 Xinghua Ji1†
Xinghua Ji1† Zejun Xing
Zejun Xing
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol., 13 December 2023
Sec. Surgical Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1249928
Background: Extraosseous myxoid chondrosarcoma (EMC) is extremely rare, and the case we report is of a particular site with partial bone destruction.
Case presentation: This case report can further strengthen the understanding of EMC and guide clinical treatment. The patient presented with a right buttock mass that was present for 1 year and that had gradually enlarged with tenderness for 6 months. The diagnosis was EMC. The interventions included puncture biopsy, surgical resection, and postoperative chemotherapy. The tumor was resected extensively, and the postoperative recovery was satisfactory. There was no recurrence or metastasis during the follow-up for 18-month.
Case presentation: The case we reported occurred in the pelvic cavity, which has not been previously reported in the literature, and there was partial bone destruction. Complete resection of the tumor was performed, and a satisfactory prognosis was achieved.
EMC accounts for almost 3% of all soft tissue sarcomas (1). It is a rare tumor with an indolent course and a high propensity for local recurrence and metastasis (2). Because EMC is very rare, the clinical manifestations are not characteristic, and early diagnosis before surgery is difficult. This article reports a case of EMC with bone destruction in the buttocks and reviews relevant literature to improve the understanding of the disease and to provide valuable information for clinical diagnosis and treatment.
Age and sex: 53-year-old female
Chief complaints: the patient presented with a mass in the buttock that had been growing in size since 2019.
History of present illness: The right buttock mass was present for 1 year and was gradually increasing in size. The clinical manifestations were symptoms of rectum and uterus compression, with discomfort, dragging sensation, and distension in the lower abdomen, and the mass inhibited defecation.
History of past illness: The patient had hypertension for 10 years, with stable blood pressure control.
Family history: None of her family members were diagnosed with carcinoma.
Physical examination: A fixed mass was present in the right buttock, with no skin changes or ulceration; the local boundary was still clear; and the patient had local tenderness. The tumor firmness and mobility were poor.
The laboratory testing showed the following: Serum red blood cell count: 3.5*1012/L; serum white blood cell count: 5.7*109/L (neutrophil percentage: 65%, lymphocyte percentage 23.7%); hemoglobin concentration: 113 g/L; hematocrit: 0.330 L/L; serum alanine aminotransferase concentration: 23.0 IU/L; serum aspartate aminotransferase: 20.0 IU/L; serum albumin concentration: 39.8 g/L; serum triglyceride concentration: 2.23 mmol/L; serum urea concentration: 5.0 mmol/L; serum creatinine concentration: 73.2 μmol/L; serum uric acid concentration: 320.3 μmol/L; alpha-fetoprotein: 1.5 ng/ml; carcinoembryonic antigen: 0.9 ng/ml; carbohydrate antigen199: 6.9 U/ml, carbohydrate antigen125: 2.2 U/ml, carbohydrate antigen153: 4.3 U/ml, and carbohydrate antigen242: 2.9.
Imaging examination: Computed tomography (CT) showed that there was a mass of cystic low-density shadow on the right side of the ischiorectal fossa, and the mass was approximately 6.95 cm×7.79 cm, with segmentations visible inside (Figure 1A). Enhanced scanning of the cyst wall and segmentations showed mild enhancement and visible right internal iliac artery branches supplying blood to the tumor. There was adjacent ischium bone destruction, and the adjacent rectum and vagina were pushed to the left (Figure 1B).
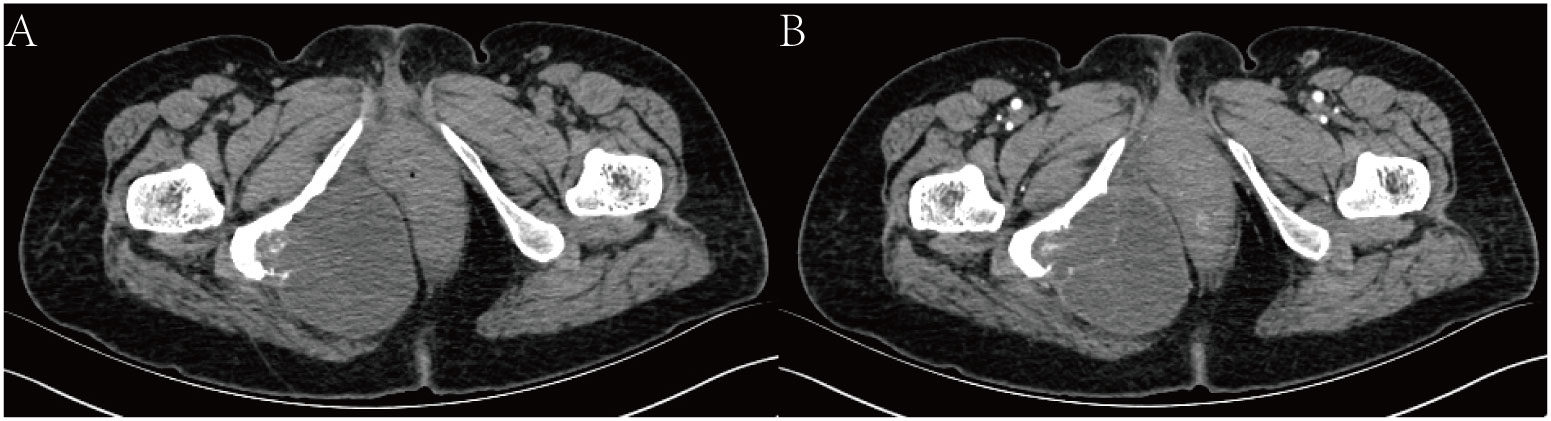
Figure 1 CT scan. (A) shows a mass of cystic low-density shadow on the right side of the ischiorectal fossa. (B) shows enhanced scanning of the cyst wall, and segmentations showed mild enhancement.
Magnetic resonance imaging (MRI) showed that there was a mass of mixed T1 and T2 signal shadows in the right ischiorectal fossa. The main body of the lesion showed equal T1 and long T2 signal shadows, and small patches of shorter T1 and equal T2 signal shadows were visible (Figures 2A, B). Diffusion-weighted imaging (DWI) and apparent diffusion coefficient (ADC) images showed a high signal (Figure 2C). The obturator internal muscle was unclear since it was involved in the lesion. The adjacent tissues were pushed to the left. The tumor was approximately 8.55 cm×7.59 cm×8.32 cm. The adjacent soft tissues showed a grid-like high signal in the fat suppression sequence (Figure 2D).
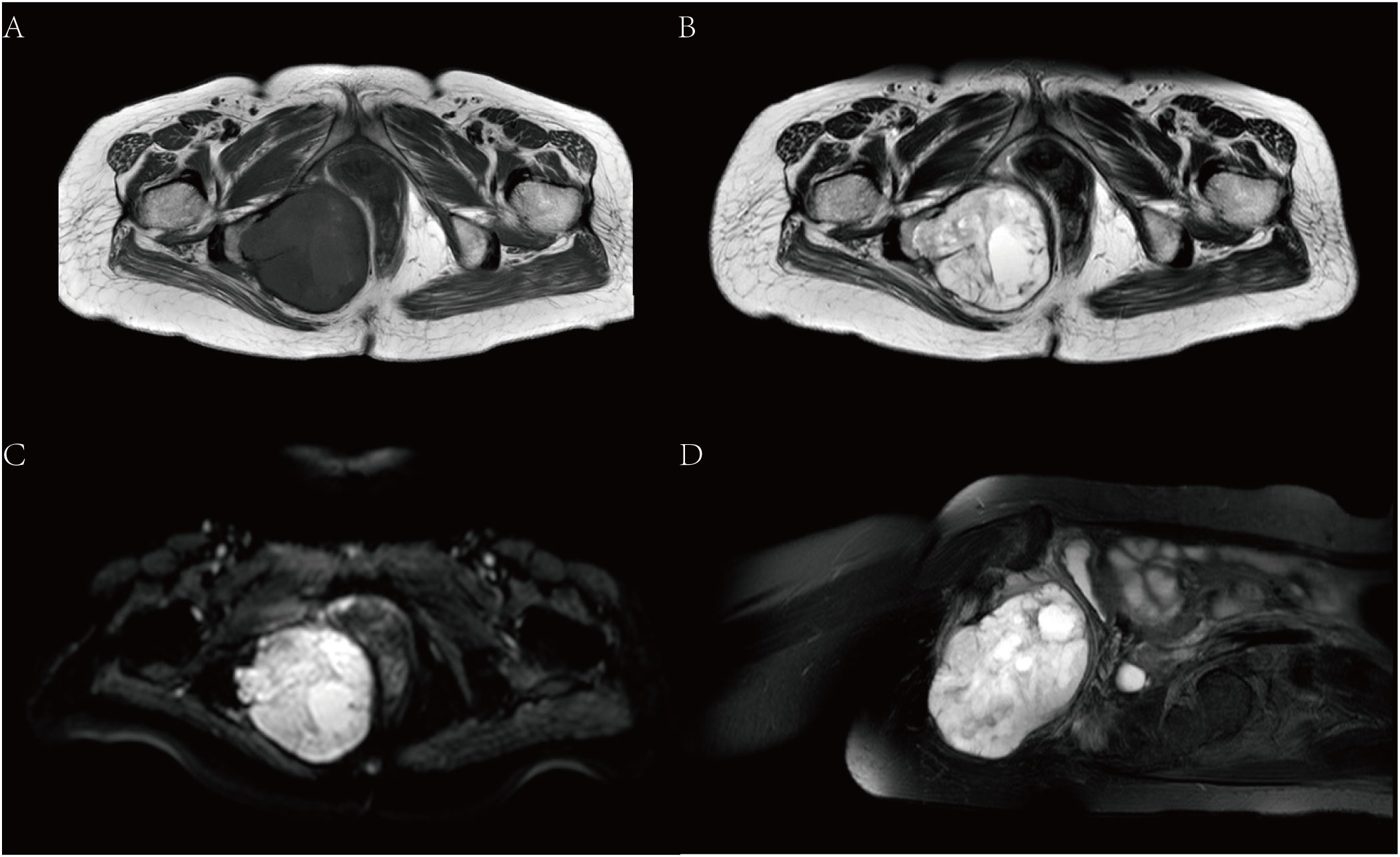
Figure 2 MRI. (A) is T1, and (B) is T2. (C) shows DWI images with high signals. (D) shows the sagittal position of the T2 fat suppression sequence.
Genetic testing: none
Final diagnosis: Immunohistochemical staining of the tumor showed positive staining for S100 and Vimentin, suspicious positivity for mesothelial cells, and negative staining for cytokeratin AE1/AE3, EMA, CK19, E-cadherin, and CK5/6. The Ki67 labeling index reached approximately 40% in some areas.
The tumor stroma showed mucinous denaturation, the tumor cells were polygonal and epithelioid, the nuclei were slightly atypia, grid-like calcification, and reactive bone formation were found within local lesions, and the bone tissues were invaded by the lesion. All the changes pointed to the diagnosis of EMC (Figures 3C, D).
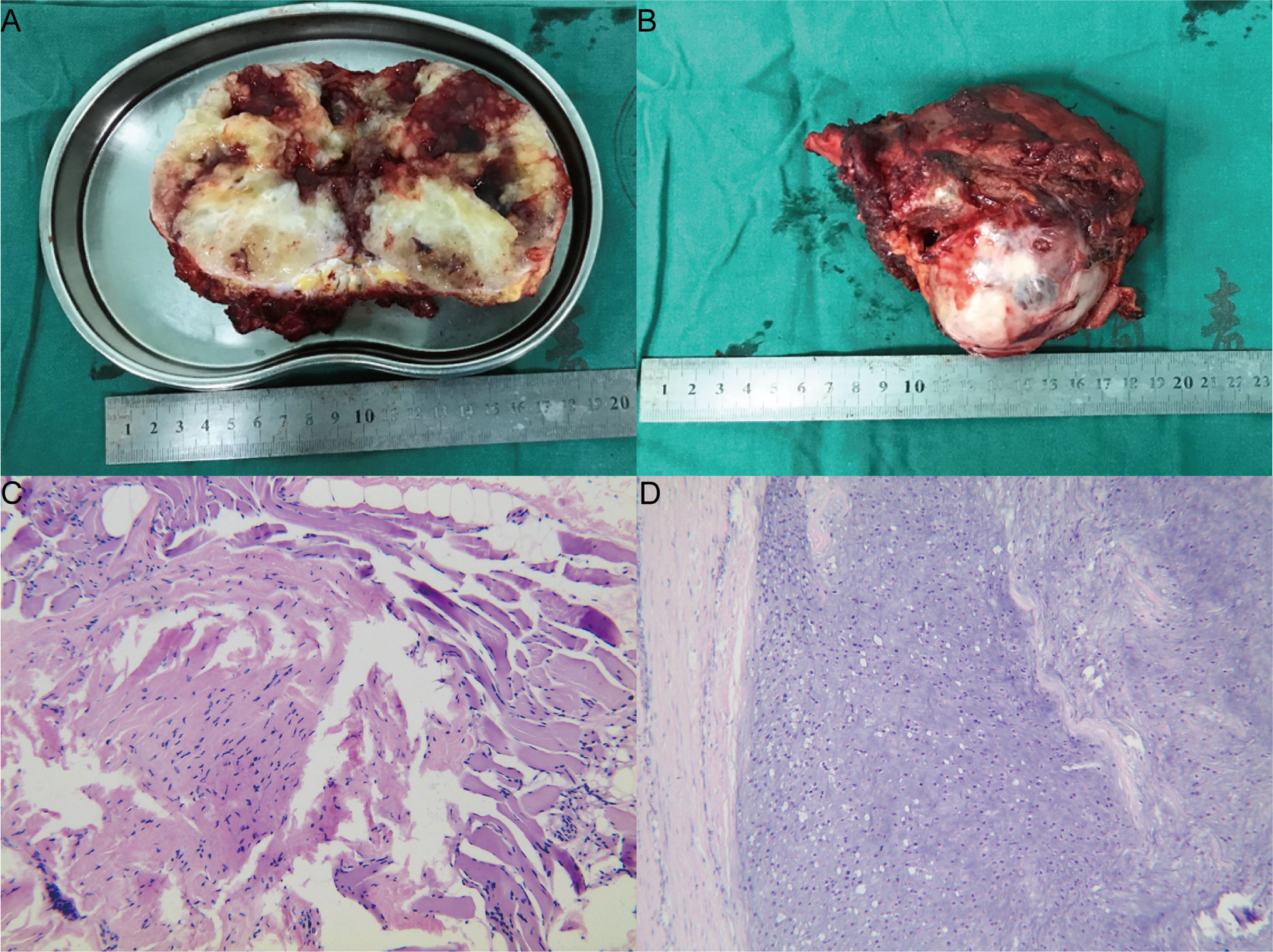
Figure 3 Pathology. (A) is a general observation, and (B) is a section observation. (C) shows the needle biopsy, and some spindle cells were visible. (D) is HE×100.
Treatment: Before the surgery, the tumor supplying artery was embolized through intervention. Subsequently, a needle biopsy of the tumor was performed, and the results showed that some spindle cells were visible (Figures 3C, D). We performed a resection of the tumor with an extended range of excision. During the surgery, we made a curved incision along the posterior midline of the sacrum at the level of S4, extending towards the right buttock tumor area. The incision was approximately 30cm long. The layers of skin, subcutaneous tissue, and deep fascia were sequentially dissected. Blunt dissection was performed from the tailbone area towards the right buttock to expose the tumor, while taking care to protect the gluteus maximus, gluteus medius, and gluteus minimus muscles. The proximal portion of the tailbone was removed first, revealing that the tumor extended anteriorly from the right buttock to the pelvic cavity, with an intact capsule. After thorough exposure of the posterior aspect of the tumor, the tumor was bluntly dissected along its margins. Exploration revealed the tumor closely related to the ischial tuberosity with significant involvement of the bone. Partial resection of the ischial tuberosity was performed to further separate the tumor deeply into the pelvic cavity. Exploration indicated that the tumor was closely related to the right pubic branch with bone involvement. To protect the surrounding tissues, partial resection of the pubic branch was performed using a saw. At this point, exploration confirmed complete separation of the tumor from the surrounding tissues, and the tumor was completely excised (Figures 3A, B). No infiltration was observed at the tumor margin. After surgery, the patient received three rounds of chemotherapy (Anlotinib, 10mg, QD, 14days) after surgery.
Outcome and follow-up: The patient recovered well after the operation, the patient’s limb function was normal, and there was no recurrence or metastasis during the 18-month follow-up after the operation (Figure 4).
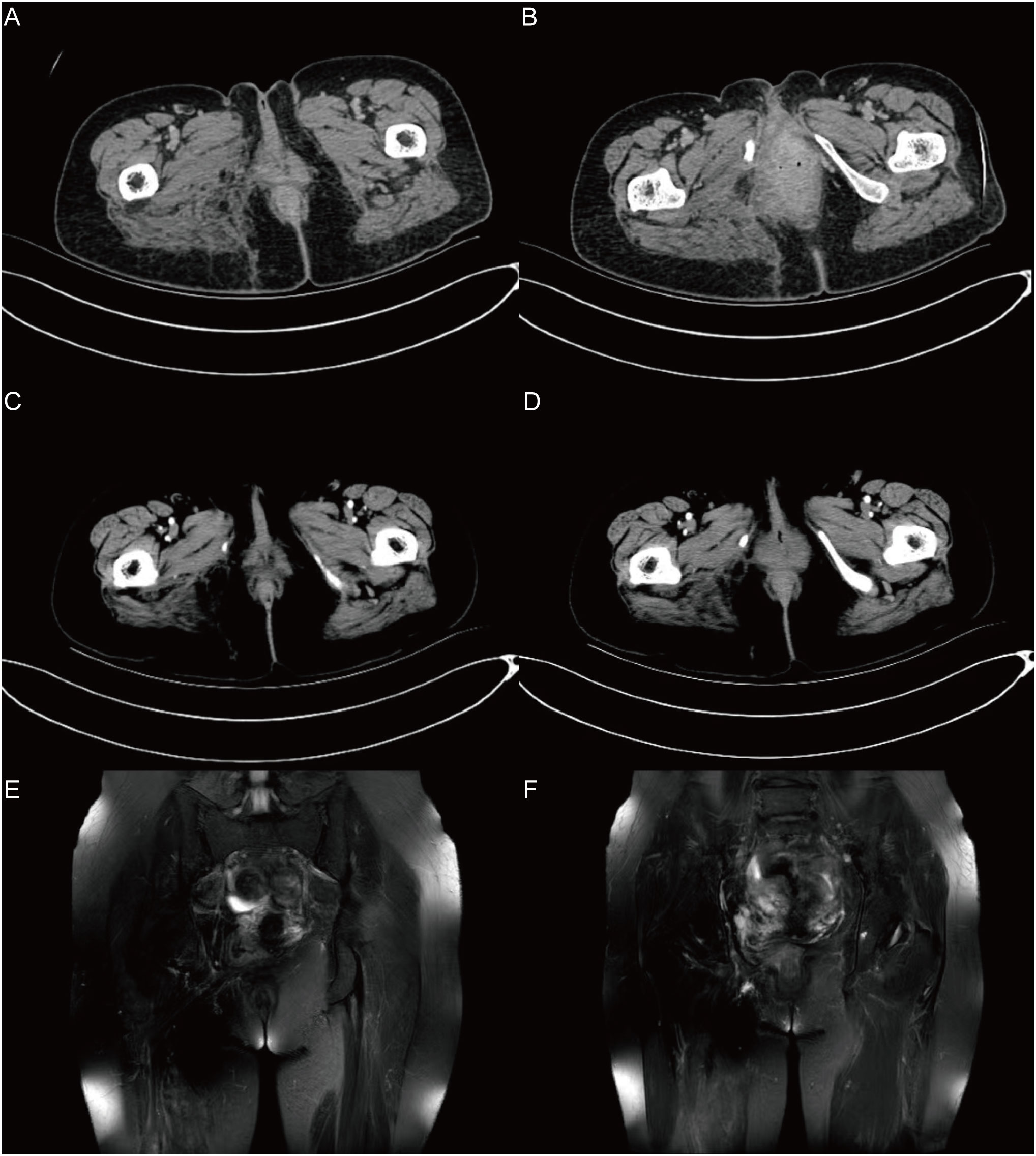
Figure 4 The follow-up results in 18 months after surgery. (A), (B) show the CT results after 1 month. (C), (D) show the CT results after 6 months. (E), (F) show the MRI results after 18 months. In the imaging examination results during the 18-month follow-up after surgery, no recurrence or metastasis was observed.
Primary EMC is a mesenchymal tumor with an undetermined differentiation direction (3). Due to its rare incidence, only 5 cases have been reported in the literature to date. We also collected information on the 5 previously reported cases of primary buttock EMC (shown in Table 1) (4–8) to perform a systematic review of this tumor type and discussing the epidemiological and radiographic features, diagnosis, treatment strategies, and prognosis of this rare disease. The case presented herein is of a 52-year-old female patient with primary buttock EMC with bone destruction. To the best of our knowledge, this is the first case of primary buttock EMC with bone destruction.
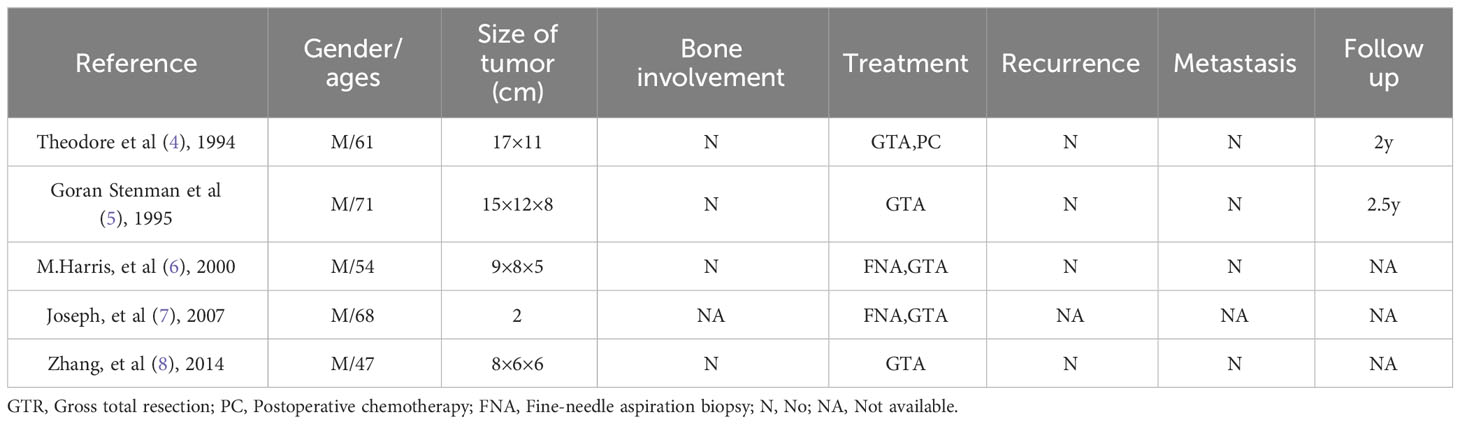
Table 1 Published and present cases of primary buttock EMC.
EMC manifests as a solitary soft tissue mass, which easily recurs after surgery and can be transferred. Usually, it does not invade bone tissue. It often occurs in the soft tissues of the proximal limbs and deep tissues of middle-aged people. The incidence in males and females is approximately 2:1. EMC is a relatively rare neoplasm with no specific findings in clinical practice. Patients commonly present with nonspecific symptoms, including tenderness and the detection of a palpable mass (9). The most common manifestation of EMC is an enlarging soft tissue mass, and some lesions are accompanied by pain and tenderness or may restrict the range of motion. This patient, a 53-year-old female, presented with a gradually enlarged mass on the right hip, with local tenderness and involvement of the pelvis; CT scanning showed that the tumor had invaded the ischial tuberosity, which is extremely rare in EMC. The tumor found in this bone is unusual since only 16 cases have been reported in the literature.
There were no specific CT findings for primitive EMC (10, 11), mostly shown as heterogeneous tumors in soft tissue. The existence of EMC is also characterized by tumors with low density (mostly 20-40 HU) on CT images and mild or no enhancement on enhanced CT images (10, 11). The average CT value of the tumor, in this case, was 20 HU, and the segmentation was slightly enhanced, which was consistent with the findings in the literature. Calcification is rarely found on the CT images of EMC. Zhang et al. retrospectively analyzed 13 EMC patients, 3 of whom showed calcification (12). EMC is characterized by the presence of a cartilage matrix on MR images. The mass showed a lobulated appearance with iso-/hypo-intensity to muscle on T1WI and hyperintensity on T2WI, and it was usually segmented into multiple lobules by multiple septa with hypointensity. The signal intensity was higher in the myxoid areas than in the solid areas, and the calcified areas appeared hypointense on T2WI. In contrast-enhanced images, peripheral, septal, or heterogeneous enhancement patterns were observed (13).
There was a large difference in the tumor sizes by the naked eye. The cut surfaces are greyish-white or grey-brown. The tumors are composed of multiple jelly-like tumor nodules. The nodules are separated by fibrous tissue, and some of them have cystic denaturation, hemorrhage, and necrosis. It can be seen under the microscope that the tumor nodules are composed of various interconnected eosinophilic spindles and oval and small round cells, and these cells are separated by a mucus-like and cartilage-like matrix in a mesh or lace shape. Positive S-100 and lack of INI-1 expression are helpful for diagnosis (14). The positivity of GFAP has been reported. Under electron microscopy, the presence of parallel microtubules and abundant REG are quite characteristic but not specific (15, 16). Certain studies have shown that they may also be positive for Leu-7 and epithelial membrane antigens. Uniformly, they are negative for SMA, and desmin (8, 17, 18). Under the electron microscope, the characteristic microtubules in the pool are arranged in bundles or parallel. In some cases, intracellular neuroendocrine granules can be seen. Therefore, EMC can express the neuroendocrine markers Syn and NSE. INSM1 could be a potential marker for the diagnosis of EMC when molecular genetic access is limited (19).
Complete surgical resection is the main treatment method for EMC. The early literature reported that EMC was not sensitive to radiotherapy and chemotherapy. With the deepening understanding of EMC, radiotherapy and chemotherapy have been found to show a certain value. Radiotherapy can reduce postoperative complications from 41% to 20% (20). Receiving external beam radiotherapy (EBRT) is associated with a cancer-specific survival benefit in localized EMC. Aggressive local therapy, including EBRT, should be considered in these patients (21). The studies in the past were aimed at improving disease control by surgical resection or radiotherapy, with some reported benefits from the tyrosine kinase inhibitor sunitinib malate in the metastatic setting (21–24). Recent research has shown that pazopanib has antitumor action with clinical value in patients with progressive and advanced EMC, which could be considered a suitable option after these patients fail to respond to first-line anthracycline-based chemotherapy (25). This provides new ideas for the treatment of EMC. Oliveira et al. (26) believe that the indicators of poor prognosis include older age, tumor diameter > 10 cm, tumor metastasis, histological anaplastic area, mitotic figures > 2/10 HPF, and Ki-67 proliferation index ≥ 10%. In this case, the Ki-67 proliferation index was ≥40%. The patient received three rounds of chemotherapy after surgery. No recurrence or metastasis occurred during the 18-month follow-up after the operation.
In conclusion, EMC is a rare malignant tumor. The case we reported occurred in the pelvic cavity, which has not been reported in the literature, and there was partial bone destruction. Complete resection of the tumor was performed, and a satisfactory prognosis was achieved. The report of this case provides theoretical support for the clinical diagnosis and treatment of EMC.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Written informed consent was obtained from the participant/patients(s) for the publication of this case report.
XJ: data acquisition, literature search, manuscript preparation, and medical management. JW: data acquisition, literature search, and manuscript preparation. XL: pathological interpretation, manuscript preparation, and editing. WZ: data acquisition and manuscript preparation. ZX: data acquisition, literature search, and manuscript preparation. All authors read and approved the final manuscript. All authors contributed to the article and approved the submitted version.
This research was supported by the Shanxi Provincial Health Committee Subjects (No. 2020011).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Al Khader A, Nsour E, Al-Zubi RB, Al Maadat HMD. Extraskeletal myxoid chondrosarcoma of the leg in a child: A case report. Medicine (2019) 98(15):e15207. doi: 10.1097/MD.0000000000015207
2. Bangerter J, Harnisch K, Chen Y, Hagedorn C, Planas-Paz L, Pauli C. Establishment, characterization and functional testing of two novel ex vivo extraskeletal myxoid chondrosarcoma (EMC) cell models. Hum Cell (2023) 36(1):446–55. doi: 10.1007/s13577-022-00818-x
3. Zambo I, Veselý K. WHO classification of tumours of soft tissue and bone 2013: the main changes compared to the 3rd edition. Ceskoslovenska patologie. (2014) 50(2):64–70.
4. Niemann TH, Bottles K, Cohen MB. Extraskeletal myxoid chondrosarcoma: Fine-needle aspiration biopsy findings. Diagn Cytopathol. (1994) 11(4):363–6. doi: 10.1002/dc.2840110410
5. Stenman G, Andersson H, Mandahl N, Meis-Kindblom J, Kindblom L. Translocation t(9;22)(q22;q12) is a primary cytogenetic abnormality in extraskeletal myxoid chondrosarcoma. Int J cancer. (1995) 62(4):398–402. doi: 10.1002/ijc.2910620407
6. Harris M, Coyne J, Tariq M, Eyden B, Atkinson M, Freemont A, et al. Extraskeletal myxoid chondrosarcoma with neuroendocrine differentiation: a pathologic, cytogenetic, and molecular study of a case with a novel translocation t(9;17)(q22;q11. 2). Am J Surg Pathol (2000) 24(7):1020–6. doi: 10.1097/00000478-200007000-00015
7. Jakowski J, Wakely P. Cytopathology of extraskeletal myxoid chondrosarcoma: report of 8 cases. Cancer (2007) 111(5):298–305. doi: 10.1002/cncr.22948
8. Zhang G, Wang A, Zhang G, Zhao S, Qu J. Extraskeletal myxoid chondrosarcoma: A report of two cases. Oncol letters. (2014) 7(4):1289–91. doi: 10.3892/ol.2014.1884
9. Brodsky C, Bucala M, Abdulfatah E, Siegel G. Extraskeletal myxoid chondrosarcoma: retrospective case series examining prognostic factors, treatment approaches, and oncologic outcomes. Am J Clin Oncol (2023) 46(4):172–7. doi: 10.1097/COC.0000000000000988
10. Gadabanahalli K, Belaval V, Bhat V, Gorur I. Primary extraskeletal myxoid chondrosarcoma of pulmonary arteries: a rare mimic of acute pulmonary thromboembolism. Interact. Cardiovasc Thorac surg. (2015) 20(4):565–6. doi: 10.1093/icvts/ivu444
11. Oike N, Ogose A, Kawashima H, Umezu H, Inagawa S. Extraskeletal myxoid chondrosarcoma arising in the femoral vein: a case report. Skeletal radiol. (2014) 43(10):1465–9. doi: 10.1007/s00256-014-1897-3
12. Zhang L, Wang R, Xu R, Qin G, Yang L. Extraskeletal myxoid chondrosarcoma: A comparative study of imaging and pathology. BioMed Res Int (2018) 2018:9684268. doi: 10.1155/2018/9684268
13. Shao R, Lao I, Wang L, Yu L, Wang J, Fan Q. Clinicopathologic and radiologic features of extraskeletal myxoid chondrosarcoma: a retrospective study of 40 Chinese cases with literature review. Ann Diagn pathol. (2016) 23:14–20. doi: 10.1016/j.anndiagpath.2016.04.004
14. Dulou R, Chargari C, Dagain A, Teriitehau C, Goasguen O, Jeanjean O, et al. Primary intracranial extraskeletal myxoid chondrosarcoma. Neurol. i neurochirurgia polska. (2012) 46(1):76–81. doi: 10.5114/ninp.2012.27176
15. Thompson L. World Health Organization classification of tumours: pathology and genetics of head and neck tumours. Ear nose throat J (2006) 85(2):74. doi: 10.1177/014556130608500201
16. Mocellin S. Extraskeletal myxoid chondrosarcoma. Soft Tissue Tumors (2021), 317–9. doi: 10.1007/978-3-030-58710-9_94
17. Wang C, You Z, Chen X, Lin J, Wu Y. Mutation of KIT in cellular extraskeletal myxoid chondrosarcoma: a case report and literature review. Diagn pathol. (2022) 17(1):42. doi: 10.1186/s13000-022-01222-7
18. Wei S, Pei J, von Mehren M, Abraham J, Patchefsky A, Cooper H. SMARCA2-NR4A3 is a novel fusion gene of extraskeletal myxoid chondrosarcoma identified by RNA next-generation sequencing. Genes Chromosomes cancer. (2021) 60(10):709–12. doi: 10.1002/gcc.22976
19. Yoshida A, Makise N, Wakai S, Kawai A, Hiraoka N. INSM1 expression and its diagnostic significance in extraskeletal myxoid chondrosarcoma. Modern Pathol (2018) 31(5):744–52. doi: 10.1038/modpathol.2017.189
20. Stacchiotti S, Pantaleo M, Astolfi A, Dagrada G, Negri T, Dei Tos A, et al. Activity of sunitinib in extraskeletal myxoid chondrosarcoma. Eur J Cancer (Oxford England: 1990). (2014) 50(9):1657–64. doi: 10.1016/j.ejca.2014.03.013
21. Kemmerer E, Gleeson E, Poli J, Ownbey R, Brady L, Bowne W. Benefit of radiotherapy in extraskeletal myxoid chondrosarcoma: A propensity score weighted population-based analysis of the SEER database. Am J Clin Oncol (2018) 41(7):674–80. doi: 10.1097/COC.0000000000000341
22. Morioka H, Takahashi S, Araki N, Sugiura H, Ueda T, Takahashi M, et al. Results of sub-analysis of a phase 2 study on trabectedin treatment for extraskeletal myxoid chondrosarcoma and mesenchymal chondrosarcoma. BMC cancer. (2016) 16:479. doi: 10.1186/s12885-016-2511-y
23. Kim A, Lim B, Choi J, Kim J. The TFG-TEC oncoprotein induces transcriptional activation of the human β-enolase gene via chromatin modification of the promoter region. Mol carcinogene. (2016) 55(10):1411–23. doi: 10.1002/mc.22384
24. Kapoor N, Shinagare A, Jagannathan J, Shah S, Krajewski K, Hornick J, et al. Clinical and radiologic features of extraskeletal myxoid chondrosarcoma including initial presentation, local recurrence, and metastases. Radiol Oncol (2014) 48(3):235–42. doi: 10.2478/raon-2014-0005
25. Stacchiotti S, Ferrari S, Redondo A, Hindi N, Palmerini E, Vaz Salgado M, et al. Pazopanib for treatment of advanced extraskeletal myxoid chondrosarcoma: a multicentre, single-arm, phase 2 trial. Lancet Oncol (2019) 20(9):1252–62. doi: 10.1016/S1470-2045(19)30319-5
Keywords: extraosseous myxoid chondrosarcoma, soft tissue sarcoma, radiography, pathology, case report
Citation: Ji X, Wei J, Li X, Zhang W and Xing Z (2023) Extraskeletal myxoid chondrosarcoma of the buttock: a case report and literature review. Front. Oncol. 13:1249928. doi: 10.3389/fonc.2023.1249928
Received: 29 June 2023; Accepted: 15 November 2023;
Published: 13 December 2023.
Edited by:
Ulrich Ronellenfitsch, Medical Faculty of the Martin-Luther-University Halle-Wittenberg, GermanyReviewed by:
Kasra Shirini, University of Maryland, United StatesCopyright © 2023 Ji, Wei, Li, Zhang and Xing. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinzheng Wei, d2p6MTU5NTcxMjUxNkAxNjMuY29t; Zejun Xing, MTg3MzUxMzA5NjVAMTYzLmNvbQ==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.