
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol., 08 January 2024
Sec. Pediatric Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1324013
This article is part of the Research TopicNew Insights into Cancer Predisposition Syndromes in Pediatric Hematology-OncologyView all 8 articles
 Francesco Fabozzi1*
Francesco Fabozzi1* Rosalba Carrozzo2
Rosalba Carrozzo2 Mariachiara Lodi1
Mariachiara Lodi1 Angela Di Giannatale1
Angela Di Giannatale1 Selene Cipri1
Selene Cipri1 Chiara Rosignoli1
Chiara Rosignoli1 Isabella Giovannoni3
Isabella Giovannoni3 Alessandra Stracuzzi3Teresa Rizza2
Alessandra Stracuzzi3Teresa Rizza2 Claudio Montante1
Claudio Montante1 Emanuele Agolini4Michela Di Nottia5
Emanuele Agolini4Michela Di Nottia5 Federica Galaverna1
Federica Galaverna1 Giada Del Baldo1
Giada Del Baldo1 Francesco Del Bufalo1
Francesco Del Bufalo1 Angela Mastronuzzi1
Angela Mastronuzzi1 Maria Antonietta De Ioris1
Maria Antonietta De Ioris1The increased availability of genetic technologies has significantly improved the detection of novel germline variants conferring a predisposition to tumor development in patients with malignant disease. The identification of variants of uncertain significance (VUS) represents a challenge for the clinician, leading to difficulties in decision-making regarding medical management, the surveillance program, and genetic counseling. Moreover, it can generate confusion and anxiety for patients and their family members. Herein, we report a 5-year-old girl carrying a VUS in the Succinate Dehydrogenase Complex Subunit C (SHDC) gene who had been previously treated for high-risk neuroblastoma and subsequently followed by the development of secondary acute myeloid leukemia. In this context, we describe how functional studies can provide additional insight on gene function determining whether the variant interferes with normal protein function or stability.
Secondary malignancies represent a major challenge in pediatric cancer patients with an incidence up to 20% among survivors (1). Myeloid neoplasms may occur from a few years to several decades after cancer treatment and they are more frequent in cancer-predisposing germline variants carriers. Therapy-related myeloid neoplasms (t-MNs) have an incidence of 0.62 per 100,000 and account for 10-20% of new diagnoses of acute myeloid leukemia/myelodysplastic syndrome in adulthood; there are few and controversial data available in the literature on t-MNs in pediatric age, although the most widely accepted hypothesis is that pathogenic germline variant (e.g., in the NF1 gene) may interact with the genotoxic effect of chemotherapeutics, inducing the development of the secondary neoplasm (2, 3).
Although all cytotoxic drugs increase the risk of developing a myeloid neoplasm, the two classes of chemotherapeutics most associated with the development of t-MNs are alkylating agents and topoisomerase II inhibitors (2).
The advances in molecular biology and genetic technologies have significantly improved the detection of germline variants in cancer patients with important implications in the management and in the decision making (4). However, the identification of these variants is not a straightforward process and the interpretation of the results is often challenging. One of the most significant burdens is represented by the Variants of Uncertain Significance (VUSs) detection, with their uncertain role and impact on the pathogenesis of neoplasms. The presence of VUSs can lead to confusion and anxiety for the individual and their family members, as well as difficulties in decision making regarding the medical management, the surveillance program and the genetic counseling. In this context, functional studies can provide additional insights regarding the possible influence of the variant on the normal protein functions and its stability.
Herein we describe a case of acute myeloid leukemia (AML) arising in a 5-year-old girl previously treated for high-risk neuroblastoma (NB).
At the onset of the second malignancy, the patient underwent genetic testing with Next Generation Sequencing (NGS) technology to identify germline genetic variants predisposing to cancer. The study found a VUS in the Succinate Dehydrogenase Complex Subunit C (SDHC) gene. Therefore, it was decided to perform a molecular study on tumor tissue, which detected the presence of the same variant in the neuroblastoma cells of the primary tumor.
Finally, a functional study was performed on the patient’s fibroblasts to clarify the potential impact and causal role of this variant.
A 3-year-old-girl was diagnosed with high-risk neuroblastoma (NB). She underwent an intensive front-line chemotherapy treatment according to European guidelines (5), without achieving a complete remission (CR) at the end of the induction phase. She received a second-line treatment including four courses of Temozolomide-Irinotecan (TEMIRI) followed by radiometabolic therapy with high-dose 131I-Metaiodobenzylguanidine (MIBG) and melphalan supported by autologous stem cell transplantation (aSCT). Subsequently, she underwent high-dose chemotherapy (Busulfan and melphalan) with a further aSCT and chemoimmunotherapy with five courses of TEMIRI and Dinutuximab beta, surgery and radiotherapy on primary tumor (see Supplementary Table 1). This very intensive treatment resulted in a CR. Unfortunately, a bone marrow evaluation during early follow-up showed the onset of secondary AML carrying t(9;11) MLL-AF9 translocation (Supplementary Figure S1).
Due to the early arising of a secondary leukemia, an NGS was performed using the Twist Custom Panel kit (clinical exome - Twist Bioscience) on NovaSeq6000 platform (Illumina), filtering for a dedicated diagnostic gene panel (ALK, APC, AXIN2, BARD1, BRCA1, BRCA2, BRIP1, CDKN1C, CHEK2, ERCC4, EZH2, FANCA, FANCB, FANCC, FANCD2, FANCE, FANCF, FANCG, FANCI, FANCL, FANCM, HRAS, KIF1B, KRAS, LZTR1, MAD2L2, NF1, PALB2, PHOX2B, PTPN11, RAF1, SDHAF2, SDHB, SDHC, SDHD, SLX4, SOS1, TP53, UBE2T, XRCC2) (6, 7). In addition, the analysis was extended filtering for a large cancer predisposition gene panel (8, 9) (Supplementary Table 2). The BaseSpace pipeline (Illumina, https://basespace.illumina.com/) and the GeneYX software (LifeMap Sciences) were used for the variant calling and annotating variants, respectively. Sequencing data were aligned to the hg19 human reference genome. Variants were examined for coverage and Qscore (minimum threshold of 30), and visualized by the Integrative Genome Viewer (IGV) (Supplementary Table 3). Exome sequencing revealed the missense variant c.197C>T (p.Ala66Val) (rs760572684) in SDHC (NM_003001.5) gene, in heterozygous state and maternally segregated (Figure 1), classified as VUS according to American College of Medical Genetics (ACMG) [BP4, PM1, PM2 criteria] (6). Conservation score (phyloP100) is -0.952. The variant is reported in GnomAD (exomes: f = 0.0000241; genomes: f = 0.0000637). The pathogenicity score is heterogeneous: meta score underlines five algorithms as uncertain prediction (BayesDel noAF, BayesDel_addAF, MetaSVM, MetaLR, REVEL) vs one benign prediction (MetaRNN). In addition, 12 individual prediction algorithms show a benign prediction (DANN, DEOGEN2, EIGEN, EIGEN PC, FATHMM-MKL, FATHMM-XF, LRT, M-CAP, MutationAssessor, PrimateAI, PROVEAN, SIFT) vs 4 uncertain (LIST-S2, Mutation Taster, MVP, SIFT4G) vs 1 pathogenic (FATHMM) (Supplementary Table 4). rs760572684 is annotated in ClinVar as a Variant of Uncertain Significance (VUS) in patients with Hereditary Cancer-Predisposing Syndrome [RCV001013940], Gastrointestinal Stromal Tumor [RCV000529986] and SDHC-Related Hereditary Paraganglioma-Pheochromocytoma Syndrome (Paragangliomas 3) [RCV000410077] and has been previously described (10). To note, the mother is in good health with mute clinical history.
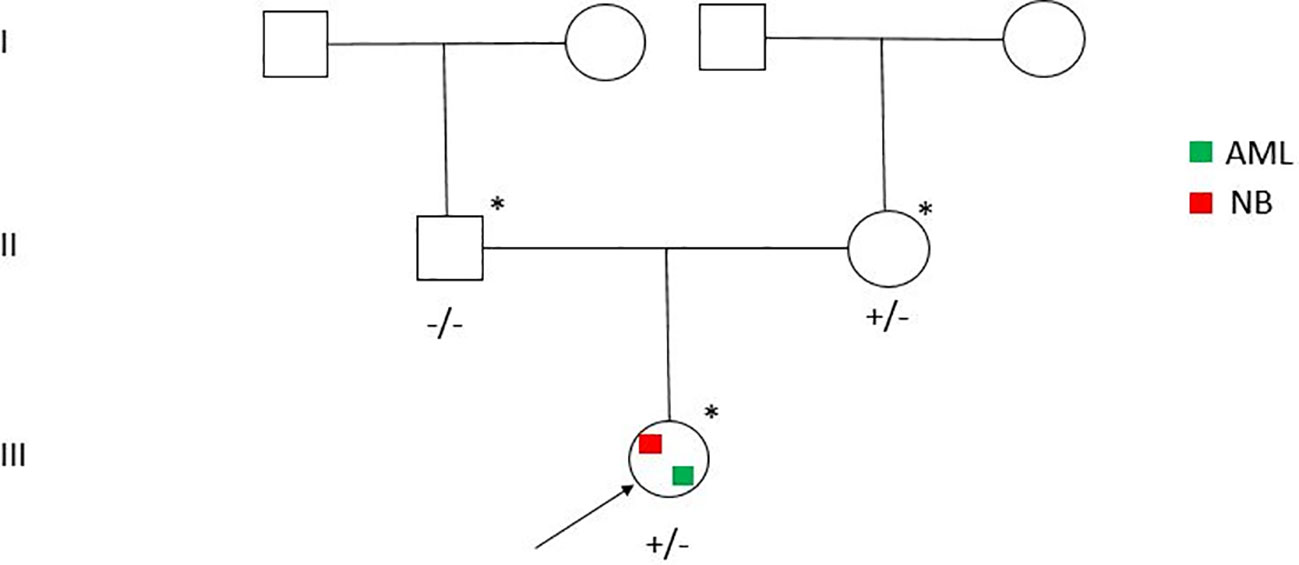
Figure 1 Pedigree of the family. Individuals with SDHC c.197C>T (p.Ala66Val) positive test are indicated by the plus sign.
In addition, the variant analysis, performed filtering on the extended gene panel, revealed a rare maternally inherited heterozygous missense variants, c.943T>C (p.Phe315Leu) in PINK1 gene (NM_032409.3). This variant can be classified as VUS according to American College of Medical Genetics (ACMG) (PP3, PM1 and PM2). The PINK1 gene is know to be associated with Parkinson disease (11) and was reported as a candidate NB susceptibility gene (12).
In view of the peculiar clinical history characterized by the occurrence of two nearly synchronous neoplasms (NB and AML) and the finding of the rs760572684, a NGS analysis on primitive tumor was performed. DNA was extracted from formalin-fixed paraffin-embedded primary tumor tissue using NucleoSpin Tissue Kit (Machery-Nagel) according to the manufacturer’s protocol, and NGS data were analyzed with Illumina TruSight Oncology 500. The molecular analysis found 5 VUSs: c.197C>T (p.A66V) on SDHC gene (the same variant found in germinal sample) (AF: 43.4%); c.1202G>A (p.R401K) on the BIRC3 gene (AF: 55.7%); c.1012G>C (p.E338Q) and c.650_651delinsAA (p.P217Q) on the EPHA3 gene (AF: 43.8% and 10.1%, respectively); and c.816A>C (p.K272N) on the MAP3K1 gene (AF: 11%). Of note, no second-hit mutation was found in the other SDHC allele.
To further investigate the possibility of a causative role of mutated SDHC, a functional analysis was performed by 3 different tests; thus, the patient underwent skin biopsy in order to obtain fibroblasts for these studies.
Fibroblasts underwent a cytochemical investigation for the enzyme succinate dehydrogenase (Figure 2); through mitochondrial respiratory chain complex V enzyme activity, we evaluated adenosine triphosphate (ATP) production in mitochondria purified from fibroblasts using different substrates: succinate, specific for succinate dehydrogenase (respiratory chain complex II) and two others specific for NADH dehydrogenase (respiratory chain complex I) (Figure 3A); by western blotting we evaluated the content of the C subunit and 2 other subunits of succinate dehydrogenase (Figure 3B). The results showed that none of the three experiments detected an impaired function in the patient’s fibroblasts compared with the controls used, allowing the activation of BS3 criteria (well-established in vitro or in vivo functional studies show no damaging effect on protein function or splicing) of the American College of Medical Genetics (ACMG) guidelines (13).
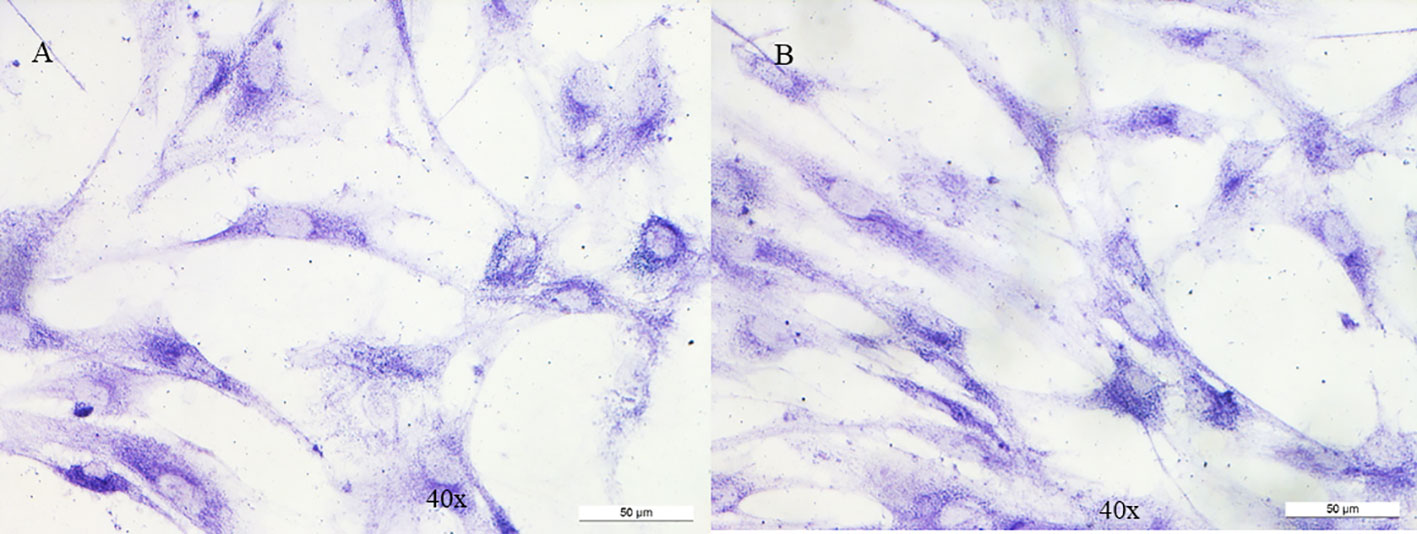
Figure 2 Cytochemistry for succinate dehydrogenase on fibroblasts. No variation is observed between control (A) and patient (B).

Figure 3 Western blotting on homogenate of fibroblasts. (A) Densitometric analysis of five experiments on three different fibroblast extractions for SDHC, SDHA and SDHB subunits. TOMM20 has been used for normalization of the referred proteins. (B) A representative experiment out of the five performed. No variation is observed between control and patient for the SDHC subunit, as well for SDHA and SDHB. Ct.1 and Ct.2: two different controls fibroblasts; Pr.1sr and Pt.2nd: two different protein extraction from patient’s fibroblasts.
Among pediatric cancers, NB is the most frequent extracranial solid tumor of childhood (5). Whereas familial forms represent only 1–2% of all cases, whole-exome sequencing approaches have identified several rare germline variants that are associated with cancer susceptibility in patients with NB who lack the classic clinical criteria for a cancer predisposition syndrome (CPS). A comprehensive list of these genes is provided by Barr and Applebaum (6). It is usually inherited in an autosomal dominant manner, with incomplete penetrance, and like retinoblastoma, conforms to the classic two-hit model hypothesized by Knudson. Today, several predisposing genes for neuroblastoma are known, and, the first of which was identified in 2004 (8). PHOX2B gene at 4p12, a neurogenesis regulatory gene, was the first gene whose role in neuroblastoma predisposition was discovered, and its mutations have been found in up to 10% of familial, often multifocal, NBs as a part of neurocristopathy syndrome, also characterized by Hirschsprung disease and congenital central hypoventilation (14).
Germline mutations in the ALK gene have also been associated with hereditary neuroblastoma cases; ALK encodes for a receptor tyrosine kinase and it is frequently mutated in NB with unfavorable biologic features (15). Another gene that has recently been associated with the hereditary form of neuroblastoma is MAX (MYC Associated Factor X) gene, which encodes for an essential transcription factor in the MYC/MAX/MXD1 axis; specifically, germline mutations in MAX are associated with overexpression of MYC, a known unfavorable prognostic factor (16). Germline mutations in TP53, SDHB, PTPN11, APC, NF1 and NKAIN2 have also been reported to occur rarely in neuroblastoma patients and thus fall within the sequenced genes in the presence of clinical conditions suggestive of cancer predisposition (multiple tumor lesions, early age of onset, family history) (17–23). In addition to these genetic variants associated with NB predisposition, there are several CPS in which the risk of NB recurrence is higher and overlaps with the risk of other malignancies. In this case, the pathway of RAS-MAPK in diseases such as RASopathies plays a leading role (24).
We identified a missense variant c.197C>T (p.Ala66Val) (rs760572684) in SDHC (NM_003001.5) gene, in heterozygous state and maternally segregated in a 3-year-old girl with a metastatic NB diagnosis and AML diagnosis soon after the end of treatment for NB.
Weaver syndrome and familial paraganglioma/pheochromocytoma have also been linked to the development of neuroblastoma, with mutations found in EZH2 and SDHB genes, respectively; the latter is a gene coding for one of the four subunits of the SDH complex involved in the mechanism of the electron transport chain and oxidative phosphorylation, which is essential for cellular ATP production (20, 25). Germline variants in the SDHA, SDHB and SDHC genes, encoding subunits of the SDH complex, have been associated with conditions characterized by the development of paragangliomas/pheochromocytomas, gastrointestinal stromal tumors and pulmonary chondromas (Carney triad and Carney-Stratakis syndrome) (26). In an article by Boikos et al., the presence of germline variants in SDHx genes in patients with Carney triad was investigated, concluding that in most cases the genetic defect remains unknown and that SDHx genes are apparently not involved in oncogenesis; moreover, although SDHx germline variants are present, the phenotype is often incomplete, suggesting the involvement of other genes, which may be contiguous with those encoding succinate dehydrogenase subunits (27).
The SDHC gene (Succinate Dehydrogenase Complex Subunit C) maps to the long arm of chromosome 1 (1q23.3) and it consists of six exons that code for a 169 amino acid (aa) polypeptide, which is one of the four subunits of succinate dehydrogenase (Figure 4) (28). Succinate dehydrogenase, also known as mitochondrial complex II, is a key heterotetrameric enzyme complex in the tricarboxylic acid cycle and aerobic respiratory chains of mitochondria. It consists of four subunits: two soluble subunits involved in electron transport, SDHA (primary catalytic subunit) and SDHB (redox subunit), and two transmembrane subunits, SDHC and SDHD, which anchor the complex in the inner mitochondrial membrane and facilitate electron transfer to the mitochondrial electron transport chain (29, 30). The oncological phenotype associated with SDHx variants is mainly due to loss of heterozygosity (LOH) events, although secondary somatic mutations are occasionally reported (31, 32). Defective activity of complex II leading to high oxygen (O2) production rate are detected in the cancer scenario, and in particular, several reports indicate that this defective activity may be caused by the pathogenic variants of the SDHC gene, that causes an increased O2 production and, consequently, an oxidative stress and genomic instability (33–35). In addition, genes involved in cellular respiration, as SDHx genes, act as tumor suppressors (30, 36, 37). SDHC variants are associated with gastrointestinal stromal tumors, paragangliomas and pheochromocytoma with an autosomal dominant transmission (31, 38–43). Insufficient data on the penetrance and phenotypic variability are reported in the literature, although some data show a lower penetrance in pathogenic variants of SDHC gene carriers compared with the SDHD pathogenic variants carriers (44–46).
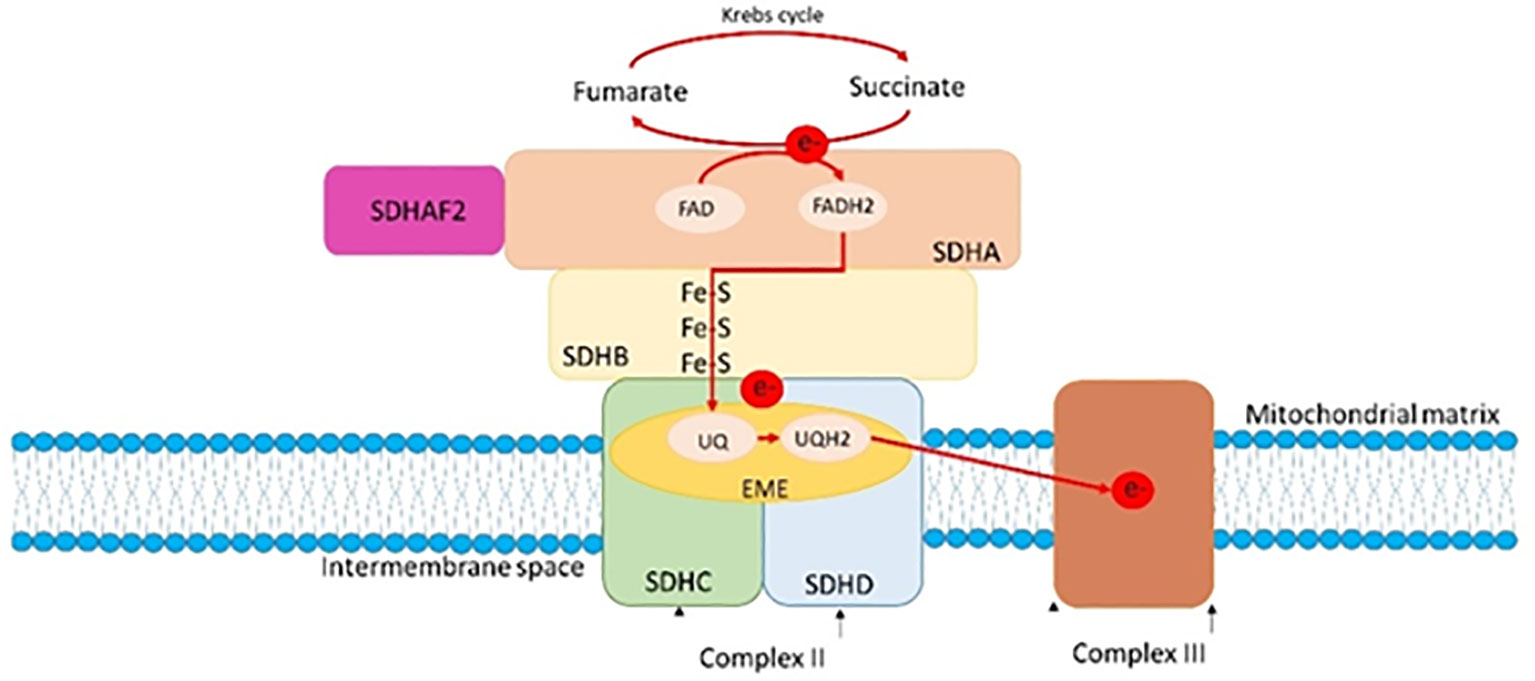
Figure 4 A: The SDH complex and its connection to the mitochondrial cristae and membranes. Taken together, the SDH subunits constitute the respiratory complex II. The hydrophilic SDHA and SDHB subunits catalyze the oxidation of succinate to fumarate as part of the tricarboxylic cycle, whereas the hydrophobic SDHD and SDHC subunits anchor the complex within the inner mitochondrial membrane. Prior to passing through Fe-S clusters in SDHB, tricarboxylic acid cycle (e-) electrons convert FAD to FADH2 in SDHA. These electrons then move on to the nearby respiratory complex III where they convert ubiquinone (Q) to ubiquinol (QH2).
Functional in vivo and in vitro studies showed that certain mutations in the SDHC gene inactivate the activity of the SDH complex in the yeast model and in the tumor tissue of patients, suggesting that these genes can act as tumor suppressor genes (47–49). In addition, studies on SDHC have shown that point mutations are associated with increased Reactive Oxygen Species (ROS) production, showing a correlation between dysfunction of all SDH complex subunits and formation of ROS (Figure 4) (50). In an elegant study by Ishii and colleagues, SDHC mutant transgenic cells showed elevated oxidative stress, increased transformation rate, tumor growth and DNA hypermutation in a mouse model (33). Another study, in which a nonsense mutation of SDHC was expressed in hamster fibroblasts, showed increased levels of ROS production, genomic instability and oxidative stress in mutant cells compared with parental cells (34). Finally, tumor formation and tumor cell proliferation may depend on the particular nature of the SDHx mutation (50).
Pathogenic and likely pathogenic variants of SDHC are splice site, frameshift and nonsense mutations, while there is a minority of missense variants (44). The variant found in our case (rs760572684) was described in the literature only in a PTEN mutation-negative patient, a 54-year-old patient with invasive breast cancer, follicular thyroid carcinoma, uterine leiomyomas, and cutaneous hemangioma with Cowden syndrome-like (10).
Of note, Trombetti and colleagues investigated the levels of SDHC variants and the oxidative mitochondrial metabolism in myeloid leukemia K562 cells over-expressing GATA-1 isoforms (51). They described a link between the levels of GATA-1S isoform (GATA Binding Protein 1 plays an important role in erythroid development by regulating the switch of fetal hemoglobin to adult hemoglobin) and SDHC alternative splicing variants (ASVs), leading to decreased of complex II activity and reduced oxidative phosphorylation efficiency, unveiling novel pro-leukemic mechanisms triggered by GATA-1S and aberrant expression of SDHC ASVs. Abnormal levels of SDHC and GATA-1S were also found in an AML patient, supporting in vitro results (51, 52). However, no SDHC germline mutations predisposing to AML have been described to date.
In our patient, no second hit mutation have been identified in the primary tumor. Furthermore, the functional study performed on the patient’s fibroblasts showed no variation between patient and control for cytochemistry analysis for succinate dehydrogenase, activity of the complex V of the mitochondrial respiratory chain on mitochondria isolated by fibroblasts and western blots on homogenate of fibroblasts, as well as SDHC, SDHA and SDHB are normal. Finally, the variant found in our patient involves a not conserved domain and we can assume that rs760572684 mutation might actually be a likely benign variant. Supporting this, the patient’s mother who presents the germline variant of the gene, is in good health and did not present history of diseases possibly related to the described mutation. Considering all, the second tumor may be more reasonably associated with iatrogenic effects of previous polichemotherapic treatment.
The case described highilights the importance of sharing new emerging gene mutations to improve the knowledge of molecular biology. Moreover, when available, the functional studies exploring the protein expression could help to define the role of VUS on the tumor history, in order to perform an appropriate follow-up and considering these mutations possible targets in a tailored therapy era. However, it should be emphasized that proband (or in this case parents) may decide to refuse to know the result of genetic testing.
The datasets presented in this study can be found in the National Center for Biotechnology Information. ClinVar; [VCV000371843.24], https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV000371843.24 and National Center for Biotechnology Information. ClinVar; [VCV002663836.1], https://www.ncbi.nlm.nih.gov/clinvar/variation/VCV002663836.1, and can be found in the article/Supplementary Material.
Ethical approval was not required for the study involving humans in accordance with the local legislation and institutional requirements. Written informed consent to participate in this study was not required from the participants or the participants’ legal guardians/next of kin in accordance with the national legislation and the institutional requirements. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
FF: Writing – original draft. RC: Writing – original draft. ML: Writing – review & editing. AD: Writing – review & editing. SC: Writing – review & editing. CR: Writing – review & editing. IG: Writing – review & editing. AS: Writing – review & editing. TR: Writing – review & editing. CM: Writing – review & editing. EA: Writing – review & editing. MD: Writing – review & editing. FG: Writing – review & editing. GD: Writing – review & editing. FD: Writing – review & editing. AM: Writing – review & editing. MD: Writing – original draft.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported also by the Italian Ministry of Health with Current Research funds.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1324013/full#supplementary-material
1. Choi DK, Helenowski I, Hijiya N. Secondary Malignancies in pediatric cancer survivors: perspectives and review of the literature. Int J Cancer (2014) 135:1764–73. doi: 10.1002/ijc.28991
2. McNerney ME, Godley LA, Le Beau MM. Therapy-related myeloid neoplasms: when genetics and environment collide. Nat Rev Cancer (2017) 17:513–27. doi: 10.1038/nrc.2017.60
3. Applebaum MA, Vaksman Z, Lee SM, Hungate EA, Henderson TO, London WB, et al. Neuroblastoma survivors are at increased risk for second Malignancies: A report from the International Neuroblastoma Risk Group Project. Eur J Cancer (2017) 72:177–85. doi: 10.1016/j.ejca.2016.11.022
4. Gröbner SN, Worst BC, Weischenfeldt J, Buchhalter I, Kleinheinz K, Rudneva VA, et al. The landscape of genomic alterations across childhood cancers. Nature (2018) 555:321–7. doi: 10.1038/nature25480
5. Chung C, Boterberg T, Lucas J, Panoff J, Valteau-Couanet D, Hero B, et al. Neuroblastoma. Pediatr Blood Cancer (2021) 68 Suppl 2:e28473. doi: 10.1002/pbc.28473
6. Barr EK, Applebaum MA. Genetic predisposition to neuroblastoma. Children (Basel) (2018) 5:E119. doi: 10.3390/children5090119
7. Kamihara J, Bourdeaut F, Foulkes WD, Molenaar JJ, Mossé YP, Nakagawara A, et al. Retinoblastoma and neuroblastoma predisposition and surveillance. Clin Cancer Res (2017) 23:e98–e106. doi: 10.1158/1078-0432.CCR-17-0652
8. Bonfiglio F, Lasorsa VA, Cantalupo S, D’Alterio G, Aievola V, Boccia A, et al. Inherited rare variants in homologous recombination and neurodevelopmental genes are associated with increased risk of neuroblastoma. EBioMedicine (2023) 87:104395. doi: 10.1016/j.ebiom.2022.104395
9. Kim J, Vaksman Z, Egolf LE, Kaufman R, Evans JP, Conkrite KL, et al. Germline pathogenic variants in neuroblastoma patients are enriched in BARD1 and predict worse survival. J Natl Cancer Inst (2023), djad183. doi: 10.1093/jnci/djad183
10. Ni Y, He X, Chen J, Moline J, Mester J, Orloff MS, et al. Germline SDHx variants modify breast and thyroid cancer risks in Cowden and Cowden-like syndrome via FAD/NAD-dependant destabilization of p53. Hum Mol Genet (2012) 21:300–10. doi: 10.1093/hmg/ddr459
11. Klein C, Djarmati A, Hedrich K, Schäfer N, Scaglione C, Marchese R, et al. PINK1, Parkin, and DJ-1 mutations in Italian patients with early-onset parkinsonism. Eur J Hum Genet (2005) 13:1086–93. doi: 10.1038/sj.ejhg.5201455
12. Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet (2013) 45:279–84. doi: 10.1038/ng.2529
13. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17:405–24. doi: 10.1038/gim.2015.30
14. Williams P, Wegner E, Ziegler DS. Outcomes in multifocal neuroblastoma as part of the neurocristopathy syndrome. Pediatrics (2014) 134:e611–616. doi: 10.1542/peds.2013-3340
15. Mossé YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature (2008) 455:930–5. doi: 10.1038/nature07261
16. Seabrook AJ, Harris JE, Velosa SB, Kim E, McInerney-Leo AM, Dwight T, et al. Multiple endocrine tumors associated with germline MAX mutations: multiple endocrine neoplasia type 5? J Clin Endocrinol Metab (2021) 106:1163–82. doi: 10.1210/clinem/dgaa957
17. Birch JM, Alston RD, McNally RJ, Evans DG, Kelsey AM, Harris M, et al. Relative frequency and morphology of cancers in carriers of germline TP53 mutations. Oncogene (2001) 20:4621–8. doi: 10.1038/sj.onc.1204621
18. Hasle H. Malignant diseases in Noonan syndrome and related disorders. Horm Res (2009) 72 Suppl 2:8–14. doi: 10.1159/000243773
19. Mutesa L, Pierquin G, Janin N, Segers K, Thomée C, Provenzi M, et al. Germline PTPN11 missense mutation in a case of Noonan syndrome associated with mediastinal and retroperitoneal neuroblastic tumors. Cancer Genet Cytogenet (2008) 182:40–2. doi: 10.1016/j.cancergencyto.2007.12.005
20. Schimke RN, Collins DL, Stolle CA. Paraganglioma, neuroblastoma, and a SDHB mutation: Resolution of a 30-year-old mystery. Am J Med Genet A (2010) 152A:1531–5. doi: 10.1002/ajmg.a.33384
21. Cascón A, Landa I, López-Jiménez E, Díez-Hernández A, Buchta M, Montero-Conde C, et al. Molecular characterisation of a common SDHB deletion in paraganglioma patients. J Med Genet (2008) 45:233–8. doi: 10.1136/jmg.2007.054965
22. Zhang J, Walsh MF, Wu G, Edmonson MN, Gruber TA, Easton J, et al. Germline mutations in predisposition genes in pediatric cancer. N Engl J Med (2015) 373:2336–46. doi: 10.1056/NEJMoa1508054
23. Romania P, Castellano A, Surace C, Citti A, De Ioris MA, Sirleto P, et al. High-resolution array CGH profiling identifies Na/K transporting ATPase interacting 2 (NKAIN2) as a predisposing candidate gene in neuroblastoma. PloS One (2013) 8:e78481. doi: 10.1371/journal.pone.0078481
24. Eleveld TF, Oldridge DA, Bernard V, Koster J, Colmet Daage L, Diskin SJ, et al. Relapsed neuroblastomas show frequent RAS-MAPK pathway mutations. Nat Genet (2015) 47:864–71. doi: 10.1038/ng.3333
25. Benn DE, Gimenez-Roqueplo A-P, Reilly JR, Bertherat J, Burgess J, Byth K, et al. Clinical presentation and penetrance of pheochromocytoma/paraganglioma syndromes. J Clin Endocrinol Metab (2006) 91:827–36. doi: 10.1210/jc.2005-1862
26. Amar L, Pacak K, Steichen O, Akker SA, Aylwin SJB, Baudin E, et al. International consensus on initial screening and follow-up of asymptomatic SDHx mutation carriers. Nat Rev Endocrinol (2021) 17:435–44. doi: 10.1038/s41574-021-00492-3
27. Boikos SA, Xekouki P, Fumagalli E, Faucz FR, Raygada M, Szarek E, et al. Carney triad can be (rarely) associated with germline succinate dehydrogenase defects. Eur J Hum Genet (2016) 24:569–73. doi: 10.1038/ejhg.2015.142
28. Satoh N, Yokoyama C, Itamura N, Miyajima-Nakano Y, Hisatomi H. Alternative splicing isoform in succinate dehydrogenase complex, subunit C causes downregulation of succinate-coenzyme Q oxidoreductase activity in mitochondria. Oncol Lett (2015) 9:330–4. doi: 10.3892/ol.2014.2699
29. van Nederveen FH, Gaal J, Favier J, Korpershoek E, Oldenburg RA, de Bruyn EMCA, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol (2009) 10:764–71. doi: 10.1016/S1470-2045(09)70164-0
30. Dalla Pozza E, Dando I, Pacchiana R, Liboi E, Scupoli MT, Donadelli M, et al. Regulation of succinate dehydrogenase and role of succinate in cancer. Semin Cell Dev Biol (2020) 98:4–14. doi: 10.1016/j.semcdb.2019.04.013
31. Hensen EF, van Duinen N, Jansen JC, Corssmit EPM, Tops CMJ, Romijn JA, et al. High prevalence of founder mutations of the succinate dehydrogenase genes in the Netherlands. Clin Genet (2012) 81:284–8. doi: 10.1111/j.1399-0004.2011.01653.x
32. Hoekstra AS, de Graaff MA, Briaire-de Bruijn IH, Ras C, Seifar RM, van Minderhout I, et al. Inactivation of SDH and FH cause loss of 5hmC and increased H3K9me3 in paraganglioma/pheochromocytoma and smooth muscle tumors. Oncotarget (2015) 6:38777–88. doi: 10.18632/oncotarget.6091
33. Ishii T, Yasuda K, Akatsuka A, Hino O, Hartman PS, Ishii N. A mutation in the SDHC gene of complex II increases oxidative stress, resulting in apoptosis and tumorigenesis. Cancer Res (2005) 65:203–9. doi: 10.1158/0008-5472.203.65.1
34. Slane BG, Aykin-Burns N, Smith BJ, Kalen AL, Goswami PC, Domann FE, et al. Mutation of succinate dehydrogenase subunit C results in increased O2.-, oxidative stress, and genomic instability. Cancer Res (2006) 66:7615–20. doi: 10.1158/0008-5472.CAN-06-0833
35. Ishii N, Ishii T, Hartman PS. The role of the electron transport SDHC gene on lifespan and cancer. Mitochondrion (2007) 7:24–8. doi: 10.1016/j.mito.2006.11.012
36. Pantaleo MA, Astolfi A, Urbini M, Nannini M, Paterini P, Indio V, et al. Analysis of all subunits, SDHA, SDHB, SDHC, SDHD, of the succinate dehydrogenase complex in KIT/PDGFRA wild-type GIST. Eur J Hum Genet (2014) 22:32–9. doi: 10.1038/ejhg.2013.80
37. Rutter J, Winge DR, Schiffman JD. Succinate dehydrogenase - Assembly, regulation and role in human disease. Mitochondrion (2010) 10:393–401. doi: 10.1016/j.mito.2010.03.001
38. Baysal BE, Willett-Brozick JE, Filho P a. A, Lawrence EC, Myers EN, Ferrell RE. An Alu-mediated partial SDHC deletion causes familial and sporadic paraganglioma. J Med Genet (2004) 41:703–9. doi: 10.1136/jmg.2004.019224
39. Schiavi F, Boedeker CC, Bausch B, Peçzkowska M, Gomez CF, Strassburg T, et al. Predictors and prevalence of paraganglioma syndrome associated with mutations of the SDHC gene. JAMA (2005) 294:2057–63. doi: 10.1001/jama.294.16.2057
40. Pasini B, McWhinney SR, Bei T, Matyakhina L, Stergiopoulos S, Muchow M, et al. Clinical and molecular genetics of patients with the Carney-Stratakis syndrome and germline mutations of the genes coding for the succinate dehydrogenase subunits SDHB, SDHC, and SDHD. Eur J Hum Genet (2008) 16:79–88. doi: 10.1038/sj.ejhg.5201904
41. McWhinney SR, Pasini B, Stratakis CA. International Carney Triad and Carney-Stratakis Syndrome Consortium. Familial gastrointestinal stromal tumors and germ-line mutations. N Engl J Med (2007) 357:1054–6. doi: 10.1056/NEJMc071191
42. Janeway KA, Kim SY, Lodish M, Nosé V, Rustin P, Gaal J, et al. Defects in succinate dehydrogenase in gastrointestinal stromal tumors lacking KIT and PDGFRA mutations. Proc Natl Acad Sci U.S.A. (2011) 108:314–8. doi: 10.1073/pnas.1009199108
43. Johnston JJ, Rubinstein WS, Facio FM, Ng D, Singh LN, Teer JK, et al. Secondary variants in individuals undergoing exome sequencing: screening of 572 individuals identifies high-penetrance mutations in cancer-susceptibility genes. Am J Hum Genet (2012) 91:97–108. doi: 10.1016/j.ajhg.2012.05.021
44. Andrews KA, Ascher DB, Pires DEV, Barnes DR, Vialard L, Casey RT, et al. Tumour risks and genotype-phenotype correlations associated with germline variants in succinate dehydrogenase subunit genes SDHB, SDHC and SDHD. J Med Genet (2018) 55:384–94. doi: 10.1136/jmedgenet-2017-105127
45. Neumann HPH, Pawlu C, Peczkowska M, Bausch B, McWhinney SR, Muresan M, et al. Distinct clinical features of paraganglioma syndromes associated with SDHB and SDHD gene mutations. JAMA (2004) 292:943–51. doi: 10.1001/jama.292.8.943
46. Bourdeau I, Grunenwald S, Burnichon N, Khalifa E, Dumas N, Binet M-C, et al. A SDHC founder mutation causes paragangliomas (PGLs) in the French Canadians: new insights on the SDHC-related PGL. J Clin Endocrinol Metab (2016) 101:4710–8. doi: 10.1210/jc.2016-1665
47. Burnichon N, Brière J-J, Libé R, Vescovo L, Rivière J, Tissier F, et al. SDHA is a tumor suppressor gene causing paraganglioma. Hum Mol Genet (2010) 19:3011–20. doi: 10.1093/hmg/ddq206
48. Smith EH, Janknecht R, Maher LJ. Succinate inhibition of alpha-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Hum Mol Genet (2007) 16:3136–48. doi: 10.1093/hmg/ddm275
49. Goffrini P, Ercolino T, Panizza E, Giachè V, Cavone L, Chiarugi A, et al. Functional study in a yeast model of a novel succinate dehydrogenase subunit B gene germline missense mutation (C191Y) diagnosed in a patient affected by a glomus tumor. Hum Mol Genet (2009) 18:1860–8. doi: 10.1093/hmg/ddp102
50. Szeto SSW, Reinke SN, Sykes BD, Lemire BD. Ubiquinone-binding site mutations in the Saccharomyces cerevisiae succinate dehydrogenase generate superoxide and lead to the accumulation of succinate. J Biol Chem (2007) 282:27518–26. doi: 10.1074/jbc.M700601200
51. Trombetti S, Sessa R, Catapano R, Rinaldi L, Lo Bianco A, Feliciello A, et al. Exploring the leukemogenic potential of GATA-1S, the shorter isoform of GATA-1: novel insights into mechanisms hampering respiratory chain complex II activity and limiting oxidative phosphorylation efficiency. Antioxid (Basel) (2021) 10:1603. doi: 10.3390/antiox10101603
Keywords: cancer predisposition syndrome, succinate dehydrogenase complex subunit C (SHDC) gene, variants of uncertain significance (VUS), neuroblastoma, acute myeloid leukemia
Citation: Fabozzi F, Carrozzo R, Lodi M, Di Giannatale A, Cipri S, Rosignoli C, Giovannoni I, Stracuzzi A, Rizza T, Montante C, Agolini E, Di Nottia M, Galaverna F, Del Baldo G, Del Bufalo F, Mastronuzzi A and De Ioris MA (2024) Case report: A safeguard in the sea of variants of uncertain significance: a case study on child with high risk neuroblastoma and acute myeloid leukemia. Front. Oncol. 13:1324013. doi: 10.3389/fonc.2023.1324013
Received: 18 October 2023; Accepted: 11 December 2023;
Published: 08 January 2024.
Edited by:
Luca Lo Nigro, Azienda Ospedaliero Universitaria Policlinico - San Marco, ItalyReviewed by:
Mario Capasso, University of Naples Federico II, ItalyCopyright © 2024 Fabozzi, Carrozzo, Lodi, Di Giannatale, Cipri, Rosignoli, Giovannoni, Stracuzzi, Rizza, Montante, Agolini, Di Nottia, Galaverna, Del Baldo, Del Bufalo, Mastronuzzi and De Ioris. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Fabozzi, ZnJhbmNlc2NvLmZhYm96emlAb3BiZy5uZXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.