 Verónica Vallejo-Ruiz
Verónica Vallejo-Ruiz Lourdes Gutiérrez-Xicotencatl
Lourdes Gutiérrez-Xicotencatl Oscar Medina-Contreras
Oscar Medina-Contreras Marcela Lizano
Marcela Lizano
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol. , 19 March 2024
Sec. Gynecological Oncology
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1356581
This article is part of the Research Topic Cervical Cancer: Updates from the Mexican National Consensus View all 10 articles
Cervical cancer (CC) is a significant health problem, especially in low-income countries. Functional studies on the human papillomavirus have generated essential advances in the knowledge of CC. However, many unanswered questions remain. This mini-review discusses the latest results on CC pathogenesis, HPV oncogenesis, and molecular changes identified through next-generation technologies. Interestingly, the percentage of samples with HPV genome integrations correlates with the degree of the cervical lesions, suggesting a role in the development of CC. Also, new functions have been described for the viral oncoproteins E5, E6, and E7, resulting in the acquisition and maintenance of cancer hallmarks, including proliferation, immune response evasion, apoptosis, and genomic instability. Remarkably, E5 oncoprotein affects signaling pathways involved in the expression of interferon-induced genes and EGFR-induced proliferation, while E6 and E7 oncoproteins regulate the DNA damage repair and cell cycle continuity pathways. Furthermore, next-generation technologies provide vast amounts of information, increasing our knowledge of changes in the genome, transcriptome, proteome, metabolome, and epigenome in CC. These studies have identified novel molecular traits associated with disease susceptibility, degree of progression, treatment response, and survival as potential biomarkers and therapeutic targets.
Cervical cancer (CC) remains a public health problem and ranks fourth in cancer mortality in women worldwide (1). The main etiologic factor for CC development is a persistent infection with high-risk (HR) human papillomavirus (HPV), responsible for almost 100% of all CC cases (2). However, some studies report that between 5 and 8% of CC cases are HPV-negative; significantly, the majority are adenocarcinomas (3, 4).
More than 200 HPV types have so far been identified. Around 15 types are classified as high-risk types, including HPV 16, 18, 31, 33, 45, 52, and 58, associated with cervical, anogenital, and oropharyngeal cancers, and HPV16 is found in approximately 60% of the CC cases (5). Low-risk HPV types, mainly types 6 and 11, commonly cause benign anogenital warts.
HPVs are small, non-enveloped viruses with an 8-kb circular double-stranded DNA contained in a 55 nm icosahedral capsid. The viral genome holds the long control region (LCR) that regulates genome replication and transcription of the early (E1-E7) and the late-expressed genes L1 and L2 (6).
HPV infection targets the cervix transformation zone, a region susceptible to the development of premalignant cervical lesions and, potentially, cancer. Initially, these lesions were categorized based on their severity and extent of atypical epithelial tissue changes, leading to the classification of cervical intraepithelial neoplasia (CIN) grades I, II, and III, as well as carcinoma in situ (CC). Later the Bethesda classification was introduced, revising the terminology. Under this system, CIN I was renamed low-grade squamous intraepithelial lesion (LSIL), whereas CIN II-III were collectively designated as high-grade SIL (HSIL). This updated classification reflects a more refined understanding of the progression and implications of HPV-related cervical lesions (7).
Through micro-wounds, HPV infects the basal cells of the stratified cervical epithelium. A mandatory cellular uptake receptor for HPV has not yet been identified; however, it has been proposed that the virus may attach to the host cell membrane via heparan sulfate proteoglycans (HSPGs) (8), α6β4 integrin complex (9), tetraspanins (10), keratinocyte and epidermal growth factor receptors (KGFR and EGFR, respectively), being EGFR signaling essential for HPV16 endocytosis (11). The virus internalizes by endocytic uptake and, sequentially, in endosomal compartments, the viral capsid binds to retromer components such as Sortin-nexin 17 and 27, helping the L2-DNA complex to escape lysosomal degradation (12, 13) to be then transported to the nucleus via dynein-mediated transport through microtubules (14). Synthesized E1 and E2 interact with the origin of replication site in the LCR. Subsequently, E2 partially represses the expression of E6 and E7; however, the small amount of oncoproteins produced is sufficient to induce a delay in differentiation, resulting in a low copy number of genome replication. Paradoxically, cell differentiation is required to activate the productive phase of the viral cycle. As the epithelium differentiates, the expression of the early viral genes, including E5 and E4, augments in the middle and upper layers, and genome amplification increases (15). E5 maintains cell proliferation and delays cell differentiation by modulating EGF/KGF receptor activities, complementing the functions of E6 and E7 (16). L1 and L2 capsid proteins are produced in the differentiated keratinocytes, where virions are assembled and released due to the disruption of the cytoskeleton promoted by the E4 (17). Most HPV infections are eliminated by the immune system, where 60% of HPV infections are cleared spontaneously within one year and 90% within two years (18). HPV infections that persist over two years exhibit an increased risk of developing cervical intraepithelial neoplasia (CIN) (19).
The determinants of HR-HPV persistent infections are not precise. Still, they may be related to the inability of the host to mount an adequate innate immune response and a robust cell-mediated immunity, as well as the ability of the viral proteins to evade immune detection (20–22). Although only 0.6% of HPV infections are known to progress to cancer, a 16-year follow-up study has shown that the risk of developing cancer is 75.4 times higher in women with persistent HR-HPV infections compared to HPV-negative women (23). It is proposed that during persistent HPV infections, mutations and chromosomal abnormalities accumulate over time, promoting integration of the viral genome into the cellular genome and contributing to cancer progression (24). Some studies report that the frequency of HPV genome integration gradually increases as cervical lesions progress, observed in 26-30% of CINI cases, 40-64% of CINII-III, and 77% of CC (25, 26).
In many cases of CC, the viral genome integration frequently occurs in the E1 and E2 genes, affecting their expression and leading to the uncontrolled expression of the oncogenes E6 and E7. The maintenance of the tumor phenotype requires the continuous expression of the E6 and E7 oncogenes (27). It has been shown that in the HPV genome of CC tumors HPV16-positive, gene losses of more than 10% occur most frequently within E1, E2, and E5 genes, with the loss of E2 in 27% of CC cases (28). However, viral integration does not occur in all CC; in some cases, HPV DNA remains as an episome (29), and methylation at E2 binding sites within the LCR has been shown to prevent E2 binding and consequently promote the continued expression of the viral oncoproteins (30).
Gene loss, duplication, or overexpression is a common feature of the HPV genome in CC (31). This is due to deletions, errors during the replication process, or mutations in genes of the HPV genome that increase the expression of the HPV gene products. HPV gene diversity and duplication have been reported in CC (32). Overexpression of E6 and E7 after HPV integration is considered the trigger for malignant progression due to cell cycle disruption and induction of genome instability; also, multiple HPV integration events have been associated with poor prognosis (31). Alterations that occur in the host genome due to the integration of HPV are fundamental in the development of CC.
The contribution of HPV to CC development is due to the transformation capacity of a variety of interactions of E6, E7, and E5 viral oncoproteins with diverse cellular proteins, which affect the normal regulation of cell signaling pathways involved in proliferation, DNA damage repair, immune system, apoptosis, and metabolism (33–35).
The viral oncoprotein E5 is a viroporine capable of increasing cell proliferation by modulating ionic homeostasis through the inhibition of the vacuolar H+ ATPase-16 kDa subunit (36). E5 regulates endosomal pH and inhibits the interaction of EGFR with c-Cbl ubiquitinase, decreasing its degradation and allowing increased mitogenic signal transduction (37). E5 negatively modulates the CDK inhibitors p27Kip-1 and p21Waf-1 (16), which allows the cell to remain in the cell cycle, maintaining viral persistence. E5 also impairs keratinocyte differentiation by downregulating KGFR and increases EGFR activity, slowing the differentiation process (38). All these events lead the cell to a continuous proliferation that could finally transform the cells (Table 1).
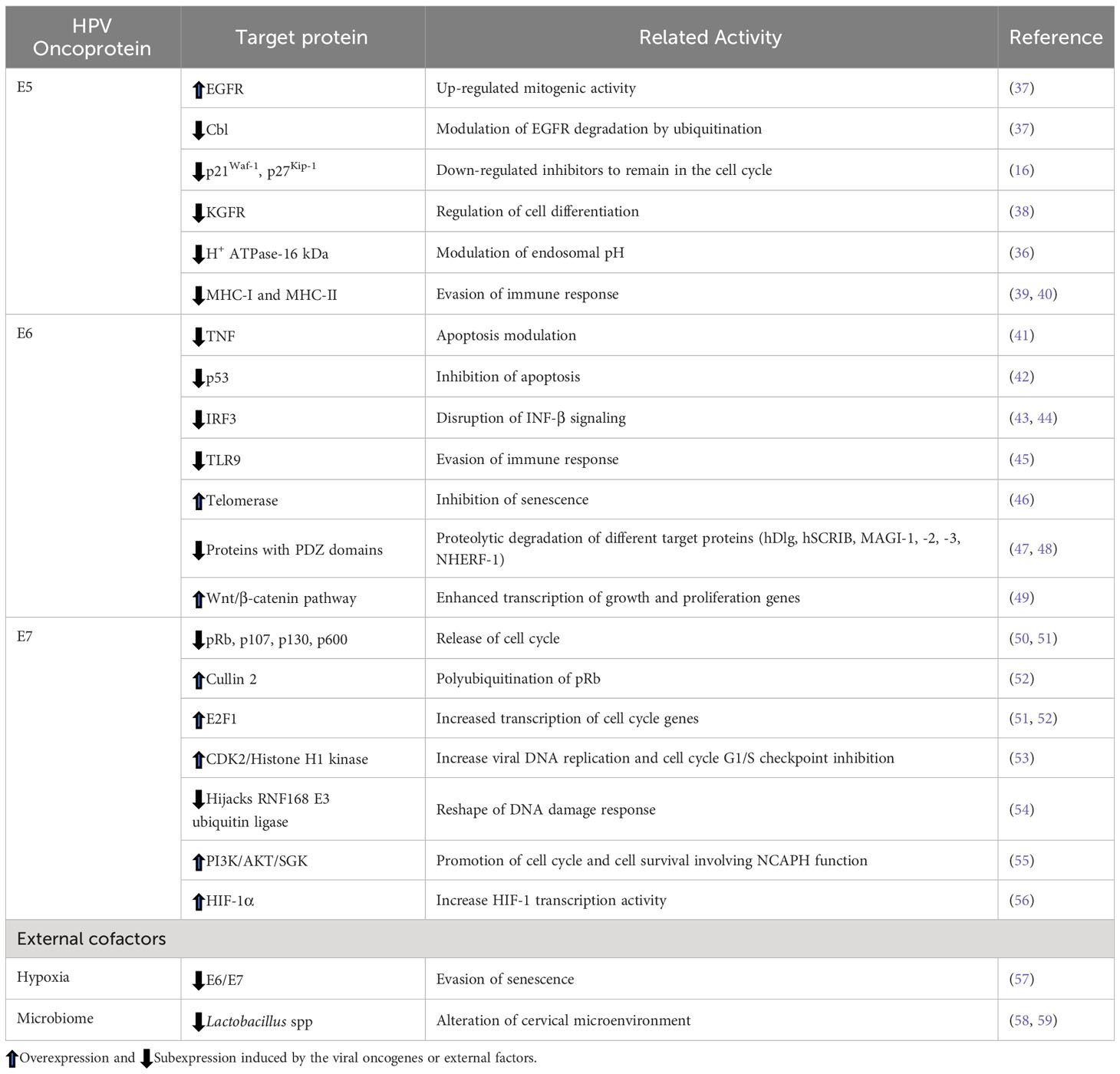
Table 1 Target proteins of HPV oncogenes.
Additionally, E5 oncoprotein interferes with the host’s immune system, promoting HPV persistence and resistance to immunotherapy; this has been demonstrated through E5 interaction with the simulator of interferon genes (STING), which suppresses interferon (IFN) signaling pathway (60). E5 obstructs the immune response by retaining MHC-I molecules in the ER and Golgi, reducing cell surface antigen presentation, and preventing viral antigen recognition and T and NK cell maturation (39). E5 also affects antigen presentation through MHC-II by preventing invariant chain degradation by inducing endosome alkalinization, thus reducing the activity of this molecule on the cell surface (39, 40). Interestingly, HPV16 E5 has been shown to suppress the expression of keratinocytes-specific IFN with the subsequent inactivation of the JAK/STAT pathway and the suppression of IFN-stimulated genes, which finally impacts the maintenance and integrity of the viral episomes (61).
One of the main targets of E6 is the tumor suppressor p53, promoting its degradation through the interaction with the ubiquitin ligase E6AP (42, 62), which induces cells to enter S-phase without arresting in G1. Moreover, E6 can stimulate the transcriptional factor OCT-4 expression, which binds to the p53 promoter recruiting the NCOR1 co-repressor, representing a new mechanism by which E6 decreases p53 levels (63). E6 has anti-apoptotic activities, which may occur by the degradation of p53, leading to the down-regulation of the proapoptotic genes PUMA and Bax and genes related to senescence modulation (64, 65). Moreover, E6 and E7 activate the transcription of Survivin and the apoptosis inhibitor c-IAP2, conferring resistance to apoptosis (66). E6 also prevents apoptotic signals through direct interaction with interferon regulatory factor 3 (IRF3), inhibiting the IFN-β1 response (43), and through hypermethylation of the promoter of the tumor suppressor death-associated protein kinase 1 (DAPK1) leading to the down-regulation of IFN genes, which consequently impairs the cellular antiviral response (44).
E6 sensitizes to radiation therapy by hijacking cellular target proteins involved in DNA damage repair, such as CHEK2, CLK2/3, ERCC3 MNAT1, PER1, RMI1, RPA1, UVSSA, and XRCC6, which promotes their colocalization in HPV replication foci, facilitating viral replication and increasing cellular genome instability (67). Additionally, E6 increases telomerase activity through the degradation of the telomerase inhibitor NFX1-91 and, in conjunction with Myc, transactivates the hTERT catalytic subunit, allowing cell immortalization due to the inhibition of senescence (46).
HR-HPV E6 proteins contain a PDZ-binding motif through which they bind proteins containing PDZ domains, sending them to degradation by ubiquitination, such as hDLG, hSCRIB, MAGI-1, -2, -3, and NHERF-1. In this condition, MAGI-2 cannot interact with PTEN to suppress AKT activation, while degradation of NHERF-1 activates the PI3K signaling pathway, thus promoting cell survival and proliferation (47, 48). In addition, degradation of NHERF1 by the E6/E6AP complex activates the Wnt/β-catenin signaling pathway, leading to the accumulation of β-catenin that induces transcription of genes that regulate cell growth and proliferation (e.g., c-Jun, c-myc, cyclin D, survivin, COX-2) (49).
Recent studies using the mouse papillomavirus (MmPV1) model demonstrated that E6 oncoprotein modulates the Notch signaling pathway by interacting with MAML1, component of the Notch pathway, affecting cell density and delaying differentiation, which allows viral persistence (68).
The E7 oncoprotein promotes cell cycle progression by sequestering and degrading the tumor suppressor protein pRB via polyubiquitination by cullin 2 (CUL2) (52), which releases the E2F transcription factor from the pRb complex, allowing the cell cycle to progress to the S-phase (50, 51). Another player in the pRb-CUL2 degradation pathway is miR-154-5p, which targets CUL2 and is down-regulated by E7 (52). Also, HR-HPV E7 oncoprotein can trigger tumorigenesis in a pRB-independent pathway by binding directly to the E2F1 transcription factor (50) and overexpressing eIF4E translation factor (69), promoting cell proliferation and migration while inhibiting apoptosis. Furthermore, E7 also modulates the G2/M cell cycle phase by upregulating the kinase activity of the histone H1 through the Cyclin A/CDK2/p107/E7 complex, which promotes HPV replication but utilizes ATM signaling to activate the p38/MK2 pathway also required for viral replication (52, 70). Moreover, E7 binds the RN1698 E3 ubiquitin ligase, hijacking its activity to promote viral replication; consequently, cellular response to DNA damage is reshaped, promoting genome instability (54). Interestingly, E7, through E2F1, promotes the expression of the NCAPH gene, which is involved in the activation of PI3K/AKT/SGK signaling and has also been implicated in proliferation, migration, invasion, epithelial-mesenchymal transition and restricts tumor formation (55). Recent findings reveal a complex interplay between PI3K/AKT/mTOR in virus-host cell communication. E6/E7 inhibits cell senescence under normoxia. Still, oxygen deprivation leads to the impairment of mTOR signaling, and hypoxic HPV-positive cells can evade senescence, although E6/E7 is down-regulated due to the activation of AKT (57).
External cofactors are also crucial for HPV-induced carcinogenesis, as is the case of the microbiome. Alterations in the cervicovaginal microbiome occur during the progression of HPV-associated lesions to CC, where an increase in resident bacteria diversity occurs along with a reduction of the resident Lactobacillus spp. Dysbiosis in the cervical microbiome influences viral persistence and is a carcinogenic co-factor (58, 59).
Table 1 shows several cellular targets of viral oncoproteins affecting biological processes.
The molecular changes in CC are not yet fully elucidated, and next-generation technologies have generated a large amount of information that has increased knowledge in this area. Bioinformatic data analysis has provided relevant information on the genome sequence, transcriptome, proteome, glycome, epigenome, etc. Several studies have revealed molecular changes that occur during the development and progression of CC and have identified potential biomarkers and molecular targets associated with susceptibility to the disease, degree of advancement, response to treatment, and survival.
CC depends largely on the genetic characteristics of the host. Koel et al. (2023) (71) performed a genome-wide association study (GWAS) to identify genomic variants associated with the full spectrum of cervical disorders, including ectropion, cervicitis, dysplasia, and CC. The variants that were mainly identified overlapped between cervicitis, dysplasia, and cancer and appeared different for ectropion. A genetic risk score (GRS) associated with CC was constructed. GRS identified people at risk of developing CC, which could be used to personalize the screening strategies for susceptible people. Most of the predictive power of the variants identified comes from the HLA region (HLA-DQA1), but it also includes regions where the closest genes are CDC42, PAX8, CLPTM1L, and ORMDL3.
During HPV carcinogenesis, somatic mutations are accumulated in the host genome. Through the whole exon-, genome- and transcriptome sequencing, driver mutations have been identified in the genomic landscape of an important number of CC samples paired with normal cervical tissue (72). Among the most frequent alterations found in CC were somatic mutations in MAPK1 (8%), EP300 (16%), FBXW7 (15%), PIK3CA (14%), PTEN (6%), NFE2L2 (4%), TP53 (5%), ERBB2 (6%) and HLA-B inactivating mutations (9%). Furthermore, in cases of cervical adenocarcinoma, the most frequent somatic mutations were found in ELF3 (13%) and CBFB (8%). Moreover, Zhou L. et al. (2022) (31) characterized virus-human integration events in CC samples, combined with the corresponding RNA-seq analysis. Among the genes frequently found disrupted by HPV integration were IL20RB, SOX14, LENG8, LENG9, CDC42EP5, CASC21, CCAT2, CASC8, and AKAP13, while genes whose expression was altered included LINC00290, LENG9, CCAT2, ARHGAP42, HNF1B, FOXD2, TMC1, IL15, and RPS6KA.
Transcriptome analyses in CC have identified changes in the expression of genes involved in cell transformation and immune response. Moreover, other RNAs involved in the disease have been identified, such as miRNAs, circRNAs, and long ncRNAs, that play essential roles in cancer pathogenesis as tumor promoters or suppressors (73–75). Salmerón-Bárcenas et al. (2023) (76) performed a bioinformatic analysis focusing on miR-182-3p, which is described in other cancers associated with chemoresistance and cancer progression. They identified miR-182-3p gene targets downregulated in CC and found that it might participate in angiogenesis and cell migration. They also proposed this miR as a potential diagnostic biomarker.
Understanding of the biological roles of circRNAs in CC progression is still under investigation, and the mechanisms by which circRNAs influence CC development and metastasis have yet to be completely elucidated. Zhang et al. (2023) (77) performed an expression analysis using microarray technology and identified differentially expressed mRNAs, miRNAs, and circRNAs in CC tissues. Their study focused on analyzing circRNAs with binding sites for miR-154-5p, a tumor suppressor in CC. hsa_circ_0000276 had the most substantial binding capacity for miR-154-5p and showed an increased expression in CC tissue. Through bioinformatic analysis, the authors showed that hsa_circ_0000276 was associated with CD47, LDHA, PDIA3, and SLC16A1, related to immune system processes; it was also reported that hsa_circ_0000276 increased proliferation and inhibited apoptosis.
Through lncRNA/mRNA microarray technology, Xin et al. (2023) (78) identified differentially expressed lncRNAs and mRNAs in HPV16 and HPV18-positive CC tissues compared with normal tissues. They found that co-expression of LINC00511-PGK1 could be important in the HPV-mediated transformation. Then, a score was proposed based on the co-expression of LINC00511 and PGK1 that predicted the overall survival of the patients with CC.
Other studies have focused on analyzing genes involved in ubiquitination, whose role in CC has yet to be determined. Hao et al. (2023) (79) identified the ubiquitination-related genes differentially expressed in CC tumors. They selected those associated with overall survival and established a prognostic gene signature that includes RBBP4, SRM, GCH1, USP14, TRAIP, CBX4, VEZF1, and TOM1 genes. Considering the importance of the anti-tumor immune response to control cancer development and progression, other groups have reported immune gene signatures. Pu et al. (2022) (80) reported an immune signature composed of 10 genes: CD96, LAG3, PDCD1, TIGIT, CD27, KLRK1, LTA, PVR, TNFRSF13C, and TNFRSF17; these genes are CD79B associated immunomodulators, that showed an independent prognostic value. A study that analyzed RNAseq databases of genes related to the glycosylation process (glycogens) showed that adenocarcinoma tumors displayed a unique glycogen expression signature. Squamous cancers showed more significant heterogeneity since six different signatures were identified related to different glycosylation pathways, such as glycosphingolipids, keratan and heparan sulfate synthesis, and glycosaminoglycan degradation (81).
Epigenetic changes have been evaluated in CC. Analyzing the Cancer Genome Atlas DNA methylation database, Yang et al, 2020 (82), identified HPV-related methylation sites in the DNA of CC tumors and classified them into clusters associated with overall survival. Aberrant mutations, amplifications, and deletions were identified in the different methylation groups, proposing a prognostic signature that allows patients to be stratified into high and low risk. Salta et al. (2023) (83) performed a meta-analysis on DNA methylation in HR-HPV-positive women with HSILs and proposed DNA methylation-based markers to discriminate lesions with a higher risk of progression to CC.
Current research on HPV has primarily focused on E6 and E7 oncogenes due to their impact on the development of CC. These genes are often found overexpressed in CC cells. Some promising approaches targeting E6 and E7 include the development of therapeutic vaccines for preventing the progress of squamous cell intraepithelial lesions to CC (84), antibodies against viral oncogenes to inhibit tumor growth (85, 86), strategies that inhibit their expression (87, 88), and immunotherapy that uses the host’s immune system to attack HPV-related cancers (89). Furthermore, E5 has gained relevance for participating in the progression and maintenance of CC. Moreover, novel bioinformatic analyses have identified vital genes, miRNAs, lncRNAs, circRNA, and signaling pathways contributing to CC progression. Some of these findings have been proposed not only as potential biomarkers but also as therapeutic targets. As research on CC continues, our knowledge of the genetic changes contributing to this disease increases, which could eventually help improve diagnostic tests and treatment options for this type of cancer.
VV-R: Writing – review & editing, Writing – original draft, Supervision, Investigation, Conceptualization. LG-X: Writing – review & editing, Writing – original draft, Investigation, Conceptualization. OM-C: Writing – review & editing, Writing – original draft, Investigation, Conceptualization. ML: Writing – review & editing, Writing – original draft, Supervision, Investigation, Conceptualization.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. CONAHCYT PRONAII-7-Virus y Cáncer 303044; National Cancer Institute of Mexico (017/007/IBI)(CEI/1144/17).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. International Agency for Research on Cancer. Cancer Today (2023). Available online at: https://gco.iarc.fr/today/home (Accessed September 13, 2023).
2. de Martel C, Plummer M, Vignat J, Franceschi S. Worldwide burden of cancer attributable to HPV by site, country and HPV type. Int J Cancer. (2017) 141:664–70. doi: 10.1002/IJC.30716
3. Burk RD, Chen Z, Saller C, Tarvin K, Carvalho AL, Scapulatempo-Neto C, et al. Integrated genomic and molecular characterization of cervical cancer. Nature. (2017) 543:378–84. doi: 10.1038/NATURE21386
4. Petry KU, Liebrich C, Luyten A, Zander M, Iftner T. Surgical staging identified false HPV-negative cases in a large series of invasive cervical cancers. Papillomavirus Res (Amsterdam Netherlands). (2017) 4:85–9. doi: 10.1016/J.PVR.2017.10.003
5. De Oliveira CM. Adaptation of alpha-papillomavirus over millennia. Acta Cytol. (2019) 63:97–9. doi: 10.1159/000492658
6. Egawa N, Egawa K, Griffin H, Doorbar J. Human papillomaviruses; epithelial tropisms, and the development of neoplasia. Viruses. (2015) 7:3863–90. doi: 10.3390/V7072802
7. Pangarkar MA. The Bethesda System for reporting cervical cytology. Cytojournal. (2022) 19. doi: 10.25259/CMAS_03_07_2021
8. Knappe M, Bodevin S, Selinka HC, Spillmann D, Streeck RE, Chen XS, et al. Surface-exposed amino acid residues of HPV16 L1 protein mediating interaction with cell surface heparan sulfate. J Biol Chem. (2007) 282:27913–22. doi: 10.1074/JBC.M705127200
9. Aksoy P, Abban CY, Kiyashka E, Qiang W, Meneses PI. HPV16 infection of HaCaTs is dependent on β4 integrin, and α6 integrin processing. Virology. (2014) 449:45–52. doi: 10.1016/J.VIROL.2013.10.034
10. Finke J, Hitschler L, Boller K, Florin L, Lang T. HPV caught in the tetraspanin web? Med Microbiol Immunol. (2020) 209:447–59. doi: 10.1007/S00430-020-00683-1
11. Bannach C, Brinkert P, Kühling L, Greune L, Schmidt MA, Schelhaas M. Epidermal growth factor receptor and abl2 kinase regulate distinct steps of human papillomavirus 16 endocytosis. J Virol. (2020) 94. doi: 10.1128/JVI.02143-19
12. Pim D, Broniarczyk J, Siddiqa A, Massimi P, Banks L. Human Papillomavirus 16 L2 Recruits both Retromer and Retriever Complexes during Retrograde Trafficking of the Viral Genome to the Cell Nucleus. J Virol. (2021) 95. doi: 10.1128/JVI.02068-20
13. Pim D, Broniarczyk J, Bergant M, Playford MP, Banks L. A novel PDZ domain interaction mediates the binding between human papillomavirus 16 L2 and sorting nexin 27 and modulates virion trafficking. J Virol. (2015) 89:10145–55. doi: 10.1128/JVI.01499-15
14. Schneider MA, Spoden GA, Florin L, Lambert C. Identification of the dynein light chains required for human papillomavirus infection. Cell Microbiol. (2011) 13:32–46. doi: 10.1111/J.1462-5822.2010.01515.X
15. Moody CA. Mechanisms by which HPV induces a replication competent environment in differentiating keratinocytes. Viruses. (2017) 9. doi: 10.3390/V9090261
16. Gutierrez-Xicotencatl L, Pedroza-Saavedra A, Chihu-Amparan L, Salazar-Piña A, Maldonado-Gama M, Esquivel-Guadarrama F. Cellular functions of HPV16 E5 oncoprotein during oncogenic transformation. Mol Cancer Res. (2021) 19:167–79. doi: 10.1158/1541-7786.MCR-20-0491
17. Doorbar J, Egawa N, Griffin H, Kranjec C, Murakami I. Human papillomavirus molecular biology and disease association. Rev Med Virol. (2015) 25 Suppl 1:2–23. doi: 10.1002/RMV.1822
18. Rodriguez AC, Schiffman M, Herrero R, Hildesheim A, Bratti C, Sherman ME, et al. Longitudinal study of human papillomavirus persistence and cervical intraepithelial neoplasia grade 2/3: critical role of duration of infection. J Natl Cancer Inst. (2010) 102:315–24. doi: 10.1093/JNCI/DJQ001
19. Ho GYF, Einstein MH, Romney SL, Kadish AS, Abadi M, Mikhail M, et al. Risk factors for persistent cervical intraepithelial neoplasia grades 1 and 2: managed by watchful waiting. J Low Genit Tract Dis. (2011) 15:268–75. doi: 10.1097/LGT.0B013E3182216FEF
20. Castro-Muñoz LJ, Rocha-Zavaleta L, Lizano M, Ramírez-Alcántara KM, Madrid-Marina V, Manzo-Merino J. Alteration of the IFN-pathway by human papillomavirus proteins: antiviral immune response evasion mechanism. Biomedicines. (2022) 10. doi: 10.3390/BIOMEDICINES10112965
21. Vanajothi R, Srikanth N, Vijayakumar R, Palanisamy M, Bhavaniramya S, Premkumar K. HPV-mediated cervical cancer: A systematic review on immunological basis, molecular biology, and immune evasion mechanisms. Curr Drug Targets. (2022) 23:782–801. doi: 10.2174/1389450123666211221160632
22. Steinbach A, Riemer AB. Immune evasion mechanisms of human papillomavirus: An update. Int J Cancer. (2018) 142:224–9. doi: 10.1002/IJC.31027
23. Chen HC, Schiffman M, Lin CY, Pan MH, You SL, Chuang LC, et al. Persistence of type-specific human papillomavirus infection and increased long-term risk of cervical cancer. J Natl Cancer Inst. (2011) 103:1387–96. doi: 10.1093/JNCI/DJR283
24. Groves IJ, Coleman N. Pathogenesis of human papillomavirus-associated mucosal disease. J Pathol. (2015) 235:527–38. doi: 10.1002/PATH.4496
25. Li W, Tian S, Wang P, Zang Y, Chen X, Yao Y, et al. The characteristics of HPV integration in cervical intraepithelial cells. J Cancer. (2019) 10:2783–7. doi: 10.7150/JCA.31450
26. Tian R, Cui Z, He D, Tian X, Gao Q, Ma X, et al. Risk stratification of cervical lesions using capture sequencing and machine learning method based on HPV and human integrated genomic profiles. Carcinogenesis. (2019) 40:1220–8. doi: 10.1093/CARCIN/BGZ094
27. Jabbar SF, Park S, Schweizer J, Berard-Bergery M, Pitot HC, Lee D, et al. Cervical cancers require the continuous expression of the human papillomavirus type 16 E7 oncoprotein even in the presence of the viral E6 oncoprotein. Cancer Res. (2012) 72:4008–16. doi: 10.1158/0008-5472.CAN-11-3085
28. Schrank TP, Kim S, Rehmani H, Kothari A, Wu D, Yarbrough WG, et al. Direct comparison of HPV16 viral genomic integration, copy loss, and structural variants in oropharyngeal and uterine cervical cancers reveal distinct relationships to E2 disruption and somatic alteration. Cancers (Basel). (2022) 14. doi: 10.3390/CANCERS14184488
29. Vinokurova S, Wentzensen N, Kraus I, Klaes R, Driesch C, Melsheimer P, et al. Type-dependent integration frequency of human papillomavirus genomes in cervical lesions. Cancer Res. (2008) 68:307–13. doi: 10.1158/0008-5472.CAN-07-2754
30. Chaiwongkot A, Vinokurova S, Pientong C, Ekalaksananan T, Kongyingyoes B, Kleebkaow P, et al. Differential methylation of E2 binding sites in episomal and integrated HPV 16 genomes in preinvasive and invasive cervical lesions. Int J Cancer. (2013) 132:2087–94. doi: 10.1002/IJC.27906
31. Zhou L, Qiu Q, Zhou Q, Li J, Yu M, Li K, et al. Long-read sequencing unveils high-resolution HPV integration and its oncogenic progression in cervical cancer. Nat Commun. (2022) 13. doi: 10.1038/S41467-022-30190-1
32. de Oliveira CM, Bravo IG, Santiago e Souza NC, Genta MLND, Fregnani JHTG, Tacla M, et al. High-level of viral genomic diversity in cervical cancers: A Brazilian study on human papillomavirus type 16. Infect Genet Evol. (2015) 34:44–51. doi: 10.1016/J.MEEGID.2015.07.002
33. Mukherjee AG, Ramesh Wanjari U, Valsala Gopalakrishnan A, Jayaraj R, Katturajan R, Kannampuzha S, et al. HPV-associated cancers: insights into the mechanistic scenario and latest updates. Med Oncol. (2023) 40. doi: 10.1007/S12032-023-02085-8
34. Ortiz-Pedraza Y, Muñoz-Bello JO, Ramos-Chávez LA, Martínez-Ramírez I, Olmedo-Nieva L, Manzo-Merino J, et al. HPV16 E6 and E7 oncoproteins stimulate the glutamine pathway maintaining cell proliferation in a SNAT1-dependent fashion. Viruses. (2023) 15. doi: 10.3390/V15020324
35. Manzo-Merino J, Contreras-Paredes A, Vázquez-Ulloa E, Rocha-Zavaleta L, Fuentes-Gonzalez AM, Lizano M. The role of signaling pathways in cervical cancer and molecular therapeutic targets. Arch Med Res. (2014) 45:525–39. doi: 10.1016/J.ARCMED.2014.10.008
36. Siddiqa A, Broniarczyk J, Banks L. Papillomaviruses and endocytic trafficking. Int J Mol Sci. (2018) 19. doi: 10.3390/IJMS19092619
37. Chen B, Zhao L, Yang R, Xu T. Advances in molecular mechanism of HPV16 E5 oncoprotein carcinogenesis. Arch Biochem Biophys. (2023) 745. doi: 10.1016/J.ABB.2023.109716
38. Wasson CW, Morgan EL, Müller M, Ross RL, Hartley M, Roberts S, et al. Human papillomavirus type 18 E5 oncogene supports cell cycle progression and impairs epithelial differentiation by modulating growth factor receptor signalling during the virus life cycle. Oncotarget. (2017) 8:103581–600. doi: 10.18632/ONCOTARGET.21658
39. Balasubramaniam SD, Balakrishnan V, Oon CE, Kaur G. Key molecular events in cervical cancer development. Medicina (Kaunas). (2019) 55. doi: 10.3390/MEDICINA55070384
40. De Freitas AC, De Oliveira THA, Barros MR, Venuti A. hrHPV E5 oncoprotein: immune evasion and related immunotherapies. J Exp Clin Cancer Res. (2017) 36. doi: 10.1186/S13046-017-0541-1
41. Cabeça TK, Abreu A de M, Andrette R, Lino V de S, Morale MG, Aguayo F, et al. HPV-mediated resistance to TNF and TRAIL is characterized by global alterations in apoptosis regulatory factors, dysregulation of death receptors, and induction of ROS/RNS. Int J Mol Sci. (2019) 20. doi: 10.3390/IJMS20010198
42. Li S, Hong X, Wei Z, Xie M, Li W, Liu G, et al. Ubiquitination of the HPV oncoprotein E6 is critical for E6/E6AP-mediated p53 degradation. Front Microbiol. (2019) 10:2483. doi: 10.3389/FMICB.2019.02483
43. Poirson J, Suarez IP, Straub M-L, Cousido-Siah A, Peixoto P, Hervouet E, et al. High-risk mucosal human papillomavirus 16 (HPV16) E6 protein and cutaneous HPV5 and HPV8 E6 proteins employ distinct strategies to interfere with interferon regulatory factor 3-mediated beta interferon expression. J Virol. (2022) 96. doi: 10.1128/JVI.01875-21
44. Yanatatsaneejit P, Chalertpet K, Sukbhattee J, Nuchcharoen I, Phumcharoen P, Mutirangura A. Promoter methylation of tumor suppressor genes induced by human papillomavirus in cervical cancer. Oncol Lett. (2020) 20:955–61. doi: 10.3892/OL.2020.11625
45. Hasan UA, Bates E, Takeshita F, Biliato A, Accardi R, Bouvard V, et al. TLR9 expression and function is abolished by the cervical cancer-associated human papillomavirus type 16. J Immunol. (2007) 178:3186–97. doi: 10.4049/JIMMUNOL.178.5.3186
46. Zhang Y, Dakic A, Chen R, Dai Y, Schlegel R, Liu X. Direct HPV E6/Myc interactions induce histone modifications, Pol II phosphorylation, and hTERT promoter activation. Oncotarget. (2017) 8:96323–39. doi: 10.18632/ONCOTARGET.22036
47. Accardi R, Rubino R, Scalise M, Gheit T, Shahzad N, Thomas M, et al. E6 and E7 from human papillomavirus type 16 cooperate to target the PDZ protein Na/H exchange regulatory factor 1. J Virol. (2011) 85:8208–16. doi: 10.1128/JVI.00114-11
48. Ganti K, Broniarczyk J, Manoubi W, Massimi P, Mittal S, Pim D, et al. The human papillomavirus E6 PDZ binding motif: from life cycle to Malignancy. Viruses. (2015) 7:3530–51. doi: 10.3390/V7072785
49. Drews CM, Case S, Pol SBV. E6 proteins from high-risk HPV, low-risk HPV, and animal papillomaviruses activate the Wnt/β-catenin pathway through E6AP-dependent degradation of NHERF1. PloS Pathog. (2019) 15. doi: 10.1371/JOURNAL.PPAT.1007575
50. Songock WK, Kim Sm, Bodily JM. The human papillomavirus E7 oncoprotein as a regulator of transcription. Virus Res. (2017) 231:56–75. doi: 10.1016/J.VIRUSRES.2016.10.017
51. Bienkowska-Haba M, Luszczek W, Zwolinska K, Scott RS, Sapp M. Genome-wide transcriptome analysis of human papillomavirus 16-infected primary keratinocytes reveals subtle perturbations mostly due to E7 protein expression. J Virol. (2020) 94. doi: 10.1128/JVI.01360-19
52. Zhao W, Liu Y, Zhang L, Ding L, Li Y, Zhang H, et al. MicroRNA-154-5p regulates the HPV16 E7-pRb pathway in Cervical Carcinogenesis by targeting CUL2. J Cancer. (2020) 11:5379–89. doi: 10.7150/JCA.45871
53. He W, Staples D, Smith C, Fisher C. Direct activation of cyclin-dependent kinase 2 by human papillomavirus E7. J Virol. (2003) 77:10566–74. doi: 10.1128/JVI.77.19.10566-10574.2003
54. Sitz J, Blanchet SA, Gameiro SF, Biquand E, Morgan TM, Galloy M, et al. Human papillomavirus E7 oncoprotein targets RNF168 to hijack the host DNA damage response. Proc Natl Acad Sci U.S.A. (2019) 116:19552–62. doi: 10.1073/PNAS.1906102116
55. Wang M, Qiao X, Cooper T, Pan W, Liu L, Hayball J, et al. HPV E7-mediated NCAPH ectopic expression regulates the carcinogenesis of cervical carcinoma via PI3K/AKT/SGK pathway. Cell Death Dis. (2020) 11. doi: 10.1038/S41419-020-03244-9
56. Bodily JM, Mehta KPM, Laimins LA. Human papillomavirus E7 enhances hypoxia-inducible factor 1-mediated transcription by inhibiting binding of histone deacetylases. Cancer Res. (2011) 71:1187–95. doi: 10.1158/0008-5472.CAN-10-2626
57. Bossler F, Kuhn BJ, Günther T, Kraemer SJ, Khalkar P, Adrian S, et al. Repression of human papillomavirus oncogene expression under hypoxia is mediated by PI3K/mTORC2/AKT signaling. MBio. (2019) 10:1–16. doi: 10.1128/MBIO.02323-18
58. Mitra A, MacIntyre DA, Lee YS, Smith A, Marchesi JR, Lehne B, et al. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci Rep. (2015) 5. doi: 10.1038/SREP16865
59. Finzer P, Küppers V, Griesser H. Dysbiotic co-factors in cervical cancer. How the microbiome influences the development of cervical intraepithelial neoplasia (CIN). Geburtshilfe Frauenheilkd. (2023) 83. doi: 10.1055/A-2044-0162
60. Miyauchi S, Kim SS, Jones RN, Zhang L, Guram K, Sharma S, et al. Human papillomavirus E5 suppresses immunity via inhibition of the immunoproteasome and STING pathway. Cell Rep. (2023) 42. doi: 10.1016/J.CELREP.2023.112508
61. Scott ML, Woodby BL, Ulicny J, Raikhy G, Orr AW, Songock WK, et al. Human papillomavirus 16 E5 inhibits interferon signaling and supports episomal viral maintenance. J Virol. (2020) 94. doi: 10.1128/JVI.01582-19
62. Martinez-Zapien D, Ruiz FX, Poirson J, Mitschler A, Ramirez J, Forster A, et al. Structure of the E6/E6AP/p53 complex required for HPV-mediated degradation of p53. Nature. (2016) 529:541–5. doi: 10.1038/NATURE16481
63. Shu S, Li Z, Liu L, Ying X, Zhang Y, Wang T, et al. HPV16 E6-Activated OCT4 Promotes Cervical Cancer Progression by Suppressing p53 Expression via Co-Repressor NCOR1. Front Oncol. (2022) 12:900856. doi: 10.3389/FONC.2022.900856
64. Celegato M, Messa L, Goracci L, Mercorelli B, Bertagnin C, Spyrakis F, et al. A novel small-molecule inhibitor of the human papillomavirus E6-p53 interaction that reactivates p53 function and blocks cancer cells growth. Cancer Lett. (2020) 470:115–25. doi: 10.1016/J.CANLET.2019.10.046
65. Zhu J, Kamara S, Wang Q, Guo Y, Li Q, Wang L, et al. Novel affibody molecules targeting the HPV16 E6 oncoprotein inhibited the proliferation of cervical cancer cells. Front Cell Dev Biol. (2021) 9:677867. doi: 10.3389/FCELL.2021.677867
66. Skelin J, Sabol I, Tomaić V. Do or die: HPV E5, E6 and E7 in cell death evasion. Pathog (Basel Switzerland). (2022) 11. doi: 10.3390/PATHOGENS11091027
67. Bruyere D, Roncarati P, Lebeau A, Lerho T, Poulain F, Hendrick E, et al. Human papillomavirus E6/E7 oncoproteins promote radiotherapy-mediated tumor suppression by globally hijacking host DNA damage repair. Theranostics. (2023) 13:1130–49. doi: 10.7150/THNO.78091
68. Saunders-Wood T, Egawa N, Zheng K, Giaretta A, Griffin HM, Doorbar J. Role of E6 in maintaining the basal cell reservoir during productive papillomavirus infection. J Virol. (2022) 96. doi: 10.1128/JVI.01181-21
69. Pang T, Wang S, Gao M, Kang H, Zhao Y, Yao Y, et al. HPV18 E7 induces the over-transcription of eIF4E gene in cervical cancer. Iran J Basic Med Sci. (2015) 18:684.
70. Anacker DC, Moody CA. Modulation of the DNA damage response during the life cycle of human papillomaviruses. Virus Res. (2017) 231:41–9. doi: 10.1016/J.VIRUSRES.2016.11.006
71. Koel M, Võsa U, Jõeloo M, Läll K, Gualdo NP, Laivuori H, et al. GWAS meta-analyses clarify the genetics of cervical phenotypes and inform risk stratification for cervical cancer. Hum Mol Genet. (2023) 32:2103–16. doi: 10.1093/HMG/DDAD043
72. Ojesina AI, Lichtenstein L, Freeman SS, Pedamallu CS, Imaz-Rosshandler I, Pugh TJ, et al. Landscape of genomic alterations in cervical carcinomas. Nature. (2014) 506:371–5. doi: 10.1038/NATURE12881
73. Pisarska J, Baldy-Chudzik K. MicroRNA-based fingerprinting of cervical lesions and cancer. J Clin Med. (2020) 9:1–24. doi: 10.3390/JCM9113668
74. Wu B, Xi S. Bioinformatics analysis of differentially expressed genes and pathways in the development of cervical cancer. BMC Cancer. (2021) 21. doi: 10.1186/S12885-021-08412-4
75. Wu M, Han Y, Gong X, Wan K, Liu Y, Zhou Y, et al. Novel insight of circRNAs in cervical cancer: potential biomarkers and therapeutic target. Front Med. (2022) 9:759928. doi: 10.3389/FMED.2022.759928
76. Salmerón-Bárcenas EG, Mendoza-Catalan MA, Ramírez-Bautista ÁU, Lozano-Santos RA, Torres-Rojas FI, Ávila-López PA, et al. Identification of mir-182-3p/FLI-1 axis as a key signaling in immune response in cervical cancer: A comprehensive bioinformatic analysis. Int J Mol Sci. (2023) 24. doi: 10.3390/IJMS24076032
77. Zhang H, Wang X, Li Y, Bai Y, Li Q, Wang S, et al. The hsa_circ_0000276-ceRNA regulatory network and immune infiltration in cervical cancer. BMC Cancer. (2023) 23. doi: 10.1186/S12885-023-10636-5
78. Xin X, Jia-Yin Y, Jun-Yang H, Rui W, Xiong-Ri K, Long-Rui D, et al. Comprehensive analysis of lncRNA-mRNA co-expression networks in HPV-driven cervical cancer reveals the pivotal function of LINC00511-PGK1 in tumorigenesis. Comput Biol Med. (2023) 159. doi: 10.1016/J.COMPBIOMED.2023.106943
79. Hao Y, Guy MM, Liu Q, Li R, Mao Z, Jiang N, et al. Construction of a prognostic model based on eight ubiquitination-related genes via machine learning and potential therapeutics analysis for cervical cancer. Front Genet. (2023) 14:1142938. doi: 10.3389/FGENE.2023.1142938
80. Pu D, Liu D, Li C, Chen C, Che Y, Lv J, et al. A novel ten-gene prognostic signature for cervical cancer based on CD79B-related immunomodulators. Front Genet. (2022) 13:933798. doi: 10.3389/FGENE.2022.933798
81. Martinez-Morales P, Cruz IM, Roa-De la Cruz L, Maycotte P, Salinas JSR, Zamora VJV, et al. Hallmarks of glycogene expression and glycosylation pathways in squamous and adenocarcinoma cervical cancer. PeerJ. (2021) 9. doi: 10.7717/PEERJ.12081
82. Yang S, Wu Y, Wang S, Xu P, Deng Y, Wang M, et al. HPV-related methylation-based reclassification and risk stratification of cervical cancer. Mol Oncol. (2020) 14:2124–41. doi: 10.1002/1878-0261.12709
83. Salta S, Lobo J, Magalhães B, Henrique R, Jerónimo C. DNA methylation as a triage marker for colposcopy referral in HPV-based cervical cancer screening: a systematic review and meta-analysis. Clin Epigenet. (2023) 15:125. doi: 10.1186/S13148-023-01537-2
84. Zhou K, Yuzhakov O, Behloul N, Wang D, Bhagat L, Chu D, et al. HPV16 E6/E7-based mRNA vaccine is therapeutic in mice bearing aggressive HPV-positive lesions. Front Immunol. (2023) 14:1213285. doi: 10.3389/FIMMU.2023.1213285
85. Jiang Z, Albanese J, Kesterson J, Warrick J, Karabakhtsian R, Dadachova E, et al. Monoclonal antibodies against human papillomavirus E6 and E7 oncoproteins inhibit tumor growth in experimental cervical cancer. Transl Oncol. (2019) 12:1289–95. doi: 10.1016/J.TRANON.2019.06.003
86. Paolini F, Amici C, Carosi M, Bonomo C, Di Bonito P, Venuti A, et al. Intrabodies targeting human papillomavirus 16 E6 and E7 oncoproteins for therapy of established HPV-associated tumors. J Exp Clin Cancer Res. (2021) 40. doi: 10.1186/S13046-021-01841-W
87. Chen Y, Jiang H, Wang T, He D, Tian R, Cui Z, et al. In vitro and in vivo growth inhibition of human cervical cancer cells via human papillomavirus E6/E7 mRNAs’ cleavage by CRISPR/Cas13a system. Antiviral Res. (2020) 178. doi: 10.1016/J.ANTIVIRAL.2020.104794
88. Sun X, Fu P, Xie L, Chai S, Xu Q, Zeng L, et al. Resveratrol inhibits the progression of cervical cancer by suppressing the transcription and expression of HPV E6 and E7 genes. Int J Mol Med. (2021) 47:335–45. doi: 10.3892/IJMM.2020.4789
Keywords: cervical cancer, human papillomavirus, E6, E7, oncogenesis
Citation: Vallejo-Ruiz V, Gutiérrez-Xicotencatl L, Medina-Contreras O and Lizano M (2024) Molecular aspects of cervical cancer: a pathogenesis update. Front. Oncol. 14:1356581. doi: 10.3389/fonc.2024.1356581
Received: 15 December 2023; Accepted: 05 March 2024;
Published: 19 March 2024.
Edited by:
Chengquan Zhao, University of Pittsburgh, United StatesReviewed by:
Larisa Litovchick, Virginia Commonwealth University, United StatesCopyright © 2024 Vallejo-Ruiz, Gutiérrez-Xicotencatl, Medina-Contreras and Lizano. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marcela Lizano, bGl6YW5vQHVuYW0ubXg=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.