
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol., 07 March 2024
Sec. Pharmacology of Anti-Cancer Drugs
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1353896
 Valérian Rivet1*
Valérian Rivet1* Vincent Sibaud2
Vincent Sibaud2 Jérémie Dion1Thibaut Volosov3Mélanie Biteau1Andréa Pastissier1Karen Delavigne1Pierre Cougoul1Odile Rauzy1Thibault Comont1
Jérémie Dion1Thibaut Volosov3Mélanie Biteau1Andréa Pastissier1Karen Delavigne1Pierre Cougoul1Odile Rauzy1Thibault Comont1Introduction: Immune checkpoint inhibitors (ICIs) are used in several advanced malignancies and may cause various immune-related adverse events (irAEs). Among them, hematological irAEs are less described. Acquired amegakaryocytic thrombocytopenia (AAT) is a rare immune hematologic disorder characterized by severe thrombocytopenia and complete absence of megakaryocytes in bone marrow.
Case presentation: Herein, we present the case of a patient in their 40s with metastatic melanoma who developed an AAT after 12 cycles of nivolumab (anti-PD1). His platelet count decreased by ≤5 × 109/l without other cytopenia. Bone marrow biopsy showed normal cellularity with a complete absence of megakaryocyte and T-CD8+ lymphocyte infiltration. Given the failure of systemic steroids, eltrombopag was started, an oral thrombopoietin receptor agonist (TPO-RA), and his platelet count subsequently increased with complete response.
Discussion: Four other cases are described on literature with the same features than non-ICI-related AAT. All cases occurred after anti-PD/PD-L1 treatment with a median onset of 5 weeks. The presentation of our case is quite different with delayed cytopenia. Both ciclosporin and TPO-RA seem to be efficient therapies.
Conclusion: TPO-RA could be preferred in oncologic patients, but safety data are still missing to define clear guidelines for immune-related AAT management.
Immune checkpoint inhibitors (ICIs) are now used in several advanced malignancies and may cause various immune-related adverse events (irAEs), including dermatological, endocrine (especially dysthyroidism and hypophysis), rheumatological, hepatic, or digestive (especially colitis) toxicities with incidences of >50%, 4%–20%, 10%, 5%–30%, and 1%–30%, respectively (1). Hematological irAEs (hem-irAEs) are quite rare with overall frequencies around 0.5% to 3.6% and 0.7% for grade ≥3 events (graded based on the National Cancer Institute Common Terminology Criteria for Adverse Events, version 5.0) (2, 3). Mortality of this hem-irAEs can be high especially for aplastic anemia and hemophagocytic lymphohistiocytosis (3). Immune thrombocytopenia (ITP) is the most frequent hematological AE, and anti-programmed death-1 (PD-1)/programmed death-ligand 1 (PD-L1) treatments are more frequently involved than anti-cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) (2, 3).
Acquired amegakaryocytic thrombocytopenia (AAT) is a rare hematologic disorder characterized by severe thrombocytopenia and complete or nearly complete absence of megakaryocytes in the bone marrow (4, 5). Usually, AAT patients are poorly responders to standard ITP therapy (corticosteroids [CS] and intravenous immunoglobulin [IVIg]) (5). Only few cases of ICI-related AAT have so far been published, and given the rarity of this entity, no strong data on clinico-biological presentation and management are available (6–8). Herein, we describe another case of AAT after nivolumab perfusion and a literature review.
Patient in their 40s with initially localized melanoma (Breslow 10 mm, BRAF V600 positive) was treated by surgery. He had a personal history of Raynaud phenomenon and circulating anti-SSa, anti-SSb and anti-centromere antibodies. He had no hematological history. On August 2021, due to recurrence with metastatic involvement in the skin and lung, he started nivolumab perfusion (480-mg flat dose on a monthly basis). After four cycles, he suffered from a localized body-hair whitening (poliosis) within a tattoo on the upper chest. On January 2022, neoplastic assessment showed complete metabolic response and immunotherapy was continued. On January 2023, after 12 cycles (around 17 months), the blood sample revealed eosinophilia (1.3 G/l, N < 0.5 G/l) without any cytopenia, considered to be due to ICI and normalized after quick steroid therapy (1 mg per kilo or 60 mg per day during 1 week and then gradually tapering off over a period of 3 weeks). ICI was discontinued. On April, the patient suffered from severe thrombocytopenia and was referred to our hospital (Figure 1). He had no bleeding. The blood finding showed normal results except for platelet count ≤5 × 109/l. No other organ disorder was suggested with not any evidence of disseminated intravascular coagulation or thrombotic microangiopathy in the laboratory data. A bone marrow (BM) smear and a BM biopsy showed normal cellularity with a complete absence of megakaryocytes associated with polyclonal T-CD8+ lymphocyte infiltration (negative clonal T-cell population assessment). There was no evidence of myelodysplastic neoplasia, myelofibrosis, or metastatic lesions (Figure 2). Cytogenetic analysis of BM showed a normal karyotype (46XY). Excluding other possible secondary diagnosis (other immune diseases, drugs, or infections), we concluded that AAT was caused by ICI. We started orally corticosteroid treatment (1 mg per kilo) on day 2, without any effect on platelet count. Platelet transfusion resulted in a moderate and transient rise in the platelet count. Finally, a treatment with eltrombopag [a thrombopoietin receptor agonist (TPO-RA)] was started on day 7 (oral 50 mg daily, increased to 75 mg after 4 days), by maintaining corticosteroids. In response, the platelet count subsequently improved after 7 days and the patient was hospital-discharged on day 15. Eltrombopag was tapered off and stopped (on day 42) after a platelet count of 653 G/l and then stabilized on a normal range. A new tumor assessment revealed a persistent complete metabolic response, and rechallenge of ICI had not yet been carried out.
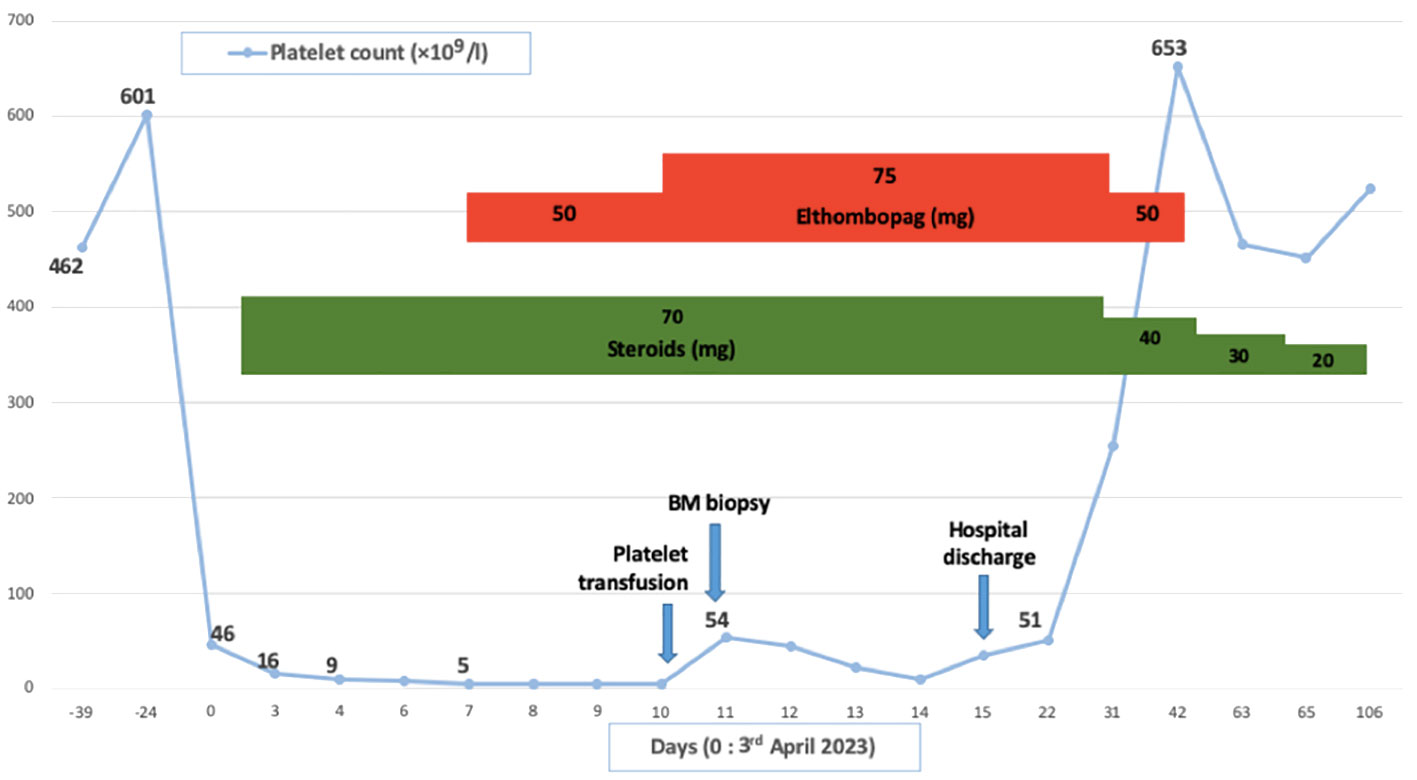
Figure 1 Clinical courses illustrating thrombocytopenia and main treatments. BM, Bone marrow.
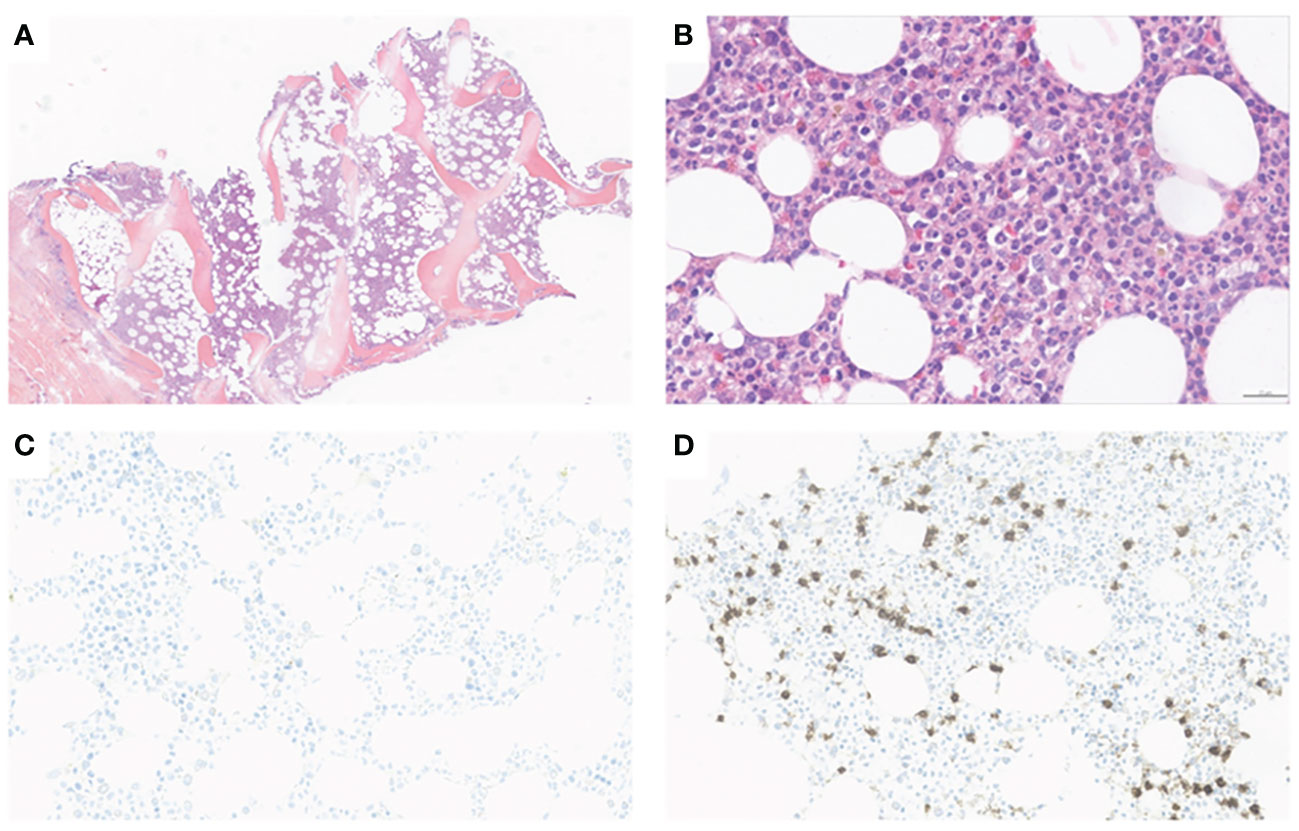
Figure 2 Bone marrow trephine biopsy. (A) Normocellular marrow (H&E, X5); (B) Absence of megacaryocytes and normal maturation pattern of erythroid and myeloid cells (H&E, X20). (C) Immunohistochemical staining with anti-CD61 confirms the absence of megacaryocytes (anti-CD61, x15). (D) Moderate neactive CD8+ T infiltrate (anti-CD8, x15).
AAT is an extremely rare cause of thrombocytopenia, but severe bleeding that is often life-threatening with a platelet count of ≤10 × 109/l is a concern in most of the cases (5, 7). The underlying pathogenic mechanism of this disease is still unknown. An autoimmune mechanism and dysregulation of humoral immunity with antibodies against TPO (9) or c-MPL (thrombopoietin receptor) was considered. T-cell-mediated processes were also suggested and supported by the presence of T-cell CD8+ in BM biopsies and by in vitro inhibition of a donor or autologous megakaryocyte (MK) colony-forming unit (CFU) by T cells of AAT patients (5, 10, 11). Response to ciclosporin also suggested a T-cytotoxicity-mediated mechanism (5). Indeed, classic therapies such as CS and IVIg are only effective on 22.4% and 5.3% of AAT patients, respectively (5). Ciclosporin (CSA) seems more efficient with an overall response around 66% of patients (5). Other treatments such as anti-thymocyte globulin or rituximab have also been reported successful (7). TPO-RA, especially eltrombopag, is already used for ITP and aplastic anemia and seems to be also effective in AAT (5, 7). However, standard treatment guidelines are still missing in this disease.
Only four other cases of AAT after ICI are available in literature (Table 1) (6–8). Large series and pharmacovigilance database study about immune-related cytopenia did not include any cases (2, 3, 12). All AAT occurred after anti-PD/PD-L1 treatment and after very few cycles of immunotherapy with a median onset of 5 weeks. The presentation of our case is quite different with delayed hem-irAEs, which appears only after 12 cycles of nivolumab perfusion. Even when AAT is induced by ICI, thrombocytopenia is severe with platelet count ≤30 × 109/l in all cases and ≤10 × 109/l in three out of five (60%) patients. Other blood lines are classically preserved in AAT. Suyama et al. described a patient with only a moderate anemia (7). The second case published by Tu et al. also suffered from other cytopenia probably caused by secondary aplastic anemia (8). BM biopsy is important to exclude blast infiltration, dysplasia, aplastic anemia, or fibrosis. It revealed in all cases an absence of megakaryocytes. The cellularity of bone marrow is variable: clear hypoplasia was found in two patients (6, 8), moderate hypocellularity (20%–30%) in one patient, and normal one in two others, including our patient (8). In a surprising way, lymphocyte T-CD8+ infiltration, classically found in BM biopsy of an AAT patient and in other hem-irAEs such as aplastic anemia, is described only in our case (3, 5, 13). Platelet transfusion was tried in three cases with effect in only 2. In our case, the effect was very transient but allows BM biopsy without any bleeding complication that may suggest an associated peripheral mechanism. The various cases available in the literature are therefore heterogeneous in clinical presentation, in other cytopenia sometimes associated with thrombopenia, and in cellularity found in BM biopsy as well as unexpected response to platelet transfusion that suggested different physiopathological processes and not only typical AAT.
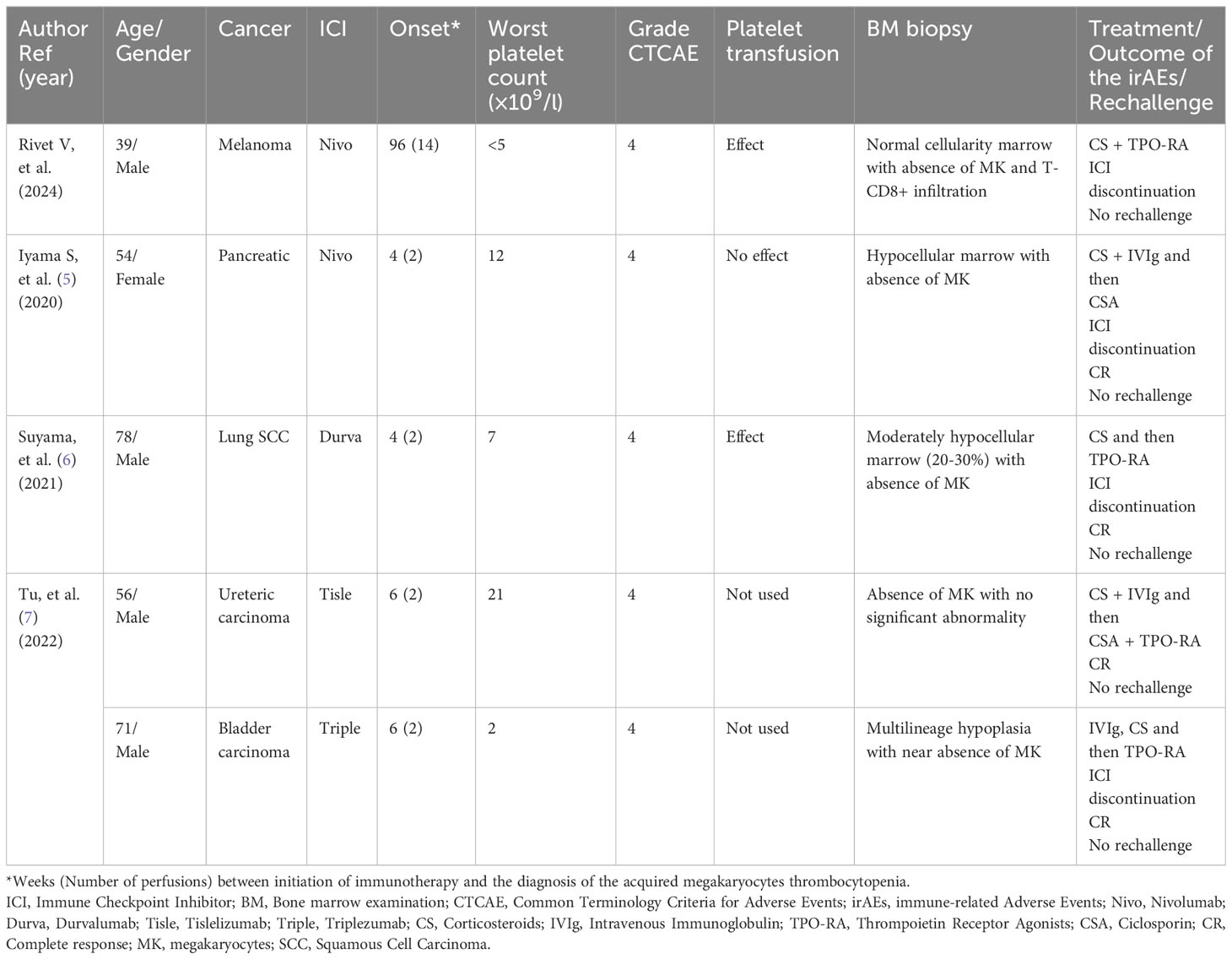
Table 1 Previously published and current case of immune checkpoint inhibitor-related acquired amegakaryocytic thrombocytopenia.
In all cases, ICI was discontinued (Table 1). According to data about non-ICI-related-AAT, CS and IVIg seem ineffective in all five AAT patients. In the case of Iyama et al., CS was nevertheless stopped after diagnosis of gastric ulcer and so response to steroid was not clear (6). CSA was used in two patients with complete response, defined as a platelet count ≥100×109/l (14): alone in one case and associated with TPO-RA in another one (6, 8). In the three other cases, complete response was obtained after TPO-RA (7, 8). Both eltrombopag (50 mg daily) and avatrombopag has led to a quick increase of platelet count (7, 8). In our patient, both eltrombopag and CS were maintained. Because of the absence of risk of life-threatening bleeding, we did not prescribe IVIg. Platelet count finally increased after dose escalation of eltrombopag at 75 mg daily. However, the platelet count did not appear affected by introduction of this treatment at 50 mg daily. In this context, a delayed response to steroids or a spontaneous correction of thrombocytopenia cannot be ruled out and efficacy of eltrombopag is uncertain. In the end, standard first-line treatments such as CS and IVIg do not appear to be effective in this entity, and TPO-RA and CS should be considered. However, for now, data seem to be more significant for TPO agonists and the onset of action may be faster with these treatments. The optimal dose of eltrombopag in AAT patients is unknown. The usual dose in ITP patients of 50 mg–75 mg daily is currently used. However, higher doses, such as those used in aplastic anemia (150 mg–225 mg daily), could improve response to treatment but would require additional data.
CSA and TPO-RA are both efficient in refractory AAT. In oncologic patients, TPO-RA could be promoted because of probably less risk of tumor progression than with CSA. Nevertheless, thrombotic risk with eltrombopag must be considered in this population (15). Finally, after immune-related cytopenia, rechallenge is rare with recurrence of the same hem-irAEs in more than 40% of patients (2). To our knowledge, no rechallenge was published in cases of AAT induced by ICI.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethical approval was not required for the study involving human samples in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
VR: Writing – original draft, Writing – review & editing, Conceptualization, Formal Analysis, Investigation, Validation. VS: Writing – review & editing. JD: Writing – review & editing. TV: Writing – review & editing. MB: Writing – review & editing. AP: Writing – review & editing. KD: Writing – review & editing. PC: Writing – review & editing. OR: Writing – review & editing. TC: Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
We thank the patient who accepted to participate in this study.
Author TC reports board, funding research, consulting, and hospitality from Novartis.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
AAT, Acquired amegakaryocytic thrombocytopenia; BM, Bone marrow; CS, Corticosteroids; CSA, Ciclosporin; ICIs, Immune Checkpoint Inhibitors; ITP, Immune thrombocytopenia; IVIg, Intravenous immunoglobulin; IrAEs, Immune-related Adverse Events; Hem-irAEs, hematological irAEs; MK, Megakaryocytes; PD-1, Anti-programmed death-1 (PD-1); PD-L1, programmed death-ligand 1 (PD-L1); CTLA-4, Anti-cytotoxic T-lymphocyte-associated protein 4; TPO-RA, Oral thrombopoietin receptor agonist.
1. Haanen J, Obeid M, Spain L, Carbonnel F, Wang Y, Robert C, et al. Management of toxicities from immunotherapy: ESMO Clinical Practice Guideline for diagnosis, treatment and follow-up. Ann Oncol. (2022) 33:1217–38. doi: 10.1016/j.annonc.2022.10.001
2. Dealanoy N, Michot JM, Comont T, Kramkimel N, Lazarovici J, Dupont R, et al. Haematological immune-related adverse events induced by anti-PD-1 or anti-PD-L1 immunotherapy: a descriptive observational study. Lancet Haematol. (2019) 6:e48–57. doi: 10.1016/S2352-3026(18)30175-3
3. Michot JM, Lazarovici J, Tieu A, Champiat S, Voisin AL, Ebbo M, et al. Haematological immune-related adverse events with immune checkpoint inhibitors, how to manage? Eur J Cancer. (2019) 122:72–90. doi: 10.1016/j.ejca.2019.07.014
4. Agarwal N, Sparh JE, Werner TL, Newton DL, Rodgers GM. Acquired amegakaryocytic thrombocytopenic purpura. Am J Hematol. (2006) 81:132–5. doi: 10.1002/ajh.20510
5. Roeser A, Moulis G, Ebbo M, Terriou L, Poullot E, Lioger B, et al. Characteristics, management and outcome of acquired amegakaryocytic thrombocytopenia. Br J Heamatol. (2022) 198:595–9. doi: 10.1111/bjh.18235
6. Iyama S, Takada K, Yoshida M, Takahashi D, Kobune M. Acquired amegakaryocytic thrombocytopenic purpura possibly induced by anti-PD-1 antibody. Ann Hematol. (2020) 99:1669–70. doi: 10.1007/s00277-020-04053-y
7. Suyama T, Hagihara M, Kubota N, Osamura Y, Shinka YMiyao N. Acquired amegakaryocytic thrombocytopenia after durvalumab administration. J Clin Exp Hematop. (2021) 61:53–7. doi: 10.3960/jslrt.20047
8. Tu X, Xue A, Wu S, Jin M, Zhao P, Zhang H. Case report: successful avatrombopag treatment for two cases of anti-PD-1 antibody-induced acquired amegakaryocytic thrombocytopenia. Front Pharmacol. (2022) 12:795884. doi: 10.3389/fphar.2021.795884
9. Kuwaka T, Hagiwara T, Kato T, Miyazaki H. Autoantibodies neutralizing thrombopoietin in a patient with a megakaryocytic thrombopenic purpura. Blood. (2000) 95:2187–8. doi: 10.1182/blood.V95.6.2187.2187
10. Kuwana M, Okazaki Y, Kajihara M. Autoantibody to c-MPL (thrombopoietin receptor) in systemic lupus erythematosus: relationship to thrombocytopenia with megakaryocytic hypoplasia. Arthritis Rheum. (2002) 46:2148–59. doi: 10.1002/art.10420
11. Gewirtz AM, Sacchetti MK, Bien R, Barry WE. Cell-mediated supression of megakaryocytopoiesis in acquired amegakaryocytic thrombocytopenic purpura. Blood. (1986) 68:619–26. doi: 10.1182/blood.V68.3.619.619
12. Martin M, Nguyen HM, Beuvon C, Bene J, Plalassin P, Atzenhoffer M, et al. Immune checkpoint inhibitor-related cytopenias: about 68 cases from the french pharmacovigilance database. Cancers. (2022) 14:5030. doi: 10.3390/cancers14205030
13. Michot JM, Vargaftig J, Leduc C, Quere C, Burroni B, Lazarovici J, et al. Immune-related bone marrow failure following anti-PD1 therapy. Eur J Cancer. (2017) 80:1–4. doi: 10.1016/j.ejca.2017.04.004
14. Rodeghiero F, Stasi R, Gernsheimer T, Michel M, Provan D, Arnold DM, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood. (2009) 113:2386–93. doi: 10.1182/blood-2008-07-162503
Keywords: acquired megakaryocytic thrombocytopenia, immune checkpoint inhibitors (ICI), immune-related adverse events (IRAE), nivolumab, eltrombopag, thrombopoietin receptor agonist (TPO-RA)
Citation: Rivet V, Sibaud V, Dion J, Volosov T, Biteau M, Pastissier A, Delavigne K, Cougoul P, Rauzy O and Comont T (2024) Immune checkpoint inhibitor-related acquired amegakaryocytosis thrombocytopenia: a case report and literature review. Front. Oncol. 14:1353896. doi: 10.3389/fonc.2024.1353896
Received: 11 December 2023; Accepted: 14 February 2024;
Published: 07 March 2024.
Edited by:
Claudia Cerella, Fondation de Recherche Cancer et Sang, LuxembourgReviewed by:
Benjamin Garmezy, Sarah Cannon Research Institute, United StatesCopyright © 2024 Rivet, Sibaud, Dion, Volosov, Biteau, Pastissier, Delavigne, Cougoul, Rauzy and Comont. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Valérian Rivet, VmFsZXJpYW4ucml2ZXRAZ21haWwuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.