
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 15 May 2024
Sec. Breast Cancer
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1342346
 Robert John Walsh1
Robert John Walsh1 Rebecca Ong2
Rebecca Ong2 Seng Wee Cheo1Peter Q.J. Low1
Seng Wee Cheo1Peter Q.J. Low1 Aishwarya Jayagopal3Matilda Lee1
Aishwarya Jayagopal3Matilda Lee1 Natalie Ngoi1Samuel G. Ow1Andrea L.A. Wong1,2Siew Eng Lim1Yi Wan Lim1Valerie Heong4Raghav Sundar1,2,5,6,7Ross A. Soo1,8Cheng Ean Chee1
Natalie Ngoi1Samuel G. Ow1Andrea L.A. Wong1,2Siew Eng Lim1Yi Wan Lim1Valerie Heong4Raghav Sundar1,2,5,6,7Ross A. Soo1,8Cheng Ean Chee1 Wei Peng Yong1,8Boon Cher Goh1,8
Wei Peng Yong1,8Boon Cher Goh1,8 Soo Chin Lee1,8
Soo Chin Lee1,8 David S.P. Tan1,2,8,9*
David S.P. Tan1,2,8,9* Joline S.J. Lim1,2,8*
Joline S.J. Lim1,2,8*Introduction: Molecular profiling of metastatic breast cancer (MBC) through the widespread use of next-generation sequencing (NGS) has highlighted actionable mutations and driven trials of targeted therapy matched to tumour molecular profiles, with improved outcomes reported using such an approach. Here, we review NGS results and treatment outcomes for a cohort of Asian MBC patients in the phase I unit of a tertiary centre.
Methods: Patients with MBC referred to a phase I unit underwent NGS via Ion AmpliSeq Cancer Hotspot v2 (ACH v2, 2014–2017) prior to institutional change to FoundationOne CDx (FM1; 2017–2022). Patients were counselled on findings and enrolled on matched therapeutic trials, where available. Outcomes for all subsequent treatment events were recorded to data cut-off on January 31, 2022.
Results: A total of 215 patients were enrolled with successful NGS in 158 patients. The PI3K/AKT/PTEN pathway was the most altered with one or more of the pathway member genes PIK3/AKT/PTEN affected in 62% (98/158) patients and 43% of tumours harbouring a PIK3CA alteration. Tumour mutational burden (TMB) was reported in 96/109 FM1 sequenced patients, with a mean TMB of 5.04 mt/Mb and 13% (12/96) with TMB ≥ 10 mt/Mb. Treatment outcomes were evaluable in 105/158 patients, with a pooled total of 216 treatment events recorded. Matched treatment was administered in 47/216 (22%) events and associated with prolonged median progression-free survival (PFS) of 21.0 weeks [95% confidence interval (CI) 11.7, 26.0 weeks] versus 12.1 weeks (95% CI 10.0, 15.4 weeks) in unmatched, with hazard ratio (HR) for progression or death of 0.63 (95% CI 0.41, 0.97; p = 0.034). In the subgroup of PIK3/AKT/PTEN-altered MBC, the HR for progression or death was 0.57 (95% CI 0.35, 0.92; p = 0.02), favouring matched treatment. Per-patient overall survival (OS) analysis (n = 105) showed improved survival for patients receiving matched treatment versus unmatched, with median OS (mOS) of 30.1 versus 11.8 months, HR = 0.45 (95% CI 0.24, 0.84; p = 0.013). Objective response rate (ORR) in the overall population was similar in matched and unmatched treatment events (23.7% versus 17.2%, odds ratio of response 1.14 95% CI 0.50, 2.62; p = 0.75).
Conclusions: Broad-panel NGS in MBC is feasible, allowing therapeutic matching, which was associated with improvements in PFS and OS.
Precision oncology has drastically changed treatment paradigms in many solid organ tumours over the last decade, driven by an exponential increase in our knowledge of somatic molecular aberrations and the ability to target key oncogenic drivers. Knowledge of such aberrations is facilitated by the widespread adoption of next-generation sequencing (NGS) for somatic profiling, allowing molecularly targeted, or “matched”, therapies to be administered to patients more frequently. Precision oncology has seen great success in non-small cell lung carcinoma (NSCLC), with current National Comprehensive Cancer Network (NCCN) and European Society for Medical Oncology (ESMO) Precision Medicine Working Group guidelines recommending testing for actionable alterations via a broad-panel approach in newly diagnosed advanced NSCLC (1, 2).
Beyond NSCLC, organ-specific approvals for targeted therapy based on somatic sequencing results exist in a growing number of settings with targets including fibroblast growth factor receptor (FGFR), RAS/RAF, the homologous recombination repair (HRR) pathway, and isocitrate dehydrogenase 1 (IDH1) (3–7). Agnostic indications are increasing, with pembrolizumab approved in 2017 for use in pre-treated tumours exhibiting microsatellite instability (MSI; MSI-high) or deficiency in mismatch repair (dMMR) proteins and more recently in cases of high tumour mutational burden (TMB ≥ 10) (8, 9). Beyond immune checkpoint inhibition, in 2022, dabrafenib combined with trametinib was approved in advanced malignancies harbouring a BRAF V600E mutation, while multiple single-arm basket trials of the TRK inhibitors larotrectinib and entrectinib in NTRK fusion-positive solid tumours led to their approval in 2018 and 2019, respectively (10–12).
Metastatic breast cancer (MBC) has traditionally had limited utility for broad-panel somatic molecular profiling, with treatment decisions driven by hormone receptor status and the presence or absence of CerbB2 overexpression/amplification. Poly (ADP-ribose) polymerase (PARP) inhibitors olaparib and talazoparib improve progression-free survival (PFS) over standard-of-care chemotherapy in germline BRCA1/2-altered human epidermal growth factor receptor 2 (HER2)-negative MBC, which accounts for approximately 10% of MBC cases (13, 14). Both agents received subsequent approval; however, to date, this does not extend to their use in patients with somatic BRCA1/2 mutations alone, though early data have suggested a potential role in this context (15).
Multiple trials have attempted to target mutations in PIK3CA, the gene encoding phosphatidylinositol 3-kinase (PI3K) catalytic subunit, which is altered in approximately 40% of MBC. Most notably in 2019, the phase III SOLAR 1 trial reported that alpelisib in combination with fulvestrant led to improved PFS over fulvestrant alone in hormone receptor positive, HER2-negative MBC harbouring hotspot mutations in PIK3CA, leading to the subsequent approval for this combination (16). Further studies have looked to target PI3K pathway alterations beyond PIK3CA. Both the phase II FAKTION and phase III CAPItello-291 trials have examined the addition of capivasertib, an oral inhibitor of AKT, to fulvestrant in patients with oestrogen receptor (ER)-positive, HER2-negative MBC after progression or relapse on an aromatase inhibitor (17, 18). The FAKTION authors reported a significant PFS and overall survival (OS) benefit favouring the novel combination; however, subgroup analysis showed that this benefit was primarily in patients considered to have PIK3/AKT/PTEN pathway alteration, with no significant benefit seen in non-altered tumours (17). Results of CAPItello-291 support a significant PFS benefit in the overall patient population with the addition of capivasertib to fulvestrant, with HR for progression or death of 0.60 [95% confidence interval (CI) 0.51, 0.71], although the magnitude of benefit was greater in those with AKT pathway alterations, HR = 0.50 (95% CI 0.38, 0.65) (18).
Phase I studies have provided evidence that molecularly matched therapy can lead to a similar benefit in refractory MBC, with several non-randomised studies showing the administration of matched therapy to be associated with improved outcomes (19–21). In a retrospective analysis including only MBC (n = 97), patients on matched clinical trials had improved PFS (HR = 0.52, p = 0.003) and OS (HR = 0.54, p < 0.001) versus those on non-matched trials (19).
Here, we report a retrospective analysis of MBC patients enrolled in the Integrated Molecular Analysis of Cancer (IMAC) study from April 28, 2014, to January 31, 2022. The IMAC study is an ongoing prospective trial using broad-panel sequencing of refractory solid-organ malignancies to identify targetable molecular alterations in the Phase I unit of the National University Cancer Institute, Singapore (NCIS). Earlier results detailing the feasibility and outcomes from April 28, 2014, to September 1, 2016, have been reported (22). We present broad-panel sequencing results as well as clinical outcomes of future treatment lines after administration of molecularly matched or unmatched therapy.
Patients with MBC enrolled in the IMAC program from April 28, 2014, to January 31, 2022, were included in the current retrospective analysis. The IMAC study enrolled patients with advanced solid organ malignancies reviewed in the Developmental Therapeutic Unit of NCIS. Available tumour tissue was obtained for NGS based on paraffin-embedded primary tumour and/or metastatic specimens, which were obtained during routine clinical care (e.g., surgery, biopsy or ascites, and pleural fluid drain). Wherever possible, fresh tumour samples were collected, with archival tumours utilised when a fresh sample was not available or technically feasible to obtain. Additionally, commencing from 2017, blood samples for circulating tumour (ct) DNA analysis were eligible for testing at the discretion of the patient’s primary physician should there be insufficient tumour tissue. Patients with successful sequencing were included in the mutational profile analysis of this current retrospective report.
For patients enrolled from April 2014 to July 2017, sequencing was via amplicon-based Ion AmpliSeq Cancer Hotspot v2 (50 genes, Appendix A), performed in our institution on Ion Torrent/PGM System (Life Technologies, Camarillo, CA, USA) as previously described (22). Patients enrolled after August 2017 to January 31, 2022, underwent tumour sequencing via FoundationOne CDx (FM1) after an institutional change in panel use. Formalin-fixed, paraffin-embedded (FFPE) tissue was processed per institutional practice, and slides were submitted to Foundation Medicine (Cambridge, MA, USA) for testing. If adequate tissue was not available in patients with no plans for further biopsy, two tubes of whole blood were submitted to Foundation Medicine for analysis under the FoundationOne Liquid CDx platform. Variants identified by FoundationOne CDx are represented as pathogenic or likely pathogenic and variants of unknown significance. For the purposes of this analysis, the variants of uncertain significance were excluded. FoundationOne CDx specimens were also simultaneously profiled for TMB as well as MSI status (23).
Patient sequencing results were discussed in the NCIS Molecular Tumour Board to identify potential matched therapies or clinical trials. Patients were counselled on outcomes of sequencing and molecular tumour board discussion and enrolled on matched trials, if available. All patients provided informed consent prior to joining a given clinical trial, all of which were approved by the appropriate local Research and Ethics Committee. If matched trials were not available in the unit, patients could undergo matched treatment off trial or unmatched treatment on or off trial after discussion with their treating oncologist.
Treatment was defined as matched if a molecular alteration detected on sequencing was targeted, or within the pathway targeted, by the administered drug. Treatment was considered non-matched if no molecular alterations were detected or if alterations detected, or the pathways within which they lie, were not targeted by administered treatment. The expression status of ER/progesterone receptor (PR) and CerbB2 was not taken into consideration. Specific scenarios included the following:
a. Anti-HER2 therapy is considered matched if ERBB2 alteration or amplification was detected on sequencing analysis regardless of immunohistochemistry results.
b. Therapeutics targeting the DNA damage response (DDR) pathway including PARP inhibitors and ATR inhibitors would be considered matched in the presence of mutations in the following list of homologous recombination-related genes previously described by Tung et al. (15): ATM, ATR, BAP1, BARD1, BLM, BRIP1, CHEK1, CHEK2, CDK12, FANCA, FANCC, FANCD2, FANCF, MRE11A, NBN, PALB2, RAD50, RAD51C, RAD51D, or WRN. Platinum therapy was not considered matched in the presence of such mutations.
c. Immunotherapy was considered matched in the presence of high TMB (≥10) or MSI-high/dMMR.
d. Patients with detected alterations (including non-hotspot PIK3CA mutations) in any of PI3K, AKT, and/or PTEN were considered as PI3K/AKT/PTEN altered, and treatment targeting this pathway was recorded as matched.
Patients enrolled on clinical trials underwent follow-up and tumour response assessment per trial protocol, while those treated outside of clinical studies were followed up per standard institution clinical practice.
Patients with successful NGS profiling were included in baseline characteristic assessment and mutational profile analysis. Patients who commenced on a new line of systemic therapy after sequencing results were available, with completion of at least one treatment cycle by data cut-off (January 31, 2022), were considered evaluable for clinical outcomes. Patients with less than one completed treatment cycle but with objective evidence of a disease progression event were deemed evaluable. Those lost to follow-up or continuing on treatment without disease progression at data cut-off were censored at the last documented clinic review.
Baseline patient characteristics were tabulated and summarised with descriptive statistics. PFS was calculated from the date of first administration of the drug to the date of radiological or clinical progression or death from any cause. Response assessment was per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 criteria. For patients with multiple sequential treatment events after sequencing, details of each individual treatment event (including matched status) were recorded and used to calculate a pooled progression-free survival analysis. The hazard ratio (HR) for risk of progression or death was calculated for this pooled assessment with appropriate frailty adjustment for shared identity.
A per-patient analysis of OS (calculated from the date of treatment start post-sequencing to death or censoring) by matched status was performed. Overall survival of patients receiving matched therapy at any point post-sequencing was compared to that of patients never receiving matched treatment post-sequencing. Clinical benefit rate was calculated as the proportion of patients with complete or partial response or more than 24 weeks of stable disease. Statistical analysis and KM plot generation was performed using STATA version 17.0 (STATA Corp., College Station, TX, USA). Oncoplot and Swimmer plot generated using R and R studio (URL https://www.R-project.org/, Boston, MA; URL http://www.rstudio.com/).
A total of 215 patients with MBC were enrolled on IMAC from April 28, 2014, to January 31, 2022. Sequencing was successful in 73% of patients (158/215), with 105 patients evaluable for treatment response (66%, 105/158). Baseline characteristics of sequenced patients (n = 158) are summarised in Table 1.
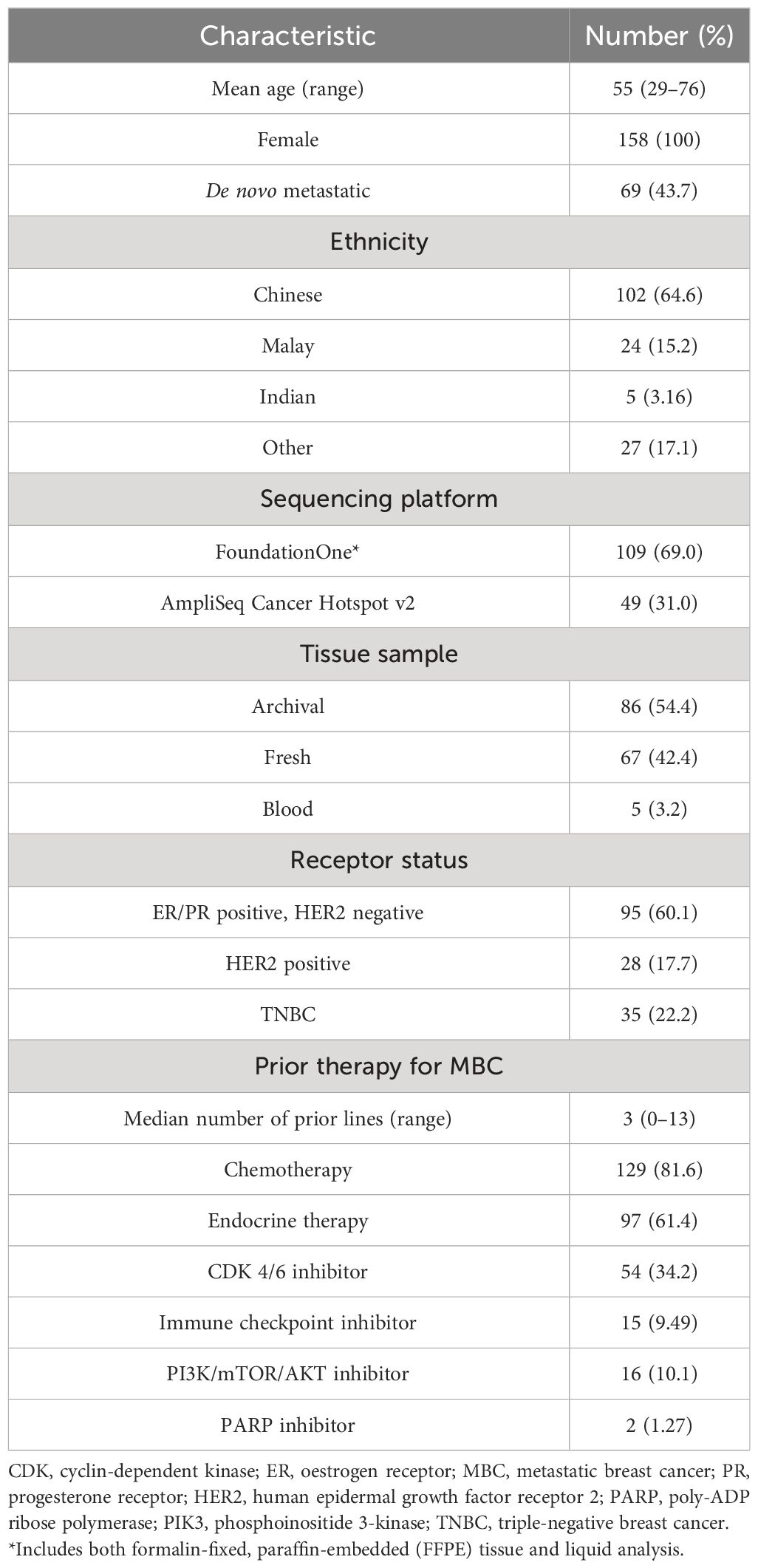
Table 1 Baseline characteristics of sequenced patients (n = 158).
Figure 1 shows an oncoplot of molecular profile results of patients with successful sequencing (n = 158). TP53 was the most commonly mutated gene (47%, 75/158 patients), with a significantly higher frequency of TP53 alterations seen in ER-negative vs. ER-positive tumours (65 vs. 38%, p = 0.001). PIK3CA alterations were seen in 43% (68/158) of patients, with 17 patients having more than one PIK3CA mutation. Of these, the hotspot mutation at PIK3CA H1047R was most common, identified in 17% (27/158) of tumours.
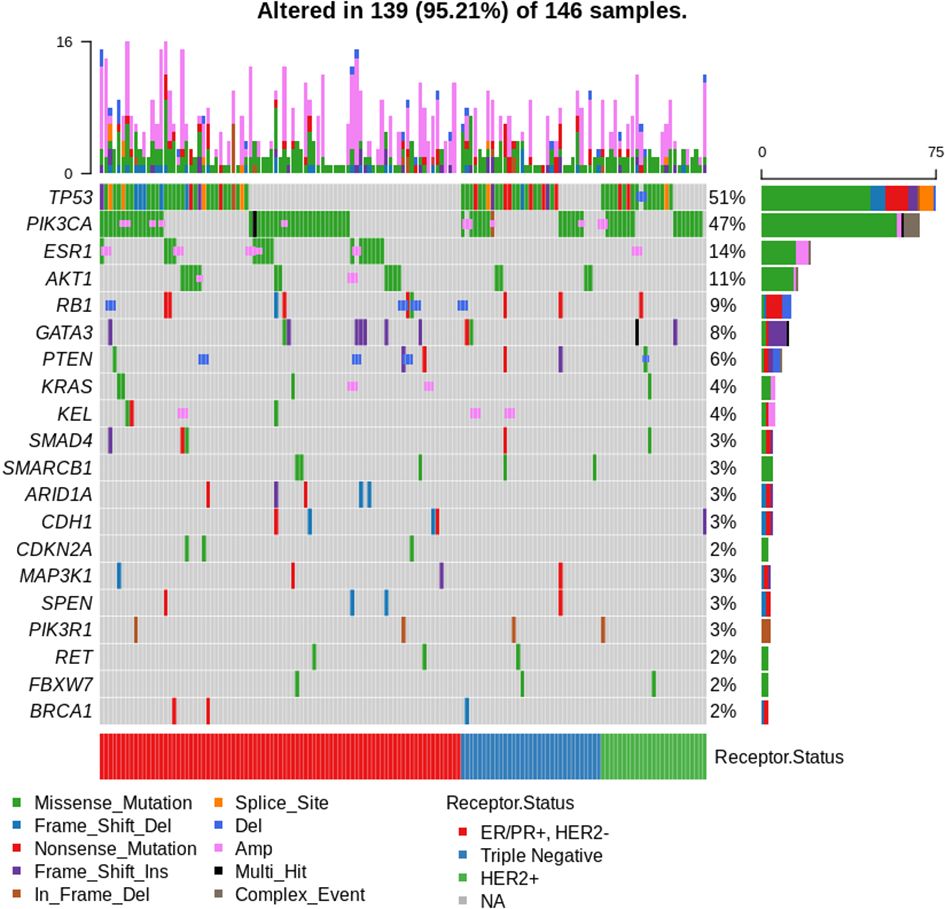
Figure 1 Oncoplot of results for patients with successful sequencing (n = 158). Top 20 most frequently mutated genes shown. A total of 12 patients had no mutations identified. Arranged by oestrogen receptor (ER), progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2) status.
The most commonly altered pathway was PI3K/AKT/PTEN, with alterations in one or more of the pathway member genes (PIK3/AKT/PTEN) seen in 62% (98/158) of patients, with no difference in frequency in ER-positive versus ER-negative tumours (p = 0.86) or sequencing performed on archival versus fresh samples (p = 0.229). Aberrations in the DDR pathway were present in 10% (16/158) of sequenced patients, with 5/158 patients harbouring BRCA1 mutations and 3/158 BRCA2 alterations. One patient had co-occurring BRCA1 and BRCA2 mutations (BRCA1 S573*, BRCA2 A938fs*21).
Of 109 patients sequenced on the FoundationOne CDx platform, TMB was reported in 96 cases, with a mean TMB of 5.04 mt/Mb (range 0.0–40 mt/Mb). High TMB (≥10 mt/Mb) was seen in 12.5% of patients (12/96). No MSI-high cases were detected.
Germline testing was performed in 26% (41/158) of sequenced patients, with pathogenic or likely pathogenic alterations identified in 8/41 cases. Patients with germline alterations who underwent sequencing with FM1 (5/8) had a corresponding somatic mutation identified in all cases. Three patients with known germline mutations in either BRCA1/2 had tumour profiling performed on the AmpliSeq Cancer Hotspot V2 test panel, which did not include BRCA1/2, and thus, somatic BRCA alterations were not identified.
A total of 105 patients (Supplementary Table 3) underwent evaluable treatment after sequencing and had a total of 216 treatment events during the follow-up period, of whom 47/216 (22%) were genomically matched. Matched events had a significantly improved median PFS of 21.0 weeks (95% CI 11.7, 26.0 weeks) versus 12.1 weeks (95% CI 10.0, 15.4 weeks) in unmatched with HR for progression of 0.63 (95% CI 0.41, 0.97; p = 0.034) (Figure 2A). Adjusting for the treatment line gave an HR = 0.59 (95% CI 0.37, 0.96; p = 0.035), favouring matched treatment. A per-patient analysis (n = 105) of OS based on the matched status of treatment post-sequencing was performed. A significant benefit was seen for patients receiving matched treatment at any point post-sequencing versus those not receiving matched treatment, with median OS (mOS) 30.1 versus 11.8 months, HR = 0.45 (95% CI 0.24, 0.84; p = 0.013) (Figure 2B). Clinical benefit rate was superior with matched therapy, 46.8% (22/47) versus 29.6% (50/169), odds ratio (OR) 2.09 (95% CI 1.08, 4.05; p = 0.028), but no significant difference was seen in terms of objective response rate (ORR) of 23.7% for matched treatment versus 17.2% for unmatched treatment, OR 1.14 (95% CI 0.50, 2.62; p = 0.752).
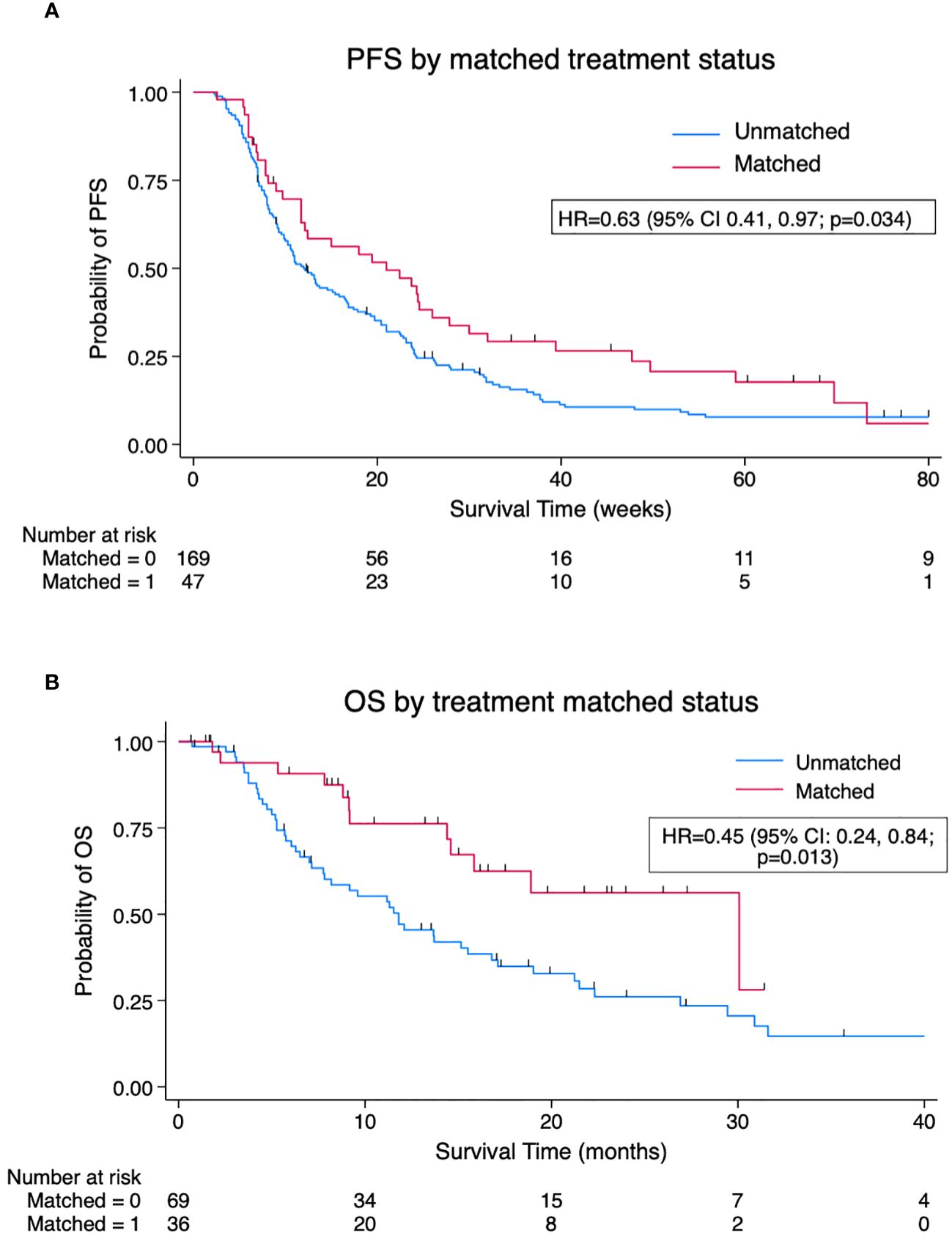
Figure 2 (A). Kaplan–Meier pooled analysis of progression-free survival (PFS) by matched versus unmatched treatment (n = 216 treatment events). (B) Kaplan–Meier per-patient analysis of overall survival (OS) by matched versus unmatched status of post-sequencing treatment (n = 105 patients). Survival favours patients receiving matched therapy at any point post-sequencing versus those never receiving matched therapy. HR, hazard ratio.
Alterations in PIK3/AKT/PTEN were seen in 64% (67/105) of sequenced patients who underwent evaluable treatment. The majority of these treated PIK3/AKT/PTEN-altered patients were HER2 negative (84%, 56/67). The 67 treated PIK3/AKT/PTEN-altered patients had 137 evaluable treatment events, of which 42/137 (31%) were genomically matched. The majority of these 42 matched events, 25/42, involved the administration of treatment targeting the PIK3/AKT/PTEN pathway (see Supplementary Table 1), while 12/42 were matched to anti-HER2 therapy in the presence of a coexisting ERBB2 amplification.
Matched treatment events in PIK3/AKT/PTEN-altered patients were associated with improved median PFS compared to unmatched treatment events, 19.4 versus 10.6 weeks, HR = 0.57 (95% CI 0.35, 0.92; p = 0.02). The difference was more pronounced in a subgroup of PIK3/AKT/PTEN altered, HER2-negative/ERBB2 non-altered patients, where matched treatment events displayed prolonged median PFS of 30.0 versus 10.7 weeks, HR = 0.48 (95% CI 0.27, 0.84; p = 0.01). Of the 42 PIK3/AKT/PTEN-altered patients matched to therapy, 10 (24%) had more than one PIK3/AKT/PTEN alteration. Patients with multiple PIK3/AKT/PTEN alterations had an improved PFS on matched therapy (median PFS not reached) versus those with single aberrations (12.4 weeks), HR = 0.27 (95% CI 0.09, 0.76; p = 0.014). The Kaplan–Meier plots are shown in Figure 3.
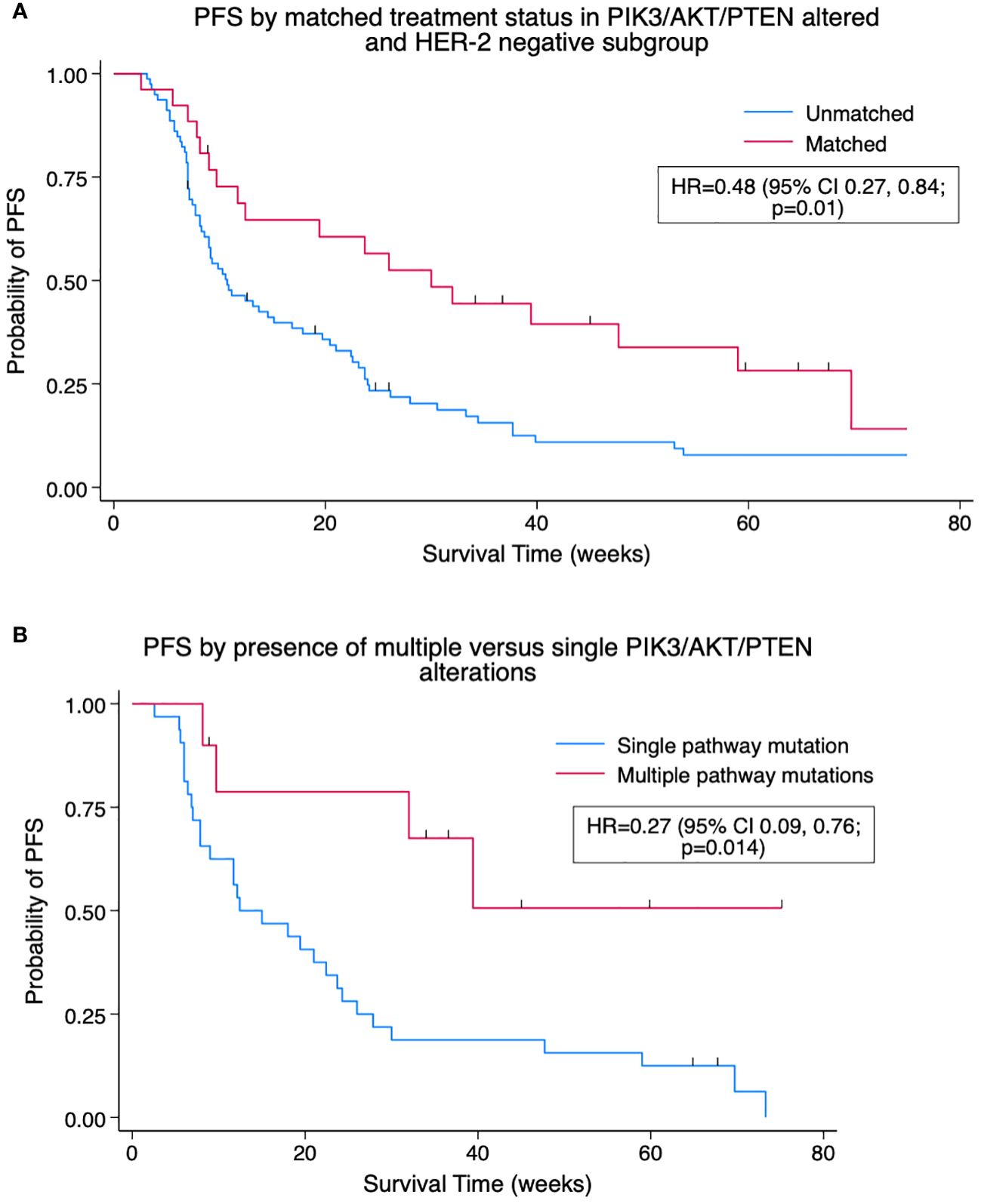
Figure 3 (A) Kaplan–Meier pooled analysis of progression-free survival (PFS) by matched treatment status in PIK3/AKT/PTEN-altered and HER2-negative subgroup. HR, hazard ratio; HER2, human epidermal growth factor receptor 2. (B) Kaplan–Meier pooled analysis of PFS by presence of multiple versus single PIK3/AKT/PTEN alterations for patients with at least one PIK3/AKT/PTEN alteration on matched therapy. HR, hazard ratio.
Of patients with DDR pathway mutations identified, 12/16 underwent evaluable treatment post-sequencing with a pooled total of 26 treatment events. There was no significant difference in PFS in patients with DDR pathway mutations (median PFS = 16.4 weeks) versus those without (median PFS = 12.4 weeks), HR = 0.71 (95% CI 0.38, 1.32, p = 0.282). Matched treatment was recorded for three patients with DDR pathway mutations. Olaparib was administered in combination with intrathecal methotrexate in a patient with germline (and corresponding somatic) BRCA1 pathogenic variant with a PFS of 59 weeks and as a single agent in a patient with germline (and somatic) PALB2 alteration for 24 weeks prior to progression. Treatment with an ATR inhibitor in the setting of a clinical trial gave a relatively short PFS of 12 weeks in a patient with somatic BRCA1 Q544* (no germline testing performed). One patient received PARPi prior to study enrolment.
Immune checkpoint inhibitors (ICIs) were administered alone or in combination to 19 patients (Supplementary Table 2), eight of whom received the combination of anti-PD-L1, AKT inhibitor, and a PARP inhibitor as part of an ongoing clinical trial. These eight patients harboured a mutation in one of PTEN/AKT and/or PIK3CA and were therefore considered matched to the use of an AKT inhibitor. In 5/19 ICI-treated patients, TMB was ≥10, and treatment was considered TMB matched. Median PFS in the 19 ICI-treated patients was 26.0 weeks (95% CI 7, -) with eight patients continuing treatment at the point of data collection (Figure 4). Analysis of this ICI-treated cohort by TMB-matched status showed no significant difference with a median PFS (mPFS) of 44.9 vs. 32.0 weeks in TMB-matched vs. unmatched patients, HR = 0.59 (95% CI 0.12, 2.94; p = 0.52).
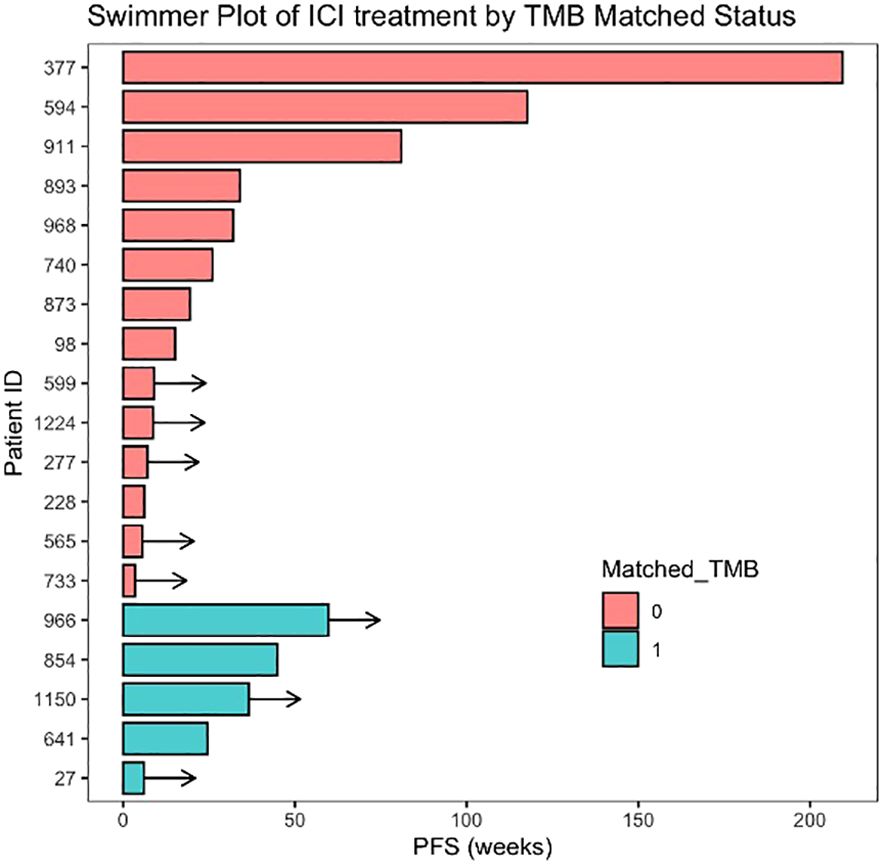
Figure 4 Swimmer plot of patients treated with immune checkpoint inhibitors (ICIs) arranged by matched status. Matched TMB = 1 represents patients matched to ICI therapy based on the presence of high tumour mutational burden (TMB ≥ 10 mt/Mb). Arrows indicate ongoing treatment at time of data cut-off.
In this retrospective study of a cohort of MBC patients from a tertiary oncology centre in Asia, we report a higher PFS for patients receiving matched versus unmatched treatment, a finding consistent with prior publications (19, 24, 25). Median PFS in the current study was 21.0 weeks in pooled treatment event analysis, a value that is difficult to compare across publications of early-phase trial cohorts due to the heterogeneous nature of such populations. A prior publication on the IMAC study in 2018, including multiple tumour types, reported a median PFS of 2.9 months, similar to the 3.2 months reported by O’Carrigan and colleagues in a pre-treated MBC cohort (19, 22). Our study also yielded a significant improvement in OS in patients receiving matched therapy at any point post-sequencing versus those who do not go on to receive any matched treatment post-sequencing, with HR = 0.45 (95% CI 0.24, 0.84; p = 0.013), highlighting that matching patients based on sequencing outcomes remain beneficial even if the matching cannot occur in the immediate setting. It is therefore vital for specialised drug development units to keep track of patients with targetable molecular mutations and update primary oncologists on new treatment options for matching when available. While OS analysis may be affected by subsequent treatment received and the presence of censored individuals in both matched and unmatched groups, it remains an encouraging result reinforcing the potential benefit of matched therapy in MBC. The frequency of clinical benefit rate (CBR) in the current study was higher for matched treatment events (OR 2.09, p = 0.028), although response rates were similar regardless of matched status at 23% (matched) versus 21% (unmatched) and were overall in keeping with expected responses for pre-treated MBC.
Of all pooled treatment events, 22% were matched based on sequencing results, with the most frequent target being the PIK3/AKT/PTEN alterations, which were present in 64% of patients. This matching frequency is lower than reported in the pan-cancer IMPACT trial (54%) and in an MBC cohort by O’Carrigan et al. (83%) but similar to that of the Moores Cancer Center PREDICT analysis (20, 26). The frequency of matching increased over time, with 11% of treatment events in the first 70 enrolled patients being matched, increasing to 22% for the second 70 patients enrolled and 35% for the most recently recruited group. This may be attributed to the developing nature of the NCIS Developmental Therapeutic Unit over the trial period and a corresponding increase in the number of potential trial options available along with increasing access to approved biomarker-selected treatments such as alpelisib in PIK3CA hotspot mutated tumours. The reported rate in the current study is also contributed by the pooling of all treatment events post-sequencing. If only the first line post-sequencing were examined, the number of patients matched to therapy was 30.5% (32/105) versus 22% if all treatment events were considered.
PIK3, AKT, and PTEN were frequently mutated in the current cohort. Currently approved therapy targeting this pathway includes alpelisib in combination with fulvestrant; however, its use is restricted to patients with HER2-negative MBC harbouring one of 11 PIK3CA hotspot mutations, based on inclusion criteria of the phase III SOLAR-1 trial (16). However, the phase II FAKTION trial points to the benefit of pathway-targeted therapy for patients with a wider span of mutations affecting PIK3/AKT/PTEN when treated with capivasertib in combination with fulvestrant (17). More recently, phase III CAPItello-291 has shown PFS benefit for unselected MBC after progression on aromatase inhibitor with the addition of capivasertib to fulvestrant with the magnitude of benefit more pronounced in AKT pathway-altered patients (18). Results from our current study support the use of matched therapy in a wider pool of PIK3/AKT/PTEN-altered cases with PFS improved in such patients receiving matched treatment compared to those on unmatched therapy (HR = 0.57, p = 0.02). Of patients receiving therapy targeting the PI3K/AKT/PTEN pathway, nine had mutations outside PIK3CA hotspot alterations, with four of nine (44%) having a PFS of ≥12 weeks on matched therapy. Of those matched to therapy, the benefit was enhanced in patients with multiple alterations in PIK3, AKT, or PTEN versus those with single mutations, with HR for progression or death of 0.27 (Figure 3B). This supports previous reports of increased ORR and PFS in patients with multiple PIK3Ca mutations while ongoing treatment with fulvestrant and taselisib (27). This highlights the potential for patients with non-hotspot mutations to benefit from treatment targeting the PI3K/AKT/PTEN pathway and the role that broad-panel NGS has in identifying such alterations.
The number of patients with mutations in the DDR pathway was low (16/158), restricting any analysis of outcomes by treatment status. One patient (IMAC 660, Supplementary Table 1) with a somatic and germline-detected PALB2 alteration had a PFS of 24 weeks with olaparib despite being heavily pre-treated (eight lines prior to therapy). Although anecdotal, this case highlights the expanded role of PARP inhibition outside of germline BRCA1/2-altered HER2-negative MBC. The benefit of PARPi outside of current approval limits has been reported in a phase I trial combining olaparib and capivasertib as well as phase II single-agent trials TBCRC-048 and Talazoparib Beyond BRCA (15, 28, 29).
We report a subgroup of patients who received ICI treatment for MBC (19/105 patients) of whom five had high TMB. The mPFS of 26 weeks in this ICI-treated group is promising when compared to those of trials of enriched subgroups, such as KEYNOTE-158, which reported an mPFS of 4.1 months in non-colorectal MSI-high cancers treated with pembrolizumab, and suggests that there remains a small group of MBC patients who can gain significant benefit from ICI (9). The role of immunotherapy in MBC is currently limited to first-line indication for pembrolizumab in combination with chemotherapy in PD-L1-positive, treatment-naïve triple-negative breast cancer (TNBC) in combination with chemotherapy, whereas its use in later-line settings as single-agent therapy has not shown significant improvement in treatment outcomes (30, 31). In the treatment-refractory setting, pembrolizumab has agnostic approval in the presence of dMMR/MSI-H or TMB ≥ 10 mt/Mb (8, 9). Numerous ongoing studies continue to evaluate the application of ICI in MBC while attempting to derive an optimal biomarker (32).
Our study has several strengths and limitations. This is one of the first reports of sequencing results and clinical outcomes in an Asian-predominant population and was set within a tertiary referral centre, which enabled access to a range of novel targeted therapies. This is of importance given the number of prospective clinical trials that are being carried out, at least in part, in Asian cancer centres and allows investigators to contextualise the molecular profile of breast cancer in this region. Sequencing was performed on validated commercial platforms ensuring robust reproducible results obtained.
The pooled analysis of treatment events risks incorporating bias where patients receive multiple subsequent treatment lines, each analysed as an individual event. This was adjusted using the frailty adjustment for shared identity in STATA analysis. The number of patients enrolled thus far has not allowed for powering to explore matching at each treatment line, which may be of interest in future studies when larger cohorts of patients with longer-term follow-up are available for analysis. The retrospective nature of the analysis risks selection bias, and low numbers of DDR pathway mutations are in part a reflection of the sample size and restrict any comparative assessment of this subgroup. The use of a more restricted sequencing panel prior to the institutional change to FM1 means that specific alterations will have been unidentified and underreported, including somatic BRCA1/2 mutations, as these genes were not included in the 50 gene panel list of ACH v2 (Supplementary Appendix A). Similarly, TMB and MSI status were not assessed by ACH v2. Mutation detection may have also been affected by tumour heterogeneity, which has been well-described in breast cancer (33, 34). The majority of patients in the current studied cohort underwent somatic sequencing on archival tissue. It is possible the mutational profile of a tumour may be significantly different at the point of study entry in comparison to the date of archival tissue acquisition due to the temporal evolution of genomic alterations. Furthermore, the spatial evolution of clones and sub-clones means that the biopsy site may have affected alterations detected in our cohort.
In conclusion, this study highlights the feasibility of somatic NGS in MBC in facilitating therapeutic matching, primarily in patients with PIK3/AKT/PTEN alterations, which were seen in the majority. Matched therapy is shown to be associated with an improvement in PFS and OS over unmatched treatment in this cohort.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
The studies involving humans were approved by National Healthcare Group Domain Specific Review Board (NHG DSRB). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
RW: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Visualization, Writing – original draft, Writing – review & editing, Resources, Software. RO: Conceptualization, Data curation, Investigation, Methodology, Project administration, Writing – original draft, Writing – review & editing. SC: Data curation, Investigation, Methodology, Writing – review & editing, Conceptualization. PL: Data curation, Investigation, Methodology, Project administration, Resources, Writing – review & editing, Conceptualization. AJ: Formal analysis, Methodology, Project administration, Resources, Software, Writing – review & editing, Conceptualization. ML: Investigation, Methodology, Project administration, Resources, Writing – review & editing, Conceptualization. NN: Data curation, Investigation, Methodology, Project administration, Resources, Writing – review & editing, Conceptualization. SO: Investigation, Methodology, Project administration, Resources, Writing – review & editing, Conceptualization. AW: Investigation, Methodology, Project administration, Resources, Writing – review & editing, Conceptualization. SEL: Investigation, Methodology, Project administration, Resources, Writing – review & editing, Conceptualization. YL: Investigation, Methodology, Project administration, Resources, Writing – review & editing, Conceptualization. VH: Data curation, Investigation, Methodology, Resources, Writing – review & editing, Conceptualization. RS: Investigation, Methodology, Resources, Writing – review & editing, Conceptualization. RS: Investigation, Methodology, Project administration, Resources, Writing – review & editing, Conceptualization. CC: Investigation, Methodology, Project administration, Resources, Writing – review & editing, Conceptualization. WY: Investigation, Methodology, Project administration, Resources, Writing – review & editing, Conceptualization. BG: Conceptualization, Investigation, Methodology, Project administration, Resources, Supervision, Writing – review & editing. SL: Conceptualization, Investigation, Methodology, Project administration, Resources, Supervision, Writing – review & editing. DT: Conceptualization, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing. JL: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This study was supported by the National Research Foundation, Singapore, and the National Medical Research Council, Singapore, under its NMRC Centre Grant Programme (NMRC/CG/M005/2017_NCIS), Clinician Scientist Award Senior Investigator Grant (CSASI21jun-0003), and NMRC Transition Award (MOH-000414).
We are grateful to the patients who participated. Interim analysis was presented in abstract form at ASCO 2020 (35).
RW reported serving on the advisory board of Pfizer and Novartis and receiving honoraria from Pfizer, AstraZeneca, Novartis, and Merck MSD and travel funding from Merck MSD outside the submitted work. NN reported honoraria from Merck, MSD, AstraZeneca, and ASGO; research funding from Cyclacel and iOnctura; and travel funding from AstraZeneca and JSGO. SO reported honoraria and consulting with Astra Zeneca, Pfizer, Novartis, Eli Lilly, and Roche. AW reported advisory board membership and/or speaker activity with honoraria with AstraZeneca, Novartis, Eisai, DKSH, Pfizer, and Roache and research funding from Otsuka Pharmaceuticals. VH reported consultancy/advisory activity with DKSH, AstraZeneca, Novartis, MSD, and Pfizer. RS reported serving on the advisory board of Bristol Myers Squibb, Merck, Eisai, Bayer, Taiho, Novartis, MSD, DKSH, and GSK; receiving honoraria from MSD, Eli Lilly, Bristol Myers Squibb, Roche, Taiho, AstraZeneca, Ipsen, and DKSH; and receiving grants from Roche, AstraZeneca, Taiho, Eisai, DKSH, Paxman Coolers, Natera, and MSD outside the submitted work. RAS reported honoraria from AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Lilly, Novartis, Pfizer, Roche/Genentech, Takeda, Yuhan, Amgen, Bayer, Merck, Merck Serono, and Puma Biotechnology; consulting or advisory role with AstraZeneca, Boehringer Ingelheim, Bristol Myers Squibb, Novartis, Pfizer, Roche/Genentech, Taiho Pharmaceutical, Yuhan, Takeda, Amgen, Lilly, Merck, Janssen, Puma Biotechnology, Merck Serono, Bayer, and Thermo Fisher Scientific; and research funding from AstraZeneca and Boehringer Ingelheim. WY reported consulting or advisory role with AbbVie/Genentech, Amgen, Bristol Myers Squibb, Ipsen, Novartis, and AstraZeneca; speakers’ bureau with Lilly, Sanofi/Aventis, Taiho Pharmaceutical, Eisai, Bayer, and MSD Oncology; and travel and accommodation expenses from Pfizer. SCL reports honoraria and consulting activity with AstraZeneca, Pfizer, Novartis, Eli Lilly, Roche, ACT Genomics, and Eisai and research funding from Taihi, Eisai, Pfizer, and ACT Genomics. JL reported advisory activity with AstraZeneca, Novartis, Roche, DKSH, Pfizer, and MSD; receiving honoraria from AstraZeneca, Novartis, Roche, DKSH, MSD, Eisai, and Pierre Fabre; receiving research funding from CTI BioPharma, Daiichi Sankyo, and Synthon Pharmaceuticals; and receiving travel grant from AstraZeneca and MSD outside the submitted work. DT is an employee of the National University Health System Singapore and reports personal fees for advisory board membership from AstraZeneca, Bayer, BioNTech Boehringer Ingelheim, Eisai, Genmab, GSK, MSD, PMV Pharma, and Roche; personal fees as an invited speaker from AstraZeneca, Eisai, GSK, Merck Serono, MSD, Roche, and Takeda; ownership of stocks/shares of Asian Microbiome Library AMiLi; institutional research grants from AstraZeneca, Bayer, Karyopharm Therapeutics, and Roche; institutional funding as coordinating PI from AstraZeneca, MSD, Eisai, Roche, and Bergen Bio; institutional funding as local PI from Roche, BioNTech, PMV Pharma, GSK, Sutro Pharma, Bayer, Byondis B.V., and Zeria Pharmaceutical Co., Ltd.; a previous non-renumerated role as Chair of the Asia Pacific Gynecologic Oncology Trials Group APGOT; a previous non-renumerated role as the Society President of the Gynecologic Cancer Group Singapore; non-renumerated membership of the Board of Directors of the GCIG; research funding from the National Medical Research Council NMRC Clinician Scientist Award Senior Investigator Grant CSASI21jun-0003, the Pangestu Family Foundation Gynaecological Cancer Research Fund; and product samples from AstraZeneca, Eisai, and MSD non-financial interest for research trials.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2024.1342346/full#supplementary-material
1. NCCN Guidelines Non-Small Cell Lung Cancer v2.2023 (2023). Available at: https://www.nccn.org/professionals/physician_gls/pdf/nscl.pdf.
2. Mosele F, Remon J, Mateo J, Westphalen CB, Barlesi F, Lolkema MP, et al. Recommendations for the use of next-generation sequencing (NGS) for patients with metastatic cancers: a report from the ESMO Precision Medicine Working Group. Ann Oncol. (2020) 31:1491–505. doi: 10.1016/j.annonc.2020.07.014
3. Loriot Y, Necchi A, Park SH, Garcia-Donas J, Huddart R, Burgess E, et al. Erdafitinib in locally advanced or metastatic urothelial carcinoma. New Engl J Med. (2019) 381:338–48. doi: 10.1056/NEJMoa1817323
4. Abou-Alfa GK, Sahai V, Hollebecque A, Vaccaro G, Melisi D, Al-Rajabi R, et al. Pemigatinib for previously treated, locally advanced or metastatic cholangiocarcinoma: a multicentre, open-label, phase 2 study. Lancet Oncol. (2020) 21:671–84. doi: 10.1016/S1470-2045(20)30109-1
5. Zhu AX, Macarulla T, Javle MM, Kelley RK, Lubner SJ, Adeva J, et al. Final overall survival efficacy results of ivosidenib for patients with advanced cholangiocarcinoma with IDH1 mutation: the phase 3 randomized clinical clarIDHy trial. JAMA Oncol. (2021) 7:1669–77. doi: 10.1001/jamaoncol.2021.3836
6. de Bono J, Mateo J, Fizazi K, Saad F, Shore N, Sandhu S, et al. Olaparib for metastatic castration-resistant prostate cancer. N Engl J Med. (2020) 382:2091–102. doi: 10.1056/NEJMoa1911440
7. Chi KN, Rathkopf DE, Smith MR, Efstathiou E, Attard G, Olmos D, et al. Phase 3 MAGNITUDE study: First results of niraparib (NIRA) with abiraterone acetate and prednisone (AAP) as first-line therapy in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) with and without homologous recombination repair (HRR) gene alterations. J Clin Oncol. (2022) 40:12–. doi: 10.1200/JCO.2022.40.6_suppl.012
8. Marabelle A, Fakih M, Lopez J, Shah M, Shapira-Frommer R, Nakagawa K, et al. Association of tumour mutational burden with outcomes in patients with advanced solid tumours treated with pembrolizumab: prospective biomarker analysis of the multicohort, open-label, phase 2 KEYNOTE-158 study. Lancet Oncol. (2020) 21:1353–65. doi: 10.1016/S1470-2045(20)30445-9
9. Marabelle A, Le DT, Ascierto PA, Di Giacomo AM, De Jesus-Acosta A, Delord JP, et al. Efficacy of pembrolizumab in patients with noncolorectal high microsatellite instability/mismatch repair-deficient cancer: results from the phase II KEYNOTE-158 study. J Clin Oncol. (2020) 38:1–10. doi: 10.1200/JCO.19.02105
10. Administration USFaD. (2022). Available at: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-dabrafenib-combination-trametinib-unresectable-or-metastatic-solid.
11. Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of larotrectinib in TRK fusion–positive cancers in adults and children. N Engl J Med. (2018) 378:731–9. doi: 10.1056/NEJMoa1714448
12. Doebele RC, Drilon A, Paz-Ares L, Siena S, Shaw AT, Farago AF, et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: integrated analysis of three phase 1-2 trials. Lancet Oncol. (2020) 21:271–82.
13. Robson M, Im SA, Senkus E, Xu B, Domchek SM, Masuda N, et al. Olaparib for metastatic breast cancer in patients with a germline BRCA mutation. N Engl J Med. (2017) 377:523–33. doi: 10.1056/NEJMoa1706450
14. Litton JK, Rugo HS, Ettl J, Hurvitz SA, Gonçalves A, Lee K-H, et al. Talazoparib in patients with advanced breast cancer and a germline BRCA mutation. N Engl J Med. (2018) 379:753–63. doi: 10.1056/NEJMoa1802905
15. Tung NM, Robson ME, Ventz S, Santa-Maria CA, Nanda R, Marcom PK, et al. TBCRC 048: phase II study of olaparib for metastatic breast cancer and mutations in homologous recombination-related genes. J Clin Oncol. (2020) 38:4274–82. doi: 10.1200/JCO.20.02151
16. André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-mutated, hormone receptor–positive advanced breast cancer. N Engl J Med. (2019) 380:1929–40.
17. Howell SJ, Casbard A, Carucci M, Ingarfield K, Butler R, Morgan S, et al. Fulvestrant plus capivasertib versus placebo after relapse or progression on an aromatase inhibitor in metastatic, oestrogen receptor-positive, HER2-negative breast cancer (FAKTION): overall survival, updated progression-free survival, and expanded biomarker analysis from a randomised, phase 2 trial. Lancet Oncol. (2022) 23:851–64. doi: 10.1016/S1470-2045(22)00284-4
18. Turner NC, Oliveira M, Howell SJ, Dalenc F, Cortés J, Gómez HL, et al. Abstract GS3-04: GS3-04 Capivasertib and fulvestrant for patients with aromatase inhibitor-resistant hormone receptor-positive/human epidermal growth factor receptor 2-negative advanced breast cancer: results from the Phase III CAPItello-291 trial. Cancer Res. (2023). doi: 10.1158/1538-7445.SABCS22-GS3-04
19. O’Carrigan B, Lim JSJ, Jalil A, Harris SJ, Papadatos-Pastos D, Banerji U, et al. Target-based therapeutic matching of phase I trials in patients with metastatic breast cancer in a tertiary referral centre. Br J Cancer. (2018) 119:922–7. doi: 10.1038/s41416-018-0290-8
20. Schwaederle M, Parker BA, Schwab RB, Daniels GA, Piccioni DE, Kesari S, et al. Precision oncology: the UC san diego moores cancer center PREDICT experience. Mol Cancer Ther. (2016) 15:743–52. doi: 10.1158/1535-7163.MCT-15-0795
21. Stockley TL, Oza AM, Berman HK, Leighl NB, Knox JJ, Shepherd FA, et al. Molecular profiling of advanced solid tumors and patient outcomes with genotype-matched clinical trials: the Princess Margaret IMPACT/COMPACT trial. Genome Med. (2016) 8:109. doi: 10.1186/s13073-016-0364-2
22. Heong V, Syn NL, Lee XW, Sapari NS, Koh XQ, Adam Isa ZF, et al. Value of a molecular screening program to support clinical trial enrollment in Asian cancer patients: The Integrated Molecular Analysis of Cancer (IMAC) Study. Int J Cancer. (2018) 142:1890–900. doi: 10.1002/ijc.31091
23. Milbury CA, Creeden J, Yip WK, Smith DL, Pattani V, Maxwell K, et al. Clinical and analytical validation of FoundationOne®CDx, a comprehensive genomic profiling assay for solid tumors. PloS One. (2022) 17:e0264138. doi: 10.1371/journal.pone.0264138
24. Schwaederle M, Zhao M, Lee JJ, Lazar V, Leyland-Jones B, Schilsky RL, et al. Association of biomarker-based treatment strategies with response rates and progression-free survival in refractory Malignant neoplasms: a meta-analysis. JAMA Oncol. (2016) 2:1452–9. doi: 10.1001/jamaoncol.2016.2129
25. Bruzas S, Kuemmel S, Harrach H, Breit E, Ataseven B, Traut A, et al. Next-generation sequencing-directed therapy in patients with metastatic breast cancer in routine clinical practice. Cancers. (2021) 13:4564. doi: 10.3390/cancers13184564
26. Tsimberidou A-M, Hong DS, Wheler JJ, Falchook GS, Janku F, Naing A, et al. Long-term overall survival and prognostic score predicting survival: the IMPACT study in precision medicine. J Hematol Oncol. (2019) 12:145. doi: 10.1186/s13045-019-0835-1
27. Hutchinson KE, Chen JW, Savage HM, Stout TJ, Schimmoller F, Cortés J, et al. Multiple PIK3CA mutation clonality correlates with outcomes in taselisib + fulvestrant-treated ER+/HER2-, PIK3CA-mutated breast cancers. Genome Med. (2023) 15:28. doi: 10.1186/s13073-023-01181-8
28. Gruber JJ, Afghahi A, Timms K, DeWees A, Gross W, Aushev VN, et al. A phase II study of talazoparib monotherapy in patients with wild-type BRCA1 and BRCA2 with a mutation in other homologous recombination genes. Nat Cancer. (2022) 3:1181–91. doi: 10.1038/s43018-022-00439-1
29. Yap TA, Kristeleit R, Michalarea V, Pettitt SJ, Lim JSJ, Carreira S, et al. Phase I trial of the PARP inhibitor olaparib and AKT inhibitor capivasertib in patients with BRCA1/2- and non–BRCA1/2-mutant cancers. Cancer Discov. (2020) 10:1528–43. doi: 10.1158/2159-8290.CD-20-0163
30. Winer EP, Lipatov O, Im SA, Goncalves A, Muñoz-Couselo E, Lee KS, et al. Pembrolizumab versus investigator-choice chemotherapy for metastatic triple-negative breast cancer (KEYNOTE-119): a randomised, open-label, phase 3 trial. Lancet Oncol. (2021) 22:499–511. doi: 10.1016/S1470-2045(20)30754-3
31. Cortes J, Rugo HS, Cescon DW, Im S-A, Yusof MM, Gallardo C, et al. Pembrolizumab plus chemotherapy in advanced triple-negative breast cancer. N Engl J Med. (2022) 387:217–26. doi: 10.1056/NEJMoa2202809
32. Nicolini A, Ferrari P, Carpi A. Immune checkpoint inhibitors and other immune therapies in breast cancer: a new paradigm for prolonged adjuvant immunotherapy. Biomedicines. (2022) 10:2511. doi: 10.3390/biomedicines10102511
33. Lüönd F, Tiede S, Christofori G. Breast cancer as an example of tumour heterogeneity and tumour cell plasticity during Malignant progression. Br J Cancer. (2021) 125:164–75. doi: 10.1038/s41416-021-01328-7
34. Li A, Keck JM, Parmar S, Patterson J, Labrie M, Creason AL, et al. Characterizing advanced breast cancer heterogeneity and treatment resistance through serial biopsies and comprehensive analytics. NPJ Precis Oncol. (2021) 5:28. doi: 10.1038/s41698-021-00165-4
Keywords: breast cancer, precision oncology, molecular profiling, next-generation sequencing (NGS), phase I
Citation: Walsh RJ, Ong R, Cheo SW, Low PQJ, Jayagopal A, Lee M, Ngoi N, Ow SG, Wong ALA, Lim SE, Lim YW, Heong V, Sundar R, Soo RA, Chee CE, Yong WP, Goh BC, Lee SC, Tan DSP and Lim JSJ (2024) Molecular profiling of metastatic breast cancer and target-based therapeutic matching in an Asian tertiary phase I oncology unit. Front. Oncol. 14:1342346. doi: 10.3389/fonc.2024.1342346
Received: 21 November 2023; Accepted: 03 April 2024;
Published: 15 May 2024.
Edited by:
Alberto Farolfi, Scientific Institute of Romagna for the Study and Treatment of Tumors (IRCCS), ItalyReviewed by:
Jiaqi Liu, National Cancer Center of China, ChinaCopyright © 2024 Walsh, Ong, Cheo, Low, Jayagopal, Lee, Ngoi, Ow, Wong, Lim, Lim, Heong, Sundar, Soo, Chee, Yong, Goh, Lee, Tan and Lim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David S.P. Tan, ZGF2aWRfc3BfdGFuQG51aHMuZWR1LnNn; Joline S.J. Lim, Y3NpbHNqakBudXMuZWR1LnNn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.