
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol. , 24 January 2024
Sec. Cancer Genetics
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1284690
 Raymond A. Isidro1,2†‡
Raymond A. Isidro1,2†‡ Anu Chittenden3‡McKenzie Walker4‡
Anu Chittenden3‡McKenzie Walker4‡ Alison Schwartz3
Alison Schwartz3 Diane R. Koeller3
Diane R. Koeller3 Connor P. Hayes4†
Connor P. Hayes4† Busra Unal4
Busra Unal4 Monica Devi Manam1†Ryan M. Buehler3Danielle K. Manning1
Monica Devi Manam1†Ryan M. Buehler3Danielle K. Manning1 Lynette M. Sholl1,2Mark S. Redston1,2Matthew B. Yurgelun2,3,5Huma Q. Rana2,3,5Judy E. Garber2,3,5
Lynette M. Sholl1,2Mark S. Redston1,2Matthew B. Yurgelun2,3,5Huma Q. Rana2,3,5Judy E. Garber2,3,5 Arezou A. Ghazani1,2,4,6*
Arezou A. Ghazani1,2,4,6*The presence of variants of uncertain significance (VUS) in DNA mismatch repair (MMR) genes leads to uncertainty in the clinical management of patients being evaluated for Lynch syndrome (LS). Currently, there is no platform to systematically use tumor-derived evidence alongside germline data for the assessment of VUS in relation to LS. We developed INT2GRATE (INTegrated INTerpretation of GeRmline And Tumor gEnomes) to leverage information from the tumor genome to inform the potential role of constitutional VUS in MMR genes. INT2GRATE platform has two components: a comprehensive evidence-based decision tree that integrates well-established clinico-genomic data from both the tumor and constitutional genomes to help inform the potential relevance of germline VUS in LS; and a web-based user interface (UI). With the INT2GRATE decision tree operating in the backend, INT2GRATE UI enables the front-end collection of comprehensive clinical genetics and tumor-derived evidence for each VUS to facilitate INT2GRATE assessment and data sharing in the publicly accessible ClinVar database. The performance of the INT2GRATE decision tree was assessed by qualitative retrospective analysis of genomic data from 5057 cancer patients with MMR alterations which included 52 positive control cases. Of 52 positive control cases with LS and pathogenic MMR alterations, 23 had all the testing parameters for the evaluation by INT2GRATE. All these variants were correctly categorized as INT2GRATE POSITIVE. The stringent INT2GRATE decision tree flagged 29 of positive cases by identifying the absence or unusual presentation of specific evidence, highlighting the conservative INT2GRATE logic in favor of a higher degree of confidence in the results. The remaining 99% of cases were correctly categorized as INCONCLUSIVE due to the absence of LS criteria and ≥1 tumor parameters. INT2GRATE is an effective platform for clinical and genetics professionals to collect and assess clinical genetics and complimentary tumor-derived information for each germline VUS in suspected LS patients. Furthermore, INT2GRATE enables the collation of integrated tumor-derived evidence relevant to germline VUS in LS, and sharing them with a large community, a practice that is needed in precision oncology.
Lynch Syndrome (LS) is a hereditary condition, most commonly associated with colorectal and endometrial cancers (CRC and EC, respectively). With an estimated population prevalence of 1:279 (1), LS is caused by heterozygous germline inactivating alterations in the DNA mismatch repair (MMR) genes MLH1, MSH2, MSH6, PMS2, deletion in EPCAM, and other rare epigenetic events (2–4). Although the diagnosis of LS requires the identification of a pathogenic germline MMR gene variant, LS is highly suspected in individuals with certain clinical features and whose tumors exhibit microsatellite instability (MSI) resulting from MMR deficiency (MMRd) (5).
Variant pathogenicity is routinely assessed according to the American College of Medical Genetics (ACMG) guidelines (6), an evidence-based system developed for constitutional Mendelian genetic disorders. This current framework for germline variant assessment does not incorporate tumor-derived evidence and a systematic, evidence-based approach for the joint interpretation of germline and tumor data does not exist. The segregation of germline and somatic variant databases further perpetuates the lack of integration of constitutional and tumor-derived information. We have previously demonstrated the value of the integrated germline and somatic framework in assessing the pathogenicity of germline variants in several cancer syndromes (7–12), and the utility of this integrated approach in the assessment of germline variants of uncertain significance (VUS) in cancer susceptibility genes (7). There is currently a need for objective assessment of VUS in high-risk individuals where germline variants might be actionable, or in other genetic settings where the identification of MMR variants might help preventative approaches.
The aim of the present study is to develop and evaluate INT2GRATE (INTegrated INTerpretation of GeRmline And Tumor gEnomes), our evidence-based decision tree for assessing germline VUS in MMR genes in patients in which LS-related CRC or EC is suspected. Using stringent criteria, INT2GRATE combines clinical, germline, and tumor-derived data to collect relevant evidence and to inform the potential relevance of germline VUS in LS. The INT2GRATE logic is coded in the back end of a web application with a friendly user interface (UI) to facilitate a convenient collation and comprehensive assessment of germline and tumor-derived evidence. The INT2GRATE UI also enables the sharing of key tumor and germline variant data in the publicly available ClinVar database by the users. Our goal is to promote integrated germline and tumor-derived assessment and foster data sharing, particularly tumor data associated with germline VUS.
Four types of well-established clinical evidence were used in the development of INT2GRATE decision tree: 1) Presence of a single germline variant in an MMR gene; 2) Qualifying clinical criteria for LS per PREMM5 (13) or Amsterdam II (14); 3) Tumor-derived genetic information, including somatic inactivation of MMR loci, BRAF p.V600E status (only for CRC), MLH1 promoter methylation, and microsatellite instability status; and 4) IHC staining of MMR proteins in tumor cells. Tumor-derived evidence is routinely used in the diagnosis of LS (5, 15). The specific logic used by INT2GRATE is detailed in Supplementary Tables 1–5. Additional information on INT2GRATE criteria and the detailed rationale for their inclusion in the decision logic are discussed in the Methods below. A diagrammatic representation of the INT2GRATE variant assessment process is shown in Figure 1.
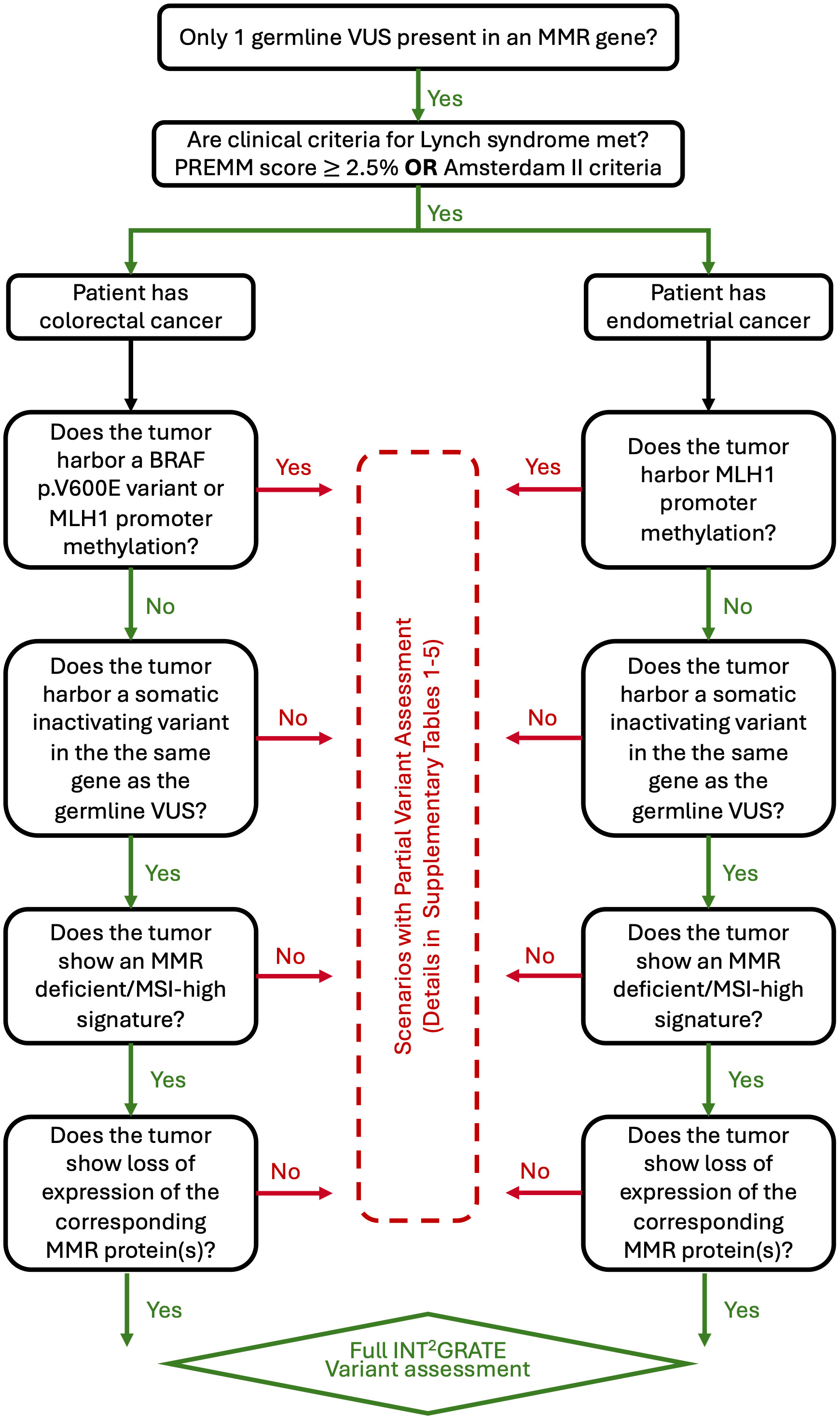
Figure 1 Diagram representing the INT2GRATE process for assessing germline variants of uncertain significance (VUS) in mismatch repair (MMR) genes. The presence of all INT2GRATE evidence enables the full assessment of a given germline MMR variant. The absence of INT2GRATE evidence leads to multiple different scenarios and partial assessment of the variant as described in Supplementary Tables 1–5. MSI denotes microsatellite instability (MSI).
INT2GRATE decision tree requires the presence of all key parameters for a full assessment of germline VUS. After assessing evidence for a given VUS, INT2GRATE renders one of three possible categories accompanied by an explanatory comment. INT2GRATE POSITIVE is designated for scenarios in which the evidence supports a likely relevance of the germline variant to LS. INT2GRATE NEUTRAL category is rendered when all evidence types in the decision tree are present, but the pattern may be unusual, or rare so that a conservative evaluation of VUS cannot be accomplished. Supplementary Tables 2–5 list details of INT2GRATE logics and comments for each gene and scenario. The conservative decision tree logic favors a higher degree of confidence in positive results. INCONCLUSIVE category is rendered when one or more key evidence is absent and INT2GRATE excludes the assessment of VUS (Supplementary Table 1).
This section describes the details of the criteria and the rationale behind the inclusion and exclusion of related evidence in the design of the INT2GRATE decision tree logic.
Pathogenic variants in an MMR gene or a 3’ deletion of the EPCAM gene establish the diagnosis of LS. Therefore, the presence of a VUS in any MMR gene is important information to incorporate into the decision algorithm for the potential role of the germline VUS. A conservative approach was taken in the design of the INT2GRATE logic. The application of INT2GRATE is contingent upon the presence of only one germline VUS in one MMR gene (Supplementary Table 1, INT2GRATE CRC Code I-I and EC Code I-I). Also, the presence of 3’ deletion of the EPCAM is an exclusion criterion for the assessment of germline VUS by INT2GRATE. These conditions exclude the possibility of potentially assessing multiple factors contributing to LS. The presence of possible cryptic rearrangements in MMR genes, not detectable by routine methods, is possible. However, in these cases, the presence of additional complementary evidence from tumor and IHC patterns would help delineate the role of the germline VUS, either by affirming or ruling out its role in LS.
The presence of clinical criteria is a requirement in the design of INT2GRATE (Supplementary Table 1, INT2GRATE CRC Code I-II and EC Code I-II). Of the criteria currently used in clinical practice, the presence of Amsterdam II (14) or PREMM5 (13) criteria was incorporated into the INT2GRATE design. This conservative approach was taken to exclude germline alterations with low penetrance. While VUS alterations with reduced penetrance may well be involved in the development of LS, large-scale data analysis will be required to elucidate their clinical significance. Clinical criteria were also used to evaluate cases in which more than one somatic variant is detected in the same MMR gene. The presence of a strong personal and/or family history with concordant tumor data is highly indicative of LS. Conversely, without such history, sporadic biallelic somatic inactivation is a likely cause of MMR-deficient tumors. Other clinical criteria were not used in the INT2GRATE design, and the rationale is discussed below.
Amsterdam II
The establishment of testing criteria relied on personal and family history of cancer. Amsterdam I criteria (also called 3-2-1) is the most stringent criteria and states that families should have at least three relatives with colorectal cancer (CRC) in two generations with one diagnosis under the age of 50 and one being a first-degree relative of the other two, and familial adenomatous polyposis excluded (16). Amsterdam II criteria were later established to include other Lynch-associated cancers, including endometrial, small bowel, ureter, or renal pelvis (14). Amsterdam II for Lynch requires at least three relatives should have a Lynch syndrome-associated cancer, one of these three relatives should be a first-degree relative of the other two, at least two successive generations should be affected, at least one relative should be diagnosed before age 50 years, and, for cases of colorectal cancer, familial adenomatous polyposis should be excluded. Because of its stringent criteria, Amsterdam II is used in the design of INT2GRATE herein, but only for the assessment of germline VUS in LS-related CRC and endometrial cancer (EC).
PREMM5
In the 2000s, as clinical testing became more readily available, data from tested LS cohorts were incorporated into different risk models. These models included MMRPredict, MMRPro, and PREMM (17–19). All three current versions are listed in the National Comprehensive Cancer Network’s Guidelines on Genetics/Familial Risk Assessment for Colorectal Cancer as viable risk calculation models for LS with a suggestion of risk score ≥ 5% as a threshold for testing. Of these, PREMM5, which has since incorporated data on all five LS genes, defines risk scores of ≥ 2.5% as acceptable scores in patients with colorectal cancer/endometrial cancer and possibly unaffected patients, with the caveat that increasing sensitivity may decrease specificity (NCCN v.1.2022). PREMM5 (13) is well-validated and easy to use on a publicly available website. Given the wide acceptance of PREMM5, this model has been incorporated into INT2GRATE as part of clinical testing criteria.
Other criteria not used in INT2GRATE design
Bethesda guidelines were developed to determine which individuals with colorectal cancer should undergo analysis through microsatellite instability testing (20). Revised guidelines included the presence of colorectal cancer under the age of 50, two or more LS-related tumors regardless of age, colorectal cancer diagnosed in patients with less than 60 years of age with MSI-H histology (tumor-infiltrating lymphocytes, Crohn’s like lymphocytic reaction, mucinous or signet-ring differentiation, or medullary growth pattern), colorectal cancer in a patient with ≥1 first-degree relative with LS-related cancer and one diagnosis before age 50, or colorectal cancer in a patient with >2 first- or second-degree relatives with LS-related cancer regardless of age. Several quantitative methods provide risk calculation for the likelihood of having LS. Early versions of these multi-variable models included the Wijnen model and the Amsterdam plus model (21, 22). Both models used the fulfillment of Amsterdam criteria in addition to two to five other characteristics, including the presence of endometrial cancer in the family. Limitations of these models include relatively small sample populations and lack of incorporation of extra-colonic cancers other than endometrial (23). Therefore, these criteria were not used in the design of the INT2GRATE.
Tumor-derived information is a key element in INT2GRATE design for the assessment of germline VUS. These criteria, currently used in clinical practice, were used in the design of INT2GRATE:
Somatic BRAF status
BRAF variant status is frequently assessed in colorectal cancer given its prognostic and treatment implications in the MMR proficient (MMRp) colorectal cancer (24–27). As a result, BRAF variant testing may be more readily available across a broader range of practice settings than multigene next-generation sequencing (NGS) panels. Similarly, some institutions routinely test for BRAF variants, instead of or in addition to MLH1 promoter methylation, in colorectal cancer with MLH1/PMS2 loss by IHC (28).
Somatic BRAF p.V600E variants are exceedingly rare in LS colorectal cancer, but are present in approximately half of sporadic mismatch repair deficient (MMRd) colorectal cancers (29, 30). The presence of BRAF p.V600E has been widely used in clinical practice to distinguish LS colorectal cancer and sporadic colorectal cancer (NCCN, 2022) (31). In keeping with clinical practice, colorectal tumors bearing BRAF p.V600E are excluded from VUS reassessment with INT2GRATE (Supplementary Table 1, INT2GRATE CRC Code I-III). Furthermore, the significance of BRAF alterations other than the p.V600E in LS is less clear. BRAF variants are rare in endometrial cancer and are not routinely assessed in the MMRd endometrial cancer (32–42). Therefore, BRAF status in endometrial cancer is not a component of INT2GRATE (Supplementary Table 1).
Somatic MLH1 methylation status
Methylation of the MLH1 promoter is the most common cause of MMRd and microsatellite instability (MSI) in both colorectal cancer and endometrial cancer and is routinely evaluated in tumors that show loss of MLH1 and PMS2 by IHC (41, 43, 44). Tumors with MMRd secondary to MLH1 promoter methylation are thought to arise sporadically rather than in the setting of LS, with rare exceptions. Whereas BRAF testing will identify approximately half of sporadic MMRd colorectal cancer cases, MLH1 promoter methylation will definitionally identify sporadic MMRd colorectal cancer. Therefore, negative MLH1 promoter methylation testing is required to proceed with this VUS reassessment approach (Supplementary Table 1, INT2GRATE CRC Code I-IV, and EC Code I-III).
Biallelic inactivation at MMR loci
Somatic inactivation at MMR loci is the expected mechanism for tumorigenesis in LS (45). If the germline variant in an MMR gene is in fact pathogenic, the remaining functional allele is often preferentially lost through an acquired inactivating single nucleotide or copy number alteration. For this reason, the presence of somatic inactivation of the wild-type allele at MMR loci is required evidence in the application of the INT2GRATE (Supplementary Table 1, INT2GRATE CRC Code I-V and EC Code I-IV). Of note, this criterion may limit the assessment of VUS in many scenarios. For instance, the somatic alteration may be cryptic or undetectable because of assay or technical limitations. This could include somatic epigenetic or structural variants not detected by standard assays. It is also worth noting that it is often unclear if the germline and somatic variants are in cis or trans; however, the somatic and germline variants are presumed to be in trans when the data collectively indicates that this is the most likely situation (e.g., a somatic variant in PMS2 is identified in a patient with a germline PMS2 VUS and isolated loss of PMS2 in tumor nuclei). Nevertheless, we have chosen to implement this rule as a required criterion in order to document biallelic inactivation as best as possible with the available findings.
MMR/MSI tumor signature
MMR deficiency manifests functionally as MSI, which can be detected by MSI polymerase chain reaction (PCR) or NGS assays. Documentation of MMRd is a requirement for this variant assessment approach (Supplementary Table 1, INT2GRATE CRC Code I-VI, and EC Code I-V). When possible, this molecular phenotype should be addressed by NGS-based assays, as they allow for the simultaneous estimation of tumor mutational burden, the detection of mutations in the MMR and BRAF genes, and the assessment of copy-number status. Whereas PCR-based assays usually examine long mononucleotide and/or dinucleotide tracts in non-coding regions, NGS-based assays can additionally assess microsatellites in coding regions targeted by the particular assay. At our institution (Brigham and Women’s Hospital, BWH), we assess for single nucleotide indels in mononucleotide tracts with a length of at least 4 nucleotides (46). Lastly, as is the case in our institution, NGS-based assays can be performed on tumor samples without paired normal tissue.
An essential component of INT2GRATE is the presence of supportive IHC data. IHC for MMR proteins is routinely used to screen cases of CRC and EC for MMRd. Loss of IHC expression of at least one MMR protein is observed in most MMRd cases and can indicate biallelic inactivation of one of the MMR proteins in certain scenarios. Therefore, evaluation of the four main MMR proteins by IHC and loss of IHC expression of at least one MMR protein are both required for this approach (Supplementary Table 1, INT2GRATE CRC Code I-VII and EC Code I-VI). These requirements limit the utility of INT2GRATE in assessing VUS that may occur in rare cases exhibiting MSI-H but intact MMR IHC (47, 48) and in cases in which only PMS2 and MSH6 IHC was performed. This conservative approach will allow for a higher degree of confidence in the results of our integrated VUS reassessment. Furthermore, the IHC pattern can support the presence of a defect in a particular MMR gene.
The INT2GRATE UI (available at https://INT2GRATE.bwh.harvard.edu/) was designed to enable an intuitive capturing of user selections, to assess VUS according to logics displayed in Supplementary Tables 1–5, and to facilitate data and knowledge sharing by providing users an option for submitting their entry and INT2GRATE results to ClinVar database. Possible scenarios were implemented in Javascript conditional logic statements that return the result based on the combination of answers selected by the user.
Design of the INT2GRATE digital UI
The digital interface for INT2GRATE is a web-based application that allows the user to assess germline VUS using a collection of relevant somatic and clinical evidence in selected cancer types. From the main page for the website (https://INT2GRATE.bwh.harvard.edu/), the user can select Lynch Syndrome-Related Colorectal Cancer or Lynch Syndrome-Related Endometrial Cancer to access their respective interfaces hosted in separate webpages of the site. Each page presents the algorithm-dependent questions (11 for Colorectal and 10 for Endometrial) via radio button selection to provide single response input. The front-end UI was designed in an HTML form to allow the user to select their entry and receive the appropriate assessment as dictated by the logical combination (Figures 2, 3). A quality control measure was implemented so that the INT2GRATE UI does not process possible entries outside of Supplementary Tables 1–5 of the current algorithm. The algorithm runs on Javascript in the client-side (i.e., user’s) browser and does not interface with a server to perform this logic. User information and entries are not sent to or stored in servers. The website is accessible through any web browser.

Figure 2 The INT2GRATE Web-Based User Interface (UI) for Lynch-Related Colorectal Cancer. INT2GRATE UI enables an intuitive collation of key evidence for the assessment of germline VUS. Upon completion of entries and pressing Submit, the assessment is performed in the backend according to the INT2GRATE logic, and an INT2GRATE category and explanation are displayed. The UI provides users with an option for downloading their entry and the INT2GRATE results and sharing them in the public ClinVar database.

Figure 3 The INT2GRATE Web-Based User Interface (UI) for Lynch-Related Endometrial Cancer. INT2GRATE UI enables an intuitive collation of key evidence for the assessment of germline VUS. Upon completion of entries and pressing Submit, the assessment is performed in the backend according to the INT2GRATE logic, and an INT2GRATE category and explanation are displayed. The UI provides users with an option for downloading their entry and the INT2GRATE results and sharing them in the public ClinVar database.
Display of the INT2GRATE results
After the completion of questions and pressing submit, responses are processed as Javascript variables according to the decision tree logic (Supplementary Tables 1–5) to determine the INT2GRATE categories and relevant comments. The response will conditionally return only one of three possible INT2GRATE categories depending on the combination of responses: a) INT2GRATE POSITIVE, b) INT2GRATE NEUTRAL, and c) INCONCLUSIVE (categories are described in the Assignment of INT2GRATE Categories section). The details of combinations that produce these three categories are outlined in Supplementary Tables 1–5. If the logical combination is not provided within the current algorithm, a response populates at the bottom of the page that indicates the logical combination falls outside of the current parameters of the algorithm.
Facilitating data sharing
After evidence collection and VUS assessment are performed and an INT2GRATE category is returned from the algorithm, the user has the option to download a synopsis of the questions and their responses for INT2GRATE POSITIVE and INT2GRATE NEUTRAL cases. The downloaded file is accessible in a spreadsheet. This file and the information captured are stored locally on the user’s computer; no information is collected by external servers or shared with external sources as a result of algorithm use.
To facilitate submission and data sharing in the ClinVar database, the submission file features several fields prompted by ClinVar for variant submission as cells in the spreadsheet, which can be directly copied into ClinVar’s submission form by the user. This synopsis will capture fields required by ClinVar including the Assertion Method, Assertion Method Citation, Citation URL, and Comment on Clinical Significance, as well as the date of download from INT2GRATE and the version of the algorithm used in the variant’s assessment on that date. The user can include additional information to the evidence summary (e.g., ACMG criteria) before submitting it to ClinVar.
Tagging downloaded files by variants
All fields on the INT2GRATE webpage are required to be filled out by the user for INT2GRATE to assess germline VUS. Additionally, the user is required to enter the specific variant being assessed in a field prompting the cDNA. The variant, gene, and cDNA will be automatically tagged to the file to be downloaded on the user’s computer to help identify different variants assessed by the user. An optional field for the protein change is provided and will be included in the file download if provided by the user.
Two clinic-based cohorts were used to evaluate the performance of INT2GRATE. The first cohort consisted of 5018 patients with MMR single nucleotide variants (SNV) and/or copy number variants (CNV) detected by the somatic OncoPanel next-generation sequencing (NGS) assay from the Center for Advanced Molecular Diagnostic at Brigham and Women’s Hospital between 2018 and 2022. For each case, tumor MSI status, tumor mutational burden, tumor type, and all reported variants in OncoPanel’s 447 genes were collected. Germline testing was performed for 348 of these individuals by Dana Farber Cancer Institute (DFCI) for clinical genetics evaluation. Thirteen of these patients were diagnosed with LS-related CRC or EC and carried a pathogenic variant in an MMR gene.
The second cohort comprised of patients evaluated at DFCI for LS between 2017 and 2022, tested by TumorNext Lynch (Ambry Genetics, CA, USA) for both germline and tumor profiles, and diagnosed with LS-related CRC or EC. Thirty-nine patients met these criteria with a pathogenic germline MMR variant. Germline and somatic genetic analysis and IHC staining were performed as described previously (9, 10, 49) and as outlined in the Supplementary Laboratory Methods.
A Comprehensive Somatic Variant Database (CSVD) was generated using the R programming language to include SNV, CNV, and structural variant data for each case, along with additional tumor-related information including tumor type and MSI and BRAF status. A second database, the Integrated Somatic, and Germline Variant Database, was generated in “R” by merging the germline variant data obtained from DFCI with the CSVD.
MLH1 germline VUS is potentially relevant to LS when each of the following criteria are met: The MLH1 VUS is the only germline alteration in MMR genes; no other germline alteration related to LS is present; clinical criteria (i.e. PREMM score or Amsterdam II) are met; somatic MLH1 promoter methylation and/or BRAF p.V600E are absent; somatic inactivation of the second MLH1 allele is documented; microsatellite instability is identified by PCR or NGS; and a compatible IHC staining pattern is present (Table 1, INT2GRATE POSITIVE CRC Codes II-I to II-IV and EC Codes II-I to II-II). Typically, these cases would show loss of MLH1 and PMS2 in tumor nuclei, as biallelic inactivation of MLH1 would result in loss of MLH1 and its dimerization partner PMS2. Isolated loss of PMS2 in tumor nuclei would also be compatible in these cases, as MLH1 alterations can also lead to retained expression of nonfunctional MLH1 with loss of PMS2 (47, 50). All INT2GRATE POSITIVE scenarios and detailed comments are outlined in Supplementary Table 2.
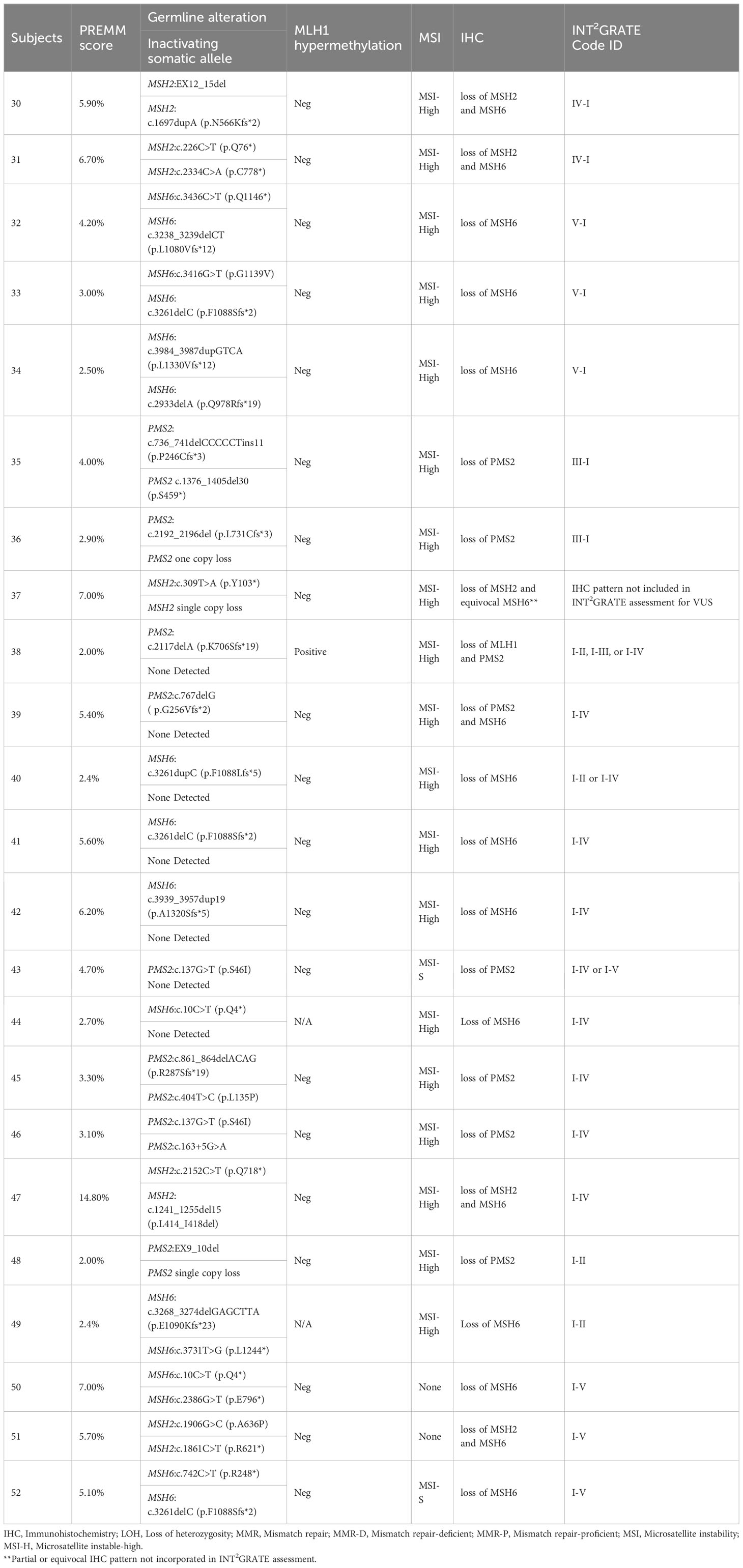
Table 1 Performance assessment of INT2GRATE in LS-related colorectal cancer.
Any unusual pattern of evidence or deviation from the above stringent criteria would render the designation of INT2GRATE NEUTRAL (i.e., the significance of VUS remains uncertain in relation to LS). The details of these scenarios and associated comments are outlined in Supplementary Table 2. For CRC, the significance of VUS remains uncertain if MLH1 promoter methylation and/or BRAF testing are positive, if results for both tests are unavailable, or if IHC shows an unusual staining pattern (i.e., isolated loss of MLH1) (Supplementary Table 2, INT2GRATE CRC Codes II-V to II-VIII). While MLH1 promoter methylation testing is preferred (see sections on BRAF and MLH1 promoter methylation status in Supplementary Methods), a negative BRAF result in the absence of MLH1 promoter methylation testing is acceptable for VUS assessment granted all other conditions are met. Confirmatory MLH1 promoter methylation testing should be considered in this scenario. For EC, the significance of VUS remains uncertain if MLH1 promoter methylation is positive, if results for this test are unavailable, or if IHC shows an unusual staining pattern (Supplementary Table 2, INT2GRATE EC Codes II-1II to II-V). Isolated loss of MLH1 is unusual; this IHC pattern would warrant a review of the IHC findings and potentially even repeat IHC.
PMS2 germline VUS is potentially relevant to LS when each of the following criteria are met: The PMS2 VUS is the only germline alteration in MMR genes; no other germline alteration related to LS is present; clinical criteria (i.e. PREMM score or Amsterdam II) are met; somatic MLH1 promoter methylation and/or BRAF p.V600E are absent; somatic inactivation of the second PMS2 allele is documented; microsatellite instability is identified by PCR or NGS; and loss of only PMS2 in tumor nuclei by IHC is present (Supplementary Table 3, INT2GRATE POSITIVE CRC Codes III-I to III-III, and EC Code III-I). In these cases, isolated loss of PMS2 by IHC would support biallelic inactivation of the PMS2 gene.
The significance of VUS remains uncertain if both MLH1 and PMS2 are lost by IHC, regardless of results for MLH1 promoter methylation and/or BRAF testing, as this pattern would suggest that the MLH1 gene is altered. (INT2GRATE NEUTRAL CRC Codes III-IV to III-VII, and EC Codes III-II to III-III).
MSH2 germline VUS is potentially relevant to LS when each of the following criteria is met: The MSH2 VUS is the only germline alteration in MMR genes; no other germline alteration related to LS is present; clinical criteria (i.e., PREMM score or Amsterdam II) are met; somatic MLH1 promoter methylation and/or BRAF p.V600E are absent; somatic inactivation of the second MSH2 allele is documented; microsatellite instability is identified by PCR or NGS; and IHC shows a compatible staining pattern (Supplementary Table 4, INT2GRATE POSITIVE CRC or EC Codes IV-I to IV-II). Typically, these cases would show loss of MSH2 and MSH6 as a result of biallelic inactivation of MSH2 and degradation of its dimerization partner. Isolated loss of MSH6 in tumor nuclei would also be compatible, as certain MSH2 alterations lead to retained MSH2 expression with loss of MSH6 (47).
The significance of VUS remains uncertain if there is an unusual IHC pattern showing an isolated loss of MSH2 (Supplementary Table 4, INT2GRATE NEUTRAL Code IV-III). In such cases, a review of the IHC findings is recommended, as MSH6 may not be uniformly lost with some IHC assays. If all MMR proteins are lost by IHC, a more detailed examination of the IHC results should be performed, as the loss of MLH1 and PMS2 could be subclonal (Supplementary Table 4, INT2GRATE NEUTRAL Code IV-III); the significance of VUS in these scenarios remains uncertain.
MSH6 germline VUS is potentially relevant to LS when each of the following criteria is met: The MSH6 VUS is the only germline alteration in MMR genes; no other germline alteration related to LS is present; clinical criteria (i.e., PREMM score or Amsterdam II) are met; somatic MLH1 promoter methylation and/or BRAF p.V600E are absent; somatic inactivation of the second MSH6 allele is documented; microsatellite instability is identified by PCR or NGS; and only MSH6 is lost in tumor nuclei by IHC (Supplementary Table 5, INT2GRATE POSITIVE CRC or EC Code V-I). Absent MSH6 on IHC would support biallelic inactivation of the MSH6 gene.
Deviations from these criteria render the designation of INT2GRATE NEUTRAL. Loss of both MSH2 and MSH6 in tumor nuclei by IHC would suggest a likely alteration in MSH2 (Supplementary Table 5, INT2GRATE NEUTRAL Code V-II). Loss of MSH6, MLH1, and PMS2 in tumor nuclei by IHC would suggest a secondary loss of MSH6 (51) (Supplementary Table 5, INT2GRATE NEUTRAL Code V-III). The significance of VUS in these scenarios remains uncertain.
The performance of INT2GRATE was assessed by a qualitative retrospective analysis of data obtained from 5057 patients from two clinic-based cohorts (n1 = 5018, and n2 = 39), including a total of 52 patients with LS-related CRC or EC and known pathogenic MMR variants that served as positive controls.
Among the 52 LS patients with known pathogenic MMR variants, 23 had all evidence or parameters for the evaluation by INT2GRATE. Each of these 23 variants was categorized as INT2GRATE POSITIVE (Tables 1, 2; Supplementary Tables 1, 2). In the positive control cohort, the personal and family history of these cases was strongly supportive of Lynch syndrome (Table 2). Demographics and Clinical Characteristics of Lynch Syndrome Subjects in Positive Control Cohorts are presented in Table 3.
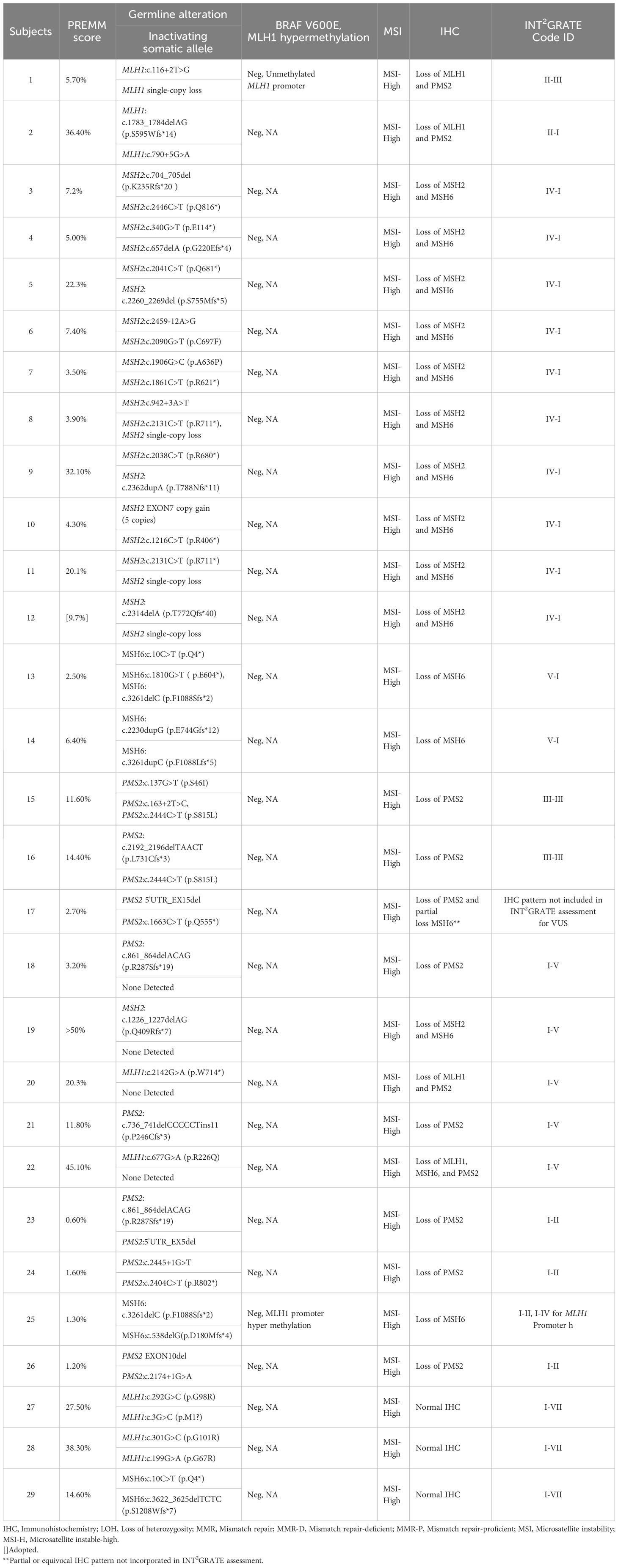
Table 2 Performance assessment of INT2GRATE in LS-related endometrial cancer.
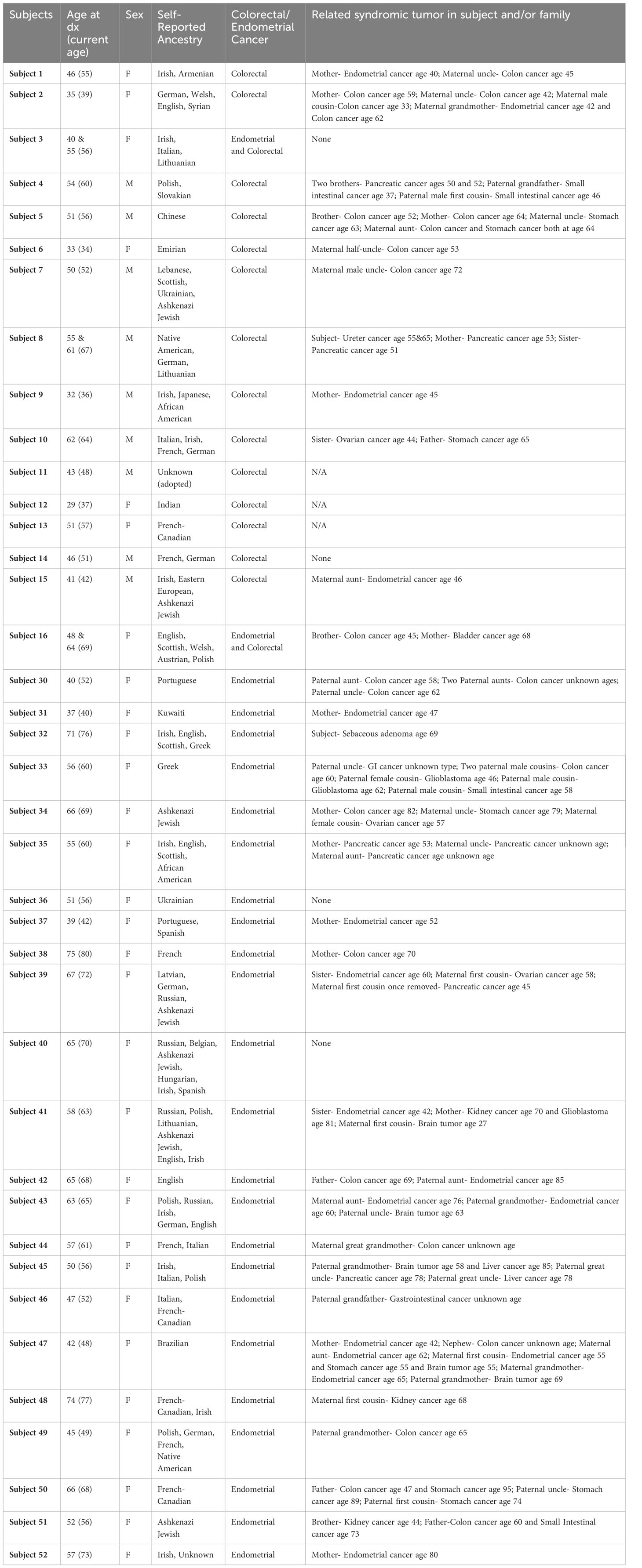
Table 3 Demographics and clinical characteristics of subjects in positive control cohorts.
The remaining 29 of 52 positive controls with known pathogenic MMR variants did not have all evidence in the INT2GRATE decision tree (Tables 1, 2; Supplementary Tables 1, 2). These included 16 variants in the CRC cancer group and 13 in the EC group. The absence of biallelic inactivation was the most frequent observation (n=15), followed by a low PREMM5 score (<2.5%) (n=8), normal, partial, or equivocal IHC (n=4), and unavailable or stable status of the MSI test (n=4) (Tables 1, 2; Supplementary Tables 1, 2). Rare pattern or absence of key evidence was flagged by the INT2GRATE assessment by assigning separate ID codes (Tables 1, 2). This highlights the conservative approach in designing INT2GRATE for the assessment of VUS in favor of a higher degree of confidence in the results.
In the remaining OncoPanel cohort, INT2GRATE logic effectively identified 5005 cases with the absence of ≥ 1 criteria, rendering a classification of INCONCLUSIVE. Overall, 3735 (74%) were MMR proficient/microsatellite stable (MMR-P/MSS) cases. A total of 1339 cases had at least one SNV in the MMR gene, of those 72% (970/1339) showed the absence of biallelic alterations at the MMR locus, and 2% (27/1339) were positive for BRAF p.V600E.
Data sharing is an essential goal of the INT2GRATE program. INT2GRATE’s user-friendly interface enabled the collection and download of all decision tree evidence in the form of questions and answers by the user. A list of evidence, the associated comment for each scenario (as described in Supplementary Tables 1-5), and a summary of responses are automatically generated as a text file for each case (Supplementary Table 6). This downloadable text file facilitates data sharing or submission of the variant’s evidence summary to ClinVar.
The standard germline sequence variant classification system (6) yields a high number of VUS, the largest type of variants reported in clinical practice. Collectively, there are 10,488 germline VUS in MLH1, MSH2, MSH6, and PMS2 in the ClinVar database to date (as of November 2023). The prevalence of VUS is disproportionately higher in underrepresented populations (52–54), partially due to information disparity in genomics, which in turn propagates health inequity in genomics. The economic, clinical, psychosocial, and emotional burden associated with VUS is well documented (55–60). While many tools and strategies aim to assess the functionality or pathogenicity of variants (61–69), none exists that systematically integrates tumor information in the assessment of germline VUS. The absence of joint databases of germline and somatic variant data further limits the utility of integrated information in cancer genetics.
INT2GRATE leverages information from the two genomes that exist in patients with cancer, germline and tumor, to inform the potential role of constitutional VUS in MMR genes. This integrated measure is an objective indication of the status of tumor progression at the molecular and cellular level and is arguably free of confounding genetic host factors and factors influenced by population genetic structure. While the majority of individuals in our study identified as having mostly European backgrounds, sharing INT2GRATE data across broad clinical settings and diverse patient populations will help measure the efficacy and utility of this germline VUS assessment tool in patients from different population backgrounds.
The INT2GRATE decision tree is designed to use well-established clinical criteria, the combination of which favors a conservative approach to assessing VUS. All scenarios that lead to the INT2GRATE POSITIVE category are those that are clinically present when a germline variant is pathogenic. It is noteworthy that INT2GRATE is not intended to re-classify VUS or change the ACMG assessment criteria. Instead, it is designed to serve as a companion tool to help clinical professionals to efficiently collect and assess tumor-derived and clinical genetic evidence for each VUS. Selected clinically reported but uncommon scenarios were also included in the decision tree, but these criteria render the designation of INT2GRATE NEUTRAL (Methods under MMR IHC Pattern and Rationale). The comments explaining the combination of evidence in these scenarios are intended to be useful to those professionals who may not have access to readily available data from large testing centers. Lastly, data sharing is a main goal of INT2GRATE. Currently, there is no platform that enables systematic collection and data sharing of integrated tumor-derived and clinical genetics evidence for VUS. Sharing both tumor and germline variant-level information will help disseminate the clinical evidence related to VUS.
In the assessment of tumor profiles, several conditions were not included in the conservative INT2GRATE decision tree. Among BRAF alterations, INT2GRATE assesses only BRAF p.V600E (not other alterations in BRAF), as it is the most common and well-characterized alteration of BRAF in MMRd CRC (32, 70). Scenarios with the absence of a somatic second hit were excluded from the design. Indeed, we report 15 LS control patients with documented pathogenic germline variants that lacked a somatic inactivating variant in the MMR gene of interest, despite loss of expression of the corresponding MMR proteins by IHC. Given the various reasons for the absence of bi-allelic inactivating of MMR genes [e.g., assay limitations, epigenetic or complex structural alterations, technical issues, insufficient coverage, low tumor purity, or poor mapping quality particularly for PMS2, or age-related LOH contribution typically observed in younger patients (13)], these scenarios were excluded from the INT2GRATE POSITIVE criteria.
The current application of INT2GRATE in LS-associated cancers is limited to CRC and EC. These two are the most common cancers (excluding skin cancer) in LS, accounting for ~39% and ~10% of all cancers diagnosed in a large prospective LS cohort, respectively (71). Most laboratories in the US employ universal MMR/MSI testing for CRC and EC, but not for other LS-associated tumors. As somatic MMR testing is incorporated into oncology practice for all tumors, the algorithm may be expanded to include other LS-associated cancers and potentially even cancers not known to be associated with LS.
The application of the PREMM5 risk model or LS clinical criteria may present an ascertainment bias in selecting VUS that meet the eligibility criteria to be evaluated by INT2GRATE. In our positive control groups, five LS patients with pathogenic variants in PMS2 or MSH6 were excluded from the assessment based on the PREMM5 criterion alone. MLH1 and MSH2 have the highest penetrance for cancer of the MMR genes, approaching cumulative risks of 70-80% for all cancers, whereas penetrance of cancer in males with MSH6 (~40%) and both males and females with PMS2 is much lower (~35%) (71). PREMM5 considers the lower penetrance of PMS2 and MSH6 but is likely affected by some selection bias due to mild phenotype. We, therefore, expect that some VUS in PMS2 and MSH6 may not reach the necessary threshold to be assessed by INT2GRATE, especially in younger individuals without the manifestation of LS. A study of a larger set of MMR variants assessed by INT2GRATE may delineate the utility of INT2GRATE in different MMR genes.
In conclusion, INT2GRATE provides a platform to systematically collect relevant data in LS, to comprehensively assess the role of germline VUS using well-established tumor and clinical evidence, and to share both the collated tumor and germline data in publicly available variant databases. As both tumor and germline testing becomes more widely available in clinical settings, sharing integrated tumor and germline genomic findings is essential for translating genomic testing results into clinical knowledge. Sharing and accessing variant-level information using INT2GRATE will help mitigate the challenges in genomic oncology imposed by VUS.
The datasets presented in this article are not readily available because of ethical and privacy restrictions. Requests to access the datasets should be directed to the corresponding author.
The studies involving humans were approved by Brigham and Women’s Hospital (Protocol #: 2004P000062) and DFCI/BWH Pop Sci: Cancer Genetics (Protocol # 22-495). The studies were conducted in accordance with the local legislation and institutional requirements. The participants provided their written informed consent to participate in this study.
RI: Data curation, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. AC: Data curation, Investigation, Methodology, Validation, Writing – original draft, Writing – review & editing. MW: Data curation, Formal analysis, Investigation, Methodology, Validation, Visualization, Writing – review & editing. AS: Data curation, Investigation, Methodology, Writing – review & editing. DK: Data curation, Investigation, Methodology, Writing – review & editing. CH: Data curation, Formal analysis, Writing – review & editing. BU: Data curation, Formal analysis, Writing – review & editing. MM: Data curation, Formal analysis, Writing – review & editing. RB: Data curation, Writing – review & editing. DM: Data curation, Methodology, Writing – review & editing. LS: Writing – review & editing. MR: Data curation, Writing – original draft, Writing – review & editing. MY: Investigation, Writing – review & editing. HR: Investigation, Writing – review & editing. JG: Investigation, Writing – review & editing. AG: Conceptualization, Data curation, Formal analysis, Investigation, Methodology, Project administration, Resources, Supervision, Validation, Visualization, Writing – original draft, Writing – review & editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1284690/full#supplementary-material
1. Win AK, Jenkins MA, Dowty JG, Antoniou AC, Lee A, Giles GG, et al. Prevalence and penetrance of major genes and polygenes for colorectal cancer. Cancer Epidemiol Biomarkers Prev (2017) 26:404–12. doi: 10.1158/1055-9965.EPI-16-0693
2. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology (2010) 138:2073–2087.e3. doi: 10.1053/j.gastro.2009.12.064
3. Rumilla K, Schowalter KV, Lindor NM, Thomas BC, Mensink KA, Gallinger S, et al. Frequency of deletions of EPCAM (TACSTD1) in MSH2-associated Lynch syndrome cases. J Mol Diagn (2011) 13:93–9. doi: 10.1016/j.jmoldx.2010.11.011
4. Kempers MJ, Kuiper RP, Ockeloen CW, Chappuis PO, Hutter P, Rahner N, et al. Risk of colorectal and endometrial cancers in EPCAM deletion-positive Lynch syndrome: a cohort study. Lancet Oncol (2011) 12:49–55. doi: 10.1016/S1470-2045(10)70265-5
5. Weiss JM, Gupta S, Burke CA, Axell L, Chen LM, Chung DC, et al. NCCN guidelines(R) insights: genetic/familial high-risk assessment: colorectal, version 1.2021. J Natl Compr Canc Netw (2021) 19:1122–32. doi: 10.1164/jnccn.2021.0048
6. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med (2015) 17:405–24. doi: 10.1038/gim.2015.30
7. Schwartz A, Manning DK, Koeller DR, Chittenden A, Isidro RA, Hayes CP, et al. An integrated somatic and germline approach to aid interpretation of germline variants of uncertain significance in cancer susceptibility genes. Front Oncol (2022) 12:942741. doi: 10.3389/fonc.2022.942741
8. Koeller DR, Manning DK, Schwartz A, Chittenden A, Hayes CP, Abraamyan F, et al. An optimized protocol for evaluating pathogenicity of VHL germline variants in patients suspected with von Hippel-Lindau syndrome: Using somatic genome to inform the role of germline variants. MethodsX (2022) 9:101761. doi: 10.1016/j.mex.2022.101761
9. Manning DK, Shivdasani P, Koeller DR, Schwartz A, Rana HQ, Garber JE, et al. Assessment of genomic alterations in non-syndromic von Hippel-Lindau: Insight from integrating somatic and germline next generation sequencing genomic data. Data Brief (2021) 39:107653. doi: 10.1016/j.dib.2021.107653
10. Rana HQ, Koeller DR, Schwartz A, Manning DK, Schneider KA, Krajewski KM, et al. Pathogenicity of VHL variants in families with non-syndromic von Hippel-Lindau phenotypes: An integrated evaluation of germline and somatic genomic results. Eur J Med Genet (2021) 64:104359. doi: 10.1016/j.ejmg.2021.104359
11. Ghazani AA, Oliver NM, St Pierre JP, Garofalo A, Rainville IR, Hiller E, et al. Assigning clinical meaning to somatic and germ-line whole-exome sequencing data in a prospective cancer precision medicine study. Genet Med (2017) 19:787–95. doi: 10.1038/gim.2016.191
12. Koeller DR, Schwartz A, Manning DK, Dong F, Lindeman NI, Garber JE, et al. Novel pathogenic germline variant of the adenomatous polyposis coli (APC) gene, p.S2627Gfs*12 identified in a mild phenotype of APC-associated polyposis: A case report. Am J Case Rep (2020) 21:e927293. doi: 10.12659/AJCR.927293
13. Kastrinos F, Uno H, Ukaegbu C, Alvero C, McFarland A, Yurgelun MB, et al. Development and validation of the PREMM(5) model for comprehensive risk assessment of lynch syndrome. J Clin Oncol (2017) 35:2165–72. doi: 10.1200/JCO.2016.69.6120
14. Vasen HF, Watson P, Mecklin JP, Lynch HT. New clinical criteria for hereditary nonpolyposis colorectal cancer (HNPCC, Lynch syndrome) proposed by the International Collaborative group on HNPCC. Gastroenterology (1999) 116:1453–6. doi: 10.1016/S0016-5085(99)70510-X
15. Pearlman R, Haraldsdottir S, de la Chapelle A, Jonasson JG, Liyanarachchi S, Frankel WL, et al. Clinical characteristics of patients with colorectal cancer with double somatic mismatch repair mutations compared with Lynch syndrome. J Med Genet (2019) 56:462–70. doi: 10.1136/jmedgenet-2018-105698
16. Vasen HF, Mecklin JP, Khan PM, Lynch HT. The international collaborative group on hereditary non-polyposis colorectal cancer (ICG-HNPCC). Dis Colon Rectum (1991) 34:424–5. doi: 10.1007/BF02053699
17. Barnetson RA, Tenesa A, Farrington SM, Nicholl ID, Cetnarskyj R, Porteous ME, et al. Identification and survival of carriers of mutations in DNA mismatch-repair genes in colon cancer. N Engl J Med (2006) 354:2751–63. doi: 10.1056/NEJMoa053493
18. Chen S, Wang W, Lee S, Nafa K, Lee J, Romans K, et al. Prediction of germline mutations and cancer risk in the Lynch syndrome. JAMA (2006) 296:1479–87. doi: 10.1001/jama.296.12.1479
19. Balmana J, Stockwell DH, Steyerberg EW, Stoffel EM, Deffenbaugh AM, Reid JE, et al. Prediction of MLH1 and MSH2 mutations in Lynch syndrome. JAMA (2006) 296:1469–78. doi: 10.1001/jama.296.12.1469
20. Rodriguez-Bigas MA, Vasen HF, Pekka-Mecklin J, Myrhoj T, Rozen P, Bertario L, et al. Rectal cancer risk in hereditary nonpolyposis colorectal cancer after abdominal colectomy. International Collaborative Group on HNPCC. Ann Surg (1997) 225:202–7. doi: 10.1097/00000658-199702000-00008
21. Wijnen JT, Vasen HF, Khan PM, Zwinderman AH, van der Klift H, Mulder A, et al. Clinical findings with implications for genetic testing in families with clustering of colorectal cancer. N Engl J Med (1998) 339:511–8. doi: 10.1056/NEJM199808203390804
22. Lipton LR, Johnson V, Cummings C, Fisher S, Risby P, Eftekhar Sadat AT, et al. Refining the Amsterdam Criteria and Bethesda Guidelines: testing algorithms for the prediction of mismatch repair mutation status in the familial cancer clinic. J Clin Oncol (2004) 22:4934–43. doi: 10.1200/JCO.2004.11.084
23. Kastrinos F, Steyerberg EW, Balmana J, Mercado R, Gallinger S, Haile R, et al. Comparison of the clinical prediction model PREMM(1,2,6) and molecular testing for the systematic identification of Lynch syndrome in colorectal cancer. Gut (2013) 62:272–9. doi: 10.1136/gutjnl-2011-301265
24. Van Cutsem E, Kohne CH, Lang I, Folprecht G, Nowacki MP, Cascinu S, et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J Clin Oncol (2011) 29:2011–9. doi: 10.1200/JCO.2010.33.5091
25. Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol (2008) 26:5705–12. doi: 10.1200/JCO.2008.18.0786
26. Pietrantonio F, Petrelli F, Coinu A, Di Bartolomeo M, Borgonovo K, Maggi C, et al. Predictive role of BRAF mutations in patients with advanced colorectal cancer receiving cetuximab and panitumumab: a meta-analysis. Eur J Cancer (2015) 51:587–94. doi: 10.1016/j.ejca.2015.01.054
27. Chen D, Huang JF, Liu K, Zhang LQ, Yang Z, Chuai ZR, et al. BRAFV600E mutation and its association with clinicopathological features of colorectal cancer: a systematic review and meta-analysis. PloS One (2014) 9:e90607. doi: 10.1371/journal.pone.0090607
28. Jin M, Hampel H, Zhou X, Schunemann L, Yearsley M, Frankel WL. BRAF V600E mutation analysis simplifies the testing algorithm for Lynch syndrome. Am J Clin Pathol (2013) 140:177–83. doi: 10.1309/AJCPB9FOVH1HGKFR
29. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature (2012) 487:330–7. doi: 10.1038/nature11252
30. Liu Y, Sethi NS, Hinoue T, Schneider BG, Cherniack AD, Sanchez-Vega F, et al. Comparative molecular analysis of gastrointestinal adenocarcinomas. Cancer Cell (2018) 33:721–735.e8. doi: 10.1016/j.ccell.2018.03.010
31. Molinari F, Signoroni S, Lampis A, Bertan C, Perrone F, Sala P, et al. BRAF mutation analysis is a valid tool to implement in Lynch syndrome diagnosis in patients classified according to the Bethesda guidelines. Tumori (2014) 100:315–20. doi: 10.1700/1578.17214
32. Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell (2018) 173:291–304.e6. doi: 10.1016/j.cell.2018.03.022
33. Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, et al. Scalable open science approach for mutation calling of tumor exomes using multiple genomic pipelines. Cell Syst (2018) 6:271–281.e7. doi: 10.1016/j.cels.2018.03.002
34. Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, et al. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell (2018) 33:676–689.e3. doi: 10.1016/j.ccell.2018.03.007
35. Gao Q, Liang WW, Foltz SM, Mutharasu G, Jayasinghe RG, Cao S, et al. Driver fusions and their implications in the development and treatment of human cancers. Cell Rep (2018) 23:227–238.e3. doi: 10.1016/j.celrep.2018.03.050
36. Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell (2018) 173:400–416.e11. doi: 10.1016/j.cell.2018.02.052
37. Sanchez-Vega F, Mina M, Armenia J, Chatila WK, Luna A, La KC, et al. Oncogenic signaling pathways in the cancer genome atlas. Cell (2018) 173:321–337.e10. doi: 10.1016/j.cell.2018.03.035
38. Bhandari V, Hoey C, Liu LY, Lalonde E, Ray J, Livingstone J, et al. Molecular landmarks of tumor hypoxia across cancer types. Nat Genet (2019) 51:308–18. doi: 10.1038/s41588-018-0318-2
39. Poore GD, Kopylova E, Zhu Q, Carpenter C, Fraraccio S, Wandro S, et al. Microbiome analyses of blood and tissues suggest cancer diagnostic approach. Nature (2020) 579:567–74. doi: 10.1038/s41586-020-2095-1
40. Ding L, Bailey MH, Porta-Pardo E, Thorsson V, Colaprico A, Bertrand D, et al. Perspective on oncogenic processes at the end of the beginning of cancer genomics. Cell (2018) 173:305–320.e10. doi: 10.1016/j.cell.2018.03.033
41. Metcalf AM, Spurdle AB. Endometrial tumour BRAF mutations and MLH1 promoter methylation as predictors of germline mismatch repair gene mutation status: a literature review. Fam Cancer (2014) 13:1–12. doi: 10.1007/s10689-013-9671-6
42. Bonneville R, Krook MA, Kautto EA, Miya J, Wing MR, Chen HZ, et al. Landscape of microsatellite instability across 39 cancer types. JCO Precis Oncol (2017) 1. doi: 10.1200/PO.17.00073
43. Chen W, Swanson BJ, Frankel WL. Molecular genetics of microsatellite-unstable colorectal cancer for pathologists. Diagn Pathol (2017) 12:24. doi: 10.1186/s13000-017-0613-8
44. Evans DG, Lalloo F, Ryan NA, Bowers N, Green K, Woodward ER, et al. Advances in genetic technologies result in improved diagnosis of mismatch repair deficiency in colorectal and endometrial cancers. J Med Genet (2022) 59:328–34. doi: 10.1136/jmedgenet-2020-107542
45. Carethers JM. Hereditary, sporadic and metastatic colorectal cancer are commonly driven by specific spectrums of defective DNA mismatch repair components. Trans Am Clin Climatol Assoc (2016) 127:81–97.
46. Papke DJ Jr., Nowak JA, Yurgelun MB, Frieden A, Srivastava A, Lindeman NI, et al. Validation of a targeted next-generation sequencing approach to detect mismatch repair deficiency in colorectal adenocarcinoma. Mod Pathol (2018) 31:1882–90. doi: 10.1038/s41379-018-0091-x
47. Hechtman JF, Rana S, Middha S, Stadler ZK, Latham A, Benayed R, et al. Retained mismatch repair protein expression occurs in approximately 6% of microsatellite instability-high cancers and is associated with missense mutations in mismatch repair genes. Mod Pathol (2020) 33:871–9. doi: 10.1038/s41379-019-0414-6
48. Bartley AN, Luthra R, Saraiya DS, Urbauer DL, Broaddus RR. Identification of cancer patients with Lynch syndrome: clinically significant discordances and problems in tissue-based mismatch repair testing. Cancer Prev Res (Phila) (2012) 5:320–7. doi: 10.1158/1940-6207.CAPR-11-0288
49. Abo RP, Ducar M, Garcia EP, Thorner AR, Rojas-Rudilla V, Lin L, et al. BreaKmer: detection of structural variation in targeted massively parallel sequencing data using kmers. Nucleic Acids Res (2015) 43:e19. doi: 10.1093/nar/gku1211
50. de Jong AE, van Puijenbroek M, Hendriks Y, Tops C, Wijnen J, Ausems MG, et al. Microsatellite instability, immunohistochemistry, and additional PMS2 staining in suspected hereditary nonpolyposis colorectal cancer. Clin Cancer Res (2004) 10:972–80. doi: 10.1158/1078-0432.ccr-0956-3
51. Shia J, Zhang L, Shike M, Guo M, Stadler Z, Xiong X, et al. Secondary mutation in a coding mononucleotide tract in MSH6 causes loss of immunoexpression of MSH6 in colorectal carcinomas with MLH1/PMS2 deficiency. Mod Pathol (2013) 26:131–8. doi: 10.1038/modpathol.2012.138
52. Ndugga-Kabuye MK, Issaka RB. Inequities in multi-gene hereditary cancer testing: lower diagnostic yield and higher VUS rate in individuals who identify as Hispanic, African or Asian and Pacific Islander as compared to European. Fam Cancer (2019) 18:465–9. doi: 10.1007/s10689-019-00144-6
53. Ricker C, Culver JO, Lowstuter K, Sturgeon D, Sturgeon JD, Chanock CR, et al. Increased yield of actionable mutations using multi-gene panels to assess hereditary cancer susceptibility in an ethnically diverse clinical cohort. Cancer Genet (2016) 209:130–7. doi: 10.1016/j.cancergen.2015.12.013
54. Caswell-Jin JL, Gupta T, Hall E, Petrovchich IM, Mills MA, Kingham KE, et al. Racial/ethnic differences in multiple-gene sequencing results for hereditary cancer risk. Genet Med (2018) 20:234–9. doi: 10.1038/gim.2017.96
55. Vos J, Otten W, van Asperen C, Jansen A, Menko F, Tibben A. The counsellees' view of an unclassified variant in BRCA1/2: recall, interpretation, and impact on life. Psychooncology (2008) 17:822–30. doi: 10.1002/pon.1311
56. Richter S, Haroun I, Graham TC, Eisen A, Kiss A, Warner E. Variants of unknown significance in BRCA testing: impact on risk perception, worry, prevention and counseling. Ann Oncol 24 Suppl 8 (2013) 24(Suppl 8):viii69–74. doi: 10.1093/annonc/mdt312
57. Hoffman-Andrews L. The known unknown: the challenges of genetic variants of uncertain significance in clinical practice. J Law Biosci (2017) 4:648–57. doi: 10.1093/jlb/lsx038
58. O'Neill SC, Rini C, Goldsmith RE, Valdimarsdottir H, Cohen LH, Schwartz MD. Distress among women receiving uninformative BRCA1/2 results: 12-month outcomes. Psychooncology (2009) 18:1088–96. doi: 10.1002/pon.1467
59. Li B, Krishnan VG, Mort ME, Xin F, Kamati KK, Cooper DN, et al. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics (2009) 25:2744–50. doi: 10.1093/bioinformatics/btp528
60. Solomon I, Harrington E, Hooker G, Erby L, Axilbund J, Hampel H, et al. Lynch syndrome limbo: patient understanding of variants of uncertain significance. J Genet Couns (2017) 26:866–77. doi: 10.1007/s10897-017-0066-y
61. Garcia FAO, de Andrade ES, Palmero EI. Insights on variant analysis in silico tools for pathogenicity prediction. Front Genet (2022) 13:1010327. doi: 10.3389/fgene.2022.1010327
62. Srinivasan P, Bandlamudi C, Jonsson P, Kemel Y, Chavan SS, Richards AL, et al. The context-specific role of germline pathogenicity in tumorigenesis. Nat Genet (2021) 53:1577–85. doi: 10.1038/s41588-021-00949-1
63. Ramroop JR, Gerber MM, Toland AE. Germline variants impact somatic events during tumorigenesis. Trends Genet (2019) 35:515–26. doi: 10.1016/j.tig.2019.04.005
64. Chanock SJ. How the germline informs the somatic landscape. Nat Genet (2021) 53:1523–5. doi: 10.1038/s41588-021-00960-6
65. Kanchi KL, Johnson KJ, Lu C, McLellan MD, Leiserson MD, Wendl MC, et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nat Commun (2014) 5:3156. doi: 10.1038/ncomms4156
66. Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res (2003) 31:3812–4. doi: 10.1093/nar/gkg509
67. Feng BJ. PERCH: A unified framework for disease gene prioritization. Hum Mutat (2017) 38:243–51. doi: 10.1002/humu.23158
68. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet Chapter 7 (2013) 76:7.20.1–7.20.41. doi: 10.1002/0471142905.hg0720s76
69. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res (2011) 39:e118. doi: 10.1093/nar/gkr407
70. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal (2013) 6:l1. doi: 10.1126/scisignal.2004088
71. Dominguez-Valentin M, Sampson JR, Seppala TT, Ten Broeke SW, Plazzer JP, Nakken S, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the Prospective Lynch Syndrome Database. Genet Med (2020) 22:15–25. doi: 10.1038/s41436-019-0596-9
Keywords: somatic and germline integration, INT2GRATE, tumor signature profile, germline VUS, Lynch syndrome
Citation: Isidro RA, Chittenden A, Walker M, Schwartz A, Koeller DR, Hayes CP, Unal B, Manam MD, Buehler RM, Manning DK, Sholl LM, Redston MS, Yurgelun MB, Rana HQ, Garber JE and Ghazani AA (2024) Development and evaluation of INT2GRATE: a platform for comprehensive assessment of the role of germline variants informed by tumor signature profile in Lynch syndrome. Front. Oncol. 13:1284690. doi: 10.3389/fonc.2023.1284690
Received: 28 August 2023; Accepted: 21 December 2023;
Published: 24 January 2024.
Edited by:
Shulan Tian, Mayo Clinic, United StatesCopyright © 2024 Isidro, Chittenden, Walker, Schwartz, Koeller, Hayes, Unal, Manam, Buehler, Manning, Sholl, Redston, Yurgelun, Rana, Garber and Ghazani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Arezou A. Ghazani, Z2hhemFuaUBmYXMuaGFydmFyZC5lZHU=; YWdoYXphbmlAYndoLmhhcnZhcmQuZWR1
†Present address: Raymond A. Isidro, Department of Pathology, Memorial Sloan Kettering Cancer Center, New York, NY, United StatesConnor P. Hayes, Center for Stem Cell & Regeneration, Baylor College of Medicine, Houston, TX, United StatesMonica Devi Manam, Division of Genetics and Genomics, Boston Children’s Hospital, Boston, MA, United States
‡These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.