 Lindor Qunaj
Lindor Qunaj Michael S. May1†
Michael S. May1† Alfred I. Neugut
Alfred I. Neugut
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol., 15 September 2023
Sec. Gastrointestinal Cancers: Colorectal Cancer
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1252516
This article is part of the Research TopicEmerging Mutations in Colorectal Cancer Development and ProgressionView all 13 articles
KRAS G12C mutations are critical in the pathogenesis of multiple cancer types, including non-small cell lung (NSCLC), pancreatic ductal adenocarcinoma (PDAC), and colorectal (CRC) cancers. As such, they have increasingly become a target of novel therapies in the management of these malignancies. However, the therapeutic success of KRAS G12C inhibitors to date has been far more limited in CRC and PDAC than NSCLC. In this review, we briefly summarize the biochemistry of KRAS targeting and treatment resistance, highlight differences in the epidemiology of various G12C-mutated cancers, and provide an overview of the published data on KRAS G12C inhibitors for various indications. We conclude with a summary of ongoing clinical trials in G12C-mutant CRC and a discussion of future directions in the management of this disease. KRAS G12C mutation, targeted therapies, colorectal cancer, non-small cell lung cancer, pancreatic cancer, drug development.
Alterations in the membrane-associated kinase (MAPK) pathway are common in various cancers. In particular, mutations in the Kirsten rat sarcoma virus gene (KRAS)—first identified as a viral oncogene in Kirsten Rat sarcoma before the discovery of the human homolog—lead to constitutive activation of its associated protein (KRAS), contributing to uncontrolled cellular proliferation and survival. KRAS mutations are among the most common activating mutations found in tumors, frequently present in non-small cell lung cancer (NSCLC), pancreatic ductal adenocarcinoma (PDAC), endometrial carcinoma, and colorectal carcinoma (CRC).
KRAS mutational status in CRC is an integral part of treatment decision-making. However, direct targeting of mutant KRAS has proven exceedingly difficult. The recent development of small molecules which directly target the G12C allele of KRAS, has been the first successful attempt at targeting KRAS in human cancer. The KRAS G12C inhibitors sotorasib and adagrasib have demonstrated efficacy as monotherapy in KRAS G12C-mutant NSCLC, where roughly 15% of patients have this mutation. The G12C allele is much less common in CRC, but there is an increasing body of evidence that KRAS G12C inhibitors can be effective in CRC alone or in combination with other therapies. The rapid pace of clinical drug development in this area has already led to important clinical gains which are likely to grow.
This paper reviews the literature on KRAS G12C mutations, with a focus on the epidemiology and prognosis of KRAS G12C within CRC and the barriers that remain to the incorporation of KRAS G12C inhibitors into clinical practice in CRC.
The endogenous activity of the MAPK pathway begins with the binding of an extracellular mitogen to a receptor tyrosine kinase (RTK). This leads to a cascade of events which ultimately triggers transcriptional changes that result in increased cellular proliferation and survival. Common mitogen-cell surface receptor pairs which initiate this pathway include epidermal growth factor (EGF) binding to EGF receptor (EGFR) and fibroblast growth factor (FGF) binding to FGF receptor (FGFR). Binding of mitogen to RTK triggers activation of tyrosine kinase activity of the RTK cytoplasmic domain. Docking proteins then bind to phosphotyrosine residues on the activated receptor. These cell-surface signals are transduced through KRAS. KRAS exists in the cellular context in two forms, an “off” state bound to guanosine diphosphate (GDP) and an “on” state bound to guanosine triphosphate (GTP). The protein contains intrinsic GTPase activity to turn itself off, and a variety of associated proteins can shift the balance between GDP-bound (GAPs, or GTPase activating proteins) and GTP-bound (GEFs, or guanine exchange factors) KRAS. In the presence of activating signals, GEFs replace GDP with GTP, allowing KRAS to turn “on.” A downstream kinase cascade, involving serial phosphorylation of downstream kinases, such as RAF, ERK and MEK, ultimately triggers transcription factor activation, leading to increased cellular proliferation, migration and survival (1–3). In the normal cellular context, the MAPK pathway is regulated by intrinsic feedback loops and negative regulators, such as Sprouty, RAS-association domain family (RASSF) proteins, and Son of Sevenless homolog 1 (SOS-1) which help to prevent aberrant activation and signaling (4).
There are extensive repositories documenting tumor mutational frequency in KRAS and other genes, including the Catalogue of Somatic Mutations in Cancer (COSMIC), The Cancer Genome Atlas (TCGA), and The International Cancer Genome Consortium (ICGC) (5). While the specific frequencies reported varies by source, activating mutations in KRAS are present in approximately 25-30% of NSCLC (6, 7), 80-95% of pancreatic ductal adenocarcinoma (PDAC) (8, 9) and 40-50% of CRC (9, 10) (Figure 1). The most common KRAS activating mutations cluster around the nucleotide-binding pocket, and involve codons 12, 13 and 61. These mutations alter the homeostatic balance between active, GTP bound KRAS and inactive, GDP-bound KRAS, either by reducing GTP hydrolysis or by increasing the rate of GTP loading. This leads to constitutive activation of the MAPK pathway (10).
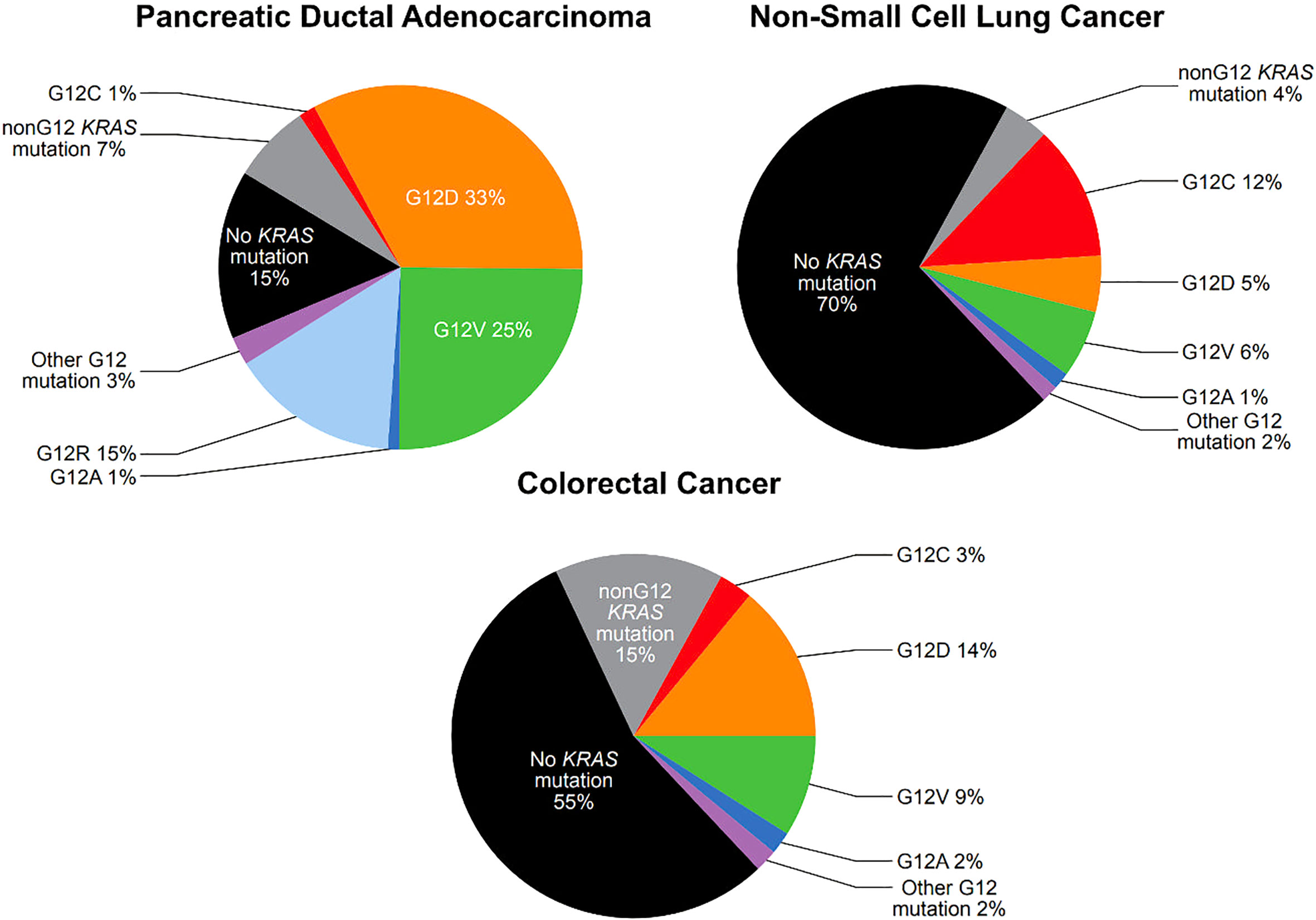
Figure 1 Prevalence of KRAS G12 mutations in pancreatic ductal adenocarcinoma, non-small cell lung cancer, and colorectal cancer (see text for citations).
Among activating KRAS mutations, KRAS G12 mutations are particularly common. KRAS G12 mutations are present in 25% of NSCLC, 78% of PDAC, and 30% of CRC (Figure 1). KRAS G12 mutations are missense mutations which result in the substitution of the amino acid glycine for another amino acid at codon 12, stabilizing KRAS in the active, GTP-bound state, though with slightly different features for each mutation (11). In KRAS G12C mutations, cysteine is substituted for glycine (12). KRAS G12C mutations are present in approximately 12% of NSCLC, 1.5% of PDAC, and 3% of CRC (Figure 1) (7, 9, 10).
KRAS G12C mutations are present in approximately 3% of CRC cases with relatively little variation in mutation rates across geographic areas. Patient cohorts of 4,632 at MD Anderson Cancer Center in Houston, TX; 2,457 in Japan; 4,897 in Brazil; and 2,559 from a Nordic cohort with CRC who underwent genomic profiling, demonstrated KRAS G12C mutational rates of 2.9%, 2.8%, 3.4%, and 3%, respectively (13–15). Within the Nordic cohort, the frequency of KRAS G12C mutations reported at eight sites in Norway, Sweden, Finland and Denmark varied from 2%-4% (15).
KRAS G12C-mutant CRC appears to occur approximately equally in men and women, and has a 60% left-sided predisposition, compared with approximately 43% in unselected CRC in the US (13, 14, 16, 17). Although some studies have reported a difference in stage at presentation and metastatic sites in KRAS G12C-mutated CRC, the preponderance of data does not support a consistent significant difference (13–15, 18, 19).
Although there is some variability in studies, most large retrospective studies have demonstrated an association between KRAS G12C mutations and worse progression-free survival (PFS) and overall survival (OS) when compared with other KRAS-mutated CRC (13, 14, 19, 20). KRAS G12V mutations may also convey a poor prognosis (19, 20). One study, which included 187 patients with KRAS G12C mutations and de novo metastatic disease, demonstrated a median PFS of 6.4 months, 3.9 months, and 3.0 months after first, second and third lines of therapy, with an OS of 25.1 months (14).
The most common mutations in CRC occur in APC, TP53, KRAS and PIK3CA (21, 22). Rates of APC and TP53 mutations do not vary consistently based on the presence or type of KRAS mutation (14, 19). PIK3CA mutations are less common in patients with KRAS G12C mutations than in patients with KRAS mutations at alternative sites (14, 19). Mutations in other genes involved in the MAPK pathway, such as BRAF and NRAS, are much less likely to be mutated in CRC cases with KRAS mutations including G12C mutations (14, 17, 19). This is consistent with molecular data from other cancers, such as NSCLC, in which KRAS mutations are almost always exclusive of other strong driver mutations, such as EGFR (23, 24).
Alterations in the microsatellite instability (MSI) pathway are of particular clinical interest as they facilitate sensitivity to immunotherapy (25, 26). MSI is caused by a deficiency of mismatch repair (dMMR) and is found in approximately 10-15% of CRC. Rates of dMMR are very low in KRAS G12C-mutant CRC (1.6% vs 2.9% in other KRAS mutants and 4.5% in all cases) (19).
KRAS G12C mutated CRC displays lower rates of CIMP pathway activation (hypermethylation) compared with KRAS WT CRC (14). Overall, these data point to KRAS G12C CRC as being a biologically distinct class of CRC with different therapeutic opportunities.
Given the high frequency of KRAS mutations across multiple tumor types, including approximately 45% of CRC, mutant KRAS has been a pharmacologic target for over three decades (27). Direct KRAS targeting has been extremely challenging, as its affinity for GTP and mostly featureless surface precluded traditional approaches of small-molecule medicinal chemistry (28). This led to a variety of secondary approaches to RAS targeting, either by attempting to target other pathway “nodes” or the translation and processing of KRAS on its way to the cell surface. The enzyme farnesyltransferase, for example, catalyzes the addition of a farnesyl lipid group to a carboxyl terminal cysteine of KRAS, enabling localization to the cell membrane (29). Farnesyltransferase inhibitors were consequently an early class of pan-RAS inhibitors which, despite many preclinical successes, failed to show success in the treatment of KRAS-mutant cancers, possibly due to alternative prenylation of KRAS (30, 31).
In the context of repeated failures to directly target KRAS, it is notable that other inhibitors of the MAPK pathway have proven central in defining treatment paradigms for CRC. The EGFR inhibitors cetuximab and panitumumab proved effective in RAS WT CRC (32, 33) and the BRAF inhibitor encorafenib demonstrated efficacy in the treatment of BRAF V600E mutant CRC (34). Confining EGFR inhibitors, which act “upstream” of KRAS, to only KRAS WT disease makes conceptual sense, but limited the usefulness of these drugs.
The first success in directly targeting KRAS grew out of the unique biochemistry of the codon 12 glycine-to-cysteine (G12C) mutation. Cysteine thiols are uniquely nucleophilic amongst amino acid side chains. This is classically seen in formation of disulfide bonds and made the cysteine residue in mutant KRAS G12C an attractive pharmacologic target (12). This property enabled disulfide-fragment-based techniques to screen hundreds of potential small-molecule targets of G12C-mutant KRAS in a mutant-specific manner, sparing WT KRAS. Building on the fact that G12C was targetable, two clinical compounds have now obtained approval: sotorasib and adagrasib—with a half-dozen or more molecules close behind.
Sotorasib was the first KRAS G12C inhibitor approved in the United States. It demonstrated efficacy and acceptable toxicity in patients with previously treated KRAS G12C mutant advanced NSCLC. In CodeBreaK100, a single-arm phase 2 trial of 126 patients, Sotorasib demonstrated an objective response rate (ORR) of 37% with a median duration of response of 11.1 months. PFS was 6.8 months and OS was 12.5 months (35). A subsequent randomized controlled trial of 345 patients demonstrated superior PFS with sotorasib than with docetaxel (5.6 vs 4.5 months; HR 0.66 with p=0.0017) and fewer grade 3 or worse adverse events (AEs) (36). Common treatment-related AEs seen with sotorasib include diarrhea, nausea, vomiting, fatigue and liver function test elevation (35, 36).
Adagrasib was evaluated in KRYSTAL-1, a phase 2 study of 116 patients with previously treated KRAS G12C mutant NSCLC. Adagrasib demonstrated similar outcomes to sotorasib in this cohort, with an ORR of 43%, median duration of response of 8.5 months, disease control rate of 80%, PFS of 6.5 months and OS of 12.6 months. Common AEs were similar (37). These two proof-of-principle trials for direct KRAS inhibition were landmarks that led to the exploration of new KRAS-targeted drugs and KRAS G12C strategies in other histologies.
Sotorasib was evaluated in a phase 1-2 trial of 38 patients with previously treated metastatic KRAS G12C-mutant PDAC, with a median of two prior lines of prior therapy. The ORR was 21%; PFS was 4.0 months and OS was 6.9 months (38)
These promising but time-limited responses have prompted further investigation into the mechanisms of KRAS G12C inhibitor resistance, and drug combinations which could help increase response rates or prolong the duration of response (39).
Early clinical studies of KRAS-targeted therapies in G12C-mutant colorectal cancer showed less activity than what is reported in NSCLC. A single-arm study of sotorasib monotherapy in previously treated KRAS G12C-mutant CRC, for instance, demonstrated an overall response rate of 9.7% across 62 evaluable patients (40). More recently, adagrasib alone was tested in a cohort of 43 CRC patients in the KRYSTAL-1 trial. The investigator-assessed objective response rate was 19%, with a median PFS of 5.6 months (4.1-8.3, 95%CI) and median OS of 19.8 months (12.5-23.0, 95%CI) (41). Safety data was comparable to the results seen in NSCLC, with the most common adverse events being diarrhea, nausea, vomiting, and fatigue (all occurring in >40% of patients).
The mechanisms underlying these discordant responses to G12C targeting in CRC and NSCLC are not clearly understood. Why do a smaller percentage of patients respond? One hypothesis is that CRC tumors undergo a significant rebound in ERK phosphorylation soon after starting KRAS inhibition, suggesting rapid development of treatment-related adaptive signaling resistance (42). KRAS-mutant CRC has also been shown to have higher levels of “upstream” receptor tyrosine kinase phosphorylation as compared to NSCLC, especially in EGFR. EGFR-mediated resistance to other targeted therapies has been seen in CRC, including with BRAF inhibition, making this a natural place to look (43).
This observation has prompted studies combining small molecule KRAS inhibitors with anti-EGFR monoclonal antibodies in CRC. CodeBreaK-101, for example, included a cohort of 40 metastatic CRC patients treated with a combination of sotorasib and panitumumab, noting an ORR of 30% (16.6-46.5, 95%CI) (44). Adagrasib was similarly evaluated with or without concurrent cetuximab in 28 heavily pretreated G12C-mutant CRC patients. Without cetuximab, ORR was 19%; with cetuximab, the ORR was 46%. Notably, the standard paradigm in CRC has been that KRAS-mutant tumors do not have responses to anti-EGFR mAb therapy. Similar results have been presented for compounds earlier in development, including GDC-6036, a G12C inhibitor being developed by Genentech, which showed an even higher ORR of 66% when combined with cetuximab (45). Overall these results suggest that this combination may receive approval concurrently, or before, monotherapy treatment for KRAS G12C CRC. Still, despite the improved responses relative to KRAS inhibition alone, these results suggest a similar discrepancy in efficacy between CRC and NSCLC patients receiving identical combinations. These trials are summarized in Table 1.
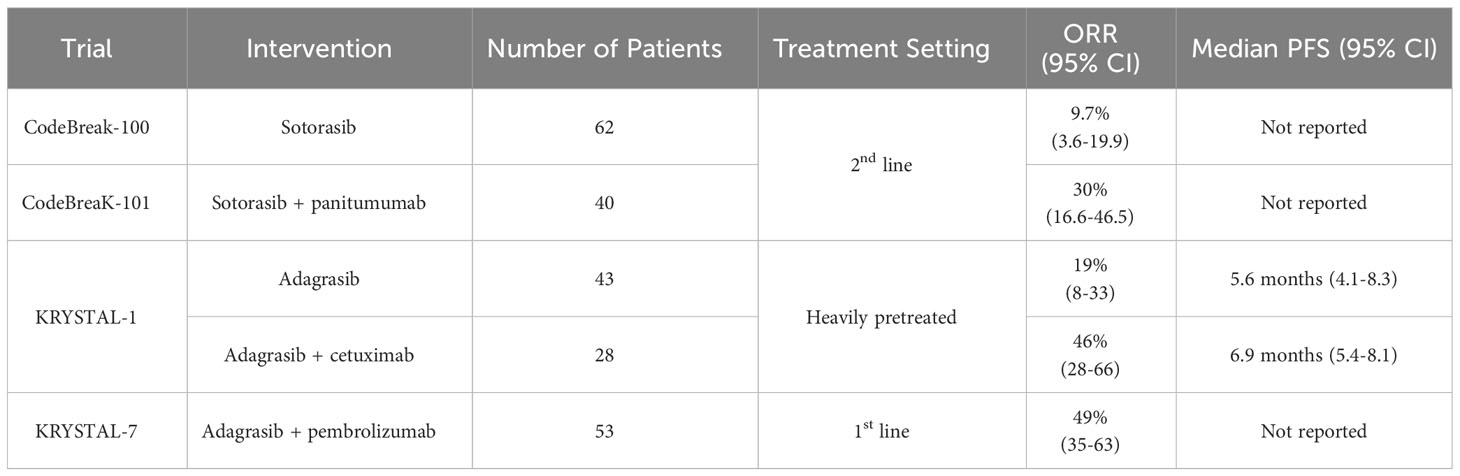
Table 1 Summary of key results from KRAS G12C inhibitor trials in CRC.
These somewhat blunted responses in CRC have not deterred continued development of new G12C inhibitors, either as monotherapy or in novel combinations for this indication. Using a search term of “G12C” for registered clinical trials yielded 76 unique entries (last updated April 27, 2023), of which 39 were trials that included patients with G12C-mutant CRC. Thirty-three studies were excluded from further review because they included only lung cancer patients, two were excluded because they were diagnostic/non-interventional studies, and one each was excluded for not being specific to G12C-mutant tumors or having KRAS mutations as an exclusion criterion. Within these exclusions, there are 12 distinct G12C inhibitors currently under investigation (Table 2).
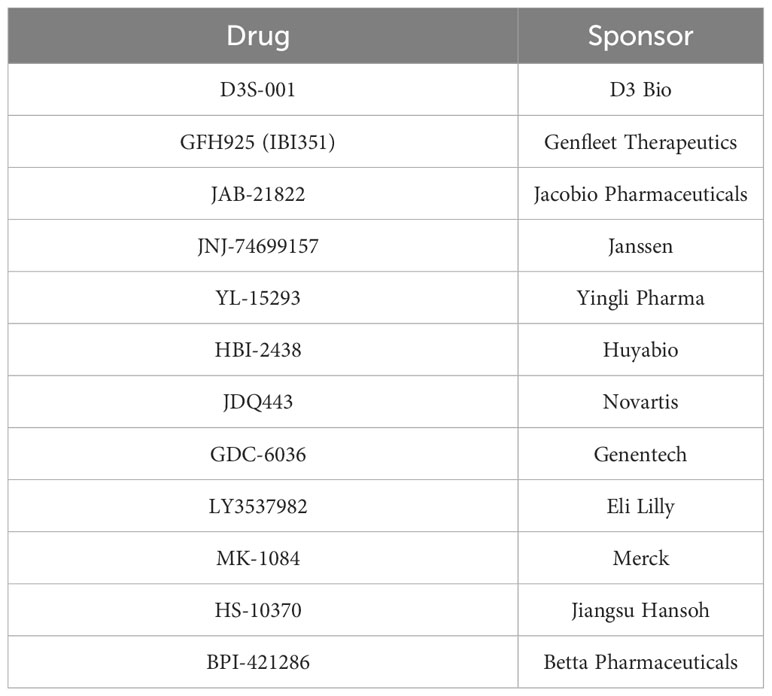
Table 2 KRAS G12C inhibitors currently in clinical development based on registered trials publicly listed on ClinicalTrials.gov.
Essentially all are KRAS-GDP covalent inhibitors. Subtle differences in pharmacology and pharmacokinetics between these compounds might translate into efficacy benefits or differing potential in combination therapy regimens. Novartis’ JDQ443, for example, binds without involving the H95 residue (46). As a result of its H95-independent mechanism, it appears to maintain activity even among tumors with G12C/H95 double KRAS mutations, which might help mitigate some acquired resistance (47). Such second-site mutations in H95 appear to occasionally mediate resistance to other KRAS G12C inhibitors, such as adagrasib (48).
Janssen’s JNJ-74699157 similarly binds near the switch II pocket through an interaction with a differing cysteine residue, suggesting it may be able to evade these resistance mechanisms as well. In a phase 1 study of 10 patients across multiple tumor types, however, zero patients had an objective response and six experienced dose-limiting muscle toxicity in the form of CPK elevations (49). In contrast, the KontRASt-01 trial of JDQ443 is ongoing, with no alarming safety signals publicly reported to date (50).
Most recently, Eli Lilly’s LY3537982 presented a preliminary read-out of its phase 1 LOXO-RAS-20001 study at the 2023 AACR Annual Meeting (51). The ORR specifically among patients with CRC (n=20) receiving LY3537982 as monotherapy was 10%, compared to 42% among those with pancreatic (n=12) or 38% in NSCLC not previously treated with a KRAS G12C inhibitor (n=8). In one cohort of 11 CRC patients, the sponsors investigated the novel agent in combination with cetuximab, demonstrating an ORR of 45%. Liver enzyme elevations were more common in this cohort. This trial was notable for including several patients who had stopped prior G12C inhibitors for toxicity. Whether that toxicity would have abated on its own or LY3537982 has a more tolerable safety profile remains to be determined in larger studies.
Apart from these investigations into novel monotherapies, there is considerable interest in combining KRAS G12C inhibitors with other targets as a way to either potentiate their clinical effect or counteract the development of resistance. While the addition of anti-EGFR antibodies was one of the earliest studied combinations, concurrently targeting BRAF, MEK, and other downstream steps in the RAS/MAPK signaling pathway holds theoretical promise as well (52). In vitro and mouse models of lung cancer, for instance, have shown that adding the mTOR inhibitor everolimus to a KRAS G12C inhibitor reduces tumor cell viability (53). Dozens of other preclinical investigations have supported combination therapies.
One prominent combination is with checkpoint inhibitors, based on the hypothesis that KRAS inhibition induces pro-inflammatory changes in the tumor microenvironment of KRAS mutated tumors. Early mouse studies combining sotorasib and anti-PD-1 therapy illustrated increased CD8+ T-cell infiltration into the tumor microenvironment, an effect not seen in subjects treated with checkpoint inhibition alone (54). This finding correlated with improved in vivo efficacy, with 9/10 mice achieving complete responses, compared to 1/10 in the monotherapy arms. Such responses in preclinical studies are typically not seen with chemotherapy-immunotherapy combinations.
In an attempt to replicate that success in humans, the KRYSTAL-7 trial tested the combination of adagrasib and pembrolizumab as a first-line therapy in patients with advanced KRAS G12C-mutant NSCLC. The preliminary results presented at the 2022 ESMO Immuno-Oncology Annual Congress featured an objective response rate of 49% (35-63, 95%CI) across 53 patients, with rates at least directionally varying by PD-L1 tumor proportion score (TPS): 59% for TPS≤50, 48% for TPS 1-49, and 30% for TPS<1 (55). Sotorasib has similarly reported results for a combination arm with pembrolizumab, but this combination led to a notably high rate of liver toxicity. The main current question is whether this liver toxicity will be a class effect or if it will be specific only to some KRAS G12C inhibitors. Trials are likely to answer this question in the coming months and years.
Several ongoing trials include combinations of existing RAS inhibitors with SHP-2 inhibitors, including Novartis’s TNO 155, BridgeBio’s BBP-398, and Revolution Medicine’s RMC-4630. A combination of BBP-398 and sotorasib had a synergistic inhibitory effect on MAPK signaling and cell viability in an in vitro model and was granted fast-track designation (56). SHP-2 is a non-receptor type protein tyrosine phosphatase encoded by the gene PTPN11. Because SHP-2 acts downstream of most receptor tyrosine kinases and is essential for RAS activation and signaling rebound, there is a preclinical rationale to support combination therapy. Monotherapy trials of SHP-2 inhibition have shown modest efficacy in KRAS-mutated tumors, especially KRAS G12C (57), but most compounds are now in development in combination with other MAP/ERK pathway inhibitors, such as the KRAS G12C inhibitors. Sotorasib combinations have started to read out (58), and further data will clarify whether this combination has value in CRC or other cancers.
There are novel molecules targeting other companion proteins of RAS for which combination therapy hypotheses are natural. One is BI-1701963, a pan-RAS inhibitor targeting the catalytic site of SOS-1. SOS-1 is a GEF that binds to GDP-bound RAS proteins and is critical for tumorigenesis; in fact, the absence of SOS-1 has been to shown to have a deleterious effect on cancer cells containing KRAS mutations (59, 60). In a phase 1 dose-escalation study of 31 patients treated with BI-1701963 monotherapy, none achieved clinical responses and only seven had at least stable disease (61).
KRAS is one of the most common and most difficult to target. The advances which led to direct KRAS G12C inhibition combined new pharmacology, chemistry, and thirty years of basic biology investigating RAS biochemistry, structural biology, and signaling adaption. The short eight years from the first disclosure of a covalent KRAS G12C binder to the demonstration of clinical efficacy and obtaining accelerated FDA approval in NSCLC is remarkable. While somewhat behind the NSCLC development, it is likely that KRAS G12C inhibitors will also receive FDA approval in CRC in the coming years.
But the efficacy for KRAS G12C inhibitors observed so far in CRC is its own evolving story that differs from other tumor types. Monotherapy response rates remain low, and it is plausible that combination therapies, such as with EGFR mAbs, will be the first approved regimens. More broadly, the duration of response for most of these molecules is relatively short.
In addition to combinations, next-generation molecules such as KRAS-GTP inhibitors may hold particular promise to increase response rates and perhaps tame some signaling-based resistance mechanisms (62). Direct pan-RAS or pan-KRAS inhibitors, which inhibit RAS regardless of mutated allele (and include on-target WT inhibition), have also been described, with lots of preclinical data and early hints of clinical efficacy (63, 64). It is conceivable that both primary and acquired resistance emergence, and resistance patterns— along with clinical efficacy—will be different for these molecules. It is also possible that toxicity will limit dosing in a clinically meaningful way, but data has yet to be shown.
There remains a great need for KRAS-directed therapy in CRC for both G12C and other alleles. With proof-of-concept activity now firmly in hand, it is likely that these therapies will become a significant part of standard CRC therapy for patients with qualifying molecular alterations in the not-distant future. The hope is that advances in patient survival, reduced progression, and perhaps even cure rates in the adjuvant setting will follow. For now, we are very much at the beginning of this frontier, rather than the end.
AN and BH conceived the study and initiated the report. LQ and MM reviewed the literature and prepared the tables and figures and did the review. All four authors contributed portions of the initial draft and all four authors reviewed and edited the manuscript and all the authors approved the final manuscript. All authors contributed to the article and approved the submitted version.
The authors acknowledge the support of NCI grant P30 CA013696. MM was supported by funding from the National Cancer Institute (R38 CA231577). LQ and MM were supported in part by a training grant from the U.S. National Cancer Institute (T32 CA203703).
AN has consulted for Ostuka, GlaxoSmithKline, United Biosource Corp., Value Analytics, Organon, and Merck. He has research funding from Otsuka and is a member of the medical advisory board of EHE Intl. BH reports consulting fees/honoraria from Amgen, Eisai, Astellas, and Boxer Capital. He has research funding from Amgen, Astellas, Ideaya, Repare Therapeutics, Monte Rosa Therapeutics, Revolution Medicines, and Roche/Genentech.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be constructed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
1. Prior IA, Lewis PD, Mattos C. A comprehensive survey of Ras mutations in cancer. Cancer Res (2012) 72(10):2457–67. doi: 10.1158/0008-5472.CAN-11-2612
2. Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer (2011) 11(11):761–74. doi: 10.1038/nrc3106
3. Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell (2014) 25(3):272–81. doi: 10.1016/j.ccr.2014.02.017
4. Casci T, Vinos J, Freeman M. Sprouty, an intracellular inhibitor of Ras signaling. Cell (1999) 96(5):655–65. doi: 10.1016/s0092-8674(00)80576-0
5. Prior IA, Hood FE, Hartley JL. The frequency of ras mutations in cancer. Cancer Res (2020) 80(14):2969–74. doi: 10.1158/0008-5472.CAN-19-3682
6. Ghimessy A, Radeczky P, Laszlo V, Hegedus B, Renyi-Vamos F, Fillinger J, et al. Current therapy of KRAS-mutant lung cancer. Cancer Metastasis Rev (2020) 39(4):1159–77. doi: 10.1007/s10555-020-09903-9
7. Judd J, Abdel Karim N, Khan H, Naqash AR, Baca Y, Xiu J, et al. Characterization of KRAS mutation subtypes in non-small cell lung cancer. Mol Cancer Ther (2021) 20(12):2577–84. doi: 10.1158/1535-7163.MCT-21-0201
8. Bailey P, Chang DK, Nones K, Johns AL, Patch AM, Gingras MC, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature (2016) 531(7592):47–52. doi: 10.1038/nature16965
9. Luo J. KRAS mutation in pancreatic cancer. Semin Oncol (2021) 48(1):10–8. doi: 10.1053/j.seminoncol.2021.02.003
10. Serebriiskii IG, Connelly C, Frampton G, Newberg J, Cooke M, Miller V, et al. Comprehensive characterization of RAS mutations in colon and rectal cancers in old and young patients. Nat Commun (2019) 10(1):3722. doi: 10.1038/s41467-019-11530-0
11. Johnson C, Burkhart DL, Haigis KM. Classification of KRAS-activating mutations and the implications for therapeutic intervention. Cancer Discov (2022) 12(4):913–23. doi: 10.1158/2159-8290.CD-22-0035
12. Ostrem JM, Peters U, Sos ML, Wells JA, Shokat KM. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature (2013) 503(7477):548–51. doi: 10.1038/nature12796
13. Chida K, Kotani D, Masuishi T, Kawakami T, Kawamoto Y, Kato K, et al. The prognostic impact of KRAS G12C mutation in patients with metastatic colorectal cancer: A multicenter retrospective observational study. Oncologist (2021) 26(10):845–53. doi: 10.1002/onco.13870
14. Henry JT, Coker O, Chowdhury S, Shen JP, Morris VK, Dasari A, et al. Comprehensive clinical and molecular characterization of KRAS (G12C)-mutant colorectal cancer. JCO Precis Oncol (2021) 5. doi: 10.1200/PO.20.00256
15. Osterlund E, Ristimaki A, Kytola S, Kuopio T, Heerva E, Muhonen T, et al. KRAS-G12C mutation in one real-life and three population-based nordic cohorts of metastatic colorectal cancer. Front Oncol (2022) 12:826073. doi: 10.3389/fonc.2022.826073
16. Meguid RA, Slidell MB, Wolfgang CL, Chang DC, Ahuja N. Is there a difference in survival between right- versus left-sided colon cancers? Ann Surg Oncol (2008) 15(9):2388–94. doi: 10.1245/s10434-008-0015-y
17. Araujo LH, Souza BM, Leite LR, Parma SAF, Lopes NP, Malta FSV, et al. Molecular profile of KRAS G12C-mutant colorectal and non-small-cell lung cancer. BMC Cancer (2021) 21(1):193. doi: 10.1186/s12885-021-07884-8
18. Schirripa M, Nappo F, Cremolini C, Salvatore L, Rossini D, Bensi M, et al. KRAS G12C metastatic colorectal cancer: specific features of a new emerging target population. Clin Colorectal Cancer (2020) 19(3):219–25. doi: 10.1016/j.clcc.2020.04.009
19. Fakih M, Tu H, Hsu H, Aggarwal S, Chan E, Rehn M, et al. Real-world study of characteristics and treatment outcomes among patients with KRAS p.G12C-mutated or other KRAS mutated metastatic colorectal cancer. Oncologist (2022) 27(8):663–74. doi: 10.1093/oncolo/oyac077
20. Hayama T, Hashiguchi Y, Okamoto K, Okada Y, Ono K, Shimada R, et al. G12V and G12C mutations in the gene KRAS are associated with a poorer prognosis in primary colorectal cancer. Int J Colorectal Dis (2019) 34(8):1491–6. doi: 10.1007/s00384-019-03344-9
21. Nguyen HT, Duong HQ. The molecular characteristics of colorectal cancer: Implications for diagnosis and therapy. Oncol Lett (2018) 16(1):9–18. doi: 10.3892/ol.2018.8679
22. Pino MS, Chung DC. The chromosomal instability pathway in colon cancer. Gastroenterology (2010) 138(6):2059–72. doi: 10.1053/j.gastro.2009.12.065
23. Cancer Genome Atlas Research N. Comprehensive molecular profiling of lung adenocarcinoma. Nature (2014) 511(7511):543–50. doi: 10.1038/nature13385
24. Unni AM, Lockwood WW, Zejnullahu K, Lee-Lin SQ, Varmus H. Evidence that synthetic lethality underlies the mutual exclusivity of oncogenic KRAS and EGFR mutations in lung adenocarcinoma. Elife (2015) 4:e06907. doi: 10.7554/eLife.06907
25. Andre T, Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in microsatellite-instability-high advanced colorectal cancer. N Engl J Med (2020) 383(23):2207–18. doi: 10.1056/NEJMoa2017699
26. Diaz LA Jr., Shiu KK, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab versus chemotherapy for microsatellite instability-high or mismatch repair-deficient metastatic colorectal cancer (KEYNOTE-177): final analysis of a randomised, open-label, phase 3 study. Lancet Oncol (2022) 23(5):659–70. doi: 10.1016/S1470-2045(22)00197-8
27. Sharma S, Saltz LB. Oral chemotherapeutic agents for colorectal cancer. Oncologist (2000) 5(2):99–107. doi: 10.1634/theoncologist.5-2-99
28. Hofmann MH, Gerlach D, Misale S, Petronczki M, Kraut N. Expanding the reach of precision oncology by drugging all KRAS mutants. Cancer Discov (2022) 12(4):924–37. doi: 10.1158/2159-8290.CD-21-1331
29. Sebti SM, Adjei AA. Farnesyltransferase inhibitors. Semin Oncol (2004) 31(1 Suppl 1):28–39. doi: 10.1053/j.seminoncol.2003.12.012
30. Sharma S, Kemeny N, Kelsen DP, Ilson D, O'Reilly E, Zaknoen S, et al. A phase II trial of farnesyl protein transferase inhibitor SCH 66336, given by twice-daily oral administration, in patients with metastatic colorectal cancer refractory to 5-fluorouracil and irinotecan. Ann Oncol (2002) 13(7):1067–71. doi: 10.1093/annonc/mdf173
31. Rao S, Cunningham D, de Gramont A, Scheithauer W, Smakal M, Humblet Y, et al. Phase III double-blind placebo-controlled study of farnesyl transferase inhibitor R115777 in patients with refractory advanced colorectal cancer. J Clin Oncol (2004) 22(19):3950–7. doi: 10.1200/JCO.2004.10.037
32. Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M, et al. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer (2012) 48(10):1466–75. doi: 10.1016/j.ejca.2012.02.057
33. Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, et al. Final results from PRIME: randomized phase III study of panitumumab with FOLFOX4 for first-line treatment of metastatic colorectal cancer. Ann Oncol (2014) 25(7):1346–55. doi: 10.1093/annonc/mdu141
34. Tabernero J, Grothey A, Van Cutsem E, Yaeger R, Wasan H, Yoshino T, et al. Encorafenib plus cetuximab as a new standard of care for previously treated BRAF V600E-mutant metastatic colorectal cancer: updated survival results and subgroup analyses from the BEACON study. J Clin Oncol (2021) 39(4):273–84. doi: 10.1200/JCO.20.02088
35. Skoulidis F, Li BT, Dy GK, Price TJ, Falchook GS, Wolf J, et al. Sotorasib for lung cancers with KRAS p.G12C mutation. N Engl J Med (2021) 384(25):2371–81. doi: 10.1056/NEJMoa2103695
36. de Langen AJ, Johnson ML, Mazieres J, Dingemans AC, Mountzios G, Pless M, et al. Sotorasib versus docetaxel for previously treated non-small-cell lung cancer with KRAS(G12C) mutation: a randomised, open-label, phase 3 trial. Lancet (2023) 401(10378):733–46. doi: 10.1016/S0140-6736(23)00221-0
37. Janne PA, Riely GJ, Gadgeel SM, Heist RS, Ou SI, Pacheco JM, et al. Adagrasib in non-small-cell lung cancer harboring a KRAS(G12C) mutation. N Engl J Med (2022) 387(2):120–31. doi: 10.1056/NEJMoa2204619
38. Strickler JH, Satake H, George TJ, Yaeger R, Hollebecque A, Garrido-Laguna I, et al. Sotorasib in KRAS p.G12C-mutated advanced pancreatic cancer. N Engl J Med (2023) 388(1):33–43. doi: 10.1056/NEJMoa2208470
39. Dunnett-Kane V, Nicola P, Blackhall F, Lindsay C. Mechanisms of resistance to KRAS(G12C) inhibitors. Cancers (Basel) (2021) 13(1). doi: 10.3390/cancers13010151
40. Fakih MG, Kopetz S, Kuboki Y, Kim TW, Munster PN, Krauss JC, et al. Sotorasib for previously treated colorectal cancers with KRAS(G12C) mutation (CodeBreaK100): a prespecified analysis of a single-arm, phase 2 trial. Lancet Oncol (2022) 23(1):115–24. doi: 10.1016/S1470-2045(21)00605-7
41. Yaeger R, Weiss J, Pelster MS, Spira AI, Barve M, Ou SI, et al. Adagrasib with or without cetuximab in colorectal cancer with mutated KRAS G12C. N Engl J Med (2023) 388(1):44–54. doi: 10.1056/NEJMoa2212419
42. Amodio V, Yaeger R, Arcella P, Cancelliere C, Lamba S, Lorenzato A, et al. EGFR blockade reverts resistance to KRAS(G12C) inhibition in colorectal cancer. Cancer Discov (2020) 10(8):1129–39. doi: 10.1158/2159-8290.CD-20-0187
43. Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D, et al. Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature (2012) 483(7387):100–3. doi: 10.1038/nature10868
44. Kuboki Y, Yaeger R, Fakih M, Strickler J, Masuishi T, Kim E, et al. 315O sotorasib in combination with panitumumab in refractory KRAS G12C-mutated colorectal cancer: Safety and efficacy for phase ib full expansion cohort. Ann Oncol (2022) 33:S680–S1.
45. Desai J, Han S-W, Lee J-S, Shacham-Shmueli E, Massarelli E, Cervantes A, et al. Abstract CT029: Phase Ib study of GDC-6036 in combination with cetuximab in patients with colorectal cancer (CRC) with KRAS G12C mutation. Cancer Res (2023) 83(8_Supplement):CT029–CT.
46. Lorthiois E, Gerspacher M, Beyer KS, Vaupel A, Leblanc C, Stringer R, et al. JDQ443, a structurally novel, pyrazole-based, covalent inhibitor of KRAS(G12C) for the treatment of solid tumors. J Med Chem (2022) 65(24):16173–203. doi: 10.1021/acs.jmedchem.2c01438
47. Weiss A, Lorthiois E, Barys L, Beyer KS, Bomio-Confaglia C, Burks H, et al. Discovery, preclinical characterization, and early clinical activity of JDQ443, a structurally novel, potent, and selective covalent oral inhibitor of KRASG12C. Cancer Discov (2022) 12(6):1500–17. doi: 10.1158/2159-8290.CD-22-0158
48. Awad MM, Liu S, Rybkin II, Arbour KC, Dilly J, Zhu VW, et al. Acquired resistance to KRAS(G12C) inhibition in cancer. N Engl J Med (2021) 384(25):2382–93. doi: 10.1056/NEJMoa2105281
49. Wang J, Martin-ROmano P, Cassier P, Johnson M, Haura E, Lenox L, et al. Phase I study of JNJ-74699157 in patients with advanced solid tumors harboring the KRAS G12C mutation. Oncologist (2022) 27(7):536–e53. doi: 10.1093/oncolo/oyab080
50. Schuler M, Tan DS, Shimizu T, Solomon B, Heist RS, de Miguel M, et al. KontRASt-01: A Phase Ib/II Dose Escalation Study of JDQ443 in Patients with advanced KRAS G12C mutated solid Tumors. Oncol Res Treat (2022) 45(Suppl 2):29.
51. Murciano-Goroff YR, Heist RS, Kuboki Y, Koyama T, Ammakkanavar NR, Hollebecque A, et al. Abstract CT028: A first-in-human phase 1 study of LY3537982, a highly selective and potent KRAS G12C inhibitor in patients with KRAS G12C-mutant advanced solid tumors. Cancer Res (2023) 83(8_Supplement):CT028–CT.
52. Ji J, Wang C, Fakih M. Targeting KRAS (G12C)-mutated advanced colorectal cancer: research and clinical developments. Onco Targets Ther (2022) 15:747–56. doi: 10.2147/OTT.S340392
53. Molina-Arcas M, Moore C, Rana S, van Maldegem F, Mugarza E, Romero-Clavijo P, et al. Development of combination therapies to maximize the impact of KRAS-G12C inhibitors in lung cancer. Sci Transl Med (2019) 11(510). doi: 10.1126/scitranslmed.aaw7999
54. Canon J, Rex K, Saiki AY, Mohr C, Cooke K, Bagal D, et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature (2019) 575(7781):217–23. doi: 10.1038/s41586-019-1694-1
55. Jänne P, Smit E, de Marinis F, Laskin J, Gomez MD, Gadgeel S, et al. LBA4 Preliminary safety and efficacy of adagrasib with pembrolizumab in treatment-naïve patients with advanced non-small cell lung cancer (NSCLC) harboring a KRASG12C mutation. Immuno-Oncol Technol (2022) 16.
56. Stice J, Donovan S, Sun Y, Kohl N, Czako B, Mseeh F, et al. (2021). BBP-398 a potent, small molecule inhibitor of SHP2, enhances the response of established NSCLC xenografts to KRAS G12C and EGFR mut inhibitors. In: AACR-NCI-EORTC Virtual International Conference on Molecular Targets and Cancer Therapeutics.
57. Koczywas M, Haura E, Janne PA, Pacheco JM, Ulahannan S, Wang JS, et al. Abstract LB001: Anti-tumor activity and tolerability of the SHP2 inhibitor RMC-4630 as a single agent in patients with RAS-addicted solid cancers. Cancer Res (2021) 81(13_Supplement):LB001–LB. doi: 10.1158/1538-7445.AM2021-LB001
58. Falchook G, MK LB, Bestvina C, Langer C, Krauss J, Strickler J, et al. (2022). Sotorasib in combination with RMC-4630, a SHP2 inhibitor. KRAS p G12C-mutated NSCLC and other solid tumors paper. In: World Conference on Lung Cancer 2022.
59. Hofmann MH, Gmachl M, Ramharter J, Savarese F, Gerlach D, Marszalek JR, et al. BI-3406, a potent and selective SOS1-KRAS interaction inhibitor, is effective in KRAS-driven cancers through combined MEK inhibition. Cancer Discov (2021) 11(1):142–57. doi: 10.1158/2159-8290.CD-20-0142
60. Jeng HH, Taylor LJ, Bar-Sagi D. Sos-mediated cross-activation of wild-type Ras by oncogenic Ras is essential for tumorigenesis. Nat Commun (2012) 3:1168. doi: 10.1038/ncomms2173
61. Johnson M, Gort E, Pant S, Lolkema M, Sebastian M, Scheffler M, et al. 524P A phase I, open-label, dose-escalation trial of BI 1701963 in patients (pts) with KRAS mutated solid tumours: a snapshot analysis. Ann Oncol (2021) 32:S591–S2. doi: 10.1016/j.annonc.2021.08.1046
62. Nichols RJ, Yang Y, Cregg J, Schulze CJ, Wang Z, Dua R, et al. RMC-6291, a next-generation tri-complex KRASG12C (ON) inhibitor, outperforms KRASG12C (OFF) inhibitors in preclinical models of KRASG12C cancers. Cancer Res (2022) 82(12_Supplement):3595–. doi: 10.1158/1538-7445.AM2022-3595
63. Wasko UN, Sastra SA, Palermo CF, Wildes D, Singh M, Olive KP. Abstract B071: Preclinical evaluation of RM-042, an orally bioavailable inhibitor of GTP-RAS, in models of pancreatic ductal adenocarcinoma. Cancer Res (2022) 82(22_Supplement):B071–B. doi: 10.1158/1538-7445.PANCA22-B071
Keywords: KRAS G12C mutation, targeted therapies, colorectal cancer, non-small cell lung cancer, pancreatic cancer, drug development
Citation: Qunaj L, May MS, Neugut AI and Herzberg BO (2023) Prognostic and therapeutic impact of the KRAS G12C mutation in colorectal cancer. Front. Oncol. 13:1252516. doi: 10.3389/fonc.2023.1252516
Received: 03 July 2023; Accepted: 28 August 2023;
Published: 15 September 2023.
Edited by:
Paul Ruff, University of the Witwatersrand, South AfricaReviewed by:
Luca Marino, Sapienza University of Rome, ItalyCopyright © 2023 Qunaj, May, Neugut and Herzberg. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benjamin O. Herzberg, Ym9oMjEwOUBjdW1jLmNvbHVtYmlhLmVkdQ==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.