 Joaquin Garcia-Solorio1†
Joaquin Garcia-Solorio1† Juan Carlos Núñez-Enriquez2†
Juan Carlos Núñez-Enriquez2† Marco Jiménez-Olivares1
Marco Jiménez-Olivares1 Janet Flores-Lujano2
Janet Flores-Lujano2 Fernanda Flores-Espino1
Fernanda Flores-Espino1 Carolina Molina-Garay1
Carolina Molina-Garay1 Alejandra Cervera3
Alejandra Cervera3 Diana Casique-Aguirre4,5
Diana Casique-Aguirre4,5 José Gabriel Peñaloza-Gonzalez6
José Gabriel Peñaloza-Gonzalez6 Ma. Del Rocío Baños-Lara7
Ma. Del Rocío Baños-Lara7 Ángel García-Soto8
Ángel García-Soto8 César Alejandro Galván-Díaz9
César Alejandro Galván-Díaz9 Alberto Olaya-Vargas9
Alberto Olaya-Vargas9 Hilario Flores Aguilar10
Hilario Flores Aguilar10 Minerva Mata-Rocha11
Minerva Mata-Rocha11 Miguel Ángel Garrido-Hernández12Juan Carlos Solís-Poblano13Nuria Citlalli Luna-Silva14Lena Sarahi Cano-Cuapio15Pierre Mitchel Aristil-Chery16Fernando Herrera-Quezada2
Miguel Ángel Garrido-Hernández12Juan Carlos Solís-Poblano13Nuria Citlalli Luna-Silva14Lena Sarahi Cano-Cuapio15Pierre Mitchel Aristil-Chery16Fernando Herrera-Quezada2 Karol Carrillo-Sanchez1
Karol Carrillo-Sanchez1 Anallely Muñoz-Rivas1Luis Leonardo Flores-Lagunes1
Anallely Muñoz-Rivas1Luis Leonardo Flores-Lagunes1 Elvia Cristina Mendoza-Caamal1
Elvia Cristina Mendoza-Caamal1 Beatriz Eugenia Villegas-Torres1
Beatriz Eugenia Villegas-Torres1 Vincent González-Osnaya1
Vincent González-Osnaya1 Elva Jiménez-Hernández17
Elva Jiménez-Hernández17 José Refugio Torres-Nava17Jorge Alfonso Martín-Trejo18
José Refugio Torres-Nava17Jorge Alfonso Martín-Trejo18 María de Lourdes Gutiérrez-Rivera19
María de Lourdes Gutiérrez-Rivera19 Rosa Martha Espinosa-Elizondo20Laura Elizabeth Merino-Pasaye21
Rosa Martha Espinosa-Elizondo20Laura Elizabeth Merino-Pasaye21 María Luisa Pérez-Saldívar2
María Luisa Pérez-Saldívar2 Silvia Jiménez-Morales22
Silvia Jiménez-Morales22 Everardo Curiel-Quesada23
Everardo Curiel-Quesada23 Haydeé Rosas-Vargas11
Haydeé Rosas-Vargas11 Juan Manuel Mejía-Arangure24,25*
Juan Manuel Mejía-Arangure24,25* Carmen Alaez-Verson1*
Carmen Alaez-Verson1*- 1Laboratorio de Diagnóstico Genómico, Instituto Nacional de Medicina Genómica (INMEGEN), Mexico City, Mexico
- 2Unidad de Investigación Médica en Epidemiología Clínica, Unidad Medica de Alta Especialidad (UMAE) Hospital de Pediatría, Centro Médico Nacional (CMN) Siglo XXI, Instituto Mexicano del Seguro Social (IMSS), Mexico City, Mexico
- 3Subdirección de Genómica Poblacional, Instituto Nacional de Medicina Genomica (INMEGEN), Mexico City, Mexico
- 4Laboratorio de Citómica del Cáncer Infantil, Centro de Investigación Biomédica de Oriente, Instituto Mexicano del Seguro Social, Delegación Puebla, Puebla, Mexico
- 5Consejo Nacional de Humanidades, Ciencias y Tecnologías (CONAHCYT), Mexico City, Mexico
- 6Servicio de Onco-Pediatría, Hospital Juárez de México, Secretaría de Salud (SSA), Mexico City, Mexico
- 7Centro de Investigación Oncológica Una Nueva Esperanza, Universidad Popular Autónoma del Estado de Puebla, Puebla, Mexico
- 8Hospital General Centro Médico La Raza, Instituto Mexicano del Seguro Social (IMSS), Mexico City, Mexico
- 9Departamento de Oncologia, Instituto Nacional de Pediatría (INP), Mexico City, Mexico
- 10Departamento de Inmunogenetica, Instituto de Diagnostico y Referencia Epidemiologicos (InDRE), Mexico City, Mexico
- 11Unidad de Investigación Médica en Genética Humana, Hospital de Pediatría, CMN Siglo XXI, Instituto Mexicano del Seguro Social (IMSS), Mexico City, Mexico
- 12Servicio de Oncohematología Pediátrica, Hospital para el Niño Poblano, Secretaría de Salud (SS), Puebla, Mexico
- 13Servicio de Oncohematología Pediátrica, Instituto Mexicano del Seguro (IMSS) Unidad Médica de Alta Especialidad (UMAE) Centro Médico Nacional (CMN) Hospital de Especialidades Dr. Manuel Ávila Camacho, Puebla, Mexico
- 14Servicio de Hemato-Oncología Pediátrica, Hospital de la Niñez Oaxaqueña "Dr. Guillermo Zárate Mijangos", Secretaria de Salud y Servicios de Salud Oaxaca (SSO), Oaxaca, Mexico
- 15Hospital Infantil de Tlaxcala, Servicio de Oncología Pediátrica, Tlaxcala, Mexico
- 16Instituto de Seguridad y Servicios Sociales de los Trabajadores al Servicio de los Poderes del Estado (ISSSTE) de Puebla, Departamento de Enseñanza e Investigació, Puebla, Mexico
- 17Servicio de Oncología, Hospital Pediátrico Moctezuma, Secretaría de Salud de la Ciudad de México (SSCDMX), Mexico City, Mexico
- 18Servicio de Hematología, Centro Médico Nacional Siglo XXI, Instituto Mexicano del Seguro Social (IMSS), Unidad Médica de Alta Especialidad (UMAE) Hospital de Pediatría “Dr. Silvestre Frenk Freund”, Mexico City, Mexico
- 19Servicio de Oncología, Centro Médico Nacional Siglo XXI, Instituto Mexicano del Seguro Social (IMSS), Unidad Médica de Alta Especialidad (UMAE) Hospital de Pediatría “Dr. Silvestre Frenk Freund”, Mexico City, Mexico
- 20Servicio de Hematología Pediátrica, Hospital General de México, Secretaría de Salud (SSA), Mexico City, Mexico
- 21Servicio de Hematología Pediátrica, Centro Médico Nacional (CMN) “20 de Noviembre”, Instituto de Seguridad Social al Servicio de los Trabajadores del Estado (ISSSTE), Mexico City, Mexico
- 22Laboratorio de Medicina de Precisión, Instituto Nacional de Medicina Genómica (INMEGEN), Mexico City, Mexico
- 23Departamento de Bioquímica, Escuela Nacional de Ciencias Biológicas, Instituto Politecnico Nacional (IPN), Mexico City, Mexico
- 24Laboratorio de Genómica Funcional del Cáncer, Instituto Nacional de Medicina Genómica (INMEGEN), Mexico City, Mexico
- 25Facultad de Medicina, Universidad Nacional Autónoma de México (UNAM), Mexico City, Mexico
Background: Recurrent genetic alterations contributing to leukemogenesis have been identified in pediatric B-cell Acute Lymphoblastic Leukemia (B-ALL), and some are useful for refining classification, prognosis, and treatment selection. IKZF1plus is a complex biomarker associated with a poor prognosis. It is characterized by IKZF1 deletion coexisting with PAX5, CDKN2A/2B, or PAR1 region deletions. The mutational spectrum and clinical impact of these alterations have scarcely been explored in Mexican pediatric patients with B-ALL. Here, we report the frequency of the IKZF1plus profile and the mutational spectrum of IKZF1, PAX5, CDKN2A/2B, and ERG genes and evaluate their impact on overall survival (OS) in a group of patients with B-ALL.
Methods: A total of 206 pediatric patients with de novo B-ALL were included. DNA was obtained from bone marrow samples at diagnosis before treatment initiation. A custom-designed next-generation sequencing panel was used for mutational analysis. Kaplan-Meier analysis was used for OS estimation.
Results: We identified the IKZF1plus profile in 21.8% of patients, which was higher than that previously reported in other studies. A significantly older age (p=0.04), a trend toward high-risk stratification (p=0.06), and a decrease in 5-year Overall Survival (OS) (p=0.009) were observed, although heterogeneous treatment protocols in our cohort would have impacted OS. A mutation frequency higher than that reported was found for IKZF1 (35.9%) and CDKN2A/2B (35.9%) but lower for PAX5 (26.6%). IKZF1MUT group was older at diagnosis (p=0.0002), and most of them were classified as high-risk (73.8%, p=0.02), while patients with CDKN2A/2BMUT had a higher leukocyte count (p=0.01) and a tendency toward a higher percentage of blasts (98.6%, >50% blasts, p=0.05) than the non-mutated patients. A decrease in OS was found in IKZF1MUT and CDKN2A/2BMUT patients, but the significance was lost after IKZF1plus was removed.
Discussion: Our findings demonstrated that Mexican patients with B-ALL have a higher prevalence of genetic markers associated with poor outcomes. Incorporating genomic methodologies into the diagnostic process, a significant unmet need in low- and mid-income countries, will allow a comprehensive identification of relevant alterations, improving disease classification, treatment selection, and the general outcome.
1 Introduction
Genomic and transcriptomic analyses performed on large cohorts of pediatric and adult B-ALL patients have identified recurrent genetic alterations and expression signatures that contribute to leukemogenesis. Some of these have clinical utility as biomarkers for refining disease classification and treatment selection. The last WHO classification for B-lymphoblastic leukemia/lymphoma comprises 13 subtypes, depending on the specific genetic alterations acquired in the leukemic cells (1). In addition to gene fusion information, specific treatment protocols currently incorporate a combined evaluation of copy number alterations (CNAs) in the selected genes. CNAs affecting one or more genes related to cell differentiation, cell cycle control, and apoptosis have been identified in approximately 71% of pediatric B-ALL cases (2). Some CNA combinations, such as the IKZF1plus profile, have been identified as adverse modifiers of childhood B-ALL prognosis (3).
IKZF1plus is a high-risk category identified by Stanulla M et al. (3) in a cohort of 991 patients with B-ALL enrolled in the International Multicenter Trial AIEOP-BFM ALL 2000. IKZF1plus was defined as “deletion of IKZF1 that co-occurred with at least one additional deletion in CDKN2A, CDKN2B (homozygous deletion only), PAX5, or PAR1 in the absence of ERG deletion”. The PAR1 region (pseudoautosomal region 1) is located in Xp22 and Yp11 and comprises the CRLF2, CSF2RA, and IL3RA genes. Patients with deletions affecting ERG were excluded from the IKZF1plus group. IKZF1plus was associated with a very poor prognosis in B-ALL patients with detectable minimal residual disease (MRD) (3). Following the initial report, several other studies have confirmed that this subgroup of patients had inferior outcomes (4–7).
IKZF1 encodes Ikaros, a zinc-finger transcription factor required for the development of all lymphoid lineages. Deletions (IKZF1DEL) and small mutations in this gene lead to the acquisition of a stem cell-like phenotype by haploinsufficiency and loss of DNA-binding capacity (8). These alterations have been reported to be independent biomarkers of adverse prognoses (9–11). Deletions in the ERG gene (ERGDEL) can suppress this negative outcome, whereas JAK-STAT activation enhances this adverse effect (12, 13). These findings support the importance of evaluating the mutational profiles of several genes rather than specific alterations in isolated genes for more accurate risk stratification.
CDKN2A and CDKN2B (CDKN2A/2B) are tumor suppressor genes located on chromosome 9p21 and encode three key cell cycle regulators, p16INK4A and p14ARF, encoded by the alternative readings of CDKN2A, and p15INK4B, encoded by CDKN2B (14). CDKN2A/2B deletions (CDKN2A/2BDEL) have been detected in approximately 20–25% of pediatric patients with B-ALL (15). It has been suggested that inactivation of CDKN2A/2B is a secondary cooperative event that plays an essential role in leukemogenesis, cell cycle regulation, chemosensitivity, and apoptosis (14). The contribution of this alteration to prognosis is controversial. Some studies support that CDKN2A/2BDEL, especially in the case of biallelic status, is associated with inferior outcomes in B-ALL (16–18), while others claim that this alteration is not a poor prognostic factor in childhood B-ALL (14, 19).
PAX5 encodes a transcription factor that participates in the development of normal B cells and in maintaining cell identity by repressing the signature genes of other lineages during differentiation (20). PAX5 is found in a physiological complex with IKZF1 and RUNX1 (21). Somatic or germline alterations that deregulate PAX5 activity may lead to B cell malignancies (22). PAX5 alterations include deletions (PAX5DEL), focal intragenic amplifications (PAX5igAMP), translocations with various partners, or point mutations. All these were identified in B-ALL at different frequencies (23). Alterations in PAX5 have been associated with inferior outcomes (2), which are worse when IKZF1 deletion is present (24).
The frequency of alterations, mutational spectrum, and clinical impact of IKZF1, PAX5, CDKN2A/2B, ERG gene mutations, and the IKZF1plus profile have not been extensively explored in Mexican pediatric B-ALL patients. A better understanding of genomic alterations occurring in B-ALL pediatric leukemia and their clinical impact is crucial for Hispanic populations such as Mexicans. High incidence rates of childhood acute leukemia have been consistently observed in the Mexican population (55.0 cases per million children under 15 years of age) (25–27). Additionally, mortality and morbidity rates are higher than those in high-income countries, particularly as a consequence of deaths related to refractory disease and treatment-related toxicity (28, 29). The identification of clinically relevant genetic alterations may contribute to more effective and personalized treatment options for these patients, potentially improving their outcomes while minimizing unnecessary treatment-related side effects and costs. Genomic alterations play a significant role in determining disease evolution and the response of cancer cells to specific treatments (30). Genetic data from ethnically diverse populations would help obtain a more comprehensive understanding of leukemia subtypes and treatment responses across diverse populations.
This study aimed to identify the frequency and heterogeneity of the IKZF1plus profile and mutational spectrum of IKZF1, PAX5, CDKN2A/2B, and ERG in a cohort of 206 Mexican pediatric patients with de novo B-ALL. The clinical impact on overall survival was also evaluated for the analyzed genes and IKZF1plus profile.
2 Materials and methods
2.1 Population
This analysis included 206 de novo B-ALL pediatric Mexican patients from 10 different states of Mexico (Supplementary Table 1A displays the number of patients by state). Patients were treated at 18 public health institutions (Supplementary Table 1B). The diagnosis was established between January 2018 and April 2023 by pediatric hematologists/oncologists according to clinical features: cell morphology, immunophenotype, and genetics, as defined by the 2008 WHO classification of lymphoid neoplasms (31). The patients had a range of 0 to 17 years, and only 3 of them were younger than one year. The risk classification was established as defined by the Children’s Oncology Group (COG) and the National Cancer Institute (NCI) (32). The presence of at least one of the following clinical features was considered as high-risk: age at diagnosis ≥10 years, WBC counts higher than 5x104 count/µL, positive minimal residual disease (MRD) on day 28, presence of nervous system infiltration, treatment failure response, and identification of any high-risk gene fusion (when that information was available). Cytogenetic results were unavailable for most patients. Before treatment initiation, 3–5 mL of bone marrow was collected into EDTA tubes at their respective medical institutions and sent to the Laboratory of Genomic Diagnosis at INMEGEN. Clinical data were collected from medical charts, including the child’s sex, age, white blood cell count in the peripheral blood, and percentage of blasts in the bone marrow at diagnosis and treatment protocol. The median follow-up of these patients was 1.4 years.
2.2 Nucleic acid extraction and quantification
DNA was isolated using the Maxwell® RSC Instrument (Promega Corporation, Madison, WI, USA). DNA concentration and quality were evaluated using a NanoDrop® 2000 spectrophotometer (Thermo Fisher Scientific Inc., Waltham, MA, USA) and a Qubit® 4 Qubit 1X dsDNA HS Assay Kit (Thermo Fisher Scientific Inc., Waltham, MA, USA).
2.3 Next-generation sequencing (Targeted DNAseq) and bioinformatics
A customized panel was designed to identify genetic alterations in CDKN2A, CDKN2B, PAX5, ERG, and IKZF1. Panel synthesis was performed using an Archer Dx (ArcherDX, Inc., Boulder, CO, USA). Library preparation was performed following the manufacturer’s instructions. Libraries were sequenced on the NextSeq 500/550 using the High Output Sequencing Reagent Kit v2.5 (300 cycles) (Illumina, Inc., San Diego, CA, USA). Bioinformatics analysis was performed using the Archer Suite Analysis v5.1.3 software (ArcherDX, Inc., Boulder, CO, USA) using the human reference genome GRCh37.p13/hg19. The bioinformatics pipeline allowed the identification of single nucleotide variations, small insertions/deletions, copy number alterations (CNAs), and structural variations (SV) (this last only for the IKZF1 gene). According to the Archer Dx bioinformatics pipeline (ArcherDX, Inc., Boulder, CO, USA), SV occurs when individual reads contain nucleotide sequences that are aligned to different genome regions. The length of the SV was determined by observing the breakpoint positions of the partners identified in the event and subtracting the differences in their genomic positions (User Manual Archer Analysis 6.0 CS001). Mutations were considered if the variant allele fraction (VAF) was ≥ 5%. A minimum 500x-fold depth coverage was required for all targeted regions in the panel.
2.4 Multiplex ligation-dependent probe amplification
NGS-predicted IKZF1, CDKN2A/2B, and PAX5 deletions were confirmed by MLPA using SALSA Probemix P335 (ALL-IKZF1) according to the manufacturer’s protocol (Holland, Amsterdam, Netherlands). This probe also allowed for the identification of PAR1 deletions, as described previously (3). The P327 iAMP21-ERG probe mix was used to confirm ERG deletions. Fragment separation was performed on a 3500 Genetic Analyzer (Thermo Fisher Scientific Inc., Waltham, MA, USA). The raw data files were imported and analyzed using Coffalyser.Net™ software (MRC Holland, Amsterdam, Netherlands).
2.5 IKZF1plus profile definition
The IKZF1plus profile was defined based on the Stanulla et al., 2018 report (3). Briefly, the patient was considered IKZF1plus positive if IKZF1DEL co-occurred with CDKN2ADEL, CDKN2BDEL (homozygous deletion only), PAX5DEL, or pseudoautosomal region 1 (PAR1) deletion. As it has been reported that ERGDEL mitigates the adverse prognosis associated with IKZF1 deletions (12), patients with ERGDEL were excluded from the IKZF1plus group. The NO-IKZF1plus group included any other patient, regardless of whether they were IKZF1MUT or IKZF1WT.
2.6 Statistical analysis
Measures of central tendency were used to describe continuous variables related to the patient’s clinical and demographic features. The χ² test or Fisher’s exact test was used to assess the relationship between categorical variables. In contrast, the nonparametric Mann–Whitney U test was used to analyze the association between continuous variables, where p <0.05 was considered statistically significant. OS was estimated using the Kaplan-Meier method. The log-rank test was used to evaluate the differences between survival distributions with a 95% confidence interval (CI). The Cox regression model was used to perform a multivariate analysis, with adjustments made for other risk factors such as sex, age at diagnosis, WBC count, percentage of blast cells, and risk stratification at diagnosis. OS was calculated from the day of diagnosis until either the last follow-up or death from any cause. Patients who did not experience any event were censored at the last follow-up visit. Those who did not attend the follow-up were censored at the date of the last known contact. Statistical calculations were performed using the RStudio software version 4.3.1, and the data were visualized using the ggplot2 package version 3.4.3.
3 Results
The clinical features of the study population are summarized in Table 1. Most patients (63.2%) were classified as high-risk. The most frequent high-risk features were age at diagnosis < 1 or ≥ 10 years, WBC counts higher than 5x104 count/µL, followed by positive minimal residual disease (MRD) on day 28. The remaining patients were classified as having a standard risk.
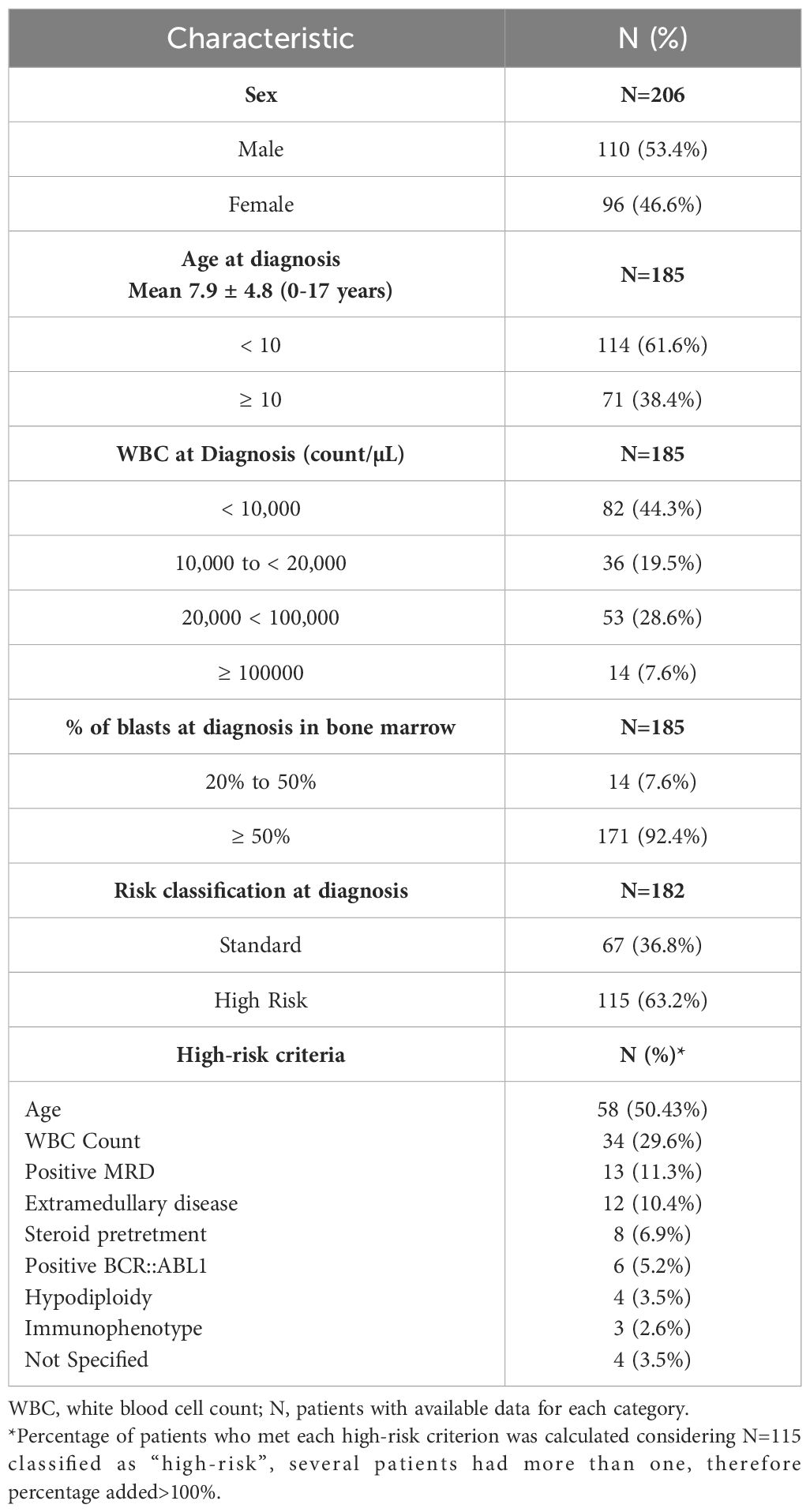
Table 1 Clinical features of the analyzed population.
This group of patients was treated using six different protocols, depending on the medical preferences and institutional resources available in each case. The number of patients per treatment protocol is presented in Supplementary Table 2.
3.1 Frequency of IKZF1plus profile in B-ALL Mexican pediatric patients
The IKZF1plus profile was present in 21.8% (45/206) of the evaluated patients. Gene deletion combinations within the IKZF1plus group are shown in Table 2. In our series, three patients were excluded from the IKZF1plus profile because of ERGDEL.
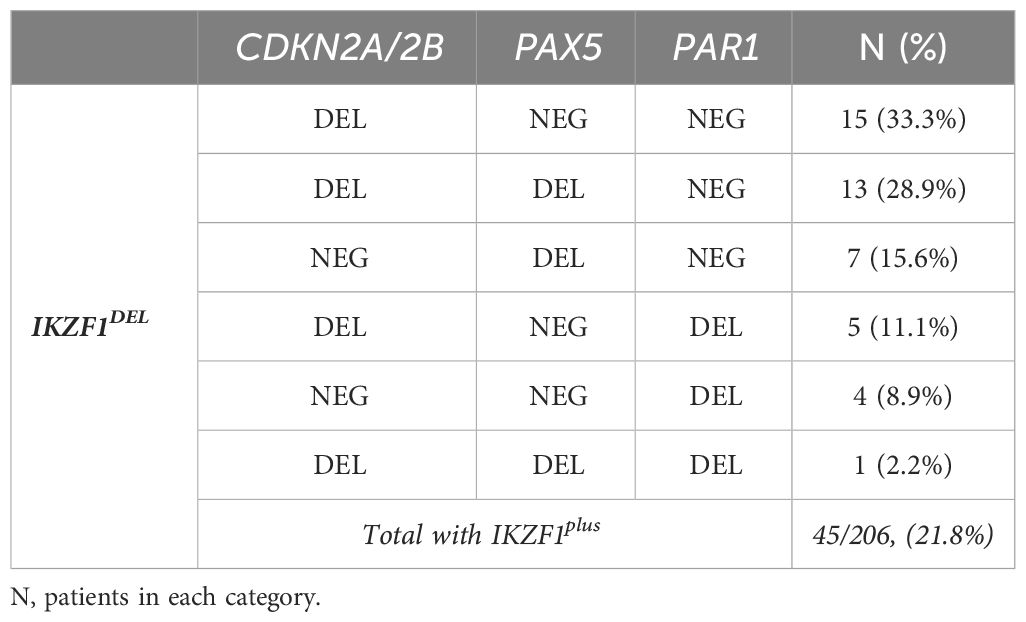
Table 2 Frequency of the different gene-deletion combinations conforming to the IKZF plus profile.
3.2 Clinical impact and overall survival analysis of the IKZF1plus profile
The patients were stratified according to the presence of IKZF1plus, and the clinical feature distribution was evaluated between the groups (Table 3). Age at diagnosis showed significant differences between the groups, with patients with IKZF1plus being older (p=0.04). Additionally, a trend toward higher risk classification was observed within the IKZF1plus group. No significant differences were found in sex distribution, percentage of bone marrow blasts, or WBC count at diagnosis between the two groups.
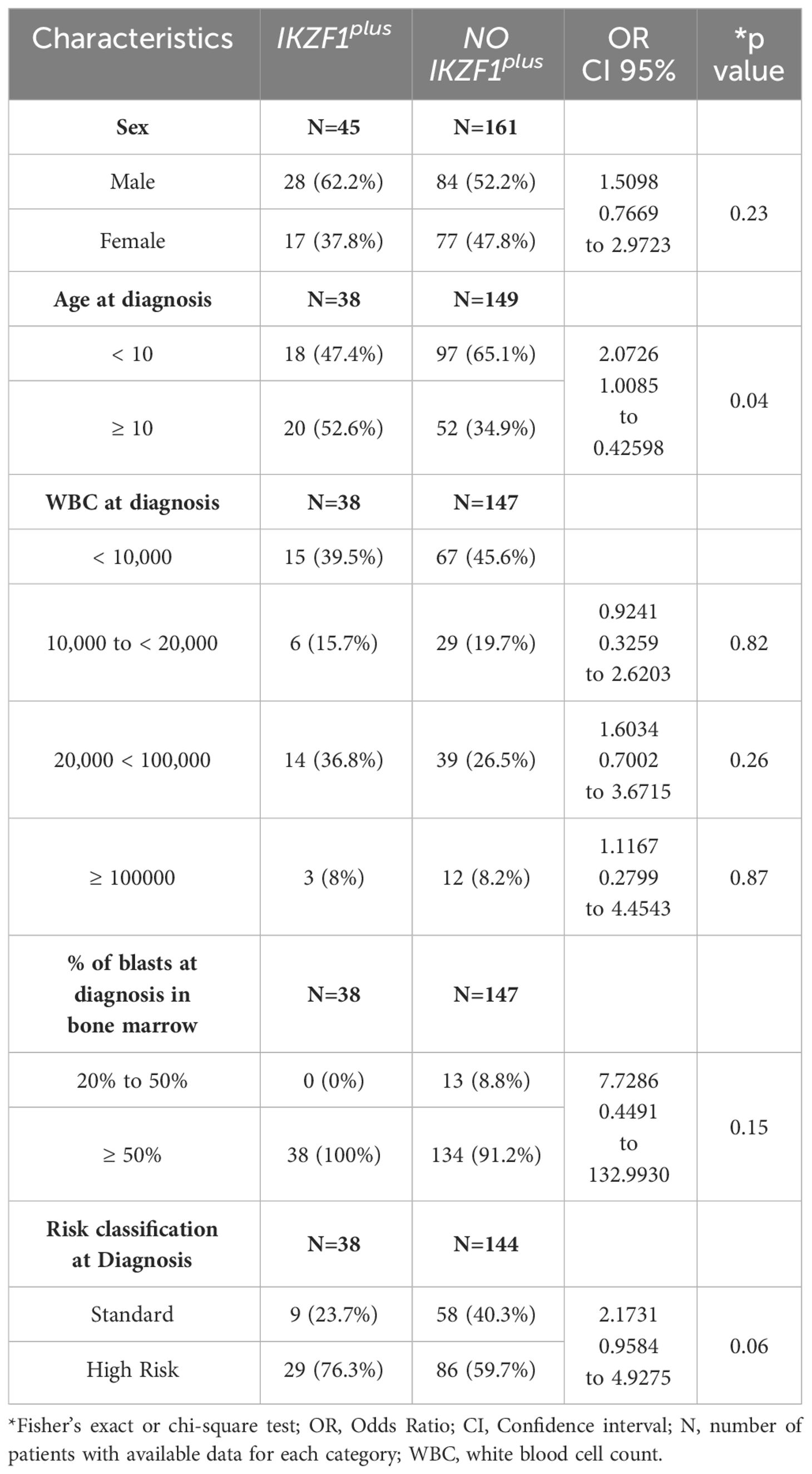
Table 3 Clinical features of patients with the IKZF1plus profile.
The impact of IKZF1plus on OS at five years was evaluated in 168 patients for whom information for OS calculation was available (35 IKZF1plus vs. 133 NO-IKZF1plus). The IKZF1plus group showed significantly reduced OS at 5 years compared to the NO-IKZF1plus group (p=0.009, Figure 1A). A significantly worse OS was also observed for the high-risk-IKZF1plus-positive subgroup when patients were stratified by the IKZF1plus status and the risk category (p=0.02, Figure 1B).
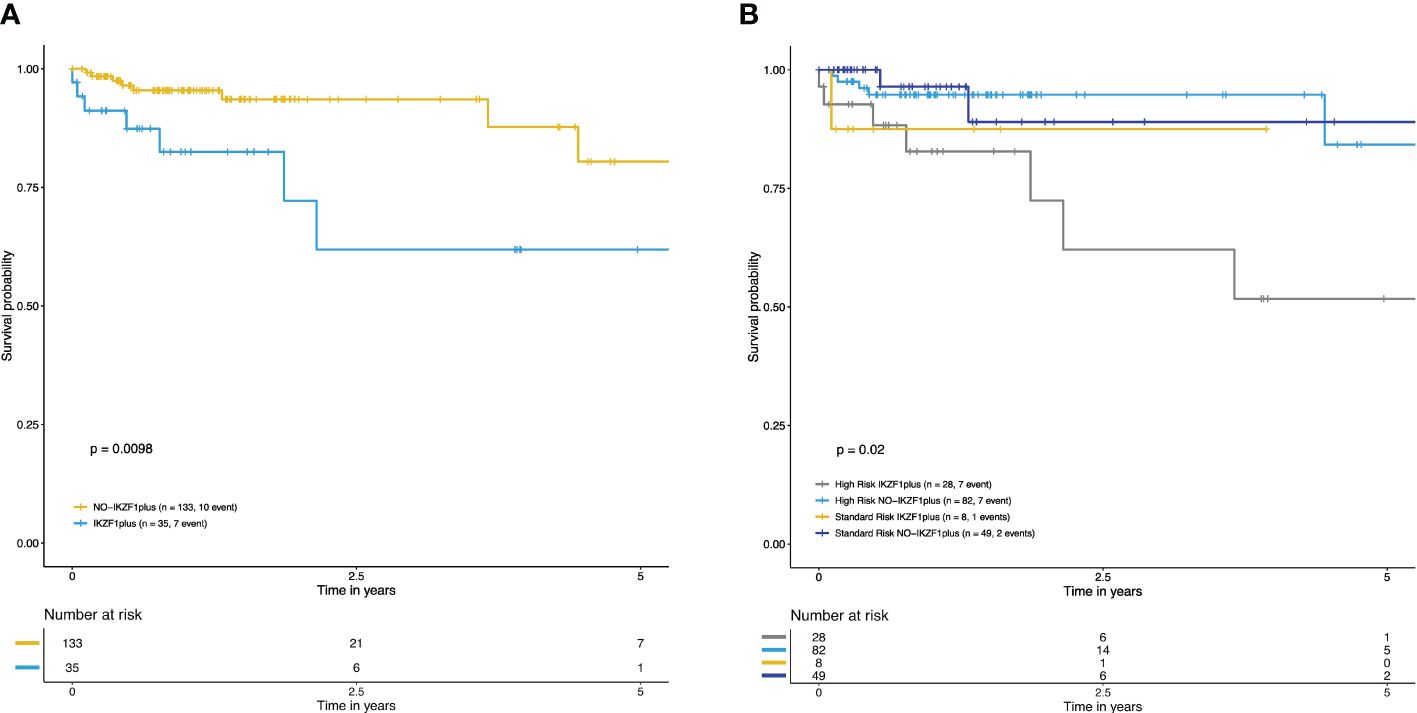
Figure 1 Overall survival curves at five years of IKZF1plus were estimated with the Kaplan–Meier method. (A) Decreased OS in patients with IKZF1plus (blue) versus NO-IKZF1plus (yellow). (B) Overall survival curves stratified according to IKZF1plus status and risk classification at diagnosis. The lowest OS was observed in the high-risk IKZF1plus positive group. Standard-risk NO-IKZF1plus patients (navy blue), high-risk NO-IKZF1plus patients (light blue), high-risk IKZF1plus (gray), standard-risk IKZF1plus (yellow).
We conducted a Cox regression multivariate analysis to examine the impact of the IKZF1plus profile, taking into account additional clinical factors such as patient age, sex, white blood cell count, percentage of blast cells, and the risk classification at the time of diagnosis. The analysis revealed that having the IKZF1plus profile significantly increases the risk of death by 3.7 times (HR = 3.7227, p = 0.02), highlighting its role as an independent poor outcome-predicting factor. The other variables included in the model did not demonstrate a statistically significant effect on the outcome.
3.3 Mutational profiles of IKZF1, PAX5, CDKN2A, CDKN2B, ERG genes and PAR1 regions in Mexican patients with B-ALL
The types of mutations identified in each gene and their frequencies are shown in Figure 2. IKZF1 was altered (IKZF1MUT) in 35.9% (74/206) of patients studied (Supplementary Table 3 for sequence mutations and Supplementary Table 4 for CNAs). Deletions were the most common alterations, representing 86.5% (64/74) of positive cases. Deletion affecting exons 4–7 (IK6 isoform) was the most common (39%, 25/64), followed by complete gene deletion (31.2%, 20/64). Moreover, a deletion spanning exons 1–8 was present in 7.9% (5/64) of cases (Figure 3A). SV was also detected in 54.7% of the patients with deletions. In contrast, only SVs were identified in seven individuals; interestingly, in six of them, the VAF was < 10%.
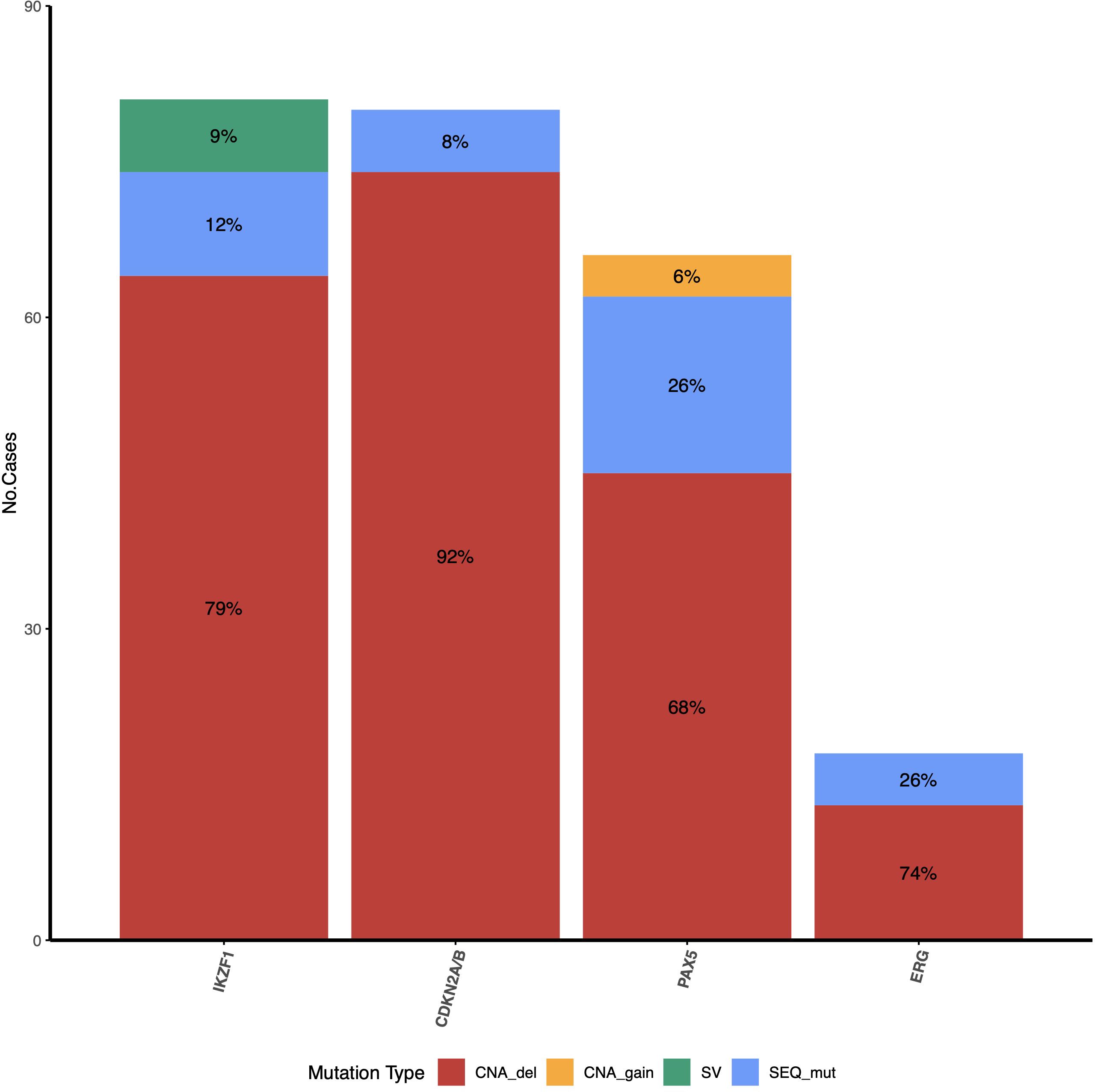
Figure 2 Frequency and type of mutations identified for IKZF1, CDKN2A/2B, PAX5, and ERG genes. For IKZF1, CNAs concomitant (red) with SV (green) were identified in some patients (Supplementary Table 4); these patients were counted only in the CNA_del group in this figure. The PAR1 region was not included because only deletions can be detected by MLPA.
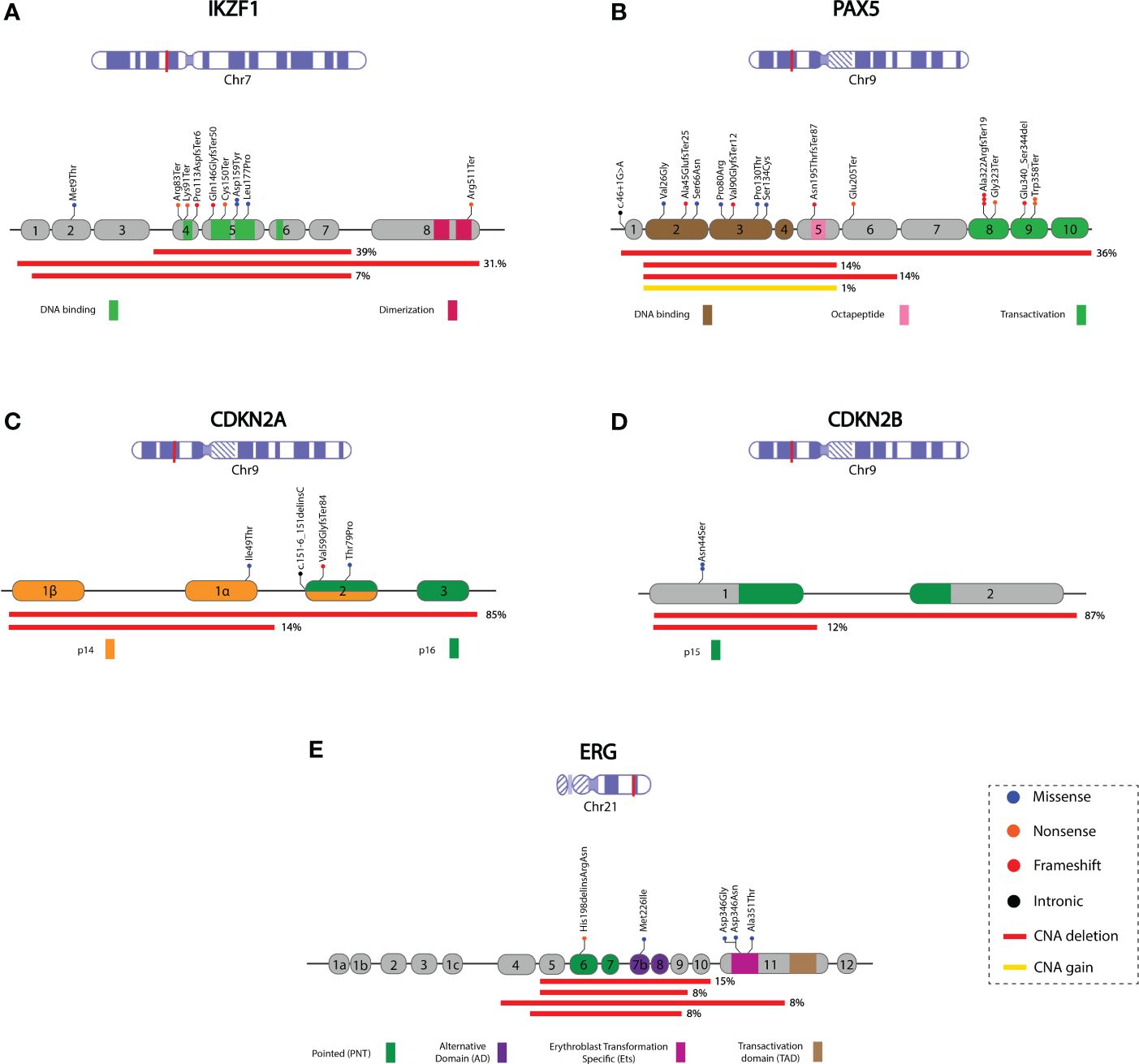
Figure 3 Schematic representation of the mutational profile identified in the IKZF1, PAX5, CDKN2A, CDKN2B, and ERG genes. Coding exons and the corresponding protein domain are shown for each gene. Red lines indicate the deleted region, and the percentage (right side) represents the proportion of cases with deletions. The remaining percentage corresponds to deleted regions with lower frequencies (not represented in the Figure). The complete deletion information is available in Supplementary Table 4. (A) IKZF1; NM_006060, (B) PAX5; NM_016734.2, the yellow line represents regions with PAX5igAMP, (C) CDKN2A; NM_000077, (D) CDKN2B; NM_004936, (E) ERG; NM_004449. Chromosome figures were taken from NCBI Bookshelf ID: NBK22266.
Sequence mutations were identified in 9 cases; in six of them, a concomitant deletion or SV was also present. Ten mutations were identified in nine cases: four missense mutations, four nonsense mutations, and two frameshift mutations, most of which were located in the DNA-binding domain coding region. In two patients, p.Asn159Tyr (N159Y) was identified without additional IKZF1 alterations.
At least one PAX5 alteration (PAX5MUT) was identified in 26.6% (55/206) of patients (Supplementary Tables 3, 4). These include CNAs caused by complete or partial gene deletions, gains, and small sequence mutations. PAX5DEL was the most common alteration, representing 75% (41/55) of PAX5MUT. In most cases, 63.4% (26/41), several exons were deleted, most commonly from exon 2 to 5 or from exon 2 to 6 (14%, 6/41 of each). In two cases, only one exon was deleted (exon 1 or exon 7). The whole gene (exons 1-10) was deleted in 36.6% (15/41) of cases. PAX5igAMP was identified in four patients; in two of them, additional genetic material was present in exons 2-5, one in exon 5, and one in the whole gene (Figure 3B; Supplementary Table 4).
A total of 17 PAX5 sequence mutations were identified in 15 patients: four nonsense mutations, five missense mutations [one p.Pro80Arg (P80R)], seven frameshifts, and one intronic variant (Supplementary Table 3). In eight of them, a second alteration in PAX5 was also present: four deletions, two sequence mutations, and two gains of material in PAX5.
CDKN2ADEL was identified in 35.92% (74/206) of the cases; in most cases (65/74), CDKN2B was entirely or partially deleted (Figures 3C, D). CDKN2A homozygous deletion was observed in 40 patients; in 30 of them, CDKN2B also showed homozygous deletion. The most frequent CDKN2ADEL comprised exons 1 to 3 and was identified in 85.1% (63/74) of the patients, followed by the deletion of exon 1, which was identified in 14.9% (11/74) of CDKN2ADEL patients. For CDKN2B, exon 1 and 2 deletions were identified in 87.7% (57/65), followed by exon 2 deletion in 12.3% (8/65) of the CDKN2B-positive cases (Supplementary Table 4). In six cases, sequence variants were identified in CDKN2A (four cases) and CDKN2B (two cases) (Supplementary Table 3).
ERGMUT was identified in 8.7% (18/206) of cases (Supplementary Tables 3, 4). Among the ERGDEL patients (13/18), involved various exons. These deletions affect the region expanding exons 2 to 11, encoding the ERG protein’s pointed domain (PNT) or alternative domain (AD) of the ERG protein. Sequencing variants were identified in five cases, three of which were located in the erythroblast transformation-specific domain (Ets) of the protein (Figure 3E, Supplementary Table 3).
Deletion of the PAR1 region was assessed by MLPA in patients with IKZF1DEL. We identified only ten patients with PAR1 deletions (Supplementary Table 4).
3.4 Clinical impact and overall survival analysis of patients with IKZF1, PAX5, CDKN2A/2B, and ERG mutations
Sex distribution, age at diagnosis, WBC count at diagnosis, percentage of blasts, and risk assignment were compared between patients positive and negative for IKZF1, PAX5, CDKN2A/2B, and ERG mutations (Supplementary Tables 5A–D). The frequency of high-risk patients and of those older than 10 years at diagnosis was significantly higher in the IKZF1MUT group (p=0.02 and p=0.002, respectively). A higher proportion (37.7%) of patients with CDKN2A/2BMUT had a WBC count in the range of 20,000–100,000 cells than CDKN2A/2BNEG patients (22.4%) (p=0.01). Additionally, a significantly higher proportion of patients had > 50% blasts at diagnosis in the CDKN2A/2BMUT (98.6%) group than in the CDKN2A/2BNEG group (89.7%) (p=0.05) (Supplementary Table 5B). In the case of PAX5MUT, a higher percentage of cases was associated with high-risk classification at diagnosis, but the difference was not statistically significant (p=0.06).
A significant decrease in OS was observed in the IKZF1MUT (p=0.02) (Supplementary Figure 1A) and CDKN2A/2BMUT (p=0.006) (Supplementary Figure 1B) groups. Significant differences (p=0.02) in OS were identified between the homozygous CDKN2AMUT and heterozygous CDKN2AMUT vs. CDKN2ANEG patients. (Supplementary Figure 1C). No significant effect on OS was observed for PAX5MUT or ERGMUT (Supplementary Figures 1D, E).
To determine whether the observed negative impact of CDKN2A/2BMUT and IKZF1MUT mutations on OS was conferred by IKZF1plus-positive patients in both groups, we excluded IKZF1plus patients from the OS analysis. After eliminating these patients, no significant differences in OS were observed between the CDKN2A/2BMUT or IKZF1MUT groups and those without mutations in these genes (Supplementary Figures 2A, B).
4 Discussion
To the best of our knowledge, this is the first study to evaluate the frequency, heterogeneity, and clinical impact of the IKZF1plus profile in an unselected group of Mexican pediatric patients with B-ALL. We also report the comprehensive mutational spectrum of IKZF1, PAX5, CDKN2A/2B, and ERG genes present at diagnosis in these patients. In contrast to most published studies that used MLPA to evaluate deletions in IKZF1, PAX5, CDKN2A/2B, and ERG genes related to the IKZF1plus profile, we used a custom-designed panel based on NGS. The bioinformatics algorithm allows the simultaneous evaluation of CNA (gain or deletions) with higher sensitivity than MLPA and of the sequence mutations present in at least 5% of the cells.
The frequency of IKZF1plus was higher (21.8%) than that reported in previous studies that analyzed patients of European ethnicities. Examples include Stanulla et al. (6%) (3), Braun et al. (2%) (4), Kicinski et al. (6%) (33), and Schwab et al. (13%) (34). Although few studies have been performed in Latin American populations, the frequencies are also lower than those detected in Argentina (9.2%) (5) and Brazil (11%, including pediatric and adult patients) (35). Using NGS, a more sensitive analysis methodology may have contributed to the higher frequency observed in our study. MLPA can only detect alterations if present in at least 25% of cells but fails to detect alterations when present in a smaller fraction of malignant cells, such as a subclone.
The two most frequent combinations, representing approximately 70% of the IKZF1plus-positive cases, comprised the simultaneous deletion of IKZF1-CDKN2A/2B or IKZF1-CDKN2A/2B-PAX5, similar to the results observed in other series (4). The high frequency of these two combinations is unsurprising given that CDKN2A/2BDEL is the most recurrently reported CNA in B-ALL, and frequently, the deletion also removes PAX5, which is located close to the CDKN2A/2B complex.
Because gene fusion information and iAMP21 were available for these patients (personal communication, the fusion data are being prepared for publication), we correlated the distribution of IKZF1plus with these alterations. The BCR::ABL1 fusion group had the highest proportion of IKZF1plus-positive patients (66.7%), followed by the iAMP21 (50%) and Ph-like groups (47%). IKZF1plus was also present in cases without these alterations but in a much lower proportion (13.7%). IKZF1plus was not identified in the ETV6::RUNX1, TCF3::PBX1, PAX5::ETV6, DUX4, or MEFD2 rearranged group of patients. The IKZF1plus molecular profile was associated with older age at diagnosis (52.6% ≥ 10 years in the IKZF1plus group vs. 34.9% in the NO-IKZF1plus cases, p=0.04) and high-risk classification at diagnosis (76.3% of IKZF1plus were classified as high-risk, p=0.06), similar to previous findings (3, 36).
According to our results, IKZF1plus had a significantly negative effect on OS. Similar results were reported by Crepinsek et al., who showed that the IKZF1plus subgroup had the lowest OS compared with IKFZ1MUT patients or IKZF1WT (36). The decline in OS observed in our patients was especially pronounced for IKZF1plus patients classified as high-risk at diagnosis compared with IKZF1plus standard-risk patients. In most studies, the negative prognostic impact of IKZF1plus on disease-free survival (DFS) was found to be limited to the subgroup of patients with positive MRD after induction (4, 33). The effect of IKZF1plus on DFS was not evaluated in our patients because the MRD information was not available. However, a more recent study that included 1,200 patients reported no statistically significant association between IKZF1plus and DFS (33).
4.1 Mutational profile of IKZF1, PAX5, CDKN2A, CDKN2B, ERG genes and PAR1 region in Mexican patients with B-ALL
The frequency of IKZF1MUT in this cohort (35.9%) was higher than that reported for Mexican patients. Ayón-Perez et al. found that 20.6% (36 cases) (37), while Rosales-Rodríguez et al. (38) detected 27% (63 cases) both using MLPA. The frequency of IKZF1MUT in our study was still higher (31.1%) than previously reported in Mexicans if only the deletions were considered in the IKZF1MUT count. IKZF1MUT frequencies in other populations were also lower: American (28.6%) (9), Brazilian (19.3%) (39), and Argentinian (9.2%) (5). The differences in the sensitivity of the methodology contributed to the increased frequency detected in our study. In most previous reports, MLPA was used; therefore, SV present in a low proportion could not be observed. In fact, seven patients in our study had SV with a VAF < 10%; concomitant deletion was not detected in these patients. Therefore, they would have been considered “negative” if MLPA had been used as the detection method.
In most patients with IKZF1DEL, deletion of exons 4–7, which encode the dominant negative IK6 isoform, was identified. Whole gene deletion, resulting in the loss of expression of the wild-type allele, was the second most frequent deletion. Four patients showed deletions in the noncoding exon 1. Deletions involving the 5’ region of IKZF1 also result in haploinsufficiency owing to a significant reduction in IKZF1 mRNA expression (40). IKZF1 is intolerant to variants that cause loss of function, according to the constraint metrics obtained from the gnomAD exome database (41).
The IKZF1N159Y mutation was present in less than 1% of our cases (two cases, 0.9%), and a similar low frequency was found in an analysis of 1988 pediatric B-ALL cases (0.4%) (42). No other IKZF1 alterations were present in these two patients, suggesting retention of the non-mutated IKZF1 allele, as previously reported in this subgroup of patients (42). This mutation is located inside the DNA-binding domain and induces nuclear mislocalization and intercellular adhesion. The transcriptomic profile of the IKZF1N159Y positives was different from that observed in other patients with alterations in IKZF1, suggesting that IKZF1N159Y could define a new subtype of B-ALL (42). The upregulation of genes involved in oncogenesis, chromatin remodeling, and signaling has been identified in patients with IKZF1N159Y (43).
IKZF1DEL was associated with older age at diagnosis, which is in accordance with the results of previous studies (9, 44). IKZF1DEL was associated with poor outcomes in several studies (9–11). In our patients, a significant decrease in OS was also observed for IKZF1MUT (p=0.02). Still, it seems to have been mainly contributed by the IKZF1plus patients present in the IKZF1MUT group since significance was lost after removing the IKZF1plus patients. In previous reports, the contribution of IKZF1plus to the poor outcome observed with IKZF1MUT was not independently evaluated.
PAX5MUT was less frequent in Mexican patients than in other ethnic groups. PAX5MUT has been found in approximately one-third of non-Hispanic B-ALL pediatric cases, according to studies performed in the USA and Netherlands (9, 45). In this study, the proportion of PAX5MUT was only 26.6%, considering all alterations (deletions, gain, and small sequence mutations). The frequency of PAX5 fusion-positive patients increased to only 27.7%, which is still lower than that reported in other studies (46). A previous study in Mexican patients found that 15.9% PAX5DEL by MLPA (38) was lower than that reported in Brazilians (25.2%) using the same methodology (39).
Monoallelic deletions affecting the paired domain were the most frequently identified PAX5 alterations, similar to the findings in other groups (9, 45). They involve small portions of the gene or cause whole-gene loss, impairing the DNA-binding capacity of the PAX5 protein. Mouse models have shown that haploinsufficiency of Pax5, caused by monoallelic deletion, confers susceptibility to B-cell transformation. However, other oncogenic events that act synergistically are required for leukemic transformation (47). In our cohort, PAX5DEL was concomitant with the BCR::ABL1, Ph-like-related fusions, and EBF1 mutations. The activation of STAT5 and mutations in EBF1, JAK3, and BCR::ABL1 have also been identified as cooperative events in PAX5DEL mice (48, 49).
Extra copies of PAX5 regions were identified in four cases, representing 1.9% of the total and 7.4% of the PAX5-positive group. An extra chromosome 9 would be present in one of them, as the other genes in chromosome 9 included in the NGS panel also showed more than two copies. No cytogenetic data were available for this patient to confirm the presence of extra Chr 9. PAX5igAMP was the most likely alteration in the three other patients, with a gain in genetic material observed for the exons encoding the DNA-binding and octapeptide domains, as reported previously (50).
PAX5igAMP has been associated with male sex, age at diagnosis older than ten years, and high-risk stratification. This alteration is frequently associated with CDKN2A/2BDEL (82%), and appears to be mutually exclusive with other significant risk-stratifying genetic lesions (50). Despite the small number of patients with PAX5igAMP in our analysis (3 cases), the clinical features were similar to those reported: three were males, 2/3 were older than ten years, and 1/3 had nine years; all were classified as high-risk at diagnosis. CDKN2A/2BDEL was present in all cases, and BCR::ABL1 was the only fusion identified (one case). Although the functional consequences of amplification have not yet been established, it has been suggested that increased copies of the DNA-binding region may alter binding to PAX5 target genes, leading to dysregulated B-cell differentiation and transformation (51). PAX5igAMP was associated with a high incidence of relapse (50). We did not evaluate the impact of PAX5igAMP on OS because of the small number of patients.
It has been suggested that some sequencing mutations may serve as initiating rather than secondary cooperative events in leukemogenesis (42). PAX5 point mutations have been identified in 7–10% of pediatric cases of B-ALL, similar to the frequency in our patients (7.3%), being the second most common PAX5 type of alteration. Fourteen mutations were identified, mainly located in the paired or transactivating domains. The most common consequence was the loss of function due to a frameshift or premature stop codon (7/14). Only seven of them have been previously reported in the COSMIC database (as of October 2023), and two previously described cases were recurrent in our patients: p.Ala322ArgfsTer19 (three cases) and p.Trp358Ter (two cases), both of which affect the transactivation domain. The frequency of PAX5P80R in our cases was lower than that previously reported. Only one case (0.5%) was identified, in contrast to the 2–6% observed in other studies (42, 52–54). In accordance with previous observations, this case was negative for fusions or iAMP21 and had an additional PAX5 loss-of-function mutation affecting the other allele. Martínez-Anaya et al. reported PAX5P80R in 1.4% of the patients; however, only Ph-like Mexican pediatric patients were analyzed (55). In seven cases with sequence mutations, both PAX5 copies were affected, with a deletion, gain, or a second point mutation in the other allele.
PAX5 alterations can function as cooperative events or germline-initiating genetic lesions in B-ALL patients (23). In most of our cases, the VAF of PAX5 sequence mutations was lower than expected, considering the percentage of blast cells present in the bone marrow sample analyzed, suggesting that PAX5 sequence mutations are secondary events that occur only in a subpopulation of malignant cells. Additionally, none of the sequencing mutations identified in our study has been reported as germline variants according to the HGMD professional database (as of October 2023) or had a VAF suggesting a germline origin.
As observed in another series of B-ALL patients, CDKN2A/2BDEL was the most frequently identified CNA (35.9%) in Mexican patients. Homozygous deletion of both genes occurred in a high proportion of the cases (40.5%). As previously reported, none of the patients had CDKN2BDEL without CDKN2ADEL (3). The frequency of CDKN2A/2BDEL was higher than that reported by Rosales-Rodríguez et al. (31.7%) in Mexican patients (38). Lower frequencies have also been reported in other Latin American and Asian populations, including Colombia (25%) (56), Brazil (31.8%) (39), China (20.2%) (15), and India (19.8%) (57). In our study, CDKN2A/2BDEL was associated with higher blast counts at diagnosis and high-risk features. Most patients with this alteration were classified as high-risk, although the difference was not statistically significant.
Sequence variants of CDKN2A/2B, including CDKN2A (1.9%) and CDKN2B (0.97%), occur in a very low proportion of patients with B-ALL. Three CDKN2A sequence variants, p.Ile49Thr, p.Val51ProfsTer89, and c.151-6_151delinsC, had a VAF close to 50% and were classified as likely pathogenic according to ACMG criteria (58). However, p.Ile49Thr is probably of germline origin since it has been previously identified in the Latino population database (f=0.00451, 1456/34586, gnomAD exomes database, as of October 2023). Although it is listed in ClinVar (ID 127523) with conflicting classifications (pathogenic vs. uncertain significance), functional studies support its role in pathogenicity (59). However, this effect appears more moderate than other known pathogenic variants of CDKN2A (60). The variant has been reported to be pathogenic in families with melanoma and pancreatic cancer (60). It has been identified several times in Mexican women with breast cancer who fulfill genetic risk criteria. Ten patients with cancer carrying this variant, identified in our laboratory, reported no family history of leukemia or other hematological malignancies during routine evaluation for cancer predisposition syndromes. No history of cancer was present in the family of the B-ALL patient carriers of this variant. The variant p.Val51ProfsTer89 produces a loss of function due to a frameshift, and c.151-6_151delinsC is a deletion insertion that affects the canonical splicing site in exon two and may cause nonsense-mediated decay. These variants are not listed in the HGMD or ClinVar databases (as of October 2023), are absent from population databases, and have not been reported as germline variants in cancer syndromes. None of the patients had a family history of cancer. As DNA obtained from non-hematopoietic tissue was unavailable, variants’ germline or somatic origins could not be clarified.
CDKN2B p.Asn44Ser was identified in two cases, with a VAF of approximately 50%. It is classified as having uncertain significance according to ACMG and has not been reported as a somatic mutation. It has a low frequency in Latino individuals. (f=0.0121%, 4/33098, gnomAD exomes, as of August 2023), and was absent from the Mexican database incorporated into the Franklin By Genoox platform (470 individuals). The allele frequency in our cohort of B-ALL patients was significantly higher than in gnomAD patients (0.4% in B-ALL vs. 0.0121% in gnomAD exomes, X2 = 22, p=2.7E-06). As both patients were from Oaxaca State, we can speculate that this variant could be frequent in this region of Mexico. However, a possible association with an increased risk of leukemia development cannot be ruled out without further investigation.
OS was significantly decreased (p=0.006) in the CDKN2A/2BMUT group, but similar to what happened for the decreased OS in the IKZF1DEL group, the significance was lost after removing the IKZF1plus patients. CDKN2A/2BDEL has been correlated with inferior 3-year event-free survival and 3-year OS rates in B-ALL patients (15). Other researchers have concluded that CDKN2A/2BDEL is associated with an increased probability of relapse and death (17, 57, 61, 62), but the contribution of IKZF1plus patients to these outcomes has not been evaluated.
No conclusive data exist regarding the contribution of CDKN2A/2ADEL heterozygous or homozygous status. In our study, CDKN2A/2BDEL heterozygous patients showed a significant decrease in OS (p=0.02) compared with homozygous and wild-type patients. However, Feng et al. (15) showed that patients with biallelic deletions had a worse 3-year EFS than those without. However, other authors have reported that homozygous CDKN2A/2BDEL is not associated with poor prognosis in childhood B-ALL (63).
In our cohort, somatic mutations in ERG were identified in 8.7% of patients, with most presenting deletions (72.2% of the ERGMUT cases) that affect one or more exons encoding the ERG protein’s pointed domain of the ERG protein or the alternative domain. All sequence variants identified in ERG are probably somatic, considering the VAF, and are located mainly in the erythroblast transformation-specific domain. According to the COSMIC database (as of October 2023), these domains are reported to be mutated in hematopoietic neoplasms.
Patients with ERGMUT had better OS, although statistical significance was not reached because of the small sample size. None of the patients with ERGDEL experienced mortality, supporting the reported association between ERGDEL and favorable outcomes (12, 13).
The present study has several limitations that must be considered when interpreting the results. It was a multicenter retrospective analysis of a heterogeneous group of patients. Treatment protocol and hospital-related factors would have influenced OS. The patients were treated according to six protocols, the only target therapy available was imatinib, and it was used only in the BCR::ABL1 positive patients. Due to the lack of molecular testing, Ph-like patients were not identified and did not receive target therapy, which would affect their OS. Economic resources and hospital infrastructure may vary among the 18 hospitals where the patients were treated. Cytogenetic/FISH characterization and MRD determination are not routinely performed in all Mexican public institutions, and this information was unavailable for these patients. Despite being one of the most extensive series evaluating molecular alterations in pediatric B-ALL, the number of patients is still small, and clinical information was not available for 100% of the patients; thus, it reduces the statistical power to detect additional differences in the distribution of clinical features between mutation-positive and mutation-negative patients or between subgroups of IKZF1plus patients or to evaluate the impact of specific mutations on OS. Finally, additional biological and socioeconomic factors that were not assessed in the present study could have contributed to the poor patient outcomes.
5 Conclusion
This study is the first to evaluate the prevalence and impact of the IKZF1plus profile on OS in Mexican pediatric patients with B-ALL using NGS. Our results showed a higher frequency than previously reported and a significantly lower OS in patients with this alteration. Our results support the notion that a high frequency of genetic markers related to poor outcomes is present in Mexican patients at the time of diagnosis. The interplay between adverse genetic features and socioeconomic challenges contributes to the higher mortality and morbidity observed in pediatric B-ALL patients in our country (64, 65). The incorporation of genomic methodologies into the diagnosis process of B-ALL, a significant unmet need in low- and mid-income countries, will allow the comprehensive identification of clinically relevant alterations, improving disease classification, treatment selection, and probably the general outcome.
Data availability statement
The data presented in this study are deposited in online repository of National Center for Biotechnology Information, ClinVar, accession number SUB13858637.
Ethics statement
The studies involving humans were approved by Comisión Nacional de Investigación Científica del Instituto Mexicano del Seguro Social (IMSS) (Number of document R-2020-785-022. The studies were conducted in accordance with the local legislation and institutional requirements. Written informed consent for participation in this study was provided by the participants’ legal guardians/next of kin.
Author contributions
JGS, JCNE, JMMA, and CAV carried out the conceptualization, data curation, formal analysis, funding acquisition, investigation, methodology, project administration, resources, software, supervision, validation, and visualization. MJO, FFE, CMG, AC, KCS, AMR, LLFL, ECMC, BEVT, VGO, ECQ, HFA, and JMMA provided technical support in methodology. JCNE, JFL, DCA, JGPG, EJH, MRBL, JRTN, AGS, JAMT, CAGD, MLGR, AOV, RMEE, LEMP, MLPS, MMR, MAGH, JCSP, NCLS, LSCC, PMAC, HRV, SJM, and FHQ provided resources such as study materials and clinical data. JGS and CAV undertook writing – original draft and writing – review & editing. Final approval of the manuscript was provided by all authors, who were accountable for all aspects of the work. JGS and JCNE share equal contributions, whereas JMMA and CAV are corresponding authors.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by Consejo Nacional de Humanidades, Ciencias y Tecnologías (CONAHCYT) grants: “Pronaii Leucemia Infantil, Proyecto Nacional de Investigación e Incidencia “: FORDECYT-PRONACES/303082/2019 and FORDECYT-PRONACES/303019/2019, FORDECYT-PRONACES/302994/2019, “Fondo Sectorial de Investigación en Salud y Seguridad Social SS/IMSS/ISSSTE-CONACYT “000000000273210 and Frontera de la Ciencia CF-2023-G-1399. JG-S received a fellowship from CONAHCYT (CVU 1042482).
Acknowledgments
The authors would like to thank the patients and their families for cooperating with this study.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer, AM, declared a shared affiliation with the author RE-E to the handling editor at the time of review.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2024.1337954/full#supplementary-material
References
1. Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBdeO, Berti E, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia. (2022) 36:1720–48. doi: 10.1038/s41375-022-01620-2
2. Steeghs EMP, Boer JM, Hoogkamer AQ, Boeree A, de Haas V, de Groot-Kruseman HA, et al. Copy number alterations in B-cell development genes, drug resistance, and clinical outcome in pediatric B-cell precursor acute lymphoblastic leukemia. Sci Rep. (2019) 9:4634. doi: 10.1038/s41598-019-41078-4
3. Stanulla M, Dagdan E, Zaliova M, Möricke A, Palmi C, Cazzaniga G, et al. IKZF1plus defines a new minimal residual disease-dependent very-poor prognostic profile in pediatric B-cell precursor acute lymphoblastic leukemia. J Clin Oncol. (2018) 36:1240–9. doi: 10.1200/JCO.2017.74.3617
4. Braun M, Pastorczak A, Sędek Ł, Taha J, Madzio J, Jatczak-Pawlik I, et al. Prognostic significance of IKZF1 deletions and IKZF1plus profile in children with B-cell precursor acute lymphoblastic leukemia treated according to the ALL-IC BFM 2009 protocol. Hematol Oncol. (2022) 40:430–41. doi: 10.1002/hon.2973
5. Felice MS, Rubio PL, Digiorge J, Barreda Frank M, Martínez CS, Guitter MR, et al. Impact of IKZF1 deletions in the prognosis of childhood acute lymphoblastic leukemia in Argentina. Cancers (Basel). (2022) 14:3283. doi: 10.3390/cancers14133283
6. Huang Y-J, Liu H-C, Jaing T-H, Wu K-H, Wang S-C, Yen H-J, et al. RAS pathway mutation is an added-value biomarker in pediatric Philadelphia-negative B-cell acute lymphoblastic leukemia with IKZF1 deletions. Pediatr Blood Cancer. (2021) 68:e28899. doi: 10.1002/pbc.28899
7. Gupta SK, Singh M, Chandrashekar PH, Bakhshi S, Trehan A, Gupta R, et al. Clinical and prognostic impact of copy number alterations and associated risk profiles in a cohort of pediatric B-cell precursor acute lymphoblastic leukemia cases treated under ICiCLe protocol. Hemasphere. (2022) 6:e782. doi: 10.1097/HS9.0000000000000782
8. Rebollo A, Schmitt C. Ikaros, Aiolos and Helios: transcription regulators and lymphoid Malignancies. Immunol Cell Biol. (2003) 81:171–5. doi: 10.1046/j.1440-1711.2003.01159.x
9. Mullighan CG, Su X, Zhang J, Radtke I, Phillips LAA, Miller CB, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. (2009) 360:470–80. doi: 10.1056/NEJMoa0808253
10. Palmi C, Valsecchi MG, Longinotti G, Silvestri D, Carrino V, Conter V, et al. What is the relevance of Ikaros gene deletions as a prognostic marker in pediatric Philadelphia-negative B-cell precursor acute lymphoblastic leukemia? Haematologica. (2013) 98:1226–31. doi: 10.3324/haematol.2012.075432
11. Waanders E, van der Velden VHJ, van der Schoot CE, van Leeuwen FN, van Reijmersdal SV, de Haas V, et al. Integrated use of minimal residual disease classification and IKZF1 alteration status accurately predicts 79% of relapses in pediatric acute lymphoblastic leukemia. Leukemia. (2011) 25:254–8. doi: 10.1038/leu.2010.275
12. Clappier E, Auclerc MF, Rapion J, Bakkus M, Caye A, Khemiri A, et al. An intragenic ERG deletion is a marker of an oncogenic subtype of B-cell precursor acute lymphoblastic leukemia with a favorable outcome despite frequent IKZF1 deletions. Leukemia. (2014) 28:70–7. doi: 10.1038/leu.2013.277
13. Zaliova M, Zimmermannova O, Dörge P, Eckert C, Möricke A, Zimmermann M, et al. ERG deletion is associated with CD2 and attenuates the negative impact of IKZF1 deletion in childhood acute lymphoblastic leukemia. Leukemia. (2014) 28:182–5. doi: 10.1038/leu.2013.282
14. Zhang W, Kuang P, Liu T. Prognostic significance of CDKN2A/B deletions in acute lymphoblastic leukaemia: a meta-analysis. Ann Med. (2019) 51:28–40. doi: 10.1080/07853890.2018.1564359
15. Feng J, Guo Y, Yang W, Zou Y, Zhang L, Chen Y, et al. Childhood acute B-lineage lymphoblastic leukemia with CDKN2A/B deletion is a distinct entity with adverse genetic features and poor clinical outcomes. Front Oncol. (2022) 12:878098. doi: 10.3389/fonc.2022.878098
16. Ampatzidou M, Papadhimitriou SI, Paisiou A, Paterakis G, Tzanoudaki M, Papadakis V, et al. The prognostic effect of CDKN2A/2B gene deletions in pediatric acute lymphoblastic leukemia (ALL): independent prognostic significance in BFM-based protocols. Diagn (Basel). (2023) 13:1589. doi: 10.3390/diagnostics13091589
17. Kathiravan M, Singh M, Bhatia P, Trehan A, Varma N, Sachdeva MS, et al. Deletion of CDKN2A/B is associated with inferior relapse free survival in pediatric B cell acute lymphoblastic leukemia. Leuk Lymph. (2019) 60:433–41. doi: 10.1080/10428194.2018.1482542
18. Braun M, Pastorczak A, Fendler W, Madzio J, Tomasik B, Taha J, et al. Biallelic loss of CDKN2A is associated with poor response to treatment in pediatric acute lymphoblastic leukemia. Leuk Lymph. (2017) 58:1162–71. doi: 10.1080/10428194.2016.1228925
19. Kim M, Yim S-H, Cho N-S, Kang S-H, Ko D-H, Oh B, et al. Homozygous deletion of CDKN2A (p16, p14) and CDKN2B (p15) genes is a poor prognostic factor in adult but not in childhood B-lineage acute lymphoblastic leukemia: a comparative deletion and hypermethylation study. Cancer Genet Cytogenet. (2009) 195:59–65. doi: 10.1016/j.cancergencyto.2009.06.013
20. Horcher M, Souabni A, Busslinger M. Pax5/BSAP maintains the identity of B cells in late B lymphopoiesis. Immunity. (2001) 14:779–90. doi: 10.1016/s1074-7613(01)00153-4
21. Okuyama K, Strid T, Kuruvilla J, Somasundaram R, Cristobal S, Smith E, et al. PAX5 is part of a functional transcription factor network targeted in lymphoid leukemia. PloS Genet. (2019) 15:e1008280. doi: 10.1371/journal.pgen.1008280
22. Hyde RK, Liu PP. Germline PAX5 mutations and B cell leukemia. Nat Genet. (2013) 45:1104–5. doi: 10.1038/ng.2778
23. Jia Z, Gu Z. PAX5 alterations in B-cell acute lymphoblastic leukemia. Front Oncol. (2022) 12:1023606. doi: 10.3389/fonc.2022.1023606
24. Li Z, Lee SHR, Chin WHN, Lu Y, Jiang N, Lim EH, et al. Distinct clinical characteristics of DUX4- and PAX5-altered childhood B-lymphoblastic leukemia. Blood Adv. (2021) 5:5226–38. doi: 10.1182/bloodadvances.2021004895
25. Giddings BM, Whitehead TP, Metayer C, Miller MD. Childhood leukemia incidence in California: High and rising in the Hispanic population. Cancer. (2016) 122:2867–75. doi: 10.1002/cncr.30129
26. Pérez-Saldivar ML, Fajardo-Gutiérrez A, Bernáldez-Ríos R, Martínez-Avalos A, Medina-Sanson A, Espinosa-Hernández L, et al. Childhood acute leukemias are frequent in Mexico City: descriptive epidemiology. BMC Cancer. (2011) 11:355. doi: 10.1186/1471-2407-11-355
27. Flores-Lujano J, Duarte-Rodríguez DA, Jiménez-Hernández E, Martín-Trejo JA, Allende-López A, Peñaloza-González JG, et al. Persistently high incidence rates of childhood acute leukemias from 2010 to 2017 in Mexico City: A population study from the MIGICCL. Front Public Health. (2022) 10:918921. doi: 10.3389/fpubh.2022.918921
28. Martín-Trejo JA, Núñez-Enríquez JC, Fajardo-Gutiérrez A, Medina-Sansón A, Flores-Lujano J, Jiménez-Hernández E, et al. Early mortality in children with acute lymphoblastic leukemia in a developing country: the role of malnutrition at diagnosis. A multicenter cohort MIGICCL study. Leuk Lymph. (2017) 58:898–908. doi: 10.1080/10428194.2016.1219904
29. Núñez-Enríquez JC, Bárcenas-López DA, Hidalgo-Miranda A, Jiménez-Hernández E, Bekker-Méndez VC, Flores-Lujano J, et al. Gene expression profiling of acute lymphoblastic leukemia in children with very early relapse. Arch Med Res. (2016) 47:644–55. doi: 10.1016/j.arcmed.2016.12.005
30. Lee SHR, Yang W, Gocho Y, John A, Rowland L, Smart B, et al. Pharmacotypes across the genomic landscape of pediatric acute lymphoblastic leukemia and impact on treatment response. Nat Med. (2023) 29:170–9. doi: 10.1038/s41591-022-02112-7
31. Campo E, Swerdlow SH, Harris NL, Pileri S, Stein H, Jaffe ES. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. (2011) 117:5019–32. doi: 10.1182/blood-2011-01-293050
32. PDQ Pediatric Treatment Editorial Board. Childhood Acute Lymphoblastic Leukemia Treatment (PDQ®): Health Professional Version (2023). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK65763/ (Accessed January 17, 2023).
33. Kicinski M, Arfeuille C, Grardel N, Bakkus M, Caye-Eude A, Plat G, et al. The prognostic value of IKZF1plus in B-cell progenitor acute lymphoblastic leukemia: Results from the EORTC 58951 trial. Pediatr Blood Cancer. (2023) 70:e30313. doi: 10.1002/pbc.30313
34. Schwab C, Cranston RE, Ryan SL, Butler E, Winterman E, Hawking Z, et al. Integrative genomic analysis of childhood acute lymphoblastic leukaemia lacking a genetic biomarker in the UKALL2003 clinical trial. Leukemia. (2023) 37:529–38. doi: 10.1038/s41375-022-01799-4
35. Maciel ALT, Barbosa TdaC, Blunck CB, Wolch K, MaChado AdeAL, da Costa ES, et al. IKZF1 deletions associate with CRLF2 overexpression leading to a poor prognosis in B-cell precursor acute lymphoblastic leukaemia. Transl Oncol. (2022) 15:101291. doi: 10.1016/j.tranon.2021.101291
36. Crepinsek K, Marinsek G, Kavcic M, Prelog T, Kitanovski L, Jazbec J, et al. Clinical impacts of copy number variations in B-cell differentiation and cell cycle control genes in pediatric B-cell acute lymphoblastic leukemia: a single centre experience. Radiol Oncol. (2021) 56:92–101. doi: 10.2478/raon-2021-0050
37. Ayón-Pérez MF, Pimentel-Gutiérrez HJ, Durán-Avelar MdeJ, Vibanco-Pérez N, Pérez-Peraza VM, Pérez-González ÓA, et al. IKZF1 gene deletion in pediatric patients diagnosed with acute lymphoblastic leukemia in Mexico. Cytogenet Genome Res. (2019) 158:10–6. doi: 10.1159/000499641
38. Rosales-Rodríguez B, Núñez-Enríquez JC, Velázquez-Wong AC, González-Torres C, Gaytán-Cervantes J, Jiménez-Hernández E, et al. Copy number alterations are associated with the risk of very early relapse in pediatric B-lineage acute lymphoblastic leukemia: A nested case-control MIGICCL study. Arch Med Res. (2021) 52:414–22. doi: 10.1016/j.arcmed.2020.12.013
39. Barbosa TC, Terra-Granado E, Quezado Magalhães IM, Neves GR, Gadelha A, Guedes Filho GE, et al. Frequency of copy number abnormalities in common genes associated with B-cell precursor acute lymphoblastic leukemia cytogenetic subtypes in Brazilian children. Cancer Genet. (2015) 208:492–501. doi: 10.1016/j.cancergen.2015.06.003
40. Morel G, Deau M-C, Simand C, Caye-Eude A, Arfeuille C, Ittel A, et al. Large deletions of the 5’ region of IKZF1 lead to haploinsufficiency in B-cell precursor acute lymphoblastic leukaemia. Br J Haematol. (2019) 186:e155–9. doi: 10.1111/bjh.15994
41. Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. (2016) 536:285–91. doi: 10.1038/nature19057
42. Gu Z, Churchman ML, Roberts KG, Moore I, Zhou X, Nakitandwe J, et al. PAX5-driven subtypes of B-progenitor acute lymphoblastic leukemia. Nat Genet. (2019) 51:296–307. doi: 10.1038/s41588-018-0315-5
43. Li J-F, Dai Y-T, Lilljebjörn H, Shen S-H, Cui B-W, Bai L, et al. Transcriptional landscape of B cell precursor acute lymphoblastic leukemia based on an international study of 1,223 cases. Proc Natl Acad Sci U.S.A. (2018) 115:E11711–20. doi: 10.1073/pnas.1814397115
44. Vrooman LM, Blonquist TM, Harris MH, Stevenson KE, Place AE, Hunt SK, et al. Refining risk classification in childhood B acute lymphoblastic leukemia: results of DFCI ALL Consortium Protocol 05-001. Blood Adv. (2018) 2:1449–58. doi: 10.1182/bloodadvances.2018016584
45. Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, et al. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. (2007) 446:758–64. doi: 10.1038/nature05690
46. Kuiper RP, Schoenmakers EFPM, van Reijmersdal SV, Hehir-Kwa JY, van Kessel AG, van Leeuwen FN, et al. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. (2007) 21:1258–66. doi: 10.1038/sj.leu.2404691
47. Dang J, Wei L, de Ridder J, Su X, Rust AG, Roberts KG, et al. PAX5 is a tumor suppressor in mouse mutagenesis models of acute lymphoblastic leukemia. Blood. (2015) 125:3609–17. doi: 10.1182/blood-2015-02-626127
48. Martín-Lorenzo A, Auer F, Chan LN, García-Ramírez I, González-Herrero I, Rodríguez-Hernández G, et al. Loss of pax5 exploits sca1-BCR-ABLp190 susceptibility to confer the metabolic shift essential for pB-ALL. Cancer Res. (2018) 78:2669–79. doi: 10.1158/0008-5472.CAN-17-3262
49. Heltemes-Harris LM, Willette MJL, Ramsey LB, Qiu YH, Neeley ES, Zhang N, et al. Ebf1 or Pax5 haploinsufficiency synergizes with STAT5 activation to initiate acute lymphoblastic leukemia. J Exp Med. (2011) 208:1135–49. doi: 10.1084/jem.20101947
50. Schwab C, Nebral K, Chilton L, Leschi C, Waanders E, Boer JM, et al. Intragenic amplification of PAX5: a novel subgroup in B-cell precursor acute lymphoblastic leukemia? Blood Adv. (2017) 1:1473–7. doi: 10.1182/bloodadvances.2017006734
51. Jean J, Kovach AE, Doan A, Oberley M, Ji J, Schmidt RJ, et al. Characterization of PAX5 intragenic tandem multiplication in pediatric B-lymphoblastic leukemia by optical genome mapping. Blood Adv. (2022) 6:3343–6. doi: 10.1182/bloodadvances.2021006328
52. Passet M, Boissel N, Sigaux F, Saillard C, Bargetzi M, Ba I, et al. PAX5 P80R mutation identifies a novel subtype of B-cell precursor acute lymphoblastic leukemia with favorable outcome. Blood. (2019) 133:280–4. doi: 10.1182/blood-2018-10-882142
53. Bastian L, Schroeder MP, Eckert C, Schlee C, Tanchez JO, Kämpf S, et al. PAX5 biallelic genomic alterations define a novel subgroup of B-cell precursor acute lymphoblastic leukemia. Leukemia. (2019) 33:1895–909. doi: 10.1038/s41375-019-0430-z
54. Jung M, Schieck M, Hofmann W, Tauscher M, Lentes J, Bergmann A, et al. Frequency and prognostic impact of PAX5 p.P80R in pediatric acute lymphoblastic leukemia patients treated on an AIEOP-BFM acute lymphoblastic leukemia protocol. Genes Chromosomes Cancer. (2020) 59:667–71. doi: 10.1002/gcc.22882
55. Martínez-Anaya D, Moreno-Lorenzana D, Reyes-León A, Juárez-Figueroa U, Dean M, Aguilar-Hernández MM, et al. Characterization of philadelphia-like pre-B acute lymphoblastic leukemia: experiences in mexican pediatric patients. Int J Mol Sci. (2022) 23:9587. doi: 10.3390/ijms23179587
56. Martinez C, Yunis LK, Cabrera E, García J, Uribe G, Quintero E, et al. Association between early risk factors and CDKN2A/B deletion in pediatric patients with acute lymphoblastic leukemia in a pediatric cancer center in Colombia. Pediatr Hematol Oncol J. (2022) 7:1–3. doi: 10.1016/j.phoj.2022.03.001
57. Agarwal M, Bakhshi S, Dwivedi SN, Kabra M, Shukla R, Seth R. Cyclin dependent kinase inhibitor 2A/B gene deletions are markers of poor prognosis in Indian children with acute lymphoblastic leukemia. Pediatr Blood Cancer. (2018) 65:e27001. doi: 10.1002/pbc.27001
58. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
59. Walker GJ, Gabrielli BG, Castellano M, Hayward NK. Functional reassessment of P16 variants using a transfection-based assay. Int J Cancer. (1999) 82:305–12. doi: 10.1002/(sici)1097-0215(19990719)82:2<305::aid-ijc24>3.0.co;2-z
60. Ranade K, Hussussian CJ, Sikorski RS, Varmus HE, Goldstein AM, Tucker MA, et al. Mutations associated with familial melanoma impair p16INK4 function. Nat Genet. (1995) 10:114–6. doi: 10.1038/ng0595-114
61. Graf Einsiedel H, Taube T, Hartmann R, Wellmann S, Seifert G, Henze G, et al. Deletion analysis of p16(INKa) and p15(INKb) in relapsed childhood acute lymphoblastic leukemia. Blood. (2002) 99:4629–31. doi: 10.1182/blood.v99.12.4629
62. Kees UR, Burton PR, Lü C, Baker DL. Homozygous deletion of the p16/MTS1 gene in pediatric acute lymphoblastic leukemia is associated with unfavorable clinical outcome. Blood. (1997) 89:4161–6. doi: 10.1182/blood.V89.11.4161
63. Mirebeau D, Acquaviva C, Suciu S, Bertin R, Dastugue N, Robert A, et al. The prognostic significance of CDKN2A, CDKN2B and MTAP inactivation in B-lineage acute lymphoblastic leukemia of childhood. Results of the EORTC studies 58881 and 58951. Haematologica. (2006) 91:881–5.
64. Castro-Ríos A, Reyes-Morales H, Pelcastre-Villafuerte BE, Rendón-Macías ME, Fajardo-Gutiérrez A. Socioeconomic inequalities in survival of children with acute lymphoblastic leukemia insured by social security in Mexico: a study of the 2007-2009 cohorts. Int J Equity Health. (2019) 18:40. doi: 10.1186/s12939-019-0940-3
Keywords: IKZF1plus, IKZF1 gene mutation, CDKN2A/2B gene mutation, PAX5 gene mutation, PAR1 deletions, pediatric B-ALL, overall survival
Citation: Garcia-Solorio J, Núñez-Enriquez JC, Jiménez-Olivares M, Flores-Lujano J, Flores-Espino F, Molina-Garay C, Cervera A, Casique-Aguirre D, Peñaloza-Gonzalez JG, Baños-Lara MDR, García-Soto Á, Galván-Díaz CA, Olaya-Vargas A, Aguilar HF, Mata-Rocha M, Garrido-Hernández MÁ, Solís-Poblano JC, Luna-Silva NC, Cano-Cuapio LS, Aristil-Chery PM, Herrera-Quezada F, Carrillo-Sanchez K, Muñoz-Rivas A, Flores-Lagunes LL, Mendoza-Caamal EC, Villegas-Torres BE, González-Osnaya V, Jiménez-Hernández E, Torres-Nava JR, Martín-Trejo JA, Gutiérrez-Rivera MdL, Espinosa-Elizondo RM, Merino-Pasaye LE, Pérez-Saldívar ML, Jiménez-Morales S, Curiel-Quesada E, Rosas-Vargas H, Mejía-Arangure JM and Alaez-Verson C (2024) IKZF1plus is a frequent biomarker of adverse prognosis in Mexican pediatric patients with B-acute lymphoblastic leukemia. Front. Oncol. 14:1337954. doi: 10.3389/fonc.2024.1337954
Received: 13 November 2023; Accepted: 29 February 2024;
Published: 03 April 2024.
Edited by:
Michele Redell, Baylor College of Medicine, United StatesReviewed by:
Jose Manuel Vagace Valero, University Hospital of Badajoz, SpainAdolfo Martinez, General Hospital of Mexico, Mexico
Kristen Kurtz, Baylor College of Medicine, United States
Copyright © 2024 Garcia-Solorio, Núñez-Enriquez, Jiménez-Olivares, Flores-Lujano, Flores-Espino, Molina-Garay, Cervera, Casique-Aguirre, Peñaloza-Gonzalez, Baños-Lara, García-Soto, Galván-Díaz, Olaya-Vargas, Aguilar, Mata-Rocha, Garrido-Hernández, Solís-Poblano, Luna-Silva, Cano-Cuapio, Aristil-Chery, Herrera-Quezada, Carrillo-Sanchez, Muñoz-Rivas, Flores-Lagunes, Mendoza-Caamal, Villegas-Torres, González-Osnaya, Jiménez-Hernández, Torres-Nava, Martín-Trejo, Gutiérrez-Rivera, Espinosa-Elizondo, Merino-Pasaye, Pérez-Saldívar, Jiménez-Morales, Curiel-Quesada, Rosas-Vargas, Mejía-Arangure and Alaez-Verson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Manuel Mejía-Arangure, am1lamlhQGlubWVnZW4uZ29iLm14; Carmen Alaez-Verson, Y2FsYWV6QGlubWVnZW4uZ29iLm14
†These authors have contributed equally to this work