
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Oncol., 02 February 2024
Sec. Hematologic Malignancies
Volume 14 - 2024 | https://doi.org/10.3389/fonc.2024.1339584
This article is part of the Research TopicRecent Advances in Diagnosis, Prognosis, and Therapy of Oncogenic Virus-driven TumorsView all 7 articles
 Eleonora Vecchio1*
Eleonora Vecchio1* Rossana Marino2
Rossana Marino2 Selena Mimmi1Camilla Canale2
Selena Mimmi1Camilla Canale2 Carmen Caiazza2
Carmen Caiazza2 Alessandro Arcucci3Maria Rosaria Ruocco2Marco Schiavone4Gianluca Santamaria1Camillo Palmieri1
Alessandro Arcucci3Maria Rosaria Ruocco2Marco Schiavone4Gianluca Santamaria1Camillo Palmieri1 Enrico Iaccino1Massimo Mallardo2Ileana Quinto1
Enrico Iaccino1Massimo Mallardo2Ileana Quinto1 Giuseppe Fiume1*
Giuseppe Fiume1*Rituximab is a commonly used chemotherapeutic drug for patients with aggressive lymphomas, such as non-Hodgkin’s lymphoma (NHL). Currently, the combination of Rituximab and chemotherapy (R-CHOP) stands as the most prevalent first-line therapy for NHL. Nevertheless, the development of new therapeutic approaches remains imperative. An increasing body of evidence highlights a novel role for IBTK in tumorigenesis and cancer growth. In this study, we aim to broaden our understanding of IBTK’s function in B-lymphoma, with a particular focus on its impact on the expression of the oncogene MYC. Here, we assessed the effects of combining Rituximab with IBTK silencing on cell viability through cell cycle analysis and Annexin V assays in vitro. Furthermore, we leveraged the transplantability of Eμ-myc lymphomas to investigate whether the inhibition of IBTK could elicit anti-tumor effects in the treatment of lymphomas in vivo. Our data suggests that IBTK silencing may serve as an effective anti-tumor agent for aggressive B-Lymphomas, underscoring its role in promoting apoptosis when used in combination with Rituximab, both in in vitro and in vivo settings.
Mounting evidence supports a novel role for IBTK in cell survival and tumor growth. IBTK exerts pleiotropic effects, impacting multiple pathways, including protein turnover as a substrate receptor of the Cullin 3 Ubiquitin ligase complex (CRL3IBTK) (1), and RNA metabolism, where it modifies RNA splicing in a cell-type-specific manner (2). Recent emerging research has further confirmed that silencing IBTK has the potential to induce apoptosis in hematological malignancies. In chronic lymphocytic leukemia (CLL), the overexpression of IBTK has been correlated with disease progression and proved essential for B cell survival under stress induced by chemotherapeutic agents (3). In the context of MYC-driven lymphomagenesis, the loss of IBTK primarily induces B-cell apoptosis and delays tumor onset (4). Previously, our research has demonstrated that IBTK haploinsufficiency also influences the tumor microenvironment in a mouse model of MYC-driven B-cell lymphoma (5). Recently, we demonstrated that silencing IBTK reduces cell viability and increases apoptosis in malignant B cells. Furthermore, our study revealed that IBTK upregulates the oncogene MYC expression, resulting in decreased survival of malignant B cells. This effect is mediated through the promotion of GSK3β ubiquitylation and proteasomal degradation, which occurs via the β-catenin axis (6). As a result, considerable attention has been directed toward exploring the potential therapeutic value of IBTK in aggressive B lymphomas. However, it’s worth noting that the impact of IBTK on the growth of MYC-driven B lymphomas had not been previously reported.
The tumorigenic potential of MYC often coincides with genetic and epigenetic alterations in hematological malignancies. Lymphomas encompass a highly diverse group of neoplasms that arise from the clonal expansion of B cells, T cells, or natural killer (NK) cells (7). Non-Hodgkin lymphoma (NHL) accounts for 90% of these cases. In Western countries, the most prevalent aggressive NHL subtypes include diffuse large B-cell lymphoma (DLBCL), Mantle Cell lymphoma (MCL), and Burkitt lymphoma. Regarding treatment options, Rituximab, in combination with chemotherapy (cyclophosphamide, doxorubicin, vincristine, and prednisone) or R-CHOP (8), has become the standard of care for adults with B-cell cancers, including patients with diffuse large B-cell lymphoma and Burkitt’s lymphoma (8). However, the emergence of rituximab resistance has become a significant challenge in the treatment of NHL patients (9), underscoring the need for the development of new therapeutic regimens. In this current study, our aim was to investigate the impact of combining Rituximab treatment with IBTK silencing on the growth of MYC-driven B lymphoma. We sought to enhance the effectiveness of anti-CD20 therapy in NHL.
Eμ-myc transgenic mice (TgN(IghMyc)22Bri/J) were obtained from The Jackson Laboratory (Bar Harbor, Maine; USA). Ibtk−/− were obtained as previously described (4). Both Eμ-myc transgenic mice and Ibtk−/− mice were congenic with C57BL/6 J mice. Eμ-myc trans-genic mice were crossed with Ibtk−/− or Ibtk+/− mice to generate Ibtk+/+ Eμ-myc and Ibtk−/− Eμ-myc littermates. The Eμ-myc transgene was detected by genomic PCR amplification of 600-bp product as described (10), while genotyping for Ibtk and βgeo genes was performed as previously described (4). Mice were monitored daily for signs of morbidity and tumor development. Moribund mice and mice with obvious tumors were sacrificed, and single-cell suspensions were obtained from tumor tissues and frozen in 10% DMSO for in vivo treatment.
To transplant Eμ-myc lymphomas to C57BL/6 mice, we thawed, washed, and counted viably frozen Eμ-myc lymphoma cells deriving from Ibtk+/+Eµ-myc and Ibtk-/-Eµ-myc mice. We suspended the lymphoma cells in RPMI media (GIBCO) and injected 1×106 lymphoma cells by the subcutaneous (s.c.) route into C57BL/6J mice, as previously described (11, 12). We monitored recipient mice daily, and as soon as each tumor volume reached about 100 mm3, the mice were randomly assigned to two groups (five mice per group): Vehicle, Rituximab 10 mg/kg (clinical formulation; Roche Diagnostics, Basel-Switzerland). Mice were treated by intraperitoneal injection of Vehicle or Rituximab (11, 12). The tumor volumes were determined by measuring length (L) and width (W) and then calculating volume (V = Length x Width2/2) at the indicated time points. At the end of treatment, mice were sacrificed, and the tumors were removed and weighed.
HEK293T, in addition to the Burkitt lymphoma cell lines Ramos, Raji, and Daudi, were purchased from Sigma‐Aldrich. It is noteworthy that Ramos cells are EBV-negative, whereas both Raji and Daudi are EBV-positive cell lines. Ramos, Raji and Daudi cells were grown in RPMI (Thermo Fisher Scientific, Waltham, MA, USA). HEK293T cells were grown in Dulbecco’s Modified Eagle Medium (DMEM; Thermo Fisher Scientific, Waltham, MA, USA). Cell culture media were supplemented with 10% fetal bovine serum (FBS), 2 mM L‐glutamine, 1 mM Na‐pyruvate, 50 mM 2βmercaptoethanol, 100 U/mL penicillin, and 100 μg/mL streptomycin; all reagents were purchased from Thermo Fisher Scientific. The plasmids pCMV6‐IBtkα‐FLAG and pCMV6 were from OriGene Technologies, Inc. (Rockville, MD, USA). The lentiviral constructs expressing the short hairpin RNA against IBtkα (shIBTK) or control short hairpin RNA (shCNTL) (TRCN0000082575 and SHC002, respectively) were from MISSION® (Sigma‐Aldrich, St. Louis, MO, USA).
HEK293T cells were transfected with plasmids using Lipofectamine 2000 (Thermo Fisher Scientific), according to the manufacturer’s protocol. Lentiviral particles were produced by transfection of HEK 293T cells, as previously described (6). Briefly, HEK293Tcells (1 × 106) were transfected with pCMV‐dR8.91 (5 μg) and pCMV‐VSVG (5 μg) together with shIBTK (10 μg) or shCNTL (10 μg); 48 h post‐transfection, cell supernatant was collected, filtered through 0.22 μm sterile filter, and used for spinoculation in the presence of 8 μg/mL polybrene. For IBtkα silencing, Ramos cells (3 × 106) were transduced with lentiviral particles (500 ng of p24) expressing shIBTK or shCNTL. Twenty‐four hours later, transduced cells were subjected to selection with puromycin (1.5 μg/mL and 0.2 μg/mL, respectively) for 48 h. When required, cells were treated with the RITUXIMAB (Roche Di-agnostics; stock solution 10mg/mL).
Annexin V‐based apoptotic assay was performed as previously described (4). Briefly, Ramos, Raji, and Daudi cells (1 × 106) were stained with FITC‐conjugated Annexin V and propidium iodide (PI) using the Annexin V‐FITC kit (Miltenyi Biotech). Data were collected by flow cytometry. Cell cycle analysis was performed as previously described (13).
Cells were lysed in modified RIPA buffer (10 mM Tris‐HCl, pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Igepal). Protein samples were subjected to electrophoresis on Nupage 4–12% polyacrylamide gel (Life Technologies), and then transferred onto a nitrocellulose membrane (GE Healthcare). Antibodies were: anti‐Myc (#5605; Cell Signaling Technology), and anti‐GAPDH (sc‐47724; Santa‐Cruz Biotechnology, Dallas, TX, USA).
Statistical analysis was performed by the two‐tailed unpaired Student’s t-test using the GraphPad Prism® software package. Statistical significance was determined by p < 0.05.
Rituximab stands as one of the most frequently utilized chemotherapeutic drugs in patients with aggressive lymphomas, particularly non-Hodgkin’s lymphoma (NHL) (14, 15). Currently, the foremost frontline therapy for NHL involves the combination of Rituximab with chemotherapy (cyclophosphamide, doxorubicin, vincristine, and prednisone), commonly referred to as R-CHOP (16). Nonetheless, this clinical standard underscores the necessity for the development of novel therapeutic approaches.
Recognizing that the acquisition of rituximab resistance often leads to chemotherapy failure in NHL patients (17), we conducted an investigation into the effects of combining Rituximab with IBTK silencing on cell viability. For our in vitro experiments, we employed the Ramos cell line as a model of NHL cells due to their relative resistance to rituximab therapy (17).
To achieve this, we transduced Ramos cells with lentiviral particles carrying short hairpin RNA for IBTK silencing (SHI) or a control short hairpin (SHC), followed by treatment with rituximab (10 μg/mL). Remarkably, our observations revealed that IBTK silencing in the presence of rituximab (10 μg/mL) substantially increased the subG1 phase (from 7.65% to 22.8%, p value < 0.005) and reduced the S phase (from 37.5% to 23.9%, p value < 0.05) (Figure 1A). These findings indicate a synergistic effect between IBTK depletion and Rituximab treatment on cell viability.
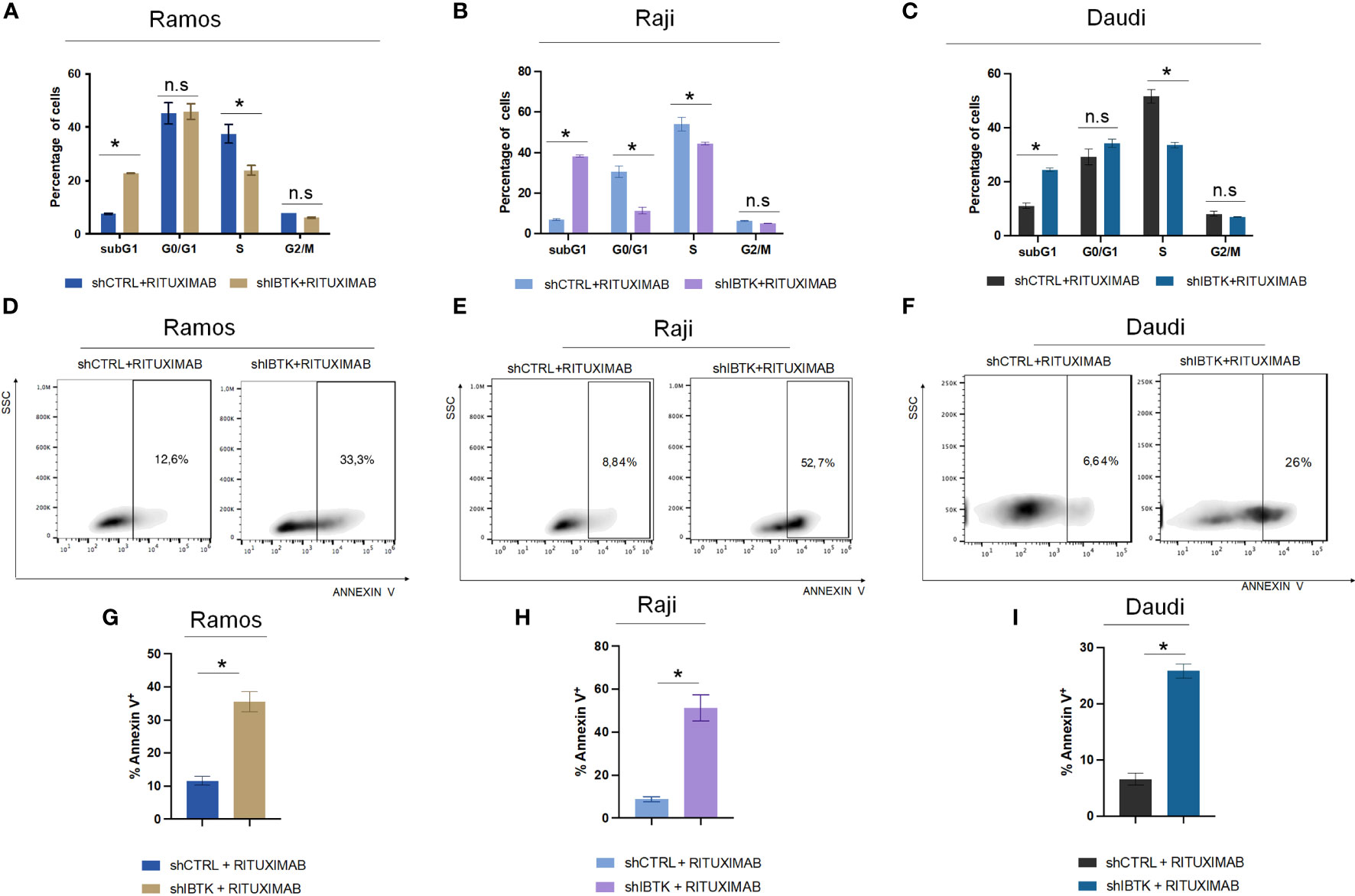
Figure 1 IBTK silencing strongly enhances the pro-apoptotic activity of Rituximab on a panel of B-lymphoma cells in vitro. Ramos, Raji and Daudi cells (1x106) were transduced with lentiviral particles (500 ng of p24) expressing short hairpins controls (shCTRL) or directed against IBTK mRNA (shIBTK). Next, cells were treated with or without Rituximab (10 µg/mL) for 48-hours. (A–C) shCTRL or shIBTK cells were labeled, fixed then stained with PI/RNase staining solution, and cell cycle analysis was performed by flow cytometry. The phases of cell cycle were evaluated by using the Watson pragmatic model. Values (mean ± SE, n = 3 for each group of samples) are shown. The bar indicates a statistically significant difference according Student’s t test. (D–F) Representative density plot of Annexin V binding assay of in vitro cultured shCTRL or shIBTK cells treated with Rituximab or left untreated. (G–I) Bar diagrams showing the quantification of apoptotic cells by Annexin V binding assay. Values (mean ± SE, n = 3 for each group of samples) are shown. The bar indicates a statistically significant difference according Student’s t test. Statistically significant difference was calculated according to Student’s t test and represented as asterisk for p value < 0.05. n.s. is for not statistically significant.
To corroborate the obtained results, we analyzed the cell cycle of NHL cell lines, Raji and Daudi in presence or absence of IBTK silencing treated with rituximab (10 μg/mL). In Raji cell line, we found that the combination of IBTK silencing and Rituximab strongly increased the subG1 phase (from 6.95% to 38.3%, p value < 0.05) along with a reduction of the percentage of cells in G0/G1 phase (from 30.9% to 12.25%, p value < 0.05), in S phase (from 56.55% to 44.75%, p value < 0.05) (Figure 1B). Consistent with the results obtained in the other NHL cell lines, in Daudi cell, we observed that the synergistic effect of IBTK silencing and Rituximab caused an increase of percentage of cell in subG1 phase (from 11.1% to 24.4%, p value < 0.005) along with a reduction of the percentage of cells in S phase (from 51.7% to 33.57%, p value < 0.05), (Figure 1C).
Subsequently, we investigated whether the synergistic effect resulting from the depletion of IBTK in combination with Rituximab treatment was attributed to the induction of apoptosis. Employing the Annexin V assay, we observed that the combination of IBTK silencing and Rituximab significantly increased the percentage of Annexin V-positive cells to 35.83%, whereas the apoptotic rate of Ramos cells treated with Rituximab alone was 11.65% (Figures 1D, 1G). Consistent with this result, the analysis of apoptosis by Annexin V assay, in Raji and Daudi cell lines silenced or not for IBTK and treated with Rituximab (10 mg/mL) showed an increase of the percentage of Annexin V-positive cells from 8.78% (SHC) to 51,23% (SHI) for Raji (Figures 1E, H) and from 6.61% (SHC) to 25.87% (SHI) for Daudi cells (Figures 1F, I). These results indicate consistency in the trends observed with the Ramos cell line, suggesting a broader applicability of our findings across different NHL cell types (Figure 1).
Given previous studies indicating MYC’s involvement in regulating cell cycle inhibitors during the early subG1 phase, influencing the transition from the G1 to the S phase (18), we sought to determine whether the combined effect of IBTK silencing and Rituximab treatment affected MYC protein levels. As anticipated, our findings demonstrated a more pronounced reduction in MYC protein levels in Ramos cells when both IBTK depletion and Rituximab treatment were employed, compared to either IBTK depletion or Rituximab treatment alone (Figures 2A, B).
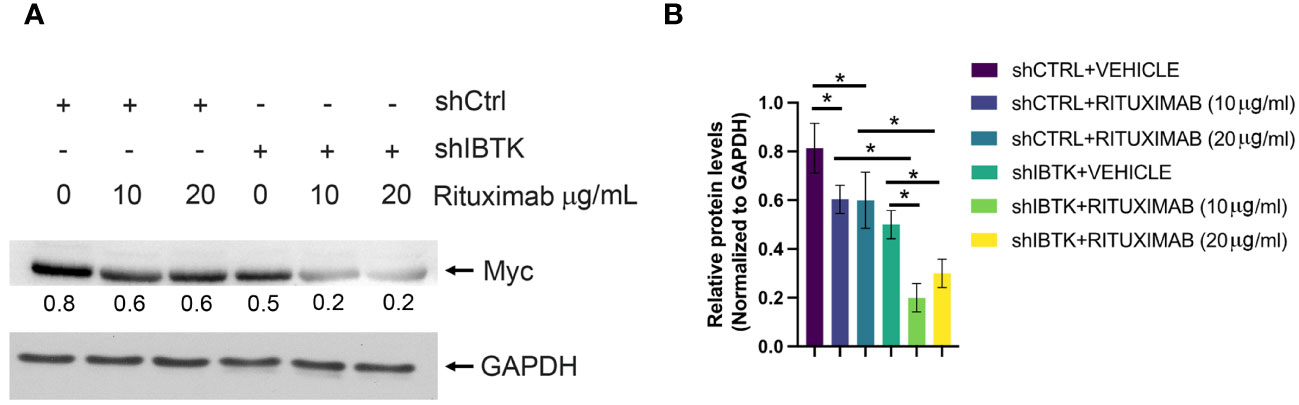
Figure 2 IBTK silencing with Rituximab treatment significatively reduces Myc protein expression on B-lymphoma cells in vitro. (A) Whole cell extracts (30 µg) from shCTRL or shIBTK Ramos, cells treated with Rituximab (10 µg/mL) or left untreated for 48 hours were separated by 12% SDS–PAGE and analyzed by western blotting using anti-MYC and anti-GAPDH antibodies. (B) Densitometric values of the Myc protein bands were normalized to GAPDH bands. Mean values ± SE are shown for three independent experiments. Statistically significant difference was calculated according to Student’s t test and represented as asterisk for p value < 0.05.
Collectively, these data suggest that the combination of IBTK silencing and Rituximab treatment induces a cell cycle block at the G1-S phase, which is associated with an increase in apoptotic rates, likely attributable to the reduction in MYC protein content (Figures 1A-C, 2A, B).
As IBTK silencing demonstrated inhibitory effects on B-lymphoma growth in vitro, we sought to investigate whether the loss of IBTK could exert anti-tumor effects in the treatment of lymphomas in an in vivo setting.
To address this, we employed the transplantability of Eµ-myc lymphomas, a preclinical mouse model of NHL (19), into immunocompetent C57BL/6 background strain mice. Specifically, we subcutaneously injected lymphoma cells (1x106 cells) derived from Ibtk+/+ Eµ-myc and Ibtk-/- Eµ-myc lymphomas into C57BL/6 wild-type mice. When each tumor volume reached approximately 100 mm3, which occurred about three days later, we administered vehicle or Rituximab intraperitoneally at a dose of 10 mg/kg.
Notably, we observed a significant reduction in both tumor volume and weight for tumors derived from Ibtk-/- Eµ-myc in comparison to those arising from Ibtk+/+ Eµ-myc. Furthermore, the combined absence of IBTK and Rituximab treatment demonstrated a synergistic effect in reducing both tumor volume and weight (p value < 0.001) (Figures 3A–C).
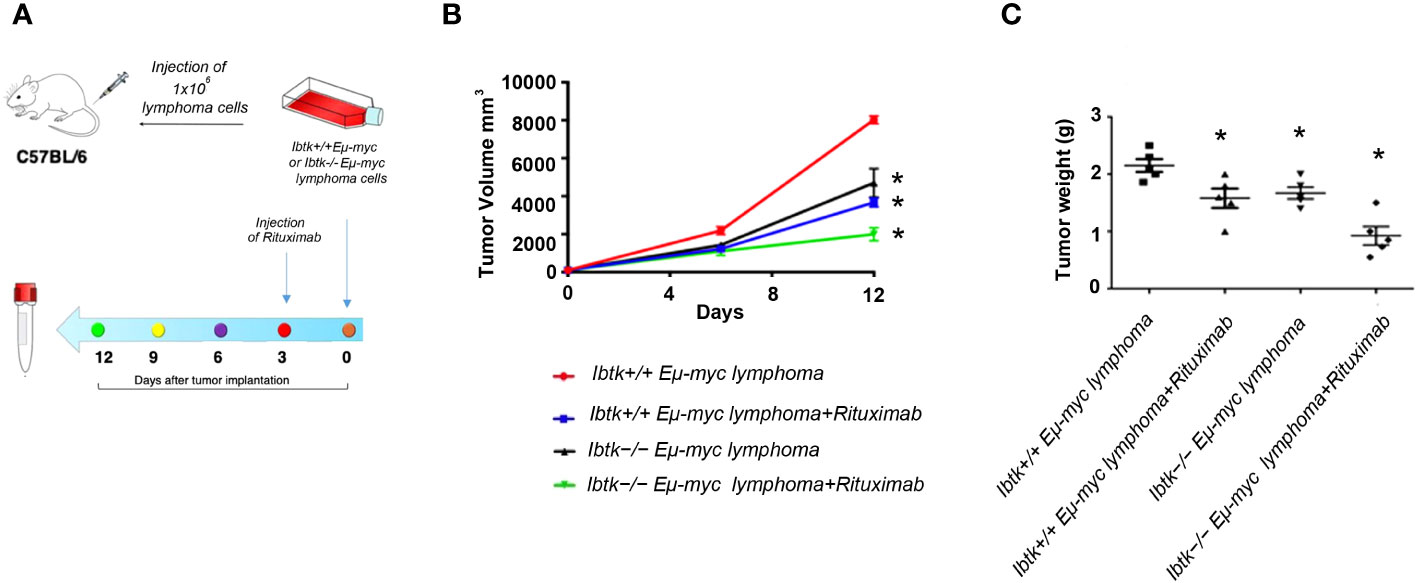
Figure 3 Effects of IBTK silencing combined with Rituximab treatment on in vivo model of B-lymphomas. (A) Workflow of the experimental design of in vivo analysis (B) Tumor growth curves of lymphoma cells (1x106), deriving from Ibtk+/+Emu-myc or Ibtk-/-Emu-myc lymphomas, subcutaneously injected into C57BL/6 wild type mice. When each tumor volume reached about 100 mm3, approximately three days after tumor engraftments, recipient mice were intraperitoneally treated with rituximab (10 mg/kg) or vehicle for indicated time. Values (mean ± SE, n = 5 for each group of samples) are shown. The asterisk indicates a statistically significant difference according Student’s t test (p < 0.01). (C) Weights of tumors deriving from subcutaneous injection of Ibtk+/+Emu-myc or Ibtk-/-Emu-myc lymphoma cells (1x106) after treatment with Rituximab (10 mg/kg) or vehicle for six days. Values (mean ± SE, n = 5 for each group of samples) are shown. The bar indicates a statistically significant difference according Student’s t test. Statistically significant difference was calculated according to Student’s t test and represented as asterisk for p value < 0.05.
The treatment of aggressive lymphomas, particularly non-Hodgkin’s lymphoma (NHL), represents a significant clinical challenge due to the emergence of resistance to Rituximab, a commonly employed chemotherapeutic agent (14, 15). The current standard of care for NHL involves the combination of Rituximab with chemotherapy, known as R-CHOP (16). Nevertheless, the development of rituximab resistance frequently leads to chemotherapy failure in NHL patients (17). To address this challenge, we embarked on a comprehensive investigation into the potential therapeutic benefits of combining Rituximab with IBTK (Inhibitor of Bruton Tyrosine Kinase) silencing, focusing on the enhancement of pro-apoptotic activity. Our in vitro results demonstrated that IBTK silencing in combination with Rituximab significantly increased the subG1 phase of cell cycle distribution and reduced the S phase. This shift towards a higher proportion of cells in the subG1 phase signifies an increase in apoptotic cells (17). The synergistic effect observed in our experiments underscores the potential of IBTK silencing to enhance the pro-apoptotic activity of Rituximab. Numerous studies have established that MYC deregulation is one of the most important events for aggressive B-lymphoma malignant transformation (20, 21). Overexpression of MYC is associated with high growth rates in vivo and in cell culture experiments, driving quiescent cells into the cell cycle (22). In contrast, low levels of MYC are associated with nondividing and differentiated cells (23). Previous studies showed that IBTK is a potential transcriptional target of MYC in aggressive MYC-driven B cell lymphomas (4, 24).
The observation of IBTK silencing-dependent apoptosis of cancerous B cells was consistent with previous observations in mouse embryonic fibroblasts (24), in DeFew and MEC-1 human B cell lines (3), and mouse pre-cancerous B-lymphoma cells (4). Interestingly, in our recent work, we identified as a direct interactor of IBTK, GSK3β (6), an essential component of the β-catenin destruction complex (25). β-catenin pathway is reported to be involved in the progression of many types of cancers, such as leukemia (26–28), myeloma (28, 29), and several subtypes of lymphoma (30, 31). Our study showed that down-regulation of IBTK could significantly inactivate β-catenin signaling through the stabilization of GSK3β which promotes β-catenin degradation, thus preventing the nuclear accumulation of β-catenin as well as the transcriptional activation of its target genes MYC, CCDN1, and CD44 (6).
Moving from our promising in vitro results to an in vivo setting, we utilized a preclinical mouse model of NHL to investigate the potential anti-tumor effects of IBTK loss. Our results demonstrate a significant reduction in both tumor volume and weight when IBTK was absent in conjunction with rituximab treatment. The synergistic reduction in tumor growth, highlighted in our study, is a noteworthy finding and underscores the potential clinical significance of combining IBTK silencing with Rituximab.
Long-term exposure of the tumor to certain drugs can lead to drug resistance and multi-drug combination is a promising approach to overcome drug resistance (32, 33). Intriguingly, we found that the combination of IBTK silencing with Rituximab treatment induces a cycle block at the G1-S phase associated with the reduction of MYC protein content and concomitantly to an increase of apoptotic rate. In addition, we found that IBTK loss and Rituximab treatment act synergistically in reducing tumor volume and weight in the Eµ-myc mouse model of non-Hodgkin’s lymphoma (NHL) (34). While our results offer promising insights, several limitations warrant consideration. First of all, our study predominantly focused on B-NHL cell lines and only one mouse model of B-NHL, not fully capturing the heterogeneity of NHL in clinical populations. We assume that we will also extend to test the combined approach in more complex and immunocompetent preclinical models, such as humanized mice. The consistency of the trends observed in our models (Ramos, Raji and Daudi cell lines) suggests a broader applicability of our findings across different NHL cell types (Figure 1). Of course, further investigations are needed to validate the translational potential of our findings. Our study presents compelling evidence that the combination of IBTK silencing with Rituximab enhances pro-apoptotic activity in NHL cells, both in vitro and in vivo. This holds great promise for addressing rituximab resistance and advancing the development of innovative therapeutic regimens for aggressive lymphomas. Future studies should aim to further elucidate the underlying mechanisms and expand the clinical applicability of this approach. Primary concern will be a consideration of regulatory approval pathways for precision medicine approaches, including early-phase studies in cohorts selected based on the IBTK profile. In addition, the evaluation of combinations with other targeted therapies, such as inhibitors of the PI3K/AKT pathway or CD20 signaling, and the assessment of the synergistic impact with immunotherapy, given its growing relevance in lymphoma, will be integral to our ongoing investigations.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study protocol was approved by the Bioethical Committee of the University Magna Graecia of Catanzaro. The animal experiments were carried out in accordance with the protocol n.794/2016‐PR, approved by the Italian Ministry of Health. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent was obtained from the owners for the participation of their animals in this study.
EV: Conceptualization, Data curation, Investigation, Methodology, Writing – original draft, Validation. RM: Methodology, Writing – original draft. SM: Data curation, Methodology, Writing – review & editing. CCan: Methodology, Writing – original draft. CCai: Methodology, Writing – review & editing. AA: Formal analysis, Writing – review & editing. MRR: Methodology, Writing – review & editing. MS: Data curation, Methodology, Writing – review & editing. GS: Data curation, Writing - review & editing. CP: Data curation, Writing – review & editing. EI: Data curation, Writing – review & editing. MM: Data curation, Formal analysis, Writing – review & editing. IQ: Conceptualization, Supervision, Validation, Visualization, Writing – review & editing. GF: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methodology, Resources, Supervision, Writing – original draft, Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This works was supported by the grants from Ministero dell’Istruzione, dell’Università e della Ricerca to GF (FFARB) and to IQ (PRIN).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Pisano A, Ceglia S, Palmieri C, Vecchio E, Fiume G, Laurentiis A, et al. CRL3IBTK regulates the tumor suppressor pdcd4 through ubiquitylation coupled to proteasomal degradation. J Biol Chem (2015) 290(22):13958–71. doi: 10.1074/jbc.M114.634535
2. Fiume G, Scialdone A, Rizzo F, Filippo De MR, Laudanna C, Albano F, et al. IBTK differently modulates gene expression and RNA splicing in heLa and K562 cells. Int J Mol Sci (2016) 17(11). doi: 10.3390/ijms17111848
3. Albano F, Chiurazzi F, Mimmi S, Vecchio E, Pastore A, Cimmino C, et al. The expression of inhibitor of bruton's tyrosine kinase gene is progressively up regulated in the clinical course of chronic lymphocytic leukaemia conferring resistance to apoptosis. Cell Death Dis (2018) 9(1):13. doi: 10.1038/s41419-017-0026-3
4. Vecchio E, Golino G, Pisano A, Albano F, Falcone C, Ceglia S, et al. IBTK contributes to B-cell lymphomagenesis in Emu-myc transgenic mice conferring resistance to apoptosis. Cell Death Dis (2019) 10(4):320. doi: 10.1038/s41419-019-1557-6
5. Vecchio E, Fiume G, Mignogna C, Iaccino E, Mimmi S, Maisano D, et al. IBTK haploinsufficiency affects the tumor microenvironment of myc-driven lymphoma in E-myc mice. Int J Mol Sci (2020) 21(3). doi: 10.3390/ijms21030885
6. Vecchio E, Nistico N, Golino G, Iaccino E, Maisano D, Mimmi S, et al. IBtkalpha Activates the beta-Catenin-Dependent Transcription of MYC through Ubiquitylation and Proteasomal Degradation of GSK3beta in Cancerous B Cells. Int J Mol Sci (2022) 23(4). doi: 10.3390/ijms23042044
7. Jaffe ES. Diagnosis and classification of lymphoma: Impact of technical advances. Semin Hematol (2019) 56(1):30–6. doi: 10.1053/j.seminhematol.2018.05.007
8. Minard-Colin V, Auperin A, Pillon M, Burke GAA, Barkauskas DA, Wheatley K, et al. Rituximab for high-risk, mature B-cell non-hodgkin's lymphoma in children. N Engl J Med (2020) 382(23):2207–19. doi: 10.1056/NEJMoa1915315
9. Sabo A, Kress TR, Pelizzola M, Pretis S, Gorski MM, Tesi A, Kress TR, Pelizzola M, Pretis S, Gorski MM, Tesi A, et al. Selective transcriptional regulation by Myc in cellular growth control and lymphomagenesis. Nature (2014) 511(7510):488–92. doi: 10.1038/nature13537
10. Lin CJ, Nasr Z, Premsrirut PK, Porco JA Jr, Hippo Y, Lowe SW, et al. Targeting synthetic lethal interactions between Myc and the eIF4F complex impedes tumorigenesis. Cell Rep (2012) 1(4):325–33. doi: 10.1016/j.celrep.2012.02.010
11. Dalle S, Dupire S, Brunet-Manquat S, Reslan L, Plesa A, Dumontet C. In vivo model of follicular lymphoma resistant to rituximab. Clin Cancer Res (2009) 15(3):851–7. doi: 10.1158/1078-0432.CCR-08-1685
12. Hicks SW, Lai KC, Gavrilescu LC, Yi Y, Sikka S, Shah P, et al. The antitumor activity of IMGN529, a CD37-targeting antibody-drug conjugate, is potentiated by rituximab in non-hodgkin lymphoma models. Neoplasia (2017) 19(9):661–71. doi: 10.1016/j.neo.2017.06.001
13. Scicchitano S, Vecchio E, Battaglia AM, Oliverio M, Nardi M, Procopio A, et al. The double-edged sword of oleuropein in ovarian cancer cells: from antioxidant functions to cytotoxic effects. Int J Mol Sci (2023) 24(1). doi: 10.3390/ijms24010842
14. Rava M, A, Nicoli P, Gritti I, Donati G, Doni M, et al. Therapeutic synergy between tigecycline and venetoclax in a preclinical model of MYC/BCL2 double-hit B cell lymphoma. Sci Transl Med (2018) 10(426). doi: 10.1126/scitranslmed.aan8723
15. Smith MR. Rituximab (monoclonal anti-CD20 antibody): mechanisms of action and resistance. Oncogene (2003) 22(47):7359–68. doi: 10.1038/sj.onc.1206939
16. Coiffier B, Thieblemont C, Neste Den Van E, Lepeu G, Plantier I, Castaigne S, et al. Long-term outcome of patients in the LNH-98.5 trial, the first randomized study comparing rituximab-CHOP to standard CHOP chemotherapy in DLBCL patients: a study by the Groupe d'Etudes des Lymphomes de l'Adulte. Blood (2010) 116(12):2040–5. doi: 10.1182/blood-2010-03-276246
17. Deyab M, Elbanani A, Tabal S, Geriani H, Lamami Y, Bredan A, et al. Rituximab synergizes with hydroxyurea or vincristine in the killing of Ramos Burkitt's lymphoma B cell line. Cancer Biother Radiopharm (2014) 29(2):87–90. doi: 10.1089/cbr.2013.1557
18. Yap CS, Peterson AL, Castellani G, Sedivy JM, Neretti N. Kinetic profiling of the c-Myc transcriptome and bioinformatic analysis of repressed gene promoters. Cell Cycle (2011) 10(13):2184–96. doi: 10.4161/cc.10.13.16249
19. Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, et al. The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid Malignancy in transgenic mice. Nature (1985) 318(6046):533–8. doi: 10.1038/318533a0
20. Ott G, Rosenwald A, Campo E. Understanding MYC-driven aggressive B-cell lymphomas: pathogenesis and classification. Hematol Am Soc Hematol Educ Program (2013) 2013:575–83. doi: 10.1182/asheducation-2013.1.575
21. Basso K, Dalla-Favera R. Germinal centres and B cell lymphomagenesis. Nat Rev Immunol (2015) 15(3):172–84. doi: 10.1038/nri3814
22. Oster SK, Ho CS, Soucie EL, Penn LZ. The myc oncogene: MarvelouslY Complex. Adv Cancer Res (2002) 84:81–154. doi: 10.1016/S0065-230X(02)84004-0
23. Spotts GD, Hann SR. Enhanced translation and increased turnover of c-myc proteins occur during differentiation of murine erythroleukemia cells. Mol Cell Biol (1990) 10(8):3952–64. doi: 10.1128/mcb.10.8.3952-3964.1990
24. Baird TD, Palam LR, Fusakio ME, Willy JA, Davis CM, McClintick JN, et al. Selective mRNA translation during eIF2 phosphorylation induces expression of IBTKalpha. Mol Biol Cell (2014) 25(10):1686–97. doi: 10.1091/mbc.e14-02-0704
25. Ruiduo C, Ying D, Qiwei W. CXCL9 promotes the progression of diffuse large B-cell lymphoma through up-regulating beta-catenin. BioMed Pharmacother (2018) 107:689–95. doi: 10.1016/j.biopha.2018.07.171
26. Roman-Gomez J, Cordeu L, Agirre X, Jimenez-Velasco A, Jose-Eneriz San E, Garate L, et al. Epigenetic regulation of Wnt-signaling pathway in acute lymphoblastic leukemia. Blood (2007) 109(8):3462–9. doi: 10.1182/blood-2006-09-047043
27. Chim CS, Liang R, Fung TK, Choi CL, Kwong YL. Epigenetic dysregulation of the death-associated protein kinase/p14/HDM2/p53/Apaf-1 apoptosis pathway in multiple myeloma. J Clin Pathol (2007) 60(6):664–9. doi: 10.1136/jcp.2006.038331
28. Kim H, Moon HW, Hur M, Yun YM, Park CM, Lee MH. Variant Burkitt-type translocation (8;22)(q24;q11) in plasma cell myeloma. Korean J Hematol (2011) 46(2):135–8. doi: 10.5045/kjh.2011.46.2.135
29. Chim CS, Pang R, Fung TK, Choi CL, Liang R. Epigenetic dysregulation of Wnt signaling pathway in multiple myeloma. Leukemia (2007) 21(12):2527–36. doi: 10.1038/sj.leu.2404939
30. Liu JJ, Dai XJ, Xu Y, Liu PQ, Zhang Y, Liu XD, et al. Inhibition of lymphoma cell proliferation by peroxisomal proliferator-activated receptor-gamma ligands via Wnt signaling pathway. Cell Biochem Biophys (2012) 62(1):19–27. doi: 10.1007/s12013-011-9253-x
31. Su N, Wang P, Li Y. Role of Wnt/beta-catenin pathway in inducing autophagy and apoptosis in multiple myeloma cells. Oncol Lett (2016) 12(6):4623–9. doi: 10.3892/ol.2016.5289
32. Catalano A, Iacopetta D, Ceramella J, Scumaci D, Giuzio F, Saturnino C, et al. Multidrug resistance (MDR): A widespread phenomenon in pharmacological therapies. Molecules (2022) 27(3). doi: 10.3390/molecules27030616
33. Wang X, Zhang H, Chen X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist (2019) 2(2):141–60. doi: 10.20517/cdr.2019.10
Keywords: rituximab, non-Hodgkin’s lymphoma, MYC, IBTK, apoptosis
Citation: Vecchio E, Marino R, Mimmi S, Canale C, Caiazza C, Arcucci A, Ruocco MR, Schiavone M, Santamaria G, Palmieri C, Iaccino E, Mallardo M, Quinto I and Fiume G (2024) Enhanced pro-apoptotic activity of rituximab through IBTK silencing in non-Hodgkin lymphoma B-cells. Front. Oncol. 14:1339584. doi: 10.3389/fonc.2024.1339584
Received: 17 November 2023; Accepted: 17 January 2024;
Published: 02 February 2024.
Edited by:
Lorenzo Leoncini, University of Siena, ItalyReviewed by:
Eleni Anastasiadou, Sapienza University of Rome, ItalyCopyright © 2024 Vecchio, Marino, Mimmi, Canale, Caiazza, Arcucci, Ruocco, Schiavone, Santamaria, Palmieri, Iaccino, Mallardo, Quinto and Fiume. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eleonora Vecchio, ZWxlb25vcmF2ZWNjaGlvQHVuaWN6Lml0; Giuseppe Fiume, Zml1bWVAdW5pY3ouaXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.