 Zhe Chen
Zhe Chen Suying Zheng
Suying Zheng Weiying Feng
Weiying Feng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 06 December 2023
Sec. Hematologic Malignancies
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1308869
This article is part of the Research TopicReviews in Hematologic Malignancies: 2023View all 15 articles
Cell death is a complex process required to maintain homeostasis and occurs when cells are damage or reach end of life. As research progresses, it is apparent that necrosis and apoptosis do not fully explain the whole phenomenon of cell death. Therefore, new death modalities such as autophagic cell death, and ferroptosis have been proposed. In recent years, ferroptosis, a new type of non-apoptotic cell death characterized by iron-dependent lipid peroxidation and reactive oxygen species (ROS) accumulation, has been receiving increasing attention. Ferroptosis can be involved in the pathological processes of many disorders, such as ischemia-reperfusion injury, nervous system diseases, and blood diseases. However, the specific mechanisms by which ferroptosis participates in the occurrence and development of leukemia still need to be more fully and deeply studied. In this review, we present the research progress on the mechanism of ferroptosis and its role in leukemia, to provide new theoretical basis and strategies for the diagnosis and treatment of clinical hematological diseases.
The term “ferroptosis” was coined in 2012, when screens for small-molecule compounds capable of inhibiting the growth of RAS-mutant cancer cells were performed. In the 1950s, Harry Eagle et al. found that cysteine-deficient cells had a different pattern of cell death than those caused by other amino acid deficiencies. In the 1970s, a cysteine-dependent liver cell death involving glutathione (GSH) depletion was reported. At the same time, Shiro et al. found that alpha-tocopherol, an inhibitor of lipid peroxidation, saved cell death from GSH and cysteine deficiency. Ursini et al. isolated an enzyme named glutathione peroxidase 4 (GPX4) in 1982, which can inhibit iron-catalyzed lipid peroxidation. GPX4 protects cell death related to lipid peroxidation and oxidative stress. Dolma et al. discovered in 2003 that a small molecule compound named erastin could target the inhibition of RAS expressing tumor cells. Erastin induced death cell showed no apoptotic features and could not be inhibited by apoptosis inhibitors, suggesting new non-apoptotic cell death form. In 2012, Dixon et al. coined the term “ferroptosis” as erastin induced cell death. Ferroptosis refers to an iron-dependent form of regulatory cell death caused by lipid peroxide overload on the cell membrane. This is a new kind of cell death, which is different from the traditional forms of autophagy, apoptosis, necrosis, and other cell death. Morphologically, mitochondrial volume decreases, density increases, mitochondrial crest disappears, and lipid reactive oxygen species (ROS) increases in the cytoplasm (1). The fatal accumulation of lipid peroxides is a fundamental feature of ferroptosis and involves the confrontation between ferroptosis production and ferroptosis defense systems in cells. Ferroptosis occurs when its promotion of cellular activity significantly exceeds the antioxidant buffer provided by the ferroptosis defense system (2–4).
Ferroptosis is affected by a range of different genes including multiple cancer-related signaling pathways which have been shown to participate in ferroptosis. For example, p53 and BRCA1-related protein 1 (BAP1) induce ferroptosis in tumor cells through multiple signaling pathways, which act as a natural barrier to cancer development (5, 6). Oncogene-mediated or oncogene-signal-mediated ferroptosis avoidance contributes to tumor occurrence, progression, metastasis, and treatment resistance regulation (7, 8). Conversely, the unique metabolism of cancer cells, their high load of ROS, and their specific mutations make some of these cells inherently susceptible to ferroptosis, thus exposing therapeutic targets for certain cancer types (9–11). With the continuous development of research, ferroptosis has been confirmed to be closely related to the occurrence of tumors, respiratory system, cardiovascular system, nervous system, ischemia reperfusion injury, and other diseases. Recent studies have shown that ferroptosis also plays an important role in the development and progression of hematological diseases, especially leukemia. The present study mainly describes the role of ferroptosis in leukemia, research progress and provides new targets and new ideas for the diagnosis and treatment of leukemia.
Ferroptosis is caused by the accumulation of lipid peroxidation, leading to the destruction of membrane structures. The prerequisite for ferroptosis is polyunsaturated fatty acids -containing phospholipids (PUFA-PLs) synthesis with peroxidation. Sensitivity to ferroptosis is regulated by several factors, including GSH and REDOX regulatory systems, such as System Xc-, GPX4 regulation, CoQ10-NAD (P) pathway, glutamine metabolic pathway, and NRF2 regulation (Figure 1). In this section, there are mainly describes Ferroptosis prerequisites, Ferroptosis defense mechanisms, and Upregulation of ferroptosis defenses.
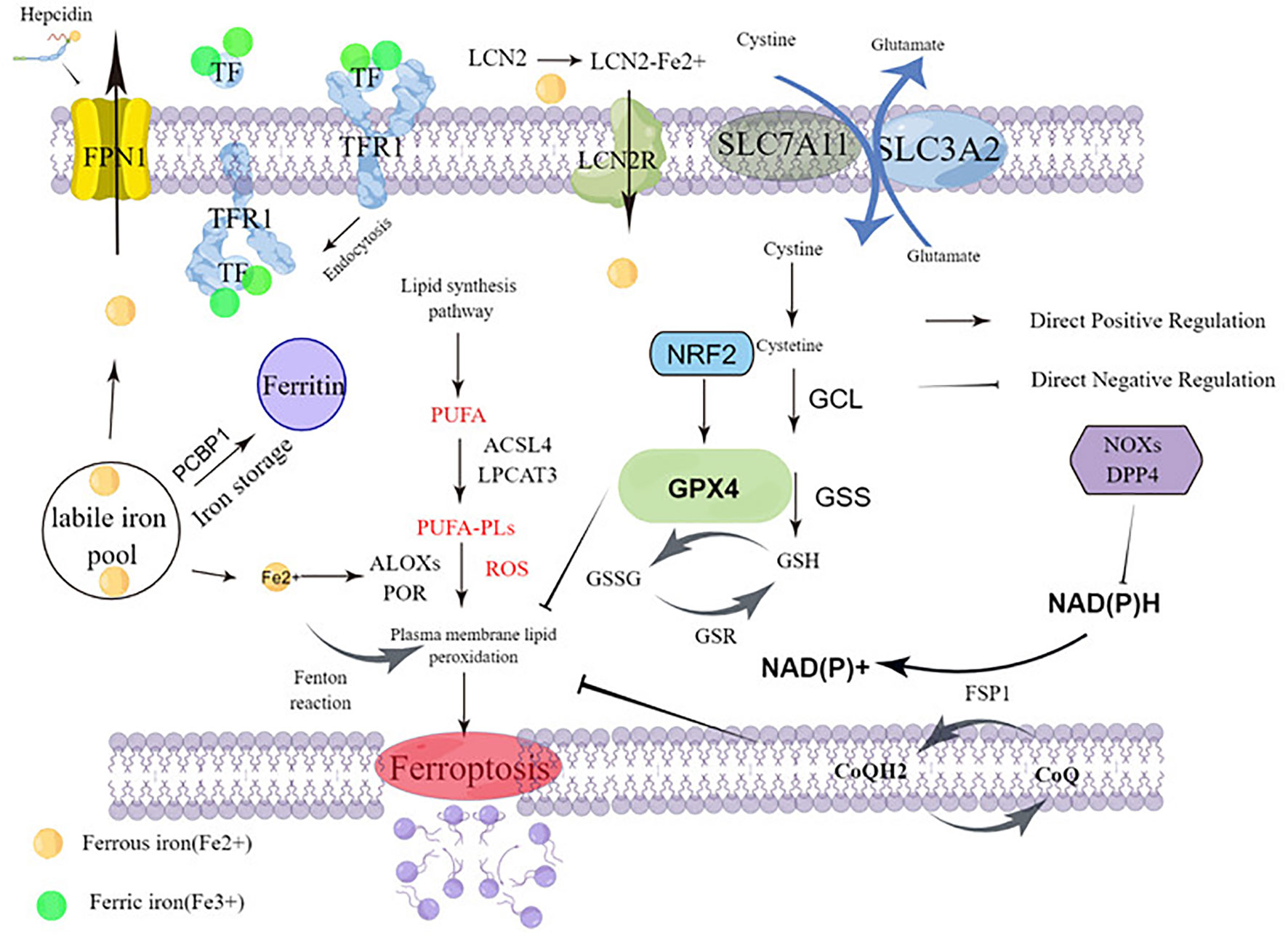
Figure 1 Molecular mechanisms of ferroptosis.
The crux of ferroptosis execution is PUFA-PLs synthesis with peroxidation. As outlined in this section, PUFA-PL synthesis and peroxidation, Iron metabolism, and Mitochondrial metabolism constitute the main prerequisites driving ferroptosis.
PUFA-PL synthesis and peroxidation: The key to triggering ferroptosis is the catalytic oxidation of phospholipids containing PUFA into polyunsaturated fatty acids, which leads to the fatal accumulation of lipid peroxides on the cell membrane and subsequent membrane rupture, resulting in ferroptosis. It is the main prerequisite of ferroptosis. Acyl-coenzyme A (CoA) synthetase long chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are critical mediators of PUFA-PL synthesis (12, 13). ACSL4 and LPCAT3 play an important role in the biosynthesis and remodeling of phosphatidylethanolamine (PE), which can activate PUFAs and affect transmembrane properties (14). ACSL4 catalyzes the linking of free PUFAs, which include arachidonic and adrenal acid to CoA forming PUFA-CoAs such as arachidonic acid-CoA or adrenal acid -CoA. These products are then subsequently re-esterified by LPCAT3 and incorporated into PLs to form PUFA-PLs which include arachidonic acid-phosphatidylethanolamine or adrenic acid-phosphatidylethanolamine (Figure 1).
Acetyl-coa carboxylase (ACC) catalyzes the carboxylation of acetyl-CoA to produce malonyl CoA, which is required for synthesis of some PUFAs (15, 16). Human cytochrome P450 redox reductase (POR) mediated or arachidonic lipoxygenase (ALOXs) mediated enzymatic reactions have also been shown to promote lipid peroxidation (17, 18) (Figure 1). POR’s ability to promote lipid peroxidation appears to be indirect, through the production of H2O2 (18). The ALOX gene plays an important role in driving ferroptosis. The mammalian ALOX family, consisting of six members (ALOXE3, ALOX5, ALOX12, ALOX12B, ALOX15, and ALOX15B), which play a context-dependent role in driving ferroptosis. For example, spermidine/spermidine n1-acetyltransferase 1 (SAT1), a target gene for tumor protein p53 (TP53), mediates the expression of ALOX15 (but not ALOX5 and ALOX12) and is involved in TP53-mediated ferroptosis (3). Interestingly, other studies have cast doubt on the role of ALOX genes in ferroptosis (19). ALOX12 does not depend on GPX4 and ALOX15 binds to phosphatidylethanolamine binding protein 1 (PEBP1), mediating RSL3-induced ferroptosis in bronchial epithelial cells, renal epithelial cells, and neuronal cells (20).
Iron metabolism: Iron is a key nutrient involved in ATP production via the mitochondrial chain complex, DNA synthesis in the process of ribonucleic acid reductase, oxygen transport, antioxidant defense (peroxidase and catalase), oxygen sensitive factors such as hypoxia-inducer factor-HiIF - and proline hydroxylase, and many other enzymes. Nutrient iron exists mainly as iron ions, which can be reduced by iron reductase. Systemic iron homeostasis is maintained by a balance of iron uptake, recycling, and loss. Iron mainly comes from food intake and the elimination of aging red blood cells, existing as Fe2+ and Fe3+. Ferrous ions are internalized into intestinal cells by active transport mechanisms in the gastrointestinal tract. Iron can also be internalized into the blood through the basolateral membrane through ferroportin 1 (FPN1; the only known iron exporter), iron through the binding of ferriferous carriers to lipidin-2 (LCN2), and subsequent endocytosis returned into the cell (21, 22). However, Fe3+ combines with transferrin (TF) on the cell membrane to form TF-Fe3+, which is finally combined with transferrin receptor 1 (TFR1) and is swallowed in vivo. Excess iron is stored in the liver primarily through ferritin (FTH and FTL). High iron levels cause the liver system to secrete hepcidin, the most relevant regulator of iron metabolism in the system. Hepcidin is a protein of iron transport from cells which binds to ferritin transporters on iron-storage cells such as intestinal epithelial cells and macrophages. This leads to internalization and degradation of the hepcidin-transporter complex, which effectively shuts down nutrient iron uptake and iron release from internal iron stores. Expression of hepcidin is controlled by a regulatory feedback mechanism of active erythropoiesis: erythropoiet-derived erythroferone (ERFE), growth differentiation factor 11 (GDF11), growth differentiation factor 15 (GDF15), and twisted gastrin protein homology 1 (TWSG1) have been shown to affect liver hepcidin secretion. Interestingly, leukemia cells require more iron than normal cells. In particular, cancer patients require a large number of red blood cell transfusions due to normal dyserythrogenesis and anemia caused by chemotherapy, and excess iron is common in leukemia patients.
Excess iron and reactive oxygen species (ROS) catalyze production and promote malignant transformation of hematopoietic stem cells through niacinamide adenine dinucleotide phosphate oxidase (NOX) and subsequent glutathione (GSH) consumption (23).
Iron’s ability to gain and lose electrons between its oxidized Fe3+ and Fe2+ forms allow it to participate in radical generation reactions. Among these processes is the Fenton reaction, where ferrous iron contributes an electron to hydrogen peroxide to produce hydroxyl radicals that induce ROS production. Abnormal iron accumulation and subsequent excess ROS levels produce oxidative stress, induce DNA, protein, or lipid damage, and even lead to cell death. It is important to note that these oxidation actions of iron can promote the development of tumors and are thought to be necessary for the development of cancer (24). Nuclear receptor coactivator 4 (NCOA4) is the target of the ferritin transporter, which mediates ferritinophagy, a selective autophagy that degrades ferritin by lysosomes. Selective autophagy degrades ferritin. Ferritinophagy increases free iron in cells. Iron pools can be coated by lysosomes via NCOA4, and then degrade and release a large amount of Fe2+, which increases the sensitivity of cells to ferroptosis (25). Inhibition of ferritin macrophages mediated by NCOA4 increases iron storage and limits iron cell apoptosis in ferroptosis (25, 26)(Figure 1).
Mitochondrial metabolism: Overexpression of ferritin mitochondria (FTMT), an iron-storage protein in mitochondria whose primary function is to provide energy for cells through oxidative phosphorylation, is a major site of iron metabolism and ROS production which inhibits erastin-induced ferroptosis in neuroblastoma cells (27). This suggests that FTMT has a widespread anti-iron declining effect. Iron chaperone PCBP1 delivers Fe2+ to ferritin, thereby limiting ferroptosis in hepatocytes (28). The role of mitochondria in the biosynthetic pathway of cell metabolism also contributes to ferroptosis. Ferroptosis requires tricarboxylic acid (TCA) cycling (29) and various non-fusion reactions in mitochondria. These reactions may drive ferroptosis by promoting ROS, ATP, and/or PUFA-PL production (30, 31).
The imbalance between injury and defense signals eventually leads to cell death. Ferroptosis defence mechanisms involve cellular antioxidant systems that directly neutralize lipid peroxides. As discussed below, there are mainly introduction the following two systems.
The GPX4–GSH system: GPX4 belongs to the GPX protein family (32, 33) and is the only GPX member capable of converting PL hydroperoxides into PL alcohols (34, 35). GPX4 is a key regulator of ferroptosis, and inhibition of its activity leads to the accumulation of lipid peroxides in cells, which signal ferroptosis in cells. Down-regulation of GPX4 increased susceptibility to ferroptosis, while up-regulation inhibited ferroptosis (36). GPX4 consists of three subtypes with different subcellular localization, namely cytoplasmic GPX4, mitochondrial GPX4, and nuclear GPX4. These isomers are encoded by the same GPX4 gene and have different transcription start sites, resulting in the n-terminal GPX4 protein of the mitochondrial or nuclear localization sequence. Only cytoplasmic GPX4 has a protective effect against ferroptosis (37). Cytoplasmic GPX4 re-expression significantly inhibited GPX4-deletion induced cell death in mouse embryonic fibroblasts. The expression or activity of GPX4 is controlled by selenium and glutathione (38, 39). Reducing glutathione (GSH), for GPX4 is a thiol-containing tripeptide derived from glycine, glutamic acid, and cysteine, of which cysteine is the rate-limiting precursor. GSH is the most abundant reducing agent in mammalian cells and is a cofactor of many enzymes. For glutathione synthesis, it is mainly formed by the redox of cysteine through the xc-/cystine/glutamate transporter (Figure 1).
Cystine glutamate transporter (system Xc-) is an amino acid reverse transporter widely distributed in the phospholipid bilayer and is an important part of the cellular antioxidant system. System Xc- is composed of solute carrier family 7 member 11 (SLC7A11) solute carrier family 3 member 2 (SLC3A2), an amino acid reverse transporter that can transfer cystine into cells and glutamate 1:1. Most cancer cells acquire intracellular cysteine-mediated cystine uptake (the oxydimer form of cysteine) primarily through the Xc- system, followed by reduction of cystine to cysteine in the cytoplasm (40, 41). Through the exchange of system Xc- (40), cysteine and glutamic acid are transported inside and outside the cell, and then participate in the synthesis of GSH. Inhibition of cysteine absorption can inhibit the activity of system Xc-, which can affect the synthesis of GSH and ultimately inhibit the activity of GPX, leading to the decline of cellular antioxidant capacity, lipid ROS accumulation, and inducing ferroptosis. Overexpression of apoptosis-inducing factor associated 2 (AIFM2) may eliminate GPX4 to inhibit ferroptosis (42) (Figure 1).
Seven members of solute vector family 11 (SLC7A11; Also called xCT) (43) is a transporter subunit in the system Xc-. The expression or activity of SLC7A11 is regulated by many factors, such as TP53 (6), NRF2 (44), BRCA1-related protein 1 (BAP1) (5), and BECN1 (45). Inhibition of SLC7A11 by small molecule compounds (such as erastin) can cause glutathione depletion to trigger ferroptosis (46). After GPX4 inactivation (10), some cancer cell lines remain resistant to ferroptosis, suggesting the presence of additional ferroptosis defense mechanisms (Figure 1).
CoQH2 system: Some recent studies suggest that ferroptosis defense systems can be divided into two main parts, GPX4 system and CoQH2 system. CoQH2 is an endogenous ferroptosis inhibitor, which has antioxidant effect in cell membrane and can reduce the oxidative damage of cell membrane. In addition to GPX4 system, DHODH/FSP1/CoQ pathway is another key inhibitory mechanism for lipid peroxidation and ferroptosis. DHODH is an enzyme involved in pyrimidine synthesis, which can reduce ubiquinone (CoQ) to ubiquinol (CoQH2) in the mitochondrial inner membrane. When GPX4 is dramatically inactivated, the flux through DHODH (3) increases significantly, leading to enhanced CoQH2 production, neutralizing lipid peroxidation, and defense against ferroptosis in mitochondria. The inactivation of mitochondrial GPX4 and DHODH releases powerful mitochondrial lipid peroxidation and triggers intense ferroptosis. Cytoplasmic GPX4 was also found to be significantly localized in the mitochondrial membrane gap (37) (Figure 1).
As a major suppressor of ferroptosis, FSP1 was originally described as a p53 response gene and therefore was originally called p53 response gene 3 (PRG3). FSP1, also known as AIFM2, is localized in the plasma membrane (as well as other subcellular compartments), and its plasma membrane localization appears to be necessary and sufficient to function FSP1’s role in inhibiting ferroptosis (47, 48). Doll et al. and Bersuker et al. found that FSP1 inhibits lipid peroxidation and ferroptosis by reducing CoQ (or its partially oxidized product hemihydroquinone) to CoQ2. This may directly reduce lipid radicals to terminate lipid autoxidation, or indirectly via regenerating oxidized α-tocopheryl radical (Vitamin E), a powerful natural antioxidant (47, 48). FSP1 acts as a NADPH-dependent CoQ redox enzyme, and can catalyze CoQ10 regeneration depending on NADPH, thereby improving the ability of free radical capture to protect cells, and also has a protective effect against ferroptosis caused by GPX4 deletion. This protective effect of CoQ reveals why some cells and tissues, such as highly metabolically active liver cells, contain large amounts of extracellular CoQ, which is inconsistent with its typical role in the mitochondrial electron transport chain. CoQ is synthesized mainly in the mitochondria (49), but detected in non-mitochondrial membranes, including the plasma membrane (50). Unanswered questions remain about the potential role of other CoQH2 producing mitochondrial enzymes in the regulation of ferroptosis.
Despite the prevailing importance of GPX4 and CoQH2 for limiting ferroptosis, both the signal pathways and the tumor microenvironment influence the function of ferroptosis in tumorigenesis and tumor therapy.This section focuses on the role of these two pathways that inhibit ferroptosis.
Hippo–YAP signaling in ferroptosis: The Hippo -Yap pathway is involved in a variety of biological functions, including cell proliferation and organ size control (51). Wu et al. demonstrated the role of intercellular interactions and intracellular NF2-YAP signaling in dictating ferroptosis, which can promote the survival of GPX4 knockout cells (9). Because YAP targets include several regulators of ferroptosis, including transferrin receptors ACSL4, TFR1, and possibly other genes, susceptibility to ferroptosis depends on Hippo pathway activity, with increased susceptibility in response to Hippo inhibition and YAP activation (9). Yang et al. found that in renal cell carcinoma (RCC), Transcription Regulator 1 (TAZ) is abundantly expressed and regulates ferroptosis through Epithelial Membrane Protein 1 (EMP1)-NOX4 (52).
Nuclear factor E2 erythroid 2-like-2 (NRF2): The NRF2 transcription pathway can up-regulate the expression of antioxidant genes or cell protective genes in various oxidative stress processes. As a major regulator of antioxidant defense, transcription factor NRF2 (53, 54) controls the transcription of many genes involved in GPX4-GSH-mediated ferroptosis defense. Sun et al. report that NRF2 plays a central role in protecting hepatocellular carcinoma (HCC) cells against ferroptosis, and NRF2 signaling is up-regulated in many human cancer types (55) (Figure 1).
Ferroptosis in the tumor microenvironment: Recent studies have also shown that the tumor microenvironment (TME), which is a multicellular environment, includes the extracellular matrix, immune cells, blood vessels, tumor cells, and other cells. In particular, immune cells determine whether ferroptosis in tumor cells will occur. CD8+ cytotoxic T cells are the main agents of anti-tumor immunity in the TME (56), secreting interferon-γ (IFN-γ) and subsequently inhibiting cystine uptake by cancer cells via down-regulation of SLC7A11 expression, thereby increasing lipid peroxidation and ferroptosis in tumors. Ferroptotic cancer cells can release several immunostimulatory signals, such as high mobility group box 1 (HMGB1) (57), calreticulin (58), ATP (59), and phosphatidylethanolamine (60). These factors can promote dendritic cell maturation, increasing the efficiency of macrophages in the phagocytosis of ferroptotic cancer cells, and further enhance the infiltration of CD8+ T cells into tumors.
Immunotherapy, combined with induction of ferroptosis, is a promising therapeutic approach. Drijvers et al. (61) found that Acyl-CoA synthetase ACSL4 mediates GPX4 inhibitor-induced sensitivity changes and ferroptosis in activated CD8+ T cells. CD8+ T cells can inhibit tumor cells by inducing iron decay and pyrosis (62, 63).
Leukemia comprises a group of heterogeneous hematopoietic stem/progenitor cell malignancies characterized by abnormal proliferation of primitive cells in the bone marrow that interfere with normal blood cell production. Its occurrence involves multiple gene changes including the transferrin receptor 1 gene, the hemochromatosis (HFE) gene, and several genes related to iron metabolism.
At present, chemotherapy, immunotherapy, and hematopoietic stem cell transplantation (HSCT) are still the main treatments for leukemia. Despite advances in treatment, the results remain disheartening. Relapse or refractory disease and resistance to chemotherapy are the main reasons for treatment failure. Overcoming drug resistance is a major challenge in cancer treatment. Combination therapy prevents drug resistance by combining drugs with different targets, modes of action, and distribution of side effects in the body to reduce toxicity (64).
As a newly discovered programmed cell death mode, ferroptosis is regulated by multiple pathways such as lipid metabolism, mitochondrial metabolism and iron metabolism. Through this new death mode, it provides a new idea for improving the prognosis of leukemia patients. However, leukemia cells seem to be able to escape oxidative stress and reduce ferroptosis through some mechanisms.
Acute myeloid leukemia (AML) is a clonal hematopoietic disease caused by a variety of genetic and epigenetic impairments, characterized by impaired differentiation and uncontrolled proliferation, with varying prognoses (65). The incidence of AML increases with age, with a mortality rate of over 90% (66) at diagnosis after age 65. Ferroptosis provides a new idea for the treatment of AML, and a variety of drugs have been shown to induce ferroptosis.
Sorafenib has been approved as a tyrosine kinase inhibitor for the treatment of liver, kidney, and thyroid cancers for more than 15 years, and has recently been shown to be effective in AML patients with FLT3-ITD mutations (67). Concurrently, it also inhibits system Xc- and thus induces ferroptosis (46). Imatinib Mesylate (IMA) (68)down-regulates the expression of NRF2 and up-regulates the expression of p53 and TFR. These results provided compelling evidence that ferroptosis participates in IMA-induced cardiotoxicity. Ferroptosis could be regarded as a target to protect against cardiotoxicity in IMA-exposed patients.
APR-246 (69) is a novel drug for the treatment of TP53-mutant AML. Its main mechanism of action is to promote the binding of p53 mutants to DNA targets to reactivate the transcriptional activity of p53 and exert tumor inhibitory effects. APR-246 (70) increases oxidative stress by depleting GSH and inhibiting thioredoxin reductase, leading to the accumulation of ROS and further promoting tumor cell death. Birsen et al. (71) found that the observed early p53-independent cell death induced by APR-246 is ferroptosis.
NEAT1 (72) is bound to cytoplasmic disheveled 2 (DVL2) and tripartite motif containing 56 (TRIM56), which promotes the degradation of DVL2 and inhibits Wnt signaling, inhibiting the self-renewal of AML stem cells. Zhang et al. (73) found that ferroptosis inducers erastin and RSL3 increased NEAT1 expression by promoting the binding of p53 to the NEAT1 promoter. Induced NEAT1 promoted the expression of MIOX by competitively binding to miR-362-3p. MIOX increased ROS production and decreased the intracellular levels of NADPH and GSH, resulting in enhanced erastin- and RSL3-induced ferroptosis.
Aldo-keto reductase family 1 member C2 (AKR1C2), suppressor of cytokine signaling1 (SOCS1) (74), dipetidyl peptidase-4 (DPP4) (75), and human immunodeficiency virus type I enhancer-binding protein zinc finger 3(HIVEP3) (76) can be used as predictive adverse prognosis. Long non-coding RNAs (lncrnas) (77) associated with ferroptosis have also been shown to accurately predict the prognosis of AML and optimize treatment strategies for AML.
Acetaldehyde dehydrogenase 3a2 (Aldh3a2) (78) is l-gmp dependent and not seen in n-gmp. It protects AML cells from oxidative cell death, and Aldh3a2 inhibition improves leukemia outcomes in vivo without compromising normal hematopoiesis. Aldh3a2 inhibition combined with ferroptosis inducer or standard AML induction chemotherapy deserves further consideration as a cancer treatment.
High mobility base Box 1 (HMGB1) (79) is a transcription factor involved in the process of chromatin remodeling, DNA recombination, and repair. HMGB1 is found in the cytoplasm and, via translocation, is expressed on the cell surface membrane or diffuse in the extracellular space. This can be caused by various cellular stressors, causing HMGB1 to migrate from the nucleus to the cytoplasm in response to erastin in HL-60/NRASQ61L cell lines and acts as a positive regulator of ferroptosis, possibly enhancing resistance to anticancer therapy.
At present, a variety of drugs have been confirmed to promote or inhibit ferroptosis in AML cells, but there is still a lack of large-scale studies, and further research is still needed to support the development. There are still many challenges in the clinical application of ferroptosis in the treatment of leukemia.
Acute lymphoblastic leukemia (ALL) is a common malignant disease of the blood system, which manifests as abnormal clonal proliferation of naive or immature T and B lymphocytes. These cells will infiltrate bone marrow, blood, or other tissues and organs, causing abnormal hematopoietic function of bone marrow and immune dysfunction. Vincristine (VCR) is often used as a treatment for ALL (80). Studies have found that VCR promotes ferroptosis by enhancing the expression of lncRNA LINC00618 and inhibiting the transcription of SLC7A11, suggesting that ferroptosis is involved in the mechanism of action of VCR.
RSL3 is an inducer of ferroptosis that binds and inactivates GPX4, mediating ferroptosis regulated by GPX4 (81). Probst et al. treated ALL cell lines with RSL3 causing lipid peroxidation, ROS production, and cell death (82). Hydnocarpin D (HD) can trigger ferroptosis through the accumulation of lipid ROS and the reduction of GSH and GPX4, while the inhibition of autophagy prevents the ferroptosis (83). PAQR3 (progestin and adipoQ receptor family member 3) is involved in the occurrence of many tumors as a tumor suppressor and can inhibit the proliferation of human leukemia cells and induce cell apoptosis (84). Jin et al. found that PAQR3 inhibits cell proliferation and aggravates ferroptosis in ALL by regulating the stability of NRF2. Hong et al. (85, 86) demonstrated the critical role of ferroptosis in Philadelphia chromosome negative (Ph-neg) B-ALL patients, with sorafenib potentially improving survival in high-risk Ph-neg B-ALL patients.
Artesunate (ART), a semi-synthetic water-soluble derivative of Artemisia annua L., is a natural product extracted from artemisia annua L. Apoptosis induced by ART corresponded to the activation of caspase-8/9/3. The expression of Bcl-xL, Bcl-2, myeloid leukemia-1, survivin, X-linked apoptosis inhibitor protein, and apoptosis inhibitor 1/2 were decreased, with increased expression of Bak. ART increased the activation of intracellular ROS and DNA damage marker gamma-H2Ax. In the ATLL mouse model, intraperitoneal injection of ART reduced tumor burden (87).
Poricoic acid A (PAA) (88) strongly reduced the cell viability of T-ALL cell lines. Mitochondrial dysfunction was also elevated by PAA, along with enhanced cellular reactive oxygen species (ROS) production. PAA treatments provoked ferroptosis in T-ALL cells with reduced glutathione (GSH) levels and elevated malonaldehyde (MDA) contents. As a new mode of regulatory cell death, amplification of ferroptosis effect may be a new idea for drug development and disease treatment.
Chronic lymphocytic leukemia (CLL) is a disease with different genetic characteristics and treatment responses. CLL is characterized by the cloning and proliferation of mature CD5-positive B cells in the blood, bone marrow, lymph nodes, and spleen, resulting in immune system decline, organ dysfunction, and slow progressive systemic failure and depletion.
Ferroptosis is less well studied in the CLL field. SLC7A11 is the main functional subunit of system Xc- to transport cystine into cells to synthesize GSH. Inhibition of SLC7A11 expression can induce ferroptosis. The expression of SLC7A11 is low in CLL compared to the high expression level of SLC7A11 in other systemic solid tumors. This will lead to an increase in intracellular ROS, and as CLL cells are more prone to oxidative stress, they may be sensitive to ferroptosis inducers.
Ferroptosis is an autophagy-dependent form of cell death. BECN1 affects the occurrence and progression of autophagy, and its repeated allelic deletion and expression variation have been reported in tumors (89). Gong et al. (90) proposed a novel FPS model for prognostic prediction of CLL and established nine ferroptosis genes associated with CLL prognosis.
Chronic myelogenous leukemia (CML) is a hematopoietic malignancy caused by reciprocal translocation of Philadelphia chromosomes 9 and 22. Ferroptosis has been less studied in the field of CML. Cysteine metabolism plays a key role in cancer cell survival in the study of CML related fields. Cysteine deficiency has been reported to inhibit tumor growth and induce ferroptosis in pancreatic cancer cells. It has also been reported that cysteine depletion can induce ferroptosis in CML cells in vitro, and thioredoxin reductase 1 (TXNRD1) (91), which is related to cell redox metabolism, is a key factor regulating ferroptosis.
Ferroptosis, as a newly discovered form of programmed cell death, has a broad prospect in tumor therapy. We have systematically and comprehensively illustrated the relationship between ferroptosis and leukemia, and found that ferroptosis plays an important role in disease progression. PUFA-PL synthesis and peroxidation, intracellular ROS levels, and homeostasis of various metabolic pathways can affect cell sensitivity to ferroptosis, thus inducing ferroptosis in blood cells. Iron accumulation and lipid peroxidation may be considered as intermediate events, but they are not the final executors of ferroptosis. Leukemia cells seem to escape oxidative stress through certain mechanisms, such as the upregulation of ferroptosis defenses and ferroptosis defense mechanisms, which reduce the occurrence of ferroptosis. However, the study of ferroptosis in hematological diseases is still in the early stage, and its specific mechanism needs to be further studied. At present, most studies affect the activity of antioxidants such as GPX4 through exogenous ferroptosis inducers, causing the accumulation of ROS and thus promoting ferroptosis. There are few large studies on ferroptosis inducers in the treatment of leukemia. The pathogenesis of leukemia is complex, and often involves multiple pathways and targets. The previous multi-drug combination chemotherapy with cytotoxic drugs could easily cause serious adverse consequences such as bone marrow suppression and immune destruction. Novel target inhibitors related to ferroptosis, or their combination with existing cytotoxic agents, may further enhance the efficacy of existing single agents, delay drug resistance and improve prognosis. Although the treatment of leukemia with ferroptosis inducers is not mature at present, but it still has high research value.
In summary, we are currently in the middle of an important phase in the development of ferroptosis research. The occurrence and development of ferroptosis, its transcriptional regulation mechanism, and the development of effective regulatory targeted drugs are of utmost importance, providing a new direction for clinical diagnosis and treatment of blood diseases. New treatments based on ferroptosis will be developed and put into clinical use soon, guided by specific biomarkers and a precise assessment of a patient’s background.
ZC: Writing – review & editing. SZ: Data curation, Writing – original draft. JH: Data curation, Writing – original draft. LF: Writing – original draft. JF: Funding acquisition, Writing – review & editing. ZZ: Methodology, Writing – review & editing. PH: Writing – original draft. WF: Writing – review & editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. Sponsored by Zhejiang Provincial Natural Science Foundation of China (LBY23H080002), the Zhejiang Provincial Program for the Cultivation of High-level Innovative Health Talents.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell (2017) 171(2):273–85. doi: 10.1016/j.cell.2017.09.021
2. Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell (2018) 172(3):409–22.e21. doi: 10.1016/j.cell.2017.11.048
3. Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, et al. DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature (2021) 593(7860):586–90. doi: 10.1038/s41586-021-03539-7
4. Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol (2020) 16(12):1351–60. doi: 10.1038/s41589-020-0613-y
5. Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol (2018) 20(10):1181–92. doi: 10.1038/s41556-018-0178-0
6. Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature (2015) 520(7545):57–62. doi: 10.1038/nature14344
7. Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin-Sandoval MS, et al. Lymph protects metastasizing melanoma cells from ferroptosis. Nature (2020) 585(7823):113–8. doi: 10.1038/s41586-020-2623-z
8. Zhang R, Liu Y, You J, et al. Tanshinone IIA inhibits ischemia-reperfusion-induced inflammation, ferroptosis and apoptosis through activation of the PI3K/Akt/mTOR pathway. Hum Exp Toxicol (2023) 42:9603271231180864. doi: 10.1177/09603271231180864
9. Wu J, Minikes AM, Gao M, Bian H, Li Y, Stockwell BR, et al. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature (2019) 572(7769):402–6. doi: 10.1038/s41586-019-1426-6
10. Qi D, Peng M. Ferroptosis-mediated immune responses in cancer. Front Immunol (2023) 14(1188365). doi: 10.3389/fimmu.2023.1188365
11. Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature (2020) 585(7826):603–8. doi: 10.1038/s41586-020-2732-8
12. Kagan VE, Tyurina YY, Vlasova II, Kapralov AA, Amoscato AA, Anthonymuthu TS, et al. Redox epiphospholipidome in programmed cell death signaling: catalytic mechanisms and regulation. Front Endocrinol (2020) 11(628079). doi: 10.3389/fendo.2020.628079
13. Xue Q, Kang R, Klionsky DJ, Tang D, Liu J, Chen X. Copper metabolism in cell death and autophagy. Autophagy (2023) 19(8):2175–95. doi: 10.1080/15548627.2023.2200554
14. Bouchaoui H, Mahoney-Sanchez L, Garçon G, Berdeaux O, Alleman LY, Devos D, et al. ACSL4 and the lipoxygenases 15/15B are pivotal for ferroptosis induced by iron and PUFA dyshomeostasis in dopaminergic neurons. Free Radical Biol Med (2023) 195:145–157. doi: 10.1016/j.freeradbiomed.2022.12.086
15. Qi Y, Zhang X, Wu Z, Tian M, Chen F, Guan W, et al. Ferroptosis regulation by nutrient signalling. Nutr Res Rev (2022) 35(2):282–94. doi: 10.1017/S0954422421000226
16. Li C, Dong X, Du W, Shi X, Chen K, Zhang W, et al. LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal Transduct Target Ther (2020) 5(1):187. doi: 10.1038/s41392-020-00297-2
17. Cui S, Simmons G Jr., Vale G, Deng Y, Kim J, Kim H, et al. FAF1 blocks ferroptosis by inhibiting peroxidation of polyunsaturated fatty acids. Proc Natl Acad Sci U States America (2022) 119(17):e2107189119. doi: 10.1073/pnas.2107189119
18. Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J, et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol Cell (2021) 81(2):355–69.e10. doi: 10.1016/j.molcel.2020.11.024
19. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol (2014) 16(12):1180–91. doi: 10.1038/ncb3064
20. Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell (2017) 171(3):628–41.e26. doi: 10.1016/j.cell.2017.09.044
21. Chi Y, Remsik J, Kiseliovas V, Derderian C, Sener U, Alghader M, et al. Cancer cells deploy lipocalin-2 to collect limiting iron in leptomeningeal metastasis. Sci (New York NY) (2020) 369(6501):276–82. doi: 10.1126/science.aaz2193
22. Jończy A, Mazgaj R, Smuda E, Żelazowska B, Kopeć Z, Starzyński RR, et al. The role of copper in the regulation of ferroportin expression in macrophages. Cells (2021) 10(9):2259. doi: 10.3390/cells10092259
23. Hole PS, Zabkiewicz J, Munje C, Newton Z, Pearn L, White P, et al. Overproduction of NOX-derived ROS in AML promotes proliferation and is associated with defective oxidative stress signaling. Blood (2013) 122(19):3322–30. doi: 10.1182/blood-2013-04-491944
24. Battaglia AM, Chirillo R, Aversa I, Sacco A, Costanzo F, Biamonte F. Ferroptosis and cancer: mitochondria meet the "Iron Maiden" Cell death. Cells (2020) 9(6):1505. doi: 10.3390/cells9061505
25. Chappell G, Geer M, Gatza E, Braun T, Churay T, Brisson J, et al. Maintenance sorafenib in FLT3-ITD AML following allogeneic HCT favorably impacts relapse and overall survival. Bone Marrow Transplant (2019) 54(9):1518–20. doi: 10.1038/s41409-019-0493-5
26. Fang Y, Chen X, Tan Q, Zhou H, Xu J, Gu Q. Inhibiting ferroptosis through disrupting the NCOA4-FTH1 interaction: A new mechanism of action. ACS Cent Sci (2021) 7(6):980–9. doi: 10.1021/acscentsci.0c01592
27. Wang YQ, Chang SY, Wu Q, Gou YJ, Jia L, Cui YM, et al. The protective role of mitochondrial ferritin on erastin-induced ferroptosis. Front Aging Neurosci (2016) 8. doi: 10.3389/fnagi.2016.00308
28. Protchenko O, Baratz E, Jadhav S, Li F, Shakoury-Elizeh M, Gavrilova O, et al. Iron chaperone poly rC binding protein 1 protects mouse liver from lipid peroxidation and steatosis. Hepatol (Baltimore Md) (2021) 73(3):1176–93. doi: 10.1002/hep.31328
29. Friedman JR, Nunnari J. Mitochondrial form and function. Nature (2014) 505(7483):335–43. doi: 10.1038/nature12985
30. Zheng D, Liu J, Piao H, Zhu Z, Wei R, Liu K. ROS-triggered endothelial cell death mechanisms: Focus on pyroptosis, parthanatos, and ferroptosis. Front Immunol (2022) 13. doi: 10.3389/fimmu.2022.1039241
31. Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, et al. Role of mitochondria in ferroptosis. Mol Cell (2019) 73(2):354–63.e3. doi: 10.1016/j.molcel.2018.10.042
32. Wu F, Du Y, Yang J, Shao B, Mi Z, Yao Y, et al. Peroxidase-like active nanomedicine with dual glutathione depletion property to restore oxaliplatin chemosensitivity and promote programmed cell death. ACS nano (2022) 16(3):3647–63. doi: 10.1021/acsnano.1c06777
33. BRIGELIUS-Flohé R, Flohé L. Regulatory phenomena in the glutathione peroxidase superfamily. Antioxid Redox Signaling (2020) 33(7):498–516. doi: 10.1089/ars.2019.7905
34. Seibt TM, Proneth B, Conrad M. Role of GPX4 in ferroptosis and its pharmacological implication. Free Radical Biol Med (2019) 133:144–52. doi: 10.1016/j.freeradbiomed.2018.09.014
35. Ursini F, Maiorino M. Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radical Biol Med (2020) 152:175–185. doi: 10.1016/j.freeradbiomed.2020.02.027
36. Wei J, Xie Q, Liu X, Wan C, Wu W, Fang K, et al. Identification the prognostic value of glutathione peroxidases expression levels in acute myeloid leukemia. Ann Trans Med (2020) 8(11):678. doi: 10.21037/atm-20-3296
37. Gan B. Mitochondrial regulation of ferroptosis. J Cell Biol (2021) 220(9):e202105043. doi: 10.1083/jcb.202105043
38. Jankowski CSR, Rabinowitz JD. Selenium modulates cancer cell response to pharmacologic ascorbate. Cancer Res (2022) 82(19):3486–98. doi: 10.1158/0008-5472.CAN-22-0408
39. Xu C, Sun S, Johnson T, Qi R, Zhang S, Zhang J, et al. The glutathione peroxidase Gpx4 prevents lipid peroxidation and ferroptosis to sustain Treg cell activation and suppression of antitumor immunity. Cell Rep (2021) 35(11):109235. doi: 10.1016/j.celrep.2021.109235
40. Koppula P, Zhang Y, Zhuang L, Gan B. Amino acid transporter SLC7A11/xCT at the crossroads of regulating redox homeostasis and nutrient dependency of cancer. Cancer Commun (London England) (2018) 38(1):12. doi: 10.1186/s40880-018-0288-x
41. Liu X, Olszewski K, Zhang Y, Lim EW, Shi J, Zhang X, et al. Cystine transporter regulation of pentose phosphate pathway dependency and disulfide stress exposes a targetable metabolic vulnerability in cancer. Nat Cell Biol (2020) 22(4):476–86. doi: 10.1038/s41556-020-0496-x
42. Wei X, Yi X, Zhu XH, Jiang DS. Posttranslational modifications in ferroptosis. Oxid Med Cell Longevity (2020) 2020:8832043. doi: 10.1155/2020/8832043
43. Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell (2021) 12(8):599–620. doi: 10.1007/s13238-020-00789-5
44. Dong H, Qiang Z, Chai D, Peng J, Xia Y, Hu R, et al. Nrf2 inhibits ferroptosis and protects against acute lung injury due to intestinal ischemia reperfusion via regulating SLC7A11 and HO-1. Aging (2020) 12(13):12943–59. doi: 10.18632/aging.103378
45. Song X, Zhu S, Chen P, Hou W, Wen Q, Liu J, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system X(c)(-) activity. Curr Biol CB (2018) 28(15):2388–99.e5. doi: 10.1016/j.cub.2018.05.094
46. Wang L, Liu Y, Du T, Yang H, Lei L, Guo M, et al. ATF3 promotes erastin-induced ferroptosis by suppressing system Xc(). Cell Death Differ (2020) 27(2):662–75. doi: 10.1038/s41418-019-0380-z
47. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature (2019) 575(7784):688–92. doi: 10.1038/s41586-019-1705-2
48. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature (2019) 575(7784):693–8. doi: 10.1038/s41586-019-1707-0
49. Guerra RM, Pagliarini DJ. Coenzyme Q biochemistry and biosynthesis. Trends Biochem Sci (2023) 48(5):463–76. doi: 10.1016/j.tibs.2022.12.006
50. Santoro MM. The antioxidant role of non-mitochondrial CoQ10: mystery solved! Cell Metab (2020) 31(1):13–5. doi: 10.1016/j.cmet.2019.12.007
51. Wang M, Dai M, Wang D, Xiong W, Zeng Z, Guo C. The regulatory networks of the Hippo signaling pathway in cancer development. J Cancer (2021) 12(20):6216–30. doi: 10.7150/jca.62402
52. Yang WH, Ding CC, Sun T, Rupprecht G, Lin CC, Hsu D, et al. The hippo pathway effector TAZ regulates ferroptosis in renal cell carcinoma. Cell Rep (2019) 28(10):2501–8.e4. doi: 10.1016/j.celrep.2019.07.107
53. Wei R, Zhao Y, Wang J, Yang X, Li S, Wang Y, et al. Tagitinin C induces ferroptosis through PERK-Nrf2-HO-1 signaling pathway in colorectal cancer cells. Int J Biol Sci (2021) 17(11):2703–17. doi: 10.7150/ijbs.59404
54. Hsieh CH, Hsieh HC, Shih FS, Wang PW, Yang LX, Shieh DB, et al. An innovative NRF2 nano-modulator induces lung cancer ferroptosis and elicits an immunostimulatory tumor microenvironment. Theranostics (2021) 11(14):7072–91. doi: 10.7150/thno.57803
55. Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatol (Baltimore Md) (2016) 63(1):173–84. doi: 10.1002/hep.28251
56. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature (2019) 569(7755):270–4. doi: 10.1038/s41586-019-1170-y
57. Chen R, Zou J, Kang R, Tang D. The redox protein HMGB1 in cell death and cancer. Antioxid Redox Signaling (2023). doi: 10.1089/ars.2023.0007
58. Yu B, Choi B, Li W, Kim DH. Magnetic field boosted ferroptosis-like cell death and responsive MRI using hybrid vesicles for cancer immunotherapy. Nat Commun (2020) 11(1):3637. doi: 10.1038/s41467-020-17380-5
59. Efimova I, Catanzaro E, Van Der Meeren L, Turubanova VD, Hammad H, Mishchenko TA, et al. Vaccination with early ferroptotic cancer cells induces efficient antitumor immunity. J Immunother Cancer (2020) 8(2):e001369. doi: 10.1136/jitc-2020-001369
60. Luo X, Gong HB, Gao HY, Wu YP, Sun WY, Li ZQ, et al. Oxygenated phosphatidylethanolamine navigates phagocytosis of ferroptotic cells by interacting with TLR2. Cell Death Differ (2021) 28(6):1971–89. doi: 10.1038/s41418-020-00719-2
61. Drijvers JM, Gillis JE, Muijlwijk T, Nguyen TH, Gaudiano EF, Harris IS, et al. Pharmacologic screening identifies metabolic vulnerabilities of CD8(+) T cells. Cancer Immunol Res (2021) 9(2):184–99. doi: 10.1158/2326-6066.CIR-20-0384
62. Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y, et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell (2022) 40(4):365–78.e6. doi: 10.1016/j.ccell.2022.02.003
63. Wang Q, Wang Y, Ding J, Wang C, Zhou X, Gao W, et al. A bioorthogonal system reveals antitumour immune function of pyroptosis. Nature (2020) 579(7799):421–6. doi: 10.1038/s41586-020-2079-1
64. Gurnari C, Voso MT, Maciejewski JP, Visconte V. From bench to bedside and beyond: therapeutic scenario in acute myeloid leukemia. Cancers (2020) 12(2):357. doi: 10.3390/cancers12020357
65. Krali O, Palle J, Bäcklin CL, Abrahamsson J, Norén-Nyström U, Hasle H, et al. DNA methylation signatures predict cytogenetic subtype and outcome in pediatric acute myeloid leukemia (AML). Genes (Basel) (2021) 12(6):895. doi: 10.3390/genes12060895
66. Horikoshi N, Cong J, Kley N, Shenk T. Isolation of differentially expressed cDNAs from p53-dependent apoptotic cells: activation of the human homologue of the Drosophila peroxidasin gene. Biochem Biophys Res Commun (1999) 261(3):864–9. doi: 10.1006/bbrc.1999.1123
67. Pollard JA, Alonzo TA, Gerbing R, Brown P, Fox E, Choi J, et al. Sorafenib in combination with standard chemotherapy for children with high allelic ratio FLT3/ITD+ Acute myeloid leukemia: A report from the children's oncology group protocol AAML1031. J Clin Oncol (2022) 40(18):2023–35. doi: 10.1200/JCO.21.01612
68. Song C, Li D, Zhang J, Zhao X. Role of ferroptosis in promoting cardiotoxicity induced by Imatinib Mesylate via down-regulating Nrf2 pathways in vitro and in vivo. Toxicol Appl Pharmacol (2022) 435:115852. doi: 10.1016/j.taap.2021.115852
69. Cluzeau T, Sebert M, Rahmé R, Cuzzubbo S, Lehmann-Che J, Madelaine I, et al. Eprenetapopt plus azacitidine in TP53-mutated myelodysplastic syndromes and acute myeloid leukemia: A phase II study by the groupe francophone des myélodysplasies (GFM). J Clin Oncol (2021) 39(14):1575–83. doi: 10.1200/JCO.20.02342
70. Xin Q, Ji Q, Zhang Y, Ma W, Tian B, Liu Y, et al. Aberrant ROS served as an acquired vulnerability of cisplatin-resistant lung cancer. Oxid Med Cell Longevity (2022) 2022:1112987. doi: 10.1155/2022/1112987
71. Birsen R, Larrue C, Decroocq J, Johnson N, Guiraud N, Gotanegre M, et al. APR-246 induces early cell death by ferroptosis in acute myeloid leukemia. Haematologica (2022) 107(2):403–16. doi: 10.3324/haematol.2020.259531
72. Yan H, Wang Z, Sun Y, Hu L, Bu P. Cytoplasmic NEAT1 suppresses AML stem cell self-renewal and leukemogenesis through inactivation of Wnt signaling. Advanced Sci (Weinheim Baden-Wurttemberg Germany) (2021) 8(22):e2100914. doi: 10.1002/advs.202100914
73. Zhang Y, Luo M, Cui X, O'Connell D, Yang Y. Long noncoding RNA NEAT1 promotes ferroptosis by modulating the miR-362-3p/MIOX axis as a ceRNA. Cell Death Differ (2022) 29(9):1850–63. doi: 10.1038/s41418-022-00970-9
74. Zhou F, Chen B. Prognostic significance of ferroptosis-related genes and their methylation in AML. Hematol (Amsterdam Netherlands) (2021) 26(1):919–30. doi: 10.1080/16078454.2021.1996055
75. Wei J, Nai GY, Dai Y, Huang XJ, Xiong MY, Yao XY, et al. Dipetidyl peptidase-4 and transferrin receptor serve as prognostic biomarkers for acute myeloid leukemia. Ann Trans Med (2021) 9(17):1381. doi: 10.21037/atm-21-3368
76. Zhang X, Zhang X, Liu K, Li W, Wang J, Liu P, et al. HIVEP3 cooperates with ferroptosis gene signatures to confer adverse prognosis in acute myeloid leukemia. Cancer Med (2022) 11(24):5050–65. doi: 10.1002/cam4.4806
77. Zheng Z, Wu W, Lin Z, Liu S, Chen Q, Jiang X, et al. Identification of seven novel ferroptosis-related long non-coding RNA signatures as a diagnostic biomarker for acute myeloid leukemia. BMC Med Genomics (2021) 14(1):236. doi: 10.1186/s12920-021-01085-9
78. Yusuf RZ, Saez B, Sharda A, van Gastel N, Yu VWC, Baryawno N, et al. Aldehyde dehydrogenase 3a2 protects AML cells from oxidative death and the synthetic lethality of ferroptosis inducers. Blood (2020) 136(11):1303–16. doi: 10.1182/blood.2019001808
79. Ye F, Chai W, Xie M, Yang M, Yu Y, Cao L, et al. HMGB1 regulates erastin-induced ferroptosis via RAS-JNK/p38 signaling in HL-60/NRAS(Q61L) cells. Am J Cancer Res (2019) 9(4):730–9.
80. Wang Z, Chen X, Liu N, Shi Y, Liu Y, Ouyang L, et al. A nuclear long non-coding RNA LINC00618 accelerates ferroptosis in a manner dependent upon apoptosis. Mol Ther J Am Soc Gene Ther (2021) 29(1):263–74. doi: 10.1016/j.ymthe.2020.09.024
81. Li S, He Y, Chen K, Sun J, Zhang L, He Y, et al. RSL3 drives ferroptosis through NF-κB pathway activation and GPX4 depletion in glioblastoma. Oxid Med Cell Longevity (2021) 2021:2915019. doi: 10.1155/2021/2915019
82. Probst L, Dächert J, Schenk B, Fulda S. Lipoxygenase inhibitors protect acute lymphoblastic leukemia cells from ferroptotic cell death. Biochem Pharmacol (2017) 140:41–52. doi: 10.1016/j.bcp.2017.06.112
83. Lou S, Hong H, Maihesuti L, Gao H, Zhu Z, Xu L, et al. Inhibitory effect of hydnocarpin D on T-cell acute lymphoblastic leukemia via induction of autophagy-dependent ferroptosis. Exp Biol Med (Maywood NJ) (2021) 246(13):1541–53. doi: 10.1177/15353702211004870
84. Xu Y, Deng N, Wang X, Chen Y, Li G, Fan H. RKTG overexpression inhibits proliferation and induces apoptosis of human leukemia cells via suppression of the ERK and PI3K/AKT signaling pathways. Oncol Lett (2017) 14(1):965–70. doi: 10.3892/ol.2017.6182
85. Jin L, Tong L. PAQR3 inhibits proliferation and aggravates ferroptosis in acute lymphoblastic leukemia through modulation Nrf2 stability. Immun Inflammation Dis (2021) 9(3):827–39. doi: 10.1002/iid3.437
86. Hong Y, Zhang L, Tian X, Xiang X, Yu Y, Zeng Z, et al. Identification of immune subtypes of Ph-neg B-ALL with ferroptosis related genes and the potential implementation of Sorafenib. BMC Cancer (2021) 21(1):1331. doi: 10.1186/s12885-021-09076-w
87. Ishikawa C, Senba M, Mori N. Evaluation of artesunate for the treatment of adult T-cell leukemia/lymphoma. Eur J Pharmacol (2020) 872:172953. doi: 10.1016/j.ejphar.2020.172953
88. Chen L, Fang W, Liu J, Qi X, Zhao L, Wang Y, et al. Poricoic acid A (PAA) inhibits T-cell acute lymphoblastic leukemia through inducing autophagic cell death and ferroptosis. Biochem Biophys Res Commun (2022) 608:108–15. doi: 10.1016/j.bbrc.2022.03.105
89. Zhou B, Liu J, Kang R, Klionsky DJ, Kroemer G, Tang D. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol (2020) 66:89–100. doi: 10.1016/j.semcancer.2019.03.002
90. Gong H, Li H, Yang Q, Zhang G, Liu H, Ma Z, et al. A ferroptosis molecular subtype-related signature for predicting prognosis and response to chemotherapy in patients with chronic lymphocytic leukemia. BioMed Res Int (2022) 2022:5646275. doi: 10.1155/2022/5646275
Keywords: programmed cell death, ferroptosis, leukemia, reactive oxygen species, immunotherapy, iron metabolism
Citation: Chen Z, Zheng S, Han J, Fu L, Fu J, Zhang Z, Hong P and Feng W (2023) Molecular mechanisms of ferroptosis and its roles in leukemia. Front. Oncol. 13:1308869. doi: 10.3389/fonc.2023.1308869
Received: 07 October 2023; Accepted: 24 November 2023;
Published: 06 December 2023.
Edited by:
Gabor Mikala, Central Hospital of Southern Pest, HungaryReviewed by:
Ferenc Uher, Central Hospital of Southern Pest, HungaryCopyright © 2023 Chen, Zheng, Han, Fu, Fu, Zhang, Hong and Feng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Weiying Feng, ZmVuZ3dlaXlpbmcxOTk3QDEyNi5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.