 Peiyi Qin1,2
Peiyi Qin1,2 Yuanfu Qi
Yuanfu Qi- 1First Clinical Medical College, Shandong University of Traditional Chinese Medicine, Jinan, China
- 2Shandong College of Traditional Chinese Medicine, Yantai, Shandong, China
- 3Department of Orthopedics, First Affiliated Hospital of Dalian Medical University, Dalian, China
- 4Department of Oncology, Affiliated Hospital of Shandong University of Traditional Chinese Medicine, Jinan, China
Lung cancer is the leading cause of cancer-related mortality worldwide, with non-small cell lung cancer (NSCLC) being the predominant type. The roles of autophagy and apoptosis in NSCLC present a dual and intricate nature. Additionally, autophagy and apoptosis interconnect through diverse crosstalk molecules. Owing to their multitargeting nature, safety, and efficacy, natural products have emerged as principal sources for NSCLC therapeutic candidates. This review begins with an exploration of the mechanisms of autophagy and apoptosis, proceeds to examine the crosstalk molecules between these processes, and outlines their implications and interactions in NSCLC. Finally, the paper reviews natural products that have been intensively studied against NSCLC targeting autophagy and apoptosis, and summarizes in detail the four most retrieved representative drugs. This paper clarifies good therapeutic effects of natural products in NSCLC by targeting autophagy and apoptosis and aims to promote greater consideration by researchers of natural products as candidates for anti-NSCLC drug discovery.
1 Introduction
Lung cancer currently holds the distinction of being the malignancy with the highest rates of mortality globally (1, 2). Approximately 85% of lung cancer cases are classified as non-small cell lung cancer (NSCLC), encompassing adenocarcinoma, squamous carcinoma, large cell carcinoma, among others (3). Studies indicate that the 5-year survival rate for NSCLC in stages III-IV is under 15% (4). Treatment modalities for this cancer typically encompass surgery, radiation, chemotherapy, targeted therapy, and immunotherapy. Surgery combined with platinum-based adjuvant chemotherapy is the first choice for early-stage NSCLC. Yet, it has been reported that over 81% of clinically diagnosed NSCLC patients are ineligible for surgical intervention (5). In cases of advanced NSCLC, it is mostly treated with gemcitabine or docetaxel in combination with platinum drugs. However, the prognosis remains poor due to factors like drug resistance, radio resistance, propensity for recurrence, and pronounced tumor aggressiveness (6–9). Acquired resistance is an urgent problem in cancer treatment, and combination therapies can help to minimize the risk of resistance. Studies have shown that the combination of decitabine and retinoic acid increases sensitivity and therapeutic efficacy in rapidly proliferating and slowly migrating NSCLC (10). The advent of targeted therapies and immunotherapy has provided a glimmer of hope for NSCLC patients. Nevertheless, these treatments are beneficial only for individuals with specific biological markers and are limited by side effects, drug resistance, and high costs (11, 12). Thus, there exists an urgent need to discover more effective, safer, and affordable therapeutic agents.
Inducing tumor cell death is the primary objective in tumor therapy, with the main modes including programmed death (apoptosis, autophagic death), necrosis, pyroptosis, ferroptosis, among others (13). Apoptosis and autophagic cell death are particularly significant in cancer treatment due to their non-inflammatory nature and minimal side effects on the body (14). Autophagy exhibits a tumor-suppressive function by eliminating tumor-inducing molecules in cancer’s early stages. However, in the progression phase, it can support tumor growth by supplying energy to stressed tumor cells under conditions like nutrient scarcity (15). Antitumor drugs can excessively upregulate autophagy-related genes, leading to autophagic death of tumor cells and impeding tumor growth (16). It has also been observed that autophagy can shield tumor cells from radiation and chemotherapy, promoting tolerability and resistance, thereby promoting tumor growth (17). Inhibition of cytoprotective autophagy is considered a promising modality for tumor therapy. In addition to cytoprotective autophagy and cytotoxicity autophagy, David A Gewirtz has proposed two other forms of autophagy, one is a nonprotective form of autophagy, in which the cell apparently performs autophagy-mediated degradation but autophagy inhibition does not result in a significant alteration of chemotherapy or radiation sensitivity and the other is cytostatic autophagy, which results in cell growth inhibition and reduced clonogenicity (16). Therefore, the interplay between autophagy and tumors is bidirectional and intricate, influenced by tissue type, tumor development stage, autophagic activity level, and therapeutic choices (18). Inducing apoptosis in tumor cells has been a focal point in cancer treatment research (19). Moreover, numerous studies reveal complex crosstalk between autophagy and apoptosis. Their interaction and mutual regulation during cancer progression and treatment pose significant challenges in cancer therapy (20). In this context, thorough research into autophagy, apoptosis, and their interplay is vital for effective cancer treatment, including for NSCLC.
Natural products are pharmacological components extracted, isolated, and optimized from nature. Their role in extracting bioactive compounds has demonstrated considerable promise in cancer therapy. Reports indicate that various natural products can influence tumor cells through multiple pathways, including apoptosis, autophagy, cell proliferation, migration/invasion, angiogenesis, and metastasis (21). Statistics show that approximately 47% of antitumor drugs originate from natural compounds (22, 23). Notable anti-cancer drugs like camptothecin, paclitaxel, and vincristine have been effectively utilized in clinical settings, demonstrating significant efficacy (24). The advantages of natural products include low toxicity, high efficiency, multi-targeting capabilities, and affordability. Currently, the therapeutic role of natural products in NSCLC is receiving increasing recognition.
Although autophagy and apoptosis in NSCLC have been extensively explored, a summary of natural products targeting these pathways in NSCLC is lacking. This review initially introduces the mechanisms of autophagy and apoptosis, analyzes the crosstalk molecules between them, and then outlines their roles and interactions in NSCLC. Finally, we reviewed the natural products that target autophagy and apoptosis in NSCLC and summarized the main four of them in detail. This review aims to provide insights for future NSCLC patient treatments.
2 Autophagy
2.1 Overview of autophagy
The term “autophagy” derives from the Greek words, “auto” meaning self, and “phagy” meaning to eat, defining it as a self-degrading process essential for organ maintenance and energy turnover during injury (25). As an evolutionarily ancient and highly conserved catabolic process, autophagy is classified into macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA), based on the method of substrate delivery to lysosomes (26). Generally, when referring to autophagy, macroautophagy is implied. Macroautophagy serves as a cellular defense mechanism, maintaining homeostasis by degrading and recycling misfolded proteins and damaged organelles (27). Notably, there is substantial evidence suggesting autophagy could be a form of cell death. Excessive autophagy may lead to extensive degradation of cytoplasm and organelles, resulting in type II programmed cell death. Autophagy is further categorized into selective and non-selective types based on the specificity of the degraded substrate. Selective autophagy targets specific components such as intracellular pathogens, damaged organelles, and misfolded protein aggregates, with mitochondrial autophagy being the most extensively studied (28). Non-selective autophagy, in contrast, randomly degrades cytoplasmic substances, including a majority of cytoplasm and organelles. This review will predominantly address macroautophagy, hereafter referred to as autophagy.
2.2 The autophagic process
Macroautophagy, hereafter referred to as autophagy, encompasses a series of signaling cascades involving various autophagy-associated genes, proteins, and compounds (29–31). As illustrated in Figure 1, this process includes the induction, nucleation, elongation, maturation, and closure of phagosomes, ultimately leading to autophagosome formation and their fusion with lysosomes.
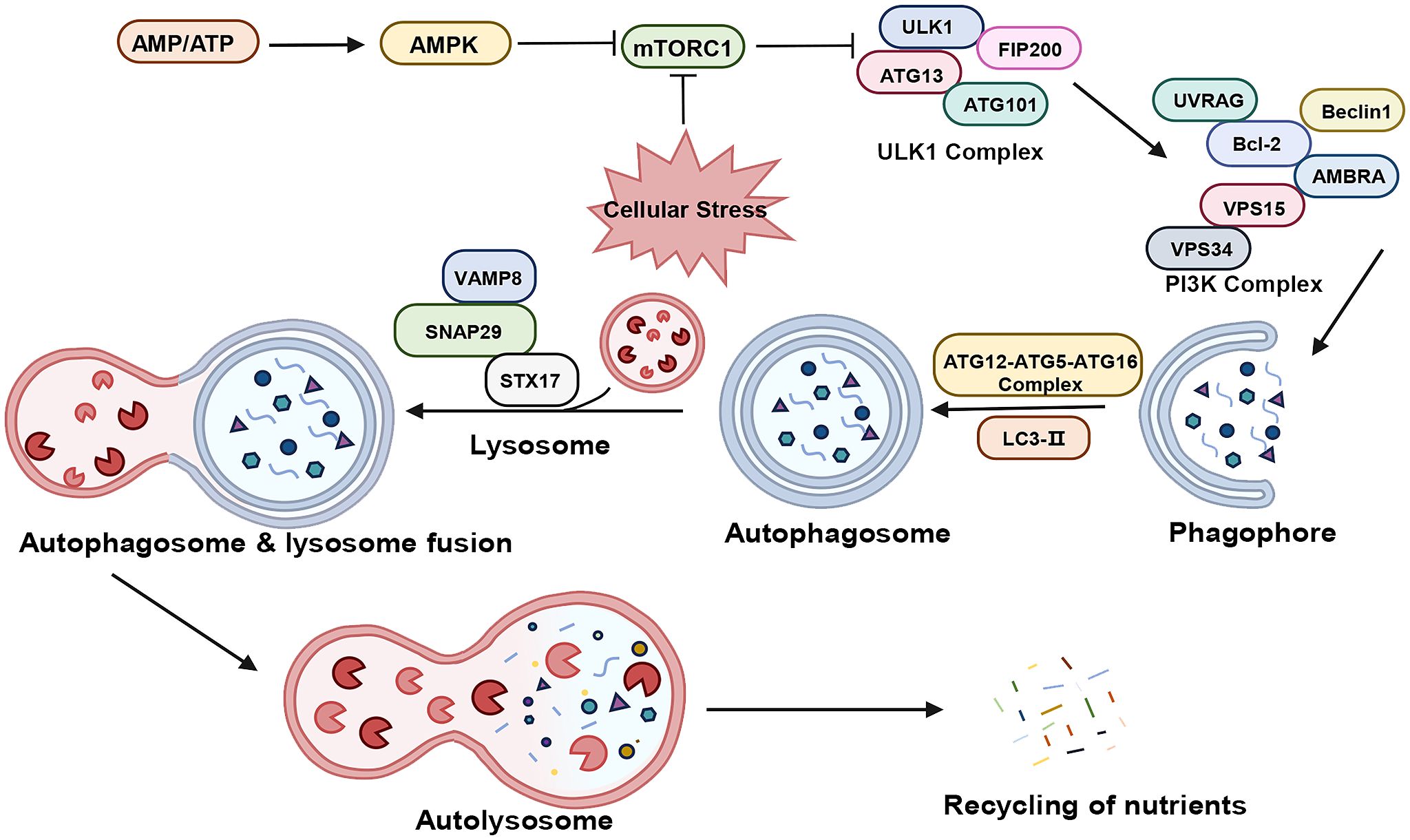
Figure 1 The process of autophagy. “→” represents promotion; “⟂” represents inhibition. AMP, adenosine monophosphate; ATP, adenosine triphosphate; AMPK, AMP-activated protein kinase; mTORC1, mammalian target of rapamycin complex 1; ULKI, Unc-51-like kinase 1; FIP200, FAK family kinase-interacting protein of 200 kDa; ATG, Autophagy Related Protein; UVRAG, ultraviolet radiation resistance-associated gene; Bcl-2, B-Cell Lymphoma 2; AMBRA, autophagy and beclin 1 regulator; Vps34, Vacuolar Protein Sorting 34: Vps15, Vacuolar Protein Sorting 15; PI3K, Phosphatidyl Inositol-3 Kinase; LC3, Light Chain Kinase 3; STX17, syntaxin 17; SNAP29, Synaptosomal-associated protein 29; VAMP8, vesicle-associated membrane protein 8.
In the physiological state, the cell maintains a very low level of basal autophagy. At this time, there is sufficient intracellular energy, Mammalian target of rapamycin complex 1 (mTORC1) is in an activated state and phosphorylates Autophagy Related Protein (ATG) 13, thus inhibiting cellular autophagy. Under stress such as hypoxia, the reduction in ATP levels activates AMP-activated protein kinase (AMPK), which phosphorylates tuberous sclerosis complex 2 (TSC2) to inhibit mTORC1 activity (32, 33). the activity of mTORC1 is inhibited, the phosphorylation level of ATG13 decreases, and the dephosphorylated ATG13 forms a complex with Unc-51-like kinase 1 (ULK1) and interacts with FAK family kinase-interacting protein of 200 kDa (FIP200) to produce the ATG13-ULK1-FIP200 complex, and induces downstream autophagosome nucleation and elongation (34–36). The nucleation process is closely related to the Vacuolar Protein Sorting 34 (Vps34) - Beclin-1 complex, which also contains Vacuolar Protein Sorting 15 (Vps15), and together they act in the nucleation of membrane vesicles and mediate the formation of pre-autophagosomal structure (PAS) (37, 38). The extension of mammalian autophagosome mainly depends on two ubiquitination-like systems: the binding process of ATG12 and the modification process of Light Chain Kinase 3 (LC3) (39, 40). Binding process of ATG12: ATG12 is first activated by ATG7, then transporter and bound to ATG5 via ATG10, and then bound to ATG16, generating a multibody complex of ATG12-ATG5-ATG16. This complex is localized on the outer membrane surface of the preautophagosome structure and participates in the expansion of the outer membrane of the preautophagosome (41). The process of LC3 modification: after the formation of LC3 precursor, it is processed into cytoplasmic soluble LC3-I by ATG4, and then covalently linked to phosphatidylethanolamine (PE) to become lipid-soluble LC3-PE (LC3-II) by ATG7 and ATG3, and participates in the membrane elongation. LC3-II is able to bind to newly formed membrane until the formation of autophagic lysosome formation. Therefore, LC3-II is commonly used as a marker for autophagy formation and is an important multi-signaling regulatory protein localized on the membrane of autophagic vesicles (42, 43). In addition, p62/SQSM1 is involved in autophagosome formation and promotes translocation of the mTORC1 complex to the lysosomal surface, where it is selectively encapsulated into the autophagosome and subsequently degraded by protein hydrolases in autophagic lysosomes (44). Thus, the expression of p62/SQSM1 protein showed a negative correlation with autophagic activity. Subsequently, the Soluble n-Ethylmaleimide Sensitive Factor Attachment Protein Receptor (SNARE) protein family orchestrates the binding of the autophagosome with the lysosome, forming the autophagolysosome (45). Lastly, lysosomal acid hydrolases decompose the cargoes within the autophagosome, with the breakdown products reabsorbed for recycling.
2.3 The four faces of autophagy in NSCLC
In early NSCLC, autophagy suppresses tumor initiation by removing damaged organelles and misfolded proteins, thus preserving cell homeostasis. Inhibition of cellular autophagy promotes tumor cell growth, showing that cellular autophagy inhibits tumorigenesis (46). However, in mid-to-late stages, autophagy is generally considered to facilitate NSCLC progression by supplying nutrients and energy to cancer cells, enabling their adaptation to various stress states. At this stage, elevated autophagy levels are positively associated with increased cancer cell invasiveness, advanced tumor stages, and reduced survival (47). In addition to the cytoprotective (cell survival) and cytotoxic (cell death) roles of autophagy described above. Bai et al. suggested that autophagy also exists in cytostatic (cell growth arrest) and nonprotective (no contribution to cell death or survival) forms in tumor cells (48).
The four faces in the process of tumor therapy: Firstly, Tumor cell autophagy protects tumor cells from damage caused by chemotherapy and radiotherapy and promotes their metastasis, invasion and drug resistance. Studies reveal that autophagy contributes to chemotherapy resistance in NSCLC, whereas autophagy inhibition can counteract tumor resistance and progression (49). Li et al. demonstrated that protein DICER, through its interaction with let-7i-5p, activates autophagy in NSCLC, thereby promoting cisplatin resistance (50). Yuqing Chen et al. confirmed that overexpression of miR-142-3p enhances chemotherapy sensitivity in NSCLC by suppressing autophagy (51). Secondly, autophagy can in turn induce programmed apoptosis or autophagic death in tumor cells because of the effects of antitumor drugs. PFAP, a newly identified protein from Pleurotus ferulae lanzi, a Chinese medicinal herb, has shown therapeutic potential in NSCLC. In vivo and in vitro studies demonstrated that PFAP induces autophagy in A549 cells via the AMPK/mTOR pathway, significantly reducing the growth of xenograft tumors in nude mice (52). Similarly, Paris saponin VII (PSVII), extracted from Trillium tschonoskii Maxim and studied by Xiang et al., activates autophagy through the AMPK/mTOR signaling pathway, inhibiting the proliferation of NSCLC cells and exhibiting anti-cancer effects (53). Thirdly, Sharma et al. demonstrated a novel cytostatic form of autophagy induced by vitamin D and its analog, EB 1089, which sensitizes NSCLC cells to radiotherapy. This effect is potentially mediated by VDR, TP53, and AMPK, contributing to an enhanced response to radiation in NSCLC by 1,25-D3 and EB1089 (54). Fourthly, Patel et al. confirmed that inhibition of autophagy to overcome radiotherapy resistance might only be effective if autophagy serves a cytoprotective role. In NSCLC, autophagy can manifest as nonprotective, and p53 does not reliably indicate autophagy’s functional status, suggesting that the inclusion of autophagy inhibitors in treatments requires careful screening (55).
3 Apoptosis
3.1 Overview of apoptosis
The term “apoptosis” originates from the Greek “apó” (from) and “ptósi” (fall), signifying “to fall off” as leaves from a tree or petals from a flower. Kerr et al. first described the apoptotic morphology of cells in the 1970s (56). Apoptosis, also known as type I programmed cell death, is an active, physiological cellular death process, driven by internal genetic mechanisms under certain physiological or pathological conditions. Triggered by intracellular and/or extracellular signals, apoptosis involves a sequence of morphological changes, such as cell shrinkage, chromatin condensation, nuclear fragmentation, and plasma membrane blistering, culminating in apoptotic body formation (56, 57). Apoptosis is crucial in maintaining internal environmental stability and various systemic physiological functions (58), Abnormal apoptosis is implicated in the development of tumors, neurodegenerative diseases, cardiovascular disorders, and autoimmune diseases (59).
3.2 The apoptotic cascades
As illustrated in Figure 2, the apoptotic process encompasses interactions among various proteins, signal transducers, and pathway cascades (60–62).
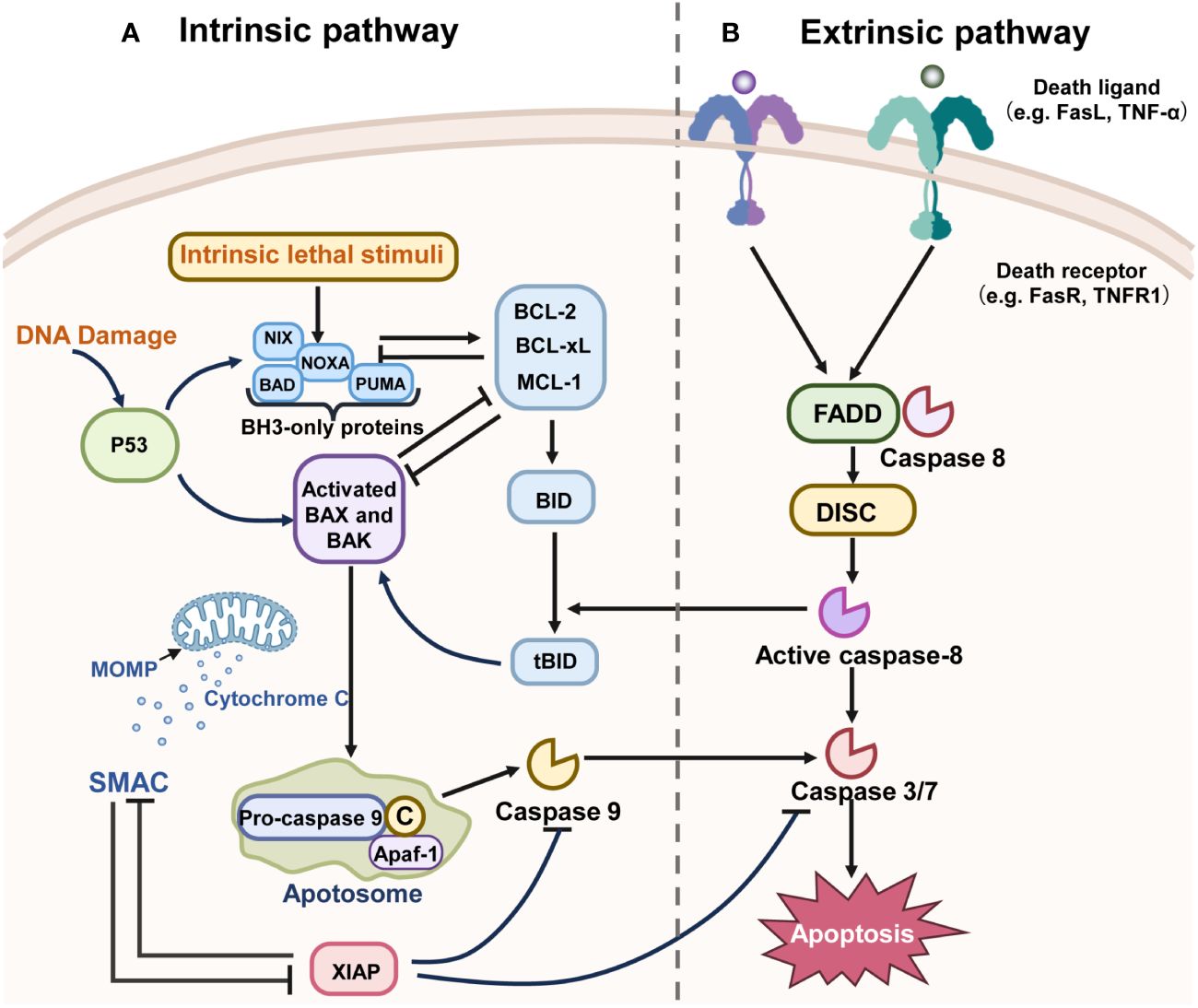
Figure 2 The mechanisms of the two main pathways of apoptosis. “→” represents promotion; “⟂” represents inhibition. (A) The intrinsic pathway of apoptosis is regulated by the BCL-2 family of proteins and can be initiated by various stimuli. For example, BH3-only proteins such as PUMA and NOXA are activated directly through transcriptional regulation by P53. These BH3-only proteins activate the pro-apoptotic proteins Bax or Bak, inhibiting the anti-apoptotic proteins like Bcl-2, BCL-XL, and MCL-1. The activation of Bax or Bak leads to the initiation of MOMP, which in turn activates the cascade, culminating in cell apoptosis. X-linked inhibitor of apoptosis (XIAP), a member of the Inhibitor of Apoptosis Protein (IAP) family that is often overexpressed in malignancies, functions to inhibit caspase activation and can be inactivated by SMAC. (B) The death receptor pathway is initiated by the activation of caspase-8, Fas, TNF, and TNF receptors, which recruit death domain proteins such as FADD and TRADD, as well as caspases, to form death-inducing signaling complexes (DISC). Other downstream molecules have been activated by DISC and lead to apoptosis.
The extrinsic apoptotic pathway, or death receptor pathway, is triggered by external stimuli. It induces apoptosis through interactions between death ligands and receptors. Death receptors, belonging to the tumor necrosis factor family, include Tumor Necrosis Factor Receptor Type 1 (TNFR1) and Fas receptor. In this pathway, binding of a death receptor to specific ligands like Tumor Necrosis Factor (TNF) or Tumor Necrosis Factor-Related Apoptosis Inducing Ligand (TRAIL) transmits the death signal into the cytoplasm, activating the intracellular apoptotic pathway (63–65). The interaction of the death receptor and ligand recruits TNFR1-Associated Death Domain (TRADD), Fas-Associated Death Domain Protein (FADD), Tumor Necrosis Factor Receptor-Associated Factor 2 (TRAF2), and Caspase-8, forming the death-inducing signaling complex (DISC). DISC initiates Protease-8 activation, which in turn triggers downstream effector proteins, thereby inducing apoptosis (66, 67).
The intrinsic apoptotic pathway, also known as the mitochondrial pathway, is initiated by mitochondrial outer membrane permeabilization (MOMP) mediated by the B-Cell Lymphoma 2 (Bcl-2) protein family under conditions like DNA damage, energy starvation, and hypoxia (67). The Bcl-2 family, pivotal in regulating endogenous apoptosis, is categorized based on the number of Bcl-2 homology (BH) domains (68). Pro-survival proteins contain BH1-BH4 domains (e.g., Bcl-XL, Bcl-2, Mcl-1), while pro-apoptotic proteins possess BH1-BH3 domains (e.g., Bax, Bak) (69). BH3 proteins detect DNA damage and signal apoptosis. Upon their migration to the mitochondrial membrane, they activate pro-apoptotic proteins Bax or Bak, or inhibit anti-apoptotic proteins like Bcl-2 and Myeloid Cell Leukemia 1 (Mcl-1) (70). Activation of pro-apoptotic proteins in mitochondria leads to MOMP, followed by cytochrome c release into the cytoplasm. Cytochrome c binds with Apoptotic Protease Activating Factor 1 (APAF-1) to form an apoptotic complex, activating Caspase 9’s precursor, which in turn activates Caspase 3 and Caspase 7, inducing a caspase cascade and triggering cell apoptosis (71). In summary, intrinsic apoptosis involves three key stages (1): Bcl-2 family protein interactions (2), Bax/Bak-mediated MOMP, (3) activation of apoptotic caspases (72). Both mitochondrial and death receptor pathways rely on caspase activation (73).
3.3 The roles of apoptosis in NSCLC
The emergence of cancer from normal tissue cells involves genomic instability and/or inflammation. One natural defense mechanism cancer cell employs to promote their growth and survival is the inhibition of apoptosis (74). Therefore, inducing apoptosis has been identified as a potential cancer treatment strategy (56).
In NSCLC treatment, numerous drugs work by triggering apoptosis in cancer cells. It is reported that Erufosine, a novel alkylphosphocholine, exhibits anti-tumor effects on NSCLC cells by inducing apoptosis and cell cycle arrest (75). Natural products also play a role in NSCLC treatment by modulating apoptosis. Lobocrassin B, a membrane-type compound from Chassophyllum soft coral, has shown potential as an anti-lung cancer agent. It increases levels of Bax, cleaved Caspase-3, -8 and-9, and decreases Bcl-2 in CL1-5 and H520 cells, effectively inducing lung cancer cell death through the mitochondria-dependent apoptosis pathway (76). Isoflavones (Biochanin A), phytoestrogens found in legumes, have been observed to induce lung cancer cell cycle arrest and apoptosis in A549 and 95D cells by regulating cell cycle-related protein expression and regulating the Bcl-2 and Caspase-3 pathways (77). Similarly, Lotus leaf flavone upregulates Caspase-3, -9, and Bax, and downregulates Bcl-2 expression in A549 cells, inhibiting cell proliferation and inducing apoptosis without harming normal cells (78). Additionally, the flavonoid vitexin induces apoptosis in A549 cells, downregulates the Bcl-2/Bax ratio, and enhances caspase-3 expression, promoting apoptosis (79).
Paradoxically, Kerr et al. observed that “spontaneous and persistent cell death is an inherent characteristic of malignant tumors”. Induction of apoptosis is often considered an important way to prevent and treat cancer. However, apoptosis may also lead to unexpected results and may even promote cancer. In the apoptotic pathway, cleavage of cysteine asparaginase activates calcium-independent phospholipase A2 (iPLA2), which produces prostaglandin E2 (PGE2) that enhances proliferation. Also in this process, apoptosis provides support for proliferating cells by removing tumor cells that respond to treatment, leading to tumor regeneration and chemoresistance (80, 81). These findings suggest a dual role for apoptosis in tumor regulation, highlighting that targeting apoptosis alone in treatment may inadvertently promote cancer progression or recurrence. Therefore, in cancer therapy, particularly NSCLC, it is crucial to consider other intracellular regulatory mechanisms. A comprehensive approach that includes differentiation and apoptosis could effectively regulate NSCLC.
4 Crosstalk between autophagy and apoptosis
4.1 Linking autophagy and apoptosis
Autophagy and apoptosis, two highly conserved evolutionary processes, collaborate to maintain cell homeostasis under stress. Despite differing physiological mechanisms, they are closely interconnected, sharing several key regulatory factors (82). The interplay between autophagy and apoptosis in NSCLC is primarily governed by Beclin-1, ATG5-ATG12, and caspase family proteins. This review focuses on a few regulatory factors in their crosstalk, with additional targets outlined in Table 1.
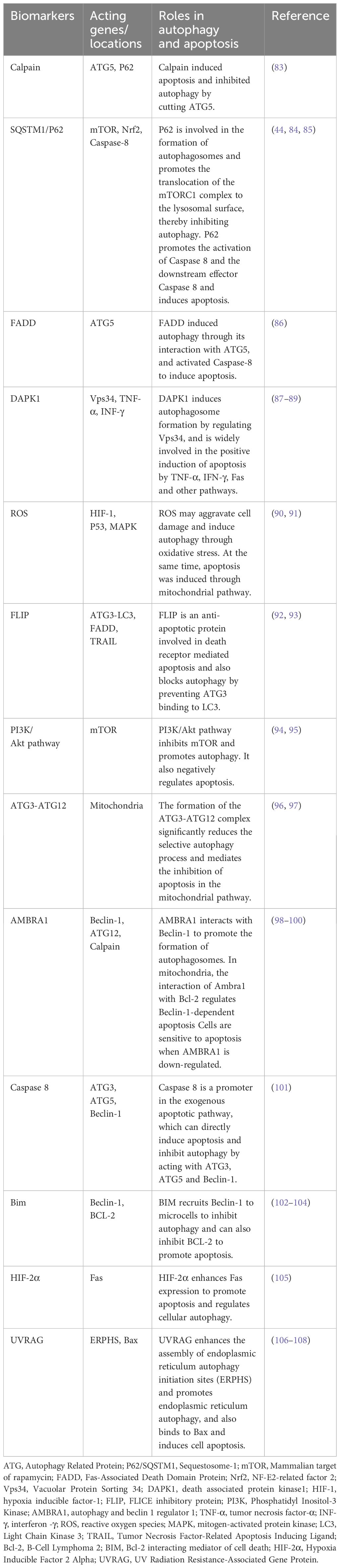
Table 1 List of target biomarkers regulating the autophagy-apoptosis crosstalk.
4.1.1 Beclin-1 and Bcl-2
The Beclin-1 and Bcl-2 interaction plays a crucial role in the autophagy-apoptosis crosstalk (109). Beclin-1, the mammalian homologue of yeast ATG6/Vacuolar Protein Sorting 30 (Vps30) and also known as the BECN1 gene, is recognized as a tumor suppressor gene. Bcl-2 family proteins, including anti-apoptotic proteins (Bcl-2, Bcl-xl, etc.) and pro-apoptotic proteins (BH3-only proteins, Bax, Bak, etc.), regulate MOMP in apoptosis’ intrinsic pathway (110). Beclin-1, pivotal in autophagy nucleation, is essential for autophagy regulation (111). As a central protein, Beclin-1 assembles cofactors to form the BECN1 (beclin 1) - phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3) - phosphatidylinositol 3-kinase regulatory subunit 4 (PIK3R4) complex, initiating the autophagy protein cascade, and holds dual roles in NSCLC autophagy regulation (112). Beclin-1, also a BH3-only protein, regulates both autophagy and apoptosis by binding with the Bcl-2 anti-apoptotic protein. Bcl-2, sharing the BH3 domain with Beclin-1, can bind to Beclin-1 via this domain, inhibiting autophagy when complexed together. Autophagy is induced when Bax competitively binds Bcl-2, dissociating Beclin-1 (101, 113, 114). Additionally, stressors like nutrient deficiency can activate c-Jun N-terminal Kinase 1 (JNK1), leading to Bcl-2 phosphorylation and subsequent autophagy promotion (115).
4.1.2 ATG
Autophagy involves various conserved autophagy-associated genes, with ATG5 and ATG12 being crucial not only for autophagosome formation but also in apoptosis (116). ATG5, instrumental in autophagic vacuole membrane elongation, combines with ATG12 to form the ATG12-ATG5 conjugate (116). In apoptotic cells, Calpain cleaves ATG5, producing truncated fragments that migrate to mitochondria and interact with Bcl-XL, inducing cytochrome C release and triggering apoptosis (83). Rubinstein et al. identified ATG12 as ATG5’s binding partner, contributing to autophagosome extension. ATG12 also plays a role in apoptosis, possibly by binding and inactivating anti-apoptotic Bcl-2 and Mcl-1 proteins (117). Additionally, ATG4D is a key mediator in autophagy and apoptosis crosstalk. ATG4, a cysteine protease originating from yeast, facilitates the attachment of ATG8 to phagocytic vesicles by cleaving ATG8 ubiquitin-like proteins’ C-terminus. Research indicates that Caspase-3 cleaves ATG4D, prompting mitochondria-targeted apoptosis (118). These findings suggest that ATG5, ATG12, and ATG4 may act as critical switch proteins between autophagy and apoptosis.
4.1.3 Caspases
Caspases, a group of cysteine proteases, are primary facilitators of apoptosis and also influence the crosstalk between autophagy and apoptosis (119, 120). Apoptotic caspases are categorized into initiators (Caspase 2, -8, -9, and -10) and effectors (Caspase -3, -6, and -7) (121, 122). These enzymes can degrade autophagy proteins, thereby inhibiting autophagy and ending cellular self-protection. ATG3, crucial in LC3 lipidation and autophagosome formation, can be cleaved by Caspase-8, impeding autophagy and promoting apoptosis (98). Additionally, activated caspases can convert autophagy protein fragments into pro-apoptotic fragments, triggering apoptosis (120). Calpain, a widely conserved family of calcium-sensitive cysteine proteases present in cytoplasm and mitochondria (123), plays a significant role. As previously mentioned, Calpain cleaves ATG5 to generate a fragment that translocate to mitochondria and promotes cytochrome C release, sensitizing cells to apoptosis. Mitochondrial autophagy (mitophagy) is the gradual accumulation of mitochondrial DNA mutations in response to stresses such as reactive oxygen species (ROS) stress, resulting in a decrease in intracellular mitochondrial membrane potential and damage to depolarization, and ultimately leading to cell death (124, 125). Calpain also degrades ATG5, thereby inhibiting mitochondrial autophagy in selective autophagy processes (83). Caspase-3, a critical terminal enzyme in apoptosis, also affects autophagy. It inhibits autophagy by cleaving Beclin-1, and the cleaved Beclin-1 moves to mitochondria, facilitating cytochrome C release from the mitochondrial outer membrane, which promotes apoptosis (126). Initiator Caspase-9, part of the intrinsic apoptosis pathway, can interact with ATG7 to initiate autophagy (92). Thus, caspases are integral in regulating both apoptosis and autophagy.
4.1.4 P53
The transcription factor P53, encoded by the TP53 gene, is a crucial tumor suppressor involved in cell cycle regulation, DNA repair, and apoptosis (127). Under normal, unstressed conditions, P53 is maintained at low levels, marked for degradation by E3 ubiquitin ligase murine double minute 2 (MDM2) (128). However, in response to stressors like DNA damage, oxidative stress, or nutrient deficiency, P53 activates, leading to cell cycle arrest, apoptosis, or senescence (129). Although wild-type TP53 can repair DNA damage and induce tumor cell apoptosis, mutations in the TP53 gene are present in over half of all cancer cases (130). P53 is a well-known apoptosis inducer, promoting transcription of pro-apoptotic genes Bax, and suppressing anti-apoptotic gene Bcl-2, thus triggering apoptosis (131). Beyond apoptosis, P53 also regulates autophagy. Cytoplasmic P53 can inhibit autophagy by interacting with autophagy protein FIP200, thereby competitively impeding autophagy (121, 132). As a transcription factor, P53 can enhance autophagy in the nucleus by activating upstream regulatory factors of mTOR. This involves two primary pathways: the first regulating autophagy via damage factor Damage-Regulated Autophagy Modulator (DRAM), and the second through AMP-activated protein kinase (AMPK) activation. Nuclear P53 transcriptionally activates the DRAM to boost autophagy (133). Additionally, nuclear P53 can activate AMPK, tuberous sclerosis complex 1 (TSC1) and TSC2, subsequently inhibiting mTOR and promoting autophagy (134). In conclusion, P53 plays a vital role in modulating the interplay between autophagy and apoptosis.
4.2 Main effects of interaction between autophagy and apoptosis in NSCLC
Under normal conditions, autophagy typically precedes apoptosis, occurring in response to various cellular stresses (135). Ideally, both autophagy and apoptosis exert anti-tumor effects: autophagy by eliminating tumor cells and apoptosis by preventing their survival. However, increasing evidence suggests that autophagy can enhance cancer cell survival and confer chemotherapy resistance in certain cancers (136). Thus, autophagy’s role in cancer development is dual: on one hand, it can inhibit apoptosis and promote tumor cell growth through protective autophagy; on the other, excessive autophagy leads to type II programmed cell death, or “ACD”. “ACD” is characterized by abundant autophagosomes and autophagolysosomes in the cytoplasm, extensive cytoplasmic degradation, but an intact nucleus, typically not reliant on Caspase family activity (137, 138). The concept of autophagy-related cell death is debated, as autophagy-mediated apoptosis and a distinct cell death mechanism (independent of apoptosis and necrosis) are all referred to as “ACD” (139). Whether autophagy promotes or inhibits apoptosis depends on the cell type, stress nature, and stress duration (140). The interaction between autophagy and apoptosis in NSCLC is complex, with their relationship categorized into three main types: promotional, synergistic, and antagonistic.
4.2.1 Promoting effect
Over-activated autophagy, leading to extensive degradation of cellular components by lysosomes, can trigger cell death (141). Autophagy-induced apoptosis can occur directly or via shared regulatory factors (142). TNF-α, a pro-inflammatory cytokine primarily produced by macrophages and monocytes, is implicated in normal inflammatory responses and immune cell regulation. In human neutrophils, autophagy-mediated TNF-α can induce early apoptosis (143), indicating that autophagy might prompt apoptosis through common regulators like TNF-α. Additionally, studies have shown that inhibiting autophagy with 3-MA also reduces cell apoptosis (144). Bruceine D (BD), a steroidal compound, induces apoptosis and autophagy in A549 and NCI-H292 cells through reactive oxygen species (ROS) production and mitogen-activated protein kinase (MAPK) pathway regulation; here, autophagy inhibition diminishes BD-induced apoptosis (145).
4.2.2 Synergy effect
Autophagy and apoptosis can have a synergistic effect, either occurring simultaneously or compensating for each other’s deficiency. For instance, during radiotherapy under nutrient deprivation, increased autophagy levels supply ATP to cells while releasing apoptotic signals, leading to cancer cell apoptosis (146). In cells lacking apoptosis ability, endoplasmic reticulum stress-induced apoptosis is suppressed, but persistent autophagy can cause oxidative damage and ultimately induce apoptosis (147). transmembrane protein 100 (TMEM100), a potential tumor suppressor, enhances autophagy and synergistically induces apoptosis in NSCLC cells (148). Licarin A, extracted from nutmeg, activates a synergistic effect of autophagy and apoptosis, culminating in NSCLC cell death (149). It was demonstrated that the Heat shock protein 90 (HSP90) inhibitor DPB could inhibit the growth of A549 cells by co-inducing apoptosis and autophagy to exert anti-NSCLC effects (150). The above studies suggest that apoptosis and autophagy have complementary synergistic roles and work together to promote cell death.
4.2.3 Antagonism effect
Under normal physiological conditions, autophagy primarily serves as a cell protection mechanism to avert cell death (151). In cancer, rapid, exponential growth often leads to tumor cells experiencing starvation and hypoxia. Autophagy, in response to genomic instability and/or inflammatory reactions, can adapt tumor cells to these stressors by degrading mitochondria. Cellular degradation of mitochondria through mitochondrial autophagy reduces ROS by eliminating depolarized mitochondria for cytoprotective purposes. Mitochondrial autophagy prevents apoptosis by reducing MOMP and decreasing the release of cytochrome C (152). This protective autophagy, however, can contribute to chemotherapy resistance, impeding cancer treatment (17). Chloroquine, a widely used autophagy inhibitor, has been shown to inhibit bevacizumab-induced autophagy, promoting apoptosis and inhibiting tumor cell proliferation (153). Research indicates that inhibiting autophagy can enhance cisplatin-induced apoptosis (154). Almonertinib, a third-generation epidermal growth factor receptor (EGFR) -targeting drug, induces both apoptosis and autophagy in NSCLC cells by promoting ROS production. The autophagy induced is cytoprotective; thus, blocking autophagy promotes apoptosis (155). The Pinocchio protein has been found to inhibit autophagy, thereby promoting apoptosis and inhibiting proliferation in A549 cells, exhibiting anti-NSCLC effects (156). Additionally, the expression of the brain-expressed X-linked 2 (BEX2) gene promote mitochondrial autophagy in NSCLC cells and inhibits cell apoptosis (157). The antagonism between autophagy and apoptosis is common in cancer, often leading to treatment-resistant cancers. Hence, targeting autophagy inhibition to enhance tumor cell apoptosis is considered an effective anticancer therapy strategy.
5 Effects of four natural products regulating autophagy and apoptosis on NSCLC
Extensive research indicates that various natural products can therapeutically influence NSCLC by modulating autophagy and apoptosis (21, 158, 159). Natural products such as camptothecin, vincristine and paclitaxel have been successfully used in clinical applications, and the search for active ingredients with anti-tumor activity and low toxicity from natural products has become a hot research topic for researchers in recent years. The mechanisms of anti-NSCLC activity of natural products from different sources in recent years have focused on inducing apoptosis, blocking the cell cycle, regulating cellular autophagy, promoting histone acetylation, inhibiting hypoxia-induced processes, increasing the level of ROS, inhibiting invasion and metastasis, and reversing drug resistance (160). In this article, we review the natural products that are currently being studied in NSCLC therapy with a focus on the regulation of autophagy and apoptosis. Through searching we found that dihydroartemisinin, curcumin, baicalein and ginsenoside are the most studied and have potential clinical applications. we summarized these four natural products in detail, with additional natural products outlined in Table 2.
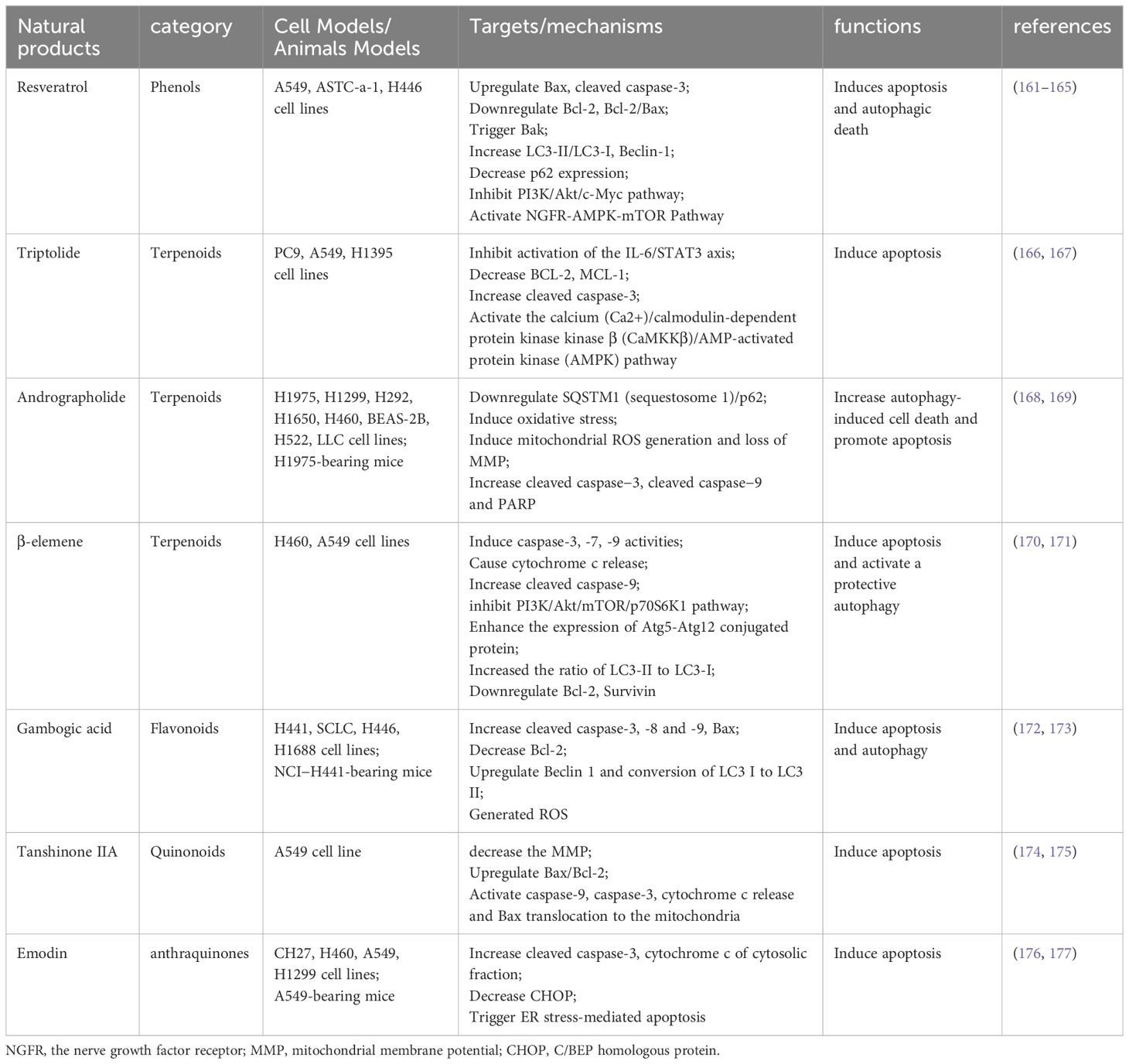
Table 2 Effects of other natural products on autophagy and apoptosis in NSCLC.
5.1 Dihydroartemisinin
Dihydroartemisinin (DHA), derived from artemisinin, is primarily known as a frontline antimalarial drug developed in China. Beyond its anti-malarial properties, an increasing body of research reports DHA’s anticancer activity against a range of cancers in vivo and in vitro. Its anticancer mechanisms encompass inhibition of proliferation, induction of apoptosis, reduction of tumor metastasis and angiogenesis, enhancement of immune function, and induction of autophagy and endoplasmic reticulum stress (178). Due to its established low toxicity and safety, DHA has been extensively studied in NSCLC treatment. Table 3 summarizes DHA’s effects on autophagy and apoptosis in NSCLC.
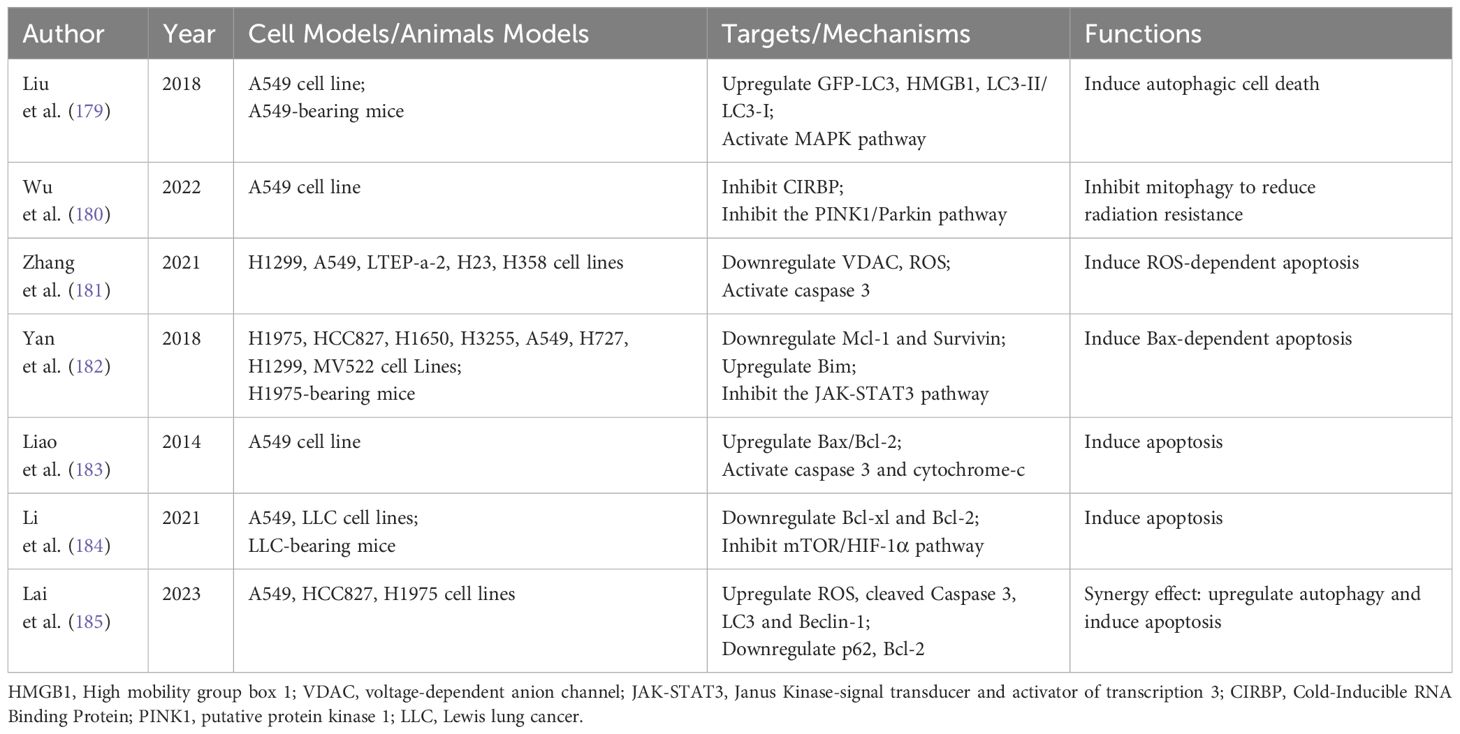
Table 3 Effects of DHA on autophagy and apoptosis in NSCLC.
5.1.1 Regulation of autophagy
Autophagy can either facilitate tumor survival or lead to autophagic cell death during tumor progression and therapy. This process is influenced by the levels of signaling molecules activated by therapeutic agents (186). Liu et al. discovered that DHA-37, a novel derivative of DHA, initiated MAPK signaling by upregulating High mobility group box 1 (HMGB1) in A549 cells. This activation led to excessive autophagic cell death, making autophagy a key factor in A549 cell mortality (179). Radiotherapy, a primary treatment for NSCLC, often encounters challenges with radiation resistance, a significant factor in residual and recurring cancer post-treatment. Research indicates that DHA can inhibit mitophagy, potentially decreasing radiation resistance and enhancing the therapeutic effect against NSCLC (180).
5.1.2 Regulation of apoptosis
Numerous studies have established that DHA can significantly induce apoptosis in NSCLC cells in vitro and in vivo. Zhang et al. found that DHA exhibits anti-NSCLC activity by inducing ROS-dependent apoptosis (181). Another study highlighted that DHA effectively inhibited signal transducer and activator of transcription 3 (STAT3) phosphorylation. Inactivation of STAT3 led to reduced levels of Mcl-1 and Survivin, thereby exerting a synergistic anticancer effect and inducing apoptosis in NSCLC cells (182). Moreover, it has been reported that DHA induces apoptosis in the A549 lung cancer cell line, evidenced by an increased Bax/Bcl-2 ratio and elevated levels of active caspase-3 and cytochrome-c (183). Additionally, both in vivo and in vitro experiments demonstrate that DHA reduces the expression of the mTOR/HIF-1α signaling pathway and induces apoptosis in NSCLC (184).
5.1.3 Regulation of autophagy and apoptosis
DHA plays a crucial role in enhancing anticancer effects and reversing drug resistance in NSCLC by regulating both autophagy and apoptosis. One study suggests that DHA treatment significantly increases the sensitivity of A549-GR cells to gefitinib by elevating apoptosis and upregulating autophagy, thereby effectively controlling tumor progression (185).
5.2 Curcumin
Curcumin, a polyphenol derived from the traditional Chinese medicine Curcuma longa, has shown anti-inflammatory, antioxidant, and anti-tumor properties (187). Its application in cancer treatment, particularly in NSCLC, has gained increasing interest. Research indicates that Curcumin can modulate various programmed cell death mechanisms, including autophagy, apoptosis, pyroptosis, and ferroptosis (188). Table 4 summarizes Curcumin’s impact on autophagy and apoptosis in NSCLC.
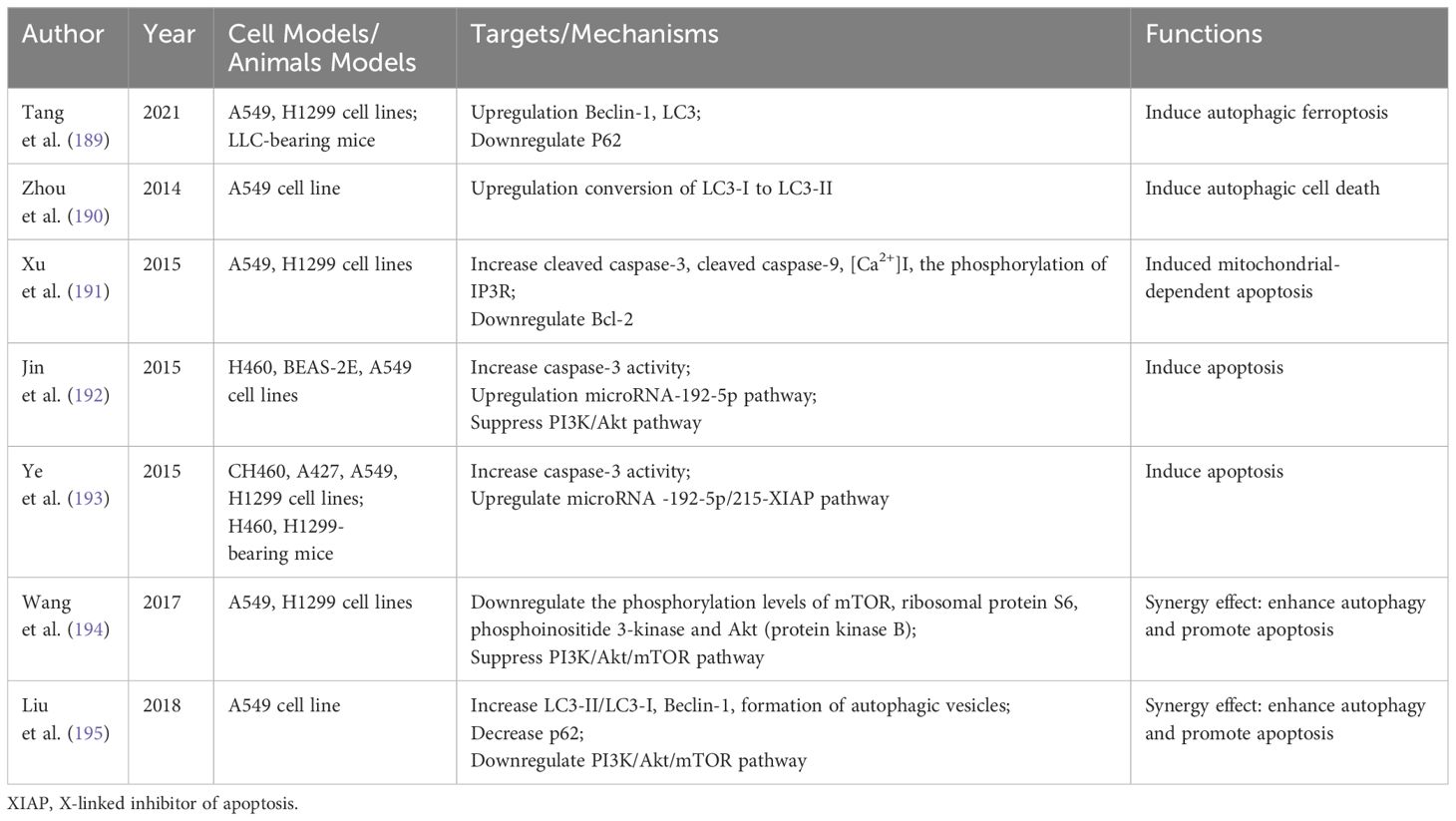
Table 4 Effects of Curcumin on autophagy and apoptosis in NSCLC.
5.2.1 Regulation of autophagy
Curcumin, a natural autophagy modulator, has the capacity to initiate autophagy in various cancers, including NSCLC (196). Tang et al. discovered that Curcumin caused mitochondrial membrane rupture and a decrease in mitochondrial cristae, enhanced autolysosome formation, increased the levels of Beclin-1 and LC3, reduced P62 levels, and induced ferroptosis by activating autophagy in NSCLC (189). Furthermore, a study revealed that hydrazinobenzoylcurcumin, a synthetic derivative of Curcumin, induces the conversion of LC3-I to LC3-II in A549 cells and enhances the fusion of autophagosomes with lysosomes, leading to autophagic cell death in cancer cells (190).
5.2.2 Regulation of apoptosis
Curcumin has been demonstrated to trigger apoptosis in NSCLC through various mechanisms. Treatment with Curcumin decreased Bcl-2 expression and increased inositol 1,4,5-trisphosphate receptor (IP3R) phosphorylation in a concentration-dependent manner. This elevation led to increased intracellular free calcium levels, with calcium overload subsequently inducing mitochondrial-dependent apoptosis in lung cancer cells (191). Additional studies have shown that Curcumin induces apoptosis in human NSCLC cells by increasing caspase-3 activity and upregulating the miR-192-5p signaling pathway (192, 193).
5.2.3 Regulation of autophagy and apoptosis
The PI3K/Akt/mTOR pathway is crucial for cell proliferation, apoptosis, autophagy, metabolism, and cell cycle progression, representing a key survival pathway often dysregulated in various human cancers (197). Multiple studies have demonstrated that Curcumin enhances autophagy and promotes apoptosis by downregulating the PI3K/Akt/mTOR pathway (194, 195).
5.3 Baicalein
Baicalein, a flavonoid extracted from the roots of Scutellaria baicalensis, has demonstrated substantial anti-tumor activity in recent in vivo and in vitro experiments, showing particular preventive and therapeutic effects on NSCLC (198). It has a wide anticancer spectrum, affects multiple cellular actions, and exhibits complex and diverse mechanisms, including the regulation of cell proliferation, metastasis, apoptosis, and autophagy (199). Baicalein is emerging as a novel anti-cancer agent for NSCLC treatment. Table 5 summarizes the effects of baicalein on autophagy and apoptosis in NSCLC.
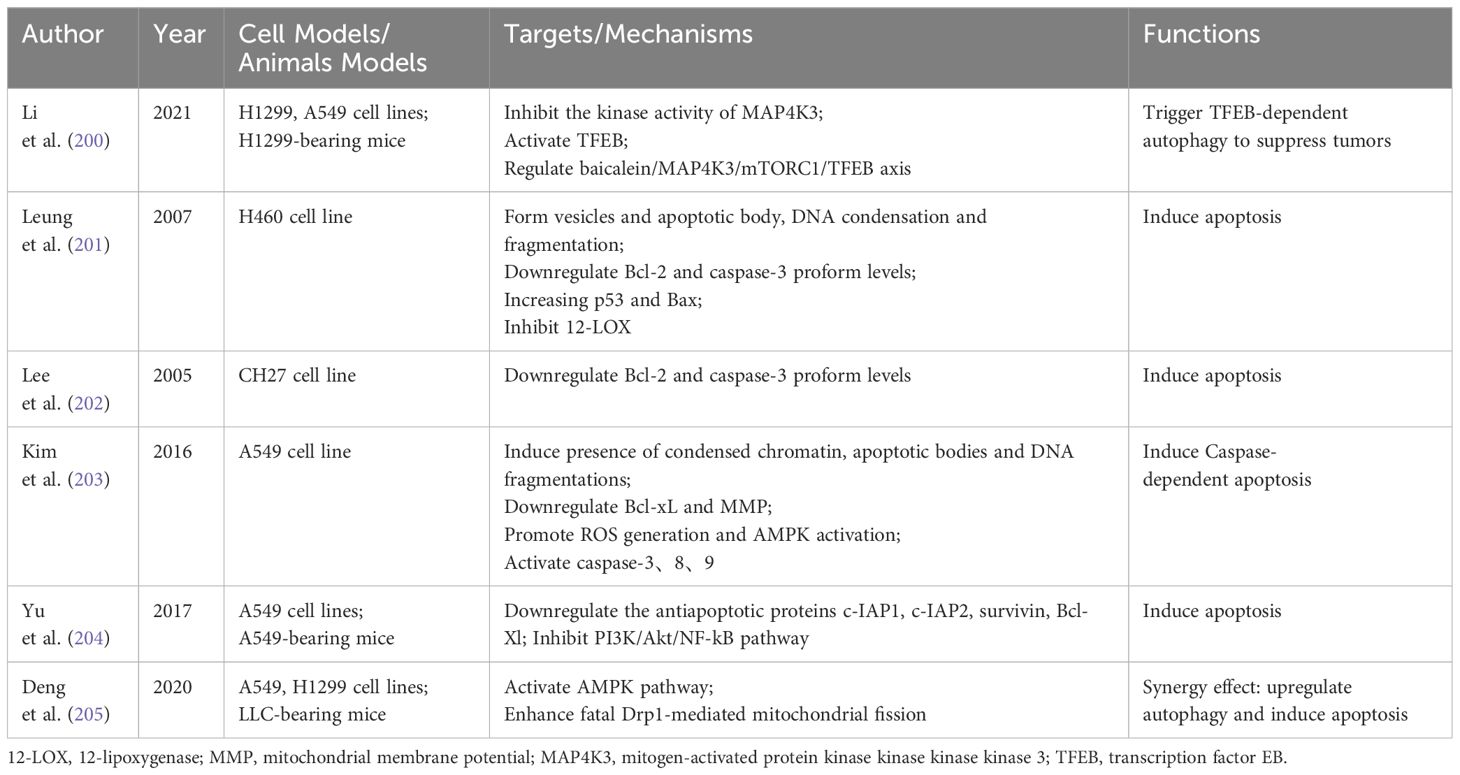
Table 5 Effects of baicalein on autophagy and apoptosis in NSCLC.
5.3.1 Regulation of autophagy
mitogen-activated protein kinase kinase kinase kinase 3 (MAP4K3) is a key regulator of mammalian cell growth and autophagy. Baicalein can directly bind to MAP4K3, leading to its inactivation. The degradation of MAP4K3 triggers transcription factor EB (TFEB)-dependent autophagy, causing arrest in lung cancer cell proliferation and effectively inhibiting tumor growth. Thus, baicalein regulates autophagy in NSCLC cells via the MAP4K3/mTORC1/TFEB axis (200).
5.3.2 Regulation of apoptosis
Bcl-2 and caspase-3 are crucial in determining cellular apoptosis. Studies indicate that baicalein induces apoptosis in NSCLC cells by downregulating Bcl-2 and Caspase-3 proform (201, 202). Another research found that baicalein’s apoptotic activity might be linked to a caspase-dependent cascade reaction (203). Additionally, baicalein promotes apoptosis in tumor cells by inhibiting NF-kB-mediated anti-apoptotic proteins, including c-IAP1, c-IAP2, survivin, and Bcl-xL, via the PI3K/Akt/NF-kB pathway. This inhibition significantly enhances NSCLC chemosensitivity to cisplatin (204).
5.3.3 Regulation of autophagy and apoptosis
Recent research indicates that baicalein modulates apoptosis and autophagy in NSCLC via the AMPK/Drp1/mitochondrial fission axis. Baicalein activates the AMPK pathway and intensifies fatal dynamin-related protein 1 (Drp1)-mediated mitochondrial fission. This process leads to the loss of mitochondrial membrane potential and the release of cytochrome c and apoptosis-inducing factor from mitochondria to cytoplasm. Such actions stimulate the combined anticancer effects of autophagy and apoptosis (205).
5.4 Ginsenoside
Ginseng is highly valued as both a medicinal and food ingredient, with ginsenosides being its most crucial constituents. Extensive research has focused on ginsenosides in cancer chemoprevention and therapy, identifying about 200 different ginsenosides in ginseng. Notably, ginsenosides Rk1, Rk3, Rg3, Rh2, and CK have been recognized for their potent anticancer effects and are currently at the forefront of scientific investigation (206). Their mechanisms are primarily associated with cell cycle arrest, apoptosis, proliferation, invasion, metastasis, angiogenesis, autophagy, and immune response (207). Table 6 summarizes the effects of ginsenosides on autophagy and apoptosis in NSCLC.
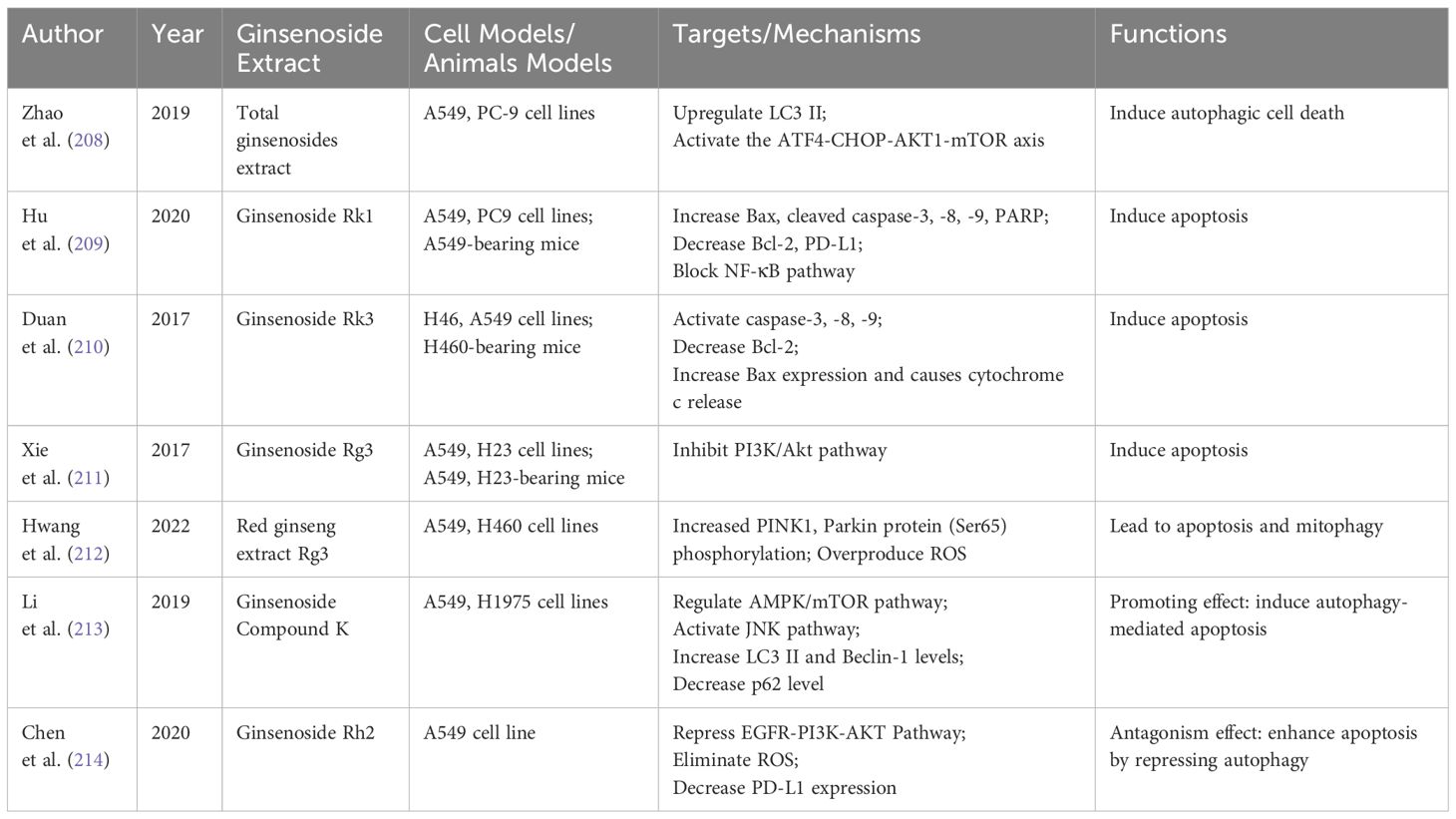
Table 6 Effects of ginsenoside on autophagy and apoptosis in NSCLC.
5.4.1 Regulation of autophagy
Autophagic cell death, classified as a type II programmed death, is an effective strategy for inducing tumor cell death in lung cancer. One study demonstrated that total ginsenosides extract (TGS) could enhance autophagy flux and induce autophagic cell death in NSCLC cells. This is achieved by activating endoplasmic reticulum stress, with further investigations indicating the involvement of the Activating Transcription Factor 4 (ATF4)- C/EBP-homologous protein (CHOP)-AKT1-mTOR axis in this process (208).
5.4.2 Regulation of apoptosis
Numerous studies have validated the role of ginsenosides in inducing apoptosis in NSCLC. A study revealed that ginsenoside Rk1 increases the protein expression of Bax, cleaved caspase-3, -8, -9, and poly ADP-ribose polymerase (PARP), while decreasing Bcl-2 expression. It also inhibits the NF-KB signaling pathway and reduces the high expression of PD-L1, thus significantly promoting apoptosis in NSCLC cells (209). Another research showed that ginsenoside Rk3 triggers apoptosis in H460 and A549 cells through death receptor-mediated, mitochondria-dependent pathways (210). Xie et al. reported that ginsenoside Rg3 significantly reduces cell viability and induces apoptosis by inhibiting the PI3K/Akt signaling pathway in A549 and H23 cells both in vitro and in vivo (211).
5.4.3 Regulation of autophagy and apoptosis
Red ginseng, a steamed form of ginseng, has been shown to promote lung cancer cell apoptosis and mitophagy. This is achieved through the activation of ROS and the PINK1-Parkin signaling pathway, as reported in a study focusing on Rg3-enriched red ginseng extract (212). Another study found that a metabolite of ginsenoside, Compound K (C-K), induces autophagy-mediated apoptosis in A549 and H1975 cells by regulating the AMPK/mTOR and JNK signaling pathways (213). Platinum-based drugs are first-line treatments for NSCLC, but cisplatin-induced productive autophagy can lead to drug resistance. Reversing this resistance is thus a key strategy in NSCLC treatment. Chen et al. discovered that ginsenoside Rh2 enhances cisplatin-induced apoptosis in A549 cells by repressing autophagy, significantly hindering NSCLC progression (214).
6 Problems and prospects
This paper initially introduced the mechanisms of autophagy and apoptosis, analyzed the crosstalk molecules between them, and then summarized their roles in NSCLC. Finally, we reviewed natural products that have been intensively studied against NSCLC targeting autophagy and apoptosis, and summarized in detail the four most retrieved representative drugs. Previous results and studies indicate that in NSCLC, natural products can regulate many target proteins on autophagy and apoptosis signaling pathways, inducing autophagic death and apoptosis in tumor cells. They also stimulate the synergistic and promotional effects of autophagy and apoptosis to co-induce tumor cell death. Additionally, natural products enhance apoptosis in tumor cells while reversing drug resistance by downregulating autophagy.
In conclusion, natural products effectively and reasonably regulate autophagy and apoptosis in NSCLC, exhibiting significant potential as a novel therapeutic strategy. Their sources are diverse, offering multiple targets, high safety, and effectiveness as anticancer agents. The data indicate that numerous natural products have been discovered through fundamental and preclinical studies for treatment of NSCLC. Despite this, there has not been a corresponding increase in the number of drugs used in the clinical setting, and possible reasons for this include the following six areas. Firstly, the discovery of new pharmacologically active prototypes in natural products is slow. Researchers should employ modern screening techniques to explore, extract, and identify natural products, assess their efficacy, safety, and bioavailability, and standardize promising anticancer natural products. Secondly, repositioning existing drugs for new applications is a growing trend in drug research and development, offering cost and time savings and resource optimization. Yet, the exploitation of new applications and anticancer domains for natural products is currently limited. Thirdly, Single natural product therapies have limited efficacy in treating and preventing cancer. literature on the combination of natural products is sparse. Researchers should focus on multi-molecule combination therapies using natural products, studying their synergistic anticancer effects. Additionally, natural products can serve as chemosensitizers, enhancing the effects of various chemotherapeutic agents and radiotherapy. And fourth, natural products exhibit side effects akin to other substances. For example, the side effects of camptothecin include bone marrow suppression, nausea, vomiting, stomatitis, abdominal pain, fatigue, diarrhea, peripheral neuropathy and hair loss. Therefore, the development of relevant technologies to aid in selecting the optimal therapeutic dose, design rational dosage forms, and alleviating their side effects and toxicity to the host is critical. Fifth, while numerous studies on natural products for cancer treatment have been documented, the majority have focused on in vitro experiments, with limited in vivo data and therefore restricted potential efficacy in human subjects. Researchers should carry out relevant in vivo experimental studies and gradually advance toward clinic trial. At last, Natural product active ingredients often face challenges like poor stability, low water solubility, rapid decomposition and metabolism, and low bioavailability, which can limit their clinical applications.
Excitingly, the advancement of novel drug delivery systems utilizing material technology and nanotechnology heralds a promising new research arena for anticancer drugs. The use of nanoscale materials such as nanoparticles, micelles, liposomes, microspheres, and microcapsules to physically or chemically encapsulate or link natural products and their derivatives for drug delivery presents a potential solution to overcome the shortcomings of natural products. Therapeutic strategies that integrate the benefits of modern delivery technologies with traditional medicines are poised to reshape the landscape of anticancer drugs in the coming decade.
Author contributions
PQ: Writing – original draft, Conceptualization, Writing – review & editing. QL: Writing – original draft, Conceptualization, Data curation. QZ: Data curation, Writing – original draft. RD: Data curation, Writing – original draft. YQ: Writing – review & editing, Conceptualization.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported in part by grants from the Program of Science and Technology of Traditional Chinese Medicine in Shandong Province [Q-2022078].
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA Cancer J Clin. (2022) 72:7–33. doi: 10.3322/caac.21708
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. (2021) 71:209–49. doi: 10.3322/caac.21660
3. Nasim F, Sabath BF, Eapen GA. Lung cancer. Med Clin North Am. (2019) 103:463–73. doi: 10.1016/j.mcna.2018.12.006
4. Lanuti M, Hong HJ, Ali S, Stock C, Temel J, Mathisen D, et al. Observations in lung cancer over multiple decades: an analysis of outcomes and cost at a single high-volume institution. Eur J Cardiothorac Surg. (2014) 46:254–61. doi: 10.1093/ejcts/ezt611
5. Lachgar A, Tazi MA, Afif M, Er-Raki A, Kebdani T, Benjaafar N. Lung cancer: Incidence and survival in Rabat, Morocco. Rev Epidemiol Sante Publique. (2016) 64:391–5. doi: 10.1016/j.respe.2016.02.012
6. Rachiglio AM, Esposito Abate R, Sacco A, Pasquale R, Fenizia F, Lambiase M, et al. Limits and potential of targeted sequencing analysis of liquid biopsy in patients with lung and colon carcinoma. Oncotarget. (2016) 7:66595–605. doi: 10.18632/oncotarget.10704
7. Tan CS, Gilligan D, Pacey S. Treatment approaches for EGFR-inhibitor-resistant patients with non-small-cell lung cancer. Lancet Oncol. (2015) 16:e447–59. doi: 10.1016/s1470-2045(15)00246-6
8. Herbst RS, Morgensztern D, Boshoff C. The biology and management of non-small cell lung cancer. Nature. (2018) 553:446–54. doi: 10.1038/nature25183
9. Wang M, Herbst RS, Boshoff C. Toward personalized treatment approaches for non-small-cell lung cancer. Nat Med. (2021) 27:1345–56. doi: 10.1038/s41591-021-01450-2
10. Pelos G, Riester M, Pal J, Myacheva K, Moneke I, Rotondo JC, et al. Fast proliferating and slowly migrating non-small cell lung cancer cells are vulnerable to decitabine and retinoic acid combinatorial treatment. Int J Cancer. (2024) 154:1029–42. doi: 10.1002/ijc.34783
11. Cukier P, Santini FC, Scaranti M, Hoff AO. Endocrine side effects of cancer immunotherapy. Endocr Relat Cancer. (2017) 24:T331–t347. doi: 10.1530/erc-17-0358
12. Kroschinsky F, Stölzel F, von Bonin S, Beutel G, Kochanek M, Kiehl M, et al. New drugs, new toxicities: severe side effects of modern targeted and immunotherapy of cancer and their management. Crit Care. (2017) 21:89. doi: 10.1186/s13054-017-1678-1
13. Peng F, Liao M, Qin R, Zhu S, Peng C, Fu L, et al. Regulated cell death (RCD) in cancer: key pathways and targeted therapies. Signal Transduct Target Ther. (2022) 7:286. doi: 10.1038/s41392-022-01110-y
14. Sen S. Programmed cell death: concept, mechanism and control. Biol Rev Camb Philos Soc. (1992) 67:287–319. doi: 10.1111/j.1469-185X.1992.tb00727.x
15. Chavez-Dominguez R, Perez-Medina M, Lopez-Gonzalez JS, Galicia-Velasco M, Aguilar-Cazares D. The double-edge sword of autophagy in cancer: From tumor suppression to pro-tumor activity. Front Oncol. (2020) 10:578418. doi: 10.3389/fonc.2020.578418
16. Gewirtz DA. The four faces of autophagy: implications for cancer therapy. Cancer Res. (2014) 74:647–51. doi: 10.1158/0008-5472.Can-13-2966
17. Li YJ, Lei YH, Yao N, Wang CR, Hu N, Ye WC, et al. Autophagy and multidrug resistance in cancer. Chin J Cancer. (2017) 36:52. doi: 10.1186/s40880-017-0219-2
18. White E. The role for autophagy in cancer. J Clin Invest. (2015) 125:42–6. doi: 10.1172/jci73941
19. Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol. (2020) 17:395–417. doi: 10.1038/s41571-020-0341-y
20. Sorice M. Crosstalk of autophagy and apoptosis. Cells. (2022) 11:1479. doi: 10.3390/cells11091479
21. Naeem A, Hu P, Yang M, Zhang J, Liu Y, Zhu W, et al. Natural products as anticancer agents: Current status and future perspectives. Molecules. (2022) 27:8367. doi: 10.3390/molecules27238367
22. Fontana F, Raimondi M, Marzagalli M, Di Domizio A, Limonta P. The emerging role of paraptosis in tumor cell biology: Perspectives for cancer prevention and therapy with natural compounds. Biochim Biophys Acta Rev Cancer. (2020) 1873:188338. doi: 10.1016/j.bbcan.2020.188338
23. Garg N, Luzzatto-Knaan T, Melnik AV, Caraballo-Rodríguez AM, Floros DJ, Petras D, et al. Natural products as mediators of disease. Nat Prod Rep. (2017) 34:194–219. doi: 10.1039/C6NP00063K
24. Cragg GM, Newman DJ. Plants as a source of anti-cancer agents. J Ethnopharmacol. (2005) 100:72–9. doi: 10.1016/j.jep.2005.05.011
25. Glick D, Barth S, Macleod KF. Autophagy: cellular and molecular mechanisms. J Pathol. (2010) 221:3–12. doi: 10.1002/path.2697
26. De Meyer GR, Grootaert MO, Michiels CF, Kurdi A, Schrijvers DM, Martinet W. Autophagy in vascular disease. Circ Res. (2015) 116:468–79. doi: 10.1161/circresaha.116.303804
27. Bolliet V, Labonne J, Olazcuaga L, Panserat S, Seiliez I. Modeling of autophagy-related gene expression dynamics during long term fasting in European eel (Anguilla Anguilla). Sci Rep. (2017) 7:17896. doi: 10.1038/s41598-017-18164-6
28. White E, Mehnert JM, Chan CS. Autophagy, metabolism, and cancer. Clin Cancer Res. (2015) 21:5037–46. doi: 10.1158/1078-0432.Ccr-15-0490
29. Levy JMM, Towers CG, Thorburn A. Targeting autophagy in cancer. Nat Rev Cancer. (2017) 17:528–42. doi: 10.1038/nrc.2017.53
30. Søreng K, Neufeld TP, Simonsen A. Membrane trafficking in autophagy. Int Rev Cell Mol Biol. (2018) 336:1–92. doi: 10.1016/bs.ircmb.2017.07.001
31. Nakatogawa H. Mechanisms governing autophagosome biogenesis. Nat Rev Mol Cell Biol. (2020) 21:439–58. doi: 10.1038/s41580-020-0241-0
32. Shaw RJ. LKB1 and AMP-activated protein kinase control of mTOR signaling and growth. Acta Physiol (Oxf). (2009) 196:65–80. doi: 10.1111/j.1748-1716.2009.01972.x
33. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. (2003) 115:577–90. doi: 10.1016/s0092-8674(03)00929-2
34. Azad MB, Chen Y, Henson ES, Cizeau J, McMillan-Ward E, Israels SJ, et al. Hypoxia induces autophagic cell death in apoptosis-competent cells through a mechanism involving BNIP3. Autophagy. (2008) 4:195–204. doi: 10.4161/auto.5278
35. Ambrosio S, Majello B. Autophagy roles in genome maintenance. Cancers (Basel). (2020) 12:1793. doi: 10.3390/cancers12071793
36. Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. (2011) 147:728–41. doi: 10.1016/j.cell.2011.10.026
37. Boukhalfa A, Roccio F, Dupont N, Codogno P, Morel E. When the autophagy protein ATG16L1 met the ciliary protein IFT20. Autophagy. (2021) 17:1791–3. doi: 10.1080/15548627.2021.1935004
38. Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. (2008) 456:259–63. doi: 10.1038/nature07416
39. Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat Cell Biol. (2007) 9:1102–9. doi: 10.1038/ncb1007-1102
40. Nedelsky NB, Todd PK, Taylor JP. Autophagy and the ubiquitin-proteasome system: collaborators in neuroprotection. Biochim Biophys Acta. (2008) 1782:691–9. doi: 10.1016/j.bbadis.2008.10.002
41. Duan Z, Shi Y, Lin Q, Hamaï A, Mehrpour M, Gong C. Autophagy-associated immunogenic modulation and its applications in cancer therapy. Cells. (2022) 11:2324. doi: 10.3390/cells11152324
42. Klionsky DJ, Abdelmohsen K, Zughaier SM. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. (2016) 12:1–222. doi: 10.1080/15548627.2015.1100356
43. Nakatogawa H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays Biochem. (2013) 55:39–50. doi: 10.1042/bse0550039
44. Duran A, Amanchy R, Linares JF, Joshi J, Abu-Baker S, Porollo A, et al. p62 is a key regulator of nutrient sensing in the mTORC1 pathway. Mol Cell. (2011) 44:134–46. doi: 10.1016/j.molcel.2011.06.038
45. Viret C, Faure M. Regulation of syntaxin 17 during autophagosome maturation. Trends Cell Biol. (2019) 29:1–3. doi: 10.1016/j.tcb.2018.10.003
46. Russell RC, Guan KL. The multifaceted role of autophagy in cancer. EMBO J. (2022) 41:e110031. doi: 10.15252/embj.2021110031
47. Ren Y, Cao L, Wang L, Zheng S, Zhang Q, Guo X, et al. Autophagic secretion of HMGB1 from cancer-associated fibroblasts promotes metastatic potential of non-small cell lung cancer cells via NFκB signaling. Cell Death Dis. (2021) 12:858. doi: 10.1038/s41419-021-04150-4
48. Bai Z, Peng Y, Ye X, Liu Z, Li Y, Ma L. Autophagy and cancer treatment: four functional forms of autophagy and their therapeutic applications. J Zhejiang Univ Sci B. (2022) 23:89–101. doi: 10.1631/jzus.B2100804
49. Lee JG, Shin JH, Shim HS, Lee CY, Kim DJ, Kim YS, et al. Autophagy contributes to the chemo-resistance of non-small cell lung cancer in hypoxic conditions. Respir Res. (2015) 16:138. doi: 10.1186/s12931-015-0285-4
50. Li C, Chen L, Song W, Peng B, Zhu J, Fang L. DICER activates autophagy and promotes cisplatin resistance in non-small cell lung cancer by binding with let-7i-5p. Acta Histochem. (2021) 123:151788. doi: 10.1016/j.acthis.2021.151788
51. Chen Y, Zhou X, Qiao J, Bao A. MiR-142-3p overexpression increases chemo-sensitivity of NSCLC by inhibiting HMGB1-mediated autophagy. Cell Physiol Biochem. (2017) 41:1370–82. doi: 10.1159/000467896
52. Liu MH, Liu F, Ng TB, Liu ZK. New fungal protein from Pleurotus ferulae lanzi induces AMPK-mediated autophagy and G1-phase cell cycle arrest in A549 lung cancer cells. Int J Biol Macromol. (2023) 244:125453. doi: 10.1016/j.ijbiomac.2023.125453
53. Xiang YC, Shen J, Si Y, Liu XW, Zhang L, Wen J, et al. a direct activator of AMPK, induces autophagy and exhibits therapeutic potential in non-small-cell lung cancer. Chin J Nat Med. (2021) 19:195–204. doi: 10.1016/s1875-5364(21)60021-3
54. Sharma K, Goehe RW, Di X, Hicks MA, Torti SV, Torti FM, et al. Gewirtz: A novel cytostatic form of autophagy in sensitization of non-small cell lung cancer cells to radiation by vitamin D and the vitamin D analog, EB 1089. Autophagy. (2014) 10:2346–61. doi: 10.4161/15548627.2014.993283
55. Patel NH, Xu J, Saleh T, Wu Y, Lima S, Gewirtz DA. Influence of nonprotective autophagy and the autophagic switch on sensitivity to cisplatin in non-small cell lung cancer cells. Biochem Pharmacol. (2020) 175:113896. doi: 10.1016/j.bcp.2020.113896
56. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer. (1972) 26:239–57. doi: 10.1038/bjc.1972.33
57. Green DR, Llambi F. Cell death signaling. Cold Spring Harb Perspect Biol. (2015) 7:235–56. doi: 10.1101/cshperspect.a006080
58. Peng YT, Chen P, Ouyang RY, Song L. Multifaceted role of prohibiting in cell survival and apoptosis. Apoptosis. (2015) 20:1135–49. doi: 10.1007/s10495-015-1143-z
59. Rudin CM, Thompson CB. Apoptosis and disease: regulation and clinical relevance of programmed cell death. Annu Rev Med. (1997) 48:267–81. doi: 10.1146/annurev.med.48.1.267
60. Mathew SJ, Haubert D, Krönke M, Leptin M. Looking beyond death: a morphogenetic role for the TNF signaling pathway. J Cell Sci. (2009) 122:1939–46. doi: 10.1242/jcs.044487
61. Galluzzi L, Kepp O, Kroemer G. Mitochondria: master regulators of danger signaling. Nat Rev Mol Cell Biol. (2012) 13:780–8. doi: 10.1038/nrm3479
62. Yuan S, Akey CW. Apoptosome structure, assembly, and procaspase activation. Structure. (2013) 21:501–15. doi: 10.1016/j.str.2013.02.024
63. Strasser A, Jost PJ, Nagata S. The many roles of FAS receptor signaling in the immune system. Immunity. (2009) 30:180–92. doi: 10.1016/j.immuni.2009.01.001
64. Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. (2001) 11:372–7. doi: 10.1016/s0962-8924(01)02064-5
65. Shen HM, Pervaiz S. TNF receptor superfamily-induced cell death: redox-dependent execution. FASEB J. (2006) 20:1589–98. doi: 10.1096/fj.05-5603rev
66. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. (2007) 35:495–516. doi: 10.1080/01926230701320337
67. Lavrik IN, Golks A, Krammer PH. Caspases: pharmacological manipulation of cell death. J Clin Invest. (2005) 115:2665–72. doi: 10.1172/jci26252
68. Brunelle JK, Letai A. Control of mitochondrial apoptosis by the Bcl-2 family. J Cell Sci. (2009) 122:437–41. doi: 10.1242/jcs.031682
69. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. (2014) 15:49–63. doi: 10.1038/nrm3722
70. Shamas-Din A, Kale J, Leber B, Andrews DW. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb Perspect Biol. (2013) 5:a008714. doi: 10.1101/cshperspect.a008714
71. Shang HS, Shih YL, Lee CH, Hsueh SC, Liu JY, Liao NC, et al. Sulforaphane-induced apoptosis in human leukemia HL-60 cells through extrinsic and intrinsic signal pathways and altering associated genes expression assayed by cDNA microarray. Environ Toxicol. (2017) 32:311–28. doi: 10.1002/tox.22237
72. Li K, van Delft MF, Dewson G. Too much death can kill you: inhibiting intrinsic apoptosis to treat disease. EMBO J. (2021) 40:e107341. doi: 10.15252/embj.2020107341
73. Taylor RC, Cullen SP, Martin SJ. Apoptosis: controlled demolition at the cellular level. Nat Rev Mol Cell Biol. (2008) 9:231–41. doi: 10.1038/nrm2312
74. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
75. Abdik H. Antineoplastic effects of erufosine on small cell and non-small cell lung cancer cells through induction of apoptosis and cell cycle arrest. Mol Biol Rep. (2022) 49:2963–71. doi: 10.1007/s11033-022-07117-6
76. Sheng H, Lv W, Zhu L, Wang L, Wang Z, Han J, et al. Liriopesides B induces apoptosis and cell cycle arrest in human non−small cell lung cancer cells. Int J Mol Med. (2020) 46:1039–50. doi: 10.3892/ijmm.2020.4645
77. Li Y, Yu H, Han F, Wang M, Luo Y, Guo X. Biochanin A induces S phase arrest and apoptosis in lung cancer cells. BioMed Res Int. (2018) 2018:3545376. doi: 10.1155/2018/3545376
78. Jia XB, Zhang Q, Xu L, Yao WJ, Wei L. Lotus leaf flavonoids induce apoptosis of human lung cancer A549 cells through the ROS/p38 MAPK pathway. Biol Res. (2021) 54:7. doi: 10.1186/s40659-021-00330-w
79. Liu X, Jiang Q, Liu H, Luo S. Vitexin induces apoptosis through mitochondrial pathway and PI3K/Akt/mTOR signaling in human non-small cell lung cancer A549 cells. Biol Res. (2019) 52:7. doi: 10.1186/s40659-019-0214-y
80. Kurtova AV, Xiao J, Mo Q, Pazhanisamy S, Krasnow R, Lerner SP, et al. Blocking PGE2-induced tumor repopulation abrogates bladder cancer chemoresistance. Nature. (2015) 517:209–13. doi: 10.1038/nature14034
81. Ichim G, Tait SW. A fate worse than death: apoptosis as an oncogenic process. Nat Rev Cancer. (2016) 16:539–48. doi: 10.1038/nrc.2016.58
82. Young MM, Kester M, Wang HG. Sphingolipids: regulators of crosstalk between apoptosis and autophagy. J Lipid Res. (2013) 54:5–19. doi: 10.1194/jlr.R031278
83. Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. (2006) 8:1124–32. doi: 10.1038/ncb1482
84. Jiang T, Harder B, Rojo de la Vega M, Wong PK, Chapman E, Zhang DD. p62 links autophagy and Nrf2 signaling. Free Radic Biol Med. (2015) 88:199–204. doi: 10.1016/j.freeradbiomed.2015.06.014
85. Fairlie WD, Tran S, Lee EF. Crosstalk between apoptosis and autophagy signaling pathways. Int Rev Cell Mol Biol. (2020) 352:115–58. doi: 10.1016/bs.ircmb.2020.01.003
86. Lin CF, Chien SY, Chen CL, Hsieh CY, Tseng PC, Wang YC. IFN-γ Induces mimic extracellular trap cell death in lung epithelial cells through autophagy-regulated DNA damage. J Interferon Cytokine Res. (2016) 36:100–12. doi: 10.1089/jir.2015.0011
87. Singh P, Ravanan P, Talwar P. Death associated protein kinase 1 (DAPK1): A regulator of apoptosis and autophagy. Front Mol Neurosci. (2016) 9:46. doi: 10.3389/fnmol.2016.00046
88. Elbadawy M, Usui T, Yamawaki H, Sasaki K. Novel functions of death-associated protein kinases through mitogen-activated protein kinase-related signals. Int J Mol Sci. (2018) 19:3031. doi: 10.3390/ijms19103031
89. Bialik S, Kimchi A. The death-associated protein kinases: structure, function, and beyond. Annu Rev Biochem. (2006) 75:189–210. doi: 10.1146/annurev.biochem.75.103004.142615
90. Li L, Tan J, Miao Y, Lei P, Zhang Q. ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol. (2015) 35:615–21. doi: 10.1007/s10571-015-0166-x
91. Scherz-Shouval R, Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends Cell Biol. (2007) 17:422–7. doi: 10.1016/j.tcb.2007.07.009
92. Li M, Gao P, Zhang J. Crosstalk between autophagy and apoptosis: Potential and emerging therapeutic targets for cardiac diseases. Int J Mol Sci. (2016) 17:332. doi: 10.3390/ijms17030332
93. Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, et al. Inhibition of death receptor signals by cellular FLIP. Nature. (1997) 388:190–5. doi: 10.1038/40657
94. Xu Z, Han X, Ou D, Liu T, Li Z, Jiang G, et al. Targeting PI3K/AKT/mTOR-mediated autophagy for tumor therapy. Appl Microbiol Biotechnol. (2020) 104:575–87. doi: 10.1007/s00253-019-10257-8
95. Lv Q, Zhong Z, Hu B, Yan S, Yan Y, Zhang J, et al. MicroRNA-3473b regulates the expression of TREM2/ULK1 and inhibits autophagy in inflammatory pathogenesis of Parkinson disease. J Neurochem. (2021) 157:599–610. doi: 10.1111/jnc.15299
96. Radoshevich L, Debnath J. ATG12-ATG3 and mitochondria. Autophagy. (2011) 7:109–11. doi: 10.4161/auto.7.1.13998
97. Radoshevich L, Murrow L, Chen N, Fernandez E, Roy S, Fung C, et al. ATG12 conjugation to ATG3 regulates mitochondrial homeostasis and cell death. Cell. (2010) 142:590–600. doi: 10.1016/j.cell.2010.07.018
98. Oral O, Oz-Arslan D, Itah Z, Naghavi A, Deveci R, Karacali S, et al. Cleavage of Atg3 protein by caspase-8 regulates autophagy during receptor-activated cell death. Apoptosis. (2012) 17:810–20. doi: 10.1007/s10495-012-0735-0
99. Gu W, Wan D, Qian Q, Yi B, He Z, Gu Y, et al. Ambra1 is an essential regulator of autophagy and apoptosis in SW620 cells: pro-survival role of Ambra1. PLoS One. (2014) 9:e90151. doi: 10.1371/journal.pone.0090151
100. Booth LA, Roberts JL, Dent P. The role of cell signaling in the crosstalk between autophagy and apoptosis in the regulation of tumor cell survival in response to sorafenib and neratinib. Semin Cancer Biol. (2020) 66:129–39. doi: 10.1016/j.semcancer.2019.10.013
101. Tsapras P, Nezis IP. Caspase involvement in autophagy. Cell Death Differ. (2017) 24:1369–79. doi: 10.1038/cdd.2017.43
102. Kong P, Zhu X, Geng Q, Xia L, Sun X, Chen Y, et al. The microRNA-423-3p-bim axis promotes cancer progression and activates oncogenic autophagy in gastric cancer. Mol Ther. (2017) 25:1027–37. doi: 10.1016/j.ymthe.2017.01.013
103. Su B, Wang XT, Sun YH, Long MY, Zheng J, Wu WH, et al. Ischemia/hypoxia inhibits cardiomyocyte autophagy and promotes apoptosis via the Egr-1/Bim/Beclin-1 pathway. J Geriatr Cardiol. (2020) 17:284–93. doi: 10.11909/j.issn.1671-5411.2020.05.004
104. Yang D, Okamura H, Teramachi J, Haneji T. Histone demethylase Jmjd3 regulates osteoblast apoptosis through targeting anti-apoptotic protein Bcl-2 and pro-apoptotic protein Bim. Biochim Biophys Acta. (2016) 1863:650–9. doi: 10.1016/j.bbamcr.2016.01.006
105. Nawrocki ST, Carew JS, Pino MS, Highshaw RA, Dunner K Jr., Huang P, et al. Bortezomib sensitizes pancreatic cancer cells to endoplasmic reticulum stress-mediated apoptosis. Cancer Res. (2005) 65:11658–66. doi: 10.1158/0008-5472.Can-05-2370
106. Qian X, He L, Yang J, Sun J, Peng X, Zhang Y, et al. UVRAG cooperates with cargo receptors to assemble the ER-phagy site. EMBO J. (2023) 42:e113625. doi: 10.15252/embj.2023113625
107. Oral O, Akkoc Y, Bayraktar O, Gozuacik D. Physiological and pathological significance of the molecular cross-talk between autophagy and apoptosis. Histol Histopathol. (2016) 31:479–98. doi: 10.14670/hh-11-714
108. Yuan Y, Zhang Y, Han L, Sun S, Shu Y. miR-183 inhibits autophagy and apoptosis in gastric cancer cells by targeting ultraviolet radiation resistance-associated gene. Int J Mol Med. (2018) 42:3562–70. doi: 10.3892/ijmm.2018.3871
109. Xu HD, Qin ZH. Beclin 1, bcl-2 and autophagy. Adv Exp Med Biol. (2019) 1206:109–26. doi: 10.1007/978-981-15-0602-4_5
110. Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. (1999) 402:672–6. doi: 10.1038/45257
111. Kang R, Zeh HJ, Lotze MT, Tang D. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. (2011) 18:571–80. doi: 10.1038/cdd.2010.191
112. Han T, Guo M, Gan M, Yu B, Tian X, Wang JB. TRIM59 regulates autophagy through modulating both the transcription and the ubiquitination of BECN1. Autophagy. (2018) 14:2035–48. doi: 10.1080/15548627.2018.1491493
113. Decuypere JP, Parys JB, Bultynck G. Regulation of the autophagic bcl-2/beclin 1 interaction. Cells. (2012) 1:284–312. doi: 10.3390/cells1030284
114. Pattingre S, Levine B. Bcl-2 inhibition of autophagy: a new route to cancer? Cancer Res. (2006) 66:2885–8. doi: 10.1158/0008-5472.Can-05-4412
115. Kaushal GP, Chandrashekar K, Juncos LA, Shah SV. Autophagy function and regulation in kidney disease. Biomolecules. (2020) 10:100. doi: 10.3390/biom10010100
116. Rubinstein AD, Kimchi A. Life in the balance - a mechanistic view of the crosstalk between autophagy and apoptosis. J Cell Sci. (2012) 125:5259–68. doi: 10.1242/jcs.115865
117. Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol Cell. (2011) 44:698–709. doi: 10.1016/j.molcel.2011.10.014
118. Nuñez G, Benedict MA, Hu Y, Inohara N. Caspases: the proteases of the apoptotic pathway. Oncogene. (1998) 17:3237–45. doi: 10.1038/sj.onc.1202581
119. Betin VM, Lane JD. Caspase cleavage of Atg4D stimulates GABARAP-L1 processing and triggers mitochondrial targeting and apoptosis. J Cell Sci. (2009) 122:2554–66. doi: 10.1242/jcs.046250
120. Wu H, Che X, Zheng Q, Wu A, Pan K, Shao A, et al. Caspases: a molecular switch node in the crosstalk between autophagy and apoptosis. Int J Biol Sci. (2014) 10:1072–83. doi: 10.7150/ijbs.9719
121. Riedl SJ, Shi Y. Molecular mechanisms of caspase regulation during apoptosis. Nat Rev Mol Cell Biol. (2004) 5:897–907. doi: 10.1038/nrm1496
122. Tait SW, Green DR. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat Rev Mol Cell Biol. (2010) 11:621–32. doi: 10.1038/nrm2952
123. Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. (2003) 83:731–801. doi: 10.1152/physrev.00029.2002
124. Lu Y, Li Z, Zhang S, Zhang T, Liu Y, Zhang L. Cellular mitophagy: Mechanism, roles in diseases and small molecule pharmacological regulation. Theranostics. (2023) 13:736–66. doi: 10.7150/thno.79876
125. Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation Res. (2005) 8:3–5. doi: 10.1089/rej.2005.8.3
126. Wirawan E, Vande Walle L, Kersse K, Cornelis S, Claerhout S, Vanoverberghe I, et al. Caspase-mediated cleavage of Beclin-1 inactivates Beclin-1-induced autophagy and enhances apoptosis by promoting the release of proapoptotic factors from mitochondria. Cell Death Dis. (2010) 1:e18. doi: 10.1038/cddis.2009.16
127. Kruse JP, Gu W. Modes of p53 regulation. Cell. (2009) 137:609–22. doi: 10.1016/j.cell.2009.04.050
128. Qin JJ, Li X, Hunt C, Wang W, Wang H, Zhang R. Natural products targeting the p53-MDM2 pathway and mutant p53: Recent advances and implications in cancer medicine. Genes Dis. (2018) 5:204–19. doi: 10.1016/j.gendis.2018.07.002
129. Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53-mediated tumor suppression. Nat Rev Cancer. (2014) 14:359–70. doi: 10.1038/nrc3711
130. Hu J, Cao J, Topatana W, Juengpanich S, Li S, Zhang B, et al. Targeting mutant p53 for cancer therapy: direct and indirect strategies. J Hematol Oncol. (2021) 14:157. doi: 10.1186/s13045-021-01169-0
131. Fridman JS, Lowe SW. Control of apoptosis by p53. Oncogene. (2003) 22:9030–40. doi: 10.1038/sj.onc.1207116
132. Yu X, Muñoz-Alarcón A, Ajayi A, Webling KE, Steinhof A, Langel Ü, et al. Inhibition of autophagy via p53-mediated disruption of ULK1 in a SCA7 polyglutamine disease model. J Mol Neurosci. (2013) 50:586–99. doi: 10.1007/s12031-013-0012-x
133. Gao W, Shen Z, Shang L, Wang X. Upregulation of human autophagy-initiation kinase ULK1 by tumor suppressor p53 contributes to DNA-damage-induced cell death. Cell Death Differ. (2011) 18:1598–607. doi: 10.1038/cdd.2011.33
134. Feng Z, Zhang H, Levine AJ, Jin S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc Natl Acad Sci U.S.A. (2005) 102:8204–9. doi: 10.1073/pnas.0502857102
135. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nat Rev Mol Cell Biol. (2007) 8:741–52. doi: 10.1038/nrm2239
136. Das S, Shukla N, Singh SS, Kushwaha S, Shrivastava R. Mechanism of interaction between autophagy and apoptosis in cancer. Apoptosis. (2021) 26:512–33. doi: 10.1007/s10495-021-01687-9
137. Zhang SW, Feng JN, Cao Y, Meng LP, Wang SL. Autophagy prevents autophagic cell death in Tetrahymena in response to oxidative stress. Dongwuxue Yanjiu. (2015) 36:167–73.
138. Chen Y, Li X, Guo L, Wu X, He C, Zhang S, et al. Combining radiation with autophagy inhibition enhances suppression of tumor growth and angiogenesis in esophageal cancer. Mol Med Rep. (2015) 12:1645–52. doi: 10.3892/mmr.2015.3623
139. Maiese K. The impact of aging and oxidative stress in metabolic and nervous system disorders: programmed cell death and molecular signal transduction crosstalk. Front Immunol. (2023) 14:1273570. doi: 10.3389/fimmu.2023.1273570
140. Athamneh K, Alneyadi A, Alsamri H, Alrashedi A, Palakott A, El-Tarabily KA, et al. Origanum majorana Essential Oil Triggers p38 MAPK-Mediated Protective Autophagy, Apoptosis, and Caspase-Dependent Cleavage of P70S6K in Colorectal Cancer Cells. Biomolecules. (2020) 10:412. doi: 10.3390/biom10030412
141. Denton D, Shravage B, Simin R, Mills K, Berry DL, Baehrecke EH, et al. Autophagy, not apoptosis, is essential for midgut cell death in Drosophila. Curr Biol. (2009) 19:1741–6. doi: 10.1016/j.cub.2009.08.042
142. Galluzzi L, Vicencio JM, Kepp O, Tasdemir E, Maiuri MC, Kroemer G. To die or not to die: that is the autophagic question. Curr Mol Med. (2008) 8:78–91. doi: 10.2174/156652408783769616
143. Pliyev BK, Menshikov M. Differential effects of the autophagy inhibitors 3-methyladenine and chloroquine on spontaneous and TNF-α-induced neutrophil apoptosis. Apoptosis. (2012) 17:1050–65. doi: 10.1007/s10495-012-0738-x
144. Park SH, Chi GY, Eom HS, Kim GY, Hyun JW, Kim WJ, et al. Role of autophagy in apoptosis induction by methylene chloride extracts of Mori cortex in NCI-H460 human lung carcinoma cells. Int J Oncol. (2012) 40:1929–40. doi: 10.3892/ijo.2012.1386
145. Fan J, Ren D, Wang J, Liu X, Zhang H, Wu M, et al. Bruceine D induces lung cancer cell apoptosis and autophagy via the ROS/MAPK signaling pathway in vitro and in vivo. Cell Death Dis. (2020) 11:126. doi: 10.1038/s41419-020-2317-3
146. Ito H, Daido S, Kanzawa T, Kondo S, Kondo Y. Radiation-induced autophagy is associated with LC3 and its inhibition sensitizes Malignant glioma cells. Int J Oncol. (2005) 26:1401–10. doi: 10.3892/ijo
147. Kepp O, Rajalingam K, Kimmig S, Rudel T. Bak and Bax are non-redundant during infection- and DNA damage-induced apoptosis. EMBO J. (2007) 26:825–34. doi: 10.1038/sj.emboj.7601533
148. He Q, Dong Y, Zhu Y, Ding Z, Zhang X, Wang Z, et al. TMEM100 induces cell death in non−small cell lung cancer via the activation of autophagy and apoptosis. Oncol Rep. (2021) 45:63. doi: 10.3892/or.2021.8014
149. Maheswari U, Ghosh K, Sadras SR. Licarin A induces cell death by activation of autophagy and apoptosis in non-small cell lung cancer cells. Apoptosis. (2018) 23:210–25. doi: 10.1007/s10495-018-1449-8
150. Zhao Y, Li K, Zhao B, Su L. HSP90 inhibitor DPB induces autophagy and more effectively apoptosis in A549 cells combined with autophagy inhibitors. In Vitro Cell Dev Biol Anim. (2019) 55:349–54. doi: 10.1007/s11626-019-00327-6
151. Shimizu S, Yoshida T, Tsujioka M, Arakawa S. Autophagic cell death and cancer. Int J Mol Sci. (2014) 15:3145–53. doi: 10.3390/ijms15023145
152. Saita S, Shirane M, Nakayama KI. Selective escape of proteins from the mitochondria during mitophagy. Nat Commun. (2013) 4:1410. doi: 10.1038/ncomms2400
153. Zhao Z, Xia G, Li N, Su R, Chen X, Zhong L. Autophagy inhibition promotes bevacizumab-induced apoptosis and proliferation inhibition in colorectal cancer cells. J Cancer. (2018) 9:3407–16. doi: 10.7150/jca.24201
154. Shi S, Tan P, Yan B, Gao R, Zhao J, Wang J, et al. ER stress and autophagy are involved in the apoptosis induced by cisplatin in human lung cancer cells. Oncol Rep. (2016) 35:2606–14. doi: 10.3892/or.2016.4680
155. Ge X, Zhang Y, Huang F, Wu Y, Pang J, Li X, et al. EGFR tyrosine kinase inhibitor Almonertinib induces apoptosis and autophagy mediated by reactive oxygen species in non-small cell lung cancer cells. Hum Exp Toxicol. (2021) 40:S49–s62. doi: 10.1177/09603271211030554
156. Gong H. Pinocembrin suppresses proliferation and enhances apoptosis in lung cancer cells in vitro by restraining autophagy. Bioengineered. (2021) 12:6035–44. doi: 10.1080/21655979.2021.1972779
157. Mu N, Wang Y, Li X, Du Z, Wu Y, Su M, et al. Crotonylated BEX2 interacts with NDP52 and enhances mitophagy to modulate chemotherapeutic agent-induced apoptosis in non-small-cell lung cancer cells. Cell Death Dis. (2023) 14:645. doi: 10.1038/s41419-023-06164-6
158. Mali SB. Cancer treatment: Role of natural products. Time to have serious rethink. Oral Oncol Rep. (2023) 6:100040. doi: 10.1016/j.oor.2023.100040
159. Ali Abdalla YO, Subramaniam B, Nyamathulla S, Shamsuddin N, Arshad NM, Mun KS, et al. Natural products for cancer therapy: A review of their mechanism of actions and toxicity in the past decade. J Trop Med. (2022) 2022:1–20. doi: 10.1155/2022/5794350
160. Wen T, Song L, Hua S. Perspectives and controversies regarding the use of natural products for the treatment of lung cancer. Cancer Med. (2021) 10:2396–422. doi: 10.1002/cam4.3660
161. Wang X, Wang D, Zhao Y. Effect and mechanism of resveratrol on the apoptosis of lung adenocarcinoma cell line A549. Cell Biochem Biophys. (2015) 73:527–31. doi: 10.1007/s12013-015-0696-3
162. Zhang W, Wang X, Chen T. Resveratrol induces apoptosis via a Bak-mediated intrinsic pathway in human lung adenocarcinoma cells. Cell Signal. (2012) 24:1037–46. doi: 10.1016/j.cellsig.2011.12.025
163. Fan Y, Li J, Yang Y, Zhao X, Liu Y, Jiang Y, et al. Resveratrol modulates the apoptosis and autophagic death of human lung adenocarcinoma A549 cells via a p53−dependent pathway: Integrated bioinformatics analysis and experimental validation. Int J Oncol. (2020) 57:925–38. doi: 10.3892/ijo.2020.5107
164. Li W, Li C, Ma L, Jin F. Resveratrol inhibits viability and induces apoptosis in the small−cell lung cancer H446 cell line via the PI3K/Akt/c−Myc pathway. Oncol Rep. (2020) 44:1821–30. doi: 10.3892/or.2020.7747
165. Li J, Fan Y, Zhang Y, Liu Y, Yu Y, Ma M. Resveratrol induces autophagy and apoptosis in non-small-cell lung cancer cells by activating the NGFR-AMPK-mTOR pathway. Nutrients. (2022) 14:2413. doi: 10.3390/nu14122413
166. Huang Y, Chen Z, Wang Y, Ba X, Huang Y, Shen P, et al. Triptolide exerts an anti-tumor effect on non−small cell lung cancer cells by inhibiting activation of the IL−6/STAT3 axis. Int J Mol Med. (2019) 44:291–300. doi: 10.3892/ijmm.2019.4197
167. Ren T, Tang YJ, Wang MF, Wang HS, Liu Y, Qian X, et al. Triptolide induces apoptosis through the calcium/calmodulin−dependent protein kinase kinaseβ/AMP−activated protein kinase signaling pathway in non−small cell lung cancer cells. Oncol Rep. (2020) 44:2288–96. doi: 10.3892/or.2020.7763
168. Wang XR, Jiang ZB, Xu C, Meng WY, Liu P, Zhang YZ, et al. Andrographolide suppresses non-small-cell lung cancer progression through induction of autophagy and antitumor immune response. Pharmacol Res. (2022) 179:106198. doi: 10.1016/j.phrs.2022.106198
169. Yang ES, Do Y, Cheon SY, Kim B, Ling J, Cho MK, et al. Andrographolide suppresses aerobic glycolysis and induces apoptotic cell death by inhibiting pyruvate dehydrogenase kinase 1 expression. Oncol Rep. (2023) 49:72. doi: 10.3892/or.2023.8509
170. Wang G, Li X, Huang F, Zhao J, Ding H, Cunningham C, et al. Antitumor effect of beta-elemene in non-small-cell lung cancer cells is mediated via induction of cell cycle arrest and apoptotic cell death. Cell Mol Life Sci. (2005) 62:881–93. doi: 10.1007/s00018-005-5017-3
171. Liu J, Hu XJ, Jin B, Qu XJ, Hou KZ, Liu YP. β-Elemene induces apoptosis as well as protective autophagy in human non-small-cell lung cancer A549 cells. J Pharm Pharmacol. (2012) 64:146–53. doi: 10.1111/j.2042-7158.2011.01371.x
172. Ye L, Zhou J, Zhao W, Jiao P, Ren G, Wang S. Gambogic acid-induced autophagy in nonsmall cell lung cancer NCI-H441 cells through a reactive oxygen species pathway. J Cancer Res Ther. (2018) 14:S942–s947. doi: 10.4103/0973-1482.206866
173. Huang T, Zhang H, Wang X, Xu L, Jia J, Zhu X. Gambogenic acid inhibits the proliferation of small−cell lung cancer cells by arresting the cell cycle and inducing apoptosis. Oncol Rep. (2019) 41:1700–6. doi: 10.3892/or.2018.6950
174. Chiu TL, Su CC. Tanshinone IIA induces apoptosis in human lung cancer A549 cells through the induction of reactive oxygen species and decreasing the mitochondrial membrane potential. Int J Mol Med. (2010) 25:231–6. doi: 10.3892/ijmm_00000335
175. Zhang J, Wang J, Jiang JY, Liu SD, Fu K, Liu HY. Tanshinone IIA induces cytochrome c-mediated caspase cascade apoptosis in A549 human lung cancer cells via the JNK pathway. Int J Oncol. (2014) 45:683–90. doi: 10.3892/ijo.2014.2471
176. Lee HZ. Protein kinase C involvement in aloe-emodin- and emodin-induced apoptosis in lung carcinoma cell. Br J Pharmacol. (2001) 134:1093–103. doi: 10.1038/sj.bjp.0704342
177. Su J, Yan Y, Qu J, Xue X, Liu Z, Cai H. Emodin induces apoptosis of lung cancer cells through ER stress and the TRIB3/NF-κB pathway. Oncol Rep. (2017) 37:1565–72. doi: 10.3892/or.2017.5428
178. Dai X, Zhang X, Chen W, Chen Y, Zhang Q, Mo S, et al. Dihydroartemisinin: A potential natural anticancer drug. Int J Biol Sci. (2021) 17:603–22. doi: 10.7150/ijbs.50364
179. Liu X, Wu J, Fan M, Shen C, Dai W, Bao Y, et al. Novel dihydroartemisinin derivative DHA-37 induces autophagic cell death through upregulation of HMGB1 in A549 cells. Cell Death Dis. (2018) 9:1048. doi: 10.1038/s41419-018-1006-y
180. Wu S, Li Z, Li H, Liao K. Dihydroartemisinin reduces irradiation-induced mitophagy and radioresistance in lung cancer A549 cells via CIRBP inhibition. Life (Basel). (2022) 12:1129. doi: 10.3390/life12081129
181. Zhang Q, Yi H, Yao H, Lu L, He G, Wu M, et al. Artemisinin derivatives inhibit non-small cell lung cancer cells through induction of ROS-dependent apoptosis/ferroptosis. J Cancer. (2021) 12:4075–85. doi: 10.7150/jca.57054
182. Yan X, Li P, Zhan Y, Qi M, Liu J, An Z, et al. Dihydroartemisinin suppresses STAT3 signaling and Mcl-1 and Survivin expression to potentiate ABT-263-induced apoptosis in Non-small Cell Lung Cancer cells harboring EGFR or RAS mutation. Biochem Pharmacol. (2018) 150:72–85. doi: 10.1016/j.bcp.2018.01.031
183. Liao K, Li J, Wang Z. Dihydroartemisinin inhibits cell proliferation via AKT/GSK3β/cyclinD1 pathway and induces apoptosis in A549 lung cancer cells. Int J Clin Exp Pathol. (2014) 7:8684–91.
184. Li Y, Xiao X, Wang H, Zhou Q, Jin Z, Zhang Y, et al. Integrating network pharmacology and experimental models to investigate the mechanisms of dihydroartemisinin in preventing NSCLC progression via mTOR/HIF-1α signaling. Eur J Pharmacol. (2021) 909:174411. doi: 10.1016/j.ejphar.2021.174411
185. Lai XY, Shi YM, Zhou MM. Dihydroartemisinin enhances gefitinib cytotoxicity against lung adenocarcinoma cells by inducing ROS-dependent apoptosis and ferroptosis. Kaohsiung J Med Sci. (2023) 39:699–709. doi: 10.1002/kjm2.12684
186. Chang H, Zou Z. Targeting autophagy to overcome drug resistance: further developments. J Hematol Oncol. (2020) 13:159. doi: 10.1186/s13045-020-01000-2
187. Hao M, Chu Y, Lei J, Yao Z, Wang P, Chen Z, et al. Pharmacological mechanisms and clinical applications of curcumin: update. Aging Dis. (2023) 14:716–49. doi: 10.14336/ad.2022.1101
188. Li Z, Li D, Chen R, Gao S, Xu Z, Li N. Cell death regulation: A new way for natural products to treat osteoporosis. Pharmacol Res. (2023) 187:106635. doi: 10.1016/j.phrs.2022.106635
189. Tang X, Ding H, Liang M, Chen X, Yan Y, Wan N, et al. Curcumin induces ferroptosis in non-small-cell lung cancer via activating autophagy. Thorac Cancer. (2021) 12:1219–30. doi: 10.1111/1759-7714.13904
190. Zhou GZ, Zhang SN, Zhang L, Sun GC, Chen XB. A synthetic curcumin derivative hydrazinobenzoylcurcumin induces autophagy in A549 lung cancer cells. Pharm Biol. (2014) 52:111–6. doi: 10.3109/13880209.2013.816971
191. Xu X, Chen D, Ye B, Zhong F, Chen G. Curcumin induces the apoptosis of non-small cell lung cancer cells through a calcium signaling pathway. Int J Mol Med. (2015) 35:1610–6. doi: 10.3892/ijmm.2015.2167
192. Jin H, Qiao F, Wang Y, Xu Y, Shang Y. Curcumin inhibits cell proliferation and induces apoptosis of human non-small cell lung cancer cells through the upregulation of miR-192-5p and suppression of PI3K/Akt signaling pathway. Oncol Rep. (2015) 34:2782–9. doi: 10.3892/or.2015.4258
193. Ye M, Zhang J, Zhang J, Miao Q, Yao L, Zhang J. Curcumin promotes apoptosis by activating the p53-miR-192-5p/215-XIAP pathway in non-small cell lung cancer. Cancer Lett. (2015) 357:196–205. doi: 10.1016/j.canlet.2014.11.028
194. Wang A, Wang J, Zhang S, Zhang H, Xu Z, Li X. Curcumin inhibits the development of non-small cell lung cancer by inhibiting autophagy and apoptosis. Exp Ther Med. (2017) 14:5075–80. doi: 10.3892/etm.2017.5172
195. Liu F, Gao S, Yang Y, Zhao X, Fan Y, Ma W, et al. Antitumor activity of curcumin by modulation of apoptosis and autophagy in human lung cancer A549 cells through inhibiting PI3K/Akt/mTOR pathway. Oncol Rep. (2018) 39:1523–31. doi: 10.3892/or.2018.6188
196. Shakeri A, Cicero AFG, Panahi Y, Mohajeri M, Sahebkar A. Curcumin: A naturally occurring autophagy modulator. J Cell Physiol. (2019) 234:5643–54. doi: 10.1002/jcp.27404
197. Gills JJ, Lopiccolo J, Dennis PA. Nelfinavir, a new anti-cancer drug with pleiotropic effects and many paths to autophagy. Autophagy. (2008) 4:107–9. doi: 10.4161/auto.5224
198. Chandrashekar N, Pandi A. Baicalein: A review on its anti-cancer effects and mechanisms in lung carcinoma. J Food Biochem. (2022) 46:e14230. doi: 10.1111/jfbc.14230
199. Morshed A, Paul S, Hossain A, Basak T, Hossain MS, Hasan MM, et al. Baicalein as promising anticancer agent: A comprehensive analysis on molecular mechanisms and therapeutic perspectives. Cancers (Basel). (2023) 15:2128. doi: 10.3390/cancers15072128
200. Li J, Yan L, Luo J, Tong L, Gao Y, Feng W, et al. Baicalein suppresses growth of non-small cell lung carcinoma by targeting MAP4K3. BioMed Pharmacother. (2021) 133:110965. doi: 10.1016/j.biopha.2020.110965
201. Leung HW, Yang WH, Lai MY, Lin CJ, Lee HZ. Inhibition of 12-lipoxygenase during baicalein-induced human lung nonsmall carcinoma H460 cell apoptosis. Food Chem Toxicol. (2007) 45:403–11. doi: 10.1016/j.fct.2006.08.021
202. Lee HZ, Leung HW, Lai MY, Wu CH. Baicalein induced cell cycle arrest and apoptosis in human lung squamous carcinoma CH27 cells. Anticancer Res. (2005) 25:959–64.
203. Kim HJ, Park C, Han MH, Hong SH, Kim GY, Hong SH, et al. Baicalein induces caspase-dependent apoptosis associated with the generation of ROS and the activation of AMPK in human lung carcinoma A549 cells. Drug Dev Res. (2016) 77:73–86. doi: 10.1002/ddr.21298
204. Yu M, Qi B, Xiaoxiang W, Xu J, Liu X. Baicalein increases cisplatin sensitivity of A549 lung adenocarcinoma cells via PI3K/Akt/NF-κB pathway. BioMed Pharmacother. (2017) 90:677–85. doi: 10.1016/j.biopha.2017.04.001
205. Deng X, Liu J, Liu L, Sun X, Huang J, Dong J. Drp1-mediated mitochondrial fission contributes to baicalein-induced apoptosis and autophagy in lung cancer via activation of AMPK signaling pathway. Int J Biol Sci. (2020) 16:1403–16. doi: 10.7150/ijbs.41768
206. Wang L, Zhang Y, Song Z, Liu Q, Fan D, Song X. Ginsenosides: a potential natural medicine to protect the lungs from lung cancer and inflammatory lung disease. Food Funct. (2023) 14:9137–66. doi: 10.1039/D3FO02482B
207. Li X, Chu S, Lin M, Gao Y, Liu Y, Yang S, et al. Anticancer property of ginsenoside Rh2 from ginseng. Eur J Med Chem. (2020) 203:112627. doi: 10.1016/j.ejmech.2020.112627
208. Zhao M, Chen Q, Xu W, Wang H, Che Y, Wu M, et al. Total ginsenosides extract induce autophagic cell death in NSCLC cells through activation of endoplasmic reticulum stress. J Ethnopharmacol. (2019) 243:112093. doi: 10.1016/j.jep.2019.112093
209. Hu M, Yang J, Qu L, Deng X, Duan Z, Fu R, et al. Ginsenoside Rk1 induces apoptosis and downregulates the expression of PD-L1 by targeting the NF-κB pathway in lung adenocarcinoma. Food Funct. (2020) 11:456–71. doi: 10.1039/C9FO02166C
210. Duan Z, Deng J, Dong Y, Zhu C, Li W, Fan D. Anticancer effects of ginsenoside Rk3 on non-small cell lung cancer cells: in vitro and in vivo. Food Funct. (2017) 8:3723–36. doi: 10.1039/C7FO00385D
211. Xie Q, Wen H, Zhang Q, Zhou W, Lin X, Xie D, et al. Inhibiting PI3K-AKt signaling pathway is involved in antitumor effects of ginsenoside Rg3 in lung cancer cell. BioMed Pharmacother. (2017) 85:16–21. doi: 10.1016/j.biopha.2016.11.096
212. Hwang SK, Jeong YJ, Cho HJ, Park YY, Song KH, Chang YC. Rg3-enriched red ginseng extract promotes lung cancer cell apoptosis and mitophagy by ROS production. J Ginseng Res. (2022) 46:138–46. doi: 10.1016/j.jgr.2021.05.005
213. Li C, Dong Y, Wang L, Xu G, Yang Q, Tang X, et al. Ginsenoside metabolite compound K induces apoptosis and autophagy in non-small cell lung cancer cells via AMPK-mTOR and JNK pathways. Biochem Cell Biol. (2019) 97:406–14. doi: 10.1139/bcb-2018-0226
Keywords: autophagy, apoptosis, crosstalk, therapeutics, natural products, non-small cell lung cancer (NSCLC)
Citation: Qin P, Li Q, Zu Q, Dong R and Qi Y (2024) Natural products targeting autophagy and apoptosis in NSCLC: a novel therapeutic strategy. Front. Oncol. 14:1379698. doi: 10.3389/fonc.2024.1379698
Received: 31 January 2024; Accepted: 18 March 2024;
Published: 02 April 2024.
Edited by:
Daniel P. Bezerra, Oswaldo Cruz Foundation (FIOCRUZ), BrazilReviewed by:
John Charles Rotondo, University of Ferrara, ItalyAhmed M. Elshazly, Kafrelsheikh University, Egypt
Xi Guangmin, Qilu Normal University, China
Copyright © 2024 Qin, Li, Zu, Dong and Qi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuanfu Qi, bTEzNzkzMTMyMzE3QDE2My5jb20=