabolic adaptations and aberrantly active STAT3 to inhibit breast tumor growth in vivo. Cell Death Dis. (2022) 13:1022. doi: 10.1038/s41419-022-05477-2
199. An H, Kim JY, Oh E, Lee N, Cho Y, Seo JH. Salinomycin promotes anoikis and decreases the CD44+/CD24- stem-like population via inhibition of STAT3 activation in MDA-MB-231 cells. PloS One. (2015) 10:e141919. doi: 10.1371/journal.pone.0141919
200. Bellat V, Verchere A, Ashe SA, Law B. Transcriptomic insight into salinomycin mechanisms in breast cancer cell lines: synergistic effects with dasatinib and induction of estrogen receptor beta. BMC Cancer. (2020) 20:661. doi: 10.1186/s12885-020-07134-3
201. Mesmar J, Abdallah R, Hamade K, Baydoun S, Al-Thani N, Shaito A, et al. Ethanolic extract of Origanum Syriacum L. leaves exhibits potent anti-breast cancer potential and robust antioxidant properties. Front Pharmacol. (2022) 13:994025. doi: 10.3389/fphar.2022.994025
202. Lee HH, Jung J, Moon A, Kang H, Cho H. Antitumor and anti-invasive effect of apigenin on human breast carcinoma through suppression of IL-6 expression. Int J Mol Sci. (2019) 20:3143. doi: 10.3390/ijms20133143
203. Chuang HC, Kapuriya N, Kulp SK, Chen CS, Shapiro CL. Differential anti-proliferative activities of poly(ADP-ribose) polymerase (PARP) inhibitors in triple-negative breast cancer cells. Breast Cancer Res Treat. (2012) 134:649–59. doi: 10.1007/s10549-012-2106-5
204. Palandri F, Tiribelli M, Benevolo G, Tieghi A, Cavazzini F, Breccia M, et al. Efficacy and safety of ruxolitinib in intermediate-1 IPSS risk myelofibrosis patients: Results from an independent study. Hematol Oncol. (2018) 36:285–90. doi: 10.1002/hon.2429
205. Al-Ali HK, Griesshammer M, Foltz L, Palumbo GA, Martino B, Palandri F, et al. Primary analysis of JUMP, a phase 3b, expanded-access study evaluating the safety and efficacy of ruxolitinib in patients with myelofibrosis, including those with low platelet counts. Br J Haematol. (2020) 189:888–903. doi: 10.1111/bjh.16462
206. Gerds AT, Tauchi T, Ritchie E, Deininger M, Jamieson C, Mesa R, et al. Phase 1/2 trial of glasdegib in patients with primary or secondary myelofibrosis previously treated with ruxolitinib. Leuk Res. (2019) 79:38–44. doi: 10.1016/j.leukres.2019.02.012
207. Gerds AT, Savona MR, Scott BL, Talpaz M, Egyed M, Harrison CN, et al. Determining the recommended dose of pacritinib: results from the PAC203 dose-finding trial in advanced myelofibrosis. Blood Adv. (2020) 4:5825–35. doi: 10.1182/bloodadvances.2020003314
208. Mesa RA, Vannucchi AM, Mead A, Egyed M, Szoke A, Suvorov A, et al. Pacritinib versus best available therapy for the treatment of myelofibrosis irrespective of baseline cytopenias (PERSIST-1): an international, randomised, phase 3 trial. Lancet Haematol. (2017) 4:e225–36. doi: 10.1016/S2352-3026(17)30027-3
209. Verstovsek S, Mesa R, Talpaz M, Kiladjian JJ, Harrison CN, Oh ST, et al. Retrospective analysis of pacritinib in patients with myelofibrosis and severe thrombocytopenia. Haematologica. (2022) 107:1599–607. doi: 10.3324/haematol.2021.279415
210. Mascarenhas J, Hoffman R, Talpaz M, Gerds AT, Stein B, Gupta V, et al. Pacritinib vs best available therapy, including ruxolitinib, in patients with myelofibrosis: A randomized clinical trial. JAMA Oncol. (2018) 4:652–59. doi: 10.1001/jamaoncol.2017.5818
211. Foltz L, Pica GM, Zerazhi H, Van Droogenbroeck J, Visanica S, Baez DLFE, et al. Safety and efficacy findings from the open-label, multicenter, phase 3b, expanded treatment protocol study of ruxolitinib for treatment of patients with polycythemia vera who are resistant/intolerant to hydroxyurea and for whom no alternative treatments are available. Leuk Lymphoma. (2019) 60:3493–502. doi: 10.1080/10428194.2019.1636985
212. Sylvine P, Thomas S, Pirayeh E. Infections associated with ruxolitinib: study in the French Pharmacovigilance database. Ann Hematol. (2018) 97:913–14. doi: 10.1007/s00277-018-3242-8
213. Porpaczy E, Tripolt S, Hoelbl-Kovacic A, Gisslinger B, Bago-Horvath Z, Casanova-Hevia E, et al. Aggressive B-cell lymphomas in patients with myelofibrosis receiving JAK1/2 inhibitor therapy. Blood. (2018) 132:694–706. doi: 10.1182/blood-2017-10-810739
214. Wang P, Zhou J. Proteolysis targeting chimera (PROTAC): A paradigm-shifting approach in small molecule drug discovery. Curr Top Med Chem. (2018) 18:1354–56. doi: 10.2174/1568026618666181010101922
215. Noblejas-Lopez M, Nieto-Jimenez C, Burgos M, Gomez-Juarez M, Montero JC, Esparis-Ogando A, et al. Activity of BET-proteolysis targeting chimeric (PROTAC) compounds in triple negative breast cancer. J Exp Clin Cancer Res. (2019) 38:383. doi: 10.1186/s13046-019-1387-5
216. Li G, Lin SS, Yu ZL, Wu XH, Liu JW, Tu GH, et al. A PARP1 PROTAC as a novel strategy against PARP inhibitor resistance via promotion of ferroptosis in p53-positive breast cancer. Biochem Pharmacol. (2022) 206:115329. doi: 10.1016/j.bcp.2022.115329
217. He R, Song Z, Bai Y, He S, Huang J, Wang Y, et al. Discovery of AXL degraders with improved potencies in triple-negative breast cancer (TNBC) cells. J Med Chem. (2023) 66:1873–91. doi: 10.1021/acs.jmedchem.2c01682
218. Adams CM, Mitra R, Xiao Y, Michener P, Palazzo J, Chao A, et al. Targeted MDM2 degradation reveals a new vulnerability for p53-inactivated triple-negative breast cancer. Cancer Discovery. (2023) 13:1210–29. doi: 10.1158/2159-8290.CD-22-1131
219. Li X, Zhang Z, Gao F, Ma Y, Wei D, Lu Z, et al. c-myc-targeting PROTAC based on a TNA-DNA bivalent binder for combination therapy of triple-negative breast cancer. J Am Chem Soc. (2023) 145:9334–42. doi: 10.1021/jacs.3c02619
220. Zhao C, Chen D, Suo F, Setroikromo R, Quax WJ, Dekker FJ. Discovery of highly potent HDAC8 PROTACs with anti-tumor activity. Bioorg Chem. (2023) 136:106546. doi: 10.1016/j.bioorg.2023.106546
221. Pu C, Liu Y, Deng R, Xu Q, Wang S, Zhang H, et al. Development of PROTAC degrader probe of CDK4/6 based on DCAF16. Bioorg Chem. (2023) 138:106637. doi: 10.1016/j.bioorg.2023.106637
222. Yang J, Chang Y, Tien JC, Wang Z, Zhou Y, Zhang P, et al. Discovery of a highly potent and selective dual PROTAC degrader of CDK12 and CDK13. J Med Chem. (2022) 65:11066–83. doi: 10.1021/acs.jmedchem.2c00384
223. Niu T, Li K, Jiang L, Zhou Z, Hong J, Chen X, et al. Noncovalent CDK12/13 dual inhibitors-based PROTACs degrade CDK12-Cyclin K complex and induce synthetic lethality with PARP inhibitor. Eur J Med Chem. (2022) 228:114012. doi: 10.1016/j.ejmech.2021.114012
224. Dale B, Anderson C, Park KS, Kaniskan HU, Ma A, Shen Y, et al. Targeting triple-negative breast cancer by a novel proteolysis targeting chimera degrader of enhancer of zeste homolog 2. ACS Pharmacol Transl Sci. (2022) 5:491–507. doi: 10.1021/acsptsci.2c00100
225. Wang C, Chen X, Liu X, Lu D, Li S, Qu L, et al. Discovery of precision targeting EZH2 degraders for triple-negative breast cancer. Eur J Med Chem. (2022) 238:114462. doi: 10.1016/j.ejmech.2022.114462
226. Wu Y, Xue J, Li J. Chemical degrader enhances the treatment of androgen receptor-positive triple-negative breast cancer. Arch Biochem Biophys. (2022) 721:109194. doi: 10.1016/j.abb.2022.109194
227. Pu C, Tong Y, Liu Y, Lan S, Wang S, Yan G, et al. Selective degradation of PARP2 by PROTACs via recruiting DCAF16 for triple-negative breast cancer. Eur J Med Chem. (2022) 236:114321. doi: 10.1016/j.ejmech.2022.114321
Keywords: triple-negative breast cancer, receptor tyrosine kinase, Janus Kinase 2, signal transducer and activator of transcription 3, small molecule compounds
Citation: Long L, Fei X, Chen L, Yao L and Lei X (2024) Potential therapeutic targets of the JAK2/STAT3 signaling pathway in triple-negative breast cancer. Front. Oncol. 14:1381251. doi: 10.3389/fonc.2024.1381251
Received: 03 February 2024; Accepted: 08 April 2024;
Published: 18 April 2024.
Edited by:
Sulma Mohammed, Purdue University, United StatesReviewed by:
Luis E. Arias-Romero, National Autonomous University of Mexico, MexicoManel Ben Hammouda, Duke University, United States
Copyright © 2024 Long, Fei, Chen, Yao and Lei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyong Lei, bGVpeF95b25nQDE2My5jb20=; Liang Yao, NzkwNzA5NzM4QHFxLmNvbQ==
†These authors have contributed equally to this work