 Johanna Kögl
Johanna Kögl Teresa L. Pan†
Teresa L. Pan† Alain G. Zeimet
Alain G. Zeimet
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 22 August 2024
Sec. Gynecological Oncology
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1369189
Somatic mutations within the exonuclease proofreading domain (EDM) of the DNA polymerase Pol ϵ (POLE) gene are increasingly being discovered in ovarian, colorectal, urological, and, especially, endometrial carcinoma (EC), where these are found in up to 10% of the cases. In EC, there are five confirmed pathogenic somatic POLE-EDM mutations that are located at codons 286, 411, 297, 456, and 459, and these are called “hotspot” mutations. POLE mutant tumors are ultramutated entities with a frequency of base substitution mutations that is among the highest in human tumors. Interestingly, these mutations are associated with excellent clinical outcome in EC. An additional six “non-hotspot” POLE-EDM EC mutations are also considered pathogenic, and they also confer a favorable prognosis. Currently, de-escalation of adjuvant treatment is recommended for patients with EC with stage I–II tumors involving any of these 11 EDM mutations, even in patients with other clinicopathological risk factors. The high tumor mutational burden and the consequent increased infiltration of immune cells due to the overexpression of different neoantigens are probably responsible for the improved prognosis. Ongoing studies are examining POLE hotspot mutations among many non-gynecologic tumors, although the impact of such mutations on clinical outcomes is still a topic of debate. Therapeutic modalities for these hypermutated tumors are also an important consideration, including the need for or de-escalation of adjuvant treatments and the response to immune therapy. This review addresses the critical role of POLE mutations in gynecologic oncology and oncology in general, focusing on definitions, variants, underlying pathogenic mechanisms, upcoming developments in the field, and the clinic behavior associated with such mutations.
The role of the DNA polymerase epsilon (POLE) in the correct replication of cellular DNA has been studied intensively (1). As a consequence of this research, the prognostic value of specific mutations in the POLE gene, so called “hotspot mutations,” has recently recognized, and this has revolutionized the management of endometrial and other cancers (2–4). A single missense mutation in a hotspot region of the gene can guide a clinician to reconsider the need for adjuvant therapy in cases, which, until recently, would have received treatments known to be associated with a high risk of complications (3). POLE mutant tumors are described in endometrial, ovarian, colorectal, and urological cancers (5–8). They lead to a ultramutated tumor phenotype and consistently demonstrate an excellent clinical outcome, especially in colorectal cancer (CRC) and endometrial cancer (EC) (5, 9).
Historically, standard treatment of EC consisted of hysterectomy, bilateral salpingo-oophorectomy, and pelvic lymph node dissection followed by adjuvant therapy in the form of radiotherapy and/or chemotherapy based on final histology (10). However, management of EC has become more patient-specific over the past 10 years: differences in the histo-molecular classification predict prognosis and dictate whether adjuvant therapies are required or to be avoided (10) (Figure 1).
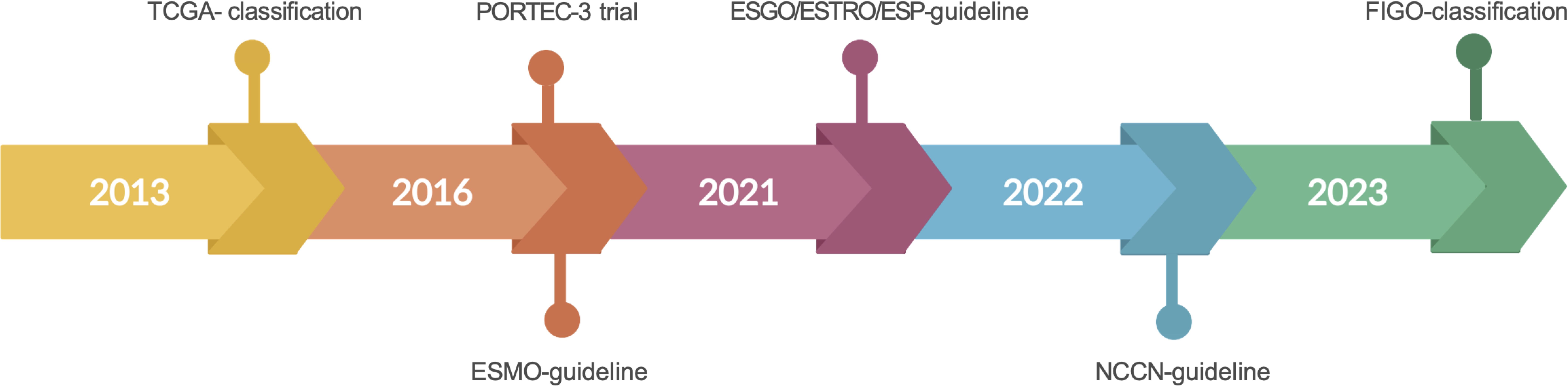
Figure 1. Major milestones in clinical practice in EC leading to substantial changes in the treatment of POLE-mutated ECs. TCGA, The Cancer Genome Atlas; ESMO, European Society for Medical Oncology; ESGO/ESTRO/ESP, European Society of Gynecological Oncology/European Society for Radiotherapy and Oncology/European Society of Pathology.
In 2013, The Cancer Genome Atlas (TCGA) identified a novel subgroup of ECs with unique mutations in POLE and associated unfavorable histomorphological features but, nevertheless, showing good survival outcomes. This new histo-molecular group was studied in the PORTEC-3 trial for patients with high-risk EC with Fédération International de Gynécologie et d’Obstétrique (FIGO) stage I–III, which investigated the benefit of adjuvant chemotherapy during and after radiotherapy over pelvic radiotherapy alone. The results of this trial showed that addition of adjuvant chemotherapy showed improved 5-year overall survival, especially in stage III patients. Crucially, molecular profiling revealed that POLE-mutated patients showed superior outcome, especially in stages I and II, irrespectively of their adjuvant treatment. This led to the author’s recommendation of de-escalation of adjuvant therapy for POLE-mutated patients (11, 12). These findings are limited by the relatively low number of POLE-mutated patients (12%) in general, and the fact that patients with stage III tumor disease showed superior outcome, if they had received a combined treatment. Furthermore, stage IV patients were not included in this trial. In 2016, the TCGA molecular classification was integrated into the ESMO (European Society for Medical Oncology) guideline, and this resulted in an updated risk classification for recurrence in stage I POLE-mutated ECs (13). The subgroup of POLE-mutated tumors was further integrated into the European Society of Gynecological Oncology (ESGO)/European Society for Radiotherapy and Oncology (ESTRO) and European Society of Pathology (ESP) guidelines for EC, resulting in specific changes in the recommendations for adjuvant treatment (3). The ESGO/ESTRO/ESP guidelines recommended that omission of adjuvant treatment should be considered for patients with stage I–II POLE-mutated EC. For the rare patients at stage III–IVA ECs with pathogenic POLE mutations, there are no reliable data on survival regarding omission of adjuvant treatment (3). In CRC as well, genetic testing for POLE was incorporated into the National Comprehensive Cancer Network guidelines in 2022 (14). Recently, this molecular classification has been integrated into the FIGO staging classification system of 2023. This shows the high clinical impact achieved by molecular characterization, including testing for POLE mutation, for optimal treatment of patients with EC. Although POLE mutant tumors tend to have a favorable outcome, there remain a number of questions to be answered: What is the physiological function and role of Pol ϵ? Is the good prognosis linked to the high mutational burden of POLE-mutated tumors? How is the correct annotation of the “POLE hotspot” mutations achieved? Is each POLE mutation pathogenic? What screening methods are available to determine POLE pathogenicity? What is the role of immunotherapy in POLE-mutated tumors and do POLE variants impact other tumor entities comparably? This review aims to discuss these fundamental questions and to highlight the current controversies related to this topic.
Accurate replication of DNA prior to cell division is essential for maintaining genomic stability and for suppressing mutagenesis and tumor development (15). The high fidelity of eukaryotic DNA replication is due to a combination of highly accurate base incorporation and 3′-5′ exonuclease proofreading by the replicative DNA polymerases Pol δ and Pol ϵ and post-replication surveillance of the newly synthesized DNA by the mismatch repair (MMR) apparatus (5). In humans, Pol ϵ belongs to the B family polymerases, comprises four subunits, and is encoded by POLE (5). The proofreading function of Pol ϵ requires highly conserved motifs in their exonuclease domain (EDM), named exo-motifs, within which lie the catalytic site residues that are essential for exonuclease activity (5, 15). Misincorporation of a base into the leading strand leads to pausing of the polymerase ϵ and, consequently, to a switch from the catalytic site to the exonuclease domain, where the incorrect base is excised and replaced by the correct base (16).
Considering the close correlation between POLE mutations and increased mutation rates, it is important to define the tumor mutational burden (TMB) when describing tumor biology. TMB indicates the number of single mutations per megabase presented in the specimen. A high TMB describes a highly mutated tissue, which could be considered to have more aggressive biological behavior. More specifically, hypermutation is defined by a mutational load of 10 or more mutations per megabase (≥10 mut/Mb) (17), whereas ultramutated tumors show a frequency of base substitution mutations that is equal to or higher than 100 mutations per megabase (ultramutation ≥100 mut/Mb) (1).
Somatic mutations within the POLE exonuclease proofreading domain (EDM) are found in 7%–12% of ECs (18), and these are always heterozygous changes (5, 15). Approximately 90% of the POLE proofreading mutations are in exons 9 and 13 and are recognized as pathogenic, i.e., driver mutations that are causal for tumor genesis by ultramutation (19). Generally, there are five common and confirmed pathogenic somatic POLE-EDM mutations that are located at codons 286, 411, 297, 456, and 459 (listed according to their decreasing prevalence). These are defined as “hotspot” mutations, but recurrent substitutions were also found in the complete TCGA EC cohort at codons 367, 424, 295, 368, 436, 444, 278, 428, 465, 352, 396, 402, 453, and 461 (decreasing frequency) (Table 1) (1). Most of these somatic substitutions lie within or close to the exo-motifs and will abolish exonuclease activity by causing perturbation of the DNA-binding pocket (8, 15). This affects protein function and subsequently increases the mutation rate (8, 15).
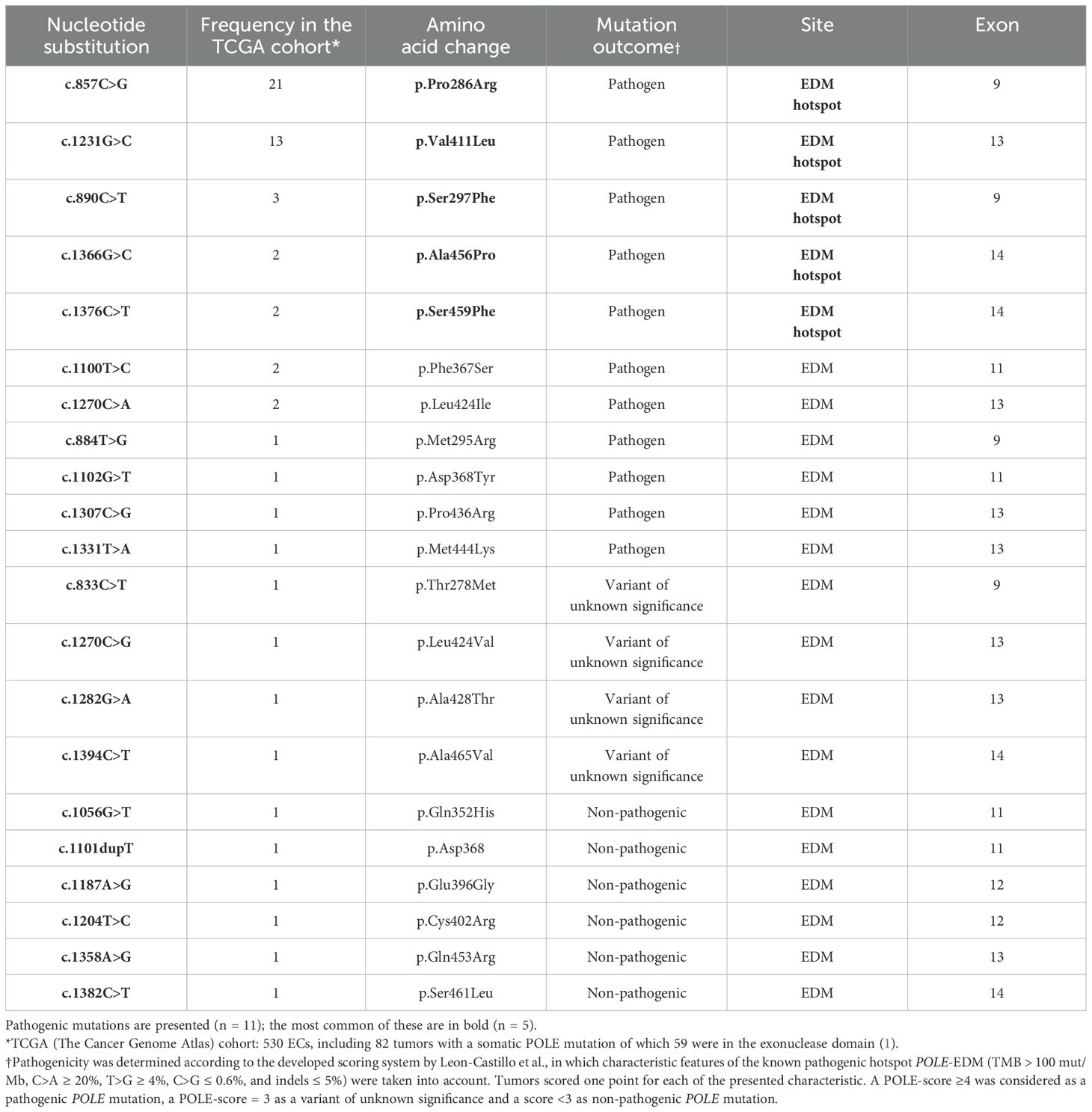
Table 1. Fifty-nine somatic POLE–exonuclease domain mutations (EDMs) in the TCGA cohort in EC with 11 pathogenic variants, listed by their frequency (1).
POLE mutant ECs are by definition ultramutated and exhibit a frequency of base substitution mutations that is among the highest in human tumors (1). In general, although TMB in POLE mutant ECs is always elevated with a median value of 268 mut/Mb, overall TMB varies not only between different hotspot mutations but also among ECs with the same hotspot mutation (1). POLE mutant ECs present distinctive features such as a strong association with endometroid histology, high grade, microsatellite stability (MSS), a low proportion of small insertion and deletion mutations (indels), and a high proportion of C>A and T>G mutations in TCT and TTT tri-nucleotide contexts. These specific biological features are described as COSMIC signature 10 (15, 18, 20). ECs with POLE hotspot mutations are associated with a high prevalence of C>A, frequently exceeding 20%, and slightly lower T>G substitutions (13%) (1). Another unique aspect is the correlation with mutations in the Mismatch Repair Genes, which are commonly referred to as microsatellite (in)stability (MSI). Endometrial tumors with POLE mutations in one of the five most common codons and MSI have a high TMB (339 mut/Mb), whereas EC tumors with non-hotspot POLE-EDM mutations and MSI have a lower TMB (median, 207 mut/Mb) (1). As expected, ECs with mutations outside the EDM and concomitant MSI status display an even lower TMB of only 48.5 mut/Mb (1). This raises the question—is the better prognosis of POLE hotspot mutant ECs linked to the concomitant high TMB? High TMB causes genomic instability, which leads to an increased neoantigen expression and activation of the immune system (21). This is associated with a better immune response, which has also been seen in other solid tumors (22) and may explain at least, in part, the favorable clinical outcome of POLE mutant tumors (21). Incorrect annotation of a POLE variant can lead to erroneous classification of an endometrial carcinoma within the POLE-mutated subgroup, and this can impact the clinical management of the patient (2). How is correct annotation of POLE pathogenicity achieved and does pathogenicity of a hotspot and non-hotspot EDM mutation differ? Of note, the tumor cell content should be determined to provide most accurate information. Analysis of the TCGA endometrial carcinoma cohort using only ECs with a known pathogenic hotspot POLE-EDM as a “truth set” allowed the development of a scoring system, with well-defined cutoff points for examining pathogenicity of POLE variants (1, 2). In order to understand the scoring system, one has to understand that pathogenicity in this sense is causal for tumor ultramutation and, thus, favors a good clinical outcome. Taking into account the characteristic features of the known pathogenic hotspot POLE-EDM (TMB > 100 mut/Mb, C>A ≥ 20%, T>G ≥ 4%, C>G ≤ 0.6%, and indels ≤ 5%), a pragmatic scoring system was developed by Leon-Castillo et al., in which tumors scored 1 point for each of the presented characteristic (1). Hotspot POLE mutation scored 3–5 points, ECs with non-hotspot POLE-EDM mutations scored ≥3 points, whereas ECs with POLE mutations outside the exonuclease domain scored ≤2 points, due to the lack of genomic alterations (1). As pathogenicity increases with recurrent mutations, recurrence was also incorporated into the described POLE-score model. Based on this model, a POLE score ≥4 was used to define pathogenicity of POLE mutations in EC (1). ECs with a POLE-score ≤2 were classified as having non-pathogenic POLE-EDM, whereas ECs with a score of 3 were classified as variant of uncertain significance (1). Considering this POLE-score, only 11 of the 21 different POLE exonuclease domain variants in the TCGA cohort qualified as pathogenic (1, 2) (Table 1). This illustrates that the presence of a POLE mutation variant alone is not sufficient for classifying an endometrial carcinoma as POLE-mutated, let alone as pathogenic. The scoring system of Leon-Castillo has not been validated on independent and larger cohorts and, therefore, does not yet represent an international standardized tool for classifying the pathogenicity of hotspot versus non-hotspot EDM and non-EDM POLE mutations and for deciding on potentially de-escalating adjuvant treatment. So far, in the majority of the relatively small early retrospective studies, only the five hotspot mutations were generally classified as POLEmut and considered as a reference category in survival analysis. This was also the case for the data analysis of the larger PORTEC-3 Trial. However, the international meta-analysis on 294 POLEmut ECs included the 11 pathogenic POLE-EDM mutations described above and revealed a recurrence rate of 3.7%. However, one case was associated with both hotspot mutations P286R and V411L (23). The still recruiting phase II, RAINBO POLEmut-BLUE Trial (NCT05255653-4), also includes the 11 mentioned pathogenic EDM mutations. This first prospective trial of POLEmut EC is investigating complete omission of adjuvant therapy in lower-risk disease and de-escalation of treatment (observation versus radiotherapy, but not chemoradiation) in higher-risk disease (2). The outcome of RAINBO-BLUE will shed light on the mutations for which a de-escalation in the adjuvant treatment can be justified without concern. However, it is unclear if POLE pathogenicity is best assessed using a scoring system, TMB, or associated MSI status. Nevertheless, as whole-genome/exome sequencing techniques become more widely available, the current list of 11 pathogenic POLE-EDMs will increase in the near future, along with the need to precisely annotate defined POLE mutations. However, more evidence will be needed before new pathogenic mutations are included among those currently known to improve prognosis and thereby affect routine treatment decisions.
Historically, EC has been classified into two subtypes (Bokhman classification) based on their clinical, endocrine, and histopathological characteristics (10, 24) In the last decade, molecular characteristics became components of Bokhman’s dualistic classification (25). However, the substantial heterogeneity of EC was not represented in this dichotomous classification (25). In 2013, analysis of TCGA identified four new genomic classes of ECs by combining information on somatic mutational burden and somatic copy number alterations (18). In recent years, Murali et al. (25) suggested that incorporation of molecular and genetic characteristics into the classification reflects tumor biology and prognostic outcome in EC more accurately. Traditionally, multiple factors such as histological subtype, G3 histology, myometrial invasion ≥50%, lymphovascular space invasion (LVSI), lymph node metastases, tumor diameter >2 cm, and presence of L1 cell adhesion molecule (L1CAM = CD171) have been identified as conferring high risk for recurrent disease (26). In more recent years, surrogate markers have been identified and incorporated in routine surgical pathology in order to allow identification of the four genomic classes of EC (2, 13). This entails sequencing of the exonuclease domain of POLE and assessment of the expression of MMR proteins (MLH1, PMS2, MSH2, and MSH6) and p53 by immunohistochemistry. This results in the current molecular classification of ECs into four new subgroups: POLEmut (ultramutated), MMR-deficient (MMRd), p53-abnormal (p53abn), and no specific molecular profile (NSMP), which represents the most heterogenous group (Table 2) (2, 13). The ProMisE (Proactive Molecular Risk Classifier for Endometrial Cancer) approach has been proven to be a reliable method for classifying tumors into the four subclassifications of TCGA. These four subgroups also show significant differences in clinical outcomes.
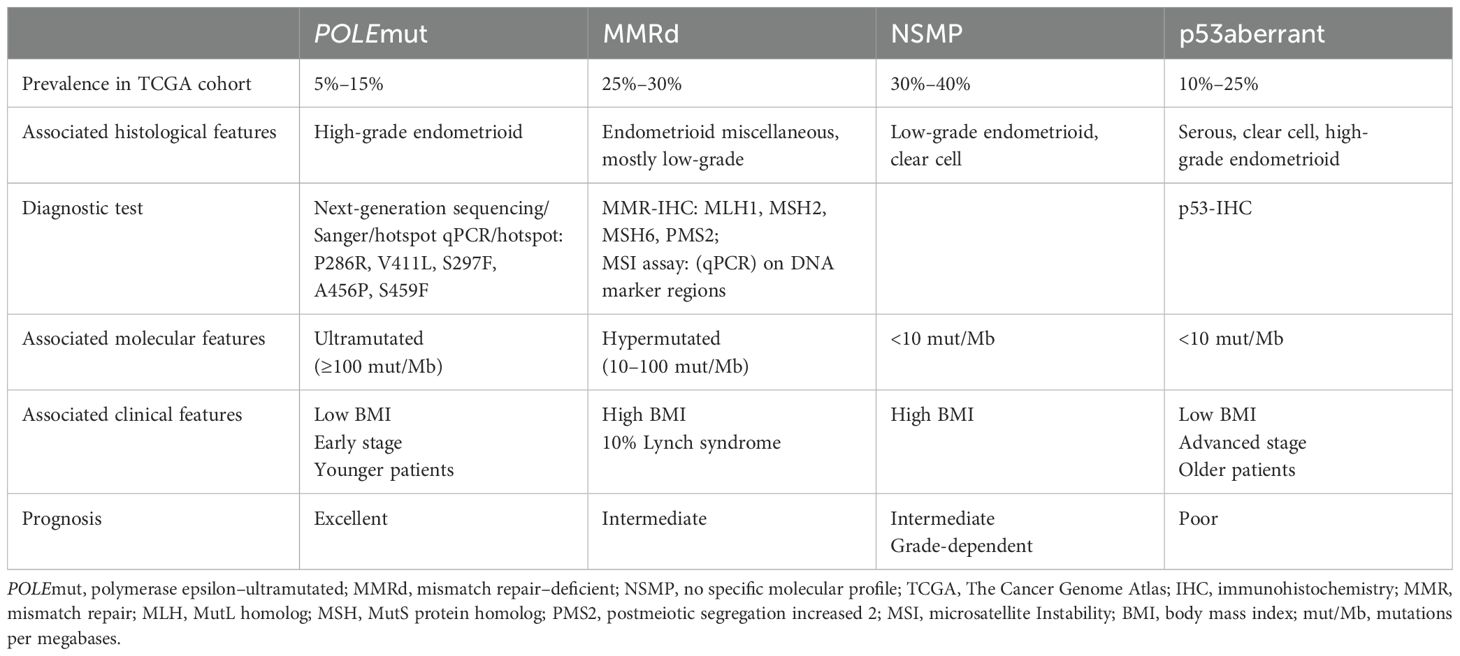
Table 2. International molecular classification of EC.
However, 3%–6% of EC tumors are referred to as multiple-classifiers, i.e., at first appearance, they belong to more than one molecular class and include those with combined POLEmut and p53abn, combined MMRd and p53abn, combined MMRd and POLEmut and a combination of all three defects (MMRd-POLEmut-p53abn) (27). Nevertheless, recently, a study has shown that multiple-classifier POLEmut-p53abn and MMRd-POLEmut can be categorized as single-classifier POLEmut and MMRd-p53abn as single-classifier MMRd EC (27). These findings result in a top-down classification hierarchy with POLEmut situated on top, followed by MMRd. In other words, if a patient’s tumor exhibits a p53abn status and a POLEmut, then no adjuvant therapy would be needed to treat such a patient in case of a stage I–II tumor. However, the current guidelines do not respect multiple classifiers yet. Furthermore, the data are yet limited on this subject.
The ESGO/ESTRO and the ESP 2021 guidelines integrated the molecular subgroups with traditional clinicopathological features into a novel risk stratification system for assessing the relative risk of recurrence and guiding treatment decisions (2, 3). The molecular characteristics have also been recently incorporated into the 2023 version of the FIGO staging classification system for EC. This new risk stratification system relies on the identification of surrogate markers that show a good relationship with clinical outcomes. However, implementation and interpretation of the surrogate markers in clinical practice remains challenging, especially for the POLE variants (2). The POLE-mutated class represents the smallest subgroup (7%) of ECs and is defined by somatic mutations in the catalytic subunit of the EDM of POLE (18, 28). Sixty percent of POLE ultramutated ECs are high-grade endometrioid lesions and 35% harbor a mutation of the TP53 gene (10). Nevertheless, among POLE multiple-classifier cases, POLEmut outweighs the other described mutational defects. The new ESGO/ESTRO/ESP guidelines recommend that patients with stage I–II POLE-mutated EC do not need adjuvant treatment, irrespective of p53abn or MMR status. Such assessments are independent of traditional high risk factors (3). Patients with EC-classified POLEmut have an excellent prognosis and are expected to benefit from a de-escalation of postoperative adjuvant treatment, whereas patients with a POLE-unrelated p53abn EC have a worse prognosis and, thus, are expected to benefit from an intensification of treatment (4). The presence of a pathogenic POLE or p53 mutation leads to a significant modification of the FIGO stage in early EC, in terms of downstaging or upstaging of disease (Table 3) (4).

Table 3. New 2023 FIGO endometrial cancer stage with molecular classification.
Thus, stage II tumors with a POLEmut are now classified as Stage IAmPOLEmut, whereas stage I tumors with a p53 mutation are classified as Stage IICmp53abn (Table 3).
Given how the presence of a POLE-EDM mutation impacts the outcome and especially the therapeutic approach in EC, this raises the question—does this impact and approach generalize to other tumor entities? The oncological literature on this topic is expanding rapidly, and, almost in every field, there is an effort to identify possible pathogenic POLE-mutated variants.
There is an increasing interest in POLE mutations in ovarian cancer (OC). Four of the five POLE hotspot mutations (P286R, S297F, V411L, and A456P) have been found in an OC cohort of 195 patients, with POLE mutations found in 1.5% of tumors. All such tumors were of the endometroid histotype and had an earlier onset with an average age at diagnosis of 48 years (6). However, Parra-Herran et al. (29) analyzed POLE-EDM mutations in ovarian clear cell cancer (OCCC) but could not detect any pathogenic POLE mutation in a total of 47 cases. Nevertheless, several variants of unknown significance were detected. Among endometroid OCs, the overall incidence of POLE mutations appears to be up to 8% with a high proportion of heterozygous POLE p.297 mutations (30). Furthermore, in endometroid OCs, POLE-mutated cases present at an early stage (75% at FIGO stage I), and none was staged FIGO III or higher. All mutations were somatic mutations at P286R and V411L, and the patients had an uneventful clinical course without recurrence. These data are extrapolated from three single center studies and are not inadequate for establishing the overall prevalence of POLE mutations among all OCs. A study by Leskela et al. (31) determined MMR, p53 and POLE-EDM in early stage endometrioid OCs based on the molecular classification used for EC. Five tumors (3%) were double classifiers, whereas most of the cohort (66%) belonged to the NSMP (no specific molecular profile) group. In the POLE-EDM–mutated group, tumors (overall 8%) were ultramutated and showed higher infiltrations of CD8-lymphocytes compared with the rest of the cohort. Although the prognosis did not differ among subgroups in the multivariate analysis, a tendency toward better prognosis in POLE-mutated and a worse prognosis in p53 abnormal tumors was noted (31).
The role of POLE-EDM mutations in endometroid OC is currently emerging in the oncological literature. It has been shown that POLE mutations are more common in endometroid OC and are associated with younger age and earlier stage at diagnosis. Whether POLE mutations also lead to a better prognosis in endometroid OCs remains unconfirmed, and there appears to be a rationale for testing patients with early onset endometroid OCs, as they could be candidates for immunotherapy.
The role of POLE-EDM mutations with regard to pathogenesis, prognosis, and therapeutic options has been widely investigated in CRC over the past few years (9). Ultramutated phenotypes with a high TMB (cutoff >150mut/Mb) can help to identify possible POLE-mutated CRCs and to guide selected screening. Furthermore, CRC POLE-mutated tumors are mainly diagnosed at relatively younger age (before 55 years) and at an early stage (14, 32–34).
Genomic studies of urothelial bladder carcinomas from the TCGA cohort have revealed a prevalence of 6.1% for POLE mutations (7). These POLE mutant urological cancers present known pathogenic hotspot mutations with a high TMB and a durable response to ICI (immune checkpoint inhibitor) therapy (35, 36).
A summary of the known POLE-EDM mutations in different tumor entities is presented in Table 4.
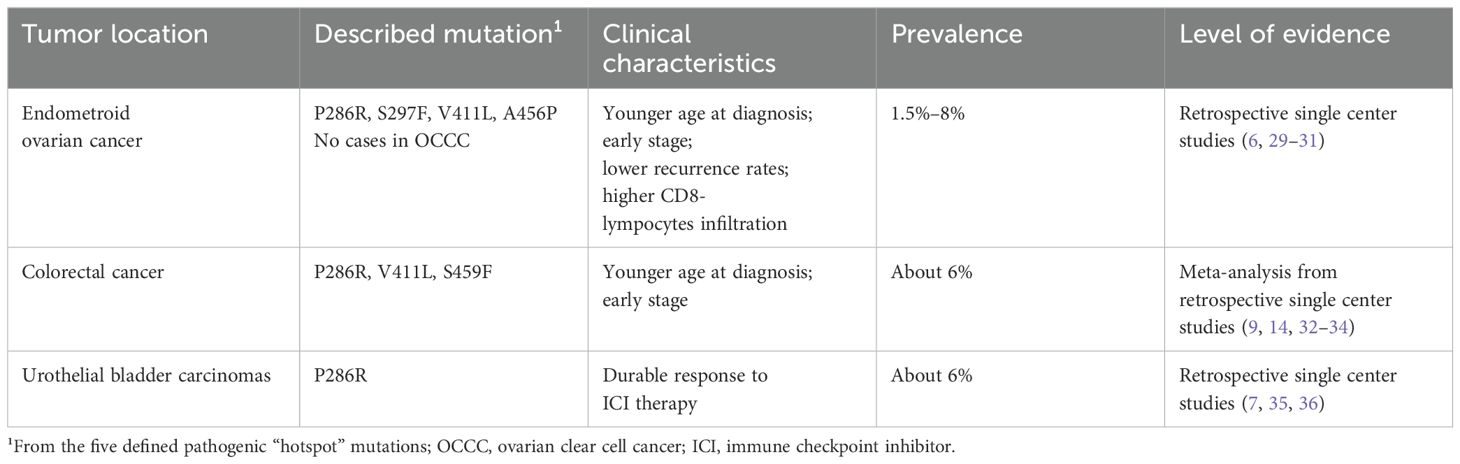
Table 4. Described pathogenic POLE-EDM mutations in different tumor entities.
Evaluation of a pathogenic POLE mutation remains challenging, as parameters and methods that allow a standard procedure in clinical practice have not been validated as yet. Whole-exome or whole-genome sequencing (WES/WGS) by Sanger or next-generation-sequencing can be used to identify POLE mutations in the exonuclease domain (exons 9–14). However, these methods are time-consuming, not widely available, and expensive; require expertise; and, therefore, limit routine use in current clinical practice. Estimation of pathogenicity of somatic POLE mutations in the absence of exome and genome sequencing has been carried out by some authors by using in silico prediction tools (1). Although this seems to be a feasible technique, the setting prognosis relies on the sequencing tools that have been used. Clinical practice requires a POLE testing method that is not only affordable, with a fast turnaround time, but also easy to interpret and implement. Therefore, sequencing methods restricted to the analysis of the hotspot POLE exonuclease domain mutations could present an alternative technique and have been developed recently by several research groups. Deveraux et al. (37, 38), for example, use a single-gene POLE hotspot SNaPshot assay in their routine prospective molecular classification of ECs. This technique involves an initial PCR amplification of the relevant gene target regions of the POLE-EDMs, followed by multiplexed single-nucleotide primer extension (38). Van den Heerik et al. (39) created a quantitative polymerase chain reaction (qPCR) assay for pathogenic POLE mutations (QPOLE). So far, there is no standardized method that allows determination of POLE mutations in ECs in clinical practice. Moreover, a workflow that reports both the molecular and histologic findings in an integrative manner is still not available. However, the integration of molecular classification together with clinicopathologic features into the ESGO/ESTRO/ESP guidelines and into the novel 2023 FIGO staging classification system shows the high clinical impact that testing of POLE mutation has in the patients’ treatment and management. This issue cannot be ignored any longer. It is critically necessary that the clinical assay used in daily practice reliably identifies POLE mutations in the hotspot POLE-EDM as their role in tumor biology and their therapeutical consequences are known. However, as new hotspot POLE-EDM mutations continue to emerge, their clinical role must be rapidly evaluated, and they should be incorporated into validated assays as appropriate. One indirect approach to identify POLE-mutated cancers is by looking at the number of tumor-infiltrating lymphocytes (TILs), as we know that a highly mutated microenvironment expresses more antigens and, therefore, activates the host’s immune system. For example, in CRC cases, MSI tumors were TIL-high (≥4 lymphocytes per high-power field), in 68% of cases with a TMB of 54 mut/Mb, whereas MSS CRCs were only TIL-high in 4.5% of cases. In contrast, MSS CRC tumors with POLE/POLD1 pathogenic variants were TIL-high in 82% of cases and had a TMB over 150 mut/Mb. These differences in tumoral immunity provide the rationale for immunotherapy (40). A possible way to screen for patients who might benefit from immunotherapy even if they are MMR-intact could be by using an immune microenvironment evaluation system such as that described by Galon et al. (41). In CRC, the Immunoscore (IS) has been shown to be a prognostic factor superior to the previous tumor, node, metastasis (TNM) classification of malignant tumors. There are ongoing validation and promotion initiatives to increase the use of IS in routine clinical settings (41). There are several ongoing clinical trials assessing the efficacy of ICI therapy for treating patients with POLE/POLD1 mutations, especially for metastatic CRC (40, 42–44). Different systems of immunoscoring have also found application in gastric and endometrial cancer (45), but further prognostic studies are needed to validate the routine use of ISs in routine clinical practice.
In EC, the ultramutated phenotype caused by POLE-EDM mutations has been shown to cause a “self-limiting” tumor progression with excellent prognosis after surgery. This has been demonstrated even without adjuvant treatment in patients previously classified as “high” or “intermediate-high” risk (10, 46). The Identification of pathogenic POLE mutations in early-stage EC also plays a crucial role when considering fertility sparing treatments (FSTs) such as hormonal therapy or hysteroscopic resection in young women (47, 48). Improving risk stratification for FST is one of the future targets, because the molecular classification and different molecular markers emerging are changing the risk profile assessment for patients. A preliminary molecular analysis of an endometrial biopsy is, therefore, necessary in patients with desire to conceive or in case of organ sparing. Pathologists should systematically perform a molecular analysis of hysteroscopic biopsy samples, including the sequencing for the presence of the POLE mutation. In OC of the endometrioid histological subtype, there is reason to believe that POLE mutations lead to a better prognosis, but further confirmation is needed (49). Although there are many molecular studies on POLE-mutated CRCs, data on the clinical implications of the POLE ultramutated phenotype are lacking. In some studies of POLE-EDM–mutated stage II CRCs, a robust intratumoral T-cell response was detected in a small subset of cases with excellent outcomes (50). However, a significantly better prognosis across all stages of CRC has not been confirmed as yet (as in the case of ECs). Therefore, the therapeutic management of CRC is currently not impacted by the presence of a POLE mutation. In other solid tumors such as pancreatic cancer, mutations in the hotspot regions of POLE are very rare events. In advanced pancreatic cancer, it is highly unlikely that POLE mutations contribute to genetic instability; therefore, POLE mutations do not serve as a relevant biomarker and should not be tested for on a regular basis (51). In a totally different oncologic entity, namely, high-grade gliomas (HGGs), a subgroup based on somatic POLE mutations has been identified. Such cases are genomically, histologically, and clinically different from the other HGGs and exhibit an improved prognosis (52). Most recently, two trials have clarified the role of ICI for the treatment of EC. These two big randomized phase III trials (RUBY and GY018) with still limited follow-up did so far not change the practice for POLE-mutated EC (53). Over the last several years, an enormous effort has been made with regard to EC to find new biomarkers that can accurately identify patients who can benefit from immunotherapy even if they are not MMR deficient. In silico analysis has proved that POLE mutant cancers display more antigenic neoepitopes than other ECs, providing a potential rational for POLE immunogenicity (54). Yet POLE-mutated tumors are currently not a recommended indication for ICIs even in advanced and metastatic EC cases. On the other hand, CRCs have recently received approval for treatment of MSI CRC with ICIs. Assessment of POLE status may help guide therapeutic decisions for tumors with high TMB and intact MMR. Recent reports have shown that, even in advanced CRC or multiresistent disease, patients can significantly benefit from ICIs, if they harbor a pathogenic POLE mutation (55–57). A case report of a high-grade CRC with POLE-EDM (P286R) mutation and TMB of 119 mut/Mb described triple-chemotherapy being ineffective, whereas ICIs had a significant impact on progression-free survival (PFS) (55). Another example involved the case of a 24-year-old male patient with an aggressive stage IV high-grade, poorly differentiated CRC, where response was complete and durable (over 48 months) with a single-agent ICI after rapidly progression with standard chemotherapy. Genetic testing of this case revealed a P286R POLE mutation and an elevated TMB of 126 mut/Mb (57). These cases highlight the interplay between genetic instability and immune-checkpoint blockade. In a case report of OC (OCCC inoperable at stage IIIB), resistant to platinum-based chemotherapy, the same P286R POLE mutation was found, and the third-line treatment attempt with a programmed cell death protein 1 (PD-1) inhibitor showed a tumor with postoperative pathologic complete response. The patient achieved a PFS of 29 months under maintenance with ICI therapy (58). Therefore, even if the ProMisE classification does not find an application in OC, the simple presence of POLE hotspot mutation can, in exceptional cases, guide for treatment with ICIs.
At the present, it is important to acknowledge that the changes in clinical treatment of POLE-mutated ECs as recommended by the ESGO/ESTRO/ESP guideline of 2021 are based on much less data than what is typically used for such profound and nearly dogmatic shifts in clinical care. Evaluation of the benefit of these clinical and therapeutic changes through prospective studies can help provide more information on this issue. Currently, prospective clinical trials, such as PORTEC-4 and TAPER, are ongoing and will shed light and yield more insights whether POLE-mutated ECs have a favorable outcome even without or de-escalated adjuvant treatment. The TAPER trial is an interventional study based on tailored adjuvant therapy in POLE-mutated and p53 wild-type/NSMP early-stage EC. Its primary objective is to determine if women with cancers with the specific molecular characteristics who underwent adequate surgery have a relative low risk (lower than 5%) of pelvic and vaginal recurrence at 3 years with no or de-escalated adjuvant treatment (59). The PORTEC-4a trial is a randomized phase III trial of molecular profile-based versus standard recommendations for adjuvant radiotherapy in stage I EC and is focused on cancers classified as high-intermediate risk according to the ESMO-ESGO-ESTRO consensus of 2015. The primary endpoint is vaginal recurrence, and other oncologic but secondary endpoints are recurrence-free and overall survival, as well as pelvic and distant recurrence. As, in this study, all patients with POLE-mutated cancers are allocated without restrictions to the “favorable molecular risk group” with omission of vaginal brachytherapy and external pelvic beam radiotherapy, this trial is very likely to extend our knowledge on prognosis of POLE-mutated cancers at least in this prespecified subset of patients. First results are expected at the end of 2024 or in the spring 2025, and study completion is set for 2028 with 550 patients enrolled (60). Lastly, the blue arm of the RAINBO trial is also focused at POLE-mutated ECs. The RAINBO program is a platform of four international clinical trials and an overarching research program, including a randomized PHASE III trial with three arms for p53-abn EC (red), MMRd EC (green), and NSMP (orange) ECs. The POLEmut-BLUE trial is a phase II trial in which the safety of de-escalation of adjuvant therapy is investigated for women with stage I–III POLEmut EC. This trial will evaluate no adjuvant therapy for lower-risk disease and no adjuvant therapy or radiotherapy alone with omission of concomitant chemotherapy for higher-risk disease. The primary endpoint of this trial will be pelvic recurrence at 3 years. This study is in the recruiting phase and main trial results are expected in 2028 (61).
A summary of relevant concluded or ongoing trials about ICIs and adjuvant treatments in EC based on molecular classification is shown in Table 5.
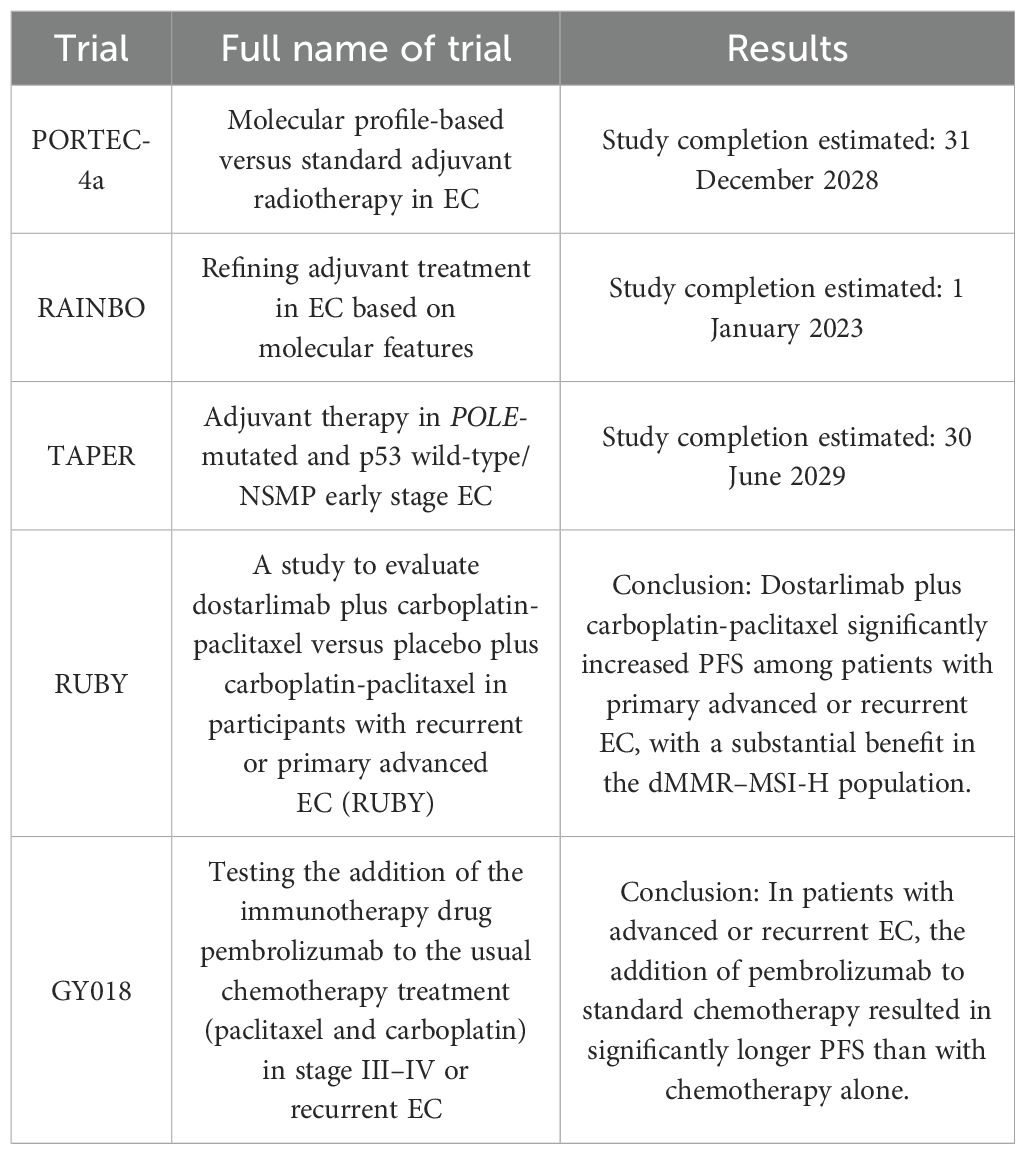
Table 5. A summary of relevant concluded or ongoing trials about ICIs and adjuvant treatments in EC based on molecular classification.
POLE mutational status in EC is of great clinical interest. It determines the prognosis of the patient, and the FIGO classification system 2023 stipulates that its presence should result in a significant de-escalation of adjuvant treatment. In the future, unresolved questions will be better answered by the results of the POLEmut-BLUE arm of the prospective phase II RAINBO trial, where even stage III patients are included. POLE-mut ECs are assigned depending on risk status either to an observational arm with complete omission of adjuvant treatment or to radiotherapy alone. We describe in detail that the sole presence of a POLE variant is not sufficient to classify a tumor as POLEmutated or to classify a POLE mutation as pathogenic. For that, the exact localization of the mutation in the POLE gene needs to be known. It has a higher probability of being pathogenic when it is located in the EDM of Pol ϵ. However, unanswered questions, such as the exact molecular pathways associated with the good prognosis of POLE mutations, remain unclear and, therefore, unmentioned. Due to inconsistencies in POLE mutation testing and its interpretation, the study investigators have advocated a concomitant TMB determination in order to underscore the pathogenicity of the POLE-EDM mutation. Performed as a routine procedure, this appears to be one of the best and easiest approaches to identify new pathogenic POLE mutations. We are hopeful that the increasing knowledge on the exact oncologic driver qualities of POLE mutations together with the outcome of mentioned prospective clinical research will enable reassured avoidance or at least de-escalation of adjuvant treatment in EC without harming patients by under- or overtreatment.
JK: Conceptualization, Writing – original draft, Writing – review & editing. TP: Conceptualization, Writing – original draft, Writing – review & editing. CM: Writing – review & editing. AZ: Conceptualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the “Verein zur Krebsforschung in der Frauenheilkunde,” an association that is exclusively financed by donation funds for cancer research in female malignancies (grant number 01722).
The authors thank Professor K.S. Joseph for reviewing the manuscript with regard to syntax and grammar issues.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. León-Castillo A, Britton H, McConechy MK, McAlpine JN, Nout R, Kommoss S, et al. Interpretation of somatic POLE mutations in endometrial carcinoma. J Pathol. (2020) 250:323–35. doi: 10.1002/path.5372
2. Léon-Castillo A. Update in the molecular classification of endometrial carcinoma. Int J Gynecol Cancer. (2023) 33:333–42. doi: 10.1136/ijgc-2022-003772
3. Concin N, Matias-Guiu X, Vergote I, Cibula D, Mirza MR, Marnitz S, et al. ESGO/ESTRO/ESP guidelines for the management of patients with endometrial carcinoma. Int J Gynecol Cancer. (2021) 31:12–39. doi: 10.1136/ijgc-2020-002230
4. Berek JS, Matias-Guiu X, Creutzberg C, Fotopoulou C, Gaffney D, Kehoe S, et al. FIGO staging of endometrial cancer: 2023. Int J Gynecol Obs. (2023) 162:383–94. doi: 10.1002/ijgo.14923
5. Rayner E, Van Gool IC, Palles C, Kearsey SE, Bosse T, Tomlinson I, et al. A panoply of errors: Polymerase proofreading domain mutations in cancer. Nat Rev Cancer. (2016) 16:71–81. doi: 10.1038/nrc.2015.12
6. Davila JI, Chanana P, Sarangi V, Fogarty ZC, Weroha SJ, Guo R, et al. Frequent POLE-driven hypermutation in ovarian endometrioid cancer revealed by mutational signatures in RNA sequencing. BMC Med Genomics. (2021) 14:165. doi: 10.1186/s12920-021-01017-7
7. Voutsadakis IA. Urothelial bladder carcinomas with high tumor mutation burden have a better prognosis and targetable molecular defects beyond immunotherapies. Curr Oncol. (2022) 29:1390–407. doi: 10.3390/curroncol29030117
8. Shinbrot E, Henninger EE, Weinhold N, Covington KR, Göksenin AY, Schultz N, et al. Exonuclease mutations in DNA polymerase epsilon reveal replication strand specific mutation patterns and human origins of replication. Genome Res. (2014) 24:1740–50. doi: 10.1101/gr.174789.114
9. Domingo E, Freeman-Mills L, Rayner E, Glaire M, Briggs S, Vermeulen L, et al. Somatic POLE proofreading domain mutation, immune response, and prognosis in colorectal cancer: a retrospective, pooled biomarker study. Lancet Gastroenterol Hepatol. (2016) 1:207–16. doi: 10.1016/S2468-1253(16)30014-0
10. Morice P, Leary A, Creutzberg C, Abu-Rustum N, Darai E. Endometrial cancer. Lancet. (2016) 387:1094–108. doi: 10.1016/S0140-6736(15)00130-0
11. Leon-Castillo A, De Boer SM, Powell ME, Mileshkin LR, Mackay HJ, Leary A, et al. Molecular classification of the PORTEC-3 trial for high-risk endometrial cancer: Impact on prognosis and benefit from adjuvant therapy. J Clin Oncol. (2020) 38:3388–97. doi: 10.1200/JCO.20.00549
12. de Boer SM, Powell ME, Mileshkin L, Katsaros D, Bessette P, Haie-Meder C, et al. Adjuvant chemoradiotherapy versus radiotherapy alone in women with high-risk endometrial cancer (PORTEC-3): patterns of recurrence and post-hoc survival analysis of a randomised phase 3 trial. Lancet Oncol. (2019) 20:1273–85. doi: 10.1016/S1470-2045(19)30395-X
13. Oaknin A, Bosse TJ, Creutzberg CL, Giornelli G, Harter P, Joly F, et al. Endometrial cancer: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up ☆. Ann Oncol. (2022) 33:860–77. doi: 10.1016/j.annonc.2022.05.009
14. Weiss JM, Gupta S, Burke CA, Axell L, Chen LM, Chung DC, et al. NCCN guidelines® Insights: genetic/familial high-risk assessment: colorectal, version 1.2021. J Natl Compr Canc Netw. (2021) 19:1122–32. doi: 10.1164/jnccn.2021.0048
15. Church DN, Briggs SEW, Palles C, Domingo E, Kearsey SJ, Grimes JM, et al. DNA polymerase ϵ and δ exonuclease domain mutations in endometrial cancer. Hum Mol Genet. (2013) 22:2820–8. doi: 10.1093/hmg/ddt131
16. Ganai RA, Bylund GO, Johansson E. Switching between polymerase and exonuclease sites in DNA polymerase ϵ. Nucleic Acids Res. (2015) 43:932–42. doi: 10.1093/nar/gku1353
17. Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R, et al. Comprehensive analysis of hypermutation in human cancer. Cell. (2017) 171:1042–1056.e10. doi: 10.1016/j.cell.2017.09.048
18. Getz G, Gabriel SB, Cibulskis K, Lander E, Sivachenko A, Sougnez C, et al. Integrated genomic characterization of endometrial carcinoma. Nature. (2013) 497:67–73. doi: 10.1038/nature12113
19. Church DN, Stelloo E, Nout RA, Valtcheva N, Depreeuw J, Ter Haar N, et al. Prognostic significance of POLE proofreading mutations in endometrial cancer. J Natl Cancer Inst. (2015) 107:402. doi: 10.1093/jnci/dju402
20. Fang H, Barbour JA, Poulos RC, Katainen R, Aaltonen LA, Wong JWH. Mutational processes of distinct POLE exonuclease domain mutants drive an enrichment of a specific TP53 mutation in colorectal cancer. PloS Genet. (2020) 16:e1008572. doi: 10.1371/journal.pgen.1008572
21. Imboden S, Nastic D, Ghaderi M, Rydberg F, Rau TT, Mueller MD, et al. Phenotype of POLE-mutated endometrial cancer. PloS One. (2019) 14:e0214318. doi: 10.1371/journal.pone.0214318
22. Strickler JH, Hanks BA, Khasraw M. Tumor mutational burden as a predictor of immunotherapy response: is more always better? Clin Cancer Res. (2021) 27:1236–41. doi: 10.1158/1078-0432.CCR-20-3054
23. McAlpine JN, Chiu DS, Nout RA, Church DN, Schmidt P, Lam S, et al. Evaluation of treatment effects in patients with endometrial cancer and POLE mutations: An individual patient data meta-analysis. Cancer. (2021) 127:2409–22. doi: 10.1002/cncr.33516
24. Bokhman JV. Two pathogenetic types of endometrial carcinoma. Gynecol Oncol. (1983) 15:10–7. doi: 10.1016/0090-8258(83)90111-7
25. Murali R, Soslow RA, Weigelt B. Classification of endometrial carcinoma: more than two types. Lancet Oncol. (2014) 15:e268–78. doi: 10.1016/S1470-2045(13)70591-6
26. Zeimet AG, Reimer D, Huszar M, Winterhoff B, Puistola U, Azim SA, et al. L1CAM in early-stage type i endometrial cancer: Results of a large multicenter evaluation. J Natl Cancer Inst. (2013) 105:1142–50. doi: 10.1093/jnci/djt144
27. León-Castillo A, Gilvazquez E, Nout R, Smit VT, McAlpine JN, McConechy M, et al. Clinicopathological and molecular characterisation of ‘multiple-classifier’ endometrial carcinomas. J Pathol. (2020) 250:312–22. doi: 10.1002/path.5373
28. Hussein YR, Weigelt B, Levine DA, Schoolmeester JK, Dao LN, Balzer BL, et al. Clinicopathological analysis of endometrial carcinomas harboring somatic POLE exonuclease domain mutations. Mod Pathol. (2015) 28:505–14. doi: 10.1038/modpathol.2014.143
29. Parra-Herran C, Bassiouny D, Lerner-Ellis J, Olkhov-Mitsel E, Ismiil N, Hogen L, et al. P53, mismatch repair protein, and POLE abnormalities in ovarian clear cell carcinoma: an outcome-based clinicopathologic analysis. Am J Surg Pathol. (2019) 43:1591–9. doi: 10.1097/PAS.0000000000001328
30. Zou Y, Liu FY, Liu H, Wang F, Li W, Huang MZ, et al. Frequent POLE1 p.S297F mutation in Chinese patients with ovarian endometrioid carcinoma. Mutat Res Mol Mech Mutagen. (2014) 761:49–52. doi: 10.1016/j.mrfmmm.2014.01.003
31. Leskela S, Romero I, Rosa-Rosa JM, Caniego-Casas T, Cristobal E, Pérez-Mies B, et al. Molecular heterogeneity of endometrioid ovarian carcinoma. Am J Surg Pathol. (2020) 44:982–90. doi: 10.1097/PAS.0000000000001478
32. Stadler ZK, Battaglin F, Middha S, Hechtman JF, Tran C, Cercek A, et al. Reliable detection of mismatch repair deficiency in colorectal cancers using mutational load in next-generation sequencing panels. J Clin Oncol. (2016) 34:2141–7. doi: 10.1200/JCO.2015.65.1067
33. Kawai T, Nyuya A, Mori Y, Tanaka T, Tanioka H, Yasui K, et al. Clinical and epigenetic features of colorectal cancer patients with somatic POLE proofreading mutations. Clin Epigenetics. (2021) 13:117. doi: 10.1186/s13148-021-01104-7
34. Hu H, Cai W, Wu D, Hu W, Wang L, et al. Ultra-mutated colorectal cancer patients with POLE driver mutations exhibit distinct clinical patterns. Cancer Med. (2021) 10:135–42. doi: 10.1002/cam4.3579
35. Lee L, Ali S, Genega E, Reed D, Sokol E, Mathew P. Aggressive-variant microsatellite-stable POLE mutant prostate cancer with high mutation burden and durable response to immune checkpoint inhibitor therapy. JCO Precis Oncol. (2018) 2:1–8. doi: 10.1200/PO.17.00097
36. Halbert B, Einstein DJ. Hot or not: tumor mutational burden (TMB) as a biomarker of immunotherapy response in genitourinary cancers. Urology. (2021) 147:119–26. doi: 10.1016/j.urology.2020.10.030
37. Devereaux KA, Steiner DF, Ho C, Gomez AJ, Gilks B, Longacre TA, et al. A multiplex SNaPshot assay is a rapid and cost-effective method for detecting POLE exonuclease domain mutations in endometrial carcinoma. Int J Gynecol Pathol. (2022) 41:541–51. doi: 10.1097/PGP.0000000000000841
38. Devereaux KA, Weiel JJ, Pors J, Steiner DF, Ho C, Charu V, et al. Prospective molecular classification of endometrial carcinomas: institutional implementation, practice, and clinical experience. Mod Pathol. (2022) 35:688–96. doi: 10.1038/s41379-021-00963-y
39. Van den Heerik ASVM, Ter Haar NT, Vermij L, Jobsen JJ, Brinkhuis M, Roothaan SM, et al. QPOLE : A quick, simple, and cheap alternative for POLE sequencing in endometrial cancer by multiplex genotyping quantitative polymerase chain reaction. JCO Glob Oncol. (2023) 9:1–11. doi: 10.1200/GO.22.00384
40. Keshinro A, Vanderbilt C, Kim JK, Firat C, Chen CT, Yaeger R, et al. Tumor-infiltrating lymphocytes, tumor mutational burden, and genetic alterations in microsatellite unstable, microsatellite stable, or mutant POLE/POLD1 colon cancer. JCO Precis Oncol. (2021) 5:817–26. doi: 10.1200/PO.20.00456
41. Galon J, Mlecnik B, Bindea G, Angell HK, Berger A, Lagorce C, et al. Towards the introduction of the ‘Immunoscore’ in the classification of Malignant tumours. J Pathol. (2014) 232:199–209. doi: 10.1002/path.4287
42. Forgó E, Gomez AJ, Steiner D, Zehnder J, Longacre TA. Morphological, immunophenotypical and molecular features of hypermutation in colorectal carcinomas with mutations in DNA polymerase ϵ (POLE). Histopathology. (2020) 76:366–74. doi: 10.1111/his.13984
43. Kim JH, Kim SY, Baek JY, Cha YJ, Ahn JB, Kim HS, et al. A phase II study of avelumab monotherapy in patients with mismatch repair-deficient/microsatellite instability-high or POLE-mutated metastatic or unresectable colorectal cancer. Cancer Res Treat. (2020) 52:1135–44. doi: 10.4143/crt.2020.218
44. Oh CR, Kim JE, Hong YS, Kim SY, Ahn JB, Baek JY, et al. Phase II study of durvalumab monotherapy in patients with previously treated microsatellite instability-high/mismatch repair-deficient or POLE-mutated metastatic or unresectable colorectal cancer. Int J Cancer. (2022) 150:2038–45. doi: 10.1002/ijc.33966
45. Jiang Y, Zhang Q, Hu Y, Li T, Yu J, Zhao L, et al. ImmunoScore signature. Ann Surg. (2018) 267:504–13. doi: 10.1097/SLA.0000000000002116
46. Meng B, Hoang LN, Mcintyre JB, Duggan MA, Nelson GS, Lee CH, et al. POLE exonuclease domain mutation predicts long progression-free survival in grade 3 endometrioid carcinoma of the endometrium. Gynecol Oncol. (2014) 134:15–9. doi: 10.1016/j.ygyno.2014.05.006
47. Ronsini C, Mosca L, Iavarone I, Nicoletti R, Vinci D, Carotenuto RM, et al. Oncological outcomes in fertility-sparing treatment in stage IA-G2 endometrial cancer. Front Oncol. (2022) 12:965029. doi: 10.3389/fonc.2022.965029
48. Cavaliere AF, Perelli F, Zaami S, D’Indinosante M, Turrini I, Giusti M, et al. Fertility sparing treatments in endometrial cancer patients: The potential role of the new molecular classification. Int J Mol Sci. (2021) 22:12248. doi: 10.3390/ijms222212248
49. Hoang LN, McConechy MK, Köbel M, Anglesio M, Senz J, Maassen M, et al. Polymerase epsilon exonuclease domain mutations in ovarian endometrioid carcinoma. Int J Gynecol Cancer. (2015) 25:1187–93. doi: 10.1097/IGC.0000000000000492
50. Mo S, Ma X, Li Y, Zhang L, Hou T, Han-Zhang H, et al. Somatic POLE exonuclease domain mutations elicit enhanced intratumoral immune responses in stage II colorectal cancer. J Immunother Cancer. (2020) 8:881. doi: 10.1136/jitc-2020-000881
51. Guenther M, Veninga V, Kumbrink J, Haas M, Westphalen CB, Kruger S, et al. POLE gene hotspot mutations in advanced pancreatic cancer. J Cancer Res Clin Oncol. (2018) 144:2161–6. doi: 10.1007/s00432-018-2746-x
52. Erson-Omay EZ, Cąglayan AO, Schultz N, Weinhold N, Omay SB, Özduman K, et al. Somatic POLE mutations cause an ultramutated giant cell high-grade glioma subtype with better prognosis. Neuro Oncol. (2015) 17:1356–64. doi: 10.1093/neuonc/nov027
53. Eskander RN, Sill MW, Beffa L, Moore RG, Hope JM, Musa FB, et al. Pembrolizumab plus chemotherapy in advanced endometrial cancer. N Engl J Med. (2023) 388:2159–70. doi: 10.1056/NEJMoa2302312
54. Van Gool IC, Eggink FA, Freeman-Mills L, Stelloo E, Marchi E, De Bruyn M, et al. POLE proofreading mutations elicit an antitumor immune response in endometrial cancer. Clin Cancer Res. (2015) 21:3347–55. doi: 10.1158/1078-0432.CCR-15-0057
55. Xiang D, Fu G, Chen Y, Chu X. Case report: POLE (P286R) mutation in a case of recurrent intestinal leakage and its treatment. Front Oncol. (2023) 13:1028179. doi: 10.3389/fonc.2023.1028179
56. Keenan BP, Van Loon K, Khilnani AD, Fidelman N, Behr SC, Atreya CE, et al. Molecular and radiological features of microsatellite stable colorectal cancer cases with dramatic responses to immunotherapy. Anticancer Res. (2021) 41:2985–92. doi: 10.21873/anticanres.15080
57. Durando ML, Menghani SV, Baumann JL, Robles DG, Day TA, Vaziri C, et al. Four-year disease-free remission in a patient with POLE mutation–associated colorectal cancer treated using anti–PD-1 therapy. JNCCN J Natl Compr Cancer Netw. (2022) 20:218–23. doi: 10.6004/JNCCN.2021.7115
58. Li S, Zhang J, Du W, Ren X, Zhang X. Pathologic complete response to immune checkpoint inhibitor in a stage IIIB ovarian clear cell carcinoma patient with POLE mutation resistant to platinum-based chemotherapy: a case report. Gland Surg. (2022) 11:1562–7. doi: 10.21037/gs-22-420
59. Tailored Adjuvant Therapy in POLE-mutated and p53-wildtype Early Stage Endometrial Cancer - Full Text View - ClinicalTrials.gov . Available online at: https://clinicaltrials.gov/study/NCT04705649 (Accessed March 13th, 2024)
60. van den Heerik ASVM, Horeweg N, Nout RA, Lutgens LCHW, van der Steen-Banasik EM, Westerveld GH, et al. PORTEC-4a: international randomized trial of molecular profile-based adjuvant treatment for women with high-intermediate risk endometrial cancer. Int J Gynecol Cancer. (2020) 30:2002–7. doi: 10.1136/ijgc-2020-001929
Keywords: POLE mutations, EDM, ultramutation, tumor mutational burden, hotspot mutations, endometrial cancer
Citation: Kögl J, Pan TL, Marth C and Zeimet AG (2024) The game-changing impact of POLE mutations in oncology—a review from a gynecologic oncology perspective. Front. Oncol. 14:1369189. doi: 10.3389/fonc.2024.1369189
Received: 11 January 2024; Accepted: 31 July 2024;
Published: 22 August 2024.
Edited by:
Louis Dubeau, University of Southern California, United StatesReviewed by:
Yosef Maruvka, Technion Israel Institute of Technology, IsraelCopyright © 2024 Kögl, Pan, Marth and Zeimet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alain G. Zeimet, YWxhaW4uemVpbWV0QGktbWVkLmFjLmF0
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.