 Domenico Ribatti
Domenico Ribatti Tiziana Annese
Tiziana Annese Roberto Tamma
Roberto Tamma- 1Department of Translational Biomedicine and Neuroscience, University of Bari Medical School, Bari, Italy
- 2Department of Medicine and Surgery, Libera Università del Mediterraneo (LUM) Giuseppe Degennaro University, Bari, Italy
Three different mechanisms of neovascularization have been described in tumor growth, including sprouting angiogenesis, intussusceptive microvascular growth and glomeruloid vascular proliferation. Tumors can also grow by means of alternative mechanisms including vascular co-option, vasculogenic mimicry, angiotropism, and recruitment of endothelial precursor cells. Vascular co-option occurs in tumors independently of sprouting angiogenesis and the non-angiogenic cancer cells are described as exploiting pre-existing vessels. Vascular co-option is more frequently observed in tumors of densely vascularized organs, including the brain, lung and liver, and vascular co-option represents one of the main mechanisms involved in metastasis, as occurs in liver and lung, and resistance to anti-angiogenic therapy. The aim of this review article is to analyze the role of vascular co-option as mechanism through which tumors develop resistance to anti-angiogenic conventional therapeutic approaches and how blocking co-option can suppress tumor growth.
Angiogenesis and alternative mode of growth of tumor vasculature
Three types of angiogenesis have been described in tumor growth: sprouting angiogenesis in which vessel outgrowth from existing vessels is initiated by specialized endothelial cells termed tip cells (1, 2), intussusceptive microvascular growth (IMG), in which the vascular network expands by insertion of newly formed columns of interstitial tissue structures (tissue pillars) into the vascular lumen (3), and glomeruloid vascular proliferation, in which small glomeruloid bodies, so-called for their morphological resemblance with the renal glomeruli, are recognizable (4) (Figure 1).
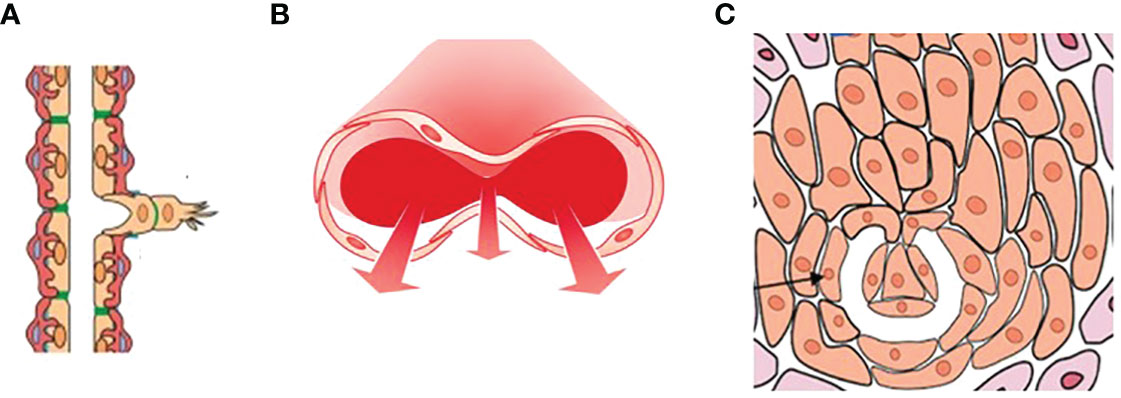
Figure 1 A drawing showing the three types of angiogenesis have been described in tumor growth: (A) sprouting angiogenesis, (B) intussusceptive microvascular growth (IMG), and (C) glomeruloid vascular proliferation. Sprouting angiogenesis involves formation and outgrowth of sprouts; IMG involves the formation of new vasculature where a pre-existing vessel splits in two; in glomeruloid vascular proliferation small glomeruloid bodies, so-called for their morphological resemblance with the renal glomeruli, are recognizable. [Reproduced from (5)].
Tumors can also grow without inducing angiogenesis, as occurs in vessel co-option or vascular co-option, vasculogenic mimicry, angiotropism, and recruitment of endothelial precursor cells (EPCs). In vasculogenic mimicry, first described in uveal melanoma (6) and subsequently in other cancers, tumor cells acquire an endothelial phenotype and form vessel-like networks. Vasculogenic mimicry can serve as a marker for tumor metastasis, a poor prognosis, worse survival, and the highest risk of cancer recurrence. Angiotropism (the pericytic-like location of tumor cells) is a marker of migration of melanoma and glioma tumor cells along the abluminal vascular surface (7). EPCs may be recruited from bone marrow mobilized by vascular endothelial growth factor A (VEGFA) or C-X-C motif chemokine 12 (CXCL12) released by tumor-infiltrating myeloid cells or cancer cells (8). However, most human tumors remain in situ without angiogenesis for a long time before they switch to an angiogenic phenotype (9).
The aim of this review article it to describe in detail through a retrospective analysis of the literature data the role of vascular co-option in the growth of primary and metastatic tumors. Moreover, the involvement of co-option in the development of resistance to conventional anti-angiogenic therapies and the possibility to overcome the resistance will be also described.
Vascular co-option
In the first work on vascular co-option the non-angiogenic cancer cells were described as “exploiting” pre-existing vessels (10). Four different histopathological growth patterns have been described in non-small cell lung cancer. In three of these patterns (basal, diffuse, and papillary), the tumors are angiogenic, whereas in the fourth pattern (alveolar), the cancer cells grow in the alveolar air space of the lung and co-opt alveolar capillaries (10) (Figure 2). Vascular co-option, described in primary and secondary (metastatic) sites (Table 1) is defined as a process in which tumor cells interact with and exploit the pre-existing vasculature of the normal tissue in which they grow without the need for vascular proliferation (16). The pre-existing vasculature can be co-opted in two ways: tumor cells replace normal epithelial cells or penetrate the stroma surrounding the blood vessels (18). The host vasculature is incorporated by the growing tumor (19). In vascular co-option, cancer preserves the well-arranged vascular architecture of the normal tissue within the tumor, and tumors utilize alternative mechanisms besides angiogenesis to obtain nutrients for growth through local tumor invasion and proliferation along co-opted vessels. Cancer cells migrate along the pre-existing vessels and infiltrate tissues between co-opted vessels (16). By means of single-cell transcriptomic analysis, it has been demonstrated that co-opted endothelial cells and pericytes are characterized by similar transcriptomic signature of quiescent healthy endothelial cells and different from tumor endothelial cell signature recognizable in angiogenic tumors, characterized by tip, proliferating, and immature endothelial cells (20). Moreover, different cell types involved in co-option, including an invasive cancer cell subtype and a M1-like macrophage subtype have been identified (20).
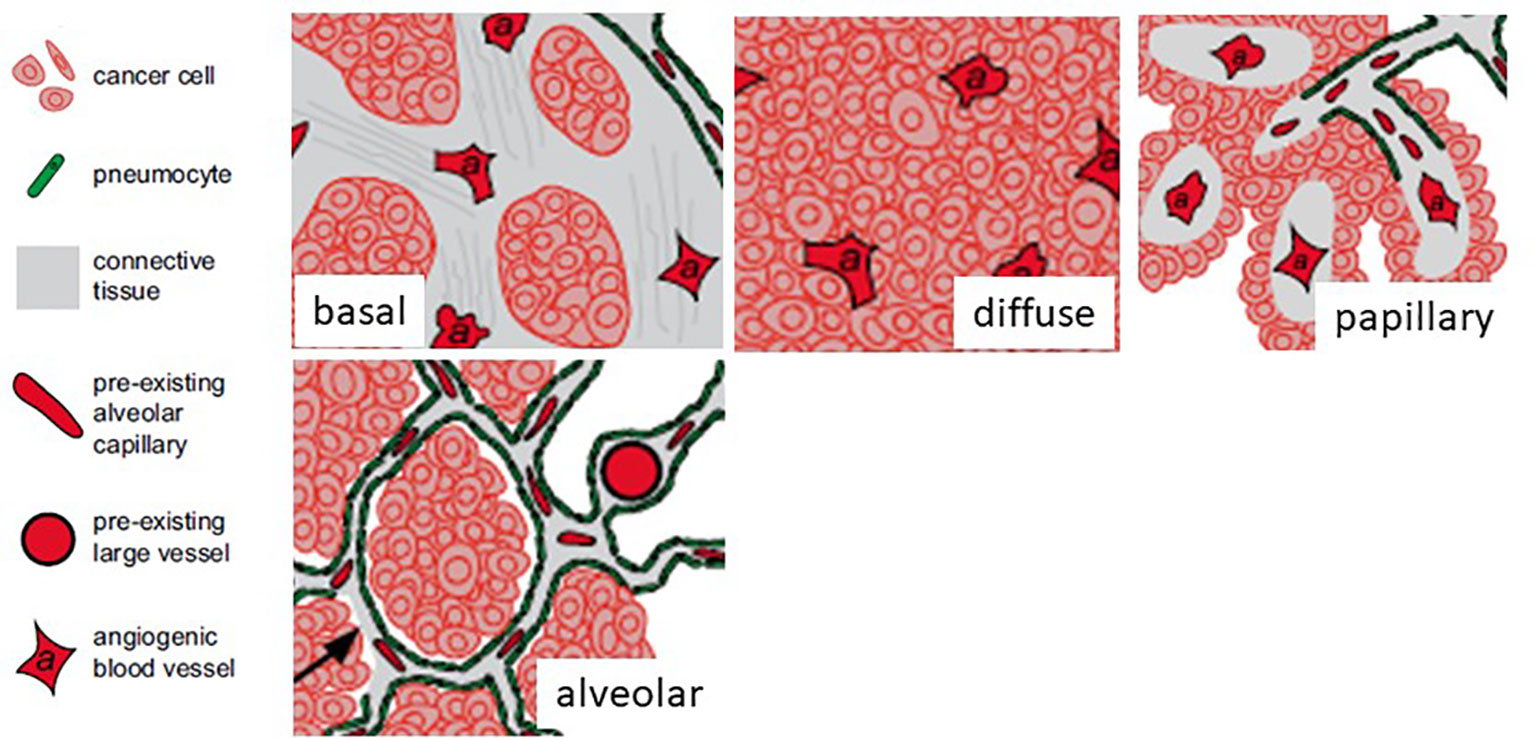
Figure 2 Four different histopathological growth patterns in non-small cell lung cancer. In three of these patterns (basal, diffuse, and papillary), the tumors are angiogenic, whereas in the fourth pattern (alveolar), the cancer cells grow in the alveolar air space of the lung and co-opt alveolar capillaries. [Modified from (11)].
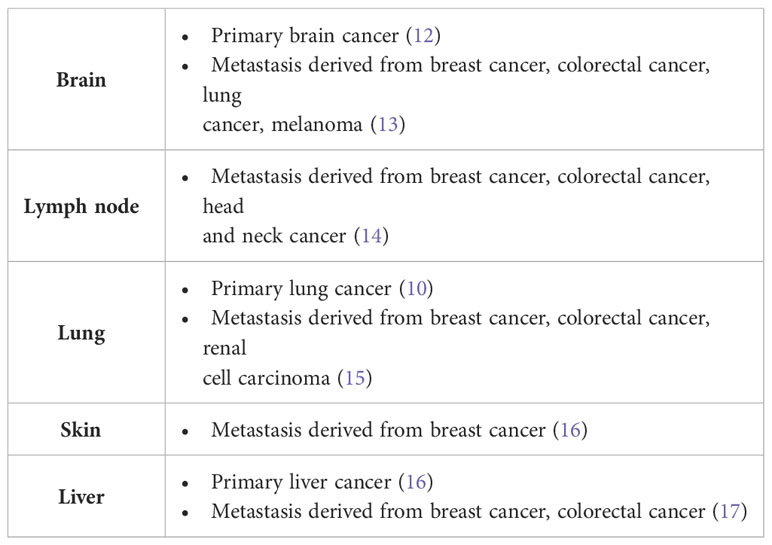
Table 1 Evidence for vascular co-option in human cancers (References between brackets).
Some tumors grow by exploiting the host vessels and initiating pre-existing blood-vessel-dependent tumor growth. These vessels then regress owing to endothelial cell apoptosis, mediated by angiopoietin- 2 (Ang-2), whereas angiogenesis occurs at the periphery of the growing tumor by the interaction of VEGF and Ang-2 (21). The co-opted vessels trigger an apoptotic cascade, probably by autocrine induction of Ang-2, followed by vessel regression resulting in tumor death. However, successful tumors overcome this vessel regression by initiating neoangiogenesis (22). Vascular co-option is more frequently observed in tumors of densely vascularized organs, including the brain, lung, and liver, where the primary tumor cells co-opt the adjacent quiescent blood vessels of the host tissue. In the brain, cancer cells can adhere to the abluminal surface of the brain vessels and grow around them or can infiltrate the brain parenchyma and migrate between brain vessels. Vessel co-option has been reported in both low- and high-grade gliomas (12). In advanced hepatocellular carcinoma, vascular co-option may interest vessels of the portal tract or liver sinusoidal vessels (16). The lymph node is also involved in nonangiogenic growth by vascular co-option (14).
Vascular co-option and metastases
Tumor cells have immediate access to blood vessels, such as when they metastasize to or are implanted within a vascularized tissue, co-opt, and often grow as cuffs around adjacent existing vessels (21). The ability of cancer cells to co-opt the vessels initiates the process through a few of them will intravasate, starting the metastatic process (23). Invasive cancer cells express Wnt7b, that promotes metastasis and vascular co-option in an experimental model of glioma (24).
The blood vessels at the interface between the tumor and surrounding tissue can be co-opted through the replacement of epithelial cells by tumor cells and/or the invasion of tumor cells into the stroma surrounding the blood vessels (25, 26). Many patients with non-angiogenic vs. angiogenic primary lung tumors developed distant lung metastases and vessel-co-option was associated with decreased overall survival, and a 3D-tumor reconstruction showed that the blood vessel immunostaining resembled a normal lung (27–29). In breast cancer lung metastases, non-angiogenic growth was present in 19% of the lung metastases (30).
Melanoma cells migrate extra vascularly in the brain metastasis along the basement membrane of the blood vessels surrounding the tumor, constituting an alternative mechanism of tumor spreading instead of intravascular dissemination (angiotropism or pericytic mimicry) (7, 31, 32). Vessel co-option occurs in other secondary brain tumors, including brain metastases of breast, colorectal, and lung cancer (13). Carcinomas that metastasize to the lung, including breast, colorectal, gastric, pancreatic, and renal cancer, may be characterized by vascular co-option (10, 15, 30, 33). Vessel co-option can occur in breast cancer metastases to many organs, including the lungs, liver, lymph nodes, skin, and brain (10, 17, 30, 34, 35).
Experimental models used for the study of vascular co-option and their impact on therapy response
Different animal models have been established to study vascular co-option. By means of intracranial transplantation it has been demonstrated that different tumor cell lines are able to co-opt pre-existing brain vessels (21). Anti-VEGF and anti-VEGF receptor 2 (VEGFR2) treatments increased the number of co-opted brain vessels after injection of human glioblastoma cell lines into nude rat striatum (36, 37). By means of intracardial transplantation into the left cardiac ventricle of anesthetized mice it has been demonstrated that serpins promote cancer cell survival and vascular co-option in brain metastasis (21). Through direct injection of tumor cells into the internal carotid artery of anesthetized mice, it has been demonstrated that VEGFA favors brain metastasis of different human melanoma cell lines to grow by means of vascular co-option without sprouting angiogenesis (38). Moreover, anti-angiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vascular co-option (39). By means of intravenous injection via jugular vein, it has been demonstrated that lung metastases co-opted the lung microvasculature (20). Moreover, by means of the same experimental model, it was shown that vascular co-option mediates resistance to anti-angiogenic therapy in lung metastasis (15). Using orthotopic injection of human hepatocellular cell lines into the livers, it has been demonstrated that vascular co-option is involved in acquired resistance to anti-angiogenic therapy (40). By means of zebrafish and chick embryo chorioallantoic membrane (CAM) assays it has been shown that connexins are involved in metastatic breast cancer and melanoma brain colonization through vascular co-option (41).
Resistance to anti-angiogenic therapies
When VEGF-targeted therapies are discontinued, the tumor vasculature is rapidly re-established (42). Resistance to anti-angiogenic therapies can be intrinsic, when it is observed at the beginning of the treatment due to inefficacy of treatment or acquired, i.e., that it affects the relapsing disease after an initial response to therapy (43, 44). The main mechanisms of resistance involve: 1) Vascular co-option (45); vasculogenic mimicry; IMG (a switch from sprouting to IMG represents an adaptive response to treatment with anti-angiogenic compounds to restore the hemodynamic and structural properties of the vasculature-enhancing tumor drug delivery and sensitivity to treatments (46). 2) Hypoxia that increases the expression of pro-angiogenic factors. The relationship between hypoxia, VEGF, and angiogenesis has significant implications for response to therapy, and hypoxic areas are refractory to chemotherapy and radiotherapy. VEGF blockade aggravates hypoxia that, in turn, upregulates the production of angiogenic factors or increases tumor invasiveness (43, 47). 3) Redundancy of the angiogenic signals, and activation of alternative signaling pathways [e.g., platelet derived growth factor/platelet derived growth factor receptor (PDGF/PDGFR); fibroblast growth factor/fibroblast growth factor receptor (FGF/FGFR); Ang2/Tie 2] (48). 4) Upregulation of other pro-angiogenic factors, including FGF2 and PDGF (48). 5) Vascular heterogeneity, and EPCs recruitment.
In metastatic colorectal cancer patients, bevacizumab treatment was associated with increased plasma levels of placental growth factor (PlGF), FGF, and PDGF prior to and along disease progression (49). Colorectal patients with poor response to bevacizumab had increased Ang2 serum levels, and VEGF and Ang2 blockade delayed tumor growth, normalized tumor vasculature, and increased survival (50, 51).
Resistance and co-option
In patients with metastatic colorectal cancer treated with neoadjuvant bevacizumab, and then underwent complete resection of the metastases, a higher number of nonangiogenic lesions was found in the viable tissue of the poor responder while prevalently angiogenic metastases were seen among the good responders (52). In two cohorts of breast cancer patients with liver metastases, one treated with bevacizumab and chemotherapy and one with chemotherapy alone prior to metastases resection, all of the metastatic lesions examined, were nonangiogenic (52). In colorectal cancer liver metastases, from three cohorts, one not receiving any neoadjuvant, a second receiving neoadjuvant chemotherapy, and a third treated with chemotherapy plus bevacizumab, upon treatment with chemotherapy and bevacizumab, the nonangiogenic lesions showed no or minimal necrosis while the angiogenic tumors had no more than 2% of viable cells (53). A similar pattern was seen in the patients treated with chemotherapy alone, the only difference being that in the angiogenic tumors, the percentage of viable tissue was higher. Untreated nonangiogenic metastases were comparable to the treated ones, while untreated angiogenic had a more variable amount of necrosis, supporting the hypothesis that nonangiogenic tumors are not only more resistant to bevacizumab but also to the neoadjuvant chemotherapy used (53).
No differences in intratumor vascularization were found in node metastases from colorectal cancer patients treated or not treated with bevacizumab (34). Antiangiogenic treatment of primary central nervous system tumors has been used for several years but, again, the results have not lived up the expectations (54). Vascular co-option mediates resistance to anti-angiogenic therapy in liver metastases (52), and in an experimental model of hepatocellular carcinoma, a switch to vascular co-option has been described as a mechanism of resistance to treatment with tyrosine kinase inhibitor sorafenib, where over time, tumor cells become more invasive which promotes co-option of liver vessels in the face of angiogenesis blockade (55).
Histological examination of patients who died after receiving treatment with cediranib, an inhibitor of VEGFR2 tyrosine kinases (56) or bevacizumab regimen (57) showed that the glioma cells were growing around preexisting vessels in a nonangiogenic fashion. Vascular co-option and progression in the absence of angiogenesis in human brain tumor samples, surgical and autoptic, have been illustrated (26, 54). Breast cancer patients treated with bevacizumab showed, by dynamic contrast-enhanced magnetic resonance imaging (DCE-MRI), three response patterns. In the first one, a decrease in K-trans values over the extent of the tumor was demonstrated, that is, decreased vascular permeability and/or vascular surface area, while the second was characterized by extensive necrosis. These two patterns indicate a response to anti-angiogenic treatment. In the third one, instead, no changes were seen, and the authors conclude that, in these lesions, the vessels were independent of VEGF. Co-option of pre-existing vascular beds in adipose tissue controls tumor growth rates and angiogenesis (58).
Metastatic lesions in stage III melanoma patients treated with bevacizumab had a mature vessel morphology and phenotype in contrast with the newly formed vessels in the relapsing disease from the patients not receiving bevacizumab (59). Lung metastases of renal cell carcinoma (RCC) can escape anti-angiogenic treatment with cediranib and gefitinib by switching phenotype and progressing in an angiogenesis-independent fashion exploiting preexisting vessel (60). Treatment of glioma with a monoclonal antibody against VEGFR2 induced co-option in quiescent cerebral vessels (37). In glioblastoma biopsies taken after antiangiogenic treatment, a pattern of infiltration around the normal brain vessels is present (26). Nonangiogenic growth and spread in the lung have also been linked to resistance to surgical treatment (61). In nonangiogenic carcinoma growing in the lung, neoplastic cells can spread by moving from one alveolar cavity to the other through the septa pores (61). Co-option of liver vessels has been demonstrated in acquired sorafenib resistance in hepatocellular carcinoma (40).
Combination therapy to overcome resistance to anti-angiogenic therapy
Dual inhibition of VEGF/PDGFR improves survival in glioblastoma (62). Bevacizumab-resistant patients exhibit an up-regulation of c-MET expression (63). The concomitant exposure to hepatocyte growth factor (HGF)/c-MET inhibitors and sunitinib abrogated angiogenesis and tumor growth (64). Application of brivanib, a dual inhibitor of FGF and VEGF pathways, in bevacizumab-resistant tumors induced an increased overall survival in breast cancer (65). Aflibercept, a dual inhibitor of VEGF and PlGF, and bevacizumab are effective in patient-derived xenograft models of colorectal cancer (66). Lenvatinib, a multiple receptor tyrosine kinase inhibitor of VEGFRs 1-3, FGFRs 1-4, KIT, PDGF receptor alpha (PDGFRα), and RET (rearranged during transfection) exerts antiproliferative and anti-angiogenic effects in hepatocellular carcinoma (67). Otherwise, both dovitinib and nintedanib (VEGF, FGF, and PDGF receptor tyrosine kinase inhibitors) are ineffective (68, 69).
Combination therapy to overcome resistance and vascular co-option
Combinatorial treatment approaches integrating anti-angiogenic therapy with blockade of vascular co-option can be considered. Anti-angiogenic therapy via VEGFR2 blockade reduced intracerebral glioblastoma growth but caused an increase in tumor migration with a vascular co-option pattern. The increase in cancer cell migration may be inhibited by combined treatment with VEGFR2 and epidermal growth factor receptor (EGFR) antibodies, as demonstrated by a decreased in vitro migration of glioblastoma cells (70).
A single agent vanucizumab, a bispecific anti-Ang2/anti-VEGFA antibody, was tested in a phase I study in adult patients with advanced solid tumors (71). Dual inhibition of Ang2 and VEGFRs normalizes tumor vasculature and prolongs survival in glioblastoma (51). Stimulation of Ang-2 expression in endothelial cells together with inhibition of VEGF signaling may inhibit vascular cooption. Signaling of Ang-2 through its receptor Tie-2 can cause sprouting angiogenesis if VEGF levels in the tumor microenvironment are high. If VEGF levels are low, Ang-2/Tie-2 signaling leads to the regression of co-opted vessels (72–74).
Inhibition of hypoxia-related macrophages and neutrophils may inhibit vascular co-option. Accumulation of lysyl oxidase like 4 (LOXL-4)-expressing neutrophils in colorectal lung cancer metastases resistant to anti-angiogenic therapy where vascular co-option is the dominant pattern of vascularization (75). M1 tumor associated macrophages (TAMs) are highly detected in vascular co-option-dependent tumors (20). Treatment with bevacizumab caused a metabolic shift toward glycolysis, promoting the vascular co-option pro-invasive phenotype in glioblastoma (76).
Concluding remarks
Many cancers can co-opt the pre-existing vasculature and, in this manner, facilitate tumor growth in situ and at distance. The existence of non-angiogenic tumors allows to predict that these tumors are non-sensitive to anti-angiogenic treatments (77). Several preclinical studies support the concept that vessel co-option can mediate intrinsic and acquired resistance to anti-angiogenic therapy, and co-option may in part explain the inefficacy of anti-angiogenic cancer therapies. In this context, blocking or inhibiting co-option might be considered as an innovative strategy in the inhibition of the growth of certain tumors, which adopt this modality of expansion, suggesting that therapeutic strategies targeting both angiogenesis and co-option might be more efficacious than targeting angiogenesis alone (73). It will be also important to consider the role of vascular co-option in the modulation of the host immune response to tumor and therefore also the efficacy of immunotherapy in the treatment of tumors (78). In this context, immune checkpoint-inhibitor treatment might induce an immune response against the co-opting cancer cells and synergize with anti-angiogenic agents (79, 80).
Author contributions
RT: Writing – review & editing. DR: Writing – original draft. TA: Writing – review & editing.
Funding
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. DR received financial support for the publication of this article (00234115-ARES, Centro Salute Ambiente, Progetto Jonico Salentino).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ausprunk DH, Folkman J. Migration and proliferation of endothelial cells in preformed and newly formed blood vessels during tumor angiogenesis. Microvascular Res (1977) 14(1):53–65. doi: 10.1016/0026-2862(77)90141-8
2. Ribatti D, Crivellato E. “Sprouting angiogenesis”, a reappraisal. Dev Biol (2012) 372(2):157–65. doi: 10.1016/j.ydbio.2012.09.018
3. Ribatti D, Djonov V. Intussusceptive microvascular growth in tumors. Cancer Lett (2012) 316(2):126–31. doi: 10.1016/j.canlet.2011.10.040
4. Sundberg C, Nagy JA, Brown LF, Feng D, Eckelhoefer IA, Manseau EJ, et al. Glomeruloid microvascular proliferation follows adenoviral vascular permeability factor/vascular endothelial growth factor-164 gene delivery. Am J Pathol (2001) 158(3):1145–60. doi: 10.1016/S0002-9440(10)64062-X
5. Ribatti D, Pezzella F. Vascular co-option and other alternative modalities of growth of tumor vasculature in glioblastoma. Front Oncol (2022) 12:874554. doi: 10.1016/S0002-9440(10)65173-5
6. Maniotis AJ, Folberg R, Hess A, Seftor EA, Gardner LM, Pe'er J, et al. Vascular channel formation by human melanoma cells in vivo and in vitro: vasculogenic mimicry. Am J Pathol (1999) 155(3):739–52. doi: 10.1111/pcmr.12120
7. Lugassy C, Péault B, Wadehra M, Kleinman HK, Barnhill RL. Could pericytic mimicry represent another type of melanoma cell plasticity with embryonic properties? Pigment Cell Melanoma Res (2013) 26(5):746–54. doi: 10.1172/JCI0214327
8. Reyes M, Dudek A, Jahagirdar B, Koodie L, Marker PH, Verfaillie CM. Origin of endothelial progenitors in human postnatal bone marrow. J Clin Invest (2002) 109(3):337–46. doi: 10.1172/JCI14327
9. Ribatti D, Nico B, Crivellato E, Roccaro AM, Vacca A. The history of the angiogenic switch concept. Leukemia (2006) 21(1):44–52. doi: 10.1016/S0959-8049(96)00377-2
10. Pezzella F, Di Bacco A, Andreola S, Nicholson AG, Pastorino U, Harris AL. Angiogenesis in primary lung cancer and lung secondaries. Eur J Cancer (1996) 32(14):2494–500. doi: 10.3171/jns.2005.103.4.0702
11. Kuczynski EA, Reynolds AR. Vessel co-option and resistance to anti-angiogenic therapies. Angiogenesis (2020) 23:55–74. doi: 10.1093/neuonc/not112
12. Bernsen H, van der Laak J, Küsters B, van der Ven A, Wesseling P. Gliomatosis cerebri: quantitative proof of vessel recruitment by cooptation instead of angiogenesis. J Neurosurg (2005) 103(4):702–6. doi: 10.1046/j.1365-2559.2001.01061.x
13. Berghoff AS, Rajky O, Winkler F, Bartsch R, Furtner J, Hainfellner JA, et al. Invasion patterns in brain metastases of solid cancers. Neuro-oncology (2013) 15(12):1664–72. doi: 10.1002/path.4845
14. Naresh KN, Nerurkar AY, Borges AM. Angiogenesis is redundant for tumour growth in lymph node metastases. Histopathology (2001) 38(5):466–70. doi: 10.1038/s41571-019-0181-9
15. Bridgeman VL, Vermeulen PB, Foo S, Bilecz A, Daley F, Kostaras E, et al. Vessel co-option is common in human lung metastases and mediates resistance to anti-angiogenic therapy in preclinical lung metastasis models. J Pathol (2017) 241(3):362–74. doi: 10.1038/sj.bjc.6601727
16. Kuczynski EA, Vermeulen PB, Pezzella F, Kerbel RS, Reynolds AR. Vessel co-option in cancer. Nat Rev Clin Oncol (2019) 16(8):469–93. doi: 10.1007/s10456-019-09690-0
17. Stessels F, Van den Eynden G, van der Auwera I, Salgado R, Van den Heuvel E, Harris AL, et al. Breast adenocarcinoma liver metastases, in contrast to colorectal cancer liver metastases, display a non-angiogenic growth pattern that preserves the stroma and lacks hypoxia. Br J cancer (2004) 90(7):1429–36. doi: 10.1002/path.4904
18. Latacz E, Caspani E, Barnhill R, Lugassy C, Verhoef C, Grunhagen D, et al. Pathological features of vessel co-option versus sprouting angiogenesis. Angiogenesis (2020) 23(1):43–54. doi: 10.1016/j.celrep.2021.109253
19. Winkler F. Hostile takeover: how tumours hijack pre-existing vascular environments to thrive. J Pathol (2017) 242(3):267–72. doi: 10.1126/science.284.5422.1994
20. Teuwen L-A, De Rooij LPMH, Cuypers A, Rohlenova K, Dumas SJ, García-Caballero M, et al. Tumor vessel co-option probed by single-cell analysis. Cell Rep (2021) 35(11):109253. doi: 10.1016/j.cell.2014.01.040
21. Holash J, Maisonpierre PC, Compton D, Boland P, Alexander CR, Zagzag D, et al. Vessel cooption, regression, and growth in tumors mediated by angiopoietins and VEGF. Science (1999) 284(5422):1994–8.
22. Valiente M, Obenauf AC, Jin X, Chen Q, Zhang XHF, Lee DJ, et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell (2014) 156(5):1002–16. doi: 10.1016/j.ccell.2018.03.020
23. Bald T, Quast T, Landsberg J, Rogava M, Glodde N, Lopez-Ramos D, et al. Ultraviolet-radiation-induced inflammation promotes angiotropism and metastasis in melanoma. Nature (2014) 507(7490):109–13. doi: 10.1038/nature13111
24. Griveau A, Seano G, Shelton SJ, Kupp R, Jahangiri A, Obernier K, et al. A glial signature and Wnt7 signaling regulate glioma-vascular interactions and tumor microenvironment. Cancer Cell (2018) 33(5):874–89.e7. doi: 10.1002/ijc.31983
25. García-Gómez P, Valiente M. Vascular co-option. Tumor Vascularization: Elsevier; (2020) p:33–47.
26. Falchetti ML, D'Alessandris QG, Pacioni S, Buccarelli M, Morgante L, Giannetti S, et al. Glioblastoma endothelium drives bevacizumab-induced infiltrative growth via modulation of PLXDC1. Int J cancer (2019) 144(6):1331–44.
27. Pastorino U, Andreola S, Tagliabue E, Pezzella F, Incarbone M, Sozzi G, et al. Immunocytochemical markers in stage I lung cancer: relevance to prognosis. J Clin Oncol (1997) 15(8):2858–65. doi: 10.1200/JCO.1997.15.8.2858
28. Adighibe O, Micklem K, Campo L, Ferguson M, Harris A, Pozos R, et al. Is nonangiogenesis a novel pathway for cancer progression? A study using 3-dimensional tumour reconstructions. Br J cancer (2006) 94(8):1176–9. doi: 10.1038/sj.bjc.6603039
29. Sardari Nia P, Colpaert C, Blyweert B, Kui B, Vermeulen P, Ferguson M, et al. Prognostic value of nonangiogenic and angiogenic growth patterns in non-small-cell lung cancer. Br J cancer (2004) 91(7):1293–300. doi: 10.1016/B978-0-12-819494-2.00004-3
30. Evidence for novel non-angiogenic pathway in breast-cancer metastasis. Breast Cancer Progression Working Party. Lancet (2000) 355(9217):1787–8. doi: 10.1038/srep23834
31. Lugassy C, Kleinman H, Barnhill R. Pericyte mimicry: an embryogenesis-derived program of extravascular tumor cell migration. Tumor Vascularization: Elsevier; (2020), 49–88.
32. Bentolila LA, Prakash R, Mihic-Probst D, Wadehra M, Kleinman HK, Carmichael TS, et al. Imaging of angiotropism/vascular co-option in a murine model of brain melanoma: implications for melanoma progression along extravascular pathways. Sci Rep (2016) 6:23834–. doi: 10.1093/jnci/djv155
33. Sardari Nia P, Hendriks J, Friedel G, Van Schil P, Van Marck E. Distinct angiogenic and non-angiogenic growth patterns of lung metastases from renal cell carcinoma. Histopathology (2007) 51(3):354–61.
34. Jeong H-S, Jones D, Liao S, Wattson DA, Cui CH, Duda DG, et al. Investigation of the lack of angiogenesis in the formation of lymph node metastases. J Natl Cancer Institute (2015) 107(9):djv155.
35. Vermeulen PB, Sardari Nia P, Colpaert C, Dirix LY, Van Marck E. Lack of angiogenesis in lymph node metastases of carcinomas is growth pattern-dependent. Histopathology (2002) 40(1):105–7. doi: 10.1046/j.1365-2559.2002.1340c.x
36. Rubenstein JL, Kim J, Ozawa T, Zhang M, Westphal M, Deen DF, et al. Anti-VEGF antibody treatment of glioblastoma prolongs survival but results in increased vascular cooption. Neoplasia (2000) 2(4):306–14. doi: 10.1038/sj.neo.7900102
37. Kunkel P, Ulbricht U, Bohlen P, Brockmann MA, Fillbrandt R, Stavrou D, et al. Inhibition of glioma angiogenesis and growth in vivo by systemic treatment with a monoclonal antibody against vascular endothelial growth factor receptor-2. Cancer Res (2001) 61(18):6624–8. doi: 10.1158/1078-0432.CCR-04-0823
38. Kusters B, Leenders WP, Wesseling P, Smits D, Verrijp K, Ruiter DJ, et al. Vascular endothelial growth factor-A(165) induces progression of melanoma brain metastases without induction of sprouting angiogenesis. Cancer Res (2002) 62(2):341–5. doi: 10.1186/s40880-016-0162-7
39. Leenders WP, Kusters B, Verrijp K, Maass C, Wesseling P, Heerschap A, et al. Antiangiogenic therapy of cerebral melanoma metastases results in sustained tumor progression via vessel co-option. Clin Cancer Res (2004) 10(18 Pt 1):6222–30. doi: 10.1242/jcs.112748
40. Kuczynski EA, Kerbel RS. Implications of vessel co-option in sorafenib-resistant hepatocellular carcinoma. Chin J cancer (2016) 35(1):97–. doi: 10.1172/JCI24612
41. Stoletov K, Strnadel J, Zardouzian E, Momiyama M, Park FD, Kelber JA, et al. Role of connexins in metastatic breast cancer and melanoma brain colonization. J Cell Sci (2013) 126(Pt 4):904–13.
42. Mancuso MR, Davis R, Norberg SM, O'Brien S, Sennino B, Nakahara T, et al. Rapid vascular regrowth in tumors after reversal of VEGF inhibition. J Clin Invest (2006) 116(10):2610–21. doi: 10.1007/s10456-014-9420-y
43. Bergers G, Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer (2008) 8(8):592–603. doi: 10.1038/nrc2442
44. Vasudev NS, Reynolds AR. Anti-angiogenic therapy for cancer: current progress, unresolved questions and future directions. Angiogenesis (2014) 17(3):471–94. doi: 10.1016/j.jhep.2006.11.021
45. Pezzella F. Mechanisms of resistance to anti-angiogenic treatments. Cancer Drug Resist (2019) 2(3):595–607. doi: 10.1016/j.ccr.2009.01.027
46. Semela D, Piguet A-C, Kolev M, Schmitter K, Hlushchuk R, Djonov V, et al. Vascular remodeling and antitumoral effects of mTOR inhibition in a rat model of hepatocellular carcinoma. J Hepatol (2007) 46(5):840–8. doi: 10.1016/j.ctrv.2011.02.002
47. Pàez-Ribes M, Allen E, Hudock J, Takeda T, Okuyama H, Viñals F, et al. Antiangiogenic therapy elicits Malignant progression of tumors to increased local invasion and distant metastasis. Cancer Cell (2009) 15(3):220–31. doi: 10.1371/journal.pone.0077117
48. Ribatti D. Novel angiogenesis inhibitors: Addressing the issue of redundancy in the angiogenic signaling pathway. Cancer Treat Rev (2011) 37(5):344–52. doi: 10.1073/pnas.1525360113
49. Lieu CH, Tran H, Jiang Z-Q, Mao M, Overman MJ, Lin E, et al. The association of alternate VEGF ligands with resistance to anti-VEGF therapy in metastatic colorectal cancer. PloS One (2013) 8(10):e77117–e. doi: 10.1073/pnas.1525349113
50. Kloepper J, Riedemann L, Amoozgar Z, Seano G, Susek K, Yu V, et al. Ang-2/VEGF bispecific antibody reprograms macrophages and resident microglia to anti-tumor phenotype and prolongs glioblastoma survival. Proc Natl Acad Sci USA (2016) 113(16):4476–81. doi: 10.1038/nm.4197
51. Peterson TE, Kirkpatrick ND, Huang Y, Farrar CT, Marijt KA, Kloepper J, et al. Dual inhibition of Ang-2 and VEGF receptors normalizes tumor vasculature and prolongs survival in glioblastoma by altering macrophages. Proc Natl Acad Sci USA (2016) 113(16):4470–5. doi: 10.1002/cjp2.100
52. Frentzas S, Simoneau E, Bridgeman VL, Vermeulen PB, Foo S, Kostaras E, et al. Vessel co-option mediates resistance to anti-angiogenic therapy in liver metastases. Nat Med (2016) 22(11):1294–302. doi: 10.1186/1471-2407-9-444
53. Lazaris A, Amri A, Petrillo SK, Zoroquiain P, Ibrahim N, Salman A, et al. Vascularization of colorectal carcinoma liver metastasis: insight into stratification of patients for anti-angiogenic therapies. J Pathol Clin Res (2018) 4(3):184–92. doi: 10.1093/jnci/djw030
54. Verhoeff JJC, van Tellingen O, Claes A, Stalpers LJA, van Linde ME, Richel DJ, et al. Concerns about anti-angiogenic treatment in patients with glioblastoma multiforme. BMC cancer (2009) 9:444–. doi: 10.1158/0008-5472.CAN-10-2602
55. Kuczynski EA, Yin M, Bar-Zion A, Lee CR, Butz H, Man S, et al. Co-option of liver vessels and not sprouting angiogenesis drives acquired sorafenib resistance in hepatocellular carcinoma. J Natl Cancer Inst (2016) 108(8). doi: 10.1093/neuonc/nop027
56. di Tomaso E, Snuderl M, Kamoun WS, Duda DG, Auluck PK, Fazlollahi L, et al. Glioblastoma recurrence after cediranib therapy in patients: lack of "rebound" revascularization as mode of escape. Cancer Res (2011) 71(1):19–28.
57. de Groot JF, Fuller G, Kumar AJ, Piao Y, Eterovic K, Ji Y, et al. Tumor invasion after treatment of glioblastoma with bevacizumab: radiographic and pathologic correlation in humans and mice. Neuro-oncology (2010) 12(3):233–42. doi: 10.1084/jem.20091846
58. Lim S, Hosaka K, Nakamura M, Cao Y. Co-option of pre-existing vascular beds in adipose tissue controls tumor growth rates and angiogenesis. Oncotarget (2016) 7(25):38282–91. doi: 10.18632/oncotarget.9436
59. Helfrich I, Scheffrahn I, Bartling S, Weis J, von Felbert V, Middleton M, et al. Resistance to antiangiogenic therapy is directed by vascular phenotype, vessel stabilization, and maturation in Malignant melanoma. J Exp Med (2010) 207(3):491–503. doi: 10.1016/j.jtho.2019.12.112
60. Kats-Ugurlu G, van Herpen C, Mulder S, Prokop M, de Waal R, Mulders P, et al. Pulmonary lymphangitis carcinomatosis of clear cell renal cell carcinoma after angiogenesis inhibition. Ann Case Rep (2018). doi: 10.2174/1568009617666171002142659
61. Yagi Y, Aly RG, Tabata K, Barlas A, Rekhtman N, Eguchi T, et al. Three-dimensional histologic, immunohistochemical, and multiplex immunofluorescence analyses of dynamic vessel co-option of spread through air spaces in lung adenocarcinoma. J Thorac Oncol (2020) 15(4):589–600. doi: 10.1158/0008-5472.CAN-10-0489
62. Massaro F, Molica M, Breccia M. Ponatinib: A review of efficacy and safety. Curr Cancer Drug Targets (2018) 18(9):847–56. doi: 10.1016/j.ccr.2012.05.037
63. Shojaei F, Lee JH, Simmons BH, Wong A, Esparza CO, Plumlee PA, et al. HGF/c-met acts as an alternative angiogenic pathway in sunitinib-resistant tumors. Cancer Res (2010) 70(24):10090–100.
64. Lu KV, Chang JP, Parachoniak CA, Pandika MM, Aghi MK, Meyronet D, et al. VEGF inhibits tumor cell invasion and mesenchymal transition through a MET/VEGFR2 complex. Cancer Cell (2012) 22(1):21–35. doi: 10.1158/1535-7163.MCT-13-0753
65. André F, Bachelot T, Campone M, Dalenc F, Perez-Garcia JM, Hurvitz SA, et al. Targeting FGFR with dovitinib (TKI258): preclinical and clinical data in breast cancer. Clin Cancer Res (2013) 19(13):3693–702. doi: 10.1158/1078-0432.CCR-13-0190
66. Chiron M, Bagley RG, Pollard J, Mankoo PK, Henry C, Vincent L, et al. Differential antitumor activity of aflibercept and bevacizumab in patient-derived xenograft models of colorectal cancer. Mol Cancer Ther (2014) 13(6):1636–44. doi: 10.1016/j.ctarc.2016.12.002
67. Vogel A, Sterneck M, Vondran F, Waidmann O, Klein I, Lindig U, et al. [The use of immuno-oncologic therapy in hepatocellular carcinoma in the context of liver transplantation. An interdisciplinary benefit/risk assessment]. Z Gastroenterol (2022) 60(2):184–91. doi: 10.1016/j.ejca.2019.07.024
68. Semrad TJ, Kim EJ, Tanaka MS, Sands J, Roberts C, Burich RA, et al. Phase II study of dovitinib in patients progressing on anti-vascular endothelial growth factor therapy. Cancer Treat Res Commun (2017) 10:21–6. doi: 10.1158/1078-0432.CCR-04-2270
69. Jones RL, Ratain MJ, O'Dwyer PJ, Siu LL, Jassem J, Medioni J, et al. Phase II randomised discontinuation trial of brivanib in patients with advanced solid tumours. Eur J Cancer (Oxford Engl 1990) (2019) 120:132–9. doi: 10.1158/1078-0432.CCR-17-1588
70. Lamszus K, Brockmann MA, Eckerich C, Bohlen P, May C, Mangold U, et al. Inhibition of glioblastoma angiogenesis and invasion by combined treatments directed against vascular endothelial growth factor receptor-2, epidermal growth factor receptor, and vascular endothelial-cadherin. Clin Cancer Res (2005) 11(13):4934–40. doi: 10.1038/nrm2639
71. Hidalgo M, Martinez-Garcia M, Le Tourneau C, Massard C, Garralda E, Boni V, et al. First-in-human phase I study of single-agent vanucizumab, A first-in-class bispecific anti-angiopoietin-2/anti-VEGF-A antibody, in adult patients with advanced solid tumors. Clin Cancer Res (2018) 24(7):1536–45. doi: 10.1158/0008-5472.CAN-08-3030
72. Augustin HG, Young Koh G, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin–Tie system. Nat Rev Mol Cell Biol (2009) 10(3):165–77. doi: 10.1016/S0002-9440(10)63998-3
73. Nasarre P, Thomas M, Kruse K, Helfrich I, Wolter V, Deppermann C, et al. Host-derived angiopoietin-2 affects early stages of tumor development and vessel maturation but is dispensable for later stages of tumor growth. Cancer Res (2009) 69(4):1324–33. doi: 10.1002/path.5449
74. Yu Q, Stamenkovic I. Angiopoietin-2 is implicated in the regulation of tumor angiogenesis. Am J Pathol (2001) 158(2):563–70. doi: 10.1007/s00401-014-1352-5
75. Palmieri V, Lazaris A, Mayer TZ, Petrillo SK, Alamri H, Rada M, et al. Neutrophils expressing lysyl oxidase-like 4 protein are present in colorectal cancer liver metastases resistant to anti-angiogenic therapy. J Pathol (2020) 251(2):213–23. doi: 10.3389/fonc.2023.1227540
76. Fack F, Espedal H, Keunen O, Golebiewska A, Obad N, Harter PN, et al. Bevacizumab treatment induces metabolic adaptation toward anaerobic metabolism in glioblastomas. Acta neuropathologica (2015) 129(1):115–31. doi: 10.3389/fonc.2022.965277
77. Pezzella F, Qian CN. Editorial: Vascular co-option and beyond for cancer biology. Front Oncol (2023) 13:1227540. doi: 10.1126/scitranslmed.aak9679
78. Cuypers A, Truong AK, Becker LM, Saavedra-Garcia P, Carmeliet P. Tumor vessel co-option: The past & the future. Front Oncol (2022) 12:965277. doi: 10.1038/s12276-020-00500-y
79. Allen E, Jabouille A, Rivera LB, Lodewijckx I, Missiaen R, Steri V, et al. Combined antiangiogenic and anti-PD-L1 therapy stimulates tumor immunity through HEV formation. Sci Transl Med (2017) 9(385):eaak9679. doi: 10.3389/fonc.2022.874554
Keywords: angiogenesis, anti-angiogenesis, resistance, tumor growth, vascular co-option
Citation: Ribatti D, Annese T and Tamma R (2023) Vascular co-option in resistance to anti-angiogenic therapy. Front. Oncol. 13:1323350. doi: 10.3389/fonc.2023.1323350
Received: 17 October 2023; Accepted: 23 November 2023;
Published: 11 December 2023.
Edited by:
Giorgio Seano, Institut Curie, FranceReviewed by:
Anca Maria Cimpean, Victor Babes University of Medicine and Pharmacy, RomaniaEva Andreuzzi, Institute for Maternal and Child Health Burlo Garofolo (IRCCS), Italy
Copyright © 2023 Ribatti, Annese and Tamma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Domenico Ribatti, ZG9tZW5pY28ucmliYXR0aUB1bmliYS5pdA==