 Sawyer Bawek
Sawyer Bawek Sayuri Gurusinghe1
Sayuri Gurusinghe1 Matthew Burwinkel
Matthew Burwinkel Amanda Przespolewski
Amanda Przespolewski
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 04 December 2024
Sec. Hematologic Malignancies
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1374963
This article is part of the Research Topic Immunotherapy and Cell Therapy for Patients with Relapsed and Refractory Acute Myeloid Leukemia View all 6 articles
Acute myeloid leukemia (AML) is a severe hematological malignancy with poor outcomes, particularly in older adults. Traditional treatment options like high-dose chemotherapy often lead to refractory or relapsed AML, with even worse outcomes. New therapies for relapsed and refractory AML are needed, and this review explores the most recent advancements in immunotherapy in AML. Checkpoint Inhibitors utilizing innate or adaptive immune targeting have shown potential to improve AML outcomes when combined with hypomethylating agents and chemotherapy. The use of adoptive cell therapy in AML demonstrates promising early data, however, there is a need for better target selection. Although early in development, both vaccine therapy as well as stimulator of interferon genes (STING) agonists have potential to enhance the innate immune response to overcome AML’s immune evasion. Immunotherapy has become a promising approach for AML treatment, especially in refractory and relapsed AML, especially in patients who are not eligible for allogeneic stem cell transplants. Future research should focus on a deeper understanding of the immune microenvironment to identify the most critical targets for optimization, as well as personalized therapeutic combination strategies. Here we present a comprehensive overview of the recent developments in immunotherapy for relapsed and refractory AML.
Acute myeloid leukemia (AML) is an aggressive hematological malignancy arising from an immature myeloid progenitor primarily impacting older adults with inferior outcomes (1). The mainstay of first-line treatment for AML has been chemotherapy (2). High-dose chemotherapy, commonly known as 7 + 3, has been the standard of care for AML for years, but it is associated with high mortality and morbidity. The current regimen of 7 + 3 induction chemotherapy is then followed by consolidation chemotherapy and/or allogeneic hematopoietic stem cell transplantation (HSCT). However, 30-40% of these patients will relapse. In patients who relapse or never respond to therapy, long-term survival is only seen in approximately 30% of patients (3). Venetoclax (Ven) has an essential role in AML as it is now approved in the front-line setting in combination with azacitidine (Aza), decitabine (Dec), or low-dose cytarabine (LDAC) for patients aged 75 years and older, or those with significant comorbidities. Elderly patients have a high induction-related mortality rate of 15-30%, mainly due to infectious complications (4–7). The present approaches highlight the need for novel therapeutic approaches, including immunotherapy. Here, we review recent developments in immunotherapy for AML. Topics reviewed include checkpoint inhibitor therapy, antibody-drug conjugates, chimeric antigen receptor-engineered T cells (CAR-T), T-cell receptor-engineered T cells (TCR-T), and dendritic cell (DC) vaccines (Figure 1).
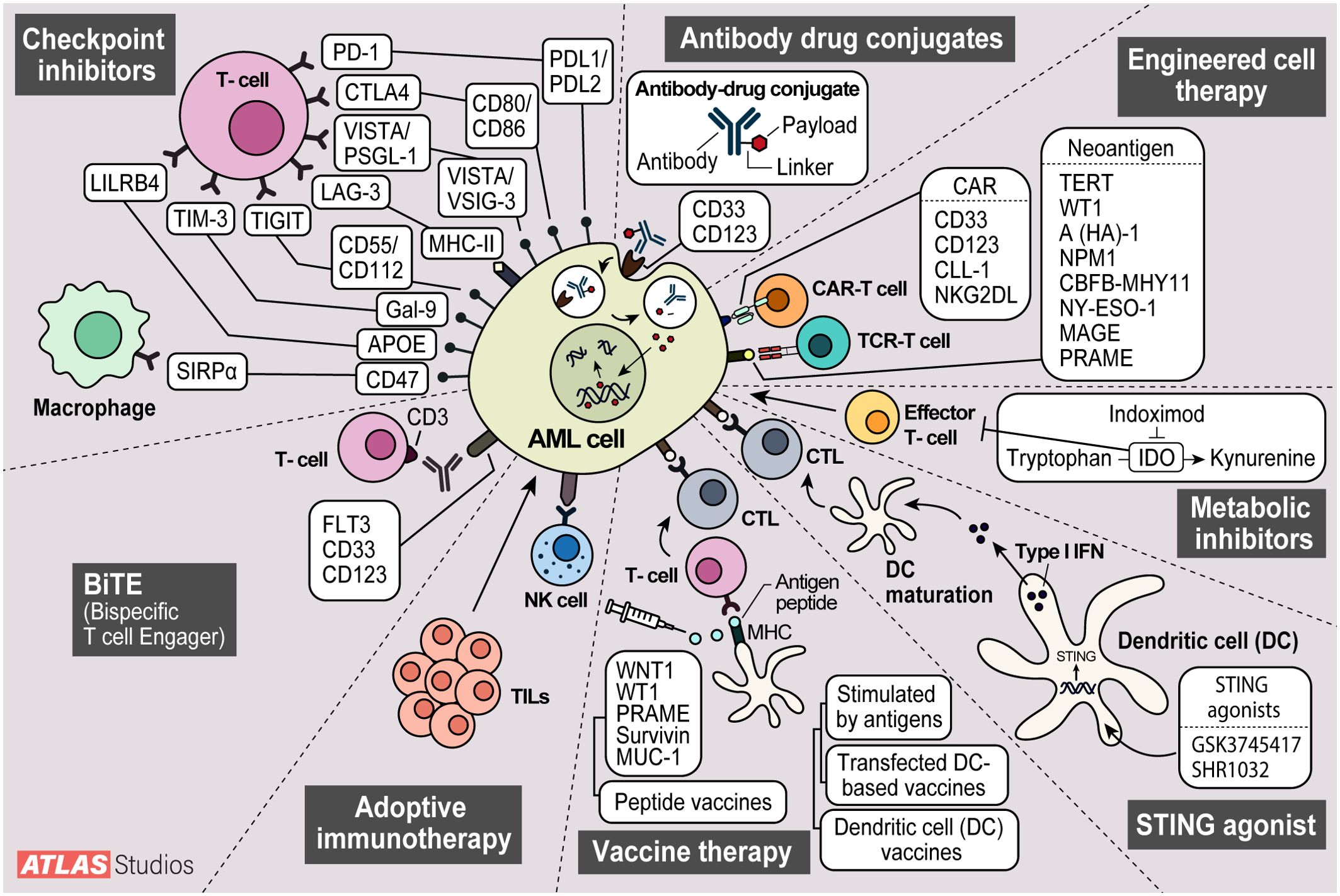
Figure 1. Modes of action of current AML therapy being investigated. 1. Checkpoint inhibitors stop cell cycle progression. 2. Antibody drug conjugates deliver toxic payloads to cancer cells. 3. Engineered T-cell therapy modifies t-cells to target cancer cells.4. Metabolic inhibitors inhibit metabolite degradation leading to build up in cancer cells and growth inhibition. 5. STING agonists elicit increased immune responses with increases in interferon. 6. Vaccines sensitize host immune cells to cancer antigens leading to increased immune surveillance. 7. Adoptive cell therapy enhances natural killer and tumor infiltrating lymphocytes anti-tumor effect. 8. Bispecific T-cell Engagers (BiTEs) offer another way to engage host t-cells with cancer cells.
Immune checkpoint inhibitors are regulatory molecules used to induce self-tolerance and prevent autoimmunity. These molecules are highly expressed in T cells. Studies have shown that AML cells express the ligands to these immune checkpoints to evade immune surveillance (8).
A knowledge of the microenvironment in AML is essential for understanding mechanisms of evading immune surveillance and therapeutic potential of targeting specific microenvironment components (9). Guo and colleagues analyzed major immune cell subsets in the AML bone marrow microenvironment and were able to predict prognosis in AML patients (9). The study analyzed subsets of mast cells, dendritic cells, monocytes and macrophages, and T cells. Exhausted T cells and immunosuppressive T cells like regulatory T cells (Treg) and other T cell subsets can be used as targets of anti-CTLA4, anti-PD1, and anti-CD25 therapies (9).
One of the most well-studied immune checkpoint axis is the programmed-cell-death-ligand 1 (PD-L1), which binds to PD-1 receptors expressed on T cells and thereby produces a negative costimulatory signal to attenuate T-cell activation (10). Several other immune checkpoint ligands have been recently explored as alternative targets for AML, and are reviewed below (Table 1) (11).
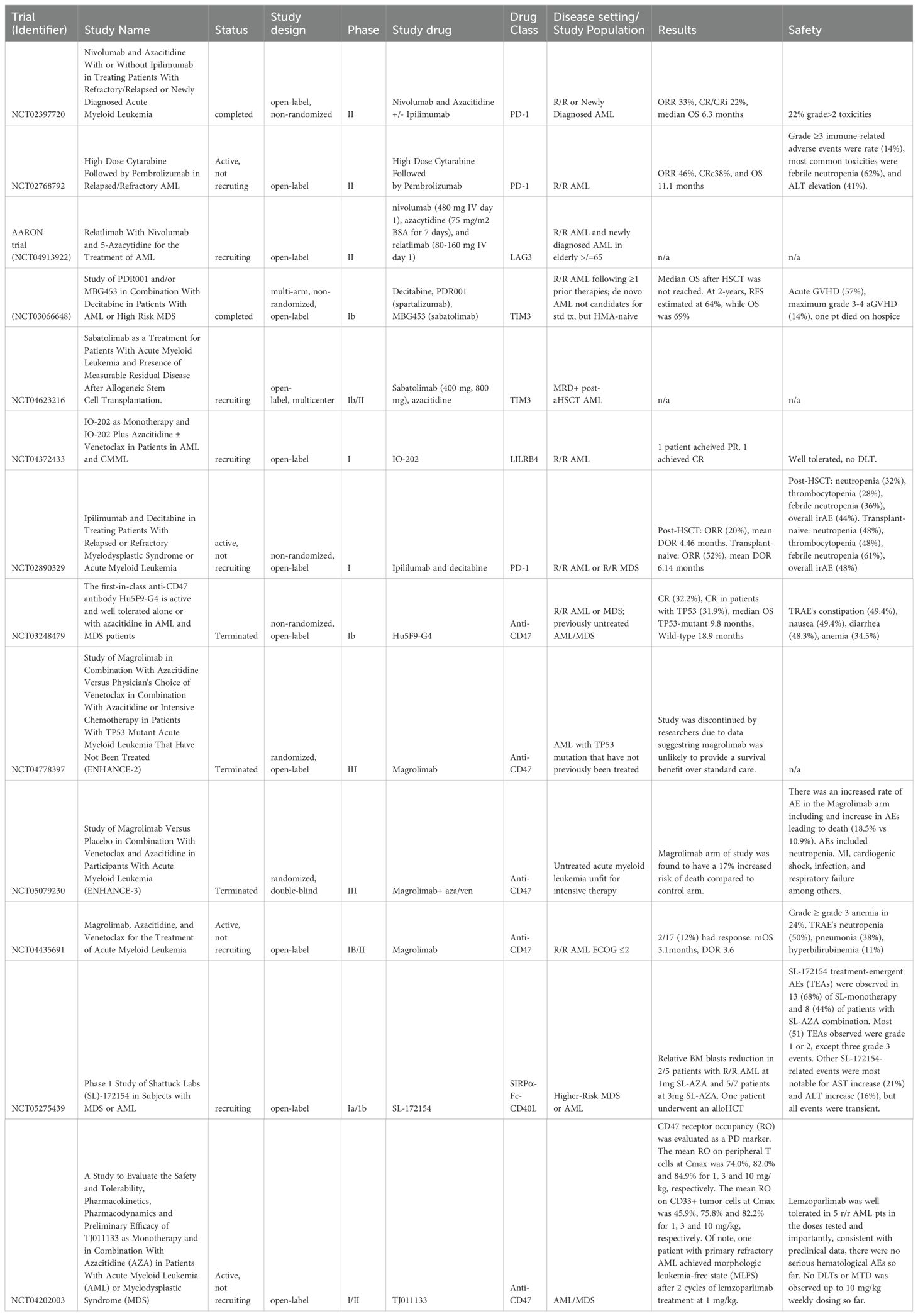
Table 1. Trials and studies using checkpoint inhibitors.
As described, PD-1, programmed death-ligand 1, is the most developed immune checkpoint inhibitor investigated in AML. Commonly used drugs in this drug class include nivolumab, pembrolizumab, and atezolizumab. Increased expression of PD-1 has been documented in subsets of populations such as relapsed/refractory (R/R) AML and demonstrates poorer outcomes. Therefore, patients with increased expression of PD-1 benefit from this immune checkpoint inhibitor therapy. Hypomethylating agents (HMA) are also used in the therapy of AML. These drugs work by inhibiting methyltransferase, altering gene expression (12). Recent studies have identified a correlation between HMA resistance and dysregulated PD-1/PD-L1 signaling. Higher levels of expression of PD-1/PD-L1 have been seen in patients with AML with no response to HMAs, which may represent a potential mechanism of resistance to this class of agents. Therefore, the efficacy of monoclonal antibodies targeting this pathway in HMA-failure myelodysplastic syndrome (MDS) or R/R AML is assessed. A nonrandomized, open-label phase II study by Daver et al. targeted PD-1 in combination with HMA in R/R AML [both HMA naïve (n=25) and HMA failure (n=45)] with nivolumab and azacitidine. They observed an overall response rate (ORR) of 33% (58% in HMA-naive and 22% in HMA-failure patients). Complete remission with incomplete count recovery (CR/CRi) was 22%, and median overall survival (OS) was 6.3 months with 22% grade >2 immune toxicities (13).
Pembrolizumab has also been evaluated in both newly diagnosed and R/R AML patients in combination with azacitidine by Gojo and colleagues (14). In the R/R cohort, the ORR was noted to be 18%, with 14% of patients experiencing CR/CRi and PR in 4% of patients with a median OS of 10.8 months for all patients. However, in newly diagnosed patients, the ORR was 59% (CR/CRi 47%; PR 12%) with a median OS of 13.1 months. Additionally, pembrolizumab has been evaluated with decitabine in R/R AML. This study was limited by a small number of patients (n=10). Stable disease was achieved by 4 patients, with 1 patient experiencing minimal residual disease and a negative CR. The median follow up was 13 months, with median OS of 7 months.
The role of PD-L1 blockade in combination with HMA in patients with AML has also been explored (15). Zeidan and colleagues randomized patients with newly diagnosed AML to receive azacitidine alone (n=64) vs. azacitidine in combination with durvalumab (n=65). The ORR was similar in both arms, with ORR of 31.3% in patients receiving azacitidine alone and ORR of 35.4% in patients receiving combination therapy. Unfortunately, no difference was observed in OS or duration of response between groups. The OS and duration of response for patients treated with single agent HMA were 13.0 months and 24.6 weeks, with the doublet cohort experiencing OS of 14.4 months and duration of response 51.7 weeks.
Lastly, intensive chemotherapy has also been evaluated in combination with anti-PD-1 therapy. A phase II single arm clinical trial conducted by Zeidner et al. examined PD-1 inhibition using pembrolizumab in combination with intensive chemotherapy (high dose cytarabine) in R/R AML. These authors demonstrated an ORR of 46% in the 37 patients treated with 38% of patients experiencing a composite complete remission rate, and a median OS of 11.1 months (15). Although much fervor has surrounded agents targeting PD-1 and PD-L1 in other malignancies due to impressive and durable responses, enthusiasm has waned for utilizing these agents in AML as a result of minimal improvement in responses and survival.
TIGIT, or T cell immunoreceptor with immunoglobulin and ITM domain; is a promising immune checkpoint (11). TIGIT is an immune checkpoint receptor part of the immunoglobulin (Ig) superfamily and is regulated by a plethora of immune cells such as NK cells, activated T cells, and regulatory T cells. CD155 and CD112 are expressed on tumor cells and antigen-presenting cells (APC) within the tumor microenvironment. TIGIT has two binding sites for these CD antigens. Studies have shown that TIGIT plays a role in regulating T and NK cell-mediated recognition. Although TIGIT inhibition alone has shown less impressive antitumor effects, dual blockade of TIGIT with PD-1 has demonstrated almost complete tumor rejection in murine tumor models, which is supportive of further investigation in patients with cancer.
Prior studies have found that the microenvironment of the leukemia bone marrow contains immunosuppressive factors promoting the evasion of leukemic stem cells from immunological surveillance. In a 2021 Xu L et al. study, findings showed higher co-expression of PD-1 and TIGIT in both CD4+ and CD8+ T cells in bone marrow samples of de novo AML patients compared to peripheral blood samples, supporting that there are increased immunosuppressive factors in bone marrow compared to peripheral blood (Table 2) (16).
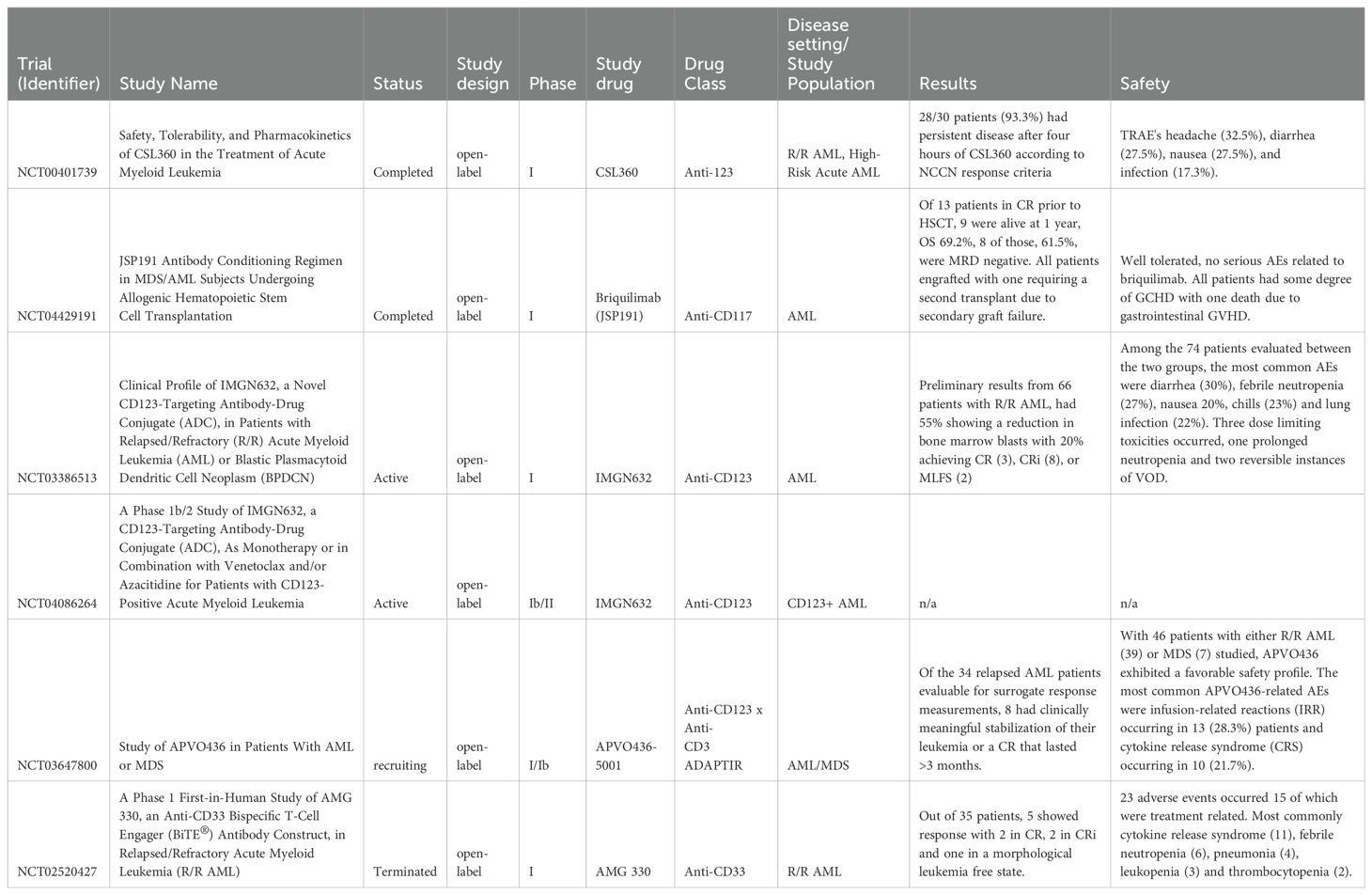
Table 2. Trials and studies using other antibody therapies such as antibody drug conjugates and bispecific t-cell engagers.
Brauneck and colleagues identified TIGIT as a target for macrophage-mediated cytotoxicity in AML. TIGIT was highly expressed on bone marrow-infiltrating immunosuppressive M2 macrophages within newly diagnosed and relapsed AML. Additionally, they found that in vitro blockade of TIGIT shifted the polarization of leukemic-associated macrophages from the M2 phenotype to the M1 phenotype. They also reported anti-CD47-mediated phagocytosis of AML cells augmented by TIGIT blockade (17).
Gournay and colleagues also demonstrated that high levels of TIGIT on CD4+ T cells three months after allogeneic HSCT was significantly associated with an increased risk of subsequent relapse in AML patients. Additionally, higher expression levels of TIGIT on CD8+ T cells were directly correlated with poorer clinical outcomes (18). These findings support that this immune checkpoint inhibitor is important in post-transplant relapse (19). Overall, TIGIT represents a potential target in AML. However, data supporting the pre-clinical or clinical activity of an agent targeting TIGIT remains to be seen.
Lymphocyte activation gene 3 (LAG3) is a co-inhibitory receptor belonging to the Ig superfamily, also known as CD223, which may be another promising immune checkpoint therapy for AML immunosuppression (20). It suppresses T cell activation and cytokine secretion and demonstrates synergism with PD-1 to inhibit an immune response. LAG3 is a promising target as it has shown efficacy against PD1-resistant AML in conjunction with PD1 therapy. Anti-LAG3 antibodies can upregulate effector T-cell activity and inhibit T-regulatory cell suppressive function.
In the 2019 study by Abdelhakim, H. et al., these authors found that AML cells promoted immunosuppression with higher expression of LAG3 on CD8+ T cells the in vitro model. Peripheral blood mononuclear cells were harvested from healthy donors and AML patients (Table 2). Anti-LAG3 antibody attenuated this immunosuppression by increasing the number of activated T cells and cytotoxic cytokines, decreasing T-reg cells, and enhancing MHC-I mediated killing of AML cells, thus supporting the inhibition of LAG3 as a means to mitigating AML immunosuppressive effect (21).
Chen, C. et al. investigated the expression pattern of LAG3 and other immune checkpoints in AML using data from de novo AML patients within the TCGA database and AML bone marrow samples (Table 2). They found that AML patients had poorer overall survival with high CTLA-4 and LAG3 expression (3-year OS 9% vs. 36% and 13% vs. 40%, respectively) (22).
Relatlimab is a monoclonal IgG4 antibody that blocks LAG-3. The Food and Drug Administration (FDA) recently approved it as a treatment for untreated advanced malignant melanoma. The AARON trial (NCT04913922) is a phase 2, open-label clinical trial within its recruiting phase to investigate the safety and efficacy of combination therapy with nivolumab, azacitidine, and relatlimab in R/R AML (Table 1) (23). The results from this clinical trial will be pivotal in understanding whether targeting LAG-3 can enhance the activity of anti-PD-1 therapy in AML when combined with HMA.
T cell immunoglobulin and mucin domain 3, TIM3, is a cell surface molecule expressed on leukemia cells (24). In CD4+ and CD8+ T cells and leukemic cells, TIM3 overexpression may lead to poor response and relapse in patients; therefore, TIM3 inhibitors are currently being investigated as potential AML treatments. Regarding acute myeloid leukemia, studies have shown that leukemia stem cells (LSC) have higher expression of TIM3 but not in healthy hematopoietic stem cells (HSC). Therefore, targeting TIM3 with antibodies or microRNA can destroy LSCs without affecting healthy HSCs. In leukemic cells of AML, TIM3 participates in an autocrine stimulatory loop, which activates the β-catenin and NF-κB pathways integral for cell survival and disease progression. Moreover, TIM3 inhibitors induce cell apoptosis in AML (24).
Additionally, Al-Amrani, S. et al. (2022) have shown that there is a higher expression of TIM3 in peripheral blood of AML cell lysates compared to healthy controls with median concentration 9.26 pg/ml and 5.1 pg/ml respectively (p=0.01) (Table 2) (25). TIM3 concentrations were also significantly elevated at diagnosis compared to remission, 9.25 pg/ml and 5.57 pg/ml, respectively (p=0.03). In bone marrow samples, TIM3 expression was higher in AML samples than in healthy controls, 10.89 pg/ml and 5.1 pg/ml, respectively, at diagnosis (p=0.04). The expression levels of TIM3 from AML bone marrow lysates at diagnosis and in remission were not significantly different (10.8 pg/ml and 5.1 pg/ml, p=0.01).
A multi-arm open-label phase 1b study (NCT03066648) investigates combination therapy with sabatolimab (MBG453), which is an anti-TIM-3 IgG4 and a hypomethylating agent (decitabine or azacitidine) (26). Preliminary findings with just sabatolimab have shown promising survival rates in R/R AML and de novo AML patients who are not candidates for standard treatment (HMA-naive) (Table 1). An analysis was performed on 28 of these patients who underwent an allogeneic HSCT. Before HSCT, 36% of patients had a complete response, and 32% had a complete molecular or complete response without hematologic recovery. 8% had a partial response or hematologic improvement, and 25% had stable disease. After allogeneic HSCT, median OS was not reached. However, the 2-year OS rate was 69%, and the 2-year recurrence-free survival rate was 64%. Regarding adverse effects, 57% experienced acute graft-vs-host disease (GVHD), 14% of which experienced grade 3/4 GVHD, where one patient died in hospice after grade 4 GVHD.
A phase 1b/2, open-label study (NCT04623216) currently recruiting aims to investigate sabotolimab monotherapy in combination with azacitidine in AML patients who received one allogeneic HSCT achieving complete remission but were minimal residual disease positive (MRD+ post-allogeneic HSCT) (Table 1) (27). Results from 21 patients showed 7 patients were in complete remission (CR) and on treatment after one year of therapy. The remaining 14 had been discontinued on treatment due to relapse, AE, or treatment change. There were toxicities, with 76% of patients experiencing any toxicity and 38% experiencing grade 3 or higher toxicities. Most commonly, neutropenia and thrombocytopenia. There were 4 patients who experienced dose limiting toxicities. A follow-up dose expansion study is planned (28). Unfortunately, although there was much enthusiasm for this compound, in January 2024, Novartis discontinued the program evaluating sabatolimab in high-risk myelodysplastic syndromes as the STIMULUS-MDS trial did not meet its primary endpoints of improved complete response rates or progression-free survival (29).
VISTA (V-domain Ig suppressor of T cell activation) is a novel immune checkpoint similar to PD-1 or CTLA-4. VISTA is highly expressed in primary AML cells and is associated with a poor prognosis (30). Based on the TCGA database, VISTA was the most correlative immune checkpoint in the overall prognosis of AML patients as it had the same higher expression as in CD34+ AML cells and CD34- myeloid cells of bone marrow. VISTA helps impede T-cell activation. Hyperactive STAT3 in AML has been associated with a high expression of VISTA. One STAT3 inhibitor, W1046, suppresses AML proliferation and survival. The inhibition of STAT3 causes a downregulation of VISTA, which allows for activation of T cells. However, the molecular mechanism of how this occurs is unknown. T-cell-mediated cytotoxicity to AML cells is also boosted through anti-VISTA mAb, and anti-VISTA mAb was also shown to prolong the survival of AML in mice. In vitro and in vivo studies have also shown that the combination of W1046 and VISTA mAB caused a drastic elevation in T cell cytotoxicity in vitro and also caused upregulation of the secretion of interferon (IFN)-y, interleukin (IL)-2, and CD4+ and CD8+ T cells. Although VISTA has been more thoroughly studied in other malignancies, the role of targeting this checkpoint as an anti-leukemic therapeutic strategy remains to be seen.
CD47 was initially discovered as an antigen in human ovarian cancer and later found to be overexpressed in hematological tumors like AML (31). CD47 supports cells in evading phagocytosis by macrophages in leukemic cancer cells, and much enthusiasm has been put behind this pathway as evidenced by multiple compounds being developed to target this checkpoint in AML (Table 1).
CD47 blocking antibodies and SIRPα-FC fusion proteins have been studied in combination with azacitidine in untreated AML patients. SL172154 is a CD47 inhibitor that has enhanced pro-phagocytic signals on leukemic stem cells/blats coupled with CD40L to enhance antigen presentation and further enhance tumor cell killing (32). A phase I dose escalation trial is evaluating SL-172154 as monotherapy vs in combination with azacitidine for patients with AML or HR-MDS. The study had 37 patients enrolled, with 27 patients having R/R AML (16 de novo; 11 secondary). SL-172154 treatment-emergent AEs (TEAs) were observed in 13 (68%) of SL-monotherapy and 8 (44%) of patients with SL-AZA combination. Most (51) TEAs observed were grade 1 or 2, except three grade 3 events. Other SL-172154-related events were most notable for AST increase (21%) and ALT increase (16%), however all events were transient.
Overall, one patient with R/R AML post 7 + 3, FLAG, and VEN/AZA achieved a Morphologic Leukemia-Free State (blast reduction 19% down to 5%) after 1 cycle 6mg/kg SL-172154 and proceeded to allogeneic HCT after cycle 2 (32). There were 12 patients with R/R AML who received SL-AZA with no objective responses observed, but the reduction in bone marrow blasts from baseline (50-75%) was seen in 2/5 patients at 1mg SL-AZA. Relative bone marrow blast reduction (35-90%) was seen in 5/7 patients at 3mg SL-AZA. One patient proceeded to allogeneic HCT.
The study demonstrated that SL-172154 is well tolerated in up to 3mg/kg monotherapy and in combination with azacitidine. There was an increase in accumulation of mature myeloid cells in BM and an increase in serum cytokines with the dose-dependent increases, which suggests a potential role for CD40 stimulation in AML (32).
Lemzoparlimab (TJ011133 or TJC4) is a differentiated anti-CD47 IgG4 antibody that targets a CD47 epitope, which allows a unique red blood cell sparing property while retaining strong anti-tumor activity (33). It is currently being evaluated in a phase I trial (NCT04202003). The unique epitope differentiates it from other CD47 axis targeting therapies. Lemzoparlimab blocks CD47-SIRPα interaction, which helps promote phagocytosis of cancer cells by macrophages and neutrophils (34).
Dosages of 1mg/kg, 3mg/kg, and 10mg/kg were used with average receptor occupancy on peripheral T cells of 74%, 82%, and 84% respectively. One patient attained morphologic leukemia-free status after two cycles of 1mg/kg, but 4/5 patients had AE, with 1/5 being a Grade 3 (35).
Hu5F9-G4 was analyzed in a clinical trial (NCT0324879) where 53% (8/15) untreated AML/MDS patients had either CR or Cri (36). Another phase Ib trial (NCT04778397), showed 56% (10/34) AML patients achieved CR/CRi to 5F9+AZA, and CR/CRi rate was greater in patients with TP53 mutations (36). Another study showed in relapsed refractory AML with prior venetoclax-naive AML, the CR/CRi rate was 63% (5/8) (37). In relapsed refractory AML with prior Venetoclax failure, the CR/CRi rate was 27% (3/13) with a median OS of 3.1 (37).
While CD47/SIRPα immune checkpoint-based immunotherapy studies have shown promising preliminary results in R/R AML, the development of CD47 monoclonal antibodies still faces safety concerns and a lack of published data on therapeutic effectiveness. One area of concern is the significant dosage requirement to achieve adequate therapeutic CD47 blockage, as a preclinical study showed that 40-60% of CD47 receptor occupancy is required for the induction of phagocytosis (38). With this in mind, magrolimab, Gilead’s CD47/SIRPa interaction blocking monoclonal antibody being investigated in their phase 3 ENHANCE-3 study, was recently terminated in February of 2024. An independent data analysis demonstrated evidence of futility within the magrolimab, azacitidine, and venetoclax arm which resulted in an increased risk of death due to infection and respiratory compromise. The FDA ultimately paused enrollment applicable to all studies of magrolimab. Gilead will not be pursuing magrolimab further in hematological cancers. With this large multi-billion-dollar study ultimately failing to demonstrate any benefit, the future of CD47 drug therapy is uncertain, even if ways are found to minimize toxicity.
LILRB4 is a member of the leukocyte immunoglobulin-like receptor family of proteins commonly seen on myeloid lineage cells. They play an important part in cell activation and differentiation. LILRB4 is seen most commonly in monocytic AML. Working through APOE, SHP-2, uPAR, and ARG1 signaling pathways, LILRB4 activation leads to downregulation of T cell activation, aiding in cancer infiltration and immune system evasion (39). Similar to other targets discussed, LILRB4 overexpression in cancer cells compared to normal cells makes it an enticing target for immune checkpoint inhibition and other therapies. NCT04372433 is an active Phase 1 trial evaluating IO-202, a humanized monoclonal antibody blocking LILRB4 from binding to APOE and fibronectin (Table 1). Part 1 of the study reported on 36 patients with R/R AML with monocytic differentiation treated with azacitidine ± Venetoclax or as monotherapy. Overall, the drug was well tolerated, with no dose-limiting toxicity observed and no study-related deaths. Unfortunately, in the monotherapy group, only one patient achieved a partial remission. In the combination group, only one patient (noted to have high LILRB4 expression) achieved CR, which was durable for 7 months as of the report. Several other patients did show blast reduction but did not achieve remission. While this trial showed limited success in general monocytic AML, patients were not screened for high LILRB4 expression. Part two of the trial will expand to look at patients with high LILRB4 expression which will hopefully yield more promising results (40). There are two other up-and-coming trials using STAR-T or CAR-T cells targeting LILRB4 that have been indexed; however, they have not started recruiting or been reported on.
CD123 is a cytokine receptor widely overexpressed in multiple hematologic cancer cells and associated with high-risk disease characteristics in adult and pediatric AML. In particular, AML cells with FLT-3 or NPM1 mutations are associated with higher levels of CD123 compared to cells with wild-type FLT3 or NPM1 (41). CD123 has become an important biomarker that can be potentially targeted in relapsed or refractory AML. CSL360 is a recombinant chimeric immunoglobulin G1 (IgG1), an anti-CD123 monoclonal antibody proven less beneficial in patients with relapsed/refractory or high-risk AML (42, 43). The phase I clinical trial (NCT00401739) assessed the efficacy and safety of CSL360 in relapsed/refractory AML cases (Table 3). The study analyzed 40 patients, with 25 of the patients having relapsed AML and 8 of the patients having refractory disease. The study found that CSL360 was well tolerated, as the most common AEs of any grade were headache (32.5%), diarrhea (27.5%), nausea (27.5%), and infection (17.3%). AEs also did not increase the frequency of the dose level escalation. The study found no anti-leukemic effects as most patients had no improvement in blast percentage in BM aspirates during the study. The study found that 28/30 patients (93.3%) had persistent disease after four hours of CSL360, according to NCCN response criteria (42).
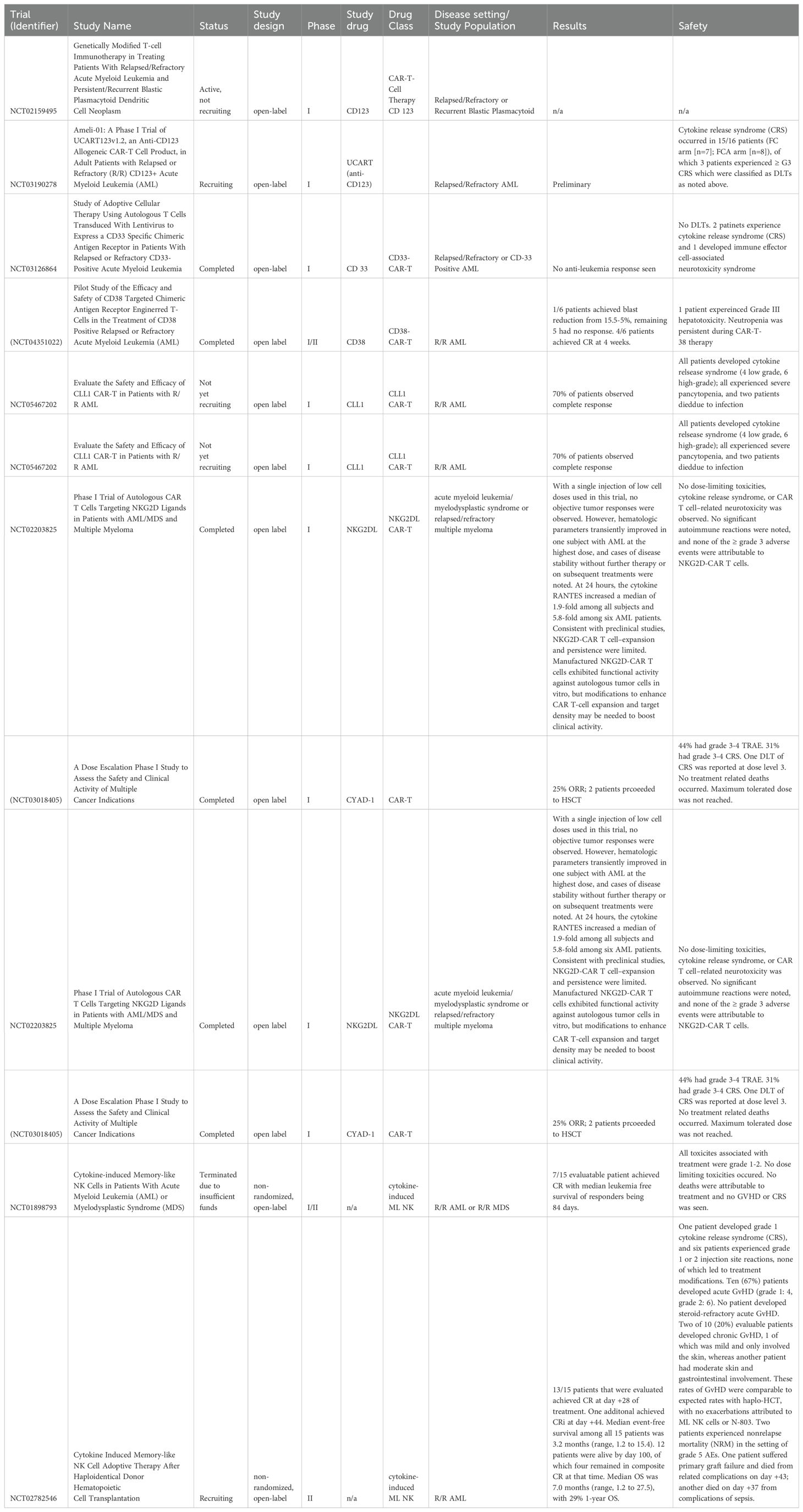
Table 3. Trials and studies using engineered cells and adoptive therapy.
CD123 antagonistic peptides assembled with nanomicelles have also been evaluated preclinically (Table 2) (16). In the study, nanomicelles were loaded with a lab-designed CD123 antagonistic peptide (mPO-6) and were investigated with CD123+ AML cell lines and a refractory AML mouse. Results showed that IV administration of mPO-6) reduced the percentage of AML cells and prolonged median survival of AML mice. It was determined that mPO-6 interferes with the axis of CD123/IL-3, which helps enhance apoptosis and prolong the median survival of AML mice.
Another preclinical study investigated SIRPα-αCD123 fusion antibodies targeting CD123 with blockade of CD47 to augment anti-CD123 activity (Table 2) (44). As reviewed previously, leukemic cells have CD47, an innate immune checkpoint upregulated on the AML cell surface. The authors developed a SIRP α-αCD123 fusion antibody that enhances the leukemic stem cell clearance by disrupting CD47/SIRPα signaling to AML. In vitro, blocking assays were used to interfere with the binding of SIRPα. The SIRP α-αCD123 fusion antibody showed increased binding and targeting of CD123+/CD47+ AML cells, even in CD47+ healthy cells. In vitro experiments also showed SIRPα-αCD123 antibodies greatly enhanced AML cell phagocytosis mediated by allogeneic and autologous macrophages. Unfortunately, due to a lack of clinical response, targeting AML via monoclonal antibody therapy has lost enthusiasm.
Antibody-drug conjugates (ADCs) combine the specificity of an antibody with a potent drug to bind and kill target cells. Targets for ADCs currently investigated in preclinical or early Phase I/II trials for AML include CLL-1, CD123 (also known as interleukin 3 receptor alpha), CXCR4, and FLT3. More studied targets like CD33 are already observing success in select patient populations.
CD33 is a receptor protein primarily expressed on leukemic blasts, and low expressions of CD33 have been seen in more complex karyotypes and translocations. Gemtuzumab ozogamicin (GO), a humanized anti-CD33 monoclonal antibody conjugated with calicheamicin, was first approved by the FDA in 2000 (45). However, the randomized phase III S0106 trial that evaluated GO versus no GO during induction and post-consolidation therapy in younger AML did not confirm promising results in the previous phase II study conducted on relapsed older adults with AML (46). The drug was withdrawn from the US market due to an unfavorable safety-efficacy profile. However, it was later placed back on the market in 2017 after multiple randomized controlled trials showed an improved OS as well as event-free survival when GO therapy is used in addition to induction/consolidation therapy in newly diagnosed AML patients with a safe toxicity profile.
The improvement of OS was further characterized when evaluating the cytogenetic profile of treated patients. One meta-analysis looked at five large, randomized trials, which analyzed GO in induction/consolidation chemotherapy in all ages of AML patients. Beneficial effects of GO were seen in favorable risk AML, along with a smaller benefit in intermediate-risk AML and no benefit in adverse-risk (47). Favorable cytogenetics saw an overall survival benefit regardless of age, 30.7% vs 34.6%. The survival benefit was mainly seen in the favorable cytogenetics (55.3% vs. 76.3%) and intermediate-risk patients (34.1% vs. 39.4%). Patients with adverse karyotypes did not see any benefit in overall survival.
Hepatic veno-occlusive disease (VOD), which is a clinical syndrome characterized by jaundice, painful hepatomegaly, and/or fluid retention, was reported in initial studies with GO in AML. Late manifestations of hepatic VOD include ascites and encephalopathy. In the meta-analysis, fractionated dosing using 3 mg/m2 was associated with less toxicity and equal efficacy (47). The data obtained from the fractionated dosing schedule led to the reapproval of GO (48).
C-type lectin-like molecule-1 (CLL-1) is primarily expressed on granulocytes and monocytes and, to a lesser degree, on precursors and HSCs. However, expression is seen in a large percentage of AMLs, including leukemic stem cells (49). Daver et al. evaluated the ADC DCLL9718S in 2021 in a phase I dose-escalation trial. DCLL9718S is a humanized monoclonal IgG1 anti-CLL1 antibody linked with two pyrrolobenzodiazepine (PBD) dimer drugs via a cleavable disulfide linker. Eighteen patients were treated in the study, and almost all participants experienced at least a grade 1 adverse event, with at least half being attributable to treatment. The most common adverse events were neutropenia and pneumonia, although no dose-limiting toxicity occurred, and no deaths occurred because of therapy. At the highest dose of 160 μg/kg, 2 patients began experiencing hepatotoxicity, although this may be intrinsic to PBD and not drugs targeting CLL-1. No patient achieved a complete or partial response. The trial was stopped, and there are no plans to move forward with the drug, given the poor tolerability and limited anti-leukemic effects observed (50). Several other groups have published their preclinical work as the potential of CLL-1 remains promising despite the setbacks seen with DCLL9718S.
CD123, also known as interleukin 3 receptor alpha, is another target with elevated expression in AML with the potential to target leukemic stem cells that been targeted by multiple groups. Kuvtun et al. have worked to change the cytotoxic drug component of ADCs. IMGN632 comprises a novel humanized anti-CD123 antibody, G4723A, linked to an alkylating agent, indolinobenzapine (IGN) pseudodimer (51). They previously had a DNA crosslinking agent, but as with several ADCs, toxicity became a limiting factor. IMGN632 showed potent activity against AML samples in vitro and in xenograft mouse models made from EOL-1, MOLM-13, and patient-derived samples (51). Substantially less toxicity to normal hematopoietic cells was seen. Daver et al. began a phase 1 clinical trial looking at IMGN632 in R/R AML and blastic plasmacytoid dendritic cell neoplasm, NCT03386513 (52). Of the 66 patients with R/R AML treated on protocol, 55% demonstrated a reduction in bone marrow blasts, with 20% achieving CR (N=3), CRi (N=8), or morphologic leukemia-free state (N=2). Among the 74 patients evaluated between the two groups, the most common AEs were diarrhea (30%), febrile neutropenia (27%), nausea 20%, chills (23%), and lung infection (22%). Three dose limiting toxicities occurred, one prolonged neutropenia and two reversible instances of VOD (53). Given the activity in the R/R setting, this combination is being explored in newly diagnosed AML patients expressing CD123 (54).
Talacotuzumab, a humanized CD123 monoclonal antibody, has shown potent in vitro ADCC against AML blasts, reducing leukemic cell growth in murine xenograft models (55). A multicenter phase 2/3 clinical study investigates talacotuzumab to determine the recommended phase 2 dose in AML patients. An open-label, randomized comparison of talacotuzumab in combination with decitabine versus decitabine alone was also investigated. The recommended phase 2 dose was 9mg/kg. CR was achieved in 15% of patients receiving combination therapy versus 11% in those receiving decitabine alone. Median OS was 5.36 months for combination therapy versus 7.26 months for decitabine alone. Therefore, combination therapy did not demonstrate improvement in efficacy versus decitabine alone.
C-X-C chemokine receptor type 4 (CXCR4) is an important receptor in homing hematopoietic cells to bone marrow and is expressed highly in many hematological malignancies, identifying another favorable immunotherapeutic target in AML. However, utilizing CXCR4 as a target does have some challenges as it is also relatively expressed on normal hematopoietic cells and other tissues. Current work on a CXCR4 ADC is all preclinical, but Costa et al. showed promising work empirically deriving optimal ADCs (56). They optimized ADCs by varying linker-payload cleavability, drug-to-antibody ratio, affinity, and Fc format to maintain a tolerable safety profile while targeting more broadly expressed proteins. Their ADC which targets CXCR4 using auristatin greatly increased survival in MV4-11 AML mouse models, among other cancers. They observed significant differences in efficacy between different constructs of the ADC. More work must be done to expand this to other ADCs and see how this translates into targeting CXCR4 in clinical settings (56).
Fms-like tyrosine kinase 3 (FLT3) is a class III receptor tyrosine kinase expressed on leukemic stem cells and blast cells of most AML patients with significantly higher expression levels than healthy tissue. Additionally, the presence of internal tandem duplication (ITD) mutation of FLT3 (FLT3-ITD) has a reportedly higher relapse risk and poorer outcome in AML patients (57). The 2023 Roas M. et al. preclinical study investigated 20D9-ADC (an ADC targeting FLT3) in vivo and in vitro models. In vitro, 20D9-ADC mediated potent cytotoxicity to FLT3 or FLT3-ITD expressing Ba/F3 cells, AML cell lines, and FLT3-ITD positive patient-derived xenograft AML cells (Table 2). It also showed no severe hematotoxicity in vitro colony formation assays when using concentrations cytotoxic in AML cell line treatment. Additionally, 20D9-ADC treatment in vivo resulted in significant tumor reduction and complete remission in AML xenograft models (57).
Bispecific T-cell Engager (BiTE) molecules function by having a single molecule interact with CD3 on T-cells and an antigen expressed by cancer cells. Like CAR-T therapies, this linkage directs a patient’s T cells to eliminate leukemic cells (58). The ideal targets for BiTE therapy are surface antigens that are selectively expressed on leukemic cells and have limited expression on normal cells. A key advantage over CAR-T is the “off-the-shelf” potential of these drugs. Unlike CAR-T, they are not processed from patient samples and can be given immediately. Blinatumomab is a CD19xCD3 bispecific BiTE molecule that has shown success in treating relapsed/refractory acute lymphoblastic leukemia. The success of blinatumomab has led to further investigation into alternative T-cell recruiting strategies in AML. Common treatment-related adverse effects include neurotoxicity, cytokine release syndrome, cytopenia, and hepatotoxicity (59).
FLT3 mutations are observed in around 25% of adult AML patients and 30% in those over 55 and are generally associated with poor outcomes (60). FLT3 also has limited expression in normal tissue, primarily found in early hematopoietic progenitors and leukemia blasts. Given this, FLT3 has been a promising target for several therapeutic options, including tyrosine kinase inhibitors and BiTEs (61). Two FLT3 BiTE molecules, one with a half-life extending moiety and one without, were analyzed by Brauchle et al. in 2020. They evaluated bulk leukemia cells from bone marrow or peripheral blood from 318 newly diagnosed or relapsed AML patients and found that 78% showed increased FLT3 protein expression. From there, they exposed FLT3 positive cell lines, such as MOLM-13, EOL-1, and MV4-11, to the BiTEs leading to cytotoxicity, whereas FLT3 negative cell lines, such as K562, showed relatively little response. Mouse xenograft models created using MOLM-13 and EOL-1 cells followed by injection of human CD3+ T cells showed a significant reduction in tumor growth with weekly FLT3 BiTE treatment. They also showed a reduction of FLT3 expression in cynomolgus monkeys with reasonable pharmacokinetics. The addition of PD-1 blockade also showed enhancement of cytotoxicity. This has been noted in other BiTE trials and is thought to be due to increased activation of T-cells inducing PD-1 expression (58).
APVO436-5001 (NCT03647800) is an ongoing phase 1b dose escalation trial looking at APVO436, a novel bispecific anti-CD123 and anti-CD3 ADAPTIR molecule. The bispecific antibody has shown anti-leukemia activity in murine xenograft models of AML (62). Nine patients with R/R AML were treated in the study; and one patient achieved a prolonged stable disease, and two had a partial response that later progressed to a complete response (62). The most common grade 3 or greater adverse events likely APVO436-related were grade 3–4 cytokine release syndrome occurring in 4 of 46 patients (8.7%), grade 3–4 anemia occurring in 2 of 46 patients (4.3%), and infusion-related reaction occurring in 2 of 26 patients (4.3%). Further evaluation of this compound is ongoing.
Flotetuzumab (MGD006) is a bispecific antibody-based molecule to CD3 and CD123 in DART format. Preclinical models have investigated flotetuzumab mediated target-effector cell association, T-cell activation and proliferation, and potent killing in AML blasts both in vitro and in vivo. A phase 1/2 clinical study of flotetuzumab in R/R AML(NCT02152956) showed that of primary induction failure patients and early relapse patients, 26.7% achieved complete remission or complete remission with partial hematologic recovery. ORR was 30.0% with a median OS of 10.2 months, a 6-month survival rate of 75%, and a 12-month survival rate 50% (63). Unfortunately, due to underwhelming responses, the development of this product was discontinued (64).
One mechanism of resistance to BiTE therapy has been a loss of tumor-associated antigens. It was hypothesized that targeting more than one tumor-associated antigen could lead to a reduced frequency of relapse (65). The study used a half-life extended CD123-FLT3 BiTE molecule in vitro, mouse xenografts, and non-human primate tolerability studies. A survival benefit of >3 weeks was seen in the xenograft model (65). Additionally, both arms of the molecule were active, as survival benefits in mice with single positive tumors could not be calculated due to the high survival. Furthermore, follow-up blood tests showed a decrease in FLT3 mRNA. Repeat dosing was also not tolerated, and cytokine release was observed. It is hypothesized that CD123 expression would be increased with the cytokine release following repeat dosing.
CD33 is expressed in >99% of AML cases, and BiTE has been effective in relapsed/refractory acute lymphoblastic leukemia. AMG 330 is a BiTE that binds to CD33 and CD3 on T cells and helps cause T-cell mediated destruction of CD33+ cells. In the presence of AML cells, AMG 330 induced the release of IFN-y, TNF, IL-2, IL-10, and IL-6 (66).
Ravandi et al. treated 35 relapsed/refractory AML patients with 12 different dose levels of AMG 330 ranging from 0.5-480 μg/d in a phase 1 study (Table 3). Of the thirty-five patients treated, thirty-one ultimately discontinued treatment. The majority of patients stopped therapy due to disease progression (n=24). Five patients discontinued therapy due to adverse events (two treatment-related), and two at the patient’s request. Overall, 23 adverse events occurred, 15 of which were treatment related. Most commonly, cytokine release syndrome (n=11), febrile neutropenia (n=6), pneumonia (n=4), leukopenia (n=3), and thrombocytopenia (n=2). Five patients demonstrated a response: two patients achieved CR, two achieved CRi, and had a morphological leukemia-free state (67).
CAR-T is of great interest in AML. However, there remains a need for identifiable targets ubiquitously expressed in AML yet dispensable (Table 4) (68).
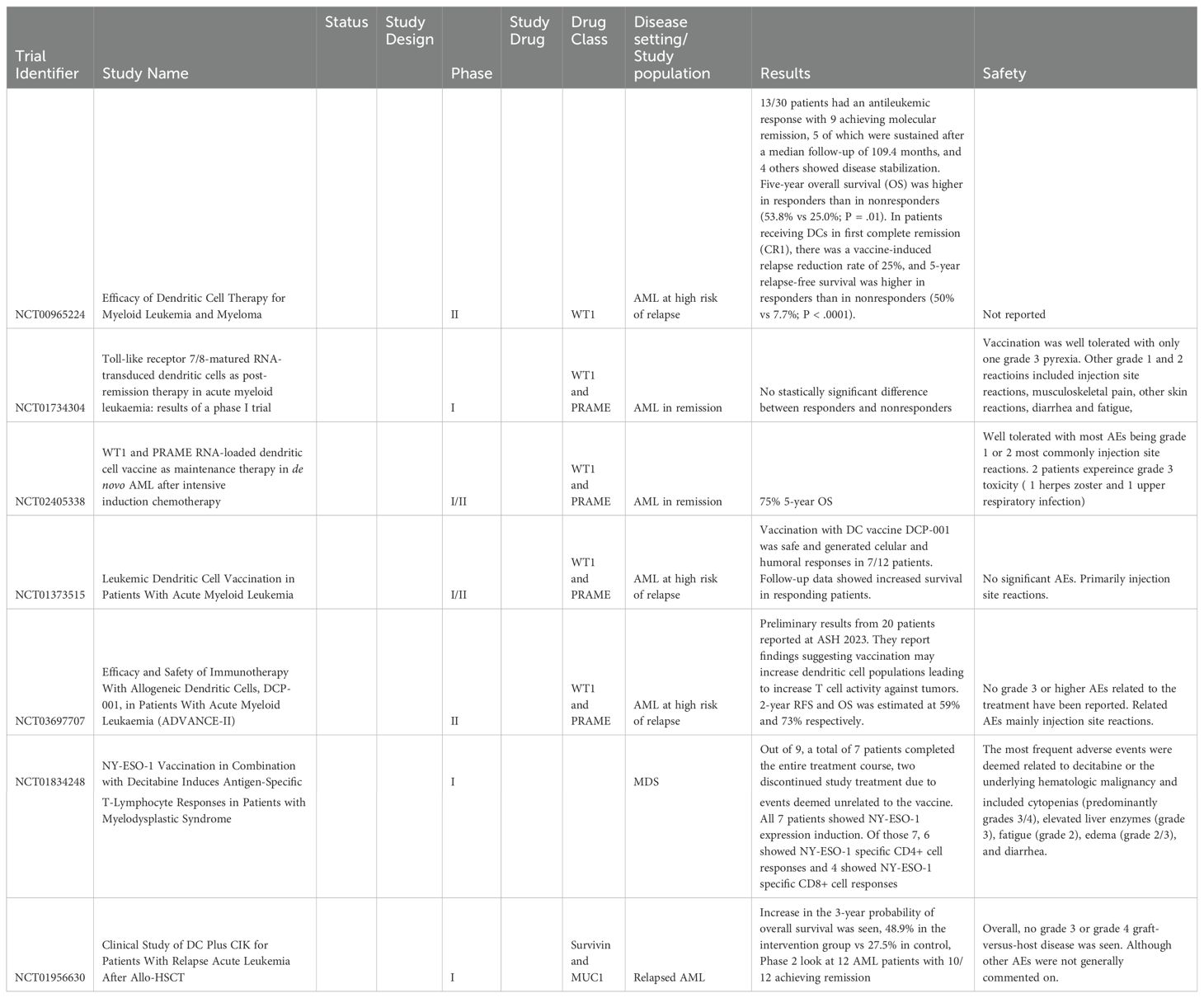
Table 4. Trials and studies using vaccine therapy.
There is a current ongoing trial (NCT02159495) where patients are being administered autologous anti-CD123 CAR-T cells for patients with CD123+ relapsed or refractory AML or CD123+ persistent or recurrent Blastic Plasmacytoid Dendritic Cell Neoplasm (69). The CD123CAR contains an anti-CD123 single-chain variable fragment, an optimized IgG4 CH2CH3 linker, a CD28 costimulatory domain, and a CD3 zeta signaling domain. The AML cohort contains six patients who have refractory AML after allogeneic HSCT and between 4-7 prior lines of therapy. Of two patients who received dose level 1, one achieved a morphologic leukemic-free state for two months. After the second infusion, blasts were reduced from 77.9% to 0.9%. Four patients received dose level 2. One of the four achieved a complete response and was transfusion-independent. The patient received another allogeneic HSCT and has remained in measurable residual disease (MRD)-negative complete response. Another patient also achieved a complete response. The other two patients did not achieve remission; however, they did have blast reductions.
Regarding safety, all toxicities were reversible. Four had grade 1 cytokine release syndrome (CRS), and one had grade 2 CRS. One developed adenoviral pneumonia, necessitating intubation. Lastly, one developed a grade 3 hypersensitivity rash. There were no dose-limiting toxicities (DLTs) and no treatment-related cytopenias. One patient with prior allogeneic HSCT developed cutaneous GVHD that was mild and recurrent after CAR-T therapy (69).
Another clinical trial of NCT03190278 is a phase I open-label trial looking at the safety and efficacy of off-the-shelf allogeneic UCART123v1.2 targeting CD123 in relapsed/refractory AML (70). There were two lymphodepletion regimen arms: one receiving fludarabine and cyclophosphamide (FC), while the other arm received fludarabine, cyclophosphamide, and alemtuzumab (FCA) to target CD52 in an attempt to reduce GVHD. Of 17 patients, four demonstrated clinical benefit. One FCA patient had greater than 90% blast reduction from 60% to 5% at day 28 post-infusion. Another FCA patient achieved a long-term MRD-negative complete response at 56 days post-infusion, which persisted for more than 12 months. The two other patients who achieved clinical benefit were part of the FC arm. One achieved blast clearance after day 14 post-infusion; however, by day 28 post-infusion, one had a rapid return of blasts. The other patient achieved a morphologic leukemic-free state. Overall, the FCA arm achieved prolonged lymphodepletion and UCART expansion compared to the FC arm. Accrual for this trial is ongoing.
NCT03126864 is a single-center, single-arm phase I trial that looked at the tolerability of 3 different doses of the autologous CD33-CAR-T cells in relapsed/refractory CD33-positive AML (71). These cells were modified to express a CD33-targeted CAR with 4-1BB and CD3ζ endo-domains, which were co-expressed with truncated human epidermal growth factor receptor (HER1t). Ten adults were enrolled, where patients received a median of 5 prior treatments, and 3 were post-allogeneic stem cell transplant patients. One patient died before receiving CAR-T. There were no DLTs at dose level 1 during the 28-day DLT assessment period. Two patients developed CRS; one patient developed immune effector cell-associated neurotoxicity syndrome (ICANS). Adverse effects from CAR-T for one patient include grade 3 tumor lysis syndrome with acute kidney injury, grade 2 mucositis, and grade 1 tachycardia. Another patient developed grade 2 orthostatic hypotension, grade 2 increased bilirubin, and grade 3 transaminitis. One patient did not experience any toxicity. All three patients who received this CAR-T therapy died due to disease progression. Despite detectable anti-CD-33 CAR-T in blood samples, no anti-leukemic responses were identified (71). The authors state that due to challenges, including the aggressive nature of relapsed/refractory AML and the need for expeditious autologous product generation, logistic, and enrollment challenges, the trial was closed to patient entry following enrollment of the eleventh patient. The sponsors have transitioned to a platform that facilitates more rapid production and in vivo expansion with a product called PRGN-3006 (NCT03927261) (72, 73).
Many of the developed CAR-T target antigens are also expressed on normal cells, which can lead to off target toxicity (74). Guo and colleagues the expression profiles of CAR-T target antigens (CD33, IL3RA, and CLEC12A) at a single-cell level, and found significant upregulation of CD33 and IL3RA compared to normal BM HSPC and CLEC12A was strongly positive in almost all AML samples (74). However, these expression levels were highly variable during treatment. Therefore, in the development of this modality of therapy it is critical for the therapeutic targets to have minimal expression on healthy tissue to avoid unnecessary adverse effects.
CD38 is an overexpressed tumor antigen in AML. Glisovec-Aplenc et al. developed autologous CD38-CAR T cells with 41BB-CD3ζ costimulatory domain in 2023 in vitro study (75). CD38 expression on activated T cells did not diminish CART-38 cell expansion or in vitro function. In xenografted mice, CART-38 rejection of AML cell lines was observed, and the overall survival of the animals was prolonged in this model. A decrease in hematologic progenitors was observed in a xenograft model of normal human hematopoiesis, which may pose a risk of decreased hematopoietic cell lines in patients in clinical trials (75). In a phase I/II (NCT04351022) clinical trial, CD38-directed CAR-T is designed to target CD38-positive R/R AML. Of the 6 patients treated in the study, two achieved CR or CR with incomplete count recovery (CRi) one week after CAR-T infusion. Four patients achieved CR or CRi 2 weeks and four weeks after CAR-T infusion. The 6-month OS rate and median OS was 50% and 12.3 months respectively. The leukemia-free survival rate and median leukemia-free survival were 50% and 10.3 months, respectively. Five patients developed grade I-II CRS and one developed grade III hepatotoxicity. No neurological toxicities were observed, but all six patients developed grade 3/4 hematological toxicities (76).
CD7 CAR-T therapy has shown significant efficacy with a tolerable safety profile in T-cell lymphoid malignancies. A phase I clinical study (NCT04938115) investigates the efficacy and safety of CD7 CAR-T (NS7CAR-T) therapy in CD7-positive R/R AML patients. After four weeks of NS7CAR-T cell infusion, 7 of 10 patients (70%) achieved CR in BM. Six of these patients achieved MRD-negative CR. Three patients showed no remission. Of the 7 patients who achieved CR, 3 patients who relapsed from prior transplants did undergo a 2nd allogeneic HSCT two months after NS7CAR-T therapy. Two of the three remained leukemia-free on day 752 and day 315, respectively. However, the remaining patient died on day 241 due to transplant-related mortality. Regarding safety, 80% of patients had mild CRS, 7 patients had grade 1 CRS, one had grade 2 CRS, and two patients had grade 3 CRS. None of the patients had neurotoxicity. One of the patients with prior allogeneic HSCT, developed mild skin GVHD after CAR-T therapy (77).
CLL-1 is a receptor highly expressed in AML cells. However, it is not found in healthy HSCs. The caveat is that CLL-1 expression is observed on all monocytes, early hematopoietic cells, and rarely non-hematological cells (78). In the 2022 Jin et al. phase 1 clinical study, 70% of R/R AML patients with CLL-1 CAR-T cells observed complete response with incomplete hematologic recovery (79). The median follow-up time was 173 days. All patients developed cytokine release syndrome (four of which were low-grade, six being high-grade). No patient developed CAR-T cell-related encephalopathy syndrome. All patients experienced severe pancytopenia, and two died due to infection from chronic agranulocytosis due to off-target toxicity.
NKG2D ligands (NKG2DL) are typically upregulated in cells that undergo malignant transformation, and other settings and are typically absent on healthy tissue (80). Additionally, NKG2DL has been reported to be expressed on AML cells and is an attractive target (80). In a phase I trial, NKG2DL was evaluated as a target for autologous CAR-T cells, with one patient experiencing a transient response (81). The THINK study is a multinational (EU/US) open-label Phase I trial that looked at CYAD-01, which is a CAR engineered with NKG2D receptor (82). The population studied is patients with R/R AML, myelodysplastic syndromes, or multiple myeloma with at least one line of treatment therapy. These patients are based in five hospitals in the USA and Belgium. Regarding efficacy, 25% achieved an objective response. Of the AML patients, three were treated with dose level 1 of CYAD-01, three were treated with dose level 2, and six were treated with dose level 3. Of the 16 patients treated, 44% had grade 3 or 4 TRAEs, and 31% had grade 3 or 4 CRS. One DLT of CRS was observed with dose level 3. No treatment-related deaths were observed.
Similar to CAR-T cells, TCR-T cells are engineered T cells. However, instead of a synthetic receptor (CAR), they express a naturally occurring T cell receptor (TCR) that targets specific tumor antigens. The targeted antigens must be highly expressed on leukemic cells but limited to no expression on healthy hematopoietic cells. If the chosen antigen is found in healthy cells, TCR-T has to be short-lived. Several tumor-associated antigens (TAA) have been identified for preclinical trials with TCR-T. These include categories: overexpressed antigens (i.e. surviving, TERT), lineage-restricted antigens (i.e., WT1), minor histocompatibility A (HA)-1, neoantigens (i.e., NPM1 and CBFB-MHY11), and cancer-testis antigen (i.e., NY-ESO-1, MAGE, PRAME) (83).
A 2017 preclinical study showed that TERT TCR-T efficiently lyses AML cells in vitro and inhibits tumor growth in the AML xenograft model (84). A 2018 preclinical study showed that HA1 TCR-T demonstrated potential killing in AML/LCL cell lines (85). A 2020 preclinical study using CBFB-MHY11 for TCR-T exhibited potent antileukemic activity against AML cells and in the xenograft model (86). Lastly, a 2020 preclinical study with HLA-DPB1 TCR demonstrated highly lysed AML in vitro and in xenograft model (87). Most recent clinical trials utilize WT1 as the TAA. A phase I clinical trial (NCT01640301) used EBV-specific donor CD8+ T cells inserted with WT1-specific TCR amongst 12 AML patients with high risk for HCT-related relapse. High-risk patients were identified by genetics, including HLA-A*0201 expression. Relapse-free survival was achieved in 100% of patients with a median of 44 months after infusion (88). Regarding safety, two patients developed grade III CRS. Eight patients developed grade III lymphopenia, and five patients developed grade IV lymphopenia. Two patients developed grade III thrombocytopenia, one exhibited grade III reduced neutrophil count, and one exhibited grade IV reduced neutrophil count. Lastly, seven patients developed grade III anemia.
Tumor-infiltrating lymphocytes infiltrate the tumor microenvironment and have the potential to recognize and kill cancer cells (89). One study investigated ex vivo isolation, expansion, and bioengineering of specific subsets of tumor-infiltrating lymphocytes (TILs) from the bone marrow of AML patients (90). Two subsets of TILS CCR7+CD95-/or and CD62L+CD45RA+ have been shown to possess anti-tumor activity (90). Researchers were able to isolate the desired TILS through the use of magnetic-activated cell sorting and flow cytometry.
Novel approaches are being analyzed to target AML while sparing normal hematopoiesis. Researchers also used bioengineering of TILs to enhance anti-tumor activity by using gene-edited hematopoietic stem and progenitor cells (HSPCs) to regenerate an antigen-negative myeloid system (91). They genetically modified TILs to express a chimeric antigen receptor (CAR) that targeted CD33, which is highly expressed in AML. The study found that in vitro differentiated CD33 KO HSPCs had reduced levels of CD33 protein expression (27% ± 4%) compared to controls (92% ± 3%) (91). Mice were injected with control, or CD33 KO HSPCs, and their human cell engraftment was followed over time in peripheral blood. It was found that mice who were injected with CD33 KO HSPC had diminished expression of CD33 sustained over time. The total number of CD45+ cells and myeloid subsets remained nearly identical between the two groups. The CAR-modified TILS showed enhanced cytotoxicity against AML cells in vitro, which suggests the potential ability to improve the efficacy of TIL-based immunotherapy in AML.
TIL-based treatments for AML are in the early stages of development with multiple possible beneficial combinational therapeutic approaches. TILs combined with hypomethylating agents and checkpoint inhibitors may enhance their anti-tumor activity.
Natural killer (NK) cells play an enormous role in innate immunity by targeting infectious agents and tumor cells. The interplay between NK cells and cytokine secretion is dictated by activating or inhibitory receptors that bind ligands on these target cells, ultimately allowing NK cell activation and killing targets (92). In AML, malignant cells evade the immune system by suppressing NK cell function. NK cell therapy is a novel immunologic approach to treatment in AML, in which suppression is prevented, allowing these cells to perform cell-specific targeted killing.
There have been challenges with harvesting NK cell lines for therapy. NK cells from donor peripheral blood mononuclear cells (PBMCs) express markers like CD16, NKG2D, NKp44, and NKp46, which allow them to recognize and kill non-self-cells such as tumor cells. Unfortunately, the quantity of NK cells in peripheral blood is low and not cost- or time-effective. Induced pluripotent stem cells are a good source of NK cells. However, they express low levels of CD16, therefore making them less cytotoxic. NK-92 cell lines are an alternative option. They are immortalized NK lymphoma cells; but have their own safety concerns (92).
CD94-NKG2A is another target for immune checkpoint therapy. It is an inhibitory receptor on NK cells and CD8+ T cells that inhibits the cytotoxic function of these respective cells (93). This receptor is inhibited by HLA-E, a non-classical MHC class I molecule found on normal and neoplastic hematopoietic cells. The 2016 Ruggeri, L. et al. study demonstrated that the humanized anti-NKG2A antibody converts NKG2A+ NK cells into effector NK cells that induce tumor cell death by killing most HLA-E+ NK resistant lymphohematopoietic cells such as dendritic cells, myeloid cells, B and T lymphocytes, and leukemic cells including AML amongst other cell types.
Recently, interest in memory NK cells has grown. These cells have differentiated in response to IL-12, IL-15, and IL-18 cytokines. These cells are specifically called cytokine-induced memory-like differentiation (ML NK) and have shown enhanced effector function and prolonged survival in various leukemic models (94). A phase I/II clinical trial investigates the best dose and side effects of activated natural NK cells in treating patients with R/R AML and R/R MDS. Chemotherapy will be given before donor NK cell infusions, which will slow the growth of cancer cells and prevent the recipient’s immune system from rejecting these donor cells. NK infusion will be from a related MHC-haploidentical donor. These modified NK cells may help strengthen the immune response to targeting and killing leukemic cells. Aldesleukin, an IL-2 cytokine, will be administered for two weeks following NK infusion to support NK growth. The study had 15 patients, 7 of whom achieved complete remission, supporting the use of allogeneic ML NK cells as therapy for relapsed AML. The caveat is that allogeneic donor NK cells were eliminated in several weeks after receiving T-cell recovery. Therefore, ML NK cell persistence could not be investigated (94).
A phase II clinical trial does study ML NK cell persistence. Patients with relapsed AML who received haploidentical hematopoietic cell transplants also received ML NK cells from their HCT donors, preventing NK cell allo-rejection. On day 28, complete remission was achieved in more than 90% of patients. Patients who have relapsed with AML after HCT and who received salvage therapy followed by T cell donor lymphocyte infusion (DLI) showed poorer outcomes. Therefore, the effects of donor ML NK infusion added to salvage therapy and T cell DLI for relapsed AML patients after HSCT were studied. The salvage therapy was fludarabine, cytarabine, and granulocyte-colony stimulating factor. T cell DLI and ML NK infusions were derived from the same HCT donor. The NK infusions were safe, and no patient experienced cytokine release syndrome. This regimen was well tolerated; only one patient exhibited mild skin GVHD. Within one month, 4 of 9 patients achieved complete remission, and 2 achieved partial remission. On day 100, two more patients reached complete remission. Additionally, with this immune-compatible therapy, ML NK cells persisted in PB and bone marrow for at least three months in most patients. These ML NK cells drawn from recipient PB, or bone marrow displayed anti-leukemic characteristics within in vitro assays and retained their phenotype over time (95).
Edited T cells are a significant area of study for CAR-T Therapy, however, this approach faces several limitations, including the availability of autologous T cells, complications with therapies, and resistance (96–98). The tumor killing ability of the T cells is also suppressed by increasing Treg subsets and senescent and exhausted T compartment (9, 99).
Research has demonstrated that mouse OP9 stromal cells expressing Notch logan DLL1 can induce T cell lineage commitment in vitro from natural mouse hematopoietic progenitor cells (100). This was later expanded with the use of human blood progenitor cells in the presence of OP9 stromal cells expressing human NOTCH1 ligand DLL1 (101). This work has illustrated the ability to generate T-cells from hematopoietic stem cells utilizing an in vitro co-culture system.
One of the future goals is to develop a method capable of generating immunocompetent and therapeutic T lymphopoiesis from unlimited and gene-editable PSCs (102). Pluripotent stem cells have a unique advantage, with efficient gene editing and self-renewal properties in vitro, making them valuable candidates for T-cell generation (103). Runx1 and Hoxa9, during the early stages of endothelial-to-hematopoietic transition to hematopoietic progenitor maturation in the PSC differentiation scheme in vitro, produced a type of induced hematopoietic progenitor cells (iHPCs) that could home to the thymus, engraft, and generate induced T cells (iT cells) with diverse TCRαβ repertoire in immunodeficient mice (102). These iT cells helped restore immune surveillance function in immunodeficient mice and also possessed anti-tumor activities in vivo when engineered to carry tumor antigen-specific TCR at the PSC stage. These findings help create a potential link to the unlimited and editable PSC source and T cell-based immunotherapy, potentially overcoming the current limitations faced by CAR-T therapies.
There is a growing focus on antileukemic vaccinations as therapy for active leukemia and prevention of relapse in high-risk patients (Table 5). Vaccine therapies are designed to elicit cellular and humoral immune responses against leukemic myeloid blasts by targeting overexpressed or specific leukemic antigens, i.e., Tumor-associated antigens (TAAs). Current TAAs targets include Wilms tumor 1 (WT1), Preferentially expressed antigen of melanoma (PRAME), Proteinase 3 (PR3), receptor for hyaluronic acid-mediated motility (RHAMM), New York esophageal squamous cell carcinoma 1 (NY-ESO-1) and mucin 1 protein (MUC1) with WT1 being the most actively studied at this time (104). While conventional peptide-based vaccines are being studied, current research on eliciting dendritic cells as key antigen-presenting cells also shows promise. Dendritic cell dysfunction is an essential method of evasion in leukemia progression. However, by taking dendritic cells and loading them with these TAAs, there is hope to close this gap in immune cancer surveillance. This approach remains early in development in AML and may require a combinatorial approach that includes agents that augment the adaptive immune system. These trials have generally shown tolerable safety profiles, with many reporting only injection site irritation. Larger phase II/III trials will be required to determine the true potential of these therapies.
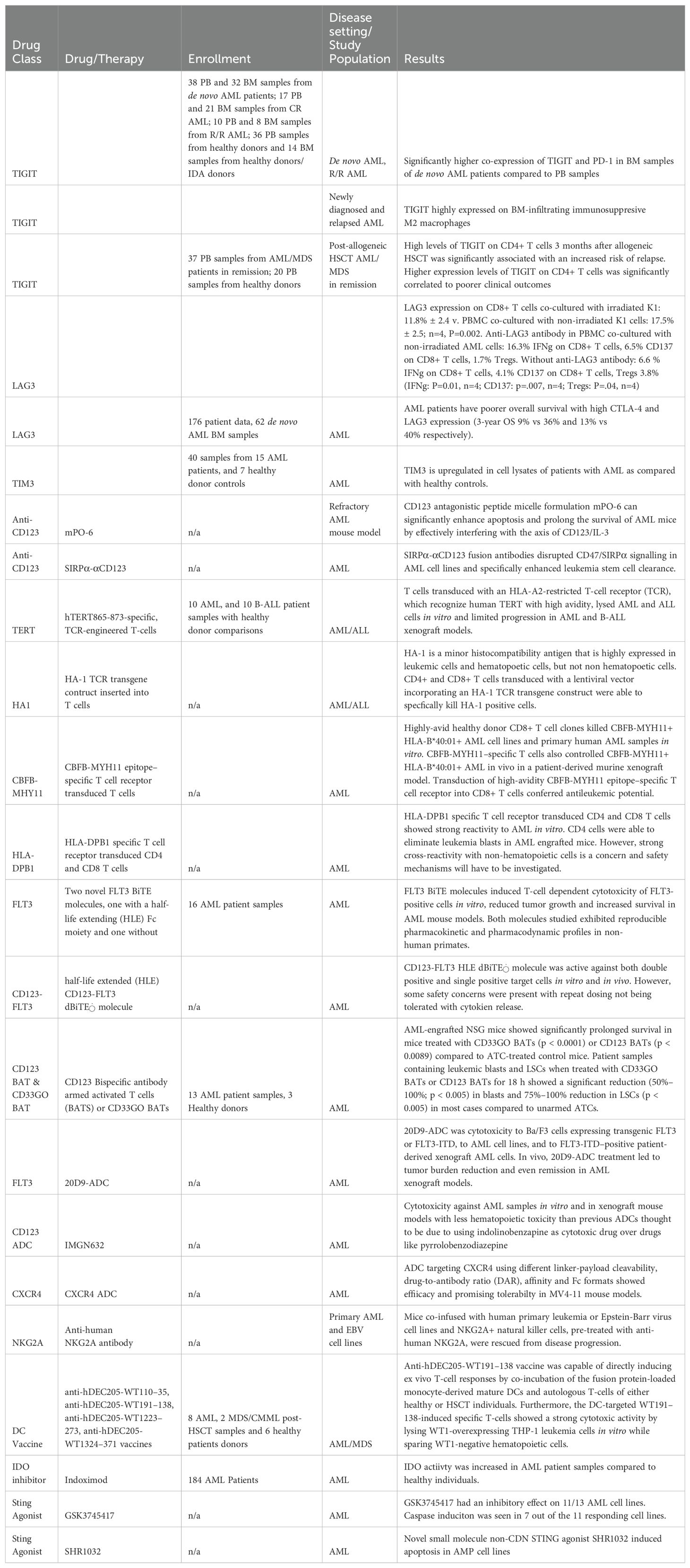
Table 5. Preclinical Studies investigating several upcoming therapy targets.
High WNT1 expression is associated with poor prognosis, which is often seen in AML. Anguille et al. reported from their phase II trial investigating dendritic cell vaccination. The study had 30 AML patients who were at increased risk for relapse were vaccinated with dendritic cells that were electroporated with WT1 mRNA. There were 13 of 30 study participants who responded to treatment, with 9 achieving molecular remission measures by normalization of WT1 levels and 4 showing disease stabilization. The five-year overall survival of the responders was 53.8% vs 25.0% in the nonresponder group (105). While this initial data is promising, larger control trials will need to be done, given that this study primarily compared nonresponders and responders with a relatively small sample size. Ogasawara et al. also studied 9 AML and 2 acute lymphoblastic leukemia (ALL) patients treated with peptide-loaded DCs in 2022. They saw an increase in progression-free survival, although no statistically significant increase in overall survival. They also observed a decrease in regulatory T cells following vaccination, suggesting DC vaccination may contribute to a reversal of immunosuppression.
Some groups have tried to increase the expression of these antigens on dendritic cells, with Dagvadori et al. creating a WTI-DEC205 fusion protein. This chimeric anti-hDEC205-WT191-138 directly induced ex-vivo responses when fusion protein-loaded DC were co-incubated with T-cells. Additionally, the co-incubated T-cells showed a robust cytotoxic activity lysing WT1-overexpressing THP-1 leukemia cells while sparing WT1-negative hematopoietic cells in vitro (104).
Several other phase 1 trials have tried combining multiple TAAs with multiple trials giving vaccines with DCs loaded with WT1 and PRAME. Lichtenegger et al. examined 10 patients treated with vaccines using WT1 and PRAME. They saw a greater immune response in those under 65, but this did not reach a statistically significant difference between responder and nonresponder OS (106). Floisand, Y et al. looked at 20 patients who had a 5-year OS of 75% but did not directly compare this to a control or nonresponder group (106). Loosdrecht et al. looked at 12 patients treated with DCP-001, now known as vididencel, a dendritic vaccine using WT1 and PRAME. They observed an immune response in 7 of the 12 patients. Patients who responded had significantly increased OS (107, 108). A follow-up study, ADVANCE-II (NCT03697707) has preliminary results from 20 patients reported at ASH 2023. They reported findings suggesting vaccinations may increase dendritic cell populations, leading to increased T-cell activity against tumors; 2-year RFS and OS were estimated at 59% and 73%, respectively (109, 110).
NY-ESO-1 has been a promising target in many cancers, given its expression profile in healthy tissue, mostly limited to embryonic development and germ cells. However, expression is seen in many cancers, and treatment with hypomethylating agents such as decitabine can enhance this expression by acting on the hypermethylated promoter of NY-ESO-1 (111). Griffith et al. looked to take advantage of this by combining decitabine with a NY-ESO-1 fusion protein vaccine in 9 MDS patients (112). A total of 7 patients completed the entire treatment course, and two discontinued study treatments due to events deemed unrelated to the vaccine. All 7 patients showed NY-ESO-1 expression induction. Of those 7, 6 showed NY-ESO-1 specific CD4+ cell responses, and 4 showed NY-ESO-1 specific CD8+ cell responses. Dendritic cells were again thought to be a key component in T-cell activation, with increased CD141Hi DCs at diagnosis being associated with immune response (112).
NCT01956630 was a 2-stage, phase 1 trial that looked at using genetically modified DC carrying TAAs Survivin and MUC1 and an RNA interference moiety to suppress SOCS1. In Stage 1, safety was assessed, with 23 patients with relapsed AL receiving the intervention and 25 receiving standard donor lymphocytes. The treatment was well tolerated, with no documented stage 3 or 4 GVHD. An increase in the 3-year OS was seen 48.9% in the intervention group vs 27.5% in the control group. Stage 2 looked at 12 relapsed AML patients, with 83% achieving complete remission (113).
Stimulator of Interferon Genes (STING) is a transmembrane protein in the endoplasmic reticulum first identified in 2008 as part of a signaling cascade leading to the production of type-1 interferon (IFN) among other inflammatory cytokines and signaling molecules (114). Type-1 IFN in sufficiently increased concentrations leads to cancer cell cycle arrest, modulation of apoptosis, and increased activity of tumor-invading lymphocytes (115). Endogenously activated by cytosolic DNA, this increased release of type-1 IFN is seen in solid tumors but is not seen in AML, suggesting a possible avenue to increase the host innate immune response to these malignancies (116).
Drugs targeting the pathway have mostly been cyclic di-nucleotide (CDN) mimics of endogenous ligands. However, these have had disappointing results thus far, with Novartis halting trials using its ADU-S100 compound due to a lack of efficacy in solid tumors. Newer small molecule agonists have generated interest with greater potency. Adam et al. demonstrated this with GSK3745417 exerting a growth inhibitory effect on 11 out of 13 tested AML cell lines (117). Further experiments showed caspase induction in 7 of those 11 cell lines, supporting an apoptotic mechanism behind the observed inhibition. The compound also blocked colony formation in AML samples from 5 donors (117). The makers, GlaxoSmithKline, are currently conducting a phase I trial to assess the therapeutic potential of this compound. Additionally, SHR1032, another non-CDN small molecule STING agonist, has generated interest in preclinical in vivo studies inducing apoptosis in AML (118).
IDO (indoleamine 2,3-dioxygenase) is an enzyme that can suppress the immune system when activated. Inhibitors of this enzyme help counteract this suppression and allow the immune system to attack cancer cells effectively (119). Studies have shown that IDO is expressed by bone marrow and peripheral blood AML blasts, while CD34+ hematopoietic precursor cells do not express the IDO protein (119). Additionally, one study showed increased IDO enzymatic activity in the blood of AML patients compared to controls (120). One study also found that IDO and FOXP3 mRNA were upregulated in AML patients. There was a positive correlation between IDO and FOXP3, indicating that IDO expression may be associated with increased Treg phenotype (121). The inhibition of IDO may be a promising approach, along with chemotherapy, in patients with upregulated IDO expression.
Indoximod is an immunometabolic adjuvant that works through suppression of master metabolic kinase mTORC1 that occurs in tryptophan-depleted cells (122). The Indoximod method of action is essential because AML cells can often experience inhibition of mTORC1 activity, which can impair T cell function and immune response (122). One study analyzed IDO-1 expression by quantifying by a novel composite IDO-1 score’ on diagnostic bone marrow biopsies of AML patients. Univariate analysis of the study found higher IDO-1 mRNA (p=0.005), higher composite IDO-1 score (p<0.0001), and multivariate model retained higher composite IDO-1 score predicted poor survival outcomes. Patients who failed induction had a higher composite IDO-1 score (p=0.01).
Indoximod has been studied in clinical trials with idarubicin/cytarabine in newly diagnosed AML. The study was an open-label, multicenter, phase 1 study (NCT02835729) that analyzed newly diagnosed AML patients treated with indoximod in combination with induction chemotherapy. Overall, 31 patients were enrolled, with 25 completing the study. The most frequent AEs were febrile neutropenia (60%) and hypoxia (16%). Overall, 21/25 (84%) of patients achieved remission (CR/CRh/CRi/CRp), and 15 of 19 (79%) in the per-protocol analysis achieved remission. All patients in the study who received HiDAC became MRD-negative. The median relapsed free and overall survival have yet to be reported. The study aimed to enhance the efficacy of chemotherapy by targeting the IDO pathway and relieving tryptophan depletion-mediated mTORC1 suppression (123). However, currently, there are no studies to our knowledge that have reported data on IDO in relapsed/refractory AML.
Over the last several years, immunotherapy has revolutionized the treatment and outcomes of cancer patients. The role of immunotherapy in the treatment of patients with AML has come to light more clearly only recently. These studies evaluating various immunotherapeutic approaches remain in the very early stages with limited numbers of patients, although the results of these studies are encouraging with signals of efficacy.
This review summarizes the most up-to-date novel advances for immunotherapies in relapsed/refractory AML. It is of particular interest for those patients ineligible for allogeneic stem cell transplantation or with relapsed or refractory disease resistant to standard cytotoxic chemotherapies. However, barriers to the evolution of immunotherapy in AML include limited AML-specific antigen expression, which can result in off-target toxicity, as well as the suppressive and dysfunctional immune milieu in heavily pre-treated AML patients. Although there is much enthusiasm for the deployment of immunotherapy in AML, based on the data generated to date, it is clear that agents targeting only the innate vs. adaptive compartments are of limited value. In addition to chemotherapy, a combinatorial approach employing agents that target both the innate and adaptive may be warranted. Furthermore, the timing of administration of immunotherapy is likely crucial, as such approaches in the relapsed/refractory setting may be hindered by insufficient T-cells, exhausted T-cells, or inadequate antigen presenting cell function. Rational cellular therapeutics, in combination with some of the more recently approved targeted agents applied earlier in the disease course, may be of particular value.
Moreover, a better understanding of the immune microenvironment within AML can better inform the development of immunotherapeutic strategies for AML. Moreover, it is crucial to identify biomarkers of response to these immunotherapeutic agents to better identify patients that may benefit from these approaches.
In summary, the complexity of AML has led to the exploration of novel immunotherapies to help identify specific targets and minimize immune escape. Despite reservations regarding low antigenicity and having long been considered a “cold” tumor, immunotherapy remains a highly promising strategy for patients with AML.
SB: Writing – original draft, Writing – review & editing. SG: Writing – original draft, Writing – review & editing. MB: Writing – original draft, Writing – review & editing. AP: Conceptualization, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hou HA, Tien HF. Genomic landscape in acute myeloid leukemia and its implications in risk classification and targeted therapies. J BioMed Sci. (2020) 27:81. doi: 10.1186/s12929-020-00674-7
2. Dohner H, Estey E, Amadori S, Appelbaum F, Büchner T, Burnett A, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. (2010) 115:453–74. doi: 10.1182/blood-2009-07-235358
3. Bolon Y, Atshan R, Allbee-Johnson M, Estrada Merly N, Lee SJ. Current use and outcome of hematopoietic stem cell transplantation: CIBMTR summary slides. (2022).
4. Atallah E, Cortes J, O'Brien S, Pierce S, Rios MB, Estey E, et al. Establishment of baseline toxicity expectations with standard frontline chemotherapy in acute myelogenous leukemia. Blood. (2007) 110:3547–51. doi: 10.1182/blood-2007-06-095844
5. Etienne A, Esterni B, Charbonnier A, Mozziconacci MJ, Arnoulet C, Coso D, et al. Comorbidity is an independent predictor of complete remission in elderly patients receiving induction chemotherapy for acute myeloid leukemia Cancer (2007) 109(7):1376–83. doi: 10.1002/cncr.22537
6. Giles FJ, Borthakur G, Ravandi F, Faderl S, Verstovsek S, Thomas D, et al. The haematopoietic cell transplantation comorbidity index score is predictive of early death and survival in patients over 60 years of age receiving induction therapy for acute myeloid leukaemia. Br J Haematol. (2007) 136:624–7. doi: 10.1111/j.1365-2141.2006.06476.x
7. Kantarjian H, O’brien S, Cortes J, Giles F, Faderl S, Jabbour E, et al. Results of intensive chemotherapy in 998 patients age 65 years or older with acute myeloid leukemia or high-risk myelodysplastic syndrome: predictive prognostic models for outcome. Cancer. (2006) 106:1090–8. doi: 10.1002/cncr.v106:5
8. Shiravand Y, Khodadadi F, Kashani SMA, Hosseini-Fard SR, Hosseini S, Sadeghirad H, et al. Immune checkpoint inhibitors in cancer therapy. Curr Oncol. (2022) 29:3044–60. doi: 10.3390/curroncol29050247
9. Guo R, Lü M, Cao F, Wu G, Gao F, Pang H, et al. Single-cell map of diverse immune phenotypes in the acute myeloid leukemia microenvironment. biomark Res. (2021) 9:15. doi: 10.1186/s40364-021-00265-0
10. Guo L, Zhang H, Chen B. Nivolumab as programmed death-1 (PD-1) inhibitor for targeted immunotherapy in tumor. J Cancer. (2017) 8:410–6. doi: 10.7150/jca.17144
11. Chauvin JM, Zarour HM. TIGIT in cancer immunotherapy. J Immunother Cancer. (2020) 8:e000957. doi: 10.1136/jitc-2020-000957
12. Gomez-Llobell M, Raindo AP, Medina JC, Centurion IG, Oregueira AM. Immune checkpoint inhibitors in acute myeloid leukemia: A meta-analysis. Front Oncol. (2022) 12:882531. doi: 10.3389/fonc.2022.882531
13. Daver N, Garcia-Manero G, Basu S, Boddu PC, Alfayez M, Cortes JE, et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: A nonrandomized, open-label, phase II study. Cancer Discovery. (2019) 9:370–83. doi: 10.1158/2159-8290.CD-18-0774
14. Gojo I, Stuart R, Webster J, Blackford A, Carols Varela J, Morrow J, et al. Multi-center phase 2 study of pembroluzimab (Pembro) and azacitidine (AZA) in patients with relapsed/refractory acute myeloid leukemia (AML) and in newly diagnosed (≥65 years) AML patients. Blood. (2019) 134:832. doi: 10.1182/blood-2019-127345
15. Zeidan AM, Boss I, Beach CL, Copeland WB, Thompson E, Fox BA, et al. A randomized phase 2 trial of azacitidine with or without durvalumab as first-line therapy for older patients with AML. Blood Adv. (2022) 6:2219–29. doi: 10.1182/bloodadvances.2021006138
16. Xu S, Zhang M, Fang X, Hu X, Xing H, Yang Y, et al. CD123 antagonistic peptides assembled with nanomicelles act as monotherapeutics to combat refractory acute myeloid leukemia. ACS Appl Mater Interf. (2022) 14:38584–93. doi: 10.1021/acsami.2c11538
17. Brauneck F, Zhang M, Fang X, Hu X, Xing H, Yang Y, et al. TIGIT blockade repolarizes AML-associated TIGIT(+) M2 macrophages to an M1 phenotype and increases CD47-mediated phagocytosis. J Immunother Cancer. (2022) 10:e004794. doi: 10.1136/jitc-2022-004794
18. Gournay V, Vallet N, Peux V, Vera K, Bordenave J, Lambert M, et al. Immune landscape after allo-HSCT: TIGIT- and CD161-expressing CD4 T cells are associated with subsequent leukemia relapse. Blood. (2022) 140:1305–21. doi: 10.1182/blood.2022015522
19. Penter L, Wu CJ. AML relapse after a TIGIT race. Blood. (2022) 140:1189–91. doi: 10.1182/blood.2022017614
20. Long L, Zhang X, Chen F, Pan Q, Phiphatwatchara P, Zeng Y, et al. The promising immune checkpoint LAG-3: from tumor microenvironment to cancer immunotherapy. Genes Cancer. (2018) 9:176–89. doi: 10.18632/genesandcancer.v9i5-6
21. Abdelhakim H, Cortez LM, Li, Meizhang BS, Braun M, Skikne BS, Lin TL, et al. LAG3 inhibition decreases AML-induced immunosuppression and improves T cell-mediated killing. Blood. (2019) 134:3605. doi: 10.1182/blood-2019-129455
22. Chen C, Liang C, Wang S, Chio CL, Zhang Y, Zeng C, et al. Expression patterns of immune checkpoints in acute myeloid leukemia. J Hematol Oncol. (2020) 13:28. doi: 10.1186/s13045-020-00853-x
23. Buecklein VL, Warm M, Spiekermann K, Schmidt C, Unterhalt M, Dver N, et al. Trial in progress: an open-label phase II study of relatlimab with nivolumab in combination with 5-azacytidine for the treatment of patients with relapsed/refractory and elderly patients with newly diagnosed acute myeloid leukemia (AARON). Blood. (2022) 140:3227–8. doi: 10.1182/blood-2022-169867
24. Rezaei M, Tan J, Zeng C, Li Y, Ganjalikhani-Hakemia M. TIM-3 in leukemia; immune response and beyond. Front Oncol. (2021) 11:753677. doi: 10.3389/fonc.2021.753677
25. Al-Amrani S, Al-Zadjali F, Jeelani Y, Al-Jabri Z, Al Bulushi MP, Al-Rawahi M, et al. TIM3 is upregulated in cell lysates of patients with acute myeloid leukemia. Blood. (2022) 140:11483–4. doi: 10.1182/blood-2022-168946
26. Brunner A, Traer E, Vey N, Scholl S, Tovar N, Porkka K, et al. Allogeneic hematopoietic cell transplantation outcomes of patients with R/R AML or higher-risk MDS treated with the TIM-3 inhibitor MBG453 (Sabatolimab) and hypomethylating agents. Blood. (2021) 138:3677–7. doi: 10.1182/blood-2021-151455
27. Sabatolimab as a Treatment for Patients With Acute Myeloid Leukemia and Presence of Measurable Residual Disease After Allogeneic Stem Cell Transplantation (2024). Available online at: https://clinicaltrials.gov/study/NCT04623216. (accessed March 15, 2024)
28. Zeiser R, Devillier R, Caterina Mico M, Valcarcel D, Call S, Niederwieser C, et al. TIM-3 inhibitor sabatolimab for patients with acute myeloid leukemia (AML) with measurable residual disease (MRD) detected after allogeneic stem cell transplantation (AlloSCT): preliminary findings from the phase ib/II stimulus-AML2 study. Blood. (2023) 142:59. doi: 10.1182/blood-2023-180876
29. Zeidan AM, Ando K, Rauzy O, Turgut M, Wang MC, Cairoli R, et al. Sabatolimab plus hypomethylating agents in previously untreated patients with higher-risk myelodysplastic syndromes (STIMULUS-MDS1): a randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Haematol. (2024) 11:e38–50. doi: 10.1016/S2352-3026(23)00333-2
30. Mo J, Den L, Peng K, Ouyang S, Ding W, Lou L, et al. Targeting STAT3-VISTA axis to suppress tumor aggression and burden in acute myeloid leukemia. J Hematol Oncol. (2023) 16:15. doi: 10.1186/s13045-023-01410-y
31. Pang WW, Pluvinage JV, Price EA, Sridhar K, Arber DA, Greenberg PL, et al. Hematopoietic stem cell and progenitor cell mechanisms in myelodysplastic syndromes. Proc Natl Acad Sci U.S.A. (2013) 110:3011–6. doi: 10.1073/pnas.1222861110
32. Daver N, Stein A, Bixby D, Chai-Ho W, Zeidner J, Maher K, et al. Safety, pharmacodynamic, and anti-tumor activity of SL-172154 as monotherapy and in combination with azacitidine (AZA) in relapsed/refractory (R/R) acute myeloid leukemia (AML) and higher-risk myelodysplastic syndromes/neoplasms (HR-MDS) patients (pts). Blood. (2023) 142:4278. doi: 10.1182/blood-2023-173991
33. Meng Z, Wang Z, Guo B, Cao W, Shen H. TJC4, a differentiated anti-CD47 antibody with novel epitope and RBC sparing properties. Blood. (2019) 134:4063. doi: 10.1182/blood-2019-122793
34. Behrens LM, van den Berg TK, van Egmond M. Targeting the CD47-SIRPalpha innate immune checkpoint to potentiate antibody therapy in cancer by neutrophils. Cancers (Basel). (2022) 14:3366. doi: 10.3390/cancers14143366
35. Qi J, Li J, Jiang B, Jiang B, Liu H, Cao X, et al. A phase I/IIa study of lemzoparlimab, a monoclonal antibody targeting CD47, in patients with relapsed and/or refractory acute myeloid leukemia (AML) and myelodysplastic syndrome (MDS): initial phase I results. Blood. (2020) 136:30–1. doi: 10.1182/blood-2020-134391
36. Sallman DA, Asch AS, Kambhampati S, Al Malki MM, Zeidner JF, et al. 330 the first-in-class anti-CD47 antibody magrolimab combined with azacitidine is well-tolerated and effective in AML patients: phase 1b results. Clinical Lymphoma Myeloma and Leukemia. Elsevier (2021) 21.
37. Daver N, Konopleva M, Maiti A, Loghavi S, Kadia TM, DiNardo CD, et al. Phase I/II study of azacitidine (AZA) with venetoclax (VEN) and magrolimab (Magro) in patients (pts) with newly diagnosed (ND) older/unfit or high-risk acute myeloid leukemia (AML) and relapsed/refractory (R/R) AML. Blood. (2022) 140:141–4. doi: 10.1182/blood-2022-170188
38. Liu J, Wang L, Zhao F, Tseng S, Narayanan C, Shura L, et al. Pre-clinical development of a humanized anti-CD47 antibody with anti-cancer therapeutic potential. PloS One. (2015) 10:e0137345. doi: 10.1371/journal.pone.0137345
39. Deng M, Gui X, Kim J, Xie L, Chen W, Li Z, et al. LILRB4 signalling in leukaemia cells mediates T cell suppression and tumour infiltration. Nature. (2018) 562:605–9. doi: 10.1038/s41586-018-0615-z
40. Dinardo C, Pollyea D, Aribi A, Jonas B, Jeyakumar D, Roboz G, et al. P536: A first-in-human phase 1 study of io-202 (Anti-lilrb4 mab) in acute myeloid leukemia (Aml) with monocytic differentiation and chronic myelomonocytic leukemia (Cmml) patients. Hemasphere. (2023) 8:e605335a. doi: 10.1097/01.HS9.0000969052.60533.5a
41. Ehninger A, Kramer M, Rollig C., Thiede C, Bornhauser M, von Bonin M, et al. Distribution and levels of cell surface expression of CD33 and CD123 in acute myeloid leukemia. Blood Cancer J. (2014) 4:e218. doi: 10.1038/bcj.2014.39
42. He SZ, Busfield S, Ritchie DS, Hertzberg MS, Durrant S, Lewis ID, et al. A Phase 1 study of the safety, pharmacokinetics and anti-leukemic activity of the anti-CD123 monoclonal antibody CSL360 in relapsed, refractory or high-risk acute myeloid leukemia. Leuk Lymph. (2015) 56:1406–15. doi: 10.3109/10428194.2014.956316
43. Lamble AJ, Kosaka Y, Laderas T, Maffit A, Kaempf A, Brady LK, et al. Reversible suppression of T cell function in the bone marrow microenvironment of acute myeloid leukemia. Proc Natl Acad Sci U.S.A. (2020) 117:14331–41. doi: 10.1073/pnas.1916206117
44. Tahk S, Vick B, Hiller B, Schmitt S, Marcinek A, Perini ED, et al. SIRPalpha-alphaCD123 fusion antibodies targeting CD123 in conjunction with CD47 blockade enhance the clearance of AML-initiating cells. J Hematol Oncol. (2021) 14:155. doi: 10.1186/s13045-021-01163-6
45. Bross PF, Beitz J, Chen G, Chen XH, Duffy E, Kieffer L, et al. Approval summary: gemtuzumab ozogamicin in relapsed acute myeloid leukemia. Clin Cancer Res. (2001) 7:1490–6.
46. Petersdorf SH, Kopecky KJ, Slovak M, Willman C, Nevill T, Brandwein J, et al. A phase 3 study of gemtuzumab ozogamicin during induction and postconsolidation therapy in younger patients with acute myeloid leukemia. Blood. (2013) 121:4854–60. doi: 10.1182/blood-2013-01-466706
47. Hills RK, Castaigne Appelbaum S, Delaunay FR, Petersdorf J, Othus S, M, et al. Addition of gemtuzumab ozogamicin to induction chemotherapy in adult patients with acute myeloid leukaemia: a meta-analysis of individual patient data from randomised controlled trials. Lancet Oncol. (2014) 15:986–96. doi: 10.1016/S1470-2045(14)70281-5
48. Jen EY, et al. FDA approval: gemtuzumab ozogamicin for the treatment of adults with newly diagnosed CD33-positive acute myeloid leukemia. Clin Cancer Res. (2018) 24:3242–6. doi: 10.1158/1078-0432.CCR-17-3179
49. Ma H, Padmanabhan IS, Parmar S, Gong Y. Targeting CLL-1 for acute myeloid leukemia therapy. J Hematol Oncol. (2019) 12:41. doi: 10.1186/s13045-019-0726-5
50. Daver N, Salhotra A, Brandwein JM, Podoltsev NA, Pollyea DA, Jurcic JG, et al. A Phase I dose-escalation study of DCLL9718S, an antibody-drug conjugate targeting C-type lectin-like molecule-1 (CLL-1) in patients with acute myeloid leukemia. Am J Hematol. (2021) 96:E175–9. doi: 10.1002/ajh.26136
51. Kovtun Y, Jones GE, Adams S, Harvey L, Audette CA, Wilhelm A, et al. A CD123-targeting antibody-drug conjugate, IMGN632, designed to eradicate AML while sparing normal bone marrow cells. Blood Adv. (2018) 2:848–58. doi: 10.1182/bloodadvances.2018017517
52. Daver N, Sweet K, Montesinos P, Wang E, Aribi A, DeAngelo D, et al. A phase 1b/2 study of IMGN632, a CD123-targeting antibody-drug conjugate (ADC), as monotherapy or in combination with venetoclax and/or azacitidine for patients with CD123-positive acute myeloid leukemia. Blood. (2020) 134:136. doi: 10.1182/blood-2020-139884
53. Daver N, Monesinos P, DeAngelo D, Wang E, Papadantonakis N, Deconinck E, et al. Clinical profile of IMGN632, a novel CD123-targeting antibody-drug conjugate (ADC), in patients with relapsed/refractory (R/R) acute myeloid leukemia (AML) or blastic plasmacytoid dendritic cell neoplasm (BPDCN). Blood. (2019) 134:734. doi: 10.1182/blood-2019-128648
54. Daver N, Monesinos P, Altman J, Wang E, Martinelli G, Roboz G, et al. Pivekimab sunirine (PVEK, IMGN632), a CD123-targeting antibody-drug conjugate, in combination with azacitidine and venetoclax in patients with newly diagnosed acute myeloid leukemia. Blood. (2023) 142:2906. doi: 10.1182/blood-2023-173413
55. Kubasch AS, Schulze F, Giagounidis A, Gotze KS, Krone J, Sockel K, et al. Single agent talacotuzumab demonstrates limited efficacy but considerable toxicity in elderly high-risk MDS or AML patients failing hypomethylating agents. Leukemia. (2020) 34:1182–6. doi: 10.1038/s41375-019-0645-z
56. Costa MJ, Kudaravalli J, Ma JT, Ho WH, Delaria K, Holz C. Optimal design, anti-tumour efficacy and tolerability of anti-CXCR4 antibody drug conjugates. Sci Rep. (2019) 9:2443. doi: 10.1038/s41598-019-38745-x
57. Roas M, Vick B, Kasper MA, Able M, Polzer H, Gerlach M, et al. Targeting FLT3 with a new-generation antibody-drug conjugate in combination with kinase inhibitors for treatment of AML. Blood. (2023) 141:1023–35. doi: 10.1182/blood.2021015246
58. Brauchle B, Goldstein RL, Karbowski CM, Henn A, Li CM, Bucklein VL, et al. Characterization of a novel FLT3 biTE molecule for the treatment of acute myeloid leukemia. Mol Cancer Ther. (2020) 19:1875–88. doi: 10.1158/1535-7163.MCT-19-1093
59. Barbullushi K, Rampi N, Serpenti F, Sciume M, Fabris S, Roberto PD, et al. Vaccination therapy for acute myeloid leukemia: where do we stand? Cancers (Basel). (2022) 14:2994. doi: 10.1158/1535-7163.MCT-19-1093
60. Kiyoi H, Kawashima N, Ishikawa Y. FLT3 mutations in acute myeloid leukemia: Therapeutic paradigm beyond inhibitor development. Cancer Sci. (2020) 111:312–22. doi: 10.1111/cas.v111.2
61. Turner AM, Lin NL, Issarachai S, Lyman SD, Broudy VC. FLT3 receptor expression on the surface of normal and Malignant human hematopoietic cells. Blood. (1996) 88:3383–90. doi: 10.1182/blood.V88.9.3383.bloodjournal8893383
62. Uckun FM, Lin TL, Mims AS, Patel P, Lee C, Shahidzadeh A, et al. A clinical phase 1B study of the CD3xCD123 bispecific antibody APVO436 in patients with relapsed/refractory acute myeloid leukemia or myelodysplastic syndrome. Cancers (Basel). (2021) 13(16):4113. doi: 10.3390/cancers13164113
63. Uy GL, Aldoss I, Foster MC, Sayre PH, Wieduwilt MJ, Advani AS, et al. Flotetuzumab as salvage immunotherapy for refractory acute myeloid leukemia. Blood. (2021) 137:751–62. doi: 10.1182/blood.2020007732
64. MacroGenics provides update on corporate progress and first quarter 2021 financial results. (2021).
65. Goldstein R, Karbowski C, Henn A, Deegen P, Wahl J, Matthes K, et al. Abstract 6313: Evaluation of a dual CD123-FLT3 BiTE molecule for acute myeloid leukemia. Cancer Res. (2022) 82:6313. doi: 10.1158/1538-7445.AM2022-6313
66. Friedrich M, Henn A, Raum T, Bajtus M, Matthes K, Hendrich L, et al. Preclinical characterization of AMG 330, a CD3/CD33-bispecific T-cell-engaging antibody with potential for treatment of acute myelogenous leukemia. Mol Cancer Ther. (2014) 13:1549–57. doi: 10.1158/1535-7163.MCT-13-0956
67. Ravandi F, Stein S, Kantarijan H, Walter R, Paschka P, Jongen-Lavrencic M, et al. A phase 1 first-in-human study of AMG 330, an anti-CD33 bispecific T-cell engager (BiTE) antibody construct, in relapsed/refractory acute myeloid leukemia (R/R AML). Blood. (2018) 25:25. doi: 10.1182/blood-2018-99-109762
68. Jacobson CA. CD19 chimeric antigen receptor therapy for refractory aggressive B-cell lymphoma. J Clin Oncol. (2019) 37:328–35. doi: 10.1200/JCO.18.01457
69. Budde L, Song J, Kim Y, Blanchard S, Wagner J, Stein A, et al. Remissions of acute myeloid leukemia and blastic plasmacytoid dendritic cell neoplasm following treatment with CD123-specific CAR T cells: A first-in-human clinical trial. Blood. (2017) 130. doi: 10.1182/blood.V130.Suppl_1.811.811
70. Sallman D, DeAngelo D, Pemmaraju N, Dinner S, Gill S, Olin R, et al. AMELI-01: A phase I trial of UCART123v1.2, an antiCD123 allogeneic CAR-T cell product, in adult patients with relapsed or refractory (R/R) CD123+Acute myeloid leukemia. Blood. (2022) 140:2371–3.
71. Tambaro FP, Singh H, Jones E, Rytting M, Mahadeo KM, Thompson P, et al. Autologous CD33-CAR-T cells for treatment of relapsed/refractory acute myelogenous leukemia. Leukemia. (2021) 35:3282–6. doi: 10.1038/s41375-021-01232-2
72. Chan T, Ma X, Carvajal-Borda F, Velez J, Plummer J, Shepard L, et al. Preclinical characterization of prgn-3006 ultracar-T™ for the treatment of AML and MDS: non-viral, multigenic autologous CAR-T cells administered one day after gene transfer. Blood. (2019) 134:2660. doi: 10.1182/blood-2019-130617
73. Sallman D, Elmariah H, Sweet K, Mishra A, Cox C, Chakaith M, et al. Phase 1/1b safety study of prgn-3006 ultracar-T in patients with relapsed or refractory CD33-positive acute myeloid leukemia and higher risk myelodysplastic syndromes. Blood. (2022) 140:10313–5. doi: 10.1182/blood-2022-169142
74. Zhang Y, Li Y, Cao W, Wang F, Xie X, Li Y, et al. Single-cell analysis of target antigens of CAR-T reveals a potential landscape of “On-target, off-tumor toxicity. Front Immunol. (2021) 12. doi: 10.3389/fimmu.2021.799206
75. Glisovic-Aplenc T, Diorio C, Chukinas JA, Veliz K, Shestova O, Shen F, et al. CD38 as a pan-hematologic target for chimeric antigen receptor T cells. Blood Adv. (2023) 7:4418–30. doi: 10.1182/bloodadvances.2022007059
76. Cui Q, Qian C, Xu N, Kang L, Dai H, Cui W, et al. CD38-directed CAR-T cell therapy: a novel immunotherapy strategy for relapsed acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. J Hematol Oncol. (2021) 14:82. doi: 10.1186/s13045-021-01092-4
77. Zhang X, Yang J, Li J, Qiu L, Hongxing L, Xiong M, et al. Naturally selected CD7-targeted chimeric antigen receptor (CAR)-T cell therapy for refractory/relapsed acute myeloid leukemia: phase I clinical trial. Blood. (2023) 142:218. doi: 10.1182/blood-2023-179086
78. Mardiana S, Gill S. CAR T cells for acute myeloid leukemia: state of the art and future directions. Front Oncol. (2020) 10:697. doi: 10.3389/fonc.2020.00697
79. Jin X, Zhang M, Sun R, Lyu H, Xiao X, Zhang X, et al. First-in-human phase I study of CLL-1 CAR-T cells in adults with relapsed/refractory acute myeloid leukemia. J Hematol Oncol. (2022) 15:88. doi: 10.1186/s13045-022-01308-1
80. Hilpert J, Gross-Hovest L, Grunebach F, Buechele C, Nuebling T, Raum T, et al. Comprehensive analysis of NKG2D ligand expression and release in leukemia: implications for NKG2D-mediated NK cell responses. J Immunol. (2012) 189:1360–71. doi: 10.4049/jimmunol.1200796
81. Baumeister SH, Murad J, Werner Daley H, Trebeden-Negre H, Gicobi JK, et al. Phase I trial of autologous CAR T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. Cancer Immunol Res. (2019) 7:100–12. doi: 10.1158/2326-6066.CIR-18-0307
82. Sallman D, Brayer J, Sagatys EM, Lonez C, Breman E, Agaugue S, et al. Remissions in relapse/refractory acute myeloid leukemia patients following treatment with NKG2D CAR-T therapy without a prior preconditioning chemotherapy. Blood. (2018) 132:902–2. doi: 10.1182/blood-2018-99-111326
83. Kang S, Li Y, Qiao J, Meng X, He Z, Gao X, et al. Antigen-specific TCR-T cells for acute myeloid leukemia: state of the art and challenges. Front Oncol. (2022) 12:787108. doi: 10.3389/fonc.2022.787108
84. Sandri S, De Sanctis F, Lamolinara A, Boschi F, Poffe O, Trovato R, et al. Effective control of acute myeloid leukaemia and acute lymphoblastic leukaemia progression by telomerase specific adoptive T-cell therapy. Oncotarget. (2017) 8:86987–7001. doi: 10.18632/oncotarget.18115
85. Dossa RG, Cunningham T, Sommermeyer D, Medina-Rodriguez I, Biernacki MA, Foster K, et al. Development of T-cell immunotherapy for hematopoietic stem cell transplantation recipients at risk of leukemia relapse. Blood. (2018) 131:108–20. doi: 10.1182/blood-2017-07-791608
86. Biernacki MA, Foster KA, Woodward KB, Coon ME, Cummings C, Cunningham TM, et al. CBFB-MYH11 fusion neoantigen enables T cell recognition and killing of acute myeloid leukemia. J Clin Invest. (2020) 130:5127–41. doi: 10.1172/JCI137723
87. Klobuch S, Hammaon K, Vatter-Leising S, Neidlinger E, Zwerger M, Wandel A, et al. HLA-DPB1 reactive T cell receptors for adoptive immunotherapy in allogeneic stem cell transplantation. Cells. (2020) 9:1264. doi: 10.3390/cells9051264
88. Chapuis AG, Egan DN, Bar M, Schmitt TM, McAfee MS, Paulson KG, et al. T cell receptor gene therapy targeting WT1 prevents acute myeloid leukemia relapse post-transplant. Nat Med. (2019) 25:1064–72. doi: 10.1038/s41591-019-0472-9
89. Jimenez-Reinoso A, Nehme-Alvarez D, Dominguez-Alonso C, Alvarez-Vallina L. Synthetic TILs: engineered tumor-infiltrating lymphocytes with improved therapeutic potential. Front Oncol. (2020) 10:593848. doi: 10.3389/fonc.2020.593848
90. Cao H, Kim DH, Howard A, Moz H, Wasnik S, Baylink DJ, et al. Ex vivo isolation, expansion and bioengineering of CCR7+CD95-/or CD62L+CD45RA+ tumor infiltrating lymphocytes from acute myeloid leukemia patients’ bone marrow. Neoplasia. (2021) 23:1252–60. doi: 10.1016/j.neo.2021.11.003
91. Kim MY, Yu KR, Kenderian SS, Ruella M, Chen S, Shin TH, et al. Genetic inactivation of CD33 in hematopoietic stem cells to enable CAR T cell immunotherapy for acute myeloid leukemia. Cell. (2018) 173:1439–1453 e19. doi: 10.1016/j.cell.2018.05.013
92. Ruggeri L, Urbani E, Andre P, Mancusi A, Tosti A, Topini F, et al. Effects of anti-NKG2A antibody administration on leukemia and normal hematopoietic cells. Haematologica. (2016) 101:626–33. doi: 10.3324/haematol.2015.135301
93. Maurer S, et al. The latest breakthroughs in immunotherapy for acute myeloid leukemia, with a special focus on NKG2D ligands. Int J Mol Sci. (2022) 23:15907. doi: 10.3390/ijms232415907
94. Parihar R. Memory NK cells to forget relapsed AML. Blood. (2022) 139:1607–8. doi: 10.1182/blood.2021014906
95. Berrien-Elliott MM, Foltz JA, Russler-Germain DA, Neal CC, Tran J, Gang M, et al. Hematopoietic cell transplantation donor-derived memory-like NK cells functionally persist after transfer into patients with leukemia. Sci Transl Med. (2022) 14:eabm1375. doi: 10.1126/scitranslmed.abm1375
96. Nie Y, Lu W, Chen D, Tu H, Guo Z, Zhou X, et al. Mechanisms underlying CD19-positive ALL relapse after anti-CD19 CAR T cell therapy and associated strategies. biomark Res. (2020) 8:18. doi: 10.1186/s40364-020-00197-1
97. Lemoine J, Ruella M, Houot R. Born to survive: how cancer cells resist CAR T cell therapy. J Hematol Oncol. (2021) 14:199. doi: 10.1186/s13045-021-01209-9
98. Hao Z, Li R, Meng L, Han Z, Hong Z. Macrophage, the potential key mediator in CAR-T related CRS. Exp Hematol Oncol. (2020) 9:15. doi: 10.1186/s40164-020-00171-5
99. Kasakovski D, Xu L, Li Y. T cell senescence and CAR-T cell exhaustion in hematological Malignancies. J Hematol Oncol. (2018) 11:91. doi: 10.1186/s13045-018-0629-x
100. Schmitt TM, Zuniga-Pflucker JC. Induction of T cell development from hematopoietic progenitor cells by delta-like-1 in vitro. Immunity. (2002) 17:749–56. doi: 10.1016/S1074-7613(02)00474-0
101. La Motte-Mohs RN, Herer E, Zuniga-Pflucker JC. Induction of T-cell development from human cord blood hematopoietic stem cells by Delta-like 1 in vitro. Blood. (2005) 105:1431–9. doi: 10.1182/blood-2004-04-1293
102. Guo R, Li R, Meng L, Han Z, Hong Z, et al. Guiding T lymphopoiesis from pluripotent stem cells by defined transcription factors. Cell Res. (2020) 30:21–33. doi: 10.1038/s41422-019-0251-7
103. Guo R, Li W, Li Y, Jian Z, Song Y. Generation and clinical potential of functional T lymphocytes from gene-edited pluripotent stem cells. Exp Hematol Oncol. (2022) 11:27. doi: 10.1186/s40164-022-00285-y
104. Dagvadorj N, Deuretzbacher A, Weisenberg D, Baumesiter E, Trebing J, Lang I, et al. Targeting of the WT1(91-138) fragment to human dendritic cells improves leukemia-specific T-cell responses providing an alternative approach to WT1-based vaccination. Cancer Immunol Immunother. (2017) 66:319–32. doi: 10.1007/s00262-016-1938-y
105. Anguille S, Van de Velde A, Smits EL, Van Tendeloo VF, Juliusson G, Cools N, et al. Dendritic cell vaccination as postremission treatment to prevent or delay relapse in acute myeloid leukemia. Blood. (2017) 130:1713–21. doi: 10.1182/blood-2017-04-780155
106. Lichtenegger FS, Schnorfeil FM, Rothe M, Deiser K, Altmann T, Bucklein VL, et al. Toll-like receptor 7/8-matured RNA-transduced dendritic cells as post-remission therapy in acute myeloid leukaemia: results of a phase I trial. Clin Transl Immunol. (2020) 9:e1117. doi: 10.1002/cti2.1117
107. van de Loosdrecht AA, van Weterin S, Santegoets SJA, Singh SK, Eeltink den Hartog CM. Y, et al. A novel allogeneic off-the-shelf dendritic cell vaccine for post-remission treatment of elderly patients with acute myeloid leukemia. Cancer Immunol Immunother. (2018) 67:1505–18. doi: 10.1007/s00262-018-2198-9
108. Jamieson C, Martinelli G, Papayannidis C, Cortes JE. Hedgehog pathway inhibitors: A new therapeutic class for the treatment of acute myeloid leukemia. Blood Cancer Discovery. (2020) 1:134–45. doi: 10.1158/2643-3230.BCD-20-0007
109. Van de Loosdrecht A, Drouet E, Platzbecker U, Holderried T, Van Elssen C, Giagounidis A, et al. Induction of cellular and humoral immune responses is associated with durable remissions in MRD+ AML-patients after maintenance treatment with an allogeneic leukemia-derived dendritic cell vaccine. Blood. (2023) 142:769. doi: 10.1182/blood-2023-185532
110. Zeeburg H, Drouet E, Platzbecker U, Holderried T, Elssen C, Giagounidis A, et al. van de loosdrecht, A, Vaccination Using an Allogeneic Leukemia-Derived Dendritic Cell Vaccine, Maintains and Improves Frequencies of Circulating Antigen Presenting Dendritic Cells Correlating with Relapse Free and Overall Survival in AML Patients. Blood. (2023) 142:2957. doi: 10.1182/blood-2023-188573
111. Srivastava P, Paluch BE, Matsuzaki J, et al. Induction of cancer testis antigen expression in circulating acute myeloid leukemia blasts following hypomethylating agent monotherapy. Oncotarget. (2016) 7:12840–56. doi: 10.18632/oncotarget.v7i11
112. Griffiths EA, Srivastava P, Matsuzaki J, Brumberger Z, Wang ES, Kocent J, et al. NY-ESO-1 vaccination in combination with decitabine induces antigen-specific T-lymphocyte responses in patients with myelodysplastic syndrome. Clin Cancer Res. (2018) 24:1019–29. doi: 10.1158/1078-0432.CCR-17-1792
113. Wang D, Huang XF, Hong B, Song XT, Hu L, Jian M, et al. Efficacy of intracellular immune checkpoint-silenced DC vaccine. JCI Insight. (2018) 3:e98368. doi: 10.1172/jci.insight.98368
114. Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. (2008) 455:674–8. doi: 10.1038/nature07317
115. Musella M, Manic G, De Mari R, Vitale I, Sistigu A. Type-I-interferons in infection and cancer: Unanticipated dynamics with therapeutic implications. Oncoimmunology. (2017) 6:e1314424. doi: 10.1080/2162402X.2017.1314424
116. Curran E, Chen X, Corrales L, Kline DE, Dubensky TW Jr, Duttagupta P, et al. STING pathway activation stimulates potent immunity against acute myeloid leukemia. Cell Rep. (2016) 15:2357–66. doi: 10.1016/j.celrep.2016.05.023
117. Adam M, Yu J, Plant R, Shelton C, Schmidt H, Yang J. Sting agonist GSK3745417 induces apoptosis, antiproliferation, and cell death in a panel of human AML cell lines and patient samples. Blood. (2022) 140:11829–9. doi: 10.1182/blood-2022-167652
118. Song C, Liu D, Liu S, Li D, Horecny I, Zhang X, et al. SHR1032, a novel STING agonist, stimulates anti-tumor immunity and directly induces AML apoptosis. Sci Rep. (2022) 12:8579. doi: 10.1038/s41598-022-12449-1
119. Curti A, Aluigi M, Pandolfi S, Ferri E, Salvestrini V, Durelli I, et al. Acute myeloid leukemia cells constitutively express the immunoregulatory enzyme indoleamine 2,3-dioxygenase. Leukemia. (2007) 21:353–5. doi: 10.1038/sj.leu.2404485
120. Corm S, Berthron C, Imbenotte M, Biggio V, Lhermitte M, Dupont C, et al. Indoleamine 2,3-dioxygenase activity of acute myeloid leukemia cells can be measured from patients’ sera by HPLC and is inducible by IFN-gamma. Leuk Res. (2009) 33:490–4. doi: 10.1016/j.leukres.2008.06.014
121. Arandi N, Ramzi M, Safaei F, and Monabati A. Overexpression of indoleamine 2,3-dioxygenase correlates with regulatory T cell phenotype in acute myeloid leukemia patients with normal karyotype. Blood Res. (2018) 53:294–8. doi: 10.5045/br.2018.53.4.294
122. Fox E, Oliver T, Rowe M, Thomas S, Zakharia Y, Gilman PB, et al. Indoximod: an immunometabolic adjuvant that empowers T cell activity in cancer. Front Oncol. (2018) 8:370. doi: 10.3389/fonc.2018.00370
Keywords: acute myeloid leukemia, immunotherapy, relapsed/refractory, cellular therapy, CAR-T
Citation: Bawek S, Gurusinghe S, Burwinkel M and Przespolewski A (2024) Updates in novel immunotherapeutic strategies for relapsed/refractory AML. Front. Oncol. 14:1374963. doi: 10.3389/fonc.2024.1374963
Received: 23 January 2024; Accepted: 11 November 2024;
Published: 04 December 2024.
Edited by:
Yan-Lai Tang, First Affiliated Hospital of Sun Yat-sen University, ChinaReviewed by:
Meng Lv, Peking University People’s Hospital, ChinaCopyright © 2024 Bawek, Gurusinghe, Burwinkel and Przespolewski. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Amanda Przespolewski, YW1hbmRhLnByemVzcG9sZXdza2lAcm9zd2VsbHBhcmsub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.