 Sumedha Saluja
Sumedha Saluja Ishu Bansal
Ishu Bansal Ruchi Bhardwaj
Ruchi Bhardwaj Mohammad Sabique Beg
Mohammad Sabique Beg Jayanth Kumar Palanichamy
Jayanth Kumar Palanichamy
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 20 March 2024
Sec. Hematologic Malignancies
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1347402
This article is part of the Research TopicGenetic and non-genetic Determinants of InflammationView all 5 articles
Hematopoiesis is a tightly regulated process that produces all adult blood cells and immune cells from multipotent hematopoietic stem cells (HSCs). HSCs usually remain quiescent, and in the presence of external stimuli like infection or inflammation, they undergo division and differentiation as a compensatory mechanism. Normal hematopoiesis is impacted by systemic inflammation, which causes HSCs to transition from quiescence to emergency myelopoiesis. At the molecular level, inflammatory cytokine signaling molecules such as tumor necrosis factor (TNF), interferons, interleukins, and toll-like receptors can all cause HSCs to multiply directly. These cytokines actively encourage HSC activation, proliferation, and differentiation during inflammation, which results in the generation and activation of immune cells required to combat acute injury. The bone marrow niche provides numerous soluble and stromal cell signals, which are essential for maintaining normal homeostasis and output of the bone marrow cells. Inflammatory signals also impact this bone marrow microenvironment called the HSC niche to regulate the inflammatory-induced hematopoiesis. Continuous pro-inflammatory cytokine and chemokine activation can have detrimental effects on the hematopoietic system, which can lead to cancer development, HSC depletion, and bone marrow failure. Reactive oxygen species (ROS), which damage DNA and ultimately lead to the transformation of HSCs into cancerous cells, are produced due to chronic inflammation. The biological elements of the HSC niche produce pro-inflammatory cytokines that cause clonal growth and the development of leukemic stem cells (LSCs) in hematological malignancies. The processes underlying how inflammation affects hematological malignancies are still not fully understood. In this review, we emphasize the effects of inflammation on normal hematopoiesis, the part it plays in the development and progression of hematological malignancies, and potential therapeutic applications for targeting these pathways for therapy in hematological malignancies.
Inflammation is one of the defense mechanisms of the body that is utilized to fight against infections and regenerate injured tissues (1). Sustained inflammatory stimuli for an extended period can lead to chronic inflammation. Chronic inflammation can promote the occurrence and development of cancer by promoting blood vessel growth, cancer cell proliferation, and tumor invasiveness (2). Inflammatory biomarkers have been linked to increased cancer risk and mortality (3), and chronic systemic low-grade inflammation is a risk factor for incident cancer (4). Inflammatory biomarkers such as the neutrophil-to-lymphocyte ratio (NLR), systemic inflammation response index (SIRI) and systemic immune-inflammation index (SII) have been found to be predictive of survival for multiple types of cancers (5). Hematopoiesis is a highly controlled process maintained by the division of quiescent, self-renewing, multipotent hematopoietic stem cells (HSCs) and lineage-specific downstream progenitors in the bone marrow (BM). Cell extrinsic signals like chemokines and cytokines activate HSCs and cells in the microenvironment that help compensate for normal cellular loss (6). Inflammation is known to play a significant role in normal hematopoiesis.
A majority of hematological malignancies result from mutations in the hematopoietic stem cells, leading to uncontrolled growth and proliferation of HSCs. While inflammation is known to play a beneficial role in immune system activation and tissue regeneration, chronic inflammation can lead to HSC damage, resulting in bone marrow failure or the development of leukemia (7).
Usually, stress or infection causes the activation of numerous signaling pathways, which increases the production of inflammatory cytokines and chemokines, which activate immune cells like B and T lymphocytes, helping to eradicate the infection. Inflammation regulates homeostasis and maintains the hematopoietic system. However, uncontrolled inflammation can also play pathogenic roles by disrupting homeostasis, which can potentially contribute to tumor development. A variety of cells of the immune response release pro-inflammatory cytokines, including IL-1β, IL-1α, TNFα, IL-6, IL-12, IFN-γ, and chemokines such as CCL2 and CXCL12, which participate in the initiation, growth, and progression of tumor and development of drug resistance (8, 9). Transcription factors involved in inflammation, such as NFκB and STAT3, are known to promote the development and progression of cancer by controlling the expression of genes involved in apoptosis, cell proliferation regulation of angiogenesis, tumor metastasis and invasion (8, 10, 11). Thus, the inflammatory elements, including cytokines, chemokines, and their receptors, play a significant role in tumorigenesis and promote the survival of tumor cells with metastatic and invasive properties (9, 12). In addition, chronic inflammation often leads to the overproduction of hematopoietic stem cells, which subsequently undergo DNA mutations, thereby leading to the development of hematological malignancies (13) (Figure 1).
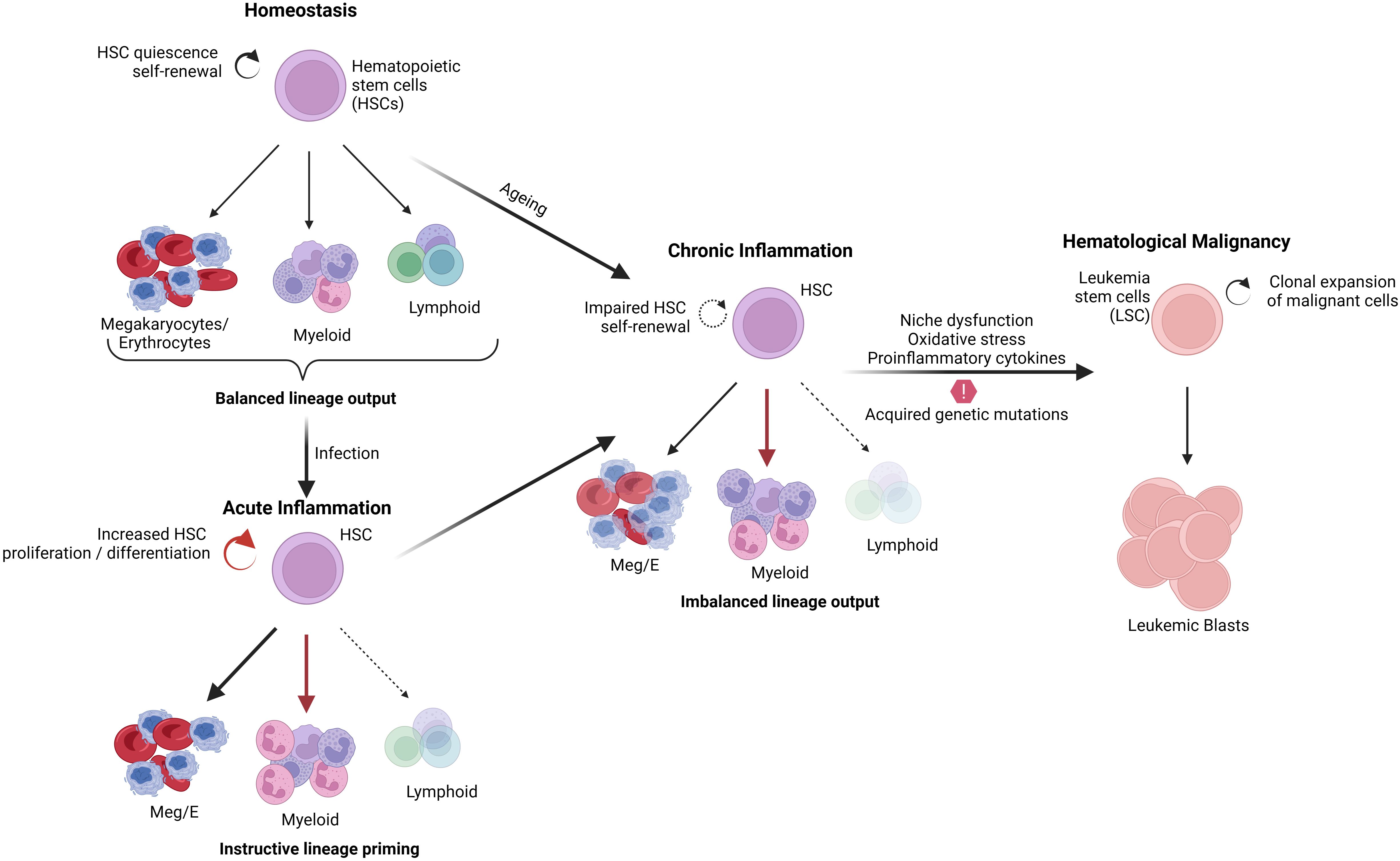
Figure 1 Effect of inflammation in normal hematopoiesis and malignant transformation.
The recent advances in molecular biology and the development of genetically modified mouse models helped unravel the various aspects of inflammation during tumorigenesis (14, 15). Therefore, it is crucial to understand the molecular mechanisms that connect inflammatory processes to tumorigenesis and metastasis, which can be targeted for therapeutic and diagnostic purposes. This review highlights the role of inflammation in normal hematopoiesis and the relevance and requirement of the inflammation process to be tightly controlled at both initiation and termination, which when lost, can lead to the development and progression of cancer.
Hematopoiesis is the formation of all blood cells and components of blood plasma hierarchically controlled by HSCs. The multipotent HSCs can give rise to all types of blood cells residing in the bone marrow (16). HSCs possess self-renewal ability, which helps maintain the stem cell pool. Although quiescent, HSCs undergo division in response to external stimuli like infections and irradiation (17). Stress conditions like inflammation can cause HSCs to undergo proliferation. During inflammation, HSCs express receptors for cytokines and chemokines that help HSCs recognize signals from immune cells to adapt their cycling and differentiation potential (18, 19). However, mutations in HSCs can lead to uncontrolled proliferation, which can cause bone marrow damage or result in hematological malignancies (17). These mutations primarily affect the self-renewal ability of HSCs, activating their proliferation and inhibiting the production of mature cells.
Somatic mutations in genes such as NPM1, which is involved in the maintenance of HSCs’ quiescence and self-renewal, and TET2, DNMT3A, involved in mediating HSCs’ differentiation, results in the transformation of HSCs thereby leading to the development of Acute Myeloid Leukemia (AML) (20). Mixed lineage leukemia (MLL) is known to be commonly rearranged in leukemia. Knockout of the MLL gene led to a reduction in HSC numbers in mice, and HSCs deficient in the MLL gene are unable to reconstitute the bone marrow of the recipient mice, signifying that the MLL gene is important for the maintenance of self-renewal ability of HSCs. Since the JAK-STAT signaling pathway activates HSCs to proliferate in response to the release of inflammatory cytokines, gain of function mutations in JAK2 non-receptor tyrosine kinase can often result in long-term activation of HSCs, sustained differentiation to the erythroid and myeloid lineages and results in AML development (21).
HSCs reside in the bone marrow within a specific microenvironment termed the HSC niche. The HSC niche comprises the extracellular matrix along with distinct cell types like mesenchymal stem cells (MSCs), osteoblasts, osteoclasts, osteolineage progenitor cells, endothelial cells (ECs) and specialized CXCL12-abundant reticular (CAR) cells and leptin receptor (LEPR) positive cells (22–28). HSCs reside in the perivascular region of sinusoids and arterioles near MSCs and ECs which are essential for maintenance of quiescence and differentiation of HSCs (29). MSCs regulate HSCs via the expression of CXCL12, angiopoietin-1 and vascular cell adhesion molecule-1 (VCAM-1) (30–33). HSCs exist in two distinct niches - endosteal and vascular niche. The endosteal niche, which contains osteoblasts, regulates the quiescence associated with HSCs and their migration to the vascular niche (34). Inside the vascular niche, that contains endothelial cells, HSCs undergo differentiation (27, 28, 35, 36). TGF-β is secreted by non-myelinating Schwann cells around the blood vessels in the bone marrow (37). Additionally, megakaryocytes around sinusoids are responsible for maintaining HSCs quiescence, through release of factors such as TGF-β along with Platelet factor (PF-4/CXCL4) and thrombopoietin (38–41). Studies with conditional deletion of CXCL12 in endosteal and vascular niche of HSCs have highlighted role of CXCL12 in the maintenance and self-renewal of HSCs. This is controlled by CXCL12 released by immature mesenchymal stem and progenitor cells, with a smaller contribution from endothelial cells (42, 43).
Hematopoietic stress conditions like systemic inflammation or infections cause HSCs to exit their quiescence and undergo proliferation and differentiation to compensate for cellular loss. The production of mature myeloid cells, including neutrophils and monocytes from HSCs, as a result of inflammation, is termed emergency myelopoiesis (25, 26, 44). The HSC niche plays a significant role in mediating the hematopoietic response to peripheral or systemic inflammation. The HSC niche secretes certain factors such as granulocyte colony-stimulating factor (G-CSF) that promote myelopoiesis during inflammation (45–47). G-CSF is the central regulator of inflammation induced emergency myelopoiesis. It is the endothelial cells which have been characterized as the main source of production of G-CSF during inflammation. During LPS-induced systemic inflammation, TLR4 signaling in endothelial cells led to elevated G-CSF synthesis, resulting in emergency granulopoiesis (7, 19, 45–47).
Inflammation induces the expression of G-CSF in ECs and IL-6 from ECs and MSCs (48). CXCL12 and Kit ligand (KITL) are required to maintain HSCs in the BM niche (27, 47). Upon inflammation, the expression of CXCL12 and KITL is downregulated (42). In order to mediate HSCs mobilization in the niche, G-CSF acts on different cell types such as MSCs, macrophages, neutrophils and osteolineage cells (49). G-CSF affects osteoblastic activity and inhibits the expression of CXCL12 directly or through functional changes in macrophages or granulocytes (48, 50). In the case of viral infections, Interferon-γ (IFN-γ) secreted by CD8+ T cells acts on MSCs in the HSC niche and leads to increased release of IL-6 by MSCs (51). In summary, HSCs undergo transient division in response to stress conditions such as inflammation to give rise to myeloid cells to compensate for cellular loss. At the same time, these HSCs further secrete pro-inflammatory cytokines such as IL-6, which will activate themselves in a paracrine or autocrine manner to mediate the expansion of HSCs in the bone marrow (52, 53).
Inflammation in the bone marrow has been reported to contribute to the development of hematological malignancies (54). Myeloid malignancies like AML, myeloproliferative neoplasms (MPN), myelodysplastic syndromes (MDS) are thought to represent a clonal disease of HSCs (55). Point mutations and mosaic chromosomal alterations increase the risk of lymphoid malignancies like chronic lymphocytic leukemia (CLL), small lymphocytic lymphoma (SLL), and diffuse large B-cell lymphoma (DLBCL) (56).
These malignant clones are called leukemia stem cells (LSCs) or leukemia initiating cells (LICs). LSCs were initially characterized in AML patients. The surface immunophenotype for LSCs is usually CD34+ CD38– CD90– along with interleukin-3 receptor (IL-3R), and CD117 positivity. These LSCs have also been identified in CML and ALL cells that carry the BCR-ABL fusion gene (54). The LSCs share surface phenotypic markers with HSCs during the evolution of leukemia. This further leads to the production of both the clonogenic leukemic progenitors and the non-clonogenic blast cells that eventually leads to full-blown leukemogenesis (57). Like HSCs, LSCs also exhibit self-renewal, quiescence, and multipotency properties but also uncontrolled proliferation (57, 58). This subpopulation of leukemia cells has properties of HSCs and, along with loss of differentiation and apoptosis, leads to cancer development. These stem cell-like features make it challenging to target LSCs and render LSCs resistant to conventional chemotherapy, thereby leading to the relapse of the disease (59).
HSCs, when mutated, can be the source of the generation of LSCs. HSCs usually have a finite lifespan. However, the self-renewing HSCs, when mutated, sustain for a long time, allowing genetic damage and malignant transformation of HSCs to LSCs (60). AML arises from multiple genetic mutations that lead to increased proliferation, survival, and impaired differentiation of hematopoietic progenitor cells (61). The most recurrent mutations in AML occur in genes such as FLT3, NPM1, CEBPA, IDH1/IDH2, DNMT3A, and RUNX1 (62). For example, internal tandem duplications in the FLT3 gene (FLT3-ITD) and mutations in the nucleophosmin (NPM1) gene are detected in approximately 30% and 50% of AML cases, respectively (63). Other common mutations include those in the DNA methyltransferase 3A (DNMT3A) gene, present in 20-30% of cases (64). The order in which these mutations are acquired can influence leukemia development. A frequent sequence is an initial DNMT3A mutation, followed by an NPM1 mutation and then FLT3-ITD mutation (60). DNMT3A mutations showed higher levels of pre-leukemic stem cells that are resistant to chemotherapy and thus further lead to leukemia development. Injection of DNMT3A mutant pre-leukemic HSCs in NSG mice demonstrated a competitive repopulation advantage over non-mutated HSCs (60). Like HSCs, the maintenance of LSCs also relies on their tumor microenvironment, termed the LSC niche. The chemokine CXCL12 binds to its receptor, CXCR4 and plays a prominent role in the homing of HSCs and LSCs in the bone marrow by mediating adherence of AML cells to stromal cells, leading to proliferation and resistance from chemotherapeutic drugs. It has been known that CXCR4 is highly expressed in AML and ALL patients and results in poor prognosis (65–67). Transforming growth factor β (TGF-β) acts as a critical regulator of quiescent G0 state in the AML and CML cells, thereby maintaining LSCs (68). Besides soluble factors, LSCs also interact with niche cells via cell-cell interactions. For example, CD44, a transmembrane glycoprotein, mediates the adhesion of LSCs to the niche and transduces intracellular signals involved in proliferation and differentiation. CD44 targeting led to the loss of migration of human and murine LSCs to the niche, leading to the eradication of LSCs (68–71).
Inflammation can trigger oncogenesis either by cell extrinsic or intrinsic mechanisms. The extrinsic mechanism is driven by external factors, including inflammatory conditions and micro-environmental factors, where a constant inflammatory state contributes to tumor initiation and progression. For instance, patients with inflammatory bowel disease have shown increased susceptibility to lymphomas, leukemias, and hepatocarcinoma. However, the cell-intrinsic pathway involves genetic alterations affecting oncogenes, tumor suppressors, and genome stability genes, which activate inflammatory pathways (such as the NFκB pathway), thereby generating an inflammatory microenvironment in tumors (72) (Figure 2).
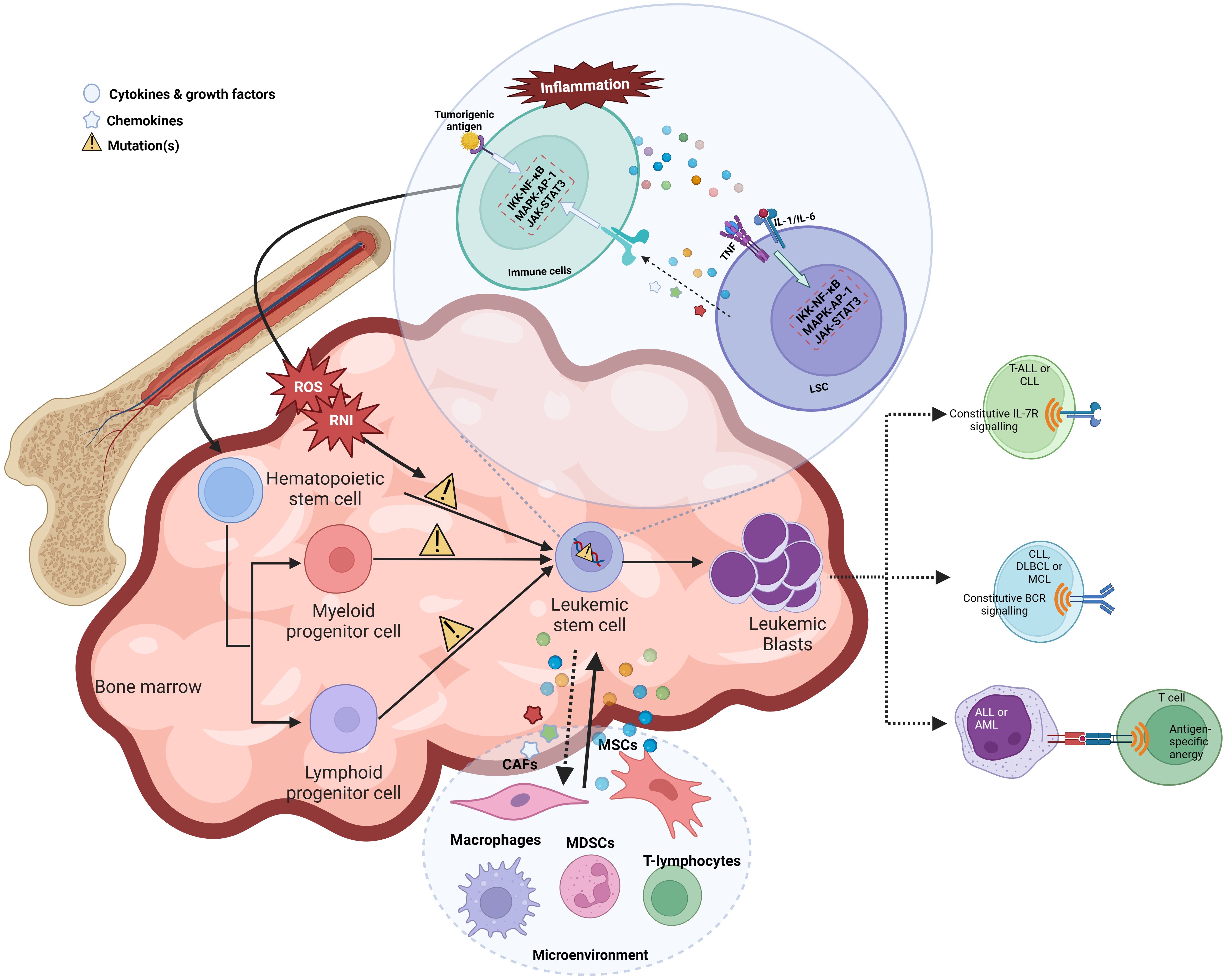
Figure 2 Chronic inflammation leads to leukemogenesis by various mechanisms.
In both malignant and inflammatory cells, NFκB is activated downstream to the TLR-MyD88 pathway (sensing microbes or tissue damage) or the inflammatory cytokines, including TNF and IL-1β (Figure 3). It has been known that NFκB is one of the primary inflammatory pathways associated with myeloid and lymphoid malignancies (73, 74). Alternatively, NFκB activation can result from genetic alterations (amplification, mutations, or deletions) in cancer cells. NFκB enhances the expression of antiapoptotic genes like BCL2, CLIP, and cIAP and increases the survival of tumor cells (75).
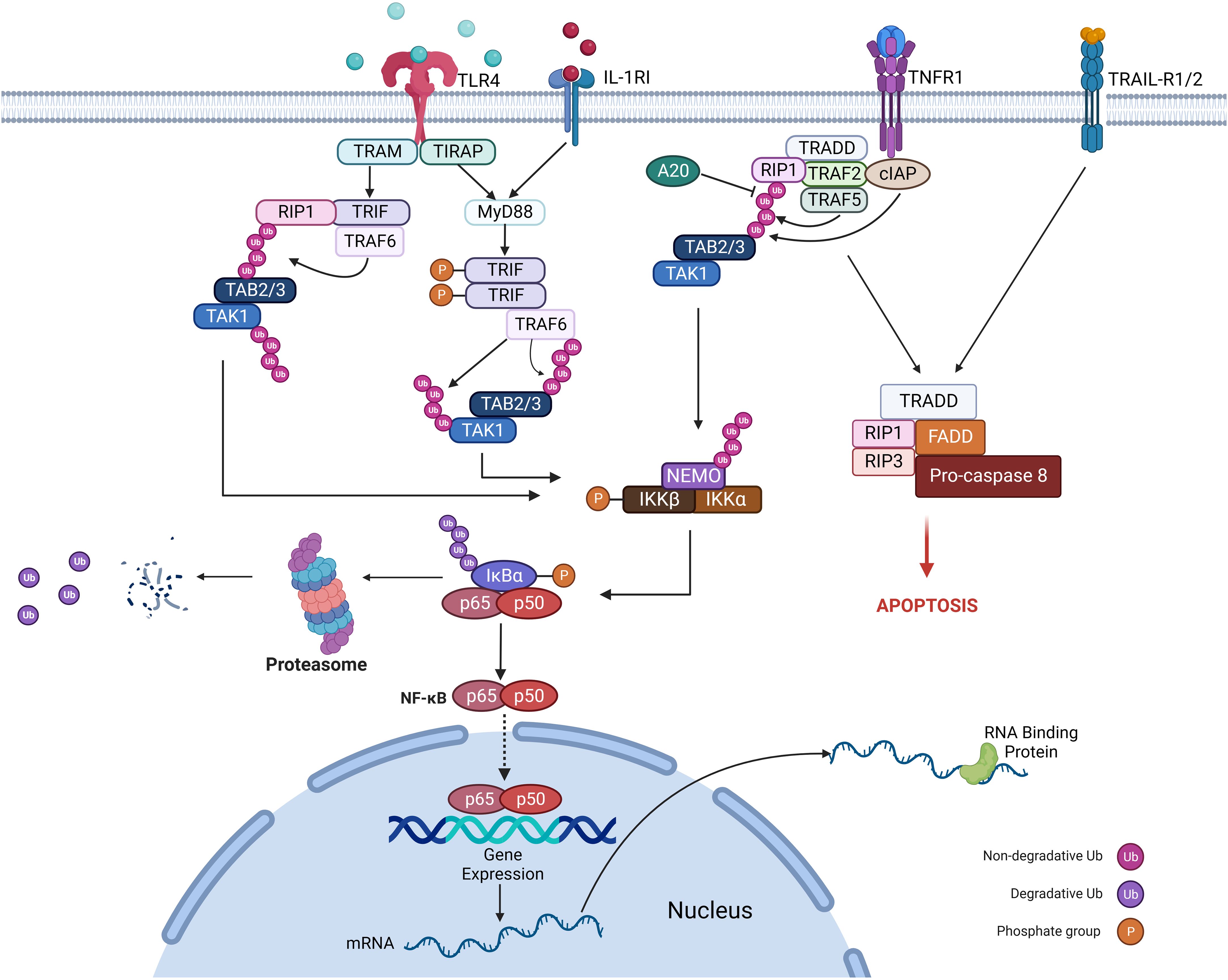
Figure 3 Mechanism of regulation of the NFκB pathway.
Inflammatory mediators like S100A8/A9 or inflammatory cytokines such as TNF-β, IL-1β, IL-6 and IFN-γ, chemokines like CCL2 and CXCL8 are seen to be upregulated in malignancies like myelodysplastic syndrome (76). These molecules contribute to carcinogenesis via the NFκB pathway and STAT3 signaling pathway. MSCs are a population of stem cells that are important in maintaining bone marrow microenvironment. MSCs also have immunoregulatory functions and maintain the immune BM microenvironment by saving the HSCs from stress stimuli. The dysregulated development of MSCs has been shown to lead to MDS induction and further AML development (77). The NFκB pathway is also upregulated in MSCs of patients with MDS. Along with NFκB, STAT3 has also been associated with cancer-related inflammation. NFκB is activated in two ways: a) IKK activity dependent (pro-inflammatory stimulus-dependent) and b) constitutive activity, which is proinflammatory stimulus-independent (73). STAT3 is a transcriptional factor which maintains constitutive expression of NFκB by acetylation of RelA. STAT3 also has a feedback inhibition on IKK activity. B16 mouse melanoma tumor cells and DU145 prostate cancer cells, showed high STAT3 activity by increasing phosphorylation of RelA protein in the presence of TNF-α. B16 melanoma cells showed a reduction in RelA phosphorylation and, thus, in NFκB activity after STAT3 knockdown (73, 78). Tumor cells have high expression of VEGF, IL-10, and IL-6; these tumor cells also highly express STAT3. High expression of STAT3 is shown to inhibit DC maturation, which in turn leads to immature DC accumulation and subsequent immunosuppression, leading to tumor cell escape. Stat3 deletion in HSCs improved DC maturation and function which induced antitumor activity in mice. Targeting STAT3 in cancer cell lines and in-vivo studies has also reduced tumor growth (79, 80).
STAT proteins are also involved in hematopoietic growth factor signal transduction. Signaling molecules activate different STATs: thrombopoietin (TPO) activates STAT3, whereas granulocyte-macrophage (GM)–CSF, TPO, and IL-3 activate STAT5. It is known that STAT1, 3 and 5 have all been overexpressed in acute and chronic leukemia (81).
A study conducted on Tet2-deficient mice showed increased IL-6 production in response to microbial infection, resulting in preleukemic myeloproliferation, signifying the critical role of inflammation in the progression of leukemia (82). A study using the MLL-AF9-induced AML mouse model showed that the leukemia cells expressed factors like TNF and CXCL12. These led to the remodeling of endosteal vessels, generating a niche that supports the overall proliferation of malignant hematopoietic clones- LSCs and a decrease in the number of normal HSCs (83).
Taken together, these studies implied the need to focus on a better understanding of the mechanisms of crosstalk between inflammation and its effect on interactions of HSCs with their niche during normal hematopoiesis and emergency myelopoiesis which may subsequently lead to the development of leukemias (Table 1).
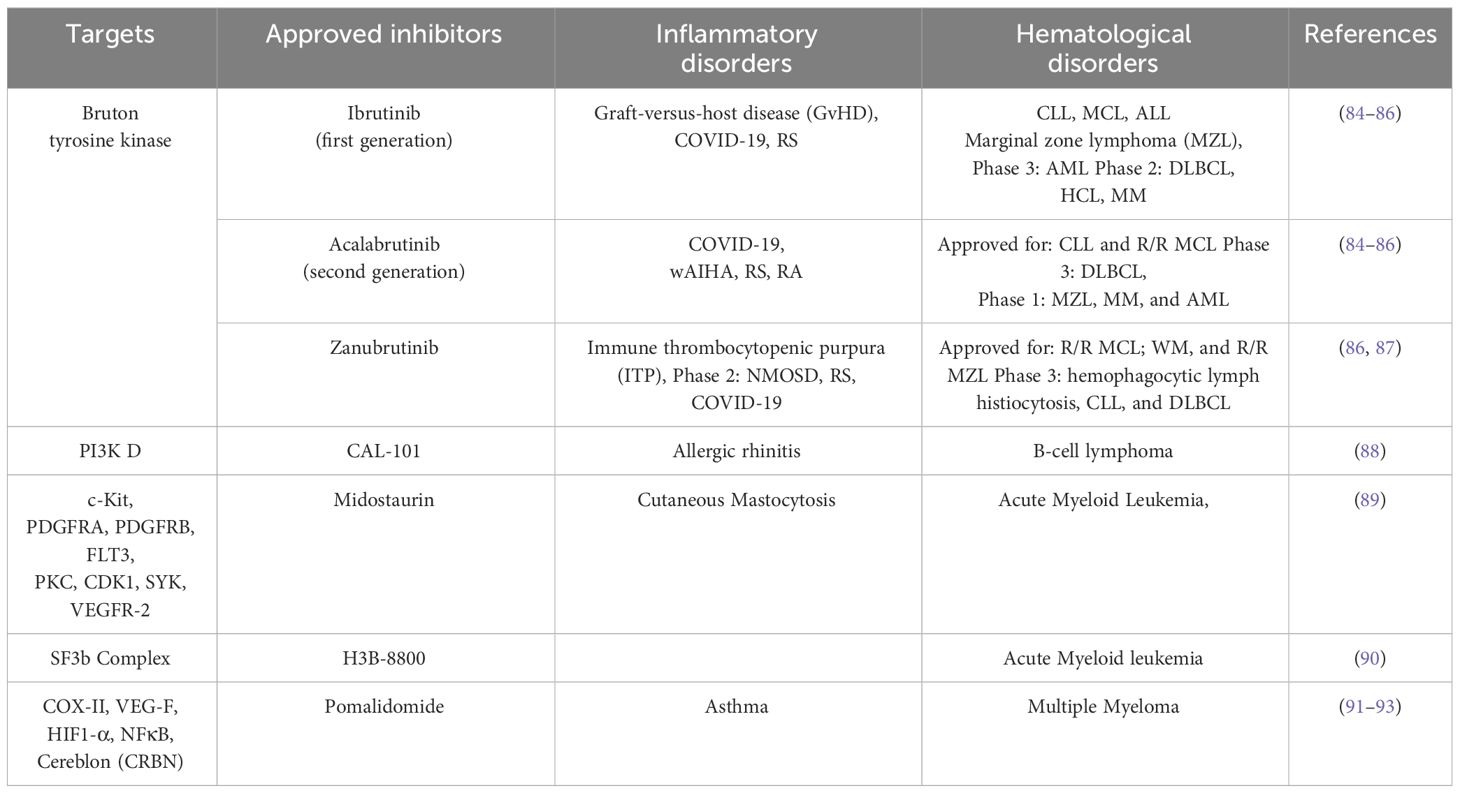
Table 1 Common targets and inhibitors in clinical trials/approved for targeting hematological and inflammatory disorders.
Myeloid malignancies result from genetic and epigenetic alterations in the myeloid progenitor cells involved in self-renewal and differentiation. Epigenetic modifications and changes in the microenvironment are known to be significant causes of these diseases. There are different types of myeloid malignancies, namely AML, Chronic Myeloid Leukemia (CML), MPNs, myelodysplastic syndrome (MDS) and chronic myelomonocytic leukemia (CMML).
AML is the most diverse hematological malignancy frequently seen in adults (94). The leukemic blasts produced by abnormal myeloid stem cells accumulate in the bone marrow, peripheral blood and other tissues, which reduces the population of normal blood cells and increases the risk of secondary infections (95, 96). Myeloproliferative neoplasms are caused by the dysfunction of multipotent hematopoietic stem cells and clonal myeloproliferation. In addition, genetic rearrangements like BCR-ABL and ETV6-RUNX1 and mutations in the tyrosine kinases like JAK kinases, Abelson (Abl) kinase, activated cdc42 (ACK) Kinases lead to abnormal proliferation (97, 98). Chronic Myeloid leukemia (CML) is usually associated with the BCR-ABL fusion gene (9;22 translocations), also known as the Philadelphia (Ph) chromosome.
Recent studies showed that alterations in the inactive HSCs and reduced interaction with BM niches could lead to their leukemic transformation and myeloid leukemia development (68, 99).
Many signaling pathways are involved in the regulation and development of normal HSCs. Proteins associated with Wnt signaling pathways help in maintaining the stemness of HSCs by regulating their quiescence and self-renewal properties (100). The regulation of HSCs' proliferation, self-renewal, and differentiation depend on various intermediates like cell cycle regulators, cyclin-dependent kinase inhibitors (CKIs), D-cyclins, p18/INK4, PTEN and many other transcription factors like HoxB4 and HoxA9 (101–103). Extrinsic regulatory pathways like Notch, TGF, Sonic Hedgehog, Smad, and Wnt also regulate HSCs' proliferation and self-renewal (103).
JunB is an essential protein responsible for regulating the proliferation and differentiation of long-term HSCs via TGF-β and Notch-Signaling pathways (101) and plays a vital role in maintaining HSCs. Its inactivation by epigenetic modifications has also been reported with the development of myeloid malignancies (104). The niche osteoblasts in CML secrete IL-1β and TNF-α pro-inflammatory cytokines, further enhancing myeloid cell proliferation and, thus, disease progression (30).
The STAT proteins are shown to be essential for myeloid differentiation. Chronic myeloid leukemia, commonly with BCR-ABL translocation, showed continuous expression of STAT3 and STAT5, leading to enhanced expression of BCL-XL, which is an anti-apoptotic BCL2 family protein (81, 105). Erythropoietin is also known to elicit phosphorylation and activate STAT5, which further helps in HSC differentiation (81, 106, 107). STAT5 inhibition led to reduced proliferation of the leukemic cells by enhancing apoptosis and inducing cell cycle arrest (108).
Tumor-associated macrophages (TAMs) are the resident macrophages in the tumor microenvironment, which promote tumorigenesis and angiogenesis, provide an immunosuppressive environment, and contribute to the poor prognosis of the disease (109). TAMs are functionally compromised. Phagocytosis inhibition happens due to the overexpression of a transmembrane protein, CD47, which interacts with the protein, signals regulatory protein alpha (SIRPα), and leads to inhibition of phagocytosis. This protein is highly expressed in LSCs, and its inhibition by anti-CD47 antibody led to an increase in phagocytosis by macrophages and increased the survival rate of myeloid mouse models (110, 111).
The cytokine TRAIL (tumor necrosis factor a-related apoptosis-inducing ligand) mediates apoptosis by caspase -8 mediated pathway. TRAIL can bind to four distinct receptors : TRAIL-R1 and TRAIL-R2 (also known as DR4 and DR5) are functional receptors which contain cytoplasmic death domains and can transduce cell death signals (112). In contrast, TRAIL-R3 and TRAIL-R4 (DcR1 and DcR2), are the truncated receptors and can block TRAIL induced apoptosis (112). It is also known that the binding of TRAIL with TRAIL-R4 leads to NFκB activation and further inflammation (113, 114). The expression of TRAIL-R1 and TRAIL-R2 is reported to be high in AML patients, and there has been evidence which shows high co-TRAIL-R3 expression linked to poor overall survival of the patients (111). TRAIL-R3 is a decoy protein which can be bypassed by targeting TRAIL-R1 and TRAIL-R2 by antibodies and thus can be used as a treatment for AML patients (112). Recombinant soluble TRAIL (rsTRAIL) has shown induction of apoptosis in cancer cell lines (115) including myeloid-leukemia cell lines (58). In addition, the activator of p53, Nutlin-3, and TRAIL enhances apoptosis in AML primary cells when wild-type p53 is present as it enhances apoptosis (115–117).
NFκB is highly expressed in AML patients and LSCs as compared to HSCs. IKKβ is a catalytic subunit of the IKβ complex, which activates the NFκB pathway. IKKβ deletion in the myeloid lineage using a LysM-Cre mouse model reduced tumor growth as well as proinflammatory cytokines without affecting apoptosis (118). IKKβ has been associated with inflammation and carcinogenesis as IKKβ activates factors like COX-2, MMP-9, MIP-2, and KC in myeloid cells, which are pro-inflammatory and linked to tumor development (118, 119).
The pro-survival protein myeloid cell leukemia (MCL-1) is an anti-apoptotic protein that regulates cell cycle progression and mitochondrial homeostasis. MCL-1 has been reported to be overexpressed in multiple myeloid malignancies like multiple myeloma and acute myeloid leukemia (120). Inflammation can contribute to overexpression of MCL-1 with inflammatory cytokines such as IL-6 and IL-8 enhancing MCL-1 transcription (121). Small molecules like AZD5991 which specifically inhibits MCL-1 showed a significant reduction in tumor growth in an OCI-AML3 mouse xenograft model. Further reduction in tumor growth was observed when the drug was administered with Venetoclax, a Bcl-2 inhibitor. Clinical trials for AZD5991 were also approved (120). Other MCL-1 inhibitors like AMG 176 (Amgen) S64315 (MIK66) are also under clinical trial for AML (120). Indisulam is a sulfonamide, targets several components of the cell cycle. It is known to target the G1 phase of the cell cycle and causes a blockade in the G1/S transition through the inhibition of the activation of both CDK2 and cyclin E (122). A phase 2 trial for Indisulam along with Idarubicin and Cytarabine was conducted which showed improved prognosis and increased survival rate in AML and high-risk MDS patients (123). E7820, another sulfonamide, is in phase II clinical trials for solid cancers (124). It acts as an inhibitor of Integrin α2 (ITGA2), which plays a key role in methotrexate-induced epithelial-mesenchymal transition (EMT) in alveolar epithelial cells (125). E7820 also selectively targets RNA splicing factor RBM39 for proteasomal degradation via DCAF15-E3-ubiquitin ligase. This action of E7820 has been observed to induce rapid loss of RBM39, accumulation of splicing errors, and growth inhibition in a DCAF15-dependent manner (125, 126). Interestingly, DCAF15 is found to be more highly expressed in Acute Myeloid Leukemia (AML) patient samples compared to normal hematopoietic progenitors. Therefore, the effects of E7820 in hematological malignancies (such as AML) are also being investigated (127).Targeting the inflammatory pathways that lead to MCL-1 overexpression may provide an alternative approach to inhibiting this anti-apoptotic protein in myeloid malignancies.
The phase I trials using anti-PD1 or anti-CTLA4 (ipilimumab) drugs as a monotherapy failed in both AML and MDS (128–130). Nivolumab, an immune checkpoint inhibitor in combination with azacitidine, a DNA methyltransferase inhibitor is under phase II trial (NCT02397720) for Refractory/Relapsed (R/R) and newly diagnosed AML patients (131).
Chimeric Antigen Receptor -T (CAR-T) cells against ligands which are expressed only on malignant cells is a new approach towards eliminating cancer. Overexpression of NKG2D ligands is seen in solid as well as hematological malignancies. However, the expression of NKG2D is seen to be absent/low in healthy tissues. CAR-T cells with a single infusion of human NKG2D were used in the phase I trial of AML, MDS and multiple myeloma patients which showed limited expansion and persistence of CAR-T cells (132).
It is known that BCR-ABL kinase leads to upregulation of activation-induced cytidine deaminase (AID) that leads to increased genetic instability. AID expression has been associated with blast crisis progression in CML and increases leukemogenesis in BCR-ABL+ B-ALL (133). It has been recently investigated that inflammation contributes enhanced expression AID through NFκB pathway and further increases malignancy in BCR-ABL+ B-ALL (134). BCR-ABL tyrosine kinase inhibitors like imatinib mesylate was the first drug approved for CML. Nilotinib (second generation inhibitor) and ponatinib (third generation inhibitor) are effective against BCR-ABL mutations like T315I, Y253H, and F317L (135–137). Omacetaxine is an inhibitor of protein translation which has been approved for CML therapy. It hinders the process of protein translation by blocking the initial elongation phase of protein synthesis. It interacts with the ribosomal A-site and impedes the precise arrangement of the side chains of amino acids in incoming aminoacyl-tRNAs. This drug degrades BCR-ABL proteins by inhibiting heat shock protein 70 in a dose dependent manner in imatinib resistant K562 cells (138). Many clinical trials with a combination of such therapies are being carried out in CML and AML patients (135).
RNA-binding proteins (RBPs) play a pivotal role in co and post transcriptional modifications. These RBPs are responsible for genetic alterations and diseases including cancers. TCGA data shows around 484 RNA-binding proteins which have been associated with myeloid malignancies. Out of these, approximately 50 percent are dysregulated in AML. There have been evidences which show dysregulation of RBPs associated with splicing in AML and hence is a potential therapeutic target. The clinical trials for H3B-8800 which targets SF3b splicing complex is one of the studies supporting development of therapeutic agents for myeloid malignancies (90, 127).
ALL is a group of malignancies of immature B or T cells that occurs predominantly in children. B-ALL constitutes 80-85%, while T-ALL accounts for 15% of pediatric to 25% of adult ALL cases (139).
B-cell acute lymphoblastic Leukemia is characterized by uncontrolled production of hematopoietic B-precursor cells. The chromosomal translocations that give rise to fusion proteins with oncogenic function and alteration in the role of B-lymphoid transcription factors such as Ikaros, E2A, EBF1 and PAX5 are known to be the causes of B-ALL. Pre-B-ALL cells have been shown to produce high levels of TNFα and IL-6, representing the inflammatory microenvironment's role in this disorder (140). The pro-inflammatory factors IL-1α, IL-1β, and TNFα were highly overproduced in supernatants derived from mononuclear cells of B-ALL patients when compared to their standard counterparts. Cytokines such as G-CSF, GM-CSF, IFNα, IL-12 and IL-7 were substantially elevated in B-ALL patients mediated by the activation of NFκB and STAT3 pathways (141). CCL2 and IL-8, chemokines that suppress normal hematopoiesis, are increased in the BM microenvironment and tend to promote the capacity of BM stromal cells to support the adhesion of ALL cells, indicating that elevated levels of CCL2 and IL8 could indirectly confer survival advantage to ALL cells (142).
Treatment of primary B-ALL patient samples with TRAIL (Apo2 ligand), an anti-cancer cytokine, showed modest apoptotic activity which was heterogeneous (143). However, TRAIL treatment of pre-B-ALL leukemia xenografts induced apoptosis in LICs and LSCs (144).
TRAIL-R1 monoclonal antibody (Mapatumumab) is in phase-II clinical trials for relapsed or refractory Non-Hodgkin's Lymphoma (NHL) as monotherapy and for multiple myeloma as combination therapy with Bortezomib. Dulanermin is recombinant TRAIL which triggers apoptosis via activation of DR4 and DR5 and is in phase III clinical trial for B-NHL patients who have progressed following rituximab therapy. Circularly permuted TRAIL (CPT) based combination therapy with Thalidomide is in phase III trials for R/R MM (145).
Furthermore, in B-ALL patients, increased peripheral levels of CXCL12 and high expression of CXCR4 on leukemic pre-B cells contribute to their proliferation, survival and homing to the BM microenvironment, which is mediated by STAT5, Rac-1 GTPase and a unique p38MAPK signaling pathway (67, 146–148). In childhood ALL, it is found that overexpression of the chemokine receptor CXCR4 on malignant acute leukemia cells is associated with extramedullary organ infiltration (149). In-vivo imaging studies of fluorescently labelled leukemic cells identified that homing of these cells to the bone marrow is dependent on the interaction of SDF-1 and its receptor CXCR4 (150). AMD3100 blocks CXCL12 binding and signaling through CXCR4 and is in phase I clinical trial for ALL (Table 2).
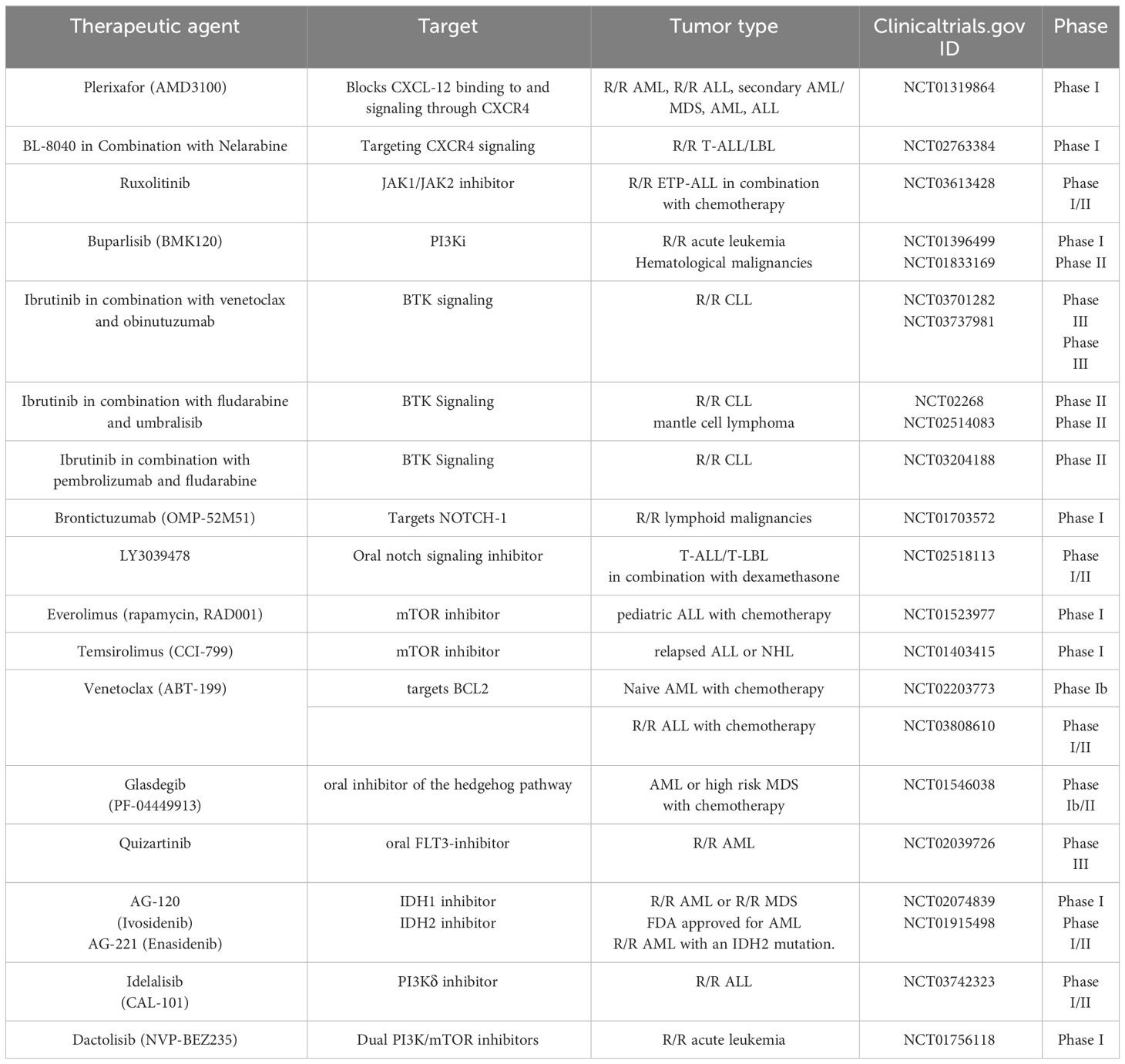
Table 2 Clinical trials investigating therapies for hematological malignancies based on targets involved in inflammation.
Mutation in IL7R on pre-B-cells is known to cause B-cell oncogenesis. In addition, several shreds of evidence indicate that the IL-7/IL-7R axis may promote lymphoid-related leukemogenesis and modulate leukemic cell responses to some antineoplastic therapies (151–154).
The ETV6-RUNX1 (TEL-AML1) fusion gene that results from t (12, 21) (p12; q21) translocation is the most frequent genetic aberration reported in childhood ALL and known to have a putative prenatal first lesion (155, 156). ETV6-RUNX1 fusion protein binds to a principal TGF-β signaling target, Smad3, and blocks the ability of TGF-β to suppress the proliferation of pre-pro-B cells, which leads to leukemogenesis (157). Activation of STAT3 in ETV6-RUNX1 positive ALL via RAC1 is responsible for the survival, proliferation, and self-renewal of leukemic cells by upregulating MYC gene (158).
Translocations in the mixed lineage leukemia (MLL) gene account for >50 fusions that may participate in transforming BM cells through the regulation of HOX genes. MLL translocations are predominantly seen in infant B-ALL (<1 year of age) and 15% of adult ALL patients (159–161). LAMP5 (a member of the lysosome-associated membrane protein (LAMP) family) is known to regulate type 1 interferon (IFN-1) and pro-inflammatory signaling downstream of TLR9 activation (162). In mixed lineage leukemia-rearranged (MLL-r) leukemia, downregulation of LAMP5 led to inhibition of NFκB signaling and increased activation of type-1 interferon signaling downstream of Toll-like receptor/interleukin 1 receptor activation in-vivo and in-vitro (163).
The expression of the BCR/ ABL1 fusion gene due to translocation t (9, 22) (q34; q11) causes 5% of pediatric and 25% of adult ALL cases (164). STAT5, the regulator of immune function is responsible for leukemic cell proliferation and survival, and its deletion results in cell cycle arrest followed by apoptosis of BCR-ABL1 positive malignant B cells (165). Patients with JAK1/2 mutations and patients with the BCR-ABL1 fusion have both been found to share similar gene expression profiles and are associated with a poor prognosis (166). IL-7R causes activation of STAT5 by activating JAK1 and JAK2; mutation in IL-Rα identified in 2-3% of B-ALL cases cause constitutive activation of JAK-STAT signaling. In addition, all B-ALL cases with JAK2 mutations overexpress CRLF2 (type I cytokine receptor subunit, also known as thymic stromal lymphopoietin receptor) (167, 168). CRLF2 directly interacts with the tyrosine kinase JAK2 and helps promote the proliferation of normal and leukemic B cells. E2A-PBX1, another fusion protein commonly seen in B-ALL expression of the WNT-16 gene, which ultimately promotes the aberrant proliferation and survival of B-lineage cells (169).
T-cell acute lymphoblastic leukemia is an aggressive blood cancer that comprises 10–15% of pediatric and ~25% of adult ALL cases, develops from the neoplastic transformation of T-cell precursors and their infiltration into BM and peripheral blood (PB) (170, 171). Aberrant Notch1 signaling plays a pivotal role in T-ALL leukemogenesis (170, 171). CNS infiltration risk is high in T-ALL patients and contributes to poor prognosis. A study on T-ALL patients and cell lines revealed that oncogenic Notch-1-induced chemokine CCR7 expression induced CNS infiltration and directional metastasis (172).
The role of IL-7 in the expansion and acceleration of leukemia progression has been revealed by engrafting T-ALL cell lines and primary T-ALL samples in immunocompromised mouse models after IL7 KO. It was shown that the IL-7/IL-7R axis causes activation of the PI3K/PKB/AKT signaling pathway resulting in downregulation of p27kip1 CDK inhibitors and upregulation of Bcl-2, promoting cell cycle progression and viability of T-ALL cells (173–175). Venetoclax (ABT-199) that targets higher BCL-2 expression is in phase-I clinical trials as monotherapy for R/R malignancies including T-ALL (NCT03236857) and in combination with Low-Intensity Chemotherapy and Venetoclax in phase I/II for R/R B or T-ALL (NCT03808610). Oligonucleotide microarray technology and pathway analysis in a study confirmed the pivotal role of IL-7 and CXCL12 in B and T-ALL (176). Activating mutations in the interleukin 7 receptor alpha chain (IL7R), Janus kinases, JAK1 or JAK3, or the Signal transducer and activator of transcription 5B (STAT5B) cause constitutive activation of JAK-STAT signaling observed in one-third of T-ALL patients (177–179).There are several ongoing clinical trials targeting the JAK/STAT pathway in T cell malignancies, which include NCT03613428, a phase I/II study combining ruxolitinib with the combination of vincristine, prednisone, and asparaginase in relapsed and refractory T-ALL (180). In T-ALL, soluble r-TRAIL failed to mediate apoptosis due to its low surface expression of death receptors DR4/DR5 in primary samples and cell lines (181).
PTEN-deficiency together with NRTK2 overexpression in T-ALL, caused activation of JAK/STAT3 and PI3K pathways, leading to aggressive disease, poor prognosis, and chemoresistance. The combined inhibition of phosphoinositide 3-kinase and STAT3 significantly suppressed the proliferation of PTEN-mutant T-ALL in culture and mouse xenografts (182). Also, array comparative genomic hybridization and sequence analysis from 44 pediatric DNA samples confirmed mutations in PI3K, PTEN or AKT ~48% T-ALL cases (183). PI3K, mTOR and PI3K/mTOR dual inhibitors including Buparlisib, Temsirolimus and Dactolisib are in clinical trials (Table 2).
CXCL12-binding receptor, commonly known as CXCR7 (CXC chemokine receptor 7), is highly expressed in T-ALL patient samples and cell lines and is responsible for T-ALL cell migration in response to CXCL12 induction (184). HTLV1-Tax (human T-cell leukemia virus, type-1 induced Tax) protein mediates HTLV1 viral-induced tumorigenesis in T-ALL by activating NFκB signaling (185). In addition, it was found that in tax-transformed cell line PX-1 is a T-ALL cell line which is transformed by the HTLV1-Tax protein. Inhibition of RelA (NFκB p65) using anti-sense oligonucleotides retarded the tumor growth of PX1 xenografts, suggest the importance of NFκB in HTLV1 associated tumors (186).
B-cell chronic lymphocytic leukemia (CLL) is characterized by aberrant accretion of mature clonal CD5+ B lymphocytes in the blood, bone marrow and lymphoid tissues. These differentiated B cells display characteristic immunophenotypes expressing CD23, CD19 and low surface membrane immunoglobulin levels. CLL is of two subtypes: unmutated-CLL which arises from a naive B cell that has encountered antigen but with insufficient stimulus to form a germinal center (GC), and IGHV mutated-CLL (M–CLL) which arises from a memory cell that, following antigen encounter, has undergone somatic hypermutation (187).
Pro-inflammatory cytokines and chemokines like IFN-γ, interleukin 6 (IL-6), IL-10, IL-8, and TNF-α are found to be significantly high in untreated CLL patients (188–190). IL-4 receptor levels are constitutively high in CLL cells (191), which stimulates the JAK/STAT pathway that protects CLL cells from chemotherapy-induced apoptosis (192). The serum of CLL patients was found to have high amounts of TNF superfamily member BAFF (B-cell activation factor of the TNF family). It is known to rescue B-CLL cells from apoptosis (193). In normal B-Cells, the binding of antigen causes signalosome activation by kinases that lead to the regulated activation of downstream NFκB, PI3K/AKT and MAP kinase pathways, which are necessary for B-cell proliferation and survival (194). In contrast, in CLL, stimulation of the BCR induces expansion of the malignant clone (187, 195). In CLL, NFκB is constitutively stimulated by various extrinsic and intrinsic stimuli, and NFκB is the critical regulator for survival and differentiation in B-cells. Antigens from the microenvironment and intra-BCR self-antigens trigger BCR signaling, leading to the recruitment of tyrosine kinases that phosphorylate the immunoreceptor tyrosine-based activation motifs (ITAMs) of Ig-α/Ig-β (196). This induces activation of Bruton's tyrosine kinase (BTK), phosphoinositide 3- kinase (PI3K), and Ras- dependent extracellular signal-regulated kinase (ERK) (197), which ultimately leads to the upregulation of NFκB which promotes CLL-B cell survival (198).
BCR signaling in CLL is heterogeneous. CLL cells from some patients do not respond to antigen engagement when IgM is used for BCR stimulation, whereas cells from other patients retain their signaling capacity (199). Unlike normal B cells that undergo apoptosis, unless they differentiate into plasma or memory cells, CLL cells represent constitutive BCR activation, which causes activation of NFκB and NFκB-regulated genes (200), induction of pro-survival signals, and production of pro-inflammatory cytokines.
CLL cells require stimulus from the microenvironment for their survival. Macrophage migration inhibitory factor (MIF), a pro-inflammatory cytokine, is overexpressed and supports tumor growth in CLL patients (201) by stimulating signaling pathways, such as MAPK, NFκB, and AKT, on binding to receptors CD74 and CXCR2/CXCR4 (202–204). In B-cells, activation of the AKT and NFκB pathways via MIF leads to the production of IL-8, leading to the up-regulation of BCL-2, which provides apoptotic resistance to blasts (205, 206). CLL disease is known for its clinical and prognostic heterogeneity, which is found to be associated with BCR encoding genes and RNA binding protein-zeta-associated protein of 70 kDa (ZAP70) (187, 195). Patients with BCR encoded by unmutated immunoglobulin variable heavy-chain genes (IGHV) (206, 207) along with ZAP70 expression (207–209) represent aggressive disease phenotype as compared to normal B cells or most CLL cases with mutated IgVH that lacks ZAP70 expression. ZAP-70 induction in CLL B cells causes activation of specific BCR-signaling molecules, including SYK, BLNK, ERK, JNK, PLCγ, and AKT kinases (210, 211), indicating ZAP-70 promotes the growth and survival of the tumor cells by stimulating BCR signaling. It is found that ZAP70 contributes to the more aggressive clinical behavior in CLL by enhancing BCR-mediated signaling through the NFκB pathway (212).
There are many drugs which target tyrosine kinases like Ibrutinib, acalabrutinib and Zanubrutinib which irreversibly inhibit Bruton’s Tyrosine kinase (BTK) by binding to the cysteine residue in its active site. BTK is a kinase that is involved in multiple signaling pathways and plays a role in B-cell and myeloid cell progression and survival and therefore becomes a therapeutic target for hematological malignancies. Ibrutinib was approved in a randomized clinical trial CLL patients including R/R CLL patients. In the phase 3 trials ibrutinib was administered in combination with obinutuzumab (anti-CD20 monoclonal antibody) which increased overall survival rate of CLL patients (213–215). Many clinical trials have taken place in using a triple combination (Ibrutinib, obinutuzumab and venetoclax) to treat high-risk CLL and R/R CLL (216–218). The cells from ibrutinib treated CLL patients showed increased expansion of CD-19 targeted CAR-T Cells (CTL019) and had also reduced the expression of PD-1 on T cells and CD200 on B cells (219). Randomized clinical trials are also taking place in CLL and SLL patients to check the efficacy of CAR-T cells targeting CD19 (autologous CART-19 cells) (NCT01747486).
Acalabrutinib has also been used in the treatment of CLL and SLL patients. It was approved as a monotherapy in 2019 even after limited efficacy (220). Phase III clinical trials are undergoing with triple combination (Acalabrutinib, obinutuzumab and venetoclax) in CLL and SLL patients (NCT03836261). It has been reported that long -term administration of acalabrutinib leads to ventricular arrhythmias and sudden deaths whereas the combined therapy decreases these incidences (221).
The effective activation and resolution of immune responses rely on the production and posttranscriptional regulation of mRNAs encoding inflammatory effector proteins. The association of RNA-binding proteins (RBPs) with mRNAs is essential in regulating their splicing, maturation, stability, and translation. In addition, several RBPs are reported to have a role in the modulation of the inflammatory response by controlling the expression of these inflammatory mRNAs and their decay.
RBPs mediate the regulation of inflammatory cytokine mRNAs like TNF-α, IL-10, and IL-6 by binding to their AU-rich elements near 3’-UTR and regulating mRNA stability, translation, and mRNA decay, thereby playing a role in inflammation-induced cancer development and progression (Figure 4).
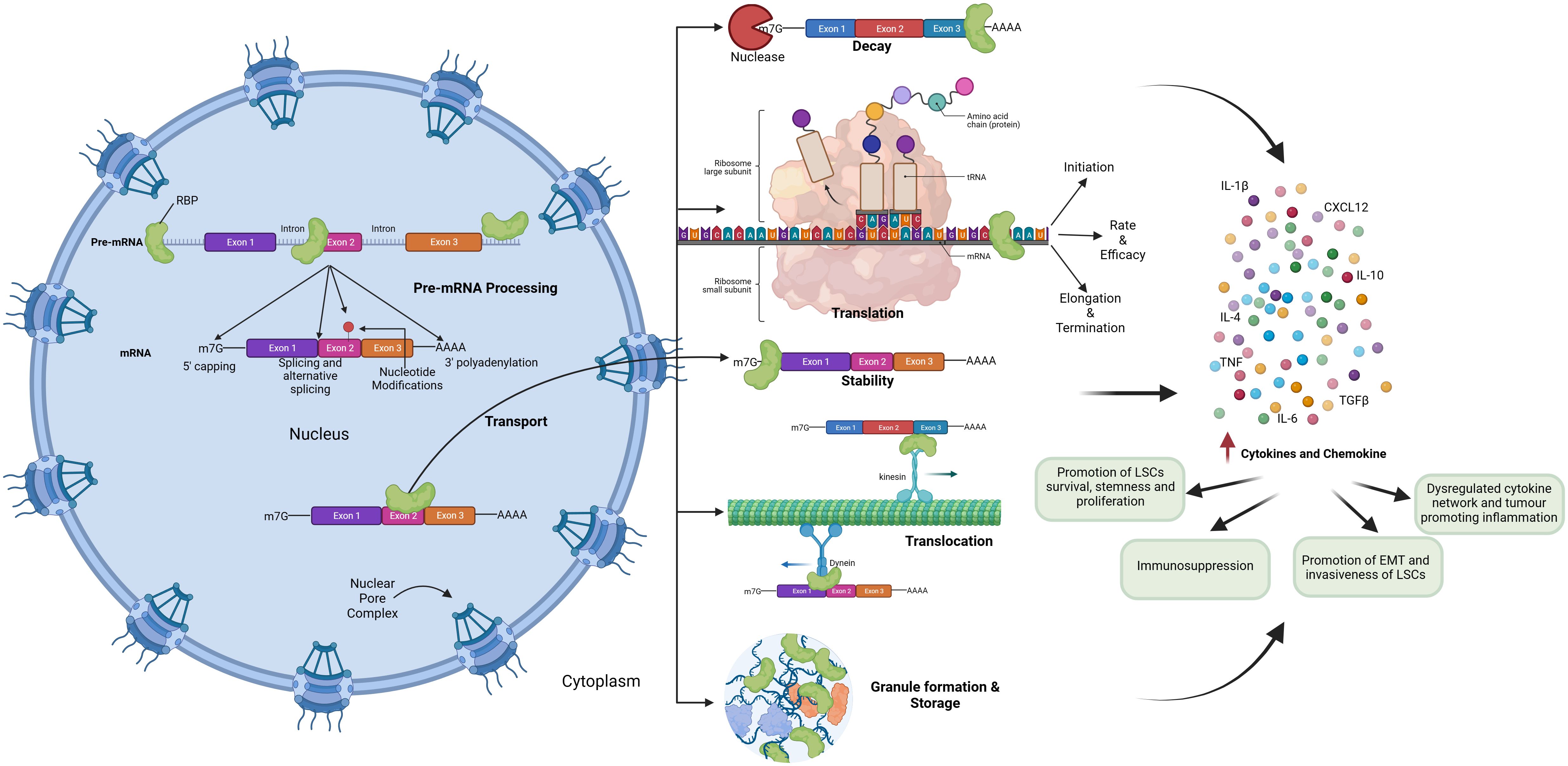
Figure 4 Role of RNA Binding Proteins in hematological malignancies by regulating inflammatory signaling.
Cytokine mRNAs such as TNF-α have shorter half-lives and undergo decay through RBPs mediated regulation of AU-rich elements (ARE) (222). The ZFP36 family of RBPs consists of three family members namely TTP, ZFNB6L1 and ZFNB6L2.
Tristetraprolin (TTP) encoded by the ZFP36 gene is a well-characterized member of the TTP family of RBPs, with its role specifically involved in the regulation of the immune system. These RBPs bind to the ARE in the 3’-UTR of target mRNAs and regulate the mRNA half-lives and decay (223, 224). The ZFP36 family plays a significant role in attenuating inflammatory responses by inhibiting the production of cytokines such as TNF-α, IL-6 and IL-10 in macrophages (225).
TTP is considered a major mRNA destabilizing protein involved in the regulation of immune cells. TTP inhibits mRNA translation and mediates decay by recruitment of mRNA destabilizing proteins such as CCR4-NOT deadenylation and decapping complexes to the TTP-bound target mRNAs. TTP functions to resolve inflammation by controlling the mRNA translation and decay of pro-inflammatory cytokines mRNAs such as TNF-α and NFκB- pathway-related signaling molecules like TNF, CSCL2, and CXCL3. Conversely, it also induces inflammation by binding to mRNAs of inflammatory inhibitors like IER3 and DUSP1, thereby mediating accurate regulation of LPS-induced inflammatory response and resolution of inflammation (226, 227).
ZFNB6L1 and ZFNB6L2 proteins act similarly to TTP and attenuate the expression of their target mRNAs. ZFP36L1 and ZFP36L2 play a crucial role in the development of B-lymphocytes by regulating quiescence. Quiescence is crucial for facilitating variable-diversity-joining (VDJ) recombination in developing B-cells and the ZFP36L1 and ZFP36L2 proteins are responsible for maintaining quiescence before the expression of the precursor B cell receptor (pre-BCR). ZFP36L1 and ZFP36L2 also restore quiescence following pre-BCR induced expansion by suppressing the expression of mRNAs such as Cyclin D3, Cyclin E2 (228, 229).
ZFNB6L1 and ZFNB6L2 regulate T-cell development by inhibiting the expression of Notch1. Double knockout of ZFNB6L1 and ZFNB6L2 in mice led to an abnormal increase in the NOTCH signaling pathway in double negative thymocytes and led to the development of T-ALL (230).
TTP primarily functions as a tumor suppressor gene in MYC-induced tumors. In a Myc-induced lymphoma model, TTP was found to be downregulated and restoration of TTP in these tumors led to the decay of mRNAs of Fst1, a pro-inflammatory cytokine and CCND1 thereby impairing the development and maintenance of lymphomas (231).
Human Antigen R, also known as ELAVL1, is one of the widely studied RBPs involved in tumorigenesis. HuR and ZFP36 share many 3’-UTR ARE binding target mRNAs. HuR, like the ZFP36 family of RBPs, binds to ARE in 3’-UTR of target mRNAs.
In contrast to the ZFP36 family, HuR increases the stability of its target mRNAs (164). HuR binds to pro-inflammatory cytokine mRNAs like COX-2, IL-2, IL-6, IL-8, IL-17, TNF-α, TGFβ and CXCL8 and increases their stability (232–236). HuR plays an indirect role in promoting Barrett’s esophagus associated carcinogenesis which is associated with chronic inflammation caused due to gastric acid reflux. HuR binds to iNOS mRNA through ARE elements at the 3’-UTR and stabilizes its mRNA thereby increasing the expression of iNOS. Inflammation induced over production of NO at the gastro-esophageal junction (GEJ) activates Caudal type homeobox (CDX2), a biomarker for Barrett’disease (237, 238).
Since HuR mainly functions to stabilize the target mRNAs, increased cytoplasmic expression of HuR has been found to be associated with various cancers such as oral, gastric, lung, breast, ovarian and renal cancers (239–244). Studies have shown that HuR expression clinically correlates with increased tumor size and higher tumor grade in breast cancer (245). Association between tumor stage and HuR expression was also seen in uterine cervical carcinoma along with non-small cell lung carcinoma (246, 247).
Apart from cancers, HuR also regulates B-cell and T-cell development in the immune system. Conditional knockout of HuR in mice models revealed that the population of pre-B-cells was reduced in bone marrow and follicular B-cells in the spleen and had significantly lower titers of serum immunoglobulins after knockout of HuR. HuR regulates splicing of mRNAs such as dihydrolipoamide S-succinyl transferase (DLST), a subunit of the 2-oxoglutarate dehydrogenase (α-KGDH) complex. Deletion of HuR led to disruption of mitochondrial metabolism and production of increased level of reactive oxygen species attributing to B-cell death (248). Thymocyte specific deletion of HuR in mice models showed that HuR is critical for T-cell development. Mice with deletion of HuR led to enlargement of thymus and loss of peripheral T- cells leading to lymphopenia (249).
HuR also has a role of polarizing macrophages to the M1 phenotype in the presence of LPS which is a systemic inflammatory stimulus. Interestingly, in a mouse model of LPS induced colitis and colorectal cancer, activated inflammatory tumor-activated macrophages from HuR-deficient mice showed increased expression of RNAs like TNF, TGF-β, IL10, Ccr2 and Ccl2. Overexpression of HuR in myeloid cells induced posttranscriptional silencing of these inflammatory cytokine mRNAs, thereby protecting mice from colon cancer development (177, 178). This demonstrates a heterogenous response of HuR to bound mRNA targets which may be tissue specific translational silencing Another myeloid specific HuR overexpression model also demonstrated a downregulation of TIA-1 and cytokines such as TNF, IL-1β, and TGFβ1 (250).
This gene is also known as CAPER/RNPC2. It plays an important role in pre-mRNA splicing and regulates steroid hormone receptor mediated transcription (251). Its higher expression is associated with several malignancies such as TNBC, non-small cell lung cancer, colorectal adenocarcinomas and AML (252). RBM39 also acts as the activator of NFκB through its interaction with transcriptional activation domain of v-rel protein. Deletion of RBM39 has been found to suppress the oncogenic activity of NFκB in lymphocytes (253), proliferation of breast cancer cells and abrogates phosphorylation of c-Jun (254). Its role has been established in multiple myeloma along the HIF1α/DARS-AS1/RBM39 axis that could be a useful target in multiple myeloma (255).
Insulin-like growth factor binding protein (IGF2BPs) are oncofetal proteins seen to be upregulated in various cancers, including different subtypes of B-ALL (256, 257). The IGF2BP family consists of three proteins that share sequence and functional homology, namely IGF2BP1, IGF2BP2, and IGF2BP3. These proteins are overexpressed during embryonic development, and re-expression is seen during the malignant transformation of cells. Overexpression of IGF2BP1 is seen in multiple epithelial tumors such as breast, pancreatic, and colon cancers (256). IGF2BP3 overexpression is also linked to numerous cervical, hepatocellular, breast and glial tumors (258). IGF2BP3 is overexpressed in the MLL translocated subtype of B- ALL, and IGF2BP1 is seen to be overexpressed in the ETV6-RUNX1 subtype of B-ALL (259, 260). This RBP family is known to influence the cytoplasmic fate of target mRNAs by regulating the translation, stabilization, location, and decay. IGF2BPs are also known to recruit mRNA stabilizers like ELAVL-1 (HuR) proteins (261).
IGF2BPs overexpression is well demonstrated in various epithelial cancers and leukemia, but the role of IGF2BPs in the induction of immune response has been recently elucidated.
One of the mechanisms of IGF2BPs mediated tumor progression is the regulation of tumor-associated inflammation. IGF2BP3 was found to bind and stabilize genes involved in the pro-inflammatory JAK/STAT signaling pathway and the ErbB signaling pathways in RS4;11 and Reh (B-ALL) cell lines. Overexpression of IGF2BP3 in the mouse bone marrow led to an expansion of progenitors belonging to all lineages (258, 262).
Similarly, IGF2BP1 targets were also identified in Reh cell line using RIP-seq and RNA-seq after IGF2BP1 knockout. The TNF-α induced NFκB pathway was one of the top targets of IGF2BP1 which was also reflected in the ETV6-RUNX1 positive B-ALL tumors (263). IGF2BP1 overexpression led to the stabilization of ubiquitin ligase receptor β-TrCP1 mRNA, which in turn caused activation of the NFκB pathway through enhanced degradation of IKBs (264). IGF2BP1 also regulates the glial cells' inflammatory responses by stabilizing the target mRNAs Gbp11 and Cp (265).
However, in mouse models of melanoma, it was observed that knockdown of IGF2BP1-3 led to an increase in the expression of pro-inflammatory interferon signaling genes like IFI44 and OAS1 reiterating the fact that RBP modulation of target mRNA half-lives is tissue or context specific (264). Similarly, in a colon cancer mouse model, it was observed that stromal expression of IGF2BP1 was critical for the inhibition of the growth of colon cancer. IGF2BP1 KO led to a global increase in pro-inflammatory cytokines and chemokines like IL-6, IL1β and MCP1 (261).
Mechanistically, target mRNA stabilization of IGF2BPs has been revealed to be dependent on an N-6-methyladenosine (m6A) RNA modification on mRNAs- IGF2BPs function as readers of m6A RNA modification present near 3’-UTRs and promote mRNA stability and translation of target mRNAs (266). IGF2BP2 functions as a regulator of macrophage phenotype. IGF2BP2 mediates the switch from M1 to M2 phenotype by binding to TSC1 and PPARα directly to regulate their expression in an m6A-dependent manner (267).
RBPs are essential for controlling post-transcriptional gene expression, which is involved in multiple aspects of RNA metabolism. RBPs play a significant role in enhancing the translation and stability of mRNAs, which in turn contributes to the development and spread of cancer. Multiple studies have revealed crucial small compounds to specifically target the interactions between RBPs and RNA. These inhibitors mainly target the RNA Binding domains of RBPs which are crucial for RBP-RNA interactions such as the RNA-recognition motif (RRM), hnRNP K homology (KH), and the zinc-finger domain (268–270). These molecules serve as valuable tools for the development of innovative therapies aimed at inhibiting the function of RBPs. The key inhibitors developed for targeting RBPs demonstrating the anticancer activity and potent promising therapeutics are summarized in Table 3.

Table 3 RBPs and their role in inflammation and hematological malignancies.
MS-444 was characterized as a small molecule by a competitive binding assay involving HuR and the ARE-RNA complex. This assay demonstrated that MS-444 inhibits the interaction between HuR and the AU rich element of the target mRNA (278). This inhibitor has been extensively studied as a potent inhibitor for in vitro and in vivo research linked to cancer, specifically in melanoma, glioma, and pancreatic carcinoma (279–281).
A novel inhibitor against HuR was discovered by confocal nano scanning screening approach, named H1N. It inhibits the adenosyl transferase activity at the 3'-terminal of the RRM3 motif in HuR, therefore blocking its contact with the target mRNA (282).
Studies have revealed that the bioactive flavonoid quercetin targets the binding of cytokine mRNAs such as TNFα and IL-6 identified through electrophoretic mobility shift assay (EMSA) (283, 284).
Another compound mitoxantrone was also screened and it prevented the formation of a stable complex between HuR and ARE of TNFα mRNA (285). Mesenchymal stem cells-based study revealed mitoxantrone also led to disruption of complex between HuR and SOX2 mRNA (286).
Using the same screening approach, DHTS (15, 16-dihydrotanshinone-I), another inhibitor of HuR was identified to target interaction with TNFα mRNA and exerted its effect in nanomolar range (287). Treatment of cells with DHTS exerted anti-cancer effects through decrease in cell growth and proliferation along with increase in cytotoxicity as seen in colon cancer cells and glioma cells (288, 289).
Fluorescence polarization assay was optimized for high throughput screening of identification of molecules that disrupts interaction of HuR to AREs of target mRNAs (290). This screening led to identification of six coumarin derivatives. CMLD-2 was the most potent HuR-ARE disruptor identified and showed anti tumor activity in breast, lung, colon and thyroid cancers (291–293).
Suramin an FD1-approved anti-trypanosomal drug, was shown to competitively bind to HuR to show anti-tumor effect in oral cancer cells (294). Trichostatin (TSA) and 5-Aza 2’deoxycytidine (AZA), known inhibitors of histone deacetylation and DNA methylation have shown to affect the nuclear-cytoplasmic translocation of HuR in order to modulate the estrogen receptor (ER) mRNA dependent on HuR leading to reduction of tumor burden in ER negative breast cancer cell lines (295) (Table 3).
Understanding the intricate interplay between inflammation, hematopoietic stem cells (HSCs), and the development of hematological malignancies is fundamental for advancing cancer immunology. This comprehension paves the way for targeted therapeutic interventions, emphasizing the importance of unravelling complexities within the tumor microenvironment.
Efforts to modulate inflammatory pathways, especially those involving NFκB and STAT3, hold promise for therapeutic advancements. Inhibiting these pathways may disrupt the pro-survival signals in cancer cells, potentially sensitizing them to conventional treatments. A strategy aimed at suppressing chronic inflammation, possibly through anti-inflammatory agents, could be explored to prevent the initiation and progression of hematological malignancies.
The advancements in precision medicine and genetic profiling make it possible to treat cancer in a more personalized manner. Identifying specific genetic alterations associated with the activation of inflammatory pathways in individual patients could lead to targeted therapies. For instance, patients with mutations in NFκB or STAT3 pathways might benefit from tailored interventions aimed at restoring normal signaling.
Immunotherapeutic strategies could be designed to harness the body’s immune system against leukemia stem cells (LSCs). Targeting surface markers such as CD44, potentially in combination with other treatments, may provide a selective approach to enhance the eradication of LSCs. Additionally, disrupting the leukemia stem cell niche, possibly through interference with CXCL12/CXCR4 signaling, could be explored to render LSCs more vulnerable to immune-mediated clearance. Understanding the dynamics of CD47-SIRPα interactions in the context of the tumor microenvironment may unveil novel strategies to overcome immunosuppression. The TRAIL pathway emerges as a potential target for inducing apoptosis in myeloid leukemia cells. Combining TRAIL-based therapies with other targeted agents, such as AKT inhibitors and p53 activators, may provide synergistic effects, leading to improved therapeutic outcomes.
Targeting the elevated pro-inflammatory cytokines (IL-1α, IL-1β, TNFα) in the microenvironment of B-ALL patients could also be explored as a therapeutic strategy. Modulating the cytokine milieu may disrupt the inflammatory support for leukemic cells and potentially enhance the efficacy of standard treatments.
Understanding the role of the tumor microenvironment in supporting malignant hematopoietic clones emphasizes the importance of modulating this niche. Innovative therapies could focus on remodeling the microenvironment to create an inhospitable terrain for LSCs while promoting the resurgence of normal HSCs. This might involve the manipulation of signaling molecules and cellular interactions within the niche.
The complexity of the interactions between inflammation, HSCs, and leukemia necessitates multidisciplinary collaboration. Integrating expertise from immunology, genetics, and oncology can facilitate a more holistic understanding of the disease mechanisms. Collaborative efforts could lead to the development of innovative treatment modalities that address both the malignant cells and their microenvironment.
The identification of specific RBPs, such as IGF2BPs, ZFP36 and HuR, as key players in modulating the inflammatory response present an opportunity for targeted therapeutic interventions. Developing drugs that selectively modulate the activity of these RBPs could offer a precise way to regulate the expression of inflammatory cytokines, potentially mitigating inflammation-associated cancer progression.
The role of insulin-like growth factor 2 binding proteins (IGF2BPs) in the regulation of tumor-associated inflammation has been elucidated in recent publications. Investigating the mechanistic aspects of how IGF2BPs influence the tumor microenvironment and immune response could reveal novel targets and methodologies for therapeutic intervention. Unravelling the specific pathways through which IGF2BPs modulate inflammation may offer new strategies for controlling cancer progression.
Investigating the potential crosstalk between RBPs and immune checkpoint molecules could provide insights into the regulation of immune responses in the tumor microenvironment. Understanding how RBPs influence the expression and function of immune checkpoint proteins, such as PD-1 and CTLA-4, may reveal additional layers of complexity in immune modulation within the context of cancer. Adopting systems biology approaches, including omics technologies, can help unravel the global impact of RBPs on the cancer transcriptome.
As research progresses, a deeper understanding of the heterogeneity in RBP expression across different cancers and individual patients may emerge. This knowledge could pave the way for personalized cancer therapies, tailoring treatment strategies based on the unique RBP profiles of patients, thus optimizing the efficacy of immunomodulatory interventions.
Translating these discoveries from the laboratory to clinical settings is crucial. Investigating the potential of RBPs as diagnostic or prognostic biomarkers could aid in stratifying patients based on their likelihood of developing inflammation-associated cancers.
SS: Conceptualization, Writing – review & editing, Writing – original draft. IB: Writing – original draft, Writing – review & editing. RB: Writing – original draft, Writing – review & editing. MB: Writing – original draft, Writing – review & editing. JP: Writing – review & editing, Conceptualization, Funding acquisition.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Wellcome Trust/DBT India Alliance Early Career Fellowship (IA/CPHE/15/1/502050) and a Science and Engineering Research Board (SERB) Core Research Grant (CRG/2021/004251) to JP. SS and IB are supported by ICMR Senior Research Fellowships. RB is supported by a DBT Junior Research Fellowship. MB is supported by a DBT Senior Research Fellowship.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Karin M, Clevers H. Reparative inflammation takes charge of tissue regeneration. Nature. (2016) 529:307–15. doi: 10.1038/nature17039
2. Qian S, Golubnitschaja O, Zhan X. Chronic inflammation: key player and biomarker-set to predict and prevent cancer development and progression based on individualized patient profiles. EPMA J. (2019) 10:365–81. doi: 10.1007/s13167-019-00194-x
3. Nøst TH, Alcala K, Urbarova I, Byrne KS, Guida F, Sandanger TM, et al. Systemic inflammation markers and cancer incidence in the UK Biobank. Eur J Epidemiol. (2021) 36:841–8. doi: 10.1007/s10654-021-00752-6
4. Van’T Klooster CC, Ridker PM, Hjortnaes J, van der Graaf Y, Asselbergs FW, Westerink J, et al. The relation between systemic inflammation and incident cancer in patients with stable cardiovascular disease: a cohort study. Eur Heart J. (2019) 40:3901–9. doi: 10.1093/eurheartj/ehz587
5. Shen YJ, Qian LQ, Ding ZP, Luo QQ, Zhao H, Xia WY, et al. Prognostic value of inflammatory biomarkers in patients with stage I lung adenocarcinoma treated with surgical dissection. Front Oncol. (2021) 11:711206. doi: 10.3389/fonc.2021.711206
6. Rieger MA, Hoppe PS, Smejkal BM, Eitelhuber AC, Schroeder T. Hematopoietic cytokines can instruct lineage choice. Science. (2009) 325:217–8. doi: 10.1126/science.1171461
7. Takizawa H, Manz MG. Impact of inflammation on early hematopoiesis and the microenvironment. Int J Hematol. (2017) 106:27–33. doi: 10.1007/s12185-017-2266-5
8. Naugler WE, Karin M. The wolf in sheep’s clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med. (2008) 14:109–19. doi: 10.1016/j.molmed.2007.12.007
9. Mantovani A, Savino B, Locati M, Zammataro L, Allavena P, Bonecchi R. The chemokine system in cancer biology and therapy. Cytokine Growth Factor Rev. (2010) 21:27–39. doi: 10.1016/j.cytogfr.2009.11.007
10. Balkwill FR, Mantovani A. Cancer-related inflammation: common themes and therapeutic opportunities. Semin Cancer Biol. (2012) 22:33–40. doi: 10.1016/j.semcancer.2011.12.005
11. Karin M. Nuclear factor-kappaB in cancer development and progression. Nature. (2006) 441:431–6. doi: 10.1038/nature04870
12. Kulbe H, Thompson R, Wilson JL, Robinson S, Hagemann T, Fatah R, et al. The inflammatory cytokine tumor necrosis factor-alpha generates an autocrine tumor-promoting network in epithelial ovarian cancer cells. Cancer Res. (2007) 67:585–92. doi: 10.1158/0008-5472.CAN-06-2941
13. Hermouet S, Bigot-Corbel E, Gardie B. Pathogenesis of myeloproliferative neoplasms: role and mechanisms of chronic inflammation. Mediators Inflamm. (2015) 2015:1–16. doi: 10.1155/2015/145293
14. Hibino S, Kawazoe T, Kasahara H, Itoh S, Ishimoto T, Sakata-Yanagimoto M, et al. Inflammation-induced tumorigenesis and metastasis. Int J Mol Sci. (2021) 22:5421. doi: 10.3390/ijms22115421
15. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
16. Pang WW, Price EA, Sahoo D, Beerman I, Maloney WJ, Rossi DJ, et al. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc Natl Acad Sci. (2011) 108:20012–7. doi: 10.1073/pnas.1116110108
17. Trumpp A, Essers M, Wilson A. Awakening dormant haematopoietic stem cells. Nat Rev Immunol. (2010) 10:201–9. doi: 10.1038/nri2726
18. Takizawa H, Boettcher S, Manz MG. Demand-adapted regulation of early hematopoiesis in infection and inflammation. Blood. (2012) 119:2991–3002. doi: 10.1182/blood-2011-12-380113
19. Boettcher S, Ziegler P, Schmid MA, Takizawa H, van Rooijen N, Kopf M, et al. Cutting edge: LPS-induced emergency myelopoiesis depends on TLR4-expressing nonhematopoietic cells. J Immunol. (2012) 188:5824–8. doi: 10.4049/jimmunol.1103253
20. Yamashita M, Dellorusso PV, Olson OC, Passegué E. Dysregulated haematopoietic stem cell behaviour in myeloid leukaemogenesis. Nat Rev Cancer. (2020) 20:365–82. doi: 10.1038/s41568-020-0260-3
21. Neubauer H, Cumano A, Müller M, Wu H, Huffstadt U, Pfeffer K. Jak2 deficiency defines an essentialDevelopmental checkpoint in definitiveHematopoiesis. Cell. (1998) 93:397–409. doi: 10.1016/S0092-8674(00)81168-X
22. Riether C, Schürch CM, Ochsenbein AF. Regulation of hematopoietic and leukemic stem cells by the immune system. Cell Death Differ. (2015) 22:187–98. doi: 10.1038/cdd.2014.89
23. Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nat Med. (2014) 20:833–46. doi: 10.1038/nm.3647
24. Wei Q, Frenette PS. Niches for hematopoietic stem cells and their progeny. Immunity. (2018) 48:632–48. doi: 10.1016/j.immuni.2018.03.024
25. Chavakis T, Mitroulis I, Hajishengallis G. Hematopoietic progenitor cells as integrative hubs for adaptation to and fine-tuning of inflammation. Nat Immunol. (2019) 20:802–11. doi: 10.1038/s41590-019-0402-5
26. Mitroulis I, Kalafati L, Hajishengallis G, Chavakis T. Myelopoiesis in the context of innate immunity. J Innate Immun. (2018) 10:365–72. doi: 10.1159/000489406
27. Mitroulis I, Kalafati L, Bornhäuser M, Hajishengallis G, Chavakis T. Regulation of the bone marrow niche by inflammation. Front Immunol. (2020) 11:1540. doi: 10.3389/fimmu.2020.01540
28. Méndez-, Bonnet D, Steensma DP, Hasserjian RP, Ghobrial IM, Gribben JG, et al. Bone marrow niches in haematological Malignancies. Nat Rev Cancer. (2020) 20:285–98. doi: 10.1038/s41568-020-0245-2
29. Kunisaki Y, Bruns I, Scheiermann C, Ahmed J, Pinho S, Zhang D, et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature. (2013) 502:637–43. doi: 10.1038/nature12612
30. Korn C, Méndez-Ferrer S. Myeloid Malignancies and the microenvironment. Blood. (2017) 129:811–22. doi: 10.1182/blood-2016-09-670224
31. Méndez-, Michurina TV, Ferraro F, Mazloom AR, MacArthur BD, Lira SA, et al. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature. (2010) 466:829–34. doi: 10.1038/nature09262
32. Sugiyama T, Kohara H, Noda M, Nagasawa T. Maintenance of the hematopoietic stem cell pool by CXCL12-CXCR4 chemokine signaling in bone marrow stromal cell niches. Immunity. (2006) 25:977–88. doi: 10.1016/j.immuni.2006.10.016
33. Arai F, Hirao A, Ohmura M, Sato H, Matsuoka S, Takubo K, et al. Tie2/angiopoietin-1 signaling regulates hematopoietic stem cell quiescence in the bone marrow niche. Cell. (2004) 118:149–61. doi: 10.1016/j.cell.2004.07.004
34. Calvi LM, Adams GB, Weibrecht KW, Weber JM, Olson DP, Knight MC, et al. Osteoblastic cells regulate the haematopoietic stem cell niche. Nature. (2003) 425:841–6. doi: 10.1038/nature02040
35. Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. (2005) 121:1109–21. doi: 10.1016/j.cell.2005.05.026
36. Kiel MJ, Radice GL, Morrison SJ. Lack of evidence that hematopoietic stem cells depend on N-cadherin-mediated adhesion to osteoblasts for their maintenance. Cell Stem Cell. (2007) 1:204–17. doi: 10.1016/j.stem.2007.06.001
37. Yamazaki S, Ema H, Karlsson G, Yamaguchi T, Miyoshi H, Shioda S, et al. Nonmyelinating schwann cells maintain hematopoietic stem cell hibernation in the bone marrow niche. Cell. (2011) 147:1146–58. doi: 10.1016/j.cell.2011.09.053
38. Zhao M, Perry JM, Marshall H, Venkatraman A, Qian P, He XC, et al. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat Med. (2014) 20:1321–6. doi: 10.1038/nm.3706
39. Nakamura-Ishizu A, Takubo K, Kobayashi H, Suzuki-Inoue K, Suda T. CLEC-2 in megakaryocytes is critical for maintenance of hematopoietic stem cells in the bone marrow. J Exp Med. (2015) 212:2133–46. doi: 10.1084/jem.20150057
40. Nakamura-Ishizu A, Takubo K, Fujioka M, Suda T. Megakaryocytes are essential for HSC quiescence through the production of thrombopoietin. Biochem Biophys Res Commun. (2014) 454:353–7. doi: 10.1016/j.bbrc.2014.10.095
41. Bruns I, Lucas D, Pinho S, Ahmed J, Lambert MP, Kunisaki Y, et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat Med. (2014) 20:1315–20. doi: 10.1038/nm.3707
42. Greenbaum A, Hsu YMS, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, et al. CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature. (2013) 495:227–30. doi: 10.1038/nature11926
43. Ding L, Morrison SJ. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature. (2013) 495:231–5. doi: 10.1038/nature11885
44. Kovtonyuk LV, Fritsch K, Feng X, Manz MG, Takizawa H. Inflamm-aging of hematopoiesis, hematopoietic stem cells, and the bone marrow microenvironment. Front Immunol. (2016) 7:502/full. doi: 10.3389/fimmu.2016.00502/full
45. Boettcher S, Gerosa RC, Radpour R, Bauer J, Ampenberger F, Heikenwalder M, et al. Endothelial cells translate pathogen signals into G-CSF-driven emergency granulopoiesis. Blood. (2014) 124(9):1393–403. doi: 10.1182/blood-2014-04-570762
46. Helbling PM, Piñeiro-Yáñez E, Gerosa R, Boettcher S, Al-Shahrour F, Manz MG, et al. Global transcriptomic profiling of the bone marrow stromal microenvironment during postnatal development, aging, and inflammation. Cell Rep. (2019) 29:3313–30. doi: 10.1016/j.celrep.2019.11.004
47. Boettcher S, Manz MG. Regulation of inflammation- and infection-driven hematopoiesis. Trends Immunol. (2017) 38:345–57. doi: 10.1016/j.it.2017.01.004
48. Semerad CL, Christopher MJ, Liu F, Short B, Simmons PJ, Winkler I, et al. G-CSF potently inhibits osteoblast activity and CXCL12 mRNA expression in the bone marrow. Blood. (2005) 106:3020–7. doi: 10.1182/blood-2004-01-0272
49. Tay J, Levesque JP, Winkler IG. Cellular players of hematopoietic stem cell mobilization in the bone marrow niche. Int J Hematol. (2017) 105:129–40. doi: 10.1007/s12185-016-2162-4
50. Winkler IG, Sims NA, Pettit AR, Barbier V, Nowlan B, Helwani F, et al. Bone marrow macrophages maintain hematopoietic stem cell (HSC) niches and their depletion mobilizes HSCs. Blood. (2010) 116:4815–28. doi: 10.1182/blood-2009-11-253534
51. Prasanna SJ, Gopalakrishnan D, Shankar SR, Vasandan AB. Pro-inflammatory cytokines, IFNγ and TNFα, influence immune properties of human bone marrow and wharton jelly mesenchymal stem cells differentially. PloS One. (2010) 5:e9016. doi: 10.1371/journal.pone.0009016
52. Pietras EM. Inflammation: a key regulator of hematopoietic stem cell fate in health and disease. Blood. (2017) 130:1693–8. doi: 10.1182/blood-2017-06-780882
53. Heazlewood SY, Neaves RJ, Williams B, Haylock DN, Adams TE, Nilsson SK. Megakaryocytes co-localise with hemopoietic stem cells and release cytokines that up-regulate stem cell proliferation. Stem Cell Res. (2013) 11:782–92. doi: 10.1016/j.scr.2013.05.007
54. Forte D, Krause DS, Andreeff M, Bonnet D, Méndez-Ferrer S. Updates on the hematologic tumor microenvironment and its therapeutic targeting. Haematologica. (2019) 104:1928–34. doi: 10.3324/haematol.2018.195396
55. Murati A, Brecqueville M, Devillier R, Mozziconacci MJ, Gelsi-Boyer V, Birnbaum D. Myeloid Malignancies: mutations, models and management. BMC Cancer. (2012) 12:304. doi: 10.1186/1471-2407-12-304
56. Niroula A, Sekar A, Murakami MA, Trinder M, Agrawal M, Wong WJ, et al. Distinction of lymphoid and myeloid clonal hematopoiesis. Nat Med. (2021) 27:1921–7. doi: 10.1038/s41591-021-01521-4
57. Huntly BJP, Gilliland DG. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer. (2005) 5:311–21. doi: 10.1038/nrc1592
58. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. (2001) 414:105–11. doi: 10.1038/35102167
59. Guzman ML, Allan JN. Concise review: leukemia stem cells in personalized medicine. Stem Cells. (2014) 32:844–51. doi: 10.1002/stem.1597
60. Shlush LI, Zandi S, Mitchell A, Chen WC, Brandwein JM, Gupta V, et al. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. (2014) 506:328–33. doi: 10.1038/nature13038
61. Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. (2015) 373:1136–52. doi: 10.1056/NEJMra1406184
62. The Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
63. Thiede C, Steudel C, Mohr B, Schaich M, Schäkel U, Platzbecker U, et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: association with FAB subtypes and identification of subgroups with poor prognosis. Blood. (2002) 99:4326–35. doi: 10.1182/blood.V99.12.4326
64. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE, et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. (2010) 363:2424–33. doi: 10.1056/NEJMoa1005143
65. Krawczyk J, O’Dwyer M, Swords R, Freeman C, Giles FJ. The role of inflammation in leukaemia. In: Aggarwal BB, Sung B, Gupta SC, editors. Inflammation and cancer. Springer Basel, Basel (2014). p. 335–60. doi: 10.1007/978-3-0348-0837-8_13
66. Spoo AC, Lübbert M, Wierda WG, Burger JA. CXCR4 is a prognostic marker in acute myelogenous leukemia. Blood. (2007) 109:786–91. doi: 10.1182/blood-2006-05-024844
67. Juarez JG, Thien M, Dela Pena A, Baraz R, Bradstock KF, Bendall LJ. CXCR4 mediates the homing of B cell progenitor acute lymphoblastic leukaemia cells to the bone marrow via activation of p38MAPK. Br J Haematol. (2009) 145:491–9. doi: 10.1111/j.1365-2141.2009.07648.x
68. Jin L, Hope KJ, Zhai Q, Smadja-Joffe F, Dick JE. Targeting of CD44 eradicates human acute myeloid leukemic stem cells. Nat Med. (2006) 12:1167–74. doi: 10.1038/nm1483
69. Krause DS, Lazarides K, von Andrian UH, Van Etten RA. Requirement for CD44 in homing and engraftment of BCR-ABL–expressing leukemic stem cells. Nat Med. (2006) 12:1175–80. doi: 10.1038/nm1489
70. Tabe Y, Shi YX, Zeng Z, Jin L, Shikami M, Hatanaka Y, et al. TGF-β-neutralizing antibody 1D11 enhances cytarabine-induced apoptosis in AML cells in the bone marrow microenvironment. PloS One. (2013) 8:e62785. doi: 10.1371/journal.pone.0062785
71. Naka K, Hoshii T, Muraguchi T, Tadokoro Y, Ooshio T, Kondo Y, et al. TGF-β–FOXO signalling maintains leukaemia-initiating cells in chronic myeloid leukaemia. Nature. (2010) 463:676–80. doi: 10.1038/nature08734
72. Mantovani A, Allavena P, Sica A, Balkwill F. Cancer-related inflammation. Nature. (2008) 454:436–44. doi: 10.1038/nature07205
73. Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. (2009) 30:1073–81. doi: 10.1093/carcin/bgp127
74. Aggarwal BB, Gehlot P. Inflammation and cancer: how friendly is the relationship for cancer patients? Curr Opin Pharmacol. (2009) 9:351–69. doi: 10.1016/j.coph.2009.06.020
75. Braun T, Carvalho G, Fabre C, Grosjean J, Fenaux P, Kroemer G. Targeting NF-jB in hematologic Malignancies. Cell Death Differ. (2006) 13:748–58. doi: 10.1038/sj.cdd.4401874
76. Fozza C, Crobu V, Isoni MA, Dore F. The immune landscape of myelodysplastic syndromes. Crit Rev Oncol Hematol. (2016) 107:90–9. doi: 10.1016/j.critrevonc.2016.08.016
77. Chen X, Li N, Weng J, Du X. Senescent mesenchymal stem cells in myelodysplastic syndrome: functional alterations, molecular mechanisms, and therapeutic strategies. Front Cell Dev Biol. (2021) 8:617466. doi: 10.3389/fcell.2020.617466
78. Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, et al. Persistently activated stat3 maintains constitutive NF-κB activity in tumors. Cancer Cell. (2009) 15:283–93. doi: 10.1016/j.ccr.2009.02.015
79. Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, et al. Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med. (2005) 11:1314–21. doi: 10.1038/nm1325
80. Yu H, Kortylewski M, Pardoll D. Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol. (2007) 7:41–51. doi: 10.1038/nri1995
81. Coffer PJ, Koenderman L, de Groot RP. The role of STATs in myeloid differentiation and leukemia. Oncogene. (2000) 19:2511–22. doi: 10.1038/sj.onc.1203479
82. Meisel M, Hinterleitner R, Pacis A, Chen L, Earley ZM, Mayassi T, et al. Microbial signals drive pre-leukaemic myeloproliferation in a Tet2-deficient host. Nature. (2018) 557:580–4. doi: 10.1038/s41586-018-0125-z
83. Duarte D, Hawkins ED, Akinduro O, Ang H, De Filippo K, Kong IY, et al. Inhibition of endosteal vascular niche remodeling rescues hematopoietic stem cell loss in AML. Cell Stem Cell. (2018) 22:64–77.e6. doi: 10.1016/j.stem.2017.11.006
84. Wen T, Wang J, Shi Y, Qian H, Liu P. Inhibitors targeting Bruton’s tyrosine kinase in cancers: drug development advances. Leukemia. (2021) 35:312–32. doi: 10.1038/s41375-020-01072-6
85. Ran F, Liu Y, Wang C, Xu Z, Zhang Y, Liu Y, et al. Review of the development of BTK inhibitors in overcoming the clinical limitations of ibrutinib. Eur J Med Chem. (2022) 229:114009. doi: 10.1016/j.ejmech.2021.114009
86. Alu A, Lei H, Han X, Wei Y, Wei X. BTK inhibitors in the treatment of hematological Malignancies and inflammatory diseases: mechanisms and clinical studies. J Hematol OncolJ Hematol Oncol. (2022) 15:138. doi: 10.1186/s13045-022-01353-w
88. Foster JG, Blunt MD, Carter E, Ward SG. Inhibition of PI3K signaling spurs new therapeutic opportunities in inflammatory/autoimmune diseases and hematological Malignancies. Pharmacol Rev. (2012) 64:1027–54. doi: 10.1124/pr.110.004051
89. Kim ES. Midostaurin: first global approval. Drugs. (2017) 77:1251–9. doi: 10.1007/s40265-017-0779-0
90. Seiler M, Yoshimi A, Darman R, Chan B, Keaney G, Thomas M, et al. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat Med. (2018) 24:497–504. doi: 10.1038/nm.4493
91. Lopez-Girona A, Mendy D, Ito T, Miller K, Gandhi AK, Kang J, et al. Cereblon is a direct protein target for immunomodulatory and antiproliferative activities of lenalidomide and pomalidomide. Leukemia. (2012) 26:2326–35. doi: 10.1038/leu.2012.119
92. Chanan-Khan AA, Swaika A, Paulus A, Kumar SK, Mikhael JR, Rajkumar SV, et al. Pomalidomide: the new immunomodulatory agent for the treatment of multiple myeloma. Blood Cancer J. (2013) 3:e143–3. doi: 10.1038/bcj.2013.38
93. Ghaderi M, Oryan S, Yousofvand N, Eidi A. Inhibition of airway contraction and inflammation by pomalidomide in a male wistar rat model of ovalbumin-induced asthma. Iran J Allergy Asthma Immunol. (2019) 18:209–17. doi: 10.18502/ijaai.v18i2.924
94. Sultan S, Zaheer HA, Irfan SM, Ashar S. Acute myeloid leukemia: clinical spectrum of 125 patients. Asian Pac J Cancer Prev. (2016) 17:369–72. doi: 10.7314/APJCP.2016.17.1.369
95. Vardiman JW, Thiele J, Arber DA, Brunning RD, Borowitz MJ, Porwit A, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. (2009) 114:937–51. doi: 10.1182/blood-2009-03-209262
96. Howard J. Myeloid Malignancies. (World Trade Center Health Program). (2014). Available at: https://www.cdc.gov/wtc/pdfs/policies/WTCHP_PP_MyeloidMalignancies_02012014-508.pdf.
97. Perricone M, Polverelli N, Martinelli G, Catani L, Ottaviani E, Zuffa E, et al. The relevance of a low JAK2 V617F allele burden in clinical practice: a monocentric study. Oncotarget. (2017) 8:37239–49. doi: 10.18632/oncotarget.v8i23
98. Siveen KS, Prabhu KS, Achkar IW, Kuttikrishnan S, Shyam S, Khan AQ, et al. Role of non receptor tyrosine kinases in hematological Malignances and its targeting by natural products. Mol Cancer. (2018) 17:31. doi: 10.1186/s12943-018-0788-y
99. Holyoake T, Jiang X, Eaves C, Eaves A. Isolation of a highly quiescent subpopulation of primitive leukemic cells in chronic myeloid leukemia. Blood. (1999) 94:2056–64. doi: 10.1182/blood.V94.6.2056
100. Fleming HE, Janzen V, Lo Celso C, Guo J, Leahy KM, Kronenberg HM, et al. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell. (2008) 2:274–83. doi: 10.1016/j.stem.2008.01.003
101. Santaguida M, Schepers K, King B, Sabnis AJ, Forsberg EC, Attema JL, et al. JunB Protects against Myeloid Malignancies by Limiting Hematopoietic Stem Cell Proliferation and Differentiation without Affecting Self-Renewal. Cancer Cell. (2009) 15:341–52. doi: 10.1016/j.ccr.2009.02.016
102. Akala OO, Clarke MF. Hematopoietic stem cell self-renewal. Curr Opin Genet Dev. (2006) 16:496–501. doi: 10.1016/j.gde.2006.08.011
103. Blank U, Karlsson G, Karlsson S. Signaling pathways governing stem-cell fate. Blood. (2008) 111:492–503. doi: 10.1182/blood-2007-07-075168
104. Yang MY, Liu TC, Chang JG, Lin PM, Lin SF. JunB gene expression is inactivated by methylation in chronic myeloid leukemia. Blood. (2003) 101:3205–11. doi: 10.1182/blood-2002-05-1598
105. Catlett-Falcone R, Landowski TH, Oshiro MM, Turkson J, Levitzki A, Savino R, et al. Constitutive activation of stat3 signaling confers resistance to apoptosis in human U266 myeloma cells. Immunity. (1999) 10:105–15. doi: 10.1016/S1074-7613(00)80011-4
106. Benekli M, Xia Z, Donohue KA, Ford LA, Pixley LA, Baer MR, et al. Constitutive activity of signal transducer and activator of transcription 3 protein in acute myeloid leukemia blasts is associated with short disease-free survival. Blood. (2002) 99:252–7. doi: 10.1182/blood.V99.1.252
107. Lin TS, Mahajan S, Frank DA. STAT signaling in the pathogenesis and treatment of leukemias. Oncogene. (2000) 19:2496–504. doi: 10.1038/sj.onc.1203486
108. Wingelhofer B, Maurer B, Heyes EC, Cumaraswamy AA, Berger-Becvar A, de Araujo ED, et al. Pharmacologic inhibition of STAT5 in acute myeloid leukemia. Leukemia. (2018) 32:1135–46. doi: 10.1038/s41375-017-0005-9
109. Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol OncolJ Hematol Oncol. (2019) 12:76. doi: 10.1186/s13045-019-0760-3
110. Jaiswal S, Jamieson CHM, Pang WW, Park CY, Chao MP, Majeti R, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. (2009) 138:271–85. doi: 10.1016/j.cell.2009.05.046
111. Majeti R, Chao MP, Alizadeh AA, Pang WW, Jaiswal S, Gibbs KD, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. (2009) 138:286–99. doi: 10.1016/j.cell.2009.05.045
112. Chamuleau MED, Ossenkoppele GJ, van Rhenen A, van Dreunen L, Jirka SMG, Zevenbergen A, et al. High TRAIL-R3 expression on leukemic blasts is associated with poor outcome and induces apoptosis-resistance which can be overcome by targeting TRAIL-R2. Leuk Res. (2011) 35:741–9. doi: 10.1016/j.leukres.2010.12.032
113. Kaufmann SH, Steensma DP. On the TRAIL of a new therapy for leukemia. Leukemia. (2005) 19:2195–202. doi: 10.1038/sj.leu.2403946
114. Zang DY, Goodwin RG, Loken MR, Bryant E, Deeg HJ. Expression of tumor necrosis factor–related apoptosis-inducing ligand, Apo2L, and its receptors in myelodysplastic syndrome: effects on in vitro hemopoiesis. Blood. (2001) 98:3058–65. doi: 10.1182/blood.V98.10.3058
115. Ashkenazi A. Targeting death and decoy receptors of the tumour-necrosis factor superfamily. Nat Rev Cancer. (2002) 2:420–30. doi: 10.1038/nrc821
116. Impicciatore G, Sancilio S, Miscia S, Di Pietro R. Nutlins and ionizing radiation in cancer therapy. Curr Pharm Des. (2010) 16:1427–42. doi: 10.2174/138161210791033932
117. Wen J, Ramadevi N, Nguyen D, Perkins C, Worthington E, Bhalla K. Antileukemic drugs increase death receptor 5 levels and enhance Apo-2L–induced apoptosis of human acute leukemia cells. Blood. (2000) 96:3900–6. doi: 10.1182/blood.V96.12.3900
118. Greten FR, Eckmann L, Greten TF, Park JM, Li ZW, Egan LJ, et al. IKKβ Links inflammation and tumorigenesis in a mouse model of colitis-associated cancer. Cell. (2004) 118:285–96. doi: 10.1016/j.cell.2004.07.013
119. Coussens LM, Werb Z. Inflammation and cancer. Nature. (2002) 420:860–7. doi: 10.1038/nature01322
120. Tron AE, Belmonte MA, Adam A, Aquila BM, Boise LH, Chiarparin E, et al. Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia. Nat Commun. (2018) 9:5341. doi: 10.1038/s41467-018-07551-w
121. Nijhuis A, Curciarello R, Mehta S, Feakins R, Bishop CL, Lindsay JO, et al. MCL-1 is modulated in Crohn’s disease fibrosis by miR-29b via IL-6 and IL-8. Cell Tissue Res. (2017) 368:325–35. doi: 10.1007/s00441-017-2576-1
122. Fukuoka K, Usuda J, Iwamoto Y, Fukumoto H, Nakamura T, Yoneda T, et al. Mechanisms of action of the novel sulfonamide anticancer agent E7070 on cell cycle progression in human non-small cell lung cancer cells. Invest New Drugs. (2001) 19:219–27. doi: 10.1023/A:1010608317361
123. Assi R, Kantarjian HM, Kadia TM, Pemmaraju N, Jabbour E, Jain N, et al. Final results of a phase 2, open-label study of indisulam, idarubicin, and cytarabine in patients with relapsed or refractory acute myeloid leukemia and high-risk myelodysplastic syndrome. Cancer. (2018) 124:2758–65. doi: 10.1002/cncr.31398
124. Milojkovic Kerklaan B, Slater S, Flynn M, Greystoke A, Witteveen PO, Megui-Roelvink M, et al. dose escalation, pharmacodynamic, pharmacokinetic, and food-effect study of α2 integrin inhibitor E7820 in patients with advanced solid tumors. Invest New Drugs. (2016) 34:329–37. doi: 10.1007/s10637-016-0344-9
125. Hülskamp MD, Kronenberg D, Stange R. The small-molecule protein ligand interface stabiliser E7820 induces differential cell line specific responses of integrin α2 expression. BMC Cancer. (2021) 21:571. doi: 10.1186/s12885-021-08301-w
126. Bewersdorf JP, Chandhok NS, Watts JM, Derkach A, Abdel-Wahab O, Stein EM, et al. E7820, an anti-cancer sulfonamide, in combination with venetoclax in patients with splicing factor mutant myeloid Malignancies: A phase II clinical trial. Blood. (2023) 142:1547–7. doi: 10.1182/blood-2023-182369
127. Wang E, Lu SX, Pastore A, Chen X, Imig J, Chun-Wei Lee S, et al. Targeting an RNA-binding protein network in acute myeloid leukemia. Cancer Cell. (2019) 35:369–384.e7. doi: 10.1016/j.ccell.2019.01.010
128. Berger R, Rotem-Yehudar R, Slama G, Landes S, Kneller A, Leiba M, et al. Phase I safety and pharmacokinetic study of CT-011, a humanized antibody interacting with PD-1, in patients with advanced hematologic Malignancies. Clin Cancer Res. (2008) 14:3044–51. doi: 10.1158/1078-0432.CCR-07-4079
129. Garcia-Manero G, Tallman MS, Martinelli G, Ribrag V, Yang H, Balakumaran A, et al. Pembrolizumab, a PD-1 inhibitor, in patients with myelodysplastic syndrome (MDS) after failure of hypomethylating agent treatment. Blood. (2016) 128:345–5. doi: 10.1182/blood.V128.22.345.345
130. Zeidan AM, Knaus HA, Robinson TM, Towlerton AMH, Warren EH, Zeidner JF, et al. A multi-center phase I trial of ipilimumab in patients with myelodysplastic syndromes following hypomethylating agent failure. Clin Cancer Res. (2018) 24:3519–27. doi: 10.1158/1078-0432.CCR-17-3763
131. Daver N, Garcia-Manero G, Basu S, Boddu PC, Alfayez M, Cortes JE, et al. Efficacy, safety, and biomarkers of response to azacitidine and nivolumab in relapsed/refractory acute myeloid leukemia: A nonrandomized, open-label, phase II study. Cancer Discov. (2019) 9:370–83. doi: 10.1158/2159-8290.CD-18-0774
132. Baumeister SH, Murad J, Werner L, Daley H, Trebeden-Negre H, Gicobi JK, et al. Phase I trial of autologous CAR-T cells targeting NKG2D ligands in patients with AML/MDS and multiple myeloma. Cancer Immunol Res. (2019) 7:100–12. doi: 10.1158/2326-6066.CIR-18-0307
133. Soverini S, Hochhaus A, Nicolini FE, Gruber F, Lange T, Saglio G, et al. BCR-ABL kinase domain mutation analysis in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors: recommendations from an expert panel on behalf of European LeukemiaNet. Blood. (2011) 118:1208–15. doi: 10.1182/blood-2010-12-326405
134. Zhang P, Qin M, Wang Y, Chen X, Miao Y, Yuan M, et al. Inflammation accelerates BCR-ABL1+ B-ALL development through upregulation of AID. Blood Adv. (2022) 6:4060–72. doi: 10.1182/bloodadvances.2021005017
135. Li S, Chen Y. Omacetaxine mepesuccinate in the treatment of intractable chronic myeloid leukemia. OncoTargets Ther. (2014) 7:177–86. doi: 10.2147/OTT.S41786
136. Weisberg E, Manley PW, Breitenstein W, Brüggen J, Cowan-Jacob SW, Ray A, et al. Characterization of AMN107, a selective inhibitor of native and mutant Bcr-Abl. Cancer Cell. (2005) 7:129–41. doi: 10.1016/j.ccr.2005.01.007
137. Frankfurt O, Licht JD. Ponatinib—A step forward in overcoming resistance in chronic myeloid leukemia. Clin Cancer Res. (2013) 19:5828–34. doi: 10.1158/1078-0432.CCR-13-0258
138. Chen Y, Hu Y, Michaels S, Segal D, Brown D, Li S. Inhibitory effects of omacetaxine on leukemic stem cells and BCR-ABL-induced chronic myeloid leukemia and acute lymphoblastic leukemia in mice. Leukemia. (2009) 23:1446–54. doi: 10.1038/leu.2009.52
139. Terwilliger T, Abdul-Hay M. Acute lymphoblastic leukemia: a comprehensive review and 2017 update. Blood Cancer J. (2017) 7:e577. doi: 10.1038/bcj.2017.53
140. Espinoza-Hernández L, Cruz-Rico J, Benítez-Aranda H, Martínez-Jaramillo G, Rodríguez-Zepeda MC, Vélez-Ruelas MA, et al. In vitro characterization of the hematopoietic system in pediatric patients with acute lymphoblastic leukemia. Leuk Res. (2001) 25:295–303. doi: 10.1016/S0145-2126(00)00141-7
141. Vilchis-Ordoñez A, Contreras-Quiroz A, Vadillo E, Dorantes-Acosta E, Reyes-López A, Quintela-Nuñez del Prado HM, et al. Bone marrow cells in acute lymphoblastic leukemia create a proinflammatory microenvironment influencing normal hematopoietic differentiation fates. BioMed Res Int. (2015) 2015:386165. doi: 10.1155/2015/386165
142. de Vasconcellos JF, Laranjeira ABA, Zanchin NIT, Otubo R, Vaz TH, Cardoso AA, et al. Increased CCL2 and IL-8 in the bone marrow microenvironment in acute lymphoblastic leukemia. Pediatr Blood Cancer. (2011) 56:568–77. doi: 10.1002/pbc.22941
143. Clodi K, Wimmer D, Li Y, Goodwin R, Jaeger U, Mann G, et al. Expression of tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptors and sensitivity to TRAIL-induced apoptosis in primary B-cell acute lymphoblastic leukaemia cells. Br J Haematol. (2000) 111:580–6. doi: 10.1111/j.1365-2141.2000.02404.x
144. Castro Alves C, Terziyska N, Grunert M, Gündisch S, Graubner U, Quintanilla-Martinez L, et al. Leukemia-initiating cells of patient-derived acute lymphoblastic leukemia xenografts are sensitive toward TRAIL. Blood. (2012) 119:4224–7. doi: 10.1182/blood-2011-08-370114
145. Snajdauf M, Havlova K, Vachtenheim J, Ozaniak A, Lischke R, Bartunkova J, et al. The TRAIL in the treatment of human cancer: an update on clinical trials. Front Mol Biosci. (2021) 8:628332. doi: 10.3389/fmolb.2021.628332
146. Juarez J, Dela Pena A, Baraz R, Hewson J, Khoo M, Cisterne A, et al. CXCR4 antagonists mobilize childhood acute lymphoblastic leukemia cells into the peripheral blood and inhibit engraftment. Leukemia. (2007) 21:1249–57. doi: 10.1038/sj.leu.2404684
147. Mowafi F, Cagigi A, Matskova L, Björk O, Chiodi F, Nilsson A. Chemokine CXCL12 enhances proliferation in pre-B-ALL via STAT5 activation. Pediatr Blood Cancer. (2008) 50:812–7. doi: 10.1002/pbc.21370
148. Freret M, Gouel F, Buquet C, Legrand E, Vannier JP, Vasse M, et al. Rac-1 GTPase controls the capacity of human leukaemic lymphoblasts to migrate on fibronectin in response to SDF-1α (CXCL12). Leuk Res. (2011) 35:971–3. doi: 10.1016/j.leukres.2011.03.011
149. Crazzolara R, Kreczy A, Mann G, Heitger A, Eibl G, Fink FM, et al. High expression of the chemokine receptor CXCR4 predicts extramedullary organ infiltration in childhood acute lymphoblastic leukaemia. Br J Haematol. (2001) 115:545–53. doi: 10.1046/j.1365-2141.2001.03164.x
150. Sipkins DA, Wei X, Wu JW, Runnels JM, Côté D, Means TK, et al. In vivo imaging of specialized bone marrow endothelial microdomains for tumour engraftment. Nature. (2005) 435:969–73. doi: 10.1038/nature03703
151. Sasson SC, Smith S, Seddiki N, Zaunders JJ, Bryant A, Koelsch KK, et al. IL-7 receptor is expressed on adult pre-B-cell acute lymphoblastic leukemia and other B-cell derived neoplasms and correlates with expression of proliferation and survival markers. Cytokine. (2010) 50:58–68. doi: 10.1016/j.cyto.2009.12.001
152. Shochat C, Tal N, Bandapalli OR, Palmi C, Ganmore I, te Kronnie G, et al. Gain-of-function mutations in interleukin-7 receptor-α (IL7R) in childhood acute lymphoblastic leukemias. J Exp Med. (2011) 208:901–8. doi: 10.1084/jem.20110580
153. Brown VI, Fang J, Alcorn K, Barr R, Kim JM, Wasserman R, et al. Rapamycin is active against B-precursor leukemia in vitro and in vivo, an effect that is modulated by IL-7-mediated signaling. Proc Natl Acad Sci USA. (2003) 100:15113–8. doi: 10.1073/pnas.2436348100
154. Williams RT, den Besten W, Sherr CJ. Cytokine-dependent imatinib resistance in mouse BCR-ABL+, Arf-null lymphoblastic leukemia. Genes Dev. (2007) 21:2283–7. doi: 10.1101/gad.1588607
155. Lilljebjörn H, Rissler M, Lassen C, Heldrup J, Behrendtz M, Mitelman F, et al. Whole-exome sequencing of pediatric acute lymphoblastic leukemia. Leukemia. (2012) 26:1602–7. doi: 10.1038/leu.2011.333
156. Zuna J, Madzo J, Krejci O, Zemanova Z, Kalinova M, Muzikova K, et al. ETV6/RUNX1 (TEL/AML1) is a frequent prenatal first hit in childhood leukemia. Blood. (2011) 117:368–9. doi: 10.1182/blood-2010-09-309070
157. Ford AM, Palmi C, Bueno C, Hong D, Cardus P, Knight D, et al. The TEL-AML1 leukemia fusion gene dysregulates the TGF-β pathway in early B lineage progenitor cells. J Clin Invest. (2009) 119:826–36. doi: 10.1172/JCI36428
158. Mangolini M, de Boer J, Walf-Vorderwülbecke V, Pieters R, den Boer ML, Williams O. STAT3 mediates oncogenic addiction to TEL-AML1 in t(12,21) acute lymphoblastic leukemia. Blood. (2013) 122:542–9. doi: 10.1182/blood-2012-11-465252
159. Meyer C, Burmeister T, Gröger D, Tsaur G, FeChina L, Renneville A, et al. The MLL recombinome of acute leukemias in 2017. Leukemia. (2018) 32:273–84. doi: 10.1038/leu.2017.213
160. for The St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project, Andersson AK, Ma J, Wang J, Chen X, Gedman AL, et al. The landscape of somatic mutations in infant MLL-rearranged acute lymphoblastic leukemias. Nat Genet. (2015) 47:330–7. doi: 10.1038/ng.3230
161. Mullighan CG. How advanced are we in targeting novel subtypes of ALL? Best Pract Res Clin Haematol. (2019) 32:101095. doi: 10.1016/j.beha.2019.101095
162. Combes A, Camosseto V, N’Guessan P, Argüello RJ, Mussard J, Caux C, et al. BAD-LAMP controls TLR9 trafficking and signalling in human plasmacytoid dendritic cells. Nat Commun. (2017) 8:913. doi: 10.1038/s41467-017-00695-1
163. Gracia-Maldonado G, Clark J, Burwinkel M, Greenslade B, Wunderlich M, Salomonis N, et al. LAMP-5 is an essential inflammatory-signaling regulator and novel immunotherapy target for mixed lineage leukemia-rearranged acute leukemia. Haematologica. (2022) 107:803–15. doi: 10.3324/haematol.2020.257451
164. Perez-Vera P, Reyes-Leon A, Fuentes-Panana EM. Signaling proteins and transcription factors in normal and Malignant early B cell development. Bone Marrow Res. (2011) 2011:502751. doi: 10.1155/2011/502751
165. Hoelbl A, Schuster C, Kovacic B, Zhu B, Wickre M, Hoelzl MA, et al. Stat5 is indispensable for the maintenance of bcr/abl-positive leukaemia. EMBO Mol Med. (2010) 2:98–110. doi: 10.1002/emmm.201000062
166. Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, et al. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. (2008) 453:110–4. doi: 10.1038/nature06866
167. Lodewijckx I, Cools J. Deregulation of the interleukin-7 signaling pathway in lymphoid Malignancies. Pharmaceuticals. (2021) 14:443. doi: 10.3390/ph14050443
168. Harvey RC, Mullighan CG, Wang X, Dobbin KK, Davidson GS, Bedrick EJ, et al. Identification of novel cluster groups in pediatric high-risk B-precursor acute lymphoblastic leukemia with gene expression profiling: correlation with genome-wide DNA copy number alterations, clinical characteristics, and outcome. Blood. (2010) 116:4874–84. doi: 10.1182/blood-2009-08-239681
169. McWhirter JR, Neuteboom STC, Wancewicz EV, Monia BP, Downing JR, Murre C. Oncogenic homeodomain transcription factor E2A-Pbx1 activates a novel WNT gene in pre-B acute lymphoblastoid leukemia. Proc Natl Acad Sci. (1999) 96:11464–9. doi: 10.1073/pnas.96.20.11464
170. Chiu PPL, Jiang H, Dick JE. Leukemia-initiating cells in human T-lymphoblastic leukemia exhibit glucocorticoid resistance. Blood. (2010) 116:5268–79. doi: 10.1182/blood-2010-06-292300
171. Nwabo Kamdje AH, Krampera M. Notch signaling in acute lymphoblastic leukemia: any role for stromal microenvironment? Blood. (2011) 118:6506–14. doi: 10.1182/blood-2011-08-376061
172. Buonamici S, Trimarchi T, Ruocco MG, Reavie L, Cathelin S, Mar BG, et al. CCR7 signalling as an essential regulator of CNS infiltration in T-cell leukaemia. Nature. (2009) 459:1000–4. doi: 10.1038/nature08020
173. Silva A, Laranjeira ABA, Martins LR, Cardoso BA, Demengeot J, Yunes JA, et al. IL-7 contributes to the progression of human T-cell acute lymphoblastic leukemias. Cancer Res. (2011) 71:4780–9. doi: 10.1158/0008-5472.CAN-10-3606
174. Barata JT, Silva A, Brandao JG, Nadler LM, Cardoso AA, Boussiotis VA. Activation of PI3K is indispensable for interleukin 7–mediated viability, proliferation, glucose use, and growth of T cell acute lymphoblastic leukemia cells. J Exp Med. (2004) 200:659–69. doi: 10.1084/jem.20040789
175. Barata JT, Cardoso AA, Nadler LM, Boussiotis VA. Interleukin-7 promotes survival and cell cycle progression of T-cell acute lymphoblastic leukemia cells by down-regulating the cyclin-dependent kinase inhibitor p27kip1. Blood. (2001) 98:1524–31. doi: 10.1182/blood.V98.5.1524
176. Tesfai Y, Ford J, Carter KW, Firth MJ, O’Leary RA, Gottardo NG, et al. Interactions between acute lymphoblastic leukemia and bone marrow stromal cells influence response to therapy. Leuk Res. (2012) 36:299–306. doi: 10.1016/j.leukres.2011.08.001
177. Vicente C, Schwab C, Broux M, Geerdens E, Degryse S, Demeyer S, et al. Targeted sequencing identifies associations between IL7R-JAK mutations and epigenetic modulators in T-cell acute lymphoblastic leukemia. Haematologica. (2015) 100:1301–10. doi: 10.3324/haematol.2015.130179
178. Vainchenker W, Constantinescu SN. JAK/STAT signaling in hematological Malignancies. Oncogene. (2013) 32:2601–13. doi: 10.1038/onc.2012.347
179. Liu C, Zhao L, Zhao J, Xu Q, Song Y, Wang H. Decreased ADAMTS-13 level is related to inflammation factors and risk stratification of acute lymphoblastic leukemia patients. Med (Baltimore). (2017) 96:e6136. doi: 10.1097/MD.0000000000006136
180. Shouse G, Nikolaenko L. Targeting the JAK/STAT pathway in T cell lymphoproliferative disorders. Curr Hematol Malig Rep. (2019) 14:570–6. doi: 10.1007/s11899-019-00545-5
181. Akahane K, Inukai T, Zhang X, Hirose K, Kuroda I, Goi K, et al. Resistance of T-cell acute lymphoblastic leukemia to tumor necrosis factor–related apoptosis-inducing ligand-mediated apoptosis. Exp Hematol. (2010) 38:885–95. doi: 10.1016/j.exphem.2010.06.014
182. Yuzugullu H, Von T, Thorpe LM, Walker SR, Roberts TM, Frank DA, et al. NTRK2 activation cooperates with PTEN deficiency in T-ALL through activation of both the PI3K–AKT and JAK–STAT3 pathways. Cell Discov. (2016) 2:1–13. doi: 10.1038/celldisc.2016.30
183. Gutierrez A, Sanda T, Grebliunaite R, Carracedo A, Salmena L, Ahn Y, et al. High frequency of PTEN, PI3K, and AKT abnormalities in T-cell acute lymphoblastic leukemia. Blood. (2009) 114:647–50. doi: 10.1182/blood-2009-02-206722
184. Melo R de CC, Longhini AL, Bigarella CL, Baratti MO, Traina F, Favaro P, et al. CXCR7 is highly expressed in acute lymphoblastic leukemia and potentiates CXCR4 response to CXCL12. PloS One. (2014) 9:e85926. doi: 10.1371/journal.pone.0085926
185. Hiscott J, Beauparlant P, Crepieux P, DeLuca C, Kwon H, Lin R, et al. Cellular and viral protein interactions regulating IκBα activity during human retrovirus infection. J Leukoc Biol. (1997) 62:82–92. doi: 10.1002/jlb.62.1.82
186. Kitajima I, Shinohara T, Bilakovics J, Brown DA, Xu X, Nerenberg M. Ablation of transplanted HTLV-I Tax-transformed tumors in mice by antisense inhibition of NF-kappa B. Science. (1992) 258:1792–5. doi: 10.1126/science.1299224
187. Chiorazzi N, Ferrarini M. B cell chronic lymphocytic leukemia: lessons learned from studies of the B cell antigen receptor. Annu Rev Immunol. (2003) 21:841–94. doi: 10.1146/annurev.immunol.21.120601.141018
188. Mahadevan D, Choi J, Cooke L, Simons B, Riley C, Klinkhammer T, et al. Gene expression and serum cytokine profiling of low stage CLL identify WNT/PCP, flt-3L/flt-3 and CXCL9/CXCR3 as regulators of cell proliferation, survival and migration. Hum Genomics Proteomics HGP. (2009) 2009:453634. doi: 10.4061/2009/453634
189. Fayad L, Keating MJ, Reuben JM, O’Brien S, Lee BN, Lerner S, et al. Interleukin-6 and interleukin-10 levels in chronic lymphocytic leukemia: correlation with phenotypic characteristics and outcome. Blood. (2001) 97:256–63. doi: 10.1182/blood.V97.1.256
190. Yoon JY, Lafarge S, Dawe D, Lakhi S, Kumar R, Morales C, et al. Association of interleukin-6 and interleukin-8 with poor prognosis in elderly patients with chronic lymphocytic leukemia. Leuk Lymphoma. (2012) 53:1735–42. doi: 10.3109/10428194.2012.666662
191. Burger JA, Burger M, Kipps TJ. Chronic lymphocytic leukemia B cells express functional CXCR4 chemokine receptors that mediate spontaneous migration beneath bone marrow stromal cells. Blood. (1999) 94:3658–67. doi: 10.1182/blood.V94.11.3658.423k11_3658_3667
192. Dietrich S, Krämer OH, Hahn E, Schäfer C, Giese T, Hess M, et al. Leflunomide induces apoptosis in fludarabine-resistant and clinically refractory CLL cells. Clin Cancer Res Off J Am Assoc Cancer Res. (2012) 18:417–31. doi: 10.1158/1078-0432.CCR-11-1049
193. Kern C, Cornuel JF, Billard C, Tang R, Rouillard D, Stenou V, et al. Involvement of BAFF and APRIL in the resistance to apoptosis of B-CLL through an autocrine pathway. Blood. (2004) 103:679–88. doi: 10.1182/blood-2003-02-0540
194. Efremov DG, Turkalj S, Laurenti L. Mechanisms of B cell receptor activation and responses to B cell receptor inhibitors in B cell Malignancies. Cancers. (2020) 12:1396. doi: 10.3390/cancers12061396
195. Stevenson FK, Caligaris-Cappio F. Chronic lymphocytic leukemia: revelations from the B-cell receptor. Blood. (2004) 103:4389–95. doi: 10.1182/blood-2003-12-4312
196. Suljagic M, Laurenti L, Tarnani M, Alam M, Malek SN, Efremov DG. Reduced expression of the tumor suppressor PHLPP1 enhances the antiapoptotic B-cell receptor signal in chronic lymphocytic leukemia B-cells. Leukemia. (2010) 24:2063–71. doi: 10.1038/leu.2010.201
197. Herman SEM, Gordon AL, Hertlein E, Ramanunni A, Zhang X, Jaglowski S, et al. Bruton tyrosine kinase represents a promising therapeutic target for treatment of chronic lymphocytic leukemia and is effectively targeted by PCI-32765. Blood. (2011) 117:6287–96. doi: 10.1182/blood-2011-01-328484
198. Zaninoni A, Imperiali FG, Pasquini C, Zanella A, Barcellini W. Cytokine modulation of nuclear factor-kappaB activity in B-chronic lymphocytic leukemia. Exp Hematol. (2003) 31:185–90. doi: 10.1016/S0301-472X(02)01046-9
199. Stevenson FK, Krysov S, Davies AJ, Steele AJ, Packham G. B-cell receptor signaling in chronic lymphocytic leukemia. Blood. (2011) 118:4313–20. doi: 10.1182/blood-2011-06-338855
200. Stadanlick JE, Kaileh M, Karnell FG, Scholz JL, Miller JP, Quinn Iii WJ, et al. Tonic B cell antigen receptor signals supply an NF-κB substrate for prosurvival BLyS signaling. Nat Immunol. (2008) 9:1379–87. doi: 10.1038/ni.1666
201. Reinart N, Nguyen PH, Boucas J, Rosen N, Kvasnicka HM, Heukamp L, et al. Delayed development of chronic lymphocytic leukemia in the absence of macrophage migration inhibitory factor. Blood. (2013) 121:812–21. doi: 10.1182/blood-2012-05-431452
202. Binsky I, Haran M, Starlets D, Gore Y, Lantner F, Harpaz N, et al. IL-8 secreted in a macrophage migration-inhibitory factor- and CD74-dependent manner regulates B cell chronic lymphocytic leukemia survival. Proc Natl Acad Sci. (2007) 104:13408–13. doi: 10.1073/pnas.0701553104
203. Bernhagen J, Krohn R, Lue H, Gregory JL, Zernecke A, Koenen RR, et al. MIF is a noncognate ligand of CXC chemokine receptors in inflammatory and atherogenic cell recruitment. Nat Med. (2007) 13:587–96. doi: 10.1038/nm1567
204. Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, et al. MIF signal transduction initiated by binding to CD74. J Exp Med. (2003) 197:1467–76. doi: 10.1084/jem.20030286
205. Gore Y, Starlets D, Maharshak N, Becker-Herman S, Kaneyuki U, Leng L, et al. Macrophage migration inhibitory factor induces B cell survival by activation of a CD74-CD44 receptor complex. J Biol Chem. (2008) 283:2784–92. doi: 10.1074/jbc.M703265200
206. Damle RN, Wasil T, Fais F, Ghiotto F, Valetto A, Allen SL, et al. Ig V Gene Mutation Status and CD38 Expression As Novel Prognostic Indicators in Chronic Lymphocytic Leukemia: Presented in part at the 40th Annual Meeting of The American Society of Hematology, held in Miami Beach, FL, December 4-8, 1998. Blood. (1999) 94:1840–7. doi: 10.1182/blood.V94.6.1840
207. Hamblin TJ, Davis Z, Gardiner A, Oscier DG, Stevenson FK. Unmutated ig VH genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood. (1999) 94:1848–54. doi: 10.1182/blood.V94.6.1848.418k05_1848_1854
208. Crespo M, Bosch F, Villamor N, Bellosillo B, Colomer D, Rozman M, et al. ZAP-70 expression as a surrogate for immunoglobulin-variable-region mutations in chronic lymphocytic leukemia. N Engl J Med. (2003) 348:1764–75. doi: 10.1056/NEJMoa023143
209. Wiestner A, Rosenwald A, Barry TS, Wright G, Davis RE, Henrickson SE, et al. ZAP-70 expression identifies a chronic lymphocytic leukemia subtype with unmutated immunoglobulin genes, inferior clinical outcome, and distinct gene expression profile. Blood. (2003) 101:4944–51. doi: 10.1182/blood-2002-10-3306
210. Chen L, Apgar J, Huynh L, Dicker F, Giago-McGahan T, Rassenti L, et al. ZAP-70 directly enhances IgM signaling in chronic lymphocytic leukemia. Blood. (2005) 105:2036–41. doi: 10.1182/blood-2004-05-1715
211. Gobessi S, Laurenti L, Longo PG, Sica S, Leone G, Efremov DG. ZAP-70 enhances B-cell–receptor signaling despite absent or inefficient tyrosine kinase activation in chronic lymphocytic leukemia and lymphoma B cells. Blood. (2006) 109:2032–9. doi: 10.1182/blood-2006-03-011759
212. Pede V, Rombout A, Vermeire J, Naessens E, Vanderstraeten H, Philippé J, et al. Expression of ZAP 70 in chronic lymphocytic leukaemia activates NF -κB signalling. Br J Haematol. (2013) 163:621–30. doi: 10.1111/bjh.12588
213. Eichhorst B, Robak T, Montserrat E, Ghia P, Niemann CU, Kater AP, et al. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann Oncol. (2021) 32:23–33. doi: 10.1016/j.annonc.2020.09.019
214. Rozkiewicz D, Hermanowicz JM, Kwiatkowska I, Krupa A, Pawlak D. Bruton’s tyrosine kinase inhibitors (BTKIs): review of preclinical studies and evaluation of clinical trials. Molecules. (2023) 28:2400. doi: 10.3390/molecules28052400
215. Sochacka-Ćwikła A, Mączyński M, Regiec A. FDA-approved drugs for hematological Malignancies—The last decade review. Cancers. (2021) 14:87. doi: 10.3390/cancers14010087
216. Rogers KA, Huang Y, Ruppert AS, Abruzzo LV, Andersen BL, Awan FT, et al. Phase II study of combination obinutuzumab, ibrutinib, and venetoclax in treatment-naïve and relapsed or refractory chronic lymphocytic leukemia. J Clin Oncol. (2020) 38:3626–37. doi: 10.1200/JCO.20.00491
217. Huber H, Edenhofer S, Von Tresckow J, Robrecht S, Zhang C, Tausch E, et al. Obinutuzumab (GA-101), ibrutinib, and venetoclax (GIVe) frontline treatment for high-risk chronic lymphocytic leukemia. Blood. (2022) 139:1318–29. doi: 10.1182/blood.2021013208
218. Rogers KA, Huang Y, Ruppert AS, Awan FT, Heerema NA, Hoffman C, et al. Phase 1b study of obinutuzumab, ibrutinib, and venetoclax in relapsed and refractory chronic lymphocytic leukemia. Blood. (2018) 132:1568–72. doi: 10.1182/blood-2018-05-853564
219. Fraietta JA, Beckwith KA, Patel PR, Ruella M, Zheng Z, Barrett DM, et al. Ibrutinib enhances chimeric antigen receptor T-cell engraftment and efficacy in leukemia. Blood. (2016) 127:1117–27. doi: 10.1182/blood-2015-11-679134
220. Eyre TA, Schuh A, Wierda WG, Brown JR, Ghia P, Pagel JM, et al. Acalabrutinib monotherapy for treatment of chronic lymphocytic leukaemia (ACE-CL-001): analysis of the Richter transformation cohort of an open-label, single-arm, phase 1–2 study. Lancet Haematol. (2021) 8:e912–21. doi: 10.1016/S2352-3026(21)00305-7
221. Bhat SA, Gambril J, Azali L, Chen ST, Rosen L, Palettas M, et al. Ventricular arrhythmias and sudden death events following acalabrutinib initiation. Blood. (2022) 140:2142–5. doi: 10.1182/blood.2022016953
222. Fu M, Blackshear PJ. RNA-binding proteins in immune regulation: a focus on CCCH zinc finger proteins. Nat Rev Immunol. (2017) 17:130–43. doi: 10.1038/nri.2016.129
223. Wang H, Ding N, Guo J, Xia J, Ruan Y. Dysregulation of TTP and HuR plays an important role in cancers. Tumor Biol. (2016) 37:14451–61. doi: 10.1007/s13277-016-5397-z
224. Zhao W, Liu M, D’Silva NJ, Kirkwood KL. Tristetraprolin Regulates Interleukin-6 Expression Through p38 MAPK-Dependent Affinity Changes with mRNA 3′ Untranslated Region. J Interferon Cytokine Res. (2011) 31:629–37. doi: 10.1089/jir.2010.0154
225. Tiedje C, Diaz-Muñoz MD, Trulley P, Ahlfors H, Laaß K, Blackshear PJ, et al. The RNA-binding protein TTP is a global post-transcriptional regulator of feedback control in inflammation. Nucleic Acids Res. (2016) 44(15):7418–40. doi: 10.1093/nar/gkw474
226. Ostareck DH, Ostareck-Lederer A. RNA-binding proteins in the control of LPS-induced macrophage response. Front Genet. (2019) 10:31. doi: 10.3389/fgene.2019.00031
227. Galloway A, Saveliev A, Łukasiak S, Hodson DJ, Bolland D, Balmanno K, et al. RNA-binding proteins ZFP36L1 and ZFP36L2 promote cell quiescence. Science. (2016) 352:453–9. doi: 10.1126/science.aad5978
228. Saveliev A, Bell SE, Turner M. Efficient homing of antibody-secreting cells to the bone marrow requires RNA-binding protein ZFP36L1. J Exp Med. (2021) 218:e20200504. doi: 10.1084/jem.20200504
229. Hodson DJ, Janas ML, Galloway A, Bell SE, Andrews S, Li CM, et al. Deletion of the RNA-binding proteins ZFP36L1 and ZFP36L2 leads to perturbed thymic development and T lymphoblastic leukemia. Nat Immunol. (2010) 11:717–24. doi: 10.1038/ni.1901
230. Rounbehler RJ, Fallahi M, Yang C, Steeves MA, Li W, Doherty JR, et al. Tristetraprolin impairs myc-induced lymphoma and abolishes the Malignant state. Cell. (2012) 150:563–74. doi: 10.1016/j.cell.2012.06.033
231. Dixon DA, Tolley ND, King PH, Nabors LB, McIntyre TM, Zimmerman GA, et al. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J Clin Invest. (2001) 108:1657–65. doi: 10.1172/JCI12973
232. Dan C, Jinjun B, Zi-Chun H, Lin M, Wei C, Xu Z, et al. Modulation of TNF -α mRNA stability by human antigen R and miR181s in sepsis-induced immunoparalysis. EMBO Mol Med. (2015) 7:140–57. doi: 10.15252/emmm.201404797
233. Shi JX, Su X, Xu J, Zhang WY, Shi Y. HuR post-transcriptionally regulates TNF-α-induced IL-6 expression in human pulmonary microvascular endothelial cells mainly via tristetraprolin. Respir Physiol Neurobiol. (2012) 181:154–61. doi: 10.1016/j.resp.2012.02.011
234. Abdelmohsen K, Gorospe M. Posttranscriptional regulation of cancer traits by HuR. Wiley Interdiscip Rev RNA. (2010) 1:214–29. doi: 10.1002/wrna.4
236. Yiakouvaki A, Dimitriou M, Karakasiliotis I, Eftychi C, Theocharis S, Kontoyiannis DL. Myeloid cell expression of the RNA-binding protein HuR protects mice from pathologic inflammation and colorectal carcinogenesis. J Clin Invest. (2012) 122:48–61. doi: 10.1172/JCI45021
237. Kusaka G. Role of nitric oxide in the pathogenesis of Barrett’s-associated carcinogenesis. World J Gastrointest Pathophysiol. (2016) 7:131. doi: 10.4291/wjgp.v7.i1.131
238. Linker K, Pautz A, Fechir M, Hubrich T, Greeve J, Kleinert H. Involvement of KSRP in the post-transcriptional regulation of human iNOS expression-complex interplay of KSRP with TTP and HuR. Nucleic Acids Res. (2005) 33:4813–27. doi: 10.1093/nar/gki797
239. Kim K, Li S, Cha J, Zhang X, Cha I. Significance of molecular markers in survival prediction of oral squamous cell carcinoma. Head Neck. (2012) 34:929–36. doi: 10.1002/hed.21856
240. Mrena J, Wiksten JP, Thiel A, Kokkola A, Pohjola L, Lundin J, et al. Cyclooxygenase-2 is an independent prognostic factor in gastric cancer and its expression is regulated by the messenger RNA stability factor huR. Clin Cancer Res. (2005) 11:7362–8. doi: 10.1158/1078-0432.CCR-05-0764
241. Wang J, Wang B, Bi J, Zhang C. Cytoplasmic HuR expression correlates with angiogenesis, lymphangiogenesis, and poor outcome in lung cancer. Med Oncol. (2011) 28:577–85. doi: 10.1007/s12032-010-9734-6
242. Denkert C, Weichert W, Winzer KJ, Müller BM, Noske A, Niesporek S, et al. Expression of the ELAV-like protein huR is associated with higher tumor grade and increased cyclooxygenase-2 expression in human breast carcinoma. Clin Cancer Res. (2004) 10:5580–6. doi: 10.1158/1078-0432.CCR-04-0070
243. Denkert C, Weichert W, Pest S, Koch I, Licht D, Köbel M, et al. Overexpression of the embryonic-lethal abnormal vision-like protein huR in ovarian carcinoma is a prognostic factor and is associated with increased cyclooxygenase 2 expression. Cancer Res. (2004) 64:189–95. doi: 10.1158/0008-5472.CAN-03-1987
244. Danilin S, Sourbier C, Thomas L, Lindner V, Rothhut S, Dormoy V, et al. Role of the RNA-binding protein HuR in human renal cell carcinoma. Carcinogenesis. (2010) 31:1018–26. doi: 10.1093/carcin/bgq052
245. Heinonen M, Bono P, Narko K, Chang SH, Lundin J, Joensuu H, et al. Cytoplasmic huR expression is a prognostic factor in invasive ductal breast carcinoma. Cancer Res. (2005) 65:2157–61. doi: 10.1158/0008-5472.CAN-04-3765
246. Wang J, Zhao W, Guo Y, Zhang B, Xie Q, Xiang D, et al. The expression of RNA-binding protein huR in non-small cell lung cancer correlates with vascular endothelial growth factor-C expression and lymph node metastasis. Oncology. (2009) 76:420–9. doi: 10.1159/000216837
247. Lim SJ, Kim HJ, Kim JY, Park K, Lee CM. Expression of huR is associated with increased cyclooxygenase-2 expression in uterine cervical carcinoma. Int J Gynecol Pathol. (2007) 26:229–34. doi: 10.1097/01.pgp.0000236946.82334.07
248. Diaz-Muñoz MD, Bell SE, Fairfax K, Monzon-Casanova E, Cunningham AF, Gonzalez-Porta M, et al. The RNA-binding protein HuR is essential for the B cell antibody response. Nat Immunol. (2015) 16:415–25. doi: 10.1038/ni.3115
249. Papadaki O, Milatos S, Grammenoudi S, Mukherjee N, Keene JD, Kontoyiannis DL. Control of thymic T cell maturation, deletion and egress by the RNA-binding protein huR. J Immunol. (2009) 182:6779–88. doi: 10.4049/jimmunol.0900377
250. Katsanou V, Papadaki O, Milatos S, Blackshear PJ, Anderson P, Kollias G, et al. HuR as a negative posttranscriptional modulator in inflammation. Mol Cell. (2005) 19:777–89. doi: 10.1016/j.molcel.2005.08.007
251. Dowhan DH, Hong EP, Auboeuf D, Dennis AP, Wilson MM, Berget SM, et al. Steroid hormone receptor coactivation and alternative RNA splicing by U2AF65-related proteins CAPERα and CAPERβ. Mol Cell. (2005) 17:429–39. doi: 10.1016/j.molcel.2004.12.025
252. Chilewski SD, Bhosale D, Dees S, Hutchinson I, Trimble R, Pontiggia L, et al. Development of CAPER peptides for the treatment of triple negative breast cancer. Cell Cycle. (2020) 19:432–47. doi: 10.1080/15384101.2020.1711579
253. Dutta J, Fan G, Gélinas C. CAPERα Is a novel rel-TAD-interacting factor that inhibits lymphocyte transformation by the potent rel/NF-κB oncoprotein v-rel. J Virol. (2008) 82:10792–802. doi: 10.1128/JVI.00903-08
254. Mercier I, Gonzales DM, Quann K, Pestell TG, Molchansky A, Sotgia F, et al. CAPER, a novel regulator of human breast cancer progression. Cell Cycle. (2014) 13:1256–64. doi: 10.4161/cc.28156
255. Tong J, Xu X, Zhang Z, Ma C, Xiang R, Liu J, et al. Hypoxia-induced long non-coding RNA DARS-AS1 regulates RBM39 stability to promote myeloma Malignancy. Haematologica. (2020) 105:1630–40. doi: 10.3324/haematol.2019.218289
256. Lederer M, Bley N, Schleifer C, Hüttelmaier S. The role of the oncofetal IGF2 mRNA-binding protein 3 (IGF2BP3) in cancer. Semin Cancer Biol. (2014) 29:3–12. doi: 10.1016/j.semcancer.2014.07.006
257. Bell JL, Wachter K, Muhleck B, Pazaitis N, Kohn M, Lederer M, et al. Insulin-like growth factor 2 mRNA-binding proteins (IGF2BPs): post-transcriptional drivers of cancer progression? Cell Mol Life Sci. (2013) 70:2657–75. doi: 10.1007/s00018-012-1186-z
258. Palanichamy JK, Tran TM, Howard JM, Contreras JR, Fernando TR, Sterne-Weiler T, et al. RNA-binding protein IGF2BP3 targeting of oncogenic transcripts promotes hematopoietic progenitor proliferation. J Clin Invest. (2016) 126:1495–511. doi: 10.1172/JCI80046
259. Sharma G, Boby E, Nidhi T, Jain A, Singh J, Singh A, et al. Diagnostic utility of IGF2BP1 and its targets as potential biomarkers in ETV6-RUNX1 positive B-cell acute lymphoblastic leukemia. Front Oncol. (2021) 11:588101. doi: 10.3389/fonc.2021.588101
260. Huang X, Zhang H, Guo X, Zhu Z, Cai H, Kong X. Insulin-like growth factor 2 mRNA-binding protein 1 (IGF2BP1) in cancer. J Hematol OncolJ Hematol Oncol. (2018) 11:88. doi: 10.1186/s13045-018-0628-y
261. Hamilton KE, Chatterji P, Lundsmith ET, Andres SF, Giroux V, Hicks PD, et al. Loss of stromal IMP1 promotes a tumorigenic microenvironment in the colon. Mol Cancer Res. (2015) 13:1478–86. doi: 10.1158/1541-7786.MCR-15-0224
262. Tran TM, Rao DS. RNA binding proteins in MLL-rearranged leukemia. Exp Hematol Oncol. (2022) 11:80. doi: 10.1186/s40164-022-00343-5
263. Sharma G, Tran TM, Bansal I, Beg MS, Bhardwaj R, Bassi J, et al. RNA binding protein IGF2BP1 synergizes with ETV6-RUNX1 to drive oncogenic signaling in B-cell Acute Lymphoblastic Leukemia. J Exp Clin Cancer Res. (2023) 42:231. doi: 10.1186/s13046-023-02810-1
264. Elcheva IA, Wood T, Chiarolanzio K, Chim B, Wong M, Singh V, et al. RNA-binding protein IGF2BP1 maintains leukemia stem cell properties by regulating HOXB4, MYB, and ALDH1A1. Leukemia. (2020) 34:1354–63. doi: 10.1038/s41375-019-0656-9
265. Ding L, Wu H, Wang Y, Li Y, Liang Z, Xia X, et al. m6A reader igf2bp1 regulates the inflammatory responses of microglia by stabilizing gbp11 and cp mRNAs. Front Immunol. (2022) 13:872252. doi: 10.3389/fimmu.2022.872252
266. Huang H, Weng H, Sun W, Qin X, Shi H, Wu H, et al. Recognition of RNA N6-methyladenosine by IGF2BP proteins enhances mRNA stability and translation. Nat Cell Biol. (2018) 20:285–95. doi: 10.1038/s41556-018-0045-z
267. Wang X, Ji Y, Feng P, Liu R, Li G, Zheng J, et al. The m6A reader IGF2BP2 regulates macrophage phenotypic activation and inflammatory diseases by stabilizing TSC1 and PPAR γ. Adv Sci. (2021) 8:2100209. doi: 10.1002/advs.202100209
268. Wu X, Xu L. The RNA-binding protein HuR in human cancer: A friend or foe? Adv Drug Deliv Rev. (2022) 184:114179. doi: 10.1016/j.addr.2022.114179
269. Majumder M, Chakraborty P, Mohan S, Mehrotra S, Palanisamy V. HuR as a molecular target for cancer therapeutics and immune-related disorders. Adv Drug Deliv Rev. (2022) 188:114442. doi: 10.1016/j.addr.2022.114442
270. Zhu TY, Hong LL, Ling ZQ. Oncofetal protein IGF2BPs in human cancer: functions, mechanisms and therapeutic potential. biomark Res. (2023) 11:62. doi: 10.1186/s40364-023-00499-0
271. Makita S, Takatori H, Nakajima H. Post-transcriptional regulation of immune responses and inflammatory diseases by RNA-binding ZFP36 family proteins. Front Immunol. (2021) 12:711633. doi: 10.3389/fimmu.2021.711633
272. Moore MJ, Blachere NE, Fak JJ, Park CY, Sawicka K, Parveen S, et al. ZFP36 RNA-binding proteins restrain T cell activation and anti-viral immunity. eLife. (2018) 7:e33057. doi: 10.7554/eLife.33057
273. Wang J, Guo Y, Chu H, Guan Y, Bi J, Wang B. Multiple functions of the RNA-binding protein huR in cancer progression, treatment responses and prognosis. Int J Mol Sci. (2013) 14:10015–41. doi: 10.3390/ijms140510015
274. Liu Y, Shen S, Yan Z, Yan L, Ding H, Wang A, et al. Expression characteristics and their functional role of IGFBP gene family in pan-cancer. BMC Cancer. (2023) 23:371. doi: 10.1186/s12885-023-10832-3
275. Du QY, Zhu ZM, Pei DS. The biological function of IGF2BPs and their role in tumorigenesis. Invest New Drugs. (2021) 39(6):1682–93. doi: 10.1007/s10637-021-01148-9
276. Xu C, Chen X, Zhang X, Zhao D, Dou Z, Xie X, et al. RNA-binding protein 39: a promising therapeutic target for cancer. Cell Death Discov. (2021) 7:214. doi: 10.1038/s41420-021-00598-7
277. Aguilar-Garrido P, Otero-Sobrino Á, Navarro-Aguadero MÁ, Velasco-Estévez M, Gallardo M. The role of RNA-binding proteins in hematological Malignancies. Int J Mol Sci. (2022) 23:9552. doi: 10.3390/ijms23179552
278. Meisner NC, Hintersteiner M, Mueller K, Bauer R, Seifert JM, Naegeli HU, et al. Identification and mechanistic characterization of low-molecular-weight inhibitors for HuR. Nat Chem Biol. (2007) 3:508–15. doi: 10.1038/nchembio.2007.14
279. Moradi F, Berglund P, Linnskog R, Leandersson K, Andersson T, Prasad CP. Dual mechanisms of action of the RNA-binding protein human antigen R explains its regulatory effect on melanoma cell migration. Transl Res. (2016) 172:45–60. doi: 10.1016/j.trsl.2016.02.007
280. Romeo C, Weber MC, Zarei M, DeCicco D, Chand SN, Lobo AD, et al. HuR contributes to TRAIL resistance by restricting death receptor 4 expression in pancreatic cancer cells. Mol Cancer Res. (2016) 14:599–611. doi: 10.1158/1541-7786.MCR-15-0448
281. Wang J, Hjelmeland AB, Nabors LB, King PH. Anti-cancer effects of the HuR inhibitor, MS-444, in Malignant glioma cells. Cancer Biol Ther. (2019) 20:979–88. doi: 10.1080/15384047.2019.1591673
282. Meisner NC, Hintersteiner M, Seifert JM, Bauer R, Benoit RM, Widmer A, et al. Terminal adenosyl transferase activity of posttranscriptional regulator huR revealed by confocal on-bead screening. J Mol Biol. (2009) 386:435–50. doi: 10.1016/j.jmb.2008.12.020
283. Ouhara K, Munenaga S, Kajiya M, Takeda K, Matsuda S, Sato Y, et al. The induced RNA-binding protein, HuR, targets 3′-UTR region of IL-6 mRNA and enhances its stabilization in periodontitis. Clin Exp Immunol. (2018) 192:325–36. doi: 10.1111/cei.13110
284. Chae MJ, Sung HY, Kim EH, Lee M, Kwak H, Chae CH, et al. Chemical inhibitors destabilize HuR binding to the AU-rich element of TNF-α mRNA. Exp Mol Med. (2009) 41:824. doi: 10.3858/emm.2009.41.11.088
285. D’Agostino VG, Adami V, Provenzani A. A novel high throughput biochemical assay to evaluate the huR protein-RNA complex formation. PloS One. (2013) 8:e72426. doi: 10.1371/journal.pone.0072426
286. Latorre E, Carelli S, Caremoli F, Giallongo T, Colli M, Canazza A, et al. Human antigen R binding and regulation of SOX2 mRNA in human mesenchymal stem cells. Mol Pharmacol. (2016) 89:243–52. doi: 10.1124/mol.115.100701
287. D’Agostino VG, Lal P, Mantelli B, Tiedje C, Zucal C, Thongon N, et al. Dihydrotanshinone-I interferes with the RNA-binding activity of HuR affecting its post-transcriptional function. Sci Rep. (2015) 5:16478. doi: 10.1038/srep16478
288. Lal P, Cerofolini L, D’Agostino VG, Zucal C, Fuccio C, Bonomo I, et al. Regulation of HuR structure and function by dihydrotanshinone-I. Nucleic Acids Res. (2017) 45:9514–27. doi: 10.1093/nar/gkx623
289. Filippova N, Yang X, Ananthan S, Sorochinsky A, Hackney JR, Gentry Z, et al. (HuR) multimerization contributes to glioma disease progression. J Biol Chem. (2017) 292:16999–7010. doi: 10.1074/jbc.M117.797878
290. Wu X, Lan L, Wilson DM, Marquez RT, Tsao Wc, Gao P, et al. Identi’cation and validation of novel small molecule disruptors of huR-mRNA interaction. ACS Chem Biol. (2015) 10(6):1476–84. doi: 10.1021/cb500851u
291. Andrade D, Mehta M, Griffith J, Oh S, Corbin J, Babu A, et al. HuR reduces radiation-induced DNA damage by enhancing expression of ARID1A. Cancers. (2019) 11:2014. doi: 10.3390/cancers11122014
292. Muralidharan R, Mehta M, Ahmed R, Roy S, Xu L, Aubé J, et al. HuR-targeted small molecule inhibitor exhibits cytotoxicity towards human lung cancer cells. Sci Rep. (2017) 7:9694. doi: 10.1038/s41598-017-07787-4
293. Allegri L, Baldan F, Roy S, Aubé J, Russo D, Filetti S, et al. The HuR CMLD-2 inhibitor exhibits antitumor effects via MAD2 downregulation in thyroid cancer cells. Sci Rep. (2019) 9:7374. doi: 10.1038/s41598-019-43894-0
294. Kakuguchi W, Nomura T, Kitamura T, Otsuguro S, Matsushita K, Sakaitani M, et al. Suramin, screened from an approved drug library, inhibits HuR functions and attenuates Malignant phenotype of oral cancer cells. Cancer Med. (2018) 7:6269–80. doi: 10.1002/cam4.1877
Keywords: inflammation, hematopoiesis, hematopoietic stem cells (HSCS), cytokines, RNA binding proteins, leukemia
Citation: Saluja S, Bansal I, Bhardwaj R, Beg MS and Palanichamy JK (2024) Inflammation as a driver of hematological malignancies. Front. Oncol. 14:1347402. doi: 10.3389/fonc.2024.1347402
Received: 01 December 2023; Accepted: 05 March 2024;
Published: 20 March 2024.
Edited by:
Rajiv Pathak, Albert Einstein College of Medicine, United StatesReviewed by:
Antonio Giovanni Solimando, University of Bari Aldo Moro, ItalyCopyright © 2024 Saluja, Bansal, Bhardwaj, Beg and Palanichamy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jayanth Kumar Palanichamy, ZHJqYXlhbnRoQGFpaW1zLmVkdQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.