
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol., 25 January 2024
Sec. Hematologic Malignancies
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1291457
This article is part of the Research TopicNovelties in Acute Myeloid Leukemia: From Biology to Clinical ApplicationsView all 13 articles
 Alessandra Sperotto1*
Alessandra Sperotto1* Maria Teresa Lupo Stanghellini2
Maria Teresa Lupo Stanghellini2 Jacopo Peccatori2
Jacopo Peccatori2 Roberta De Marchi1Simona Piemontese2Giulia Ciotti1Marco Basso1Elisabetta Pierdomenico1Paolo Fiore2
Roberta De Marchi1Simona Piemontese2Giulia Ciotti1Marco Basso1Elisabetta Pierdomenico1Paolo Fiore2 Fabio Ciceri2
Fabio Ciceri2 Michele Gottardi1
Michele Gottardi1Therapy-related myeloid neoplasms (t-MNs), which develop after cytotoxic, radiation, or immunosuppressive therapy for an unrelated disease, account for 7%–8% of acute myeloid leukemia (AML). Worse outcomes and consequently shortened survival are associated with t-MNs as compared with de novo AML. Therapy-related MNs are being reported with increasing frequency in successfully treated acute promyelocytic leukemia (APL), in particular, before the introduction of all-trans retinoic acid (ATRA) plus arsenic trioxide (ATO). Considering the high curability of APL, t-MNs represent one of the prognosis-limiting factors in this setting of leukemia. We report our experience with a patient who developed t-AML 15 years after treatment for APL. Treatment included three cycles of chemotherapy with CPX-351 (Vyxeos, Jazz Pharmaceuticals) followed, as in remission, by an allogeneic hematopoietic stem cell transplant. A review of available literature was also included.
Therapy-related myeloid neoplasms (t-MNs), including therapy-related myelodysplasia (t-MDS) and acute myeloid leukemia (t-AML), have been extensively reported after cytotoxic therapy or immunosuppressive treatment for solid tumors, lymphomas, or autoimmune disorders, more rarely after treatment for acute myeloid leukemia (1).
Traditionally subgrouped according to the previous exposure to alkylating agents, topoisomerase II inhibitors, or radiotherapy (RT), more recently, t-MN development has been associated with new agents belonging to different classes of chemotherapy (CHT) drugs, such as poly(ADP-ribose) polymerase inhibitors or purine analogs (2, 3).
Moreover, recent advances in deep sequencing techniques have significantly improved the knowledge of t-MNs over the last years, changing some of the classical views.
Acute promyelocytic leukemia (APL) is characterized by the translocations that fuse the PML gene on chromosome 15 to the RARalpha gene on chromosome 17 [t(15;17)], leading to a PML–RARalpha fusion gene; other peculiarities are the morphology of blast cells and a specific coagulopathy. Thanks to the advent of all-trans retinoic acid (ATRA) combined with anthracycline-based chemotherapy (4, 5) and/or arsenic trioxide (ATO), a cure rate higher than 70% has been achieved, even in relapsed patients (6–8). Thereby, the number of long-term survivors of this disease has increased over time. Consequently, more patients will be at risk of late complications related to antileukemic treatment.
Regarding t-MNs occurring after treatment for APL, sporadic cases have been reported in the literature, while only three major studies have assessed the incidence of t-MNs, ranging from 0.97% to 6.5% (4, 5). Moreover, only one of those studies calculated the cumulative incidence of a competing risk at a given time, resulting in approximately 2.2% at 6 years (9).
Survival in t-MNs is poor. In addition to the biology of t-MNs, the patient’s previous disease history and remission status at t-MN diagnosis are significant factors contributing to unfavorable outcomes. Also, t-MNs secondary to APL are usually difficult to treat, representing one of the prognosis-limiting factors for the curable APL disease.
We report a patient who developed t-AML 15 years after completion of maintenance therapy according to the GIMEMA AIDA2000 protocol for a previous APL still in molecular remission. A comprehensive review of the literature of previously published cases is also included.
A 46-year-old man presented in January 2005 with fatigue, dyspnea, and a history of bleeding tendency. Coagulation tests showed disseminated intravascular coagulation, and peripheral blood cell count was as follows: hemoglobin 90 g/L, white cells 66.000 × 109/L (with 60% hypergranular promyelocytes), and platelets 12.000 × 109/L. Bone marrow revealed 70% hypergranular promyelocytes, with the characteristics t(15;17)(q22;q21) in all metaphases examined; molecular biology studies (performed by reverse transcription–polymerase chain reaction (RT-PCR)) confirmed the presence of PML/RARα gene rearrangement type bcr3. A diagnosis of high-risk hypergranular APL was made. Next-generation sequencing (NGS) analysis was not performed at diagnosis of APL.
The patient was treated according to the GIMEMA AIDA2000 protocol, receiving induction treatment with oral ATRA (45 mg/m2 per day for a total of 45 days) and four doses of intravenous idarubicin (12 mg/m2 on days 2, 4, 6, and 8): a complete molecular remission was achieved on day 38. Consolidation (according to a risk-adapted strategy) consisted of three courses, as follows: one course with intravenous cytosine arabinoside (Ara-C) (1 g/m2 on days 1, 2, 3, and 4) plus idarubicin (15 mg/m2 on days 1, 2, 3, and 4) plus oral ATRA (45 mg/m2 per day for 15 days); then, intravenous mitoxantrone (10 mg/m2 on days 1, 2, 3, 4, and 5) plus etoposide (100 mg/m2 on days 1, 2, 3, 4, and 5) plus oral ATRA (45 mg/m2 per day for 15 days); finally, intravenous idarubicin (12 mg/m2 on days 1) plus Ara-C (150 mg/m2 every 8 hours on days 1, 2, 3, 4, and 5) plus 6-thioguanine (70 mg/m2 every 8 hours on days 1, 2, 3, 4, and 5) plus oral ATRA (45 mg/m2 per day for 15 days).
Then, as in molecular remission, maintenance therapy was started, consisting of intramuscular methotrexate (15 mg/m2) plus oral 6-mercaptopurine (50 mg/m2) alternating with oral ATRA (45 mg/m2 per day for 15 days) every 3 months for a total of 2 years.
Annual cytogenetic and molecular analyses were performed until December 2015, confirming molecular remission. From January 2018 to February 2020, the patient stopped his annual follow-ups. In March 2020, blood cell count revealed mild anemia (hemoglobin 120 g/L) and thrombocytopenia (platelets 111.000 × 109/L). Bone marrow analysis, performed in May 2020, confirmed molecular remission with initial cytological signs of dysplasia. Blood cell count remained stable until May 2022, when a morphological analysis of peripheral blood detected almost 10% blast cells. Bone marrow aspiration was hypercellular, showing 60% blast cells and red-cell line hyperplasia with multiple dyserythropoietic changes in erythroblasts (megaloblastic features, abnormal mitosis, and lobulated nuclei). Cytogenetic analysis revealed a complex karyotype (47, XY, +8, −2, −5, ins(mar;9)(?;q)?, del(12)(p13), +mar, inc), without t(15;17)(q22;q21). The molecular biology study was negative for PML/RARα gene rearrangement and positive for WT1 gene hyperexpression and KIT-D816V exon 17 mutation.
NGS analysis, performed using second-generation sequencing technology on an Illumina MiSeq System (Illumina, San Diego, CA, USA) high-throughput sequencing platform, showed TP53 positivity with a variant allele frequency (VAF) of 78.0%.
Treatment with CPX-351 (Vyxeos, Jazz Pharmaceuticals, Dublin, Ireland; a liposomal encapsulation of cytarabine and daunorubicin in a synergistic 5:1 drug ratio) was started on June 2022—when the patient was 63 years old. CPX-351 has a specific indication for newly diagnosed s-AML, including t-AML, and the choice of CPX-351 was also linked to the age of the patient, good performance status, and time to previous treatment.
Before starting treatment, the patient had a normal echocardiogram [left ventricular ejection fraction (LVEF) 68%] and spirometry (diffusing capacity of the lungs for carbon monoxide (DLCO) 85%) and was considered fit for an intensive chemotherapy program. A total of three cycles of CPX-351 were administered (first and second induction and then consolidation), all well tolerated.
Cytofluorimetric remission but not a complete clearance of WT1 gene hyperexpression (Figure 1) was obtained after the first CPX-351 cycle and then maintained during the other two cycles.
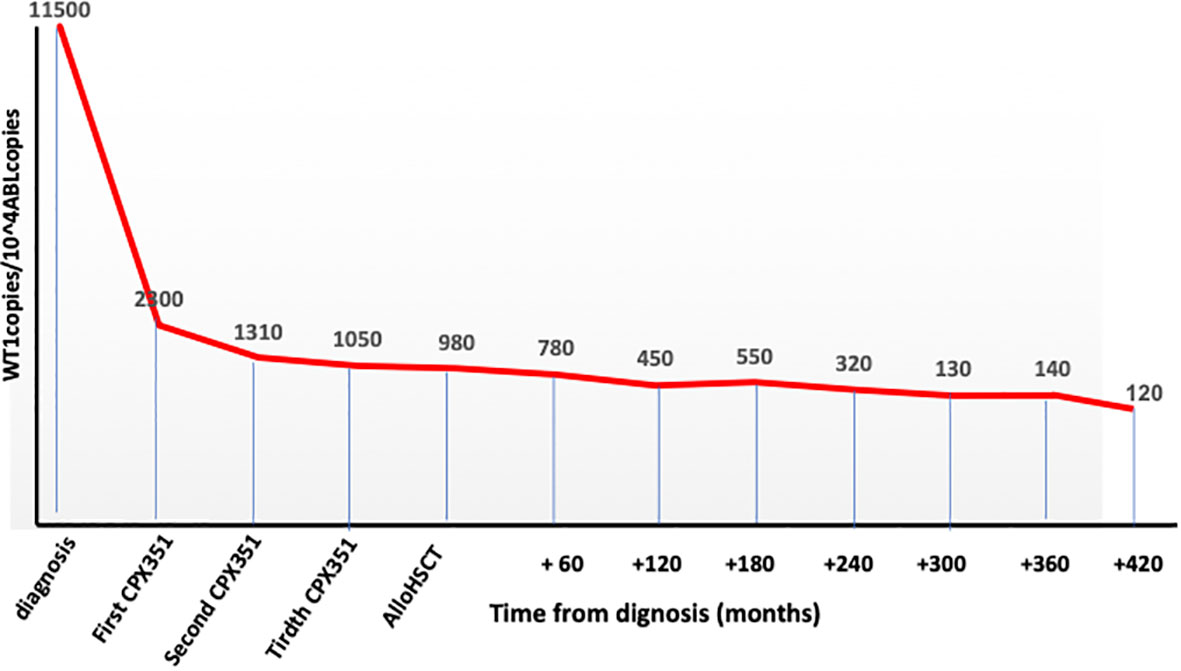
Figure 1 WT1 clearance during the whole program.
In October 2022, as still in cytofluorimetric remission but with WT1 over conventional threshold limits (Figure 1), an allogeneic hematopoietic stem cell transplant from an unrelated donor was performed. The patient was 64 years old at transplant with a hematopoietic cell transplant-specific comorbidity index (HCT-CI) score of 3 (previous leukemia) (10).
The conditioning regimen consisted of treosulfan i.v. plus fludarabine i.v.; graft versus host disease (GVHD) prophylaxis consisted of sirolimus, mycophenolate, and post-transplant cyclophosphamide. Allogeneic peripheral blood stem cells were infused on October 15, 2022.
The patient developed acute and then chronic skin GVHD, treated and resolved by steroid and extracorporeal photopheresis. Immunosuppressive treatment was completely withdrawn in March 2023. At the last follow-up—August 31, 2023—the patient was alive, with a mild chronic GVHD (mouth and skin), and in molecular remission with a full donor chimerism.
A total of 57 t-MN cases secondary to APL treatment were reported in the literature from 1992 to 2010: 44 (77.0%) patients were diagnosed with t-MDS and 13 (23.0%) with t-AML. After 2010, no other t-MN cases secondary to APL treatment were reported in the literature.
The main characteristics of the 57 patients are listed in Table 1 (t-MDS) and Table 2 (t-AML); the median age at diagnosis of APL was 51.5 years (8–73).

Table 1 Main characteristics and clinical course of patients developing t-MDS after APL therapy.
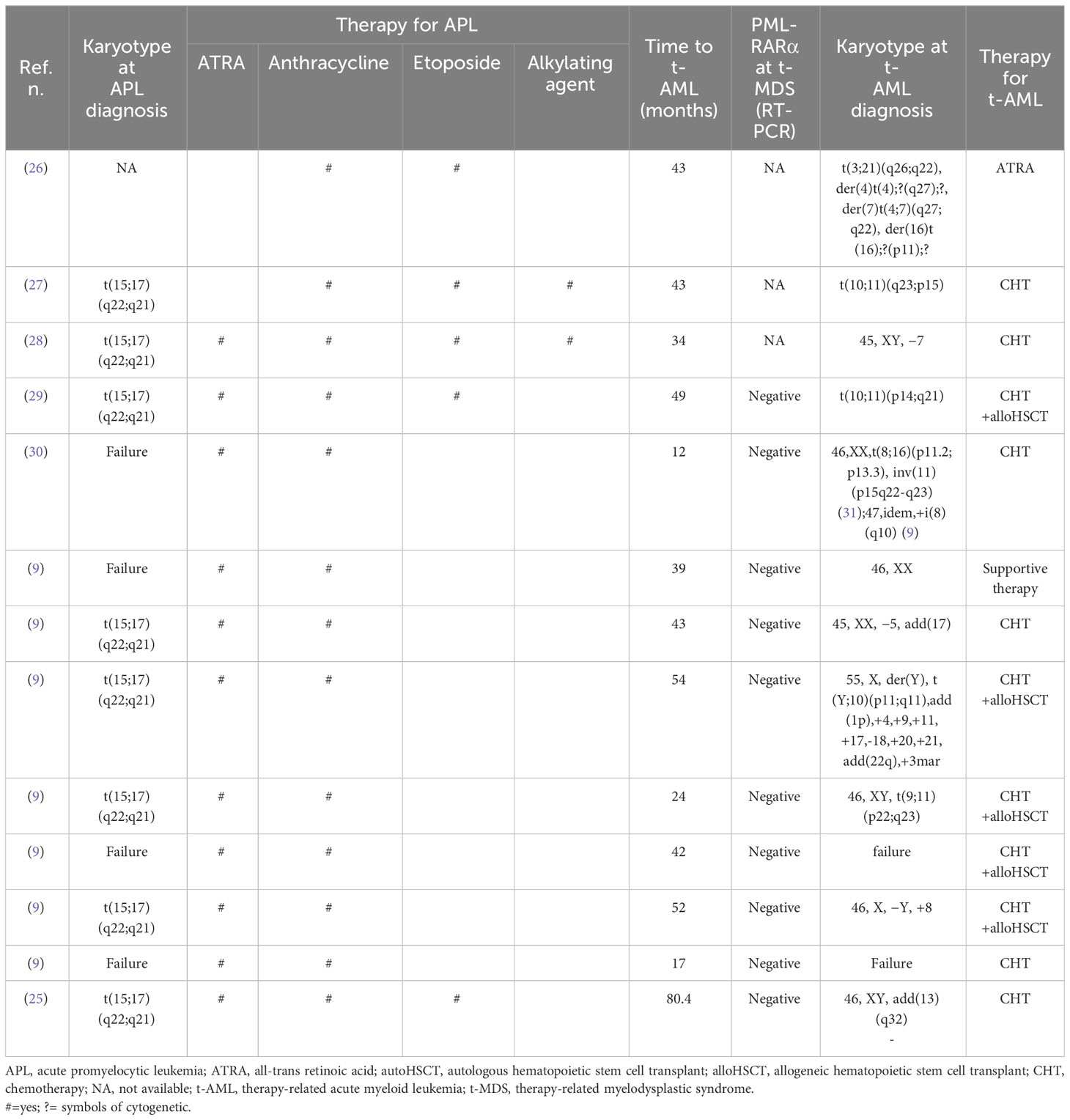
Table 2 Main characteristics and clinical course of patients developing t-AML after APL therapy.
Table 3 summarizes the clinical and treatment characteristics of the whole population.
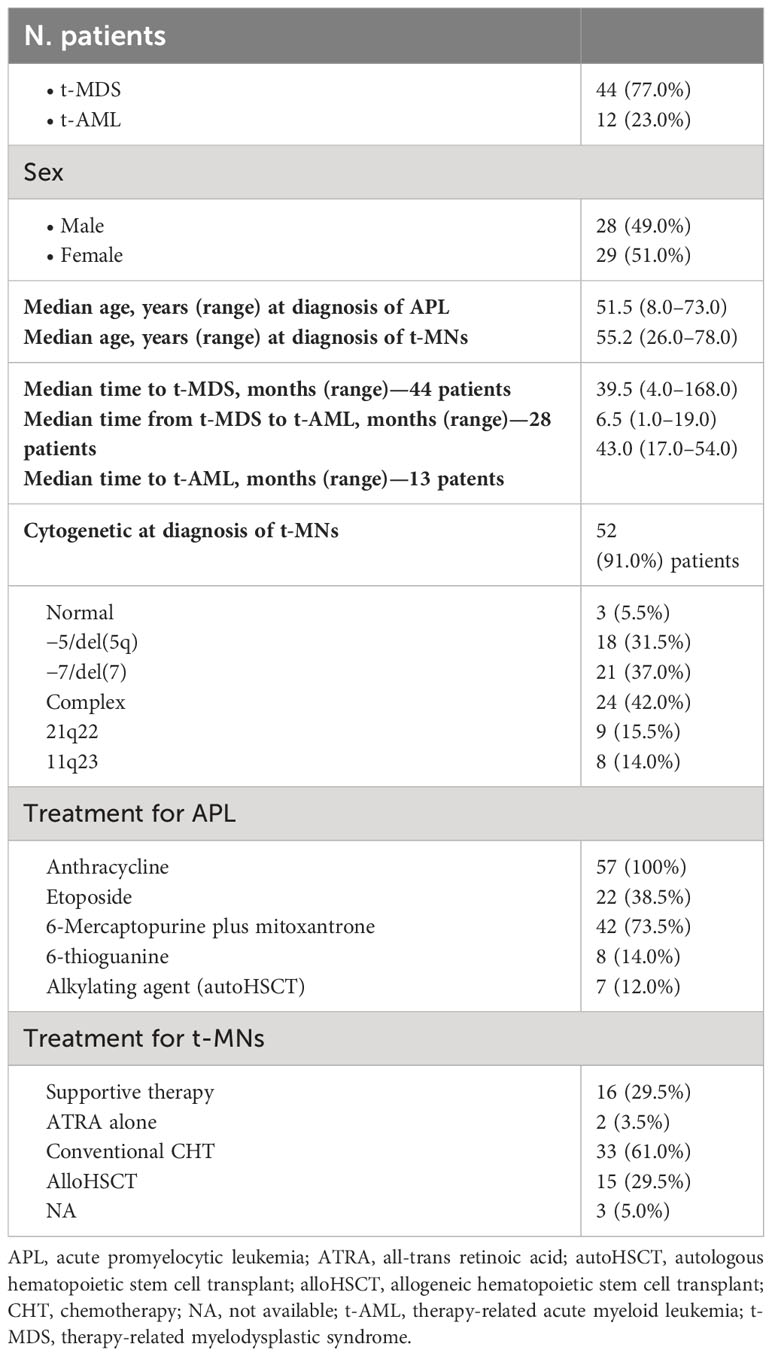
Table 3 Main characteristics: treatment and outcome of the whole population.
In all 57 patients, RT-PCR monitoring and/or cytogenetic analysis indicated molecular remission of APL at diagnosis of t-MNs. Overall, the median time from the achievement of remission to diagnosis of t-MN was 42.5 months (4–168).
No significant statistical difference between t-MDS and t-AML was observed in the time from the first complete response (CR) to the development of t-MNs (t-MDS: 39.5 months (4–168) vs. t-AML: 43 months (17–54), p = 0.07).
Using conventional karyotyping or fluorescent in situ hybridization, cytogenetic characterization was successful in 52 (91.0%) of 57 patients and was abnormal in all except three cases, with complex karyotypes (≥three independent abnormalities) observed in 24 (42.0%) patients (Table 3).
Except for three patients for whom treatment was not included in the report, in all the other 54 patients, therapy for t-MN consisted of only supportive therapy in 16 patients (29.5%) (15 MDS and one AML); all of them died after a median of 9 months from t-MN diagnosis (range, 1 to 39 months). The majority of patients (33%–61.0%) were treated with conventional chemotherapy (in one patient, an autologous stem cell transplant was performed after induction treatment): for two patients, follow-up was not available, and all the others died of progressive disease.
Allogeneic stem cell transplant (alloHSCT) was performed in 15 (29.5%) patients (three patients up-front and 11 patients after induction chemotherapy): four transplanted patients lived more than 12 months from transplant, but follow-up was not subsequently updated, while all the others died due to transplant-related mortality (five patients) or progressive disease (six patients) within 12 months from reinfusion (Table 3).
Therapy-related AML after APL treatment is a relatively infrequent (<7.0%) and late complication bearing a poor prognosis (4, 5).
Incidences reported in the largest studies ranged between 0.97% (European APL study: a series of 617 patients with a median follow-up of 51 months) and 6.5% (Italian study of 46 patients: follow-up not reported) (4, 5). Because the risk of developing t-AML continues for many years after the end of treatment, the PETHEMA group evaluated the cumulative incidence of t-AML in patients enrolled in three consecutive trials (LPA96, LPA99, and LPA2005): 918 patients were observed for a median of 77 months with a cumulative incidence of t-AML of 2.2% at 6 years, not comparable with the crude incidence of the other two studies (9).
The introduction of ATO in combination with ATRA had further reduced the incidence of t-AML in the APL setting, as reported by the Italian-German APL0406 study, where, with a follow-up of 6 years; no t-MN cases were observed in ATRA/ATO group patients vs. 1.5% in those treated with the AIDA regimen (10). Similar results have been reported from the AML17 trial (National Cancer Research Institute): with a follow-up of 5.7 years, no t-AMLs were observed in the ATRA/ATO group vs. 1.0% in the AIDA group (32).
Cytogenetic abnormalities have been largely described in patients with t-AML: a decreased prevalence of normal karyotype (<30.0%) and a prevalence of complex or unbalanced karyotypes with chromosomal deletions as compared with the de novo AML were reported (2, 21, 33–35). The combination of multiple chemo-immunotherapy agents with different mechanisms of action makes it difficult to ascribe the mutagenic potential to a single drug. Traditionally, recurrent translocations as t(15;17), t(8;21), inv(16), t(15;17), and 11q23 abnormalities were associated with topoisomerase II inhibitors, and t-AML usually developed after a latency time of 1 to 3 years (36–38). Very complex karyotypes (>5 simultaneous chromosomal abnormalities) and deletions of chromosomes 5 and 7 were usually associated with alkylating agents or radiotherapy, occurring after a latency of 5 to 7 years (2, 39, 40).
Therapy-related MDS is usually characterized by very complex karyotypes and consequently by a poor and very poor cytogenetic risk (40). Therefore, according to the Revised International Prognostic Scoring System (IPSS-R), a high prevalence of high- and very-high-risk subgroups was expected in the t-MDS setting. The IPSS-R is applicable to t-MDS and de novo MDS and reliably predicts AML transformation. As reported in the literature, 28 of the 44 (63.5%) t-MDS cases that developed after APL treatment subsequently progressed to t-AML at a median time of 6.5 months (1–19) (Tables 1, 3).
The molecular characteristics of MNs have been extensively analyzed in recent years: in more than 95% of AML and MDS, somatic mutations have been detected, without significant difference in the overall number of mutations in secondary vs. de novo subtypes. Moreover, none of the genes were exclusively mutated in t-AML. Mutations in RNA-splicing genes, epigenetic regulators genes, or cohesin complex genes were more than 90.0% specific for the diagnosis of s-AML (39, 41) and were present in only 30.0% of t-AML.
In the 57 t-MN cases that developed after APL treatment reported in the literature, in addition to an anthracycline (all the 57 patients), 22 (38.5%) patients also received etoposide, 42 (73.5%) received 6-mercaptopurine plus mitoxantrone as maintenance treatment, and eight (14.0%) received 6-thioguanine. Only seven patients (12.0%) received an alkylating agent as a part of the conditioning regimen for autologous stem cell transplantation.
Concerning cytogenetic analysis, in the 57 cases reported in the literature (Table 3), balanced translocations that involved 21q22 and 11q23 (typical breakpoints observed in t-AML occurring after administration of topoisomerase II inhibitors) were detected in nine (15.5%) and eight (14.0%) patients, respectively (Table 3). Moreover, 18 (31.5%) patients had −5/del(5q), and 21 (37.0%) had −7/del(7) abnormalities. Complex karyotypes (≥three independent abnormalities) were revealed in 24 (42.0%) patients (Table 3).
No NGS analysis was performed in the 57 t-MN cases reported in the literature, while in 11 patients (19.5%), molecular analysis by RT-PCR was reported (ref (25), Tables 1, 2).
Our patient was extensively studied by RT-PCR and NGS at diagnosis of t-AML, confirming the absence of PML/RARα gene rearrangement and presence of KIT-D816V exon −17 mutation and TP53 gene mutation, with a VAF of 78.0%.
In an independent series, mutations of TP53 were reported in 30.0% to 47.0% of cases of t-MNs, resulting in the single most frequent molecular abnormality in this setting associated with complex karyotype in almost 80.0% of cases (39, 42–44). Lindsley et al. showed that TP53 mutations define a specific subgroup of t-AML, which differs from other AMLs like s-AML, in terms of younger age, lower recurrent driver mutations, more cytogenetic abnormalities, and poor prognosis with a reduced probability of achieving response after conventional treatment (39).
KIT mutations are detected in approximately 4%–6% of adult patients with de novo AML (45, 46) and 20%–40% of adult patients with de novo core-binding factor (CBF) leukemia (47–51). Three mutational hot spots (exon 8, exon 10–11, and exon 17) have been identified in the KIT gene (37, 52–54). Of these, exon 17 (detected in our patient) represents the site of KIT mutations most strongly associated with poor prognosis.
As KIT mutations have been reported mostly in CBF-AML, most studies on KIT mutations have been limited to CBF-AML, with few studies investigating KIT mutations in t-MNs. Schnittger et al. performed a large-scale study involving almost 2,000 unselected patients with AML: among 125 t-AML patients of the series, KIT mutation was detected in only one patient, who also presented t(8;21) translocation (54). Another study on 140 patients with t-MNs reported two cases with KIT-D816V mutation, one of which had t(8;21) (55).
KIT and TP53 mutations were not detected together in any of the cases reported in the literature. Survival in t-MNs is poor when compared with that in other leukemia subtypes: until recent years, patients with t-MNs have been conventionally excluded from many clinical trials. This is particularly relevant in patients with previous APL, which is now considered a curable disease in many patients. New drugs with specific activity on secondary leukemia (including t-AML), targeting pathogenic mutations or interfering with immune mechanisms, are or will be available in the future. Our patient was treated with CPX-351 (Vyxeos, Jazz Pharmaceuticals): up to now, no other cases treated with CPX-351 and allogeneic stem cell transplant for a t-AML that developed after treatment according to GIMEMA AIDA2000 protocol have been reported in the literature.
The risk of anthracycline-induced heart failure increases as the cumulative dose administered increases: 3%–5% at 400 mg/m2 and as high as 18%–48% at 700 mg/m2 (56). However, there is a different level of risk for each patient scheduled for anthracycline therapy: patients younger than 5 years or older than 65 years, with prior or concurrent chest irradiation, pre-existing heart disease, or already known cardiovascular risk factors, have an increased risk of cardiotoxicity.
Our patient was 46 years old when he was treated according to GIMEMA AIDA2000 protocol: the anthracycline cumulative dose administered (as by protocol) was 600 mg/m2, and no concomitant cardiovascular risk factors were present at diagnosis, but unfortunately, LVEF before treatment was not available.
Before starting treatment for t-AML, our patient was 63 years old, without cardiac dysfunction (LVEF 68%), hypertension, or other cardiovascular risk factors.
As mentioned, CPX-351 is a liposomal encapsulation of cytarabine and daunorubicin: in the heart, liposomes cannot get out of the vascular space because capillaries have tight junctions. As the tendency to accumulate in the heart cells is limited, this may reduce the risk of cardiotoxicity. On the contrary, the liposomes reach high concentrations in the tumor site, leaving the circulatory system where tumor growth damages the capillaries (56).
In our patient, no cardiac dysfunction or other cardiovascular diseases were developed during the treatment for t-AML (from induction to transplant).
Of the 57 t-MN patients reported in the literature (Table 3), 15 underwent allogeneic stem cell transplant; no details about disease status at transplant and at last follow-up were reported, particularly about the molecular response. In our patient, with a high-risk genetic profile (TP53 and KIT-D816V exon −17 mutation), a molecular response was achieved with a transplant procedure and confirmed at the last follow-up. Of course, a longer follow-up would be needed for overall response and chronic GVHD assessment.
Considering the high curability of APL with excellent complete remission and long-term survival rates, it is necessary to try to reduce the incidence of t-MNs with a risk-adapted strategy and use chemotherapy-free regimens like ATO/ATRA.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
AS: Conceptualization, Writing – original draft, Writing – review & editing. MS: Investigation, Writing – review & editing. JP: Investigation, Writing – review & editing. RM: Investigation, Writing – review & editing. SP: Data curation, Writing – review & editing. GC: Data curation, Writing – review & editing. EP: Data curation, Writing – review & editing. PF: Data curation, Writing – review & editing. FC: Writing – review & editing. MG: Writing – review & editing. MB: Data curation.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Arana-Yi C, Block AW, Sait SN, Ford LS, Barcos M, Baer MR. Therapy-related myelodysplastic syndrome and acute myeloid leukemia following treatment of acute myeloid leukemia: Possible role of cytarabine. Leuk Res (2008) 32:1034–48. doi: 10.1016/j.leukres.2007.11.006
2. McNerney ME, Godley LA, Le Beau MM. Therapy-related myeloid neoplasms: when genetics and environment collide. Nat Rev Cancer (2017) 17(9):513–27. doi: 10.1038/nrc.2017.60
3. Morice PM, Leary A, Dolladille C, Chretien B, Poulain L, Gonzalez-Martin A, et al. Myelodysplastic syndrome and acute myeloid leukaemia in patients treated with PARP inhibitors: a safety meta-analysis of randomised controlled trials and a retrospective study of the WHO pharmacovigilance database. Lancet Haematol (2021) 8(2):e122–34. doi: 10.1016/S2325-3026(20)30360
4. Latagliata R, Petti MC, Fenu S, Mancini M, Spiriti MAO, Breccia M. Therapy-related myelodisplastic syndrome-acute myelogenous leukemia in patients treated for acute promyelocytic leukemia: An emerging problem. Blood (2002) 99:822–4. doi: 10.1182/blood.v99.3.822
5. Lobe I, Rigal-Huguet F, Vekhoff A, Desablens B, Bordessoule D, Mounier C, et al. Myelodisplastic syndrome after acute promyelocytic leukaemia: The European APL group experience. Leukemia (2003) 17:1600–1604. doi: 10.1038/sj.leu.2403034
6. Fenaux P, Chomienne C, Degos L. All-trans retinoic acid and chemotherapy in the treatment of acute promyelocytic leukemia. Semin Hematol (2001) . 38:13–25. doi: 10.1016/s0037-1963(01)90002-2
7. Sanz MA, Martin G, Rayon C, Esteve J, Gonzalez M, Diaz-Mediavilla J, et al. A modified AIDA protocol with anthracycline-based consolidation results in high antileukemic efficacy and reduced toxicity in newly diagnosed PML/RARalpha – positive acute promyelocytic leukemia. PETHEMA group. Blood (1999) 94:3015–21.
8. Tallman MS, Andersen JW, Schiffer CA, Appelbaum FR, Feusner JH, Woods WG, et al. All-trans retinoic acid in acute promyelocytic leukemia: a long-term outcome and prognostic factor analysis from the North American Intergroup protocol. Blood (2002) 100:4298–302. doi: 10.1182/blood-2002-02-0632
9. Montesinos P, Gonzalez JD, Gonzalez J, Rayon C, de Lisa E, Amigo ML, et al. Therapy-related myeloid neoplasms in patients with acute promyelocytic leukemia treated with all-trans-retinoic acid and anthracycline-based chemotherapy. J Clin Oncol (2010) 28:3872–9. doi: 10.1200/JCO.2010.29.2268
10. Sorror ML, Maris MB, Storb R, Baron F, Sandmaier BM, Maloney DG, et al. Hematopoietic cell transplant (HCT) – specific comorbidity index: a new tool for risk assessment before allogeneic HCT. Blood (2005) 106(8):2912–9. doi: 10.1182/blood-2005-05-2004
11. Jubashi T, Nagai K, Miyazaki Y, Nakamura H, Matsuo T, Kuriyama K, et al. A unique case of t(15;17) acute promyelocitic leukaemia (M3) developing into acute myeloblastic leukemia (M1) with t(7;21) at relapse. Br J Haematol (1993) 83:665–8. doi: 10.1111/j.1365-2141.1993.tb04709.x
12. Hatzis T, Standen GR, Howell RT, Savill C, Wagstaff M, Scott GL. Acute promyelocytic leukaemia (M3): relapse with acute myeloblastic leukaemia (M2) and dic(5;17)(q11;p11). Am J Hematol (1995) 48:40–4. doi: 10.1002/ajh.2830480108
13. Bseiso AW, Kantarjian H, Estey E. Myelodisplastic syndrome following successful therapy of acute promyelocytic leukemia. Leukemia (1997) 11:168–9. doi: 10.1038/sj.leu2400539
14. Stavroyianni N, Yataganas X, Abazis D, Pangalos C, Meletis J. Acute promyelocytic leukemia relapsing into FAB-M2 acute myeloid leukemia with trisomy 8. Cancer Genet Cytogenet (2000) 117:82–3. doi: 10.1016/s0165-4608(99)00132-6
15. Au WY, Lam CC, Ma ES, Man C, Wan T, Kwong YL. Therapy-related myelodysplastic syndrome after eradication of acute promyelocytic leukemia: cytogenetic and molecular features. Hum Pathol (2001) 32:126–9. doi: 10.1053/hupa.2001.21128
16. Pecci A, Invernizzi R. A therapy-related myelodisplastic syndrome with unusual features in a patient treated for acute promyelocytic leukemia. Haematologica (2001) 86:102–3.
17. Garcia-Manero G, Kantarjian HM, Kornblau S, Estey E. Therapy-related myelodysplastic syndreom or acute myelogenous leukemia in patients with acute promyelocytic leukemia. Leukemia (1988) 2002:16.
18. Athanasiadou A, Saloum R, Zorbas I, Tsompanakou A, Batsis I, Fassas A, et al. Therapy-related myelodysplastic syndrome with monosomy 5 and 7 following successful therapy for acute promyelocytic leukemia with anthracyclines. Leuk Lymphoma (2002) 43:2409–11. doi: 10.1080/1042819021000040143
19. Panizo C, Patino A, Lecumberri R, Calasanz MJ, Odero MD, Bendandi M, et al. Secondary Myelodisplastic syndrome after treatment for promyelocitic leukemia: clinical and genetic features of two cases. Cancer Genet Cytogenet (2003) 143:178–81. doi: 10.1016/s0165-4608(02)00859-2
20. Pawarode A, Finlay E, Sait SNJ, Barcos M, Baer MR. Isochromosome 1q in a myelodisplastic syndrome after treatment for acute promyelocytic leukemia. Cancer Gent Cytogenet (2006) 167:155–60. doi: 10.1016/j.cancergencyto.2005.11.013
21. Fianchi L, Pagano L, Piciocchi A, Candoni A, Gaidano G, Breccia M, et al. Characteristics and outcome of therapy- related myeloid neoplasms: Report from the Italian network on secondary leukemias. Am J Hematol (2015) 90:E80–E85. doi: 10.1002/ajh.23966
22. Snijder S, Mellink CHM, van der Lelie H. Translocation (2;11)(q37;q23) in therapy-related myelodysplastic syndrome after treatment for acute promyelocytic leukemia. Cancer Gent Cytogenet (2008) 180:149–52. doi: 10.1016/j.cancergencyto.2007.10.003
23. Meloni G, Diverio D, Vignetti M, Avvisati G, Capria S, Petti MC, et al. Autologous bone marrow transplantation for acute promyelocytic leukemia in second remission: prognostic relevance of pretransplant minimal residual disease assessment by reverse – transcription polymerase chain reaction of the PML/RAR alpha fusion gene. Blood (1997) 90:1321–5. doi: 10.1182/blood.V90.3.1321
24. Zompi S, Viguie F. Therapy-related acute myeloid leukemia and myelodysplasia after successful treatment of acute promyelocytic leukemia. Leuk Lymphoma (2002) 43:275–80. doi: 10.1080/10428190920006044
25. Imagawa J, Harada Y, Shimomura T, Tanaka H, Okikawa Y, Hyodo H, et al. Clinical and genetic features of therapy – related myeloid neoplasms after chemotherapy for acute promyelocytic leukemia. Blood (2010) 116:6018–22. doi: 10.1182/blood-2010-06-289389
26. Miyazaki H, Ino T, Sobue R, Kojima H, Wakita M, Nomura T, et al. Translocation (3;21)(q26;q22) in treatment-related acute leukemia secondary to acute promyelocytic leukemia. Cancer Gent Cytogenet (1994) 74:84–6. doi: 10.1016/0165-4608(94)90002-7
27. Sawada H, Morimoto H, Wake A, Yamasaki Y, Izumi Y. Therapy-related acute myeloid leukemia with t(10;11)(q23;p15) following successful chemotherapy for acute promyelocytic leukemia with t(15;17)(q22;q21). Int J Hematol (1999) 69:270–1.
28. Felice MS, Rossi J, Gallego M, Zubizarreta PA, Cygler AM, Alfaro E, et al. Acute trilineage leukemia with monosomy of chromosome 7 following an acute promyelocytic leukemia. Leuk Lymphoma (1999) 34:409–13. doi: 10.3109/10428199909050968
29. Todisco E, Testi AM, Avvisati G, Moleti ML, Cedrone M, Cimino G, et al. Therapy – related acute myelomonocytic leukemia following successful treatment for acute promyelocytic leukemia. Leukemia (1995) 9:1583–5.
30. Lee GY, Christina S, Tien SL, Ghafar AB, Hwang W, Lim LC, et al. Acute promyelocytic leukemia with PML-RARA fusion i(17q) and therapy related acute myeloid leukemia. Cancer Gent Cytogenet (2005) 159:129–36. doi: 10.1016/j.cancergencyto.2004.09.019
31. Cicconi L, Platzbecker U, Avvisati G, Paoloni F, Thiede C, Vignetti M, et al. Long-term results of all-trans retinoic acid and arsenic trioxide in non- high-risk acute promyelocytic leukemia: update of the APL0406 Italian-German randomized trial. Leukemia (2020) 34(3):914–8. doi: 10.1038/s41375-019-0589-3
32. Russell N, Burnett A, Hills R, Betteridge S, Dennis M, Jovanovic J, et al. NCRI AML Working Group. Attenuated arsenic trioxide plus ATRA therapy for newly diagnosed and relapsed APL: long-term follow-up of the AML17 trial. Blood (2018) 132:1452–4. doi: 10.1182/blood-2018-05-851824
33. Smith SM, Le Beau MM, Huo D, Karrison T, Sobecks RM, Anastasi J, et al. Clinical-cytogenetic associations in 306 patients with therapy-related myelodysplasia and myeloid leukemia: the University of Chicago series. Blood (2003) 102(1):43–52. doi: 10.1182/blood-2002-11-3343
34. Frohling S, Schlenk RF, Kayser S, Morhardt M, Benner A, Dohner K, et al. German-Austrian AML Study Group. Cytogenetics and age are major determinants of outcome in intensively treated acute myeloid leukemia patients older than 60 years: results from AMLSG trial AML HD98-B. Blood (2006) 108) 10:3280–8. doi: 10.1182/blood-2006-04-014324
35. Grimwade D, Hills RK, Moorman AV, Walker H, Chatters S, Goldstone AH, et al. National Cancer Research Institute Adult Leukaemia Working Group. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood (2010) 116:354–65. doi: 10.1182/blood-2009-11-254441
36. Kayser S, Krzykalla J, Elliott MA, Norsworthy K, Gonzales P, Hills RK, et al. Characteristics and outcome of patients with therapy-related acute promyelocytic leukemia front-line treated with or without arsenic trioxide. Leukemia (2017) 31:2347–54. doi: 10.1038/leu.2017.92
37. Paschka P, Dohner K. Core-binding factor acute myeloid leukemia: can we improve on HiDAC consolidation? Hematol (Am Soc Hematol Educ Program) (2013) 2013:209–19. doi: 10.1182/asheducation-2013.1.209
38. Rogers HJ, Wang X, Xie Y, Davis AR, Thkral B, Wang SA, et al. Comparison of therapy-related and de novo core binding factor acute myeloid leukemia: A bone marrow pathology group study. Am J Hematol (2020) 95:799–808. doi: 10.1002/ajh.25814
39. Lindsley RC, Mar BG, Mazzola E, Grauman PV, Shareef S, Allen SL, et al. Acute myeloid leukemia ontogeny is defined by distinct somatic mutations. Blood (2015) 125:1367–76. doi: 10.1182/blood-2014-11-610543
40. Kuendgen A, Nomdedeu M, Tuechler H, Garcia-Manero G, Komorkji RS, Sekeres MA, et al. Therapy-related myelodysplastic syndromes deserve specific diagnostic sub-classification and risk-stratification-an approach to classification of patients with t-MDS. Leukemia (2021) 35:835–49. doi: 10.1038/s41375-020-0917-7
41. Papaemmanuil E, Gerstung M, Malcovati L, Tauro S, Gundem G, Van Loo P. et al; Chronic Myeloid Disorders Working Group of the International Cancer Genome Consortium. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood (2013) 122:3616–3627, quiz 3699. doi: 10.1182/blood-2013-08-518886
42. Singhal D, Wee LYA, Kutyna MM, Chhetri R, Geoghegan J, Schreiber AW, et al. The mutational burden of therapy-related myeloid neoplasms is similar to primary myelodysplastic syndrome but has a distinctive distribution. Leukemia (2019) 33:2842–53. doi: 10.1038/s41375-019-0479-8
43. Lindsley RC, Saber W, Mar BG, Redd R, Wang T, Haagenson M, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation. N Engl J Med (2017) 376:536–47. doi: 10.1056/NEJMoa1611604
44. Bernard E, Nannya Y, Hasserjian RP, Devlin SM, Tuechler H, Medina-Martinez JS, et al. Implications of TP53 allelic state for genome stability, clinical presentation and outcomes in myelodysplastic syndromes [published correction appears in Nat Med. 2021;27(3):562]. Nat Med (2020) 26:1549–56. doi: 10.1038/s41591-020-1008-z
45. Ley TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med (2013) 368:2059–74. doi: 10.1056/NEJMoa1301689
46. Patel JP, Goünen M, Figueroa ME, Fernandez H, Sun Z, Racevskis J, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med (2012) 366:1079–89. doi: 10.1056/NEJMoa1112304
47. Krauth MT, Eder C, Alpermann T, Bacher U, Nadarajah N, Kern W, et al. High number of additional genetic lesions in acute myeloid leukemia with t(8;21)/RUNX1-RUNX1T1: Frequency and impact on clinical outcome. Leukemia (2014) 28:1449–58. doi: 10.1038/leu.2014.4
48. Qin YZ, Zhu HH, Jiang Q, Jiang H, Zhang LP, Xu LP, et al. Prevalence and prognostic significance of c-KIT mutations in core binding factor acute myeloid leukemia: A comprehensive large-scale study from a single Chinese center. Leuk Res (2014) 38:1435–40. doi: 10.1016/j.leukres.2014.09.017
49. Allen C, Hills RK, Lamb K, Evans C, Tinsley S, Sellar R, et al. The importance of relative mutant level for evaluating impact on outcome of KIT, FLT3 and CBL mutations in core-binding factor acute myeloid leukemia. Leukemia (2013) 27:1891–901. doi: 10.1038/leu.2013.186
50. Paschka P, Du J, Schlenk RF, Gaidzik VI, Bullinger L, Corbacioglu A, et al. Secondary genetic lesions in acute myeloid leukemia with inv(16) or t(16;16): A study of the German-Austrian AML Study Group (AMLSG). Blood (2013) 121:170–7. doi: 10.1182/blood-2012-05-431486
51. Kim HJ, Ahn HK, Jung CW, Moon JH, Park CH, Lee KO, et al. KIT D816 mutation associates with adverse outcomes in core binding factor acute myeloid leukemia, especially in the subgroup with RUNX1/RUNX1T1 rearrangement. Ann Hematol (2013) 92:163–71. doi: 10.1007/s00277-012-1580-5
52. Ishikawa Y. Molecular pathogenesis and treatment of core binding factor-acute myeloid leukemia. Jpn J Clin Hematol (2018) 59:1997–2006.
53. Kim SY, Kang JJ, Lee HH, Kang JJ, Kim B, Kim CG, et al. Mechanism of activation of human c-KIT kinase by internal tandem duplications of the juxtamembrane domain and point mutations at aspartic acid 816. Biochem Biophys Res Commun (2011) 410:224–8. doi: 10.1016/j.bbrc.2011.05.111
54. Berenstein R. Class III receptor tyrosine kinases in acute leukemia—Biological functions and modern laboratory analysis. biomark Insights (2015) 10(Suppl. 3):1–14. doi: 10.4137/BMI.S22433
55. Christiansen DH, Andersen MK, Desta F, Pedersen-Bjergaard J. Mutations of genes in the receptor tyrosine kinase (RTK)/RAS-BRAF signal transduction pathway in therapy-related myelodysplasia and acute myeloid leukemia. Leukemia (2005) 19:2232–40. doi: 10.1038/sj.leu2404009
Keywords: acute promyelocytic leukemia, therapy-related myeloid neoplasm, allogeneic hematopoietic stem cell transplantation, CPX-351, acute myeloid leukemia
Citation: Sperotto A, Stanghellini MTL, Peccatori J, De Marchi R, Piemontese S, Ciotti G, Basso M, Pierdomenico E, Fiore P, Ciceri F and Gottardi M (2024) CPX-351 and allogeneic stem cell transplant for a therapy-related acute myeloid leukemia that developed after treatment of acute promyelocytic leukemia: a case report and review of the literature. Front. Oncol. 13:1291457. doi: 10.3389/fonc.2023.1291457
Received: 09 September 2023; Accepted: 28 December 2023;
Published: 25 January 2024.
Edited by:
Anna Maria Testi, Sapienza University of Rome, ItalyReviewed by:
Nico Gagelmann, University Medical Center Hamburg-Eppendorf, GermanyCopyright © 2024 Sperotto, Stanghellini, Peccatori, De Marchi, Piemontese, Ciotti, Basso, Pierdomenico, Fiore, Ciceri and Gottardi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alessandra Sperotto, YWxlc3NhbmRyYS5zcGVyb3R0b0Bpb3YudmVuZXRvLml0
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.