
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 15 May 2024
Sec. Cancer Genetics
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1365474
This article is part of the Research TopicAdvances in Genetics and Molecular Diagnosis in Colorectal, Stomach, and Pancreatic Cancer Vol IIView all 8 articles
 Guojing Xie1†
Guojing Xie1† Qin Si1†Guangjie Zhang1,2Yu Fan1,3Qinghua Li1Ping Leng1,3Fengling Qiao1,3Simin Liang1Rong Yu1,3*
Qin Si1†Guangjie Zhang1,2Yu Fan1,3Qinghua Li1Ping Leng1,3Fengling Qiao1,3Simin Liang1Rong Yu1,3* Yingshuang Wang1,3*
Yingshuang Wang1,3*Genomic imprinting plays an important role in the growth and development of mammals. When the original imprint status of these genes is lost, known as loss of imprinting (LOI), it may affect growth, neurocognitive development, metabolism, and even tumor susceptibility. The LOI of imprint genes has gradually been found not only as an early event in tumorigenesis, but also to be involved in progression. More than 120 imprinted genes had been identified in humans. In this review, we summarized the most studied LOI of two gene clusters and 13 single genes in cancers. We focused on the roles they played, that is, as growth suppressors and anti-apoptosis agents, sustaining proliferative signaling or inducing angiogenesis; the molecular pathways they regulated; and especially their clinical significance. It is notable that 12 combined forms of multi-genes’ LOI, 3 of which have already been used as diagnostic models, achieved good sensitivity, specificity, and accuracy. In addition, the methods used for LOI detection in existing research are classified into detection of biallelic expression (BAE), differentially methylated regions (DMRs), methylation, and single-nucleotide polymorphisms (SNPs). These all indicated that the detection of imprinting genes’ LOI has potential clinical significance in cancer diagnosis, treatment, and prognosis.
Genomic imprinting has significant roles in individual growth, development, and cell differentiation in mammals (1). In this epigenetic process, a small group of genes, called imprinted genes, are expressed depending on their parental origin. Imprinting is manifested mainly as silencing of transcription when a gene is expressed by one parent and activation of transcription when it is expressed by the other parent (2). When the original imprint status of imprinted genes is lost, known as loss of imprinting (LOI), silenced alleles are abnormally activated, or active genes are suppressed. Such imprint disorders can affect growth, neurocognitive development, metabolism, and even tumor susceptibility.
Insulin-like growth factor 2 (IGF2) is among the most studied genes affected by LOI in cancers. The LOI of IGF2 gene was firstly demonstrated in wilms’ tumor (WT), a renal malignancy of childhood with an embryonic origin (3). LOI of the IGF2 has also been found in some adult somatic tumors including colorectal cancer (CRC), renal cell carcinoma (RCC), stomach adenocarcinoma (STAD), and esophageal squamous cell carcinoma (ESCC) (4–7). The LOI of imprinted genes has gradually emerged as an early event in tumorigenesis, as well as being implicated in the development of tumors (8). Some studies have reported aberrant gene imprinting status in specific cancer types, whereas others have focused on the impact of these changes on tumors.
LOI affects tumorigenesis and progression mainly through conferring resistance to apoptosis and evasion of growth suppressors, sustaining proliferative signaling, inducing angiogenesis, and activating metastasis (Figure 1). For example, IGF2 overexpression caused by LOI leads to the activation of the AKT and extracellular-regulated kinase (ERK) pathways, which promotes tumorigenesis (including cell proliferation and resistance to apoptosis) and metastasis (mainly liver metastases in CRC) (9, 10). Moreover, higher serum IGF2 concentration is associated with metastasis in CRC, and is an indicator of poor prognosis (9). In triple-negative breast cancer (TNBC), the LOI of potassium two-pore domain channel subfamily K member 9 (KCNK9) gene involving differentially methylated region (DMR) hypomethylation leads to overexpression of the gene, increasing mitochondrial membrane potential and anti-apoptotic effect (11, 12). In human hepatocellular carcinoma (HCC), hypomethylation at CpG85 has been reported to lead to an increase in levels of an alternative RB1-E2B transcript and concomitant downregulation of the RB1 main transcript in confirmed retinoblastoma (Rb) LOI, resulting in the absence of the Rb pathway and the loss of its suppressor function (13). Inhibition of transforming growth factor-β (TGF-β) signaling increases the probability of malignancy (14). Hypermethylation of the DIRAS family GTPase 3 (DIRAS3) CpG has been found to lead to LOI, resulting in a decrease in its expression, blunting the Ras or phosphatidyl-inositol-3 Kinase (PI3K) pathway (15, 16). Defects in these feedback mechanisms could enhance proliferative signaling (17). The LOI of maternally expressed 3 (MEG3) inactivates its expression, thereby enhancing angiogenesis and promoting tumorigenesis (18, 19). These findings suggest potential actionable targets for LOI genes in cancers.
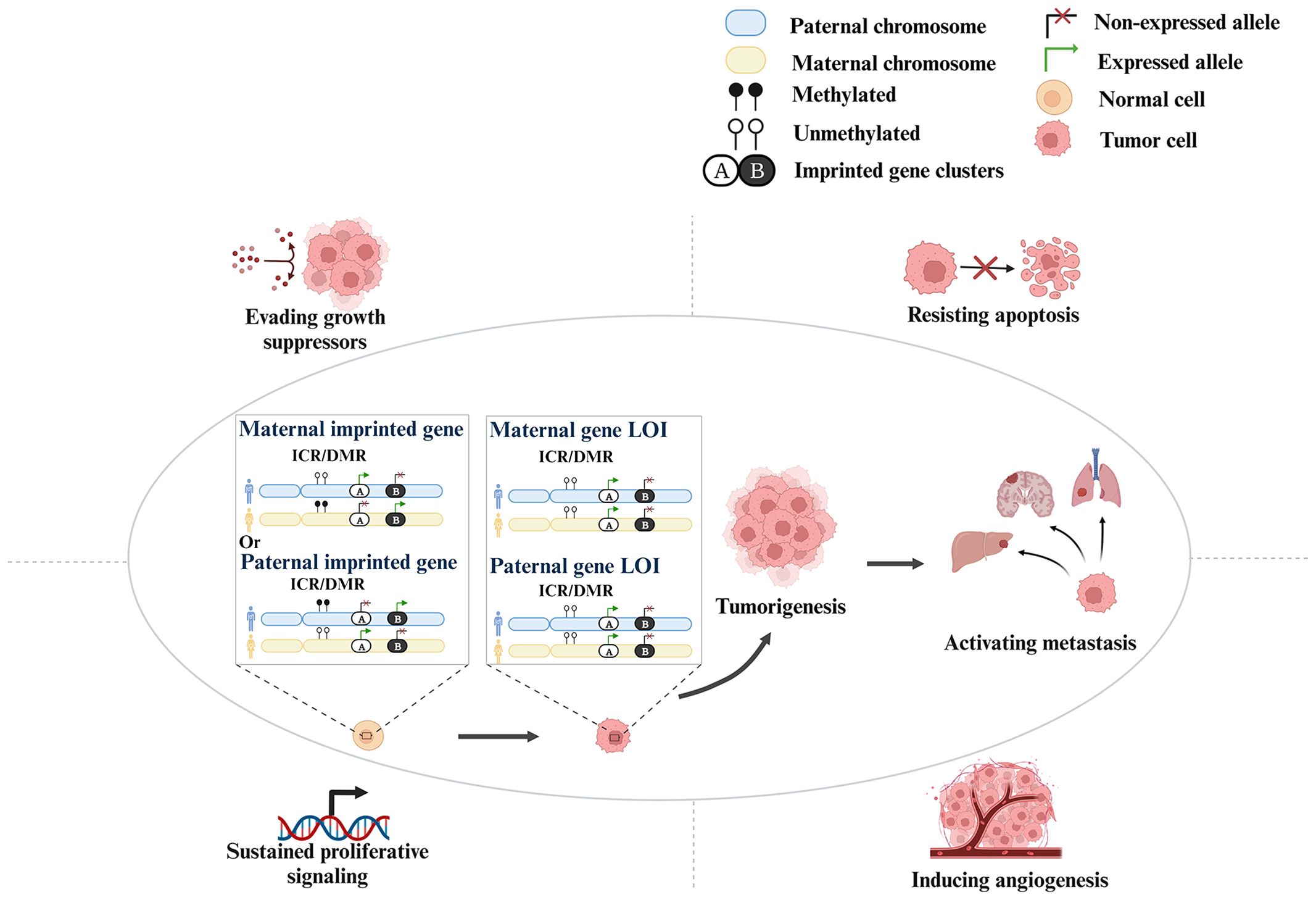
Figure 1 The molecular mechanism of LOI affecting cancers.
Disruption of the imprinting status also has implications for cancer diagnosis and prognosis. Studies have established diagnostic models using multiple imprinted genes based on the differences in allelic expression between normal, benign tumor, and cancerous tissues and have shown that these can function as efficient epigenetic biomarkers (20–22). Moreover, the presence of LOI at the delta-like non-canonical notch ligand 1 (DLK1) and MEG3 locus has been found to vary between two different histological subtypes of rhabdomyosarcoma (RMS) (23). Therefore, LOI detection represents a novel tool for cancer diagnosis. For instance, patients with esophageal adenocarcinoma (EAC) <65 years old with IGF2 LOI were found to have longer 5-year disease-free survival (DFS) (24), whereas in patients with CRC, the LOI was associated with higher overall mortality (4). These results demonstrate the importance of understanding the role of LOI in cancers and also illustrate the complexity arising from cancer tissue specificity.
More than 120 imprinted genes have been identified in humans (as displayed at geneimprint, http://www.geneimprint.com). However, there has been a lack of studies summarizing which imprinted genes are associated with cancers. Therefore, in this review, we used a systematic literature search strategy (Supplementary Figure 1A) to identify a total of 297 studies (after elimination of duplicate records). The two authors cross-checked the remaining articles, resulting in a total of 105 articles to be included in the review (the process is summarized in Supplementary Data 1, search strategy and selection criteria). These articles comprised results for 13 single genes (including two gene clusters) in 26 types of cancers and 12 combined forms of multi-gene LOI testing in 16 imprinted genes. Thus, our review provides a basis and prospective reference for the co-detection of imprinted genes and the selection of suitable biomarkers to establish novel clinical models in the future.
Imprinted genes are regulated by imprinted cluster-associated DMRs that play a critical part in maintaining parent-specific gene expression patterns known as imprinted control regions (ICRs). LOI is due to aberrant methylation in the DMRs of imprinted genes, usually loss of methylation maintenance, which produces aberrant transcripts that lead to activation of normally silent alleles. Methods for the detection of LOI have been established based on these mechanisms, such as the detection of biallelic expression (BAE), detection of DMR methylation, and detection of single-nucleotide polymorphisms (SNPs). In addition, 17 LOI detection methods are also summarized and grouped into three categories according to their principles, which will be helpful for selecting the appropriate LOI detection method in cancers.
LOI often occurs in many imprinted genes in malignancies, involving either a single imprinted gene LOI in one type of cancer or a specific cancer with multiple imprinted genes’ LOI simultaneously. Single genes, especially oncogenes or proto-oncogenes, may undergo alternations in expression when LOI occurs, subsequently affecting their biological functions in specific cancer types. Importantly, some of these genes are regulated in clusters. The chromosomal locations and regions that regulate imprinting and expression offer the potential for new therapeutic targets to be developed. Table 1 provides a summary of LOI sites, expression levels, and clinical significance of 13 single imprinted genes in 26 types of cancers, as well as epigenetically mediated mechanisms of carcinogenesis, based on the literature. In addition, multi-gene testing has shown that most gene combinations are grouped in clusters or in similar positions; some of these combinations have already been used to establish tumor diagnostic models, showing impressive potential for direct clinical applications. These gene combinations could also provide an index for future diagnostic models. Table 2 lists 12 gene combinations identified by multi-gene LOI testing, of which 3 have been established as cancer diagnostic models.
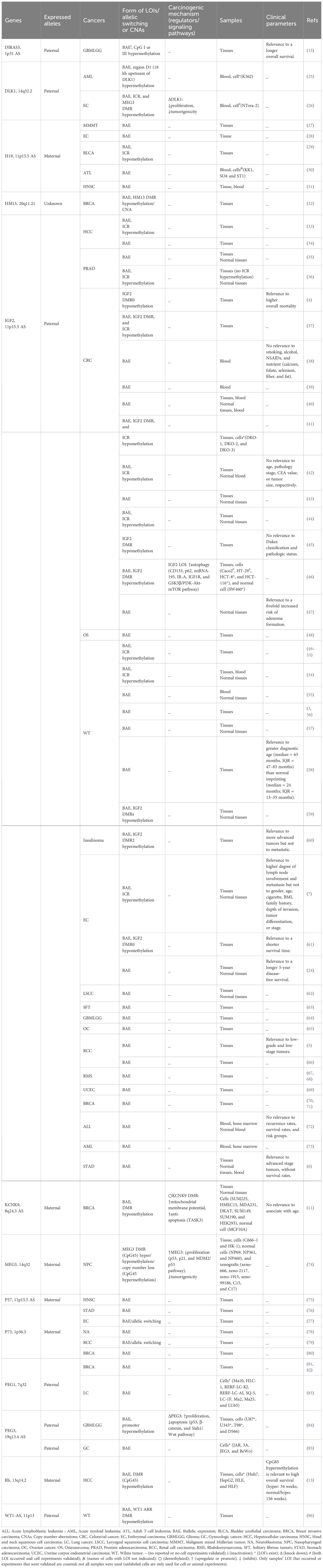
Table 1 Single-gene LOI in cancers.
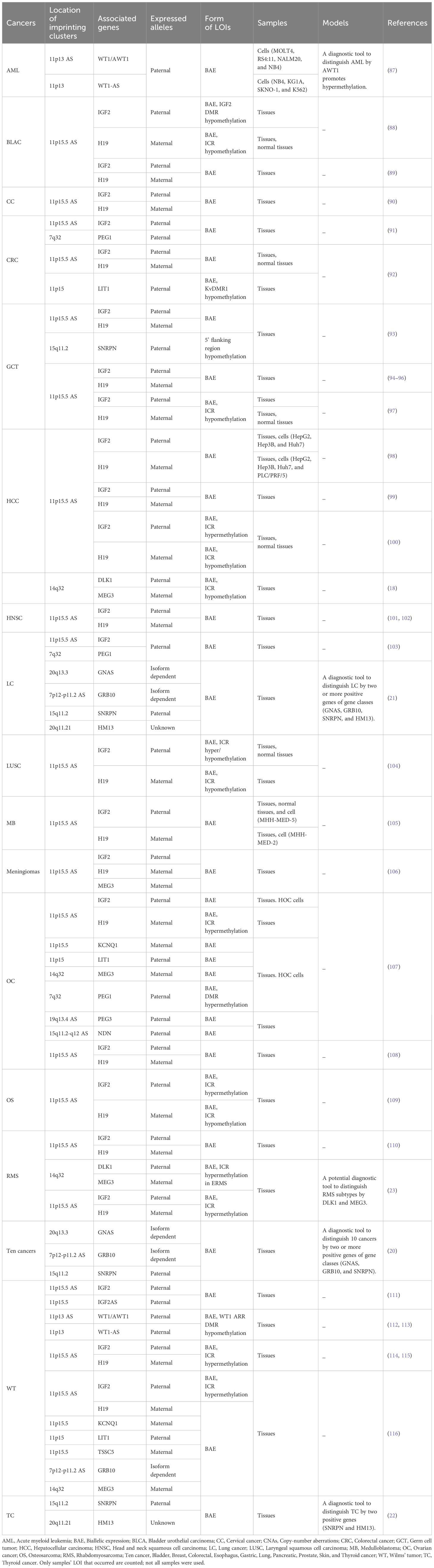
Table 2 Multiple-gene LOI in cancers.
IGF2 and H19, located on chromosome 11p15.5 in humans, are a mutually imprinted pair of genes that share a common regulatory locus (Figure 2A) (117). The IGF2 gene consists of 10 exons, and its expression is driven by five promoters (p0–p4) that possess different transcriptional activities both pre- and postnatally. In some cancer cells, four promoters (p0, p2, p3, and p4) whose IGF2 mRNA transcripts are imprinted contribute significantly to total IGF2 expression (118–120). Human H19 expressing a long non-coding RNA (lncRNA) contains six exons and two promoters. H19 DMR, also known as imprinting control region 1 (ICR1), is located between IGF2 and H19 and contains the binding sites for the epigenetic master regulator CTCF (121, 122). ICR1, IGF2 promoter-specific DMRs 0, 1, and 2, which partially overlap the IGF2 intronic and exonic sequences, the IGF2 enhancer region downstream from H19, and imprinting factor zinc finger protein 57 (ZFP57) jointly play a crucial part in maintaining normal imprinting and expression of these two genes in mammals (123–125).
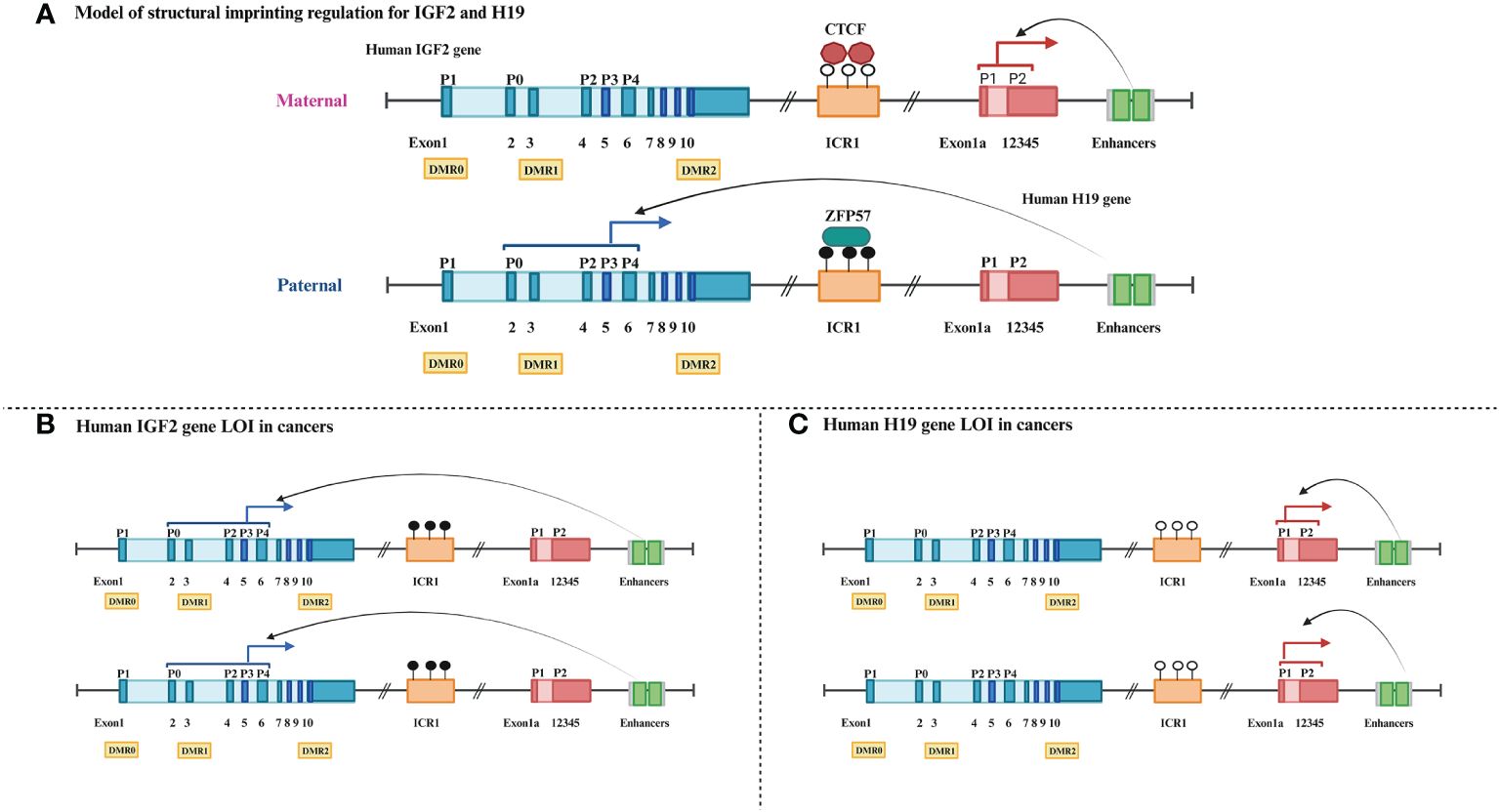
Figure 2 Schematic comparison of normal and loss of imprinting for human IGF2-H19 gene cluster. (A) Dark blue boxes: IGF2 exons, light blue boxes: IGF2 introns, P0–P4: IGF2 promoter regions, yellow rectangles: IGF2 DMRs, orange rectangle: ICR1, black circle: methylated, white circle: unmethylated, red polygons: insulator binding protein CTCF, black green rectangle: transcription element ZFP57, dark red boxes: H19 exon, light red box: H19 introns, grayish green squares: cis-remote control element enhancers. (B) Blue solid arrows: parent-specific transcripts of IGF2. (C) Red solid arrows: parent-specific transcripts of H19.
In most healthy adults, IGF2, which encodes proteins that promote fetal growth, is expressed only by the paternal allele (maternal ICR1 hypomethylation), whereas H19, which encodes an lncRNA with growth inhibitory properties, is expressed only by the maternal allele (paternal ICR1 hypermethylation) (126). This balance of expression of different parental alleles is broken when LOI occurs, routinely exhibiting opposing methylation states and biological functions, especially in the majority of patients with tumors. IGF2 LOI associated with hypermethylation of ICR1 and hypomethylation of IGF2 DMRs is prevalent and increases gene expression levels in the majority of cancers (Figure 2B) (4, 33). Moreover, ICR1 hypomethylation is also considered to be characteristic of H19 LOI and regularly results in the upregulation of H19 mRNA expression in human bladder cancer (Figure 2C) (29).
IGF2 undergoes normal imprinting changes, can act synergistically with multiple signaling pathways, and participates in physiological processes (autophagy, oncogenesis, and glycemic metabolism) of patients. It is well known that IGF2/IGF1R binding exerts cellular autophagy mediated by inhibiting the PI3K-Akt-mTOR signaling pathway in the CRC (127). Activated glycogen synthase kinase-3β (GSK3β) can inhibit B-cell lymphoma-2 (Bcl-2) as a mediating event to stimulate autophagy (128, 129). A recent study demonstrated that IGF2 LOI cancer stem cells (CSCs) were generally more prone to tumor formation and had higher levels of autophagy (CD133 with high expression and p62 with low expression) compared with maintenance of imprinting (MOI) cells in patients with CRC (46). Low expression of miRNA-195 in patients with CRC increased IGF2/IR-A binding, which more strongly promoted Akt expression and phosphorylation than IGF2/IGF1R, further decreasing GSK3β phosphorylation (46, 130). Overexpression of IGF2 related to LOI and receptor tyrosine kinase genes including DDR1, ERBB2, and FGFR1 have implicated the IGF2-INSR pathway in sphere formation of solitary fibrous tumor (SFT) (63). Hypoglycemia was also observed in SFT patients with IGF2 LOI.
Several studies have assessed the clinical value of imprinted genes in tumors. LOI and ICR/DMR methylation and alterations in expression levels due to LOI are relevant to clinical parameters, especially those related to survival and mortality. LOI of IGF2 was first identified in WT, which is a hereditary malignant embryonic tumor of infants (3), with a relatively older age at diagnosis of children with IGF2 LOI (median = 65 months, IQR = 47–83 months) (58). However, subsequent studies also found IGF2 LOI in adult somatic cell tumors, such as CRC, RCC, STAD, and ESCC. IGF2 LOI has been reported to be associated with a fivefold increased risk of adenoma formation and higher overall mortality in CRC (4, 47). IGF2 LOI appeared to predispose RCC patients to low-grade and low-stage tumors (5) and was more likely to occur in advanced STAD (6). Patients with ESCC with IGF2 LOI showed a higher degree of lymph node involvement, metastasis, and shorter survival times (7, 61). However, patients with EAC with IGF2 LOI were found to have a longer 5-year DFS (24). These not only show the importance of paying attention to LOI in cancers but also illustrate the complexity arising from cancer tissue specificity. Finally, H19 LOI has been found to be present in patients with head and neck carcinoma, and patients with high expression of H19 appeared to be more likely to experience relapse (31).
The human DLK1 gene resides in the chromosomal 14q32 region, positioned with MEG3, with which it constructs an imprinted gene cluster (NCBI reference sequence: NC_000014.9). The paternally expressed protein-coding DLK1 gene is composed of 5 exons, whereas MEG3 with 13 exons maternally expresses an lncRNA. At the DLK1-MEG3 locus, it is regulated by both the ICR and MEG3 DMR containing the CTCF binding DNA sequence, which lies among the two genes (131, 132). Aronson et al. revealed that a hierarchical and unidirectional regulation existed between the ICR and MEG3 DMR, and the dominant ICR was established as a dichotomous control element that maintained imprinting through allele-specific restriction of the DNA (de)methylation mechanism (Figure 3A) (133).

Figure 3 Schematic comparison of normal and loss of imprinting for human DLK1-MEG3 gene cluster. (A) Dark green boxes: DLK1 exons, light green boxes: DLK1 introns, dark blue rectangles: ICR (CpG Island CGI and TRE work independently on different alleles to restrict the activities of TETs and DNMTs), yellow quads in CGI: conserved tandem repeat array, black circle: methylated, white circle: unmethylated, orange trapezoid: demethylated enzyme TETs, blue cloud: methylated enzyme DNMTs, black green rectangle: transcription element ZFP57, dark yellow boxes: MEG3 exon, light yellow box: MEG3 introns. (B) Green solid arrows: parent-specific transcripts of DLK1, red letter x: absence. (C) Yellow solid arrows: parent-specific transcripts of MEG3.
DLK1 and MEG3 are methylated on the paternal allele, but unmethylated on the maternal allele, which regulates their expression in healthy individuals (134). However, in some cancer patients, the parental alleles are expressed in an imbalanced manner and usually exhibit opposite methylation states and expression. DLK1 LOI (ICR and MEG3 DMR hypermethylation) manifests as biallelic DLK1 expression and MEG3 silencing, whereas MEG3 LOI shows ICR and MEG3 DMR hypomethylation and the opposite expression trend (Figures 3B, C). In addition to LOI, allelic switching (opposite single allele expression) accompanied by gains or losses of DNA methylation primarily on IG-DMR at the DLK1-MEG3 locus had also been discovered in some patients with HCC (18). MEG3 copy number loss was found only in patients with nasopharyngeal carcinoma (NPC) whose LOI manifested as DMR hypermethylation (74). These results indicate that genetics and epigenetics may synergistically influence the vast majority of tumors.
Similar to IGF2 and H19, DLK1 and MEG3 also perform diverse biological functions in cancers. LOI was found to upregulate DKL1 mRNA expression; however, knocking down its expression would inhibit proliferation and tumorigenicity in embryonal carcinoma (EC) (26). DLK1 appears to exert a cancer-promoting role. Conversely, in glioma (GBMLGG), lower expression of MEG3 promotes not only oncogenesis, but also malignant behavior such as proliferation, migration, and tumorigenicity (135). When restored to normal expression levels, MEG3 played a tumor suppressor role suppressed by inducing a significant downward adjustment of focal adhesion kinase (FAK), vimentin, and inhibitory phosphorylation of non-receptor tyrosine kinase (SRC). Furthermore, MEG3 restoration increased levels of β-actin (an important skeletal protein), caveolin-1 (a negative growth regulator), and connexin-43, as well as activating N-myc downstream-regulated gene 1 (NDRG1), which has previously been shown to inhibit metastasis and migration in CRC (136). MEG3 also increased expression of p53 and a potent cyclin-dependent kinase inhibitor called p21, which might explain the observed enhancement of G1/S cell cycle arrest, and stimulated E3 ubiquitin ligase MDM2 production, which could represent suppressed NPC metastasis through the p53-MDM2-Slug pathway (74).
With respect to clinical applications, LOI of imprinted genes combined may appear to be useful for differentiating tumor subtypes. It has also been shown that both embryonal and alveolar rhabdomyosarcomas (ERMS and ARMS, respectively) show LOI for the DMR of the IGF2-H19 locus, while ERMS consistently shows LOI of the DMR at the DLK1-MEG3 locus (23).
Apart from the best-known genetic changes in the form of heredity, such as mutations, genomic instability, loss of heterozygosity (LOH) and copy number aberrations (CNAs) leading to the inactivation of oncogenes or proto-oncogenes, epigenetic change can also cause this phenomenon. In contrast to clustered genes, single-gene LOI exhibits BAE or dysregulation of aberrant transcripts. Alteration of an imprinting control center may lead to abnormal expression of oncogenes or tumor suppressor genes, causing different effects on promoting and suppressing cancer.
On the one hand, LOI genes that promote cancer comprise Rb, KCNK9, and paternally expressed gene 3 (PEG3). The Rb gene, a retinoblastoma susceptibility gene, was the first tumor suppressor gene to be cloned and have its full sequence determined. Anwar et al. identified that LOI (CpG85 hypomethylation) is also a novel pathway for the inactivation of Rb in HCC (13). The Rb gene expresses only the maternal gene, while the paternal gene expresses the abnormal transcript (RB1-E2B) that starts at the CpG85 island. In the absence of imprinting, levels of RB1-E2B will increase, eventually leading to decreased expression of the main transcript RB. Patients with CpG85 hypermethylation have shorter overall survival (the median survival rates for hypermethylation and normal/hypomethylation are 34 and 156 weeks, respectively). KCNK9 LOI was found due to DMR hypomethylation, which leads to overexpression of its gene product, increasing mitochondrial membrane potential and anti-apoptosis in TNBC (11, 12). Hypermethylation of the PEG3 promoter leads to LOI and decreased PEG3 mRNA expression, increasing β-catenin levels, promoting proliferation, and inhibiting p53-dependent apoptosis in human GBMLGG (84). On the other hand, the LOI gene that inhibits cancer is P73. The increased expression of P73, including that resulting from LOI, could be a partial compensatory mechanism for defective p53 in ESCC (77).
Traditional cytology and histopathology, imaging examination, and use of serum biomarkers have contributed tremendously to the early detection of cancer, but accurate diagnostic assessment of nodules and early-stage cancers with insufficient evidence of tumor morphology or abnormal metabolism remains a great clinical challenge at present (137–140). However, epigenetics may compensate for this deficiency. There is already clear evidence that epigenetic changes during carcinogenesis often precede morphological changes (141, 142). To provide reference information for more accurate tumor-specific diagnosis and precise personalized treatment in clinical settings, we summarize 12 combined forms of multi-gene LOI testing in Table 2, of which 3 types of combinations have been established as cancer diagnostic models.
Some researchers have successfully exploited a novel method, quantitative chromogenic imprinted gene in situ hybridization (QCIGISH), targeting non-coding intron nascent RNA, to directly observe BAE, multiallelic expression (MAE), and total expression (TE) at transcription sites of imprinted genes in the nucleus to select these appropriate imprinted genes for the construction, optimization, and validation of tumor diagnostic models (20). First, a diagnostic model for 10 different solid cancer types (bladder, breast, colorectal, esophageal, gastric, lung, pancreatic, prostate, skin, and thyroid cancers) was built using imprinted genes’ GNAS complex locus (GNAS), growth factor receptor bound protein 10 (GRB10), and small nuclear ribonucleoprotein polypeptide N (SNRPN) with a total sensitivity of 94%, a specificity of 92%, and an accuracy of 93% (20). Next, based on the above preliminary model, a more specific diagnostic model for grading lung cancer (LC) was also established using GNAS, GRB10, SNRPN, and histocompatibility minor 13 (HM13). This diagnostic model was highly effective in the diagnosis of both different subtypes of LC and small lung nodules, with an overall sensitivity of 99.1%, a specificity of 92.1%, and an area under the curve (AUC) of 0.99 (21). Lastly, a thyroid cancer (TC) diagnostic model through imprinted genes SNRPN and HM13 has achieved an overall diagnostic sensitivity of 100%, a specificity of 91.5%, a positive predictive value (PPV) of 96.5%, a negative predictive value (NPV) of 100%, and a diagnostic accuracy of 97.5% in a prospective validation (22).
In sum, these findings provide considerable benefits and ideas for screening or predicting appropriate tumor markers, comprehensive clinical risk assessment, and finding new epigenetic therapeutic targets. This fully reflects the importance and non-negligibility of tumor epigenetics.
Various methods have been used in the detection of imprinted gene LOI in the past three decades. In the 105 studies listed in Table 3, restriction fragment length polymorphism PCR (RFLP-PCR) was the most frequently used method, used up to 84 times (75/105) from 1993 to 2020. This was followed by bisulfite sequencing PCR (BSP) (25/105, 2003–2021) and pyrosequencing (8/105, 2007–2014). LOI arises from abnormal methylation of the DMR of imprinted genes (usually loss of methylation maintenance), which produces double alleles (aberrant transcripts leading to silencing of a normally active allele). LOI can also be discriminated based on SNPs. According to the detection objects used, the 17 methods for this purpose can be categorized into three types: (I) detection of BAE: hot-stop PCR, nest PCR, QCIGISH, RFLP-PCR, real-time quantitative reverse transcription PCR, reverse transcription PCR, and pyrosequencing; (II) detection of DMR methylation: BSP, bisulfite PCR-Luminex, combined bisulfite restriction analysis, Illumina 450 K arrays, pyrosequencing, methylation-specific PCR, NOMe-sequencing, RFLP-PCR, and the MassARRAY EpiTYPER; and (III) detection of SNPs: SNuPE assays, RNA sequencing (RNA-seq), and DNA sequencing. To make the results more credible and convincing, there is a growing trend towards the simultaneous use of multiple analytical methods with the same or different principles and away from the use of single or single-principle methods in some studies.
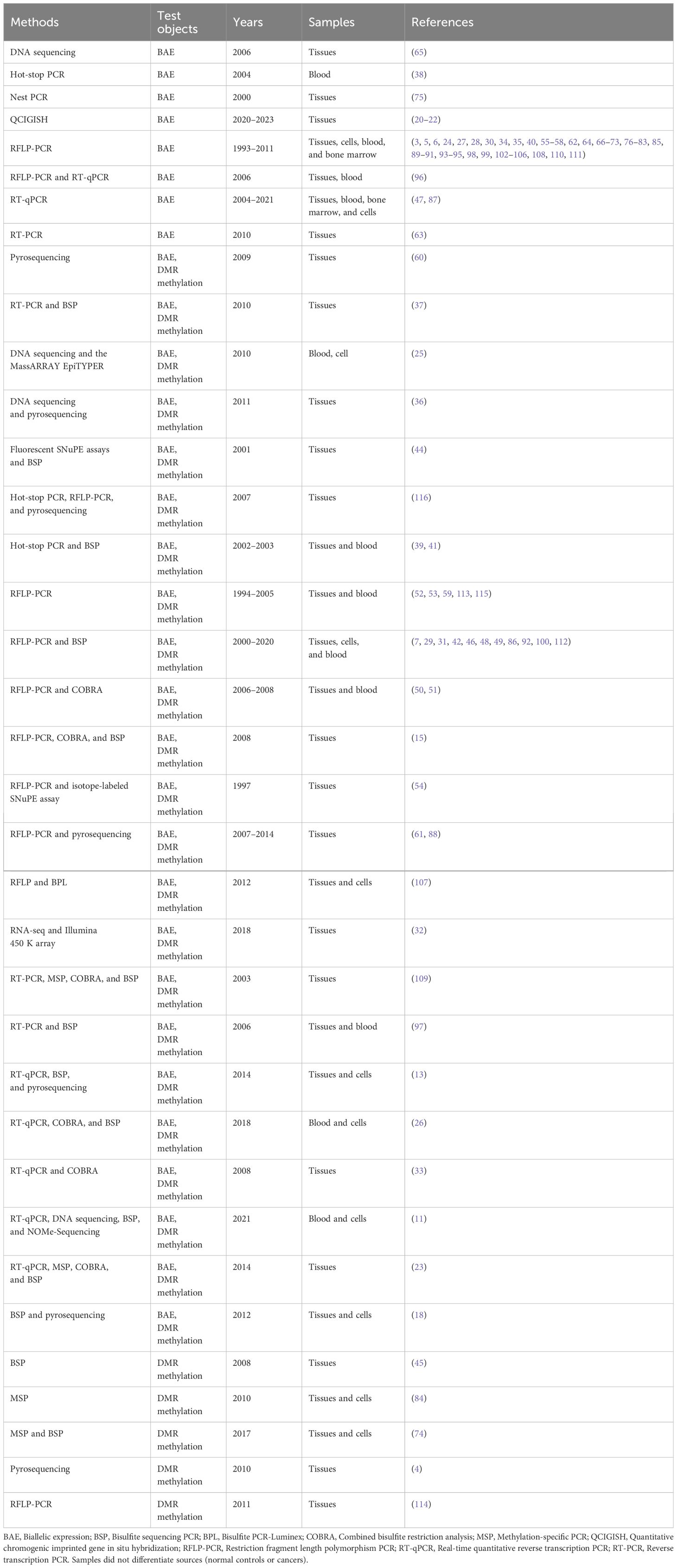
Table 3 Detection methods of LOI in cancers.
Sequencing techniques based on sulfite treatment are widely used; however, despite their convenience, their drawbacks are also increasingly obvious. Sulfite treatment may lead to severe degradation of the input DNA owing to harsh reaction conditions, which is a common problem with most sequencing methods. Chemical enzymes compensate for this defect (143). For instance, a combination of chemical enzymes such as APOBEC3A (A3A) or engineered APOBEC3C (eA3C) and sequencing technologies has achieved consistent and reliable results (144, 145). This highlights the potential of multidisciplinary combinations to lead to new approaches.
Notably, several high-throughput techniques are being used for genomic methylation and allele-specific expression (ASE), showing great promise for the analysis and detection of imprinted gene LOI in cancers. The demand for comprehensive descriptions of DNA methylation patterns has led to a diversity of DNA methylation profiling technologies, including reduced representation bisulfite sequencing (RRBS) based on bisulfite conversion, methylated DNA binding domain sequencing, methylated DNA immunoprecipitation sequencing (MeDIP-seq) based on affinity enrichment, and methylation-sensitive restriction enzyme sequencing (MRE-seq) based on endonuclease digestion that targets genomic distribution (146). Recent studies have shown that utilizing the complementary properties of MeDIP-seq and MRE-seq can provide a rapid comparative analysis of the entire methylome at a fraction of the cost of whole-genome bisulfite sequencing (WGBS) (the gold standard method for detecting methylation at single-base resolution) with higher accuracy and reproducibility than either individual method (147–149). Analysis of existing RNA-seq datasets can be used to identify ASE of imprinted genes beyond evaluation of gene expression, thereby detecting the LOI of imprinted genes (150). However, when heterogeneous populations of cells, such as cancer samples, are analyzed, only single-cell measurements allowed the detection of widespread LOI events (151). Therefore, the use of effective and appropriate data analysis methods to analyze single-cell transcriptomic data will provide a major advantage in the analysis of tumor epigenetic aberrations. For example, BrewerIX, a standardized approach for the analysis of known imprinted genes, can be used to analyze RNA-seq data from single breast cancer cells to identify LOI of imprinted genes (151). Differential allelic expression using single-cell data (DAESC), a powerful method for differential ASE analysis using single-cell RNA sequencing (scRNA-seq) from multiple individuals, is capable of analyzing genes with differential ASE in pancreatic endocrine cells from patients with type 2 diabetes and controls, taking into account the effect of allelic switching, although it is not suitable for estimating cancer cells (152). These findings suggest that establishing standardized data analysis methods and combining existing LOI methods or potential methods with different characteristics may be a viable option in the cancer field, compared with exploring new detection methods that may have unknown limitations.
In conclusion, the presence or absence of LOI in cancers can be determined by using multiple methods of the same type vertically, two or more different types of methods horizontally, or even methods that combine multiple disciplines, making the results more accurate and reliable. Furthermore, the establishment of standardized data analysis methods for high-throughput technologies, in addition to combining multiple approaches, will help to uncover more potential imprinted genes and LOI, thereby facilitating the discovery of context-specific regulatory effects in cancers. As sequencing costs decrease, these methods will also be appealing in clinical practice.
The established association between LOI and microsatellite instability (MSI) seems to provide a new epigenetic view of cancer susceptibility (40, 91), although this is complex, given the expression of imprinted genes in a parent-of-origin-specific manner. For the imprinted gene Rb, allele mutations from different parents have different effects on tumor susceptibility in hereditary retinoblastoma: if the mutation is of paternal origin, the offspring has a 12% chance of developing retinoblastoma, whereas when the mutation is of maternal origin, the offspring have a 75% chance of developing retinoblastoma (16, 153). Beyond embryonic-derived blastomas, epigenetic alterations in imprinted genes, often presenting as LOI, have been found in various somatic cancers. In addition, LOI of imprinted genes has been increasingly implicated in malignant behavior. The detection of LOI thus has potential clinical significance in cancer diagnosis, treatment, and prognosis.
Here, we have summarized 13 single-gene LOI in cancers, identifying the relevant detection sites and cancer types and considering whether they promote or inhibit functions in cancers. This provides a convenient index for co-detection of imprinted gene LOI in specific types of cancer. Moreover, as recent studies have found that aberrant gene imprinting patterns can occur together with cancer-associated CNAs (154) or allelic switching (77), we have also included these types of change in our analysis of studies (Table 1). Although the role of aberrant imprinting patterns in tumors is unquestionable, few studies have considered CNAs (1/70) or allelic switching (2/70) when reporting methylation profiles. Therefore, we suggest increasing the investigation of CNAs or allelic switching in future research to improve the accuracy of functional research on LOI genes. Coupled genes may be either clustered, as in the IGF2-H19 locus or DLK1-MEG3 locus, or non-clustered in specific cancers. In the analysis of loci for multi-gene detection panels, 12 combined forms of multi-genes were included, of which 3 gene combinations have been established as cancer diagnostic models. It is possible that more patterns may be found in the future based on the characteristics of imprinted genes in clustered LOI. There is also evidence to suggest that both the imprinting state and expression can be uncoupled in clustered genes. For instance, IGF2 LOI was not found to be coupled with downregulation of H19 expression in HCC (98, 99); in RMS, although H19 LOI was present, the imprinting state of IGF2 was maintained (110). These cases not only illustrate the complexity arising from cancer tissue specificity but also indicate an independent control mechanism for imprinting.
Notably, the role of imprinting gene LOI may vary among different tumors. For instance, the protein encoded by p73 is structurally and functionally similar to that encoded by p53, a tumor suppressor. In p53-defect ESCC, p73 was found to have elevated expression and LOI, which is speculated to be a substitute mechanism for the tumor-suppressing function (79). However, in RCC, LOI or switching of allelic expression of p73 is associated with cancer development (77). On the other hand, even if a gene undergoes LOI, its downstream pathways may differ in different tumor types. In CRC, LOI of IGF2 can enhance cell autophagy through the PI3K/Akt/mTOR pathway, whereas it might promote tumor formation through the IGF2-INSR pathway in SFT (46, 63). These findings suggest that it will be necessary for the future design of targeted LOI therapies to consider mutations of key factors in downstream pathways in different tumor types.
In the detection of imprinted genes’ LOI in cancers, although DNA methylation status changes are characteristic of LOI, their detection is distinct from that of overall DNA or promoter region methylation. Therefore, the focus should be on DMR/ICR only. In HCC, global loss of methylation and increased methylation at DLK1 and MEG3 DMR/ICR-specific sites have been simultaneously observed (18). Detection methods for LOI have evolved from qualitative to quantitative, from detecting overall CpG islands to single CpG site, and to more simplified procedures (Supplementary Figure 1B). Although we have summarized the mature LOI methods currently used in tumor detection based on the literature, when considering the depth of sequencing, sample requirements, and mutation detection, high-throughput methods such as whole-genome sequencing, whole-exon sequencing, and single-cell sequencing have great application prospects for LOI detection of imprinted genes in cancers (32, 155).
Both blood samples and tissue samples are suitable for the detection of LOI. IGF2 LOI has been found in the blood and tissues of both patients with CRC and healthy controls and may be a valuable predictive marker of an individual’s risk of carcinoma (39, 40, 47). Although blood samples are more clinically accessible, tissue samples were more commonly used in the studies reviewed here (63/70 for single-gene detection, 34/35 for multiple-gene detection). This may be because in adult cancer patients, only the imprinted genes in cancer cells are LOI, while those in somatic cells maintain their imprint. With the development of enrichment methods for circulating tumor cells, use of tumor-derived exosomes in liquid biopsies, and advances in circulating cell-free DNA (cfDNA) methylation detection methods, blood samples have greater application prospects (156–158). Blood tests may therefore be of great informative value for large-scale LOI testing in cancer-susceptible populations.
GX: Writing – original draft. QS: Writing – original draft. GZ: Writing – original draft. YF: Writing – original draft. QL: Writing – original draft. PL: Writing – original draft. FQ: Writing – original draft. SL: Writing – original draft. RY: Writing – original draft. YW: Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China (82003001), the Sichuan Science and Technology Program (2022NSFSC1584), and the Foundation of Xinglin Scholar Research Program Project of Chengdu University of TCM (QJRC2022048).
We sincerely appreciate all contributors to the references listed. Figures are created using BioRender.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2024.1365474/full#supplementary-material
1. Tucci V, Isles AR, Kelsey G, Ferguson-Smith AC, Erice Imprinting G. Genomic imprinting and physiological processes in mammals. Cell. (2019) 176:952–65. doi: 10.1016/j.cell.2019.01.043
2. Lozano-Urena A, Jimenez-Villalba E, Pinedo-Serrano A, Jordan-Pla A, Kirstein M, Ferron SR. Aberrations of genomic imprinting in glioblastoma formation. Front Oncol. (2021) 11:630482. doi: 10.3389/fonc.2021.630482
3. Ogawa O, Eccles MR, Szeto J, McNoe LA, Yun K, Maw MA, et al. Relaxation of insulin-like growth factor II gene imprinting implicated in Wilms' tumour. Nature. (1993) 362:749–51. doi: 10.1038/362749a0
4. Baba Y, Nosho K, Shima K, Huttenhower C, Tanaka N, Hazra A, et al. Hypomethylation of the IGF2 DMR in colorectal tumors, detected by bisulfite pyrosequencing, is associated with poor prognosis. Gastroenterology. (2010) 139:1855–64. doi: 10.1053/j.gastro.2010.07.050
5. Nonomura N, Nishimura K, Miki T, Kanno N, Kojima Y, Yokoyama M, et al. Loss of imprinting of the insulin-like growth factor II gene in renal cell carcinoma. Cancer Res. (1997) 57:2575–7.
6. Zuo QS, Yan R, Feng DX, Zhao R, Chen C, Jiang YM, et al. Loss of imprinting and abnormal expression of the insulin-like growth factor 2 gene in gastric cancer. Mol Carcinog. (2011) 50:390–6. doi: 10.1002/(SICI)1097-0142(19990315)85:6<1389::AID-CNCR24>3.3.CO;2-M
7. Gao T, He B, Pan Y, Gu L, Chen L, Nie Z, et al. H19 DMR methylation correlates to the progression of esophageal squamous cell carcinoma through IGF2 imprinting pathway. Clin Transl Oncol. (2014) 16:410–7. doi: 10.1007/s12094-013-1098-x
8. Ribarska T, Bastian KM, Koch A, Schulz WA. Specific changes in the expression of imprinted genes in prostate cancer–implications for cancer progression and epigenetic regulation. Asian J Androl. (2012) 14:436–50. doi: 10.1038/aja.2011.160
9. Kasprzak A, Adamek A. Insulin-like growth factor 2 (IGF2) signaling in colorectal cancer-from basic research to potential clinical applications. Int J Mol Sci. (2019) 20(19):4915. doi: 10.3390/ijms20194915
10. Murrell A, Ito Y, Verde G, Huddleston J, Woodfine K, Silengo MC, et al. Distinct methylation changes at the IGF2-H19 locus in congenital growth disorders and cancer. PloS One. (2008) 3:e1849. doi: 10.1371/journal.pone.0001849
11. Skaar DA, Dietze EC, Alva-Ornelas JA, Ann D, Schones DE, Hyslop T, et al. Epigenetic dysregulation of KCNK9 imprinting and triple-negative breast cancer. Cancers (Basel). (2021) 13(23):6031. doi: 10.3390/cancers13236031
12. Pei L, Wiser O, Slavin A, Mu D, Powers S, Jan LY, et al. Oncogenic potential of TASK3 (Kcnk9) depends on K+ channel function. Proc Natl Acad Sci U.S.A. (2003) 100:7803–7. doi: 10.1073/pnas.1232448100
13. Anwar SL, Krech T, Hasemeier B, Schipper E, Schweitzer N, Vogel A, et al. Deregulation of RB1 expression by loss of imprinting in human hepatocellular carcinoma. J Pathol. (2014) 233:392–401. doi: 10.1002/path.4376
14. Korah J, Canaff L, Lebrun JJ. The retinoblastoma tumor suppressor protein (pRb)/E2 promoter binding factor 1 (E2F1) pathway as a novel mediator of TGFβ-induced autophagy. J Biol Chem. (2016) 291:2043–54. doi: 10.1074/jbc.M115.678557
15. Riemenschneider MJ, Reifenberger J, Reifenberger G. Frequent biallelic inactivation and transcriptional silencing of the DIRAS3 gene at 1p31 in oligodendroglial tumors with 1p loss. Int J Cancer. (2008) 122:2503–10. doi: 10.1002/ijc.23409
16. Uribe-Lewis S, Woodfine K, Stojic L, Murrell A. Molecular mechanisms of genomic imprinting and clinical implications for cancer. Expert Rev Mol Med. (2011) 13:e2. doi: 10.1017/S1462399410001717
17. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. (2011) 144:646–74. doi: 10.1016/j.cell.2011.02.013
18. Anwar SL, Krech T, Hasemeier B, Schipper E, Schweitzer N, Vogel A, et al. Loss of imprinting and allelic switching at the DLK1-MEG3 locus in human hepatocellular carcinoma. PloS One. (2012) 7:e49462. doi: 10.1371/journal.pone.0049462
19. Zhou Y, Zhang X, Klibanski A. MEG3 noncoding RNA: a tumor suppressor. J Mol Endocrinol. (2012) 48:R45–53. doi: 10.1530/JME-12–0008
20. Shen R, Cheng T, Xu C, Yung RC, Bao J, Li X, et al. Novel visualized quantitative epigenetic imprinted gene biomarkers diagnose the Malignancy of ten cancer types. Clin Epigenet. (2020) 12:71. doi: 10.1186/s13148–020-00861–1
21. Zhou J, Cheng T, Li X, Hu J, Li E, Ding M, et al. Epigenetic imprinting alterations as effective diagnostic biomarkers for early-stage lung cancer and small pulmonary nodules. Clin Epigenet. (2021) 13:220. doi: 10.1186/s13148–021-01203–5
22. Xu H, Zhang Y, Wu H, Zhou N, Li X, Pineda JP, et al. High diagnostic accuracy of epigenetic imprinting biomarkers in thyroid nodules. J Clin Oncol. (2023) 41:1296–306. doi: 10.1200/JCO.22.00232
23. Schneider G, Bowser MJ, Shin DM, Barr FG, Ratajczak MZ. The paternally imprinted DLK1-GTL2 locus is differentially methylated in embryonal and alveolar rhabdomyosarcomas. Int J Oncol. (2014) 44:295–300. doi: 10.3892/ijo.2013.2153
24. Zhao R, DeCoteau JF, Geyer CR, Gao M, Cui H, Casson AG. Loss of imprinting of the insulin-like growth factor II (IGF2) gene in esophageal normal and adenocarcinoma tissues. Carcinogenesis. (2009) 30:2117–22. doi: 10.1093/carcin/bgp254
25. Khoury H, Suarez-Saiz F, Wu S, Minden MD. An upstream insulator regulates DLK1 imprinting in AML. Blood. (2010) 115:2260–3. doi: 10.1182/blood-2009–03-212746
26. Sellers ZP, Schneider G, Maj M, Ratajczak MZ. Analysis of the paternally-imprinted DLK1-MEG3 and IGF2-H19 tandem gene loci in NT2 embryonal carcinoma cells identifies DLK1 as a potential therapeutic target. Stem Cell Rev Rep. (2018) 14:823–36. doi: 10.1007/s12015–018-9838–5
27. Hashimoto K, Azuma C, Tokugawa Y, Nobunaga T, Aki TA, Matsui Y, et al. Loss of H19 imprinting and up-regulation of H19 and SNRPN in a case with Malignant mixed Mullerian tumor of the uterus. Hum Pathol. (1997) 28:862–5. doi: 10.1016/s0046–8177(97)90162–3
28. Hibi K, Nakamura H, Hirai A, Fujikake Y, Kasai Y, Akiyama S, et al. Loss of H19 imprinting in esophageal cancer. Cancer Res. (1996) 56:480–2
29. Takai D, Gonzales FA, Tsai YC, Thayer MJ, Jones PA. Large scale mapping of methylcytosines in CTCF-binding sites in the human H19 promoter and aberrant hypomethylation in human bladder cancer. Hum Mol Genet. (2001) 10:2619–26. doi: 10.1093/hmg/10.23.2619
30. Takeuchi S, Hofmann WK, Tsukasaki K, Takeuchi N, Ikezoe T, Matsushita M, et al. Loss of H19 imprinting in adult T-cell leukaemia/lymphoma. Br J Haematol. (2007) 137:380–1. doi: 10.1111/j.1365-2141.2007.06581.x
31. Esteves LI, Javaroni AC, Nishimoto IN, Magrin J, Squire JA, Kowalski LP, et al. DNA methylation in the CTCF-binding site I and the expression pattern of the H19 gene: does positive expression predict poor prognosis in early stage head and neck carcinomas? Mol Carcinog. (2005) 44:102–10. doi: 10.1002/mc.20126
32. Goovaerts T, Steyaert S, Vandenbussche CA, Galle J, Thas O, Van Criekinge W, et al. A comprehensive overview of genomic imprinting in breast and its deregulation in cancer. Nat Commun. (2018) 9:4120. doi: 10.1038/s41467–018-06566–7
33. Honda S, Arai Y, Haruta M, Sasaki F, Ohira M, Yamaoka H, et al. Loss of imprinting of IGF2 correlates with hypermethylation of the H19 differentially methylated region in hepatoblastoma. Br J Cancer. (2008) 99:1891–9. doi: 10.1038/sj.bjc.6604754
34. Montagna M, Menin C, Chieco-Bianchi L, D'Andrea E. Occasional loss of constitutive heterozygosity at 11p15.5 and imprinting relaxation of the IGFII maternal allele in hepatoblastoma. J Cancer Res Clin Oncol. (1994) 120:732–6. doi: 10.1007/BF01194272
35. Jarrard DF, Bussemakers MJ, Bova GS, Isaacs WB. Regional loss of imprinting of the insulin-like growth factor II gene occurs in human prostate tissues. Clin Cancer Res. (1995) 1:1471–8
36. Bhusari S, Yang B, Kueck J, Huang W, Jarrard DF. Insulin-like growth factor-2 (IGF2) loss of imprinting marks a field defect within human prostates containing cancer. Prostate. (2011) 71:1621–30. doi: 10.1002/pros.21379
37. Cheng YW, Idrees K, Shattock R, Khan SA, Zeng Z, Brennan CW, et al. Loss of imprinting and marked gene elevation are 2 forms of aberrant IGF2 expression in colorectal cancer. Int J Cancer. (2010) 127:568–77. doi: 10.1002/ijc.25086
38. Cruz-Correa M, Cui H, Giardiello FM, Powe NR, Hylind L, Robinson A, et al. Loss of imprinting of insulin growth factor II gene: a potential heritable biomarker for colon neoplasia predisposition. Gastroenterology. (2004) 126:964–70. doi: 10.1053/j.gastro.2003.12.051
39. Cui H, Cruz-Correa M, Giardiello FM, Hutcheon DF, Kafonek DR, Brandenburg S, et al. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science. (2003) 299:1753–5. doi: 10.1126/science.1080902
40. Cui H, Horon IL, Ohlsson R, Hamilton SR, Feinberg AP. Loss of imprinting in normal tissue of colorectal cancer patients with microsatellite instability. Nat Med. (1998) 4:1276–80. doi: 10.1038/3260
41. Hengmi C, Onyango P, Brandenburg S, Yiqian W, Hsieh CL, Feinberg AP. Loss of imprinting in colorectal cancer linked to hypomethylation of H19 and IGF2. Cancer Res. (2002) 62:6442–6
42. Tian F, Tang Z, Song G, Pan Y, He B, Bao Q, et al. Loss of imprinting of IGF2 correlates with hypomethylation of the H19 differentially methylated region in the tumor tissue of colorectal cancer patients. Mol Med Rep. (2012) 5:1536–40. doi: 10.3892/mmr.2012.833
43. Maenaka S, Hikichi T, Imai MA, Minamoto T, Kawahara E. Loss of imprinting in IGF2 in colorectal carcinoma assessed by microdissection. Oncol Rep. (2006) 15:791–5. doi: 10.3892/or
44. Nakagawa H, Chadwick RB, Peltomaki P, Plass C, Nakamura Y, de la Chapelle A. Loss of imprinting of the insulin-like growth factor II gene occurs by biallelic methylation in a core region of H19-associated CTCF-binding sites in colorectal cancer. Proc Natl Acad Sci U.S.A. (2001) 98:591–6. doi: 10.1073/pnas.98.2.591
45. Ohta M, Sugimoto T, Seto M, Mohri D, Asaoka Y, Tada M, et al. Genetic alterations in colorectal cancers with demethylation of insulin-like growth factor II. Hum Pathol. (2008) 39:1301–8. doi: 10.1016/j.humpath.2008.02.005
46. Gao T, Liu X, He B, Pan Y, Wang S. IGF2 loss of imprinting enhances colorectal cancer stem cells pluripotency by promoting tumor autophagy. Aging (Albany NY). (2020) 12:21236–52. doi: 10.18632/aging.103837
47. Woodson K, Flood A, Green L, Tangrea JA, Hanson J, Cash B, et al. Loss of insulin-like growth factor-II imprinting and the presence of screen-detected colorectal adenomas in women. J Natl Cancer Inst. (2004) 96:407–10. doi: 10.1093/jnci/djh042
48. Li Y, Meng G, Huang L, Guo QN. Hypomethylation of the P3 promoter is associated with up-regulation of IGF2 expression in human osteosarcoma. Hum Pathol. (2009) 40:1441–7. doi: 10.1016/j.humpath.2009.03.003
49. Hengmi C, Niemitz EL, Ravenel JD, Onyango P, Brandenburg SA, Lobanenkov VV, et al. Loss of Imprinting of Insulin-like Growth Factor-II in Wilms’ Tumor Commonly Involves Altered Methylation but not Mutations of CTCF or Its Binding Site. Cancer Res. (2001) 61:4947–50.
50. Haruta M, Arai Y, Sugawara W, Watanabe N, Honda S, Ohshima J, et al. Duplication of paternal IGF2 or loss of maternal IGF2 imprinting occurs in half of Wilms tumors with various structural WT1 abnormalities. Genes Chromosomes Cancer. (2008) 47:712–27. doi: 10.1002/gcc.20572
51. Satoh Y, Nakadate H, Nakagawachi T, Higashimoto K, Joh K, Masaki Z, et al. Genetic and epigenetic alterations on the short arm of chromosome 11 are involved in a majority of sporadic Wilms' tumours. Br J Cancer. (2006) 95:541–7. doi: 10.1038/sj.bjc.6603302
52. Mummert SK, Lobanenkov VA, Feinberg AP. Association of chromosome arm 16q loss with loss of imprinting of insulin-like growth factor-II in Wilms tumor. Genes Chromosomes Cancer. (2005) 43:155–61. doi: 10.1002/gcc.20176
53. Taniguchi T, Sullivan MJ, Ogawa O, Reeve AE. Epigenetic changes encompassing the IGF2/H19 locus associated with relaxation of IGF2 imprinting and silencing of H19 in Wilms tumor. Proc Natl Acad Sci U.S.A. (1995) 92:2159–63. doi: 10.1073/pnas.92.6.2159
54. Okamoto K, Morison IM, Taniguchi T, Reeve AE. Epigenetic changes at the insulin-like growth factor II/H19 locus in developing kidney is an early event in Wilms tumorigenesis. Proc Natl Acad Sci U.S.A. (1997) 94:5367–71. doi: 10.1073/pnas.94.10.5367
55. Ogawa O, Becroft DM, Morison IM, Eccles MR, Skeen JE, Mauger DC, et al. Constitutional relaxation of insulin-like growth factor II gene imprinting associated with Wilms' tumour and gigantism. Nat Genet. (1993) 5:408–12. doi: 10.1038/ng1293–408
56. Vu TH, Hoffman A. Alterations in the promoter-specific imprinting of the insulin-like growth factor-II gene in Wilms' tumor. J Biol Chem. (1996) 271:9014–23. doi: 10.1074/jbc.271.15.9014
57. Wang WH, Duan JX, Vu TH, Hoffman AR. Increased expression of the insulin-like growth factor-II gene in Wilms' tumor is not dependent on loss of genomic imprinting or loss of heterozygosity. J Biol Chem. (1996) 271:27863–70. doi: 10.1074/jbc.271.44.27863
58. Ravenel JD, Broman KW, Perlman EJ, Niemitz EL, Jayawardena TM, Bell DW, et al. Loss of imprinting of insulin-like growth factor-II (IGF2) gene in distinguishing specific biologic subtypes of Wilms tumor. J Natl Cancer Inst. (2001) 93:1698–703. doi: 10.1093/jnci/93.22.1698
59. Sullivan MJ, Taniguchi T, Jhee A, Kerr N, Reeve AE. Relaxation of IGF2 imprinting in Wilms tumours associated with specific changes in IGF2 methylation. Oncogene. (1999) 18:7527–34. doi: 10.1038/sj.onc.1203096
60. Dejeux E, Olaso R, Dousset B, Audebourg A, Gut IG, Terris B, et al. Hypermethylation of the IGF2 differentially methylated region 2 is a specific event in insulinomas leading to loss-of-imprinting and overexpression. Endocr Relat Cancer. (2009) 16:939–52. doi: 10.1677/ERC-08–0331
61. Murata A, Baba Y, Watanabe M, Shigaki H, Miyake K, Ishimoto T, et al. IGF2 DMR0 methylation, loss of imprinting, and patient prognosis in esophageal squamous cell carcinoma. Ann Surg Oncol. (2014) 21:1166–74. doi: 10.1245/s10434–013-3414–7
62. Grbesa I, Ivkic M, Pegan B, Gall-Troselj K. Loss of imprinting and promoter usage of the IGF2 in laryngeal squamous cell carcinoma. Cancer Lett. (2006) 238:224–9. doi: 10.1016/j.canlet.2005.07.003
63. Hajdu M, Singer S, Maki RG, Schwartz GK, Keohan ML, Antonescu CR. IGF2 over-expression in solitary fibrous tumours is independent of anatomical location and is related to loss of imprinting. J Pathol. (2010) 221:300–7. doi: 10.1002/path.2715
64. Uyeno S, Aoki Y, Nato M, Sagisaka K, Kayama T, Yoshimoto T, et al. IGF2 but not H19 Shows Loss of Imprinting in Human Glioma. Cancer Res. (1996) 56:5356–9.
65. Murphy SK, Huang Z, Wen Y, Spillman MA, Whitaker RS, Simel LR, et al. Frequent IGF2/H19 domain epigenetic alterations and elevated IGF2 expression in epithelial ovarian cancer. Mol Cancer Res. (2006) 4:283–92. doi: 10.1158/1541–7786.MCR-05–0138
66. Oda H, Kume H, Shimizu Y, Inoue T, Ishikawa T. Loss of imprinting of igf2 in renal-cell carcinomas. Int J Cancer. (1998) 75:343–6. doi: 10.1002/(sici)1097–0215(19980130)75:3<343::aid-ijc3>3.0.co;2–2
67. Pedone PV, Tirabosco R, Cavazzana AO, Ungaro P, Basso G, Luksch R, et al. Mono- and bi-allelic expression of insulin-like growth factor II gene in human muscle tumors. Hum Mol Genet. (1994) 3:1117–21. doi: 10.1093/hmg/3.7.1117
68. Zhan S, Shapiro DN, Helman LJ. Activation of an imprinted allele of the insulin-like growth factor II gene implicated in rhabdomyosarcoma. J Clin Invest. (1994) 94:445–8. doi: 10.1172/JCI117344
69. Roy RN, Gerulath AH, Cecutti A, Bhavnani BR. Loss of IGF-II imprinting in endometrial tumors: overexpression in carcinosarcoma. Cancer Lett. (2000) 153:67–73. doi: 10.1016/s0304–3835(00)00347–5
70. Shetty PJ, Movva S, Pasupuleti N, Vedicherlla B, Vattam KK, Venkatasubramanian S, et al. Regulation of IGF2 transcript and protein expression by altered methylation in breast cancer. J Cancer Res Clin Oncol. (2011) 137:339–45. doi: 10.1007/s00432-010-0890-z
71. Wu HK, Squire JA, Catzavelos CG, Weksberg R. Relaxation of imprinting of human insulin-like growth factor II gene, IGF2, in sporadic breast carcinomas. Biochem Biophys Res Commun. (1997) 235:123–9. doi: 10.1006/bbrc.1997.6744
72. Vorwerk P, Wex H, Bessert C, Hohmann B, Schmidt U, Mittler U. Loss of imprinting of IGF-II gene in children with acute lymphoblastic leukemia. Leuk Res. (2003) 27:807–12. doi: 10.1016/s0145–2126(03)00014–6
73. Wu HK, Weksberg R, Minden MD, Squire JA. Loss of imprinting of human insulin-like growth factor II gene, IGF2, in acute myeloid leukemia. Biochem Biophys Res Commun. (1997) 231:466–72. doi: 10.1006/bbrc.1997.6127
74. Chak WP, Lung RW, Tong JH, Chan SY, Lun SW, Tsao SW, et al. Downregulation of long non-coding RNA MEG3 in nasopharyngeal carcinoma. Mol Carcinog. (2017) 56:1041–54. doi: 10.1002/mc.22569
75. Lai S, Goepfert H, Gillenwater AM, Luna MA, El-Naggar AK. Loss of imprinting and genetic alterations of the cyclin-dependent kinase inhibitor p57KIP2 gene in head and neck squamous cell carcinoma. Clin Cancer Res. (2000) 6:3172–6
76. Min Ju K, Bum Joon P, Do Sun B, Jae Il P, Hyo Jong K, Jae Hoon P, et al. Loss of imprinting and elevated expression of wild-type p73 in human gastric adenocarcinoma. Clin Cancer Res. (2000) 6:1767–71
77. Cai YC, Yang GY, Nie Y, Wang LD, Zhao X, Song YL, et al. Molecular alterations of p73 in human esophageal squamous cell carcinomas: loss of heterozygosity occurs frequently; loss of imprinting and elevation of p73 expression may be related to defective p53. Carcinogenesis. (2000) 21:683–9. doi: 10.1093/carcin/21.4.683
78. Liu W, Mai M, Yokomizo A, Qian C, Tindall DJ, Smith DI, et al. Differential expression and allelotyping of the p73 gene in neuroblastoma. Int J Oncol. (2000) 16:181–5. doi: 10.3892/ijo
79. Mai M, Qian C, Yokomizo A, Tindall DJ, Bostwick D, Polychronakos C, et al. Loss of imprinting and allele switching of p73 in renal cell carcinoma. Oncogene. (1998) 17:1739–41. doi: 10.1038/sj.onc.1202099
80. Zaika AI, Kovalev S, Marchenko ND, Moll UM. Overexpression of the wild type p73 gene in breast cancer tissues and cell lines. Cancer Res. (1999) 59:3257–63
81. Pedersen IS, Dervan P, McGoldrick A, Harrison M, Ponchel F, Speirs V, et al. Promoter switch: a novel mechanism causing biallelic PEG1/MEST expression in invasive breast cancer. Hum Mol Genet. (2002) 11:1449–53. doi: 10.1093/hmg/11.12.1449
82. Pedersen IS, Dervan PA, Broderick D, Harrison M, Miller N, Delany E, et al. Frequent loss of imprinting of PEG1/MEST in invasive breast cancer. Cancer Res. (1999) 59:5449–51
83. Nakanishi H, Suda T, Katoh M, Watanabe A, Igishi T, Kodani M, et al. Loss of imprinting of PEG1/MEST in lung cancer cell lines. Oncol Rep. (2004) 12:1273–8. doi: 10.3892/or
84. Jiang X, Yu Y, Yang HW, Agar NY, Frado L, Johnson MD. The imprinted gene PEG3 inhibits Wnt signaling and regulates glioma growth. J Biol Chem. (2010) 285:8472–80. doi: 10.1074/jbc.M109.069450
85. Dowdy SC, Gostout BS, Shridhar V, Wu X, Smith DI, Podratz KC, et al. Biallelic methylation and silencing of paternally expressed gene 3 (PEG3) in gynecologic cancer cell lines. Gynecol Oncol. (2005) 99:126–34. doi: 10.1016/j.ygyno.2005.05.036
86. Malik K, Salpekar A, Hancock A, Moorwood K, Jackson S, Charles A, et al. Identification of differential methylation of the WT1 antisense regulatory region and relaxation of imprinting in Wilms' tumor. Cancer Res. (2000) 60:2356–60
87. Guillaumet-Adkins A, Richter J, Odero MD, Sandoval J, Agirre X, Catala A, et al. Hypermethylation of the alternative AWT1 promoter in hematological Malignancies is a highly specific marker for acute myeloid leukemias despite high expression levels. J Hematol Oncol. (2014) 7:4. doi: 10.1186/1756–8722-7–4
88. Byun HM, Wong HL, Birnstein EA, Wolff EM, Liang G, Yang AS. Examination of IGF2 and H19 loss of imprinting in bladder cancer. Cancer Res. (2007) 67:10753–8. doi: 10.1158/0008–5472.CAN-07–0329
89. Elkin M, Shevelev A, Schulze E, Tykocinsky M, Cooper M, Ariel I, et al. The expression of the imprinted H19 and IGF-2 genes in human bladder carcinoma. FEBS Lett. (1995) 374:57–61. doi: 10.1016/0014-5793(95)01074-O
90. Kim SJ, Park SE, Lee C, Lee SY, Jo JH, Kim JM, et al. Alterations in promoter usage and expression levels of insulin-like growth factor-II and H19 genes in cervical carcinoma exhibiting biallelic expression of IGF-II. Biochim Biophys Acta. (2002) 1586:307–15. doi: 10.1016/s0925–4439(01)00109–0
91. Nishihara S, Hayashida T, Mitsuya K, Schulz TC, Ikeguchi M, Kaibara N, et al. Multipoint imprinting analysis in sporadic colorectal cancers with and without microsatellite instability. Int J Oncol. (2000) 17:317–22. doi: 10.3892/ijo.17.2.317
92. Nakano S, Murakami K, Meguro M, Soejima H, Higashimoto K, Urano T, et al. Expression profile of LIT1/KCNQ1OT1 and epigenetic status at the KvDMR1 in colorectal cancers. Cancer Sci. (2006) 97:1147–54. doi: 10.1111/j.1349-7006.2006.00305.x
93. Schneider DT, Schuster AE, Fritsch MK, Hu J, Olson T, Lauer S, et al. Multipoint imprinting analysis indicates a common precursor cell for gonadal and nongonadal pediatric germ cell tumors. Cancer Res. (2001) 61:7268–76.
94. Nonomura N, Miki T, NishimuraI K, Kanno N, Kojima Y, Okuyama A. Alterd imprinting of the H19 and insulin-like growth factor ii genes in testicular tumors. J Urol. (1997) 157:1977–9. doi: 10.1016/S0022-5347(01)64913-9
95. Ross JA, Schmidt PT, Perentesis JP, Davies SM. Genomic imprinting of H19 and insulin-like growth factor-2 in pediatric germ cell tumors. Cancer. (1999) 85:1389–94. doi: 10.1002/(SICI)1097-0142(19990315)85:63.3.CO;2-M
96. Stier S, Neuhaus T, Albers P, Wernert N, Grunewald E, Forkert R, et al. Loss of imprinting of the insulin-like growth factor 2 and the H19 gene in testicular seminomas detected by real-time PCR approach. Arch Toxicol. (2006) 80:713–8. doi: 10.1007/s00204-006-0087-z
97. Kawakami T, Zhang C, Okada Y, Okamoto K. Erasure of methylation imprint at the promoter and CTCF-binding site upstream of H19 in human testicular germ cell tumors of adolescents indicate their fetal germ cell origin. Oncogene. (2006) 25:3225–36. doi: 10.1038/sj.onc.1209362
98. Kim KS, Lee YI. Biallelic expression of the H19 and IGF2 genes in hepatocellular carcinoma. Cancer Lett. (1997) 119:143–8. doi: 10.1016/s0304–3835(97)00264–4
99. Rainier S, Dobry CJ, Feinberg AP. Loss of imprinting in hepatoblastoma. Cancer Res. (1995) 55:1836–8
100. Wu J, Qin Y, Li B, He WZ, Sun ZL. Hypomethylated and hypermethylated profiles of H19DMR are associated with the aberrant imprinting of IGF2 and H19 in human hepatocellular carcinoma. Genomics. (2008) 91:443–50. doi: 10.1016/j.ygeno.2008.01.007
101. el-Naggar AK, Lai S, Tucker SA, Clayman GL, Goepfert H, Hong WK, et al. Frequent loss of imprinting at the IGF2 and H19 genes in head and neck squamous carcinoma. Oncogene. (1999) 18:7063–9. doi: 10.1038/sj.onc.1203192
102. Rainho CA, Kowalski LP, Rogatto SR. Loss of imprinting and loss of heterozygosity on 11p15.5 in head and neck squamous cell carcinomas. Head Neck. (2001) 23:851–9. doi: 10.1002/hed.1124
103. Kohda M, Hoshiya H, Katoh M, Tanaka I, Masuda R, Takemura T, et al. Frequent loss of imprinting of IGF2 and MEST in lung adenocarcinoma. Mol Carcinog. (2001) 31:184–91. doi: 10.1002/mc.1053
104. Grbesa I, Marinkovic M, Ivkic M, Kruslin B, Novak-Kujundzic R, Pegan B, et al. Loss of imprinting of IGF2 and H19, loss of heterozygosity of IGF2R and CTCF, and Helicobacter pylori infection in laryngeal squamous cell carcinoma. J Mol Med (Berl). (2008) 86:1057–66. doi: 10.1007/s00109–008-0369–4
105. Albrecht S, Waha A, Koch A, Kraus JA, Goodyer CG, Pietsch T. Variable imprinting of H19 and IGF2 in fetal cerebellum and medulloblastoma. J Neuropathol Exp Neurol. (1996) 55:1270–6. doi: 10.1097/00005072–199612000–00011
106. Muller S, Zirkel D, Westphal M, Zumkeller W. Genomic imprinting of IGF2 and H19 in human meningiomas. Eur J Cancer. (2000) 36:651–5. doi: 10.1016/s0959–8049(99)00328–7
107. Hiura H, Okae H, Kobayash H, Miyauchi N, Sato F, Sato A, et al. High-throughput detection of aberrant imprint methylation in the ovarian cancer by the bisulphite PCR-Luminex method. BMC Med Genomics. (2012) 5:8. doi: 10.1186/1755–8794-5–8
108. Chun Ling C, Sin Ming I, Danny C, Ling Chui W, Ngan SHY. Loss of imprinting of the IGF-II and H19 genes in epithelial ovarian cancer. Clin Cancer Res. (2000) 6:474–9
109. Ulaner GA, Vu TH, Li T, Hu JF, Yao XM, Yang Y, et al. Loss of imprinting of IGF2 and H19 in osteosarcoma is accompanied by reciprocal methylation changes of a CTCF-binding site. Hum Mol Genet. (2003) 12:535–49. doi: 10.1093/hmg/ddg034
110. Anderson J, Gordon A, McManus A, Shipley J, Pritchard-Jones K. Disruption of imprinted genes at chromosome region 11p15.5 in paediatric rhabdomyosarcoma. Neoplasia. (1999) 1:340–8. doi: 10.1038/sj.neo.7900052
111. Vu TH, Chuyen NV, Li T, Hoffman AR. Loss of imprinting of IGF2 sense and antisense transcripts in Wilms' tumor. Cancer Res. (2003) 63:1900–5
112. Hancock AL, Brown KW, Moorwood K, Moon H, Holmgren C, Mardikar SH, et al. A CTCF-binding silencer regulates the imprinted genes AWT1 and WT1-AS and exhibits sequential epigenetic defects during Wilms' tumourigenesis. Hum Mol Genet. (2007) 16:343–54. doi: 10.1093/hmg/ddl478
113. Dallosso AR, Hancock AL, Brown KW, Williams AC, Jackson S, Malik K. Genomic imprinting at the WT1 gene involves a novel coding transcript (AWT1) that shows deregulation in Wilms' tumours. Hum Mol Genet. (2004) 13:405–15. doi: 10.1093/hmg/ddh038
114. Hubertus J, Lacher M, Rottenkolber M, Muller-Hocker J, Berger M, Stehr M, et al. Altered expression of imprinted genes in Wilms tumors. Oncol Rep. (2011) 25:817–23. doi: 10.3892/or.2010.1113
115. Steenman MJ, Rainier S, Dobry CJ, Grundy P, Horon IL, Feinberg AP. Loss of imprinting of IGF2 is linked to reduced expression and abnormal methylation of H19 in Wilms' tumour. Nat Genet. (1994) 7:433–9. doi: 10.1038/ng0794–433
116. Bjornsson HT, Brown LJ, Fallin MD, Rongione MA, Bibikova M, Wickham E, et al. Epigenetic specificity of loss of imprinting of the IGF2 gene in Wilms tumors. J Natl Cancer Inst. (2007) 99:1270–3. doi: 10.1093/jnci/djm069
117. Baral K, Rotwein P. The insulin-like growth factor 2 gene in mammals: Organizational complexity within a conserved locus. PloS One. (2019) 14:e0219155. doi: 10.1371/journal.pone.0219155
118. Mineo R, Fichera E, Liang SJ, Fujita-Yamaguchi Y. Promoter usage for insulin-like growth factor-II in cancerous and benign human breast, prostate, and bladder tissues, and confirmation of a 10th exon. Biochem Biophys Res Commun. (2000) 268:886–92. doi: 10.1006/bbrc.2000.2225
119. Monk D, Sanches R, Arnaud P, Apostolidou S, Hills FA, Abu-Amero S, et al. Imprinting of IGF2 P0 transcript and novel alternatively spliced INS-IGF2 isoforms show differences between mouse and human. Hum Mol Genet. (2006) 15:1259–69. doi: 10.1093/hmg/ddl041
120. Li T, Chen H, Li W, Cui J, Wang G, Hu X, et al. Promoter histone H3K27 methylation in the control of IGF2 imprinting in human tumor cell lines. Hum Mol Genet. (2014) 23:117–28. doi: 10.1093/hmg/ddt405
121. Tost J, Jammes H, Dupont JM, Buffat C, Robert B, Mignot TM, et al. Non-random, individual-specific methylation profiles are present at the sixth CTCF binding site in the human H19/IGF2 imprinting control region. Nucleic Acids Res. (2007) 35:701. doi: 10.1093/nar/gkl1107
122. Freschi A, Del Prete R, Pignata L, Cecere F, Manfrevola F, Mattia M, et al. The number of the CTCF binding sites of the H19/IGF2:IG-DMR correlates with DNA methylation and expression imprinting in a humanized mouse model. Hum Mol Genet. (2021) 30:1509–20. doi: 10.1093/hmg/ddab132
123. Kurukuti S, Tiwari VK, Tavoosidana G, Pugacheva E, Murrell A, Zhao Z, et al. CTCF binding at the H19 imprinting control region mediates maternally inherited higher-order chromatin conformation to restrict enhancer access to Igf2. Proc Natl Acad Sci U.S.A. (2006) 103:10684–9. doi: 10.1073/pnas.0600326103
124. Charalambous M, Menheniott TR, Bennett WR, Kelly SM, Dell G, Dandolo L, et al. An enhancer element at the Igf2/H19 locus drives gene expression in both imprinted and non-imprinted tissues. Dev Biol. (2004) 271:488–97. doi: 10.1016/j.ydbio.2004.04.022
125. Riso V, Cammisa M, Kukreja H, Anvar Z, Verde G, Sparago A, et al. ZFP57 maintains the parent-of-origin-specific expression of the imprinted genes and differentially affects non-imprinted targets in mouse embryonic stem cells. Nucleic Acids Res. (2016) 44:8165–78. doi: 10.1093/nar/gkw505
126. Nordin M, Bergman D, Halje M, Engstrom W, Ward A. Epigenetic regulation of the Igf2/H19 gene cluster. Cell Prolif. (2014) 47:189–99. doi: 10.1111/cpr.12106
127. Sipos F, Szekely H, Kis ID, Tulassay Z, Muzes G. Relation of the IGF/IGF1R system to autophagy in colitis and colorectal cancer. World J Gastroenterol. (2017) 23:8109–19. doi: 10.3748/wjg.v23.i46.8109
128. Syed Abd Halim SA, Abd Rashid N, Woon CK, Abdul Jalil NA. Natural products targeting PI3K/AKT in myocardial ischemic reperfusion injury: A scoping review. Pharm (Basel). (2023) 16(5):739. doi: 10.3390/ph16050739
129. Domoto T, Pyko IV, Furuta T, Miyashita K, Uehara M, Shimasaki T, et al. Glycogen synthase kinase-3beta is a pivotal mediator of cancer invasion and resistance to therapy. Cancer Sci. (2016) 107:1363–72. doi: 10.1111/cas.13028
130. Li Y, Chang Q, Rubin BP, Fletcher C, Morgan TW, Mentzer SJ, et al. Insulin receptor activation in solitary fibrous tumours. J Pathol. (2007) 211:550–4. doi: 10.1002/path.2136
131. Beygo J, Kuchler A, Gillessen-Kaesbach G, Albrecht B, Eckle J, Eggermann T, et al. New insights into the imprinted MEG8-DMR in 14q32 and clinical and molecular description of novel patients with Temple syndrome. Eur J Hum Genet. (2017) 25:935–45. doi: 10.1038/ejhg.2017.91
132. Sellers ZP, Bolkun L, Kloczko J, Wojtaszewska ML, Lewandowski K, Moniuszko M, et al. Increased methylation upstream of the MEG3 promotor is observed in acute myeloid leukemia patients with better overall survival. Clin Epigenet. (2019) 11:50. doi: 10.1186/s13148-019-0643-z
133. Aronson BE, Scourzic L, Shah V, Swanzey E, Kloetgen A, Polyzos A, et al. A bipartite element with allele-specific functions safeguards DNA methylation imprints at the Dlk1-Dio3 locus. Dev Cell. (2021) 56:3052–65 e5. doi: 10.1016/j.devcel.2021.10.004
134. Kojima S, Shiochi N, Sato K, Yamaura M, Ito T, Yamamura N, et al. Epigenome editing reveals core DNA methylation for imprinting control in the Dlk1-Dio3 imprinted domain. Nucleic Acids Res. (2022) 50:5080–94. doi: 10.1093/nar/gkac344
135. Buccarelli M, Lulli V, Giuliani A, Signore M, Martini M, D'Alessandris QG, et al. Deregulated expression of the imprinted DLK1-DIO3 region in glioblastoma stemlike cells: tumor suppressor role of lncRNA MEG3. Neuro Oncol. (2020) 22:1771–84. doi: 10.1093/neuonc/noaa127
136. Quann K, Gonzales DM, Mercier I, Wang C, Sotgia F, Pestell RG, et al. Caveolin-1 is a negative regulator of tumor growth in glioblastoma and modulates chemosensitivity to temozolomide. Cell Cycle. (2013) 12:1510–20. doi: 10.4161/cc.24497
137. Rivera MP, Mehta AC, Wahidi MM. Establishing the diagnosis of lung cancer: Diagnosis and management of lung cancer, 3rd ed: American College of Chest Physicians evidence-based clinical practice guidelines. Chest. (2013) 143:e142S–e65S. doi: 10.1378/chest.12–2353
138. Sheng R, Zhang Y, Wang H, Zhang W, Jin K, Sun W, et al. A multi-center diagnostic system for intrahepatic mass-forming cholangiocarcinoma based on preoperative MRI and clinical features. Eur Radiol. (2023) 34(1):548–59. doi: 10.1007/s00330-023-10002-w
139. Zhang Y, Tan W, Wang X, Zheng X, Huang Y, Li B, et al. Metabolic biomarkers significantly enhance the prediction of HBV-related ACLF occurrence and outcomes. J Hepatol. (2023) 79:1159–71. doi: 10.1016/j.jhep.2023.07.011
140. National Lung Screening Trial Research T, Aberle DR, Adams AM, Berg CD, Black WC, Clapp JD, et al. Reduced lung-cancer mortality with low-dose computed tomographic screening. N Engl J Med. (2011) 365:395–409. doi: 10.1056/NEJMoa1102873
141. Jelinic P, Shaw P. Loss of imprinting and cancer. J Pathol. (2007) 211:261–8. doi: 10.1002/path.2116
142. Feinberg AP. The key role of epigenetics in human disease prevention and mitigation. N Engl J Med. (2018) 378:1323–34. doi: 10.1056/NEJMra1402513
143. Gabrieli T, Michaeli Y, Avraham S, Torchinsky D, Margalit S, Schutz L, et al. Chemoenzymatic labeling of DNA methylation patterns for single-molecule epigenetic mapping. Nucleic Acids Res. (2022) 50:e92. doi: 10.1093/nar/gkac460
144. Xiong J, Chen KK, Xie NB, Ji TT, Yu SY, Tang F, et al. Bisulfite-free and single-base resolution detection of epigenetic DNA modification of 5-methylcytosine by methyltransferase-directed labeling with APOBEC3A deamination sequencing. Anal Chem. (2022) 94:15489–98. doi: 10.1021/acs.analchem.2c03808
145. Wang M, Xie NB, Chen KK, Ji TT, Xiong J, Guo X, et al. Engineered APOBEC3C sequencing enables bisulfite-free and direct detection of DNA methylation at a single-base resolution. Anal Chem. (2023) 95:1556–65. doi: 10.1021/acs.analchem.2c04616
146. Li S, Tollefsbol TO. DNA methylation methods: Global DNA methylation and methylomic analyses. Methods. (2021) 187:28–43. doi: 10.1016/j.ymeth.2020.10.002
147. Li D, Zhang B, Xing X, Wang T. Combining MeDIP-seq and MRE-seq to investigate genome-wide CpG methylation. Methods. (2015) 72:29–40. doi: 10.1016/j.ymeth.2014.10.032
148. Zhang B, Zhou Y, Lin N, Lowdon RF, Hong C, Nagarajan RP, et al. Functional DNA methylation differences between tissues, cell types, and across individuals discovered using the M&M algorithm. Genome Res. (2013) 23:1522–40. doi: 10.1101/gr.156539.113
149. Harris RA, Wang T, Coarfa C, Nagarajan RP, Hong C, Downey SL, et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat Biotechnol. (2010) 28:1097–105. doi: 10.1038/nbt.1682
150. Sarel-Gallily R, Keshet G, Kinreich S, Haim-Abadi G, Benvenisty N. EpiTyping: analysis of epigenetic aberrations in parental imprinting and X-chromosome inactivation using RNA-seq. Nat Protoc. (2023) 18:3881–917. doi: 10.1038/s41596–023-00898–5
151. Martini P, Sales G, Diamante L, Perrera V, Colantuono C, Riccardo S, et al. BrewerIX enables allelic expression analysis of imprinted and X-linked genes from bulk and single-cell transcriptomes. Commun Biol. (2022) 5(1):146. doi: 10.1038/s42003–022-03087–4
152. Qi G, Strober BJ, Popp JM, Keener R, Ji H, Battle A. Single-cell allele-specific expression analysis reveals dynamic and cell-type-specific regulatory effects. Nat Commun. (2023) 14(1):6317. doi: 10.1038/s41467–023-42016–9
153. Kanber D, Berulava T, Ammerpohl O, Mitter D, Richter J, Siebert R, et al. The human retinoblastoma gene is imprinted. PloS Genet. (2009) 5:e1000790. doi: 10.1371/journal.pgen.1000790
154. Martin-Trujillo A, Vidal E, Monteagudo-Sanchez A, Sanchez-Delgado M, Moran S, Hernandez Mora JR, et al. Copy number rather than epigenetic alterations are the major dictator of imprinted methylation in tumors. Nat Commun. (2017) 8:467. doi: 10.1038/s41467–017-00639–9
155. Santoni FA, Stamoulis G, Garieri M, Falconnet E, Ribaux P, Borel C, et al. Detection of imprinted genes by single-cell allele-specific gene expression. Am J Hum Genet. (2017) 100:444–53. doi: 10.1016/j.ajhg.2017.01.028
156. Matuschek C, Bolke E, Lammering G, Gerber PA, Peiper M, Budach W, et al. Methylated APC and GSTP1 genes in serum DNA correlate with the presence of circulating blood tumor cells and are associated with a more aggressive and advanced breast cancer disease. Eur J Med Res. (2010) 15:277–86. doi: 10.1186/2047–783x-15–7-277
157. Xiao W, Dong W, Zhang C, Saren G, Geng P, Zhao H, et al. Effects of the epigenetic drug MS-275 on the release and function of exosome-related immune molecules in hepatocellular carcinoma cells. Eur J Med Res. (2013) 18:61. doi: 10.1186/2047–783X-18–61
Keywords: cancer, diagnosis, progression, prognosis, epigenetic control, neoplastic gene regulation, gene imprint, methods
Citation: Xie G, Si Q, Zhang G, Fan Y, Li Q, Leng P, Qiao F, Liang S, Yu R and Wang Y (2024) The role of imprinting genes’ loss of imprints in cancers and their clinical implications. Front. Oncol. 14:1365474. doi: 10.3389/fonc.2024.1365474
Received: 04 January 2024; Accepted: 23 April 2024;
Published: 15 May 2024.
Edited by:
Carlos A. Vaccaro, Italian Hospital of Buenos Aires, ArgentinaReviewed by:
Ildar Fakhradiyev, Kaz akh National Medical University, KazakhstanCopyright © 2024 Xie, Si, Zhang, Fan, Li, Leng, Qiao, Liang, Yu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yingshuang Wang, d2FuZ3lpbmdzaHVhbmdAY2R1dGNtLmVkdS5jbg==; Rong Yu, eXVyb25nQGNkdXRjbS5lZHUuY24=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.