 Rui Wu
Rui Wu Megan S. Lim*
Megan S. Lim*- Department of Pathology and Laboratory Medicine, Memorial Sloan Kettering Cancer Center, New York, NY, United States
Anaplastic large cell lymphomas (ALCL) encompass several distinct subtypes of mature T-cell neoplasms that are unified by the expression of CD30 and anaplastic cytomorphology. Identification of the cytogenetic abnormality t(2;5)(p23;q35) led to the subclassification of ALCLs into ALK+ ALCL and ALK- ALCL. According to the most recent World Health Organization (WHO) Classification of Haematolymphoid Tumours as well as the International Consensus Classification (ICC) of Mature Lymphoid Neoplasms, ALCLs encompass ALK+ ALCL, ALK- ALCL, and breast implant-associated ALCL (BI-ALCL). Approximately 80% of systemic ALCLs harbor rearrangement of ALK, with NPM1 being the most common partner gene, although many other fusion partner genes have been identified to date. ALK- ALCLs represent a heterogeneous group of lymphomas with distinct clinical, immunophenotypic, and genetic features. A subset harbor recurrent rearrangement of genes, including TYK2, DUSP22, and TP63, with a proportion for which genetic aberrations have yet to be characterized. Although primary cutaneous ALCL (pc-ALCL) is currently classified as a subtype of primary cutaneous T-cell lymphoma, due to the large anaplastic and pleomorphic morphology together with CD30 expression in the malignant cells, this review also discusses the pathobiological features of this disease entity. Genomic and proteomic studies have contributed significant knowledge elucidating novel signaling pathways that are implicated in ALCL pathogenesis and represent candidate targets of therapeutic interventions. This review aims to offer perspectives on recent insights regarding the pathobiological and genetic features of ALCL.
1 Introduction
Anaplastic large cell lymphomas (ALCL) refer to a heterogeneous group of CD30-positive T-cell neoplasms with diverse clinical, histologic, and genetic features. The disease group comprises approximately 15% of all peripheral T-cell lymphoma and 3 to 5% of all non-Hodgkin lymphoma (1). ALCL was first recognized in 1985 based on the large size of the neoplastic cells with uniform strong expression of CD30 (2). The recurrent chromosomal translocation t (2;5)(p23;q35), which was identified in 1994, results in a novel fusion tyrosine kinase involving the N-terminus of the nucleophosmin (NPM1) gene and the C-terminus of the anaplastic lymphoma kinase (ALK) gene (3). The chimeric protein NPM::ALK functions as an oncogenic tyrosine kinase which impacts diverse cellular signaling pathways leading to lymphoma. In addition to NPM1, over 20 distinct partner genes of ALK have been identified (Figure 1) (4). Moreover, the significantly enhanced survival of ALK+ ALCL patients and distinct genetic features rationalized its distinction from ALK- ALCLs (5, 6). Based on the recent understanding of the genetic basis of ALK- ALCL and those that occur in breast-implant ALCL, the updated 5th edition of the World Health Organization Classification of Haematolymphoid Tumours (5), and the International Consensus Classification (ICC) of Mature Lymphoid Neoplasms (7) recognize three subtypes, namely ALK+ ALCL, ALK- ALCL, breast implant-associated ALCL (BI-ALCL) (5). Further, we discuss the pathobiological features of primary cutaneous anaplastic large cell lymphoma (pc-ALCL) due to many overlapping histologic and immunophenotypic features with other ALCLs. Recent genomic studies provide an enhanced understanding of the pathobiological events in this group of intriguing neoplasms that will be summarized in this manuscript (6, 8).
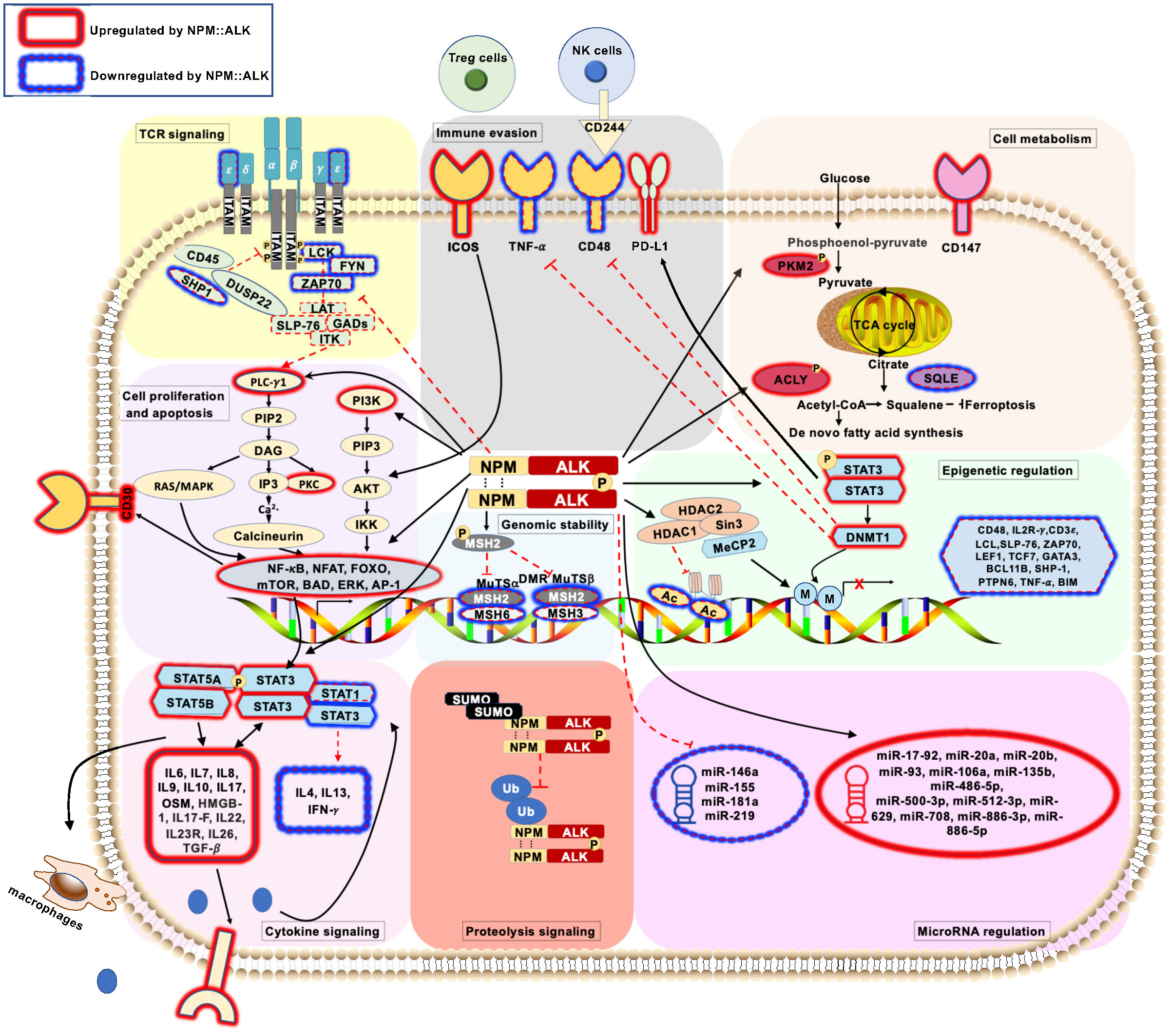
Figure 1 Schematic representation of pathogenic signaling pathways in ALK+ ALCL ( ) represents deregulated T-cell receptor signaling pathways mediated by NPM::ALK. (
) represents deregulated T-cell receptor signaling pathways mediated by NPM::ALK. ( ) represents deregulated cell proliferation and apoptosis signaling pathways mediated by NPM::ALK. (
) represents deregulated cell proliferation and apoptosis signaling pathways mediated by NPM::ALK. ( ) represents deregulated cellular metabolism signaling pathways mediated by NPM::ALK. (
) represents deregulated cellular metabolism signaling pathways mediated by NPM::ALK. ( ) represents deregulated genomic stability signaling pathways mediated by NPM::ALK. (
) represents deregulated genomic stability signaling pathways mediated by NPM::ALK. ( ) represents deregulated epigenetic regulation signaling pathway mediated by NPM::ALK. (
) represents deregulated epigenetic regulation signaling pathway mediated by NPM::ALK. ( ) represents deregulated miRNA repertoire by NPM::ALK. (
) represents deregulated miRNA repertoire by NPM::ALK. ( ) represents immune evasion mechanisms mediated by NPM::ALK. (
) represents immune evasion mechanisms mediated by NPM::ALK. ( ) represents deregulated cytokine signaling mediated by NPM::ALK. (
) represents deregulated cytokine signaling mediated by NPM::ALK. ( ) represents deregulated proteolytic signaling mediated by NPM::ALK.
) represents deregulated proteolytic signaling mediated by NPM::ALK.
2 Pathobiology of ALK+ ALCL
2.1 ALK rearrangements in ALK+ ALCL
ALK+ ALCLs, by definition, express the ALK protein, which functions as a strong driver oncogene. ALK gene rearrangements result in the expression of a novel fusion protein and constitutive activation of the ALK tyrosine kinase (9). The majority (80%) of cases express NPM::ALK as a result of t(2;5)(q23;35) involving the 3’ segment of the ALK gene on chromosome 2p23 and the 5’ segment of the nucleophosmin (NPM1) gene on chromosome 5q35. The chimeric protein NPM::ALK is composed of the N-terminal oligomerization domain of NPM1 and the C-terminal tyrosine kinase domain of ALK. Due to ligand-independent oligomerization mediated by NPM1, the catalytic domain of ALK undergoes transautophosphorylation with constitutive tyrosine kinase activity, which translates into increased intracellular signaling promoting cell proliferation, resistance to apoptosis, and oncogenic transformation (10). Apart from NPM1 as its N-terminal fusion partner, in approximately 20% of ALK+ ALCLs, ALK is fused with other genes, including tropomyosin 3 and 4 (TPM3 and TPM4) (11), TRK-fused gene (TFG) (12), 5-amino-imidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase (ATIC) (13), ring finger protein 213 (RNF213) (14), clathrin heavy chain (CLTC) (14), moesin (MSN) (15), non-muscle myosin heavy chain (MYH9) (15), TNF receptor-associated factor 1 (TRAF1) (16), eukaryotic translation elongation factor I gamma (EEF1G) (17) and the poly(A) binding protein cytoplasmic 1 (PABPC1) (18). In contrast to NPM::ALK, which is expressed in both the nucleus and cytoplasm, other ALK fusion proteins are localized in various cellular compartments (Table 1) (3, 11, 13–20). TPM3::ALK, the second most frequent fusion, is present in approximately 15% of ALK+ ALCLs. Despite all ALK rearrangements involving the same region of ALK, the downstream signaling pathways vary due to the different fusion partners (21). The pathogenetic mechanisms regulated by variant ALK fusion genes have not been explored due to the rarity of cases.
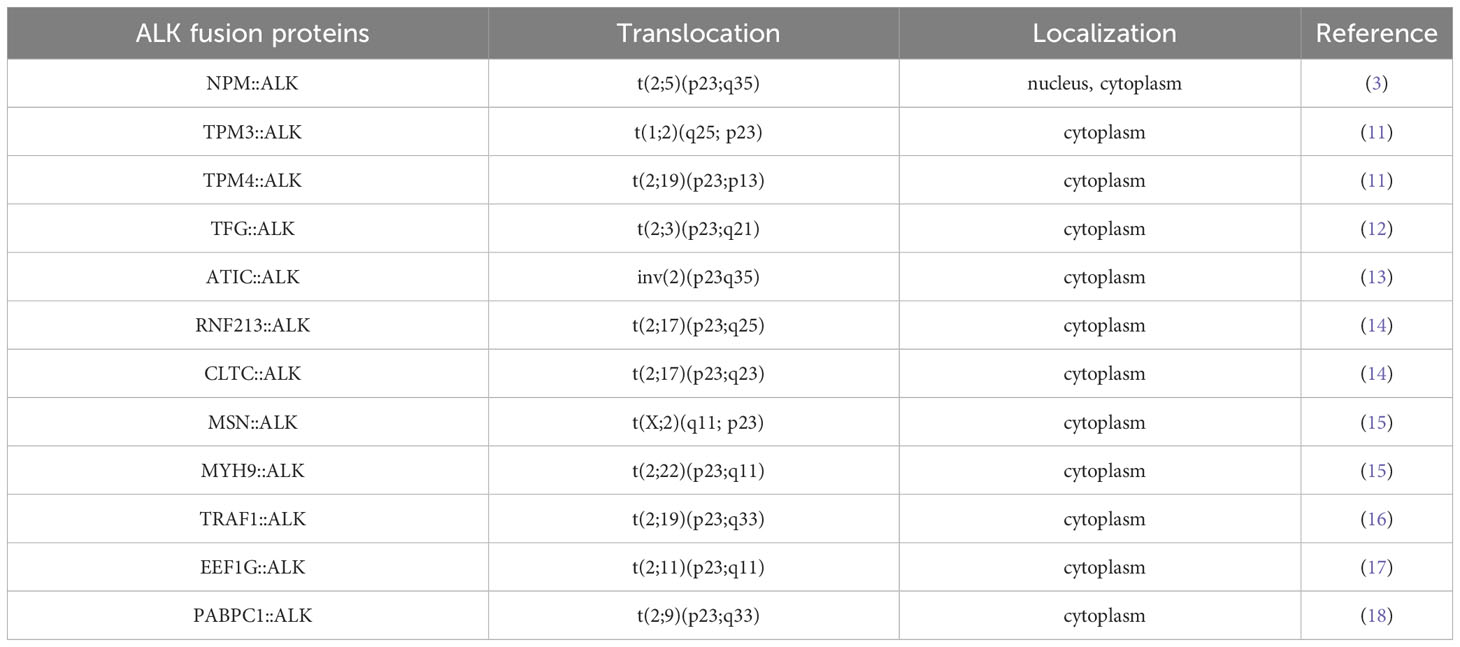
Table 1 Summary of chromosomal rearrangements in ALK+ ALCL.
2.2 Deregulated T-cell receptor signaling pathway in ALK+ ALCL
T-cell receptor (TCR) engagement triggers various cascades of signaling pathways, including phospholipase C-γ1 (PLC-γ1)-inositol triphosphate (IP3)-Ca2+-nuclear factor of activated T-cells (NFAT) pathway (22, 23), the protein kinase C (PKC)-IĸB kinase (IKK)-nuclear factor (NF)-κB pathway (24), the Ras-extracellular signal-related kinase (ERK)-activator protein (AP)-1 pathway (25), as well as the Phosphoinositide 3-kinase (PI3K)-AKT-mammalian target of rapamycin (mTOR) pathway (26). These activated signaling pathways ultimately determine cell fate through cytokine production, cell survival, cell proliferation, and differentiation. Although ALK+ ALCLs express rearranged T-cell receptors, the expression of many pivotal TCR molecules, including TCF-1, TCF-1/LEF-1, LCK, ZAP-70, LAT, NFATc1, c-Jun, c-Fos, and Syk is repressed (27). Inhibition of the kinase activity of NPM::ALK or exposure to DNA methyltransferase inhibitors rescues the expression of CD3ε, ZAP70, LAT, and SLP76, suggesting that NPM::ALK-mediated transcriptional repression occurs via DNA methylation to downregulate components of the TCR signaling cascade in ALK+ ALCL (28). Apart from this, NPM::ALK further mimics TCR-induced signal transduction by directly interacting with and phosphorylating PLC-γ1, which triggers downstream signaling cascades for cell survival (29, 30). As such, NPM::ALK promotes the proliferation and survival of malignant cells by bypassing the TCR signaling pathway (Figure 1).
In addition to promoting cell proliferation and differentiation, T-cell receptor engagement also induces activation-induced T-cell death (AICD) through Fas-mediated apoptosis to prevent the accumulation of alloreactive T-cells and the development of graft-vs-host disease (31). It has been demonstrated that FLICE-like inhibitory protein (c-FLIP) prevents neoplastic cells from undergoing Fas-mediated apoptosis in ALK+ ALCL. Specifically, exposure of ALK+ ALCL cell lines that express high levels of c-FLIP, to CH-11, a CD95/FAS agonistic antibody, alone is not able to reduce the viability of malignant cells. si-RNA-mediated knockdown of c-FLIP together with CH-11 treatment rescues Fas-mediated apoptosis by triggering downstream caspase signaling pathways in ALK+ ALCL cells (32). Further investigation is required to determine whether the loss of TCR signaling molecules contributes to reduced AICD process in ALK+ ALCL.
2.3 Deregulated cell proliferation and apoptosis in ALK+ ALCL
NPM::ALK regulates many proliferative and anti-apoptotic signaling pathways, including mitogen-activated protein (MAP) kinase (33), JAK-STAT (34, 35), PLC-γ1 (30), and PI3K-AKT (29), to promote lymphomagenesis (Figure 1). The MAP kinase signaling pathway is a major cell proliferation and survival regulator (36). NPM::ALK phosphorylates extracellular signal-regulated kinase (ERK)1/2 in a MEK1/2-dependent manner, and perturbation of MEK and ERK1/2 reduced cell proliferation and promoted cell apoptosis of ALK+ ALCL cells (37). NPM::ALK also regulates cell growth of ALK+ ALCL via activation of the PLC-γ pathway. The interaction of NPM::ALK with PLC-γ1 occurs via the tyrosine 644 residue, which is located at the C-terminus of the chimeric protein, and expression of NPM::ALKY644F abrogates PLC-γ1 phosphorylation and activation (30). The PI3K-AKT pathway has been implicated in oncogenesis for its role in cell-cycle progression. NPM::ALK constitutively activates PI3K and its downstream effector, the serine/threonine kinase AKT, and thus promotes growth and inhibits apoptosis in ALK+ALCL (38). AKT phosphorylates the Bcl2-associated death promoter (BAD), thereby suppressing apoptosis and promoting cell survival (39). Similarly, mTOR, a serine/threonine protein kinase and a key regulator of cell growth and proliferation, is also activated by NPM::ALK. Inhibition of the mTOR pathway leads to cell cycle arrest and apoptosis in ALK+ ALCL (40). Further, NPM::ALK promotes the expression of anti-apoptotic factors Bcl-xl and cell cycle-promoting cyclin-dependent kinase 4 (CDK4) and increased levels of phospho-RB to trigger cell proliferation (37). Thus, NPM::ALK regulates cell proliferation and survival while inhibiting apoptosis by orchestrating multiple signaling pathways.
2.4 Deregulated cellular metabolism in ALK+ ALCL
Neoplastic metabolic reprogramming is largely characterized by the shift from efficient energy-producing pathways to strategies for biomass production to support cell growth. In this regard, integrated analysis of the phosphoproteomic and metabolomic signature revealed that NPM::ALK signaling triggers an increase in biomass production by rerouting glycolytic intermediates (6) as well as modulation of lipid metabolism, amino acid metabolism, and nucleotide metabolism (Figure 1). Particularly, NPM::ALK-mediated phosphorylation of PKM2 at Y105, a key enzyme in aerobic glycolysis, leads to a metabolic switch promoting lymphomagenesis (6). Regarding lipid metabolism, NPM::ALK phosphorylates ATP citrate lyase (ACLY) at residue 682, which may serve as a switch to promote lipid synthesis required for cell proliferation. ACLY is a critical enzyme that catalyzes acetyl-CoA synthesis and connects vital biosynthetic pathways such as carbohydrate and lipid metabolism (41). Genetic or pharmacologic disruption in the NPM::ALK-ACLY signaling axis leads to impaired cell proliferation, impaired clonogenic potential, reduced tumor growth in an in vivo xenograft model, and attenuated lipid synthesis in ALK+ ALCL (42). NPM::ALK also modulates cancer metabolism through the downregulation of CD147, causing aberrant glycolysis and thus impairing the major energy source of tumor cells (43).
Further, metabolic alterations in cancer not only modulate the metabolic state of the cell but also impact cellular signaling and the epigenetic state. ALK+ ALCL-derived cell lines and primary tumors exhibit cholesterol auxotrophy due to reduced expression of a critical enzyme, squalene monooxygenase rendering the accumulation of squalene, a metabolite with antioxidant-like properties. Squalene monooxygenase oxidizes squalene to 2,3-oxidosqualene. Aggregation of squalene in cells prevents malignant cells from ferroptosis, which is induced by oxidative stress (44). Cholesterol auxotrophy of ALK+ ALCL can be a therapeutic vulnerability that can be utilized in combination with conventional therapies. In summary, NPM::ALK signaling orchestrates cellular metabolic reprogramming that favors lymphomagenesis.
2.5 Increased genomic instability in ALK+ ALCL
Under physiologic conditions, cells consistently encounter intracellular stress, such as reactive oxygen species (ROS) generated by cellular metabolism, and extracellular stress, such as UV light and carcinogenic chemicals. These stresses can disrupt genomic integrity. One of the hallmarks of cancer is genomic instability. It is generally accepted that NPM::ALK is required and sufficient to transform primary human T cells in a relatively short span of time. Furthermore, genetic alterations (single nucleotide variants) in ALK+ ALCL are relatively uncommon, suggesting a stable genome (45–47). In ALK+ ALCL, however, impaired DNA repair pathways (48), particularly DNA mismatch repair, may represent a mechanism by which tumor cells initiate additional genetic lesions, considering that a subset of ALK+ALCL patients develop resistance against ALK-specific treatment approaches. To initiate DNA mismatch repair, it is essential for the system to recognize DNA lesions. Two ATPase protein complexes participate in the mismatch recognition process, namely MuTSα, which identifies the base-base mismatches and small insertion/deletion, and MuTsβ, which modulates larger insertion/deletion. MuTSα is composed of MutS protein homolog 2 (MSH2) and MutS protein homolog 6 (MSH6). MuTSβ is composed of MSH2 and MutS protein homolog 3 (MSH3) (49). NPM::ALK directly binds to MSH2 and phosphorylates it at the tyrosine 238 residue. This abnormal interaction prevents MSH2 from binding to its normal partners, MSH3 and MSH6, leading to the ablation of normal MuTs complexes and consequentially impaired DNA damage repair (50). In addition, the expression of NPM::ALK in primary CD4+ T cells downregulates genes participating in DNA repair pathways (51). Thus, NPM::ALK ablates genomic stability by compromising the DNA mismatch repair process (Figure 1). Therefore, the role of NPM::ALK in promoting genomic instability by compromising the DNA repair process needs further study.
2.6 Epigenetic deregulation in ALK+ ALCL
Epigenetic modifications are heritable yet reversible covalent modifications in DNA or histones that alter the expression of genes without affecting DNA code. The most studied and significant modifications are the methylation of DNA at cytosine residues that function to repress gene expression (52) and the methylation or acetylation of distinct amino acids of the histone tail that dictate their repressive or activating properties (53). These modifications and their combinations dictate nucleosome positioning and local chromatin conformation that provide access to transcriptional regulators to modulate gene expression. DNA modifications direct histone modifications, and methylation of DNA causes steric hindrance to the transcriptional regulators (54). Moreover, the interaction of DNA with methyl-binding proteins, such as methyl CpG binding protein 2 (MeCP2), also prevents transcription factor binding at the locus causing repression of the target gene (52, 55).
NPM::ALK regulates the transcriptional silencing of many gene promoters and enhancer regions that encode tumor suppressors through its downstream effector transcription factors (Figure 1) (56). NPM::ALK activates the transcription factor STAT3, which upregulates DNA methyltransferase 1 (DNMT1) to methylate target genes for repression (57). As an example, IL-2Rγ promoter methylation is induced by NPM::ALK. NPM::ALK promotes STAT3 binding to the IL-2Rγ promoter, which then recruits DNMT1 to its promoter for its silencing (58). Notably, deleting DNMT1 abrogates lymphomagenesis, suggesting a therapeutic opportunity for targeting DNMT1 in patients who develop resistance to ALK inhibitor treatment (59). ALK+ALCL also exhibits CpG Island methylation at STAT5A, a tumor suppressor that reciprocally suppresses NPM::ALK gene expression by binding to its enhancer (60). These results show that silencing of tumor suppressor genes by DNA methylation may contribute to the neoplastic transformation of ALK+ ALCL.
In addition to DNA methylation, gene expression is regulated by the chromatin remodeling machinery, which modulates accessibility of the chromatin. Deregulation of chromatin remodelers has been demonstrated to participate in cancer development as well as lymphomagenesis (61, 62). Among them, SWI/SNF is a multi-subunit chromatin remodeling complex that uses the energy generated by ATP hydrolysis to displace or evict nucleosomes and further regulates local chromatin conformation (63). Expression of BRM-Related Gene1 (BRG1), a core component of the human SWI/SNF complex (64), is mediated by NPM::ALK. Further, the expression of BRG1 is dependent on the kinase activity of NPM::ALK. Knockdown of BRG1 in ALK+ ALCL cells results in a decrease in cell viability compared to scramble shRNA control (65). The role of other chromatin remodelers in the pathogenesis of ALK+ ALCL needs further investigation, and the relationship between the chromatin remodelers and NPM::ALK is still largely underexplored.
Loss of cellular identity is intrinsic to neoplastic transformation. ALCLs, despite originating from T-cells, exhibit downregulation of the transcriptional program that defines its T-cell phenotype. The pharmacologic treatment combining DNA demethylation and histone acetylation was insufficient to restore the T-cell phenotype in ALK+ ALCL cells. This suggests that additional stimulus is required to repress the T-cell phenotype. However, other T-cell lymphoma cells exposed to the same treatment exhibited expression of genes characteristic of ALCL (ID2, LGALS4, c-JUN) as well as loss of T-cell phenotype marked by loss of CD3, LCK, and ZAP70 expression indicating that global DNA demethylation and histone acetylation are critical for cellular reprogramming towards an ALCL-like phenotype (66).
The combinatorial pattern of DNA methylation and histone post-translational modifications (PTMs) are increasingly appreciated as epigenetic signatures of cancer subtypes. These modifications regulate cellular processes, such as cell cycle regulation, apoptosis, and DNA damage response (67–70). BCL2L11, also known as BIM (Bcl-2 interacting mediator of cell death), a Bcl-2 homology 3 (BH3)-only proapoptotic protein that belongs to the Bcl-2 family, is epigenetically silenced via the combinatorial deregulation of DNA methylation and histone acetylation in ALK+ ALCL (66, 71). Recruitments of MeCP2 and Sin3a/histone deacetylase1/2 (HDAC1/2) corepressor complex to the BIM promoter contributes to its silencing. Exposure of the DNA methylase inhibitor, 5-azacytidine, or the HDAC inhibitor, trichostatin, alone to ALK+ ALCL cells is not only able to rescue the expression of BIM at both mRNA and protein level but also increases apoptosis (71). This suggests that DNA methylation and histone acetylation together may contribute to the pathogenesis of ALK+ ALCL. In this regard, the pharmacological modulation of altered epigenetic machinery may represent novel therapeutic interventions.
Histone PTMs alone can also dictate disease-specific changes in the transcriptional program, and the pattern of histone PTMs can be utilized as a novel biomarker of disease subtypes (72). HDAC inhibitors have already been approved by the Food and Drug Administration (FDA) for the treatment of T-cell malignancies (73–75). However, the comprehensive landscape of histone PTMs, such as methylation and acetylation (66, 76), phosphorylation (77), ubiquitination (78), and sumoylation (79) for different classes of ALCL is yet to be determined. Since the current therapeutic approach of using HDAC inhibitors has been shown to cause nonselective toxicity, further understanding of the comprehensive epigenetic landscape of ALCL is warranted as it may lead to discoveries of novel histone modifications and their writers and erasers, which can be targeted for precision therapeutics (80). Evaluation of ALCL subtypes with highly sensitive proteomic approaches for histone modification analysis as well as single-cell proteomic approaches with enrichment and analysis of histone PTMs will add significant value to define the epigenetic signature of the disease.
2.7 Deregulated MicroRNA repertoire in ALK+ ALCL
It has been observed that nearly 90% of the human genome is transcriptionally active, yet only 1.4% of this transcriptome is constituted by protein-coding mRNA (81). The role of non-coding RNAs (ncRNA) is underappreciated yet critical to cell physiology and diseases, including ALCL. In ALK+ ALCL, the fusion protein NPM::ALK is associated with non-coding RNAs (ncRNAs), such as microRNAs to alter the gene expression signature of ALK+ ALCL (Figure 1) (82, 83). Along with tRNA and ribosomal RNA, the non-coding transcriptome is comprised of small nuclear RNA (snRNA), long noncoding RNA (lncRNA), and microRNA (miRNA). MicroRNAs are short, usually 20-23 nt long non-coding RNA that function by activating the RNA-induced silencing complex (RISC) against specific mRNA targets (84). miRNA array based on locked nucleic acid (LNA) technology containing 636 human and 425 murine miRNA probes performed on ALK+ ALCL cell lines identified distinct miRNA clusters from ALK+, to ALK- ALCL. These clusters are cross-validated with Npm::alk transgenic mice and primary ALK+ and ALK- ALCL to classify the miRNA unique to each disease group. These studies demonstrated strong upregulation of the miR-17-92 cluster in ALK+ ALCL and miR-155 upregulation in ALK- ALCL. Further, reduced expression of miR-101 is observed in both ALK+ ALCL and ALK- ALCL (82). Subsequent studies identified 32 miRNAs associated with ALK expression in vitro, presenting distinct miRNA expression profiles (85). These studies identify 7 miRNAs, of which 5 are upregulated (miR-512-3p, miR-886-5p, miR-886-3p, miR-708, miR-135b) and 2 downregulated (miR-146a, miR-155) in ALK+ ALCL. Another similar study identifies a distinct profile of miRNA that are specific to ALK+ or ALK- ALCL and cross-validated earlier findings. Moreover, it also identifies that miR-181a, which participates in the regulation of T-cell differentiation and TCR signaling, is significantly downregulated in ALK+ ALCL (86).
The role of exosomal miRNA in promoting disease dissemination of ALK+ ALCL has been recently reported. RNA sequencing studies identified 12 miRNAs that are significantly differentially expressed in the plasma of 20 NPM::ALK+ ALCL patients compared to healthy donors (n=5). Among these miRNAs, the level of miR-122-5p has further been validated as highly expressed in a larger cohort of ALCL patients (n=66) compared with healthy donors. Levels of miR-122-5p are elevated in late-stage (III-IV) ALCL patients compared to those with early-stage (I-II) disease. Interestingly, the expression of miR-122-5p is barely detectable in lymph nodes and other tissues but highly enriched in the liver of ALCL patients. In vitro and in vivo experiments indicate that miR-122-5p expressed in small extracellular vesicles promotes the proliferation and progression of ALCL cells (87). These mechanisms employed by miRNA using small extracellular vesicles for the pathogenesis of ALK+ ALCL may represent opportunities for discovery of novel mechanisms of disease dissemination as well as identification of prognostic biomarkers.
2.8 Immune evasion in ALK+ ALCL
Immune evasion by cancer cells is increasingly appreciated as an emerging hallmark of cancer. ALK+ ALCL cells exploit molecular mechanisms that bypass immune recognition (Figure 1). NPM::ALK-STAT3 signaling in ALK+ ALCL induces expression of transforming growth factor beta (TGF-β), IL-10, and cell surface receptor PD-L1 (CD274, B7H1), creating an immunosuppressive tumor microenvironment (88). The NPM::ALK-STAT3-DNMT1 pathway also epigenetically downregulates CD48, an immune surveillance molecule, to prevent tumor cell recognition by natural killer cells. STAT3 directly binds and methylates the promoter of CD48 in association with DNMT1. Pharmacologic inhibition of NPM::ALK, STAT3, or DNMT1 sensitizes ALK+ ALCL towards NK cell-mediated cytotoxicity in vitro. Further, expression of CD48 in ALK+ ALCL cell line increases NK cell-mediated cytotoxicity in vitro and in a xenograft mouse model (89). Similarly, NPM::ALK-STAT3 pathway induces the expression of ICOS, a member of the CD28 costimulatory receptor superfamily, by transcriptional induction, as well as suppresses the ICOS inhibitor miR-219 (90). Since ICOS engagement promotes ALK+ ALCL proliferation, it is tempting to speculate that by engaging its ligand (ICOS-L), tumor-specific ICOS subverts other critical co-stimulatory signals from immune cells, impairing cytotoxic response to tumor cells. Previous studies suggest that ALK+ ALCLs and ALK+ ALCL cell lines, do not express TNF-α as a result of promoter methylation, thus preventing its proapoptotic function on tumor cells (91). Importantly, inhibition of DNMT1 by 5’-aza-2’-deoxy-cytidine (5-ADC) rescues the expression of TNF-α mRNA and protein. Further, exogenous TNF-α expression inhibits the growth of ALK+ ALCL cell lines and induces the activation of apoptotic pathway intermediates, namely caspase 8 and caspase 3. Hence, inhibition of DNMT1 not only triggers the NK cell-mediated cytotoxicity but also promotes the proapoptotic signaling pathway in ALK+ ALCL, raising the possibility of DNA methyltransferase inhibitors as a therapeutic option for ALK+ ALCL. The observation that the serum titers of anti-ALK antibodies in patients are inversely proportional to stage stratification and progression of disease indicates that NPM::ALK protein is immunogenic and triggers a natural immune response that keeps a check on disease progression to some extent (92). Therefore, it will be important to comprehensively investigate NPM::ALK-mediated immune escape mechanisms. A better understanding of the immune evasion mechanism will help in developing potential alternative or combinatorial therapeutic interventions for ALK+ ALCL.
2.9 Deregulation of transcription factors in ALK+ ALCL
Various models have been proposed for the origin of malignant cells in ALK+ ALCL. The expression of CD4 or CD8 and CD30, along with clonal T-cell receptor (TCR) rearrangement, suggests that the malignant cells may originate from activated T cells (93), while the expression of FoxP3, IL10, and TGFβ suggests a regulatory T cell origin (88), and BATF and BATF3 expression is associated with a Th17/group 3 innate lymphoid cell origin (94). In addition, NPM::ALK-transformed CD4+ T lymphocytes and primary ALK+ ALCL biopsies share characteristics with early T cell precursors (51). Further, ALK+ ALCL cells overexpress stem cell transcription factors (OCT4, SOX2, and NANOG) and HIF2A, which regulate hematopoietic precursor differentiation and cell growth. These findings suggest that NPM::ALK signaling may trigger dedifferentiation to early thymic progenitor-like characteristics in CD30+ mature CD4+ T cells (95). In another study utilizing the RAG2-/- mice model, which lacks the machinery to produce mature T or B cells (96), it was shown that NPM::ALK is capable of promoting thymic T cell maturation and TCR-independent tumor formation, suggesting that the initial stage of ALK+ ALCL development may occur in the thymus (97).
Further, constitutive activation of STAT3 is highly prevalent in ALK+ ALCL and contributes to its pathogenesis. NPM::ALK interacts and phosphorylates STAT3 leading to its activation and nuclear translocation, where it regulates the transcription of a number of genes known to be involved in apoptosis and cell cycle progression (Figure 1) (35, 98). In ALK+ ALCL, the activation of STAT3 is multifactorial. JAK3, a major physiologic activator of STAT3, is highly activated in ALK+ ALCL lines and primary tumors (34). JAK3 interacts with NPM::ALK, and its inhibition decreases the tyrosine kinase activity of NPM::ALK (99, 100). Constitutive activation of STAT3 in ALK+ ALCL is also contributed by the downregulation of SH2 domain-containing protein tyrosine phosphatase-1 (SHP1) in ALK+ ALCL (101, 102). SHP1 interacts with JAK and NPM::ALK and dephosphorylate crucial tyrosine sites and thus inhibits the kinase activity (101, 103). ALK+ ALCL from children and adult patients exhibit loss of SHP1 at a frequency of 50% and 86%, respectively. Further, SHP1 is methylated and thus silenced in a number of ALK+ ALCL cases (101, 102).
ALK+ ALCL cells also aberrantly express multiple members of the activator protein-1 (AP-1) family of transcription factors, which includes proteins of the Jun, Fos, ATF, and Mf subfamilies (104). AP-1 family proteins regulate a wide range of cellular and biological activities, including cell cycle and proliferation, apoptosis, autophagy, and lipid synthesis (105). They also regulate cell migration and invasion as well as inflammatory response and immune cell development and activation. Studies have shown that AP-1 proteins play a pivotal role in promoting cell survival, proliferation, and suppression of AP-1 proteins can lead to apoptosis in ALK+ ALCL (94, 106, 107). Since AP-1 family proteins regulate a myriad of signaling pathways, further investigation will be required to comprehensively understand their impact on the ALK+ ALCL pathogenesis.
In addition, C/EBPβ, CCAAT enhancer binding protein, a transcription factor that belongs to the C/ECP leucine zipper transcription factor family, is highly expressed in ALK+ ALCLs (108). The overexpression of C/EBPβ is mediated through the NPM::ALK-STAT3 axis and is dependent on the kinase activity of NPM::ALK (108, 109). Moreover, NPM::ALK also fosters stability and translation of C/EBPβ mRNA via enhancing binding of AU-binding protein HuR to the 3′-UTR of C/EBPβ transcript (110). C/EBPβ modulates gene expression and miRNA levels to promote the transformation, proliferation, and survival of the malignant cells in ALK+ ALCL (111, 112). Therefore, targeting the deregulated transcription factors and the signaling pathways regulated by them may serve as novel therapeutic interventions for ALK+ ALCL.
2.10 Deregulated cytokine signaling in ALK+ ALCL
Cytokine and cytokine receptor signaling orchestrate the immune response, hematopoiesis, cell differentiation, and cell growth (113). There is an aberrant cytokine repertoire in ALK+ ALCL (Figure 1). Integrated unbiased N-glycoproteomic and transcriptomic profiling of 32 different B cell, T cell, and NK cell lymphoma cell lines has identified many cytokine receptors, including the interleukin receptor IL-R, as well as T helper (Th) receptors, expressed by ALK+ ALCL cells (8). Similarly, the level of IL-2R, Oncostatin M (OSM), IL-6, IL-8, IL-9, IL-10, IL-17a, IL-22, and soluble CD30 is decreased in either pediatric or adult ALK+ ALCL patient samples after they reached complete remission (114–116). There is a correlation between stages of the disease, presence of the minimal disseminated disease, anti-ALK antibody titers, and risk of relapse with concentrations of cytokines including IL-6, interferon-γ (IFN-γ), IFN-γ induced protein as well as sIL-2R among ALK+ ALCL pediatric patients (114). Moreover, levels of IL-6 demonstrated an independent prognostic value with a hazard ratio of 2.9 ± 0.4.
In addition, exogenous NPM::ALK expression leads to significant reductions of GM-CSF, TNF, and IL2 (51). Inhibition of NPM::ALK reduces the expression of cytokine receptor proteins, including IL-1R1, IL-1R2, IL-1RAP, IL-2RA, IL-4, IL-18RA, and IL-31RB (8). These observations suggest that constitutively activated ALK signaling contributes to deregulation of cytokine signaling.
Functional studies reveal that NPM::ALK regulates multiple JAK-STAT pathways, including IL-2/STAT5, and IL-6/STAT3 to participate in the aberrant cytokine secretion in ALK+ ALCL (8, 117). Particularly, NPM::ALK induces upregulation of STAT3 and STAT5 expression, which upregulates IL-31RB in ALK+ ALCL (118). In addition to STATs, NPM::ALK also enhances cytokine production by inducing the expression of other transcription factors, such as AP-1. AP-1 binds to promotors of multiple cytokines and thus regulates IL17F, IL22, IL26, and IL23R genes in ALK+ ALCL (94, 119).
Besides activation, NPM::ALK also deregulates the cytokine signaling pathway by suppressing transcription factor function. Among normal human endothelial cells, STAT1 is one of the major modulators of IFN-γ, which can further antagonize IL-6-mediated STAT3 activation (120). During activation, STAT1 forms a homodimer. It can also bind with STAT3 and form a heterodimer. The gene expression levels and specificities are modulated by the STAT1 homodimer vs heterodimer ratio (121). In ALK+ ALCL, NPM::ALK also downregulates STAT1 to antagonize STAT3 and further decrease the production of antitumor cytokine IFN-γ (122).
Further, epigenetic modulation also contributes to cytokine deregulation in ALK+ ALCL. It has been reported that NPM::ALK downregulates SHP1 tyrosine phosphatase, a negative modulator of multiple cytokine signaling pathways, including Epo-R, IL-4, IL-13, IL-3R, IL-2R, through STAT3-mediated upregulation of DNA methyltransferase 1 in ALK+ ALCL (102, 123, 124).
The tumor microenvironment also contributes to the formation of deregulated cytokine repertoire (125, 126). However, the composition and cross-talk between the neoplastic cells and tumor microenvironment of ALK+ ALCL need further investigation.
2.11 Deregulated proteolysis in ALK+ ALCL
Deregulated proteolysis by ubiquitination or sumoylation contributes significantly to the sustained signaling of oncogenic proteins (127, 128). The proteasomal degradation process of target proteins requires small ubiquitin binding to the substrate (127). Similarly, SUMOylation is another post-translational modification characterized by the reversible conjugation of small ubiquitin-like modifiers (SUMOs) with the target protein. SUMOylation modification often competes with ubiquitin for substrate binding and is believed to protect candidate proteins from proteasomal degradation (128). Studies suggest that the SUMOylating of NPM::ALK antagonizes its ubiquitination and subsequent degradation prolonging its oncogenic signaling (129). Further, the removal of sumoylation by SENP1 (a sentrin-specific family of proteases) promotes NPM::ALK protein turnover and ensues a decrease in cell viability, cell proliferation, and colony formation ability. It can be surmised that targeting NPM::ALK degradation may have therapeutic benefits in ALK+ ALCL that are resistant to NPM::ALK kinase inhibitors. In this regard, several efforts are underway to develop ALK protein degraders at different levels of preclinical or clinical settings (130–133).
3 Pathobiology of ALK- ALCL
ALK- ALCL is a CD30+ large T-cell lymphoma that typically affects the older population and has variable prognosis (134, 135). Currently, ALK- ALCL is subdivided into three classes, namely systemic ALCL, breast implant-associated ALCL, and primary cutaneous ALCL. Depending upon the genetic lesions acquired, the pathogenic mechanisms and disease aggressiveness may vary.
3.1 Pathobiology of systemic ALK- ALCL
In ALK- ALCL, two gene rearrangements and identified recurrent mutations subclassify ALK- ALCL into three more categories, namely fusion involving DUSP22::IRF4, fusions involving TP63 gene, and other types of ALK- ALCL (Figure 2).
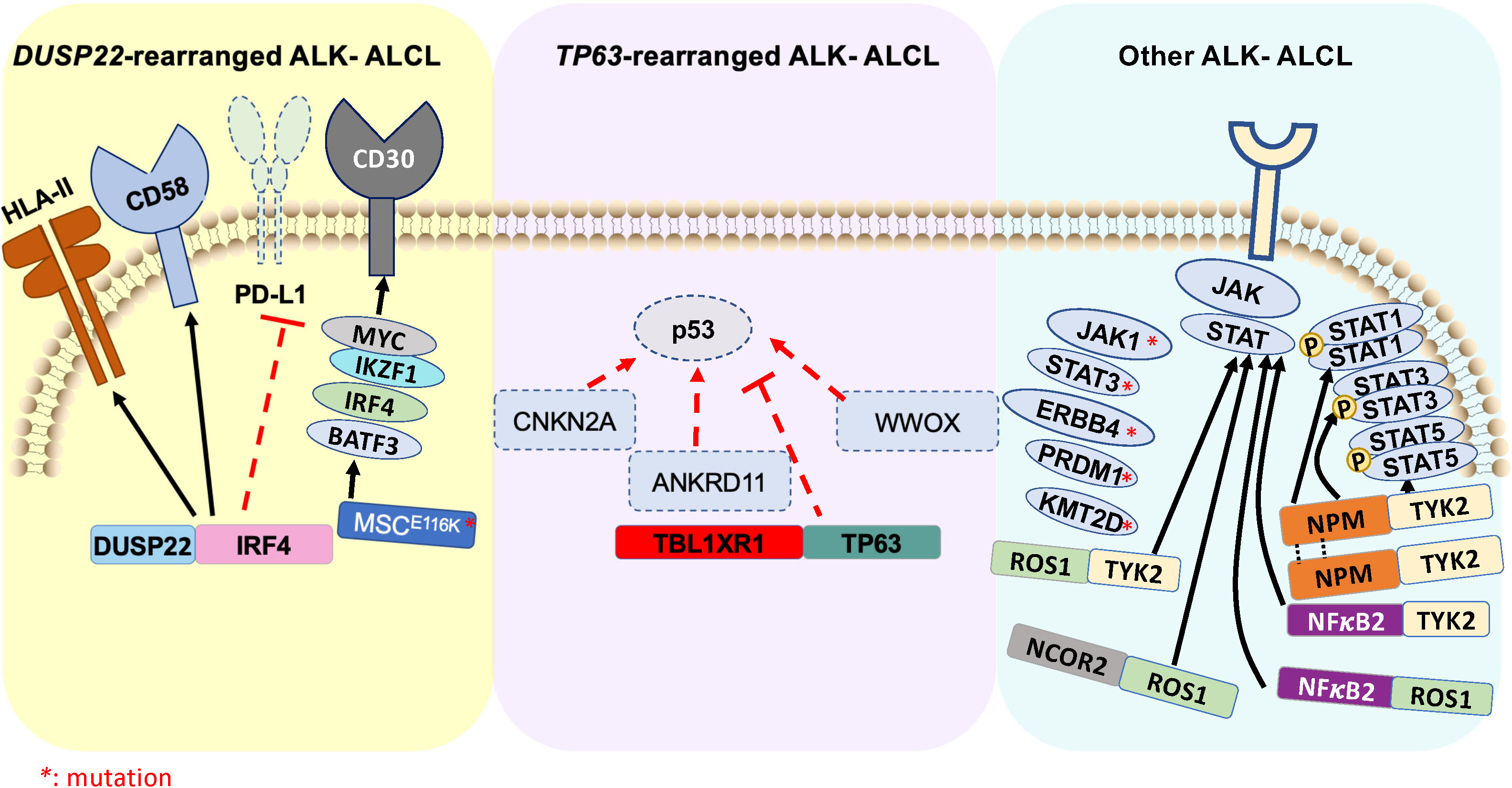
Figure 2 Schematic illustration of genomic rearrangements that contribute to pathogenesis of ALK- ALCL () represents ALK- ALCL harboring DUSP22-rearrangement and associated signaling pathways. () represents ALK- ALCL harboring TP63-rearrangement and associated signaling pathways. () represents various driver oncogenic fusions involving tyrosine kinase and transcription factors, as well as high-frequency mutations that constitute an independent subgroup of ALK- ALCL.
3.1.1 DUSP22-rearranged ALK- ALCL
Rearrangement of DUSP22 occurs near the DUSP22::IRF4 locus on 6p25.3 (136). The FRA7H fragile site on 7q32.3 is the most affected gene in the translocation t (6;7)(p25.3,q32.3). DUSP22 rearrangements are detected at a frequency of 30% in ALK- ALCL cases using fluorescence in situ hybridization (FISH). This rearrangement leads to downregulation of DUSP22, and upregulation of the microRNA miR-29 on 7q32.3 but does not affect the expression of IRF4. This subgroup lacks the expression of genes associated with JAK-STAT3 signaling, but results in overexpression of the immunogenic cancer-testis antigen, marked DNA hypomethylation, and exhibits a reduced expression of PD-L1 and high expression of CD58 and HLA class II (137). Further, a novel recurrent mutation in MSCE116K, a gene encoding musculin, has been recently identified in DUSP22 rearranged ALK- ALCL. This mutation induces the CD30-IRF4-MYC signaling axis (Figure 2) and drives cell proliferation (138). Interestingly, DUSP22 inhibits interleukin-6 (IL-6)-induced STAT3 activation, and its downregulation is another mechanism by which STAT3 signaling may be activated in ALK- ALCL (139). Notably, the 5-year overall survival of DUSP22-rearranged cases is approximately 85-90%, which is significantly higher than other ALK- ALCL (140).
3.1.2 TP63-rearranged ALK- ALCL
The TP63 gene, a member of the p53 family, is expressed either as a full-length isoform carrying a transactivator domain (TP63) or as an amino-deleted isoform (ΔNp63) (141). P63 triggers multiple signaling in cancer-specific contexts, including regulation of the cell cycle, apoptosis, stemness, and tumorigenesis (142). Approximal 8% of ALK- ALCL show rearrangement of TP63 in 3q28, frequently with TBL1XR1 as a result of an inversion (3)(q26q28) (Figure 2) (143). Rearrangements involving DUSP22 and TP63 are mutually exclusive. TP63-rearranged ALK- ALCL exhibits the worst prognosis within the ALCL subtypes, with a 5-year overall survival rate of 17%. The biological significance of the genetic rearrangement of TP63 in ALK- ALCL is yet to be determined.
3.1.3 Other types of ALK- ALCL
In ALK- ALCL, oncogenic mutations in JAK1 and/or STAT3 (Figure 2), which contribute to the consistent activation of the STAT3 signaling pathway, has been identified in nearly 20% of cases. In addition, oncogenic fusion genes involving a transcription factor and a tyrosine kinase, such as NFκB2::ROS1, NCOR2::ROS1, NFκB2::TYK2, and PABPC4::TYK2 have been identified in ALK- ALCL (144). These fusion chimeras result in increased STAT3 activity and develop ALCL phenotype via STAT3 signaling, suggesting that intercepting STAT3 activation may have a therapeutic advantage (94). A recent deep-targeted next-generation sequencing of 47 ALK+ and 35 ALK- ALCL demonstrated that, on average, ALK- ALCL harbor 4.2 mutations/patient compared to 2.6 mutations/patient for ALK+ ALCL. Among all the mutations, STAT3 and JAK1 mutations are the most frequent (26%) in ALK- ALCL. The mutations that predicted poor prognosis of ALK- ALCL includes TP53, STAT3, EPHA5, JAK1, PRDM1, LRP1B, and KMT2D (46).
Approximately 25% of ALK- ALCL expresses an oncogenic truncated ERB-B2 receptor tyrosine kinase-4 (ERBB4) that is not detected in ALK+ ALCL and PTCL-NOS and may likely form another subgroup of ALK- ALCL. ERBB4 expression is mutually exclusive of DUSP22, TP63, and ROS1 rearrangements. Pharmacologic inhibition of ERBB4 partly controls ALCL cell growth and disease progression in an ERBB4-positive patient-derived tumor graft model (145).
Hence, better understanding and targeting these rearrangements and mutation-mediated signaling pathways may serve as novel therapeutic interventions for different subtypes of ALK- ALCL.
3.2 Pathobiology of breast implant-associated ALCL
Breast implant-associated ALCL (BI-ALCL) is a distinct subtype of mature T-cell lymphoma. A persistent chronic inflammation occurring post-breast implants, particularly those with a textured outer shell, has been documented as the underlying cause of the disease (146). Cross-talk between the malignant cells and reactive cells in the microenvironment is thought to contribute to the formation of an inflammatory milieu characteristic of BI-ALCL. Elevated levels of IL-1β, IL-6, and TNF-α, the macrophage-activating cytokines, have been detected after culturing peripheral blood mononuclear cells obtained from healthy donors to the surfaces of the silicone breast implants for 4 days (147). However, no T-cell activation or specific effector cell subtype skewing has been observed. In addition, elevated expression of IL-13, IgE+ eosinophils, and mast cells in the microenvironment of primary BI-ALCL specimens suggests that allergic inflammation may contribute to the development of BI-ALCL (148).
Tumors display complex karyotypes with losses of chromosomes 1p,4q, 8p, 10p, 15, 16, 20 and gain of chromosomes 2, 9p, 12p, 19p, and 21 in BI-ALCL patients (149, 150). Targeted sequencing of 180 genes in 11 cases identified highly recurrent activating STAT3 mutations and recurrent deletions of 1p22 involving RPL5, a tumor suppressor that regulates cell proliferation. In addition, abnormalities were identified in TGF-β, PKC, WNT/β-catenin pathway, and inflammasome signaling. Amplifications involving TNFRSF11A and PDGFRA were also identified (151). Genomic profiling of BI-ALCL using a variety of sequencing platforms did not detect any genomic rearrangements involving ALK, DUSP22, and TP63, suggesting less heterogeneity in the genetic manifestation than other subtypes of ALCL (152). Predominant JAK-STAT pathway, TP53, and DNMT3A could be molecular drivers of BI-ALCL (153). JAK1 mutations were found in 13% (3/23) of cases, with the most frequent point mutation involving G1097(D, V or S) identified in 44% (4/9) of cases (152, 154, 155). The frequency of STAT3 mutations was 26% (6/23), with the most predominant mutations identified involving S614R (155). Apart from the JAK-STAT pathway, the second most frequent alterations in BI-ALCL were identified in epigenetic modifiers, including TET2, TET3, ARID4B, KDM5C, KDM6A, KMT2C/D, CHD2, CREBBP and SMARCB at the frequency of ~55-75% (150, 154). Currently, the first line of therapy involves surgical removal of the implant in combination with radiotherapy and standard chemotherapy. However, therapeutic targeting of JAK/STAT pathway and epigenetic deregulations may be considered as alternative therapeutic opportunities for BI-ALCL.
4 Pathobiology of primary cutaneous anaplastic large cell lymphoma
Primary cutaneous anaplastic large cell lymphoma (pc-ALCL) is a CD30+ lymphoproliferative disorder that manifests in the skin. The malignant cells exhibit large anaplastic and pleomorphic morphology with expression of CD30 in approximately 75% of cells. It has a relatively good prognosis in the absence of high-stage disease. The disease is currently classified as a subtype of primary cutaneous lymphoid proliferations and lymphomas that encompass a spectrum of other diseases, including lymphomatoid papulosis (LyP) (5). Morphologic features of pc-ALCL partly overlap with other diseases, such as LyP (156–158) and reactive lymphoid hyperplasia (159). Therefore, genetic characterization of the disease is critical for correct diagnosis.
The majority of pc-ALCLs lack genomic rearrangements in ALK, DUSP22, and TP63. Although unusual, ALK-positive cases with only skin lesions have been identified, the frequency of these cases and ALK fusion partners are yet to be further determined (156, 160, 161). Array comparative genomic hybridization analysis of pc-ALCL demonstrates that nearly 40% of cases exhibit chromatin imbalances targeting region encompassing genes RAF1 (3p25), CTSB (8p22), FES (15q26.1), FGFR1 (8p11), NRAS (1p13.2), MYCN (2p24.1), and CBFA2 (21q22.3) (162). Further, highly recurrent genomic loss of chromosomes 6q16-6q21, 6q27, and 13q34, as well as gain on the chromosome 7q31 and 17q, were also detected (163). In addition, a recurrent translocation involving IRF4::MUM1 at chromosome 6p25.3 was identified at the frequency of approximately 20 to 25% in pc-ALCL. However, the protein expression of IRF4 and MUM1 is also detected in systemic ALCL, and therefore, examining the expression of IRF4 and MUM1 by IHC does not reliably distinguish pc-ALCL from systemic ALCL (157). Further, we identified a novel recurrent NPM::TYK2 gene fusion in a proportion of primary cutaneous CD30+ lymphoproliferative disorders (15%), which activates STAT1/3/5 signaling and promotes cell proliferation (164). Importantly, a transgenic conditional knock-in Cd4-CreNPM::TYK2fl/fl mouse model demonstrates spontaneous development of CD30+ mature T-cell lymphoma with 90% penetrance (165). Hence, targeting TYK2 may serve as a therapeutic intervention for neoplasms harboring the NPM::TYK2 rearrangement.
5 Conclusions and future perspectives
Our understanding of ALK+ ALCLs has provided opportunities for targeted therapies such as small molecular inhibitors of ALK (crizotinib, alectinib, and ceritinib) and antibody-drug conjugates targeting the tumor-specific expression of CD30. Given that a significant fraction of patients experience relapse or refractory responses, there is a continued need for the development of novel therapeutic approaches that target aberrant signaling and/or immune evasion mechanisms. ALK- ALCLs remain a genetically heterogeneous group of mature T-cell lymphoma. The identification of gene rearrangements involving TYK2, DUSP22, TP63, and ERBB4, and genetic alterations characteristic of distinct subsets of ALK- ALCLs, will facilitate improved stratification of disease outcomes. The discovery of novel gene rearrangements within the ALK- ALCL category and their functional consequences will be crucial for precision therapeutics. BI-ALCL demonstrates a predominant role of activated JAK-STAT3 signaling as the major driver of disease partly due to recurrent point mutations in JAK1 and STAT3. Further, the contribution of epigenetic modifiers in conjunction with JAK-STAT3 signaling in the propagation of BI-ALCL has not been functionally explored and warrants further investigation. Moreover, an integrated approach of genetic, epigenetic, and proteomic profiling may offer an opportunity to identify novel therapeutic targets for ALCL. Despite studies that have identified the role of cytokine deregulation in ALCL, the composition of the microenvironment and its role in regulating tumor cell survival mechanisms remains largely unexplored.
Author contributions
RW and ML contributed to the ideas and structure of the manuscript. RW and ML wrote the review. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Vose JM, Neumann M, Harris ME. International peripheral T-cell and natural killer/T-cell lymphoma study: pathology findings and clinical outcomes. J Clin Oncol (2008) 26(25):4124–30. doi: 10.1200/JCO.2008.16.4558
2. Falini B, Stein H, Pileri S, Canino S, Farabbi R, Martelli MF, et al. Expression of lymphoid-associated antigens on Hodgkin’s and Reed-Sternberg cells of Hodgkin’s disease. An immunocytochemical study on lymph node cytospins using monoclonal antibodies. Histopathology (1987) 11(12):1229–42. doi: 10.1111/j.1365-2559.1987.tb01869.x
3. Morris SW, Kirstein MN, Valentine MB, Dittmer KG, Shapiro DN, Saltman DL, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin’s lymphoma. Science (1994) 263(5151):1281–4. doi: 10.1126/science.8122112
4. Duyster J, Bai RY, Morris SW. Translocations involving anaplastic lymphoma kinase (ALK). Oncogene (2001) 20(40 REV. ISS. 4):5623–37. doi: 10.1038/sj.onc.1204594
5. Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IB de O, Berti E, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: Lymphoid neoplasms. Leukemia (2022) 36(7):1720–48. doi: 10.1038/s41375-022-01620-2
6. McDonnell SRP, Hwang SR, Rolland D, Murga-Zamalloa C, Basrur V, Conlon KP, et al. Integrated phosphoproteomic and metabolomic profiling reveals NPM-ALK–mediated phosphorylation of PKM2 and metabolic reprogramming in anaplastic large cell lymphoma. Blood (2013) 122(6):958. doi: 10.1182/blood-2013-01-482026
7. Campo E, Jaffe ES, Cook JR, Quintanilla-Martinez L, Swerdlow SH, Anderson KC, et al. The international consensus classification of mature lymphoid neoplasms: a report from the clinical advisory committee. Blood (2022) 140(11):1229–53. doi: 10.1182/blood.2022015851
8. Rolland DCM, Basrur V, Jeon YK, McNeil-Schwalm C, Fermin D, Conlon KP, et al. Functional proteogenomics reveals biomarkers and therapeutic targets in lymphomas. Proc Natl Acad Sci (2017) 114(25):6581–6586. doi: 10.1073/pnas.1701263114
9. Andraos E, Dignac J, Meggetto F. NPM-ALK: A driver of lymphoma pathogenesis and a therapeutic target. Cancers (2021) 13:144. doi: 10.3390/cancers13010144
10. Marzec M, Kasprzycka M, Liu X, El-Salem M, Halasa K, Raghunath PN, et al. Oncogenic tyrosine kinase NPM/ALK induces activation of the rapamycin-sensitive mTOR signaling pathway. Oncogene (2007) 26(38):5606–14. doi: 10.1038/sj.onc.1210346
11. Lawrence B, Perez-Atayde A, Hibbard MK, Rubin BP, Cin PD, Pinkus JL, et al. TPM3-ALK and TPM4-ALK oncogenes in inflammatory myofibroblastic tumors. Am J Pathol (2000) 157(2):377–84. doi: 10.1016/S0002-9440(10)64550-6
12. Hernández L, Pinyol M, Hernández S, Beà S, Pulford K, Rosenwald A, et al. TRK-fused gene (TFG) is a new partner of ALK in anaplastic large cell lymphoma producing two structurally differentTFG-ALK translocations. Blood (1999) 94(9):3265–8. doi: 10.1182/blood.V94.9.3265
13. Colleoni GWB, Bridge JA, Garicochea B, Liu J, Filippa DA, Ladanyi M. ATIC-ALK: A novel variant ALK gene fusion in anaplastic large cell lymphoma resulting from the recurrent cryptic chromosomal inversion, inv(2)(p23q35). Am J Pathol (2000) 156(3):781–9. doi: 10.1016/S0002-9440(10)64945-0
14. Cools J, Wlodarska I, Somers R, Mentens N, Pedeutour F, Maes B, et al. Identification of novel fusion partners of ALK, the anaplastic lymphoma kinase, in anaplastic large-cell lymphoma and inflammatory myofibroblastic tumor. Genes Chromosomes Cancer (2002) 34(4):354–62. doi: 10.1002/gcc.10033
15. Armstrong F, Duplantier MM, Trempat P, Hieblot C, Lamant L, Espinos E, et al. Differential effects of X-ALK fusion proteins on proliferation, transformation, and invasion properties of NIH3T3 cells. Oncogene (2004) 23:36. doi: 10.1038/sj.onc.1207813
16. Feldman AL, Vasmatzis G, Asmann YW, Davila J, Middha S, Eckloff BW, et al. Novel TRAF1-ALK fusion identified by deep RNA sequencing of anaplastic large cell lymphoma. Genes Chromosomes Cancer (2013) 52(11):1097–102. doi: 10.1002/gcc.22104
17. Palacios G, Shaw TI, Li Y, Singh RK, Valentine M, Sandlund JT, et al. Novel ALK fusion in anaplastic large cell lymphoma involving EEF1G, a subunit of the eukaryotic elongation factor-1 complex. Leukemia (2017) 31(3):743–7. doi: 10.1038/leu.2016.331
18. Graetz D, Crews KR, Azzato EM, Singh RK, Raimondi S, Mason J, et al. Leukemic presentation of ALK-positive anaplastic large cell lymphoma with a novel partner, poly(A) binding protein cytoplasmic 1 (PABPC1), responding to single-agent crizotinib. Haematologica (2019) 104(5):e218. doi: 10.3324/haematol.2018.215103
19. Amano Y, Ishikawa R, Sakatani T, Ichinose J, Sunohara M, Watanabe K, et al. Oncogenic TPM3-ALK activation requires dimerization through the coiled-coil structure of TPM3. Biochem Biophys Res Commun (2015) 457(3):457–60. doi: 10.1016/j.bbrc.2015.01.014
20. Shi JH, Ling C, Wang TT, Zhang LN, Liu WW, Qin Y, et al. TRK-fused gene (TFG) regulates ULK1 stability via TRAF3-mediated ubiquitination and protects macrophages from LPS-induced pyroptosis. Cell Death Dis (2022) 13(1):93. doi: 10.1038/s41419-022-04539-9
21. Chiarle R, Voena C, Ambrogio C, Piva R, Inghirami G. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer (2008) 8(1):11–23. doi: 10.1038/nrc2291
22. Granja C, Lin LL, Yunis EJ, Relias V, Dasgupta JD. PLC gamma 1, a possible mediator of T cell receptor function. J Biol Chem (1991) 266(25):16277–80. doi: 10.1016/S0021-9258(18)55290-X
23. Gonen R, Beach D, Ainey C, Yablonski D. T cell receptor-induced activation of phospholipase C-γ1 depends on a sequence-independent function of the P-I region of SLP-76. J Biol Chem (2005) 280(9):8364–70. doi: 10.1074/jbc.M409437200
24. Li Y, Sedwick CE, Hu J, Altman A. Role for protein kinase Ctheta (PKCtheta) in TCR/CD28-mediated signaling through the canonical but not the non-canonical pathway for NF-kappaB activation. J Biol Chem (2005) 280(2):1217–23. doi: 10.1074/jbc.M409492200
25. Downward J, Graves JD, Warne PH, Rayter S, Cantrell DA. Stimulation of p21ras upon T-cell activation. Nature (1990) 346(6286):719–23. doi: 10.1038/346719a0
26. Colombetti S, Basso V, Mueller DL, Mondino A. Prolonged TCR/CD28 engagement drives IL-2-independent T cell clonal expansion through signaling mediated by the mammalian target of rapamycin. J Immunol (2006) 176(5):2730–8. doi: 10.4049/jimmunol.176.5.2730
27. Geissinger E, Sadler P, Roth S, Grieb T, Puppe B, Müller N, et al. Disturbed expression of the T-cell receptor/CD3 complex and associated signaling molecules in CD30+ T-cell lymphoproliferations. Haematologica (2010) 95(10):1697. doi: 10.3324/haematol.2009.021428
28. Ambrogio C, Martinengo C, Voena C, Tondat F, Riera L, di Celle PF, et al. NPM-ALK oncogenic tyrosine kinase controls T-cell identity by transcriptional regulation and epigenetic silencing in lymphoma cells. Cancer Res (2009) 69(22):8611–9. doi: 10.1158/0008-5472.CAN-09-2655
29. Bai RY, Ouyang T, Miething C, Morris SW, Peschel C, Duyster J. Nucleophosmin–anaplastic lymphoma kinase associated with anaplastic large-cell lymphoma activates the phosphatidylinositol 3-kinase/Akt antiapoptotic signaling pathway. Blood (2000) 96(13):4319–27. doi: 10.1182/blood.V96.13.4319
30. Bai RY, Dieter P, Peschel C, Morris SW, Duyster J. Nucleophosmin-anaplastic lymphoma kinase of large-cell anaplastic lymphoma is a constitutively active tyrosine kinase that utilizes phospholipase C-gamma to mediate its mitogenicity. Mol Cell Biol (1998) 18(12):6951–61. doi: 10.1128/MCB.18.12.6951
31. Green DR, Droin N, Pinkoski M. Activation-induced cell death in T cells. Immunol Rev (2003) 193:70–81. doi: 10.1034/j.1600-065X.2003.00051.x
32. Oyarzo MP, Medeiros LJ, Atwell C, Feretzaki M, Leventaki V, Drakos E, et al. c-FLIP confers resistance to FAS-mediated apoptosis in anaplastic large-cell lymphoma. Blood (2006) 107(6):2544–7. doi: 10.1182/blood-2005-06-2601
33. Staber PB, Vesely P, Haq N, Ott RG, Funato K, Bambach I, et al. The oncoprotein NPM-ALK of anaplastic large-cell lymphoma induces JUNB transcription via ERK1/2 and JunB translation via mTOR signaling. Blood (2007) 110(9):3374–83. doi: 10.1182/blood-2007-02-071258
34. Lai R, Rassidakis G, Lin Q, Atwell C, Medeiros L, Amin H. Jak3 activation is significantly associated with ALK expression in anaplastic large cell lymphoma. Hum Pathol (2005) 36(9):939–44. doi: 10.1016/j.humpath.2005.07.011
35. Zamo A, Chiarle R, Piva R, Howes J, Fan Y, Chilosi M, et al. Anaplastic lymphoma kinase (ALK) activates Stat3 and protects hematopoietic cells from cell death. Oncogene (2002) 21(7):1038–47. doi: 10.1038/sj.onc.1205152
36. Wei Z, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res (2002) 12:1. doi: 10.1038/sj.cr.7290105
37. Marzec M, Kasprzycka M, Liu X, Raghunath PN, Wlodarski P, Wasik MA. Oncogenic tyrosine kinase NPM/ALK induces activation of the MEK/ERK signaling pathway independently of c-Raf. Oncogene (2007) 26:6. doi: 10.1038/sj.onc.1209843
38. Slupianek A, Nieborowska-Skorska M, Hoser G, Morrione A, Majewski M, Xue L, et al. Role of phosphatidylinositol 3-kinase-Akt pathway in nucleophosmin/anaplastic lymphoma kinase-mediated lymphomagenesis. Cancer Res (2001) 61(5):2194–2199
39. Datta SR, Dudek H, Xu T, Masters S, Haian F, Gotoh Y, et al. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell (1997) 91(2):231–41. doi: 10.1016/S0092-8674(00)80405-5
40. Gao J, Yin M, Zhu Y, Gu L, Zhang Y, Li Q, et al. Prognostic significance and therapeutic potential of the activation of anaplastic lymphoma kinase/protein kinase B/mammalian target of rapamycin signaling pathway in anaplastic large cell lymphoma. BMC Cancer (2013) 13(1):471. doi: 10.1186/1471-2407-13-471
41. Icard P, Wu Z, Fournel L, Coquerel A, Lincet H, Alifano M. ATP citrate lyase: A central metabolic enzyme in cancer. Cancer Lett (2020) 471:125–34. doi: 10.1016/j.canlet.2019.12.010
42. Basappa J, ElAzzouny MA, Rolland DCM, Sahasrabuddhe AA, Ma K, Bazilevsky GA, et al. Tyrosine phosphorylation is critical for ACLY activity in lipid metabolism and cancer. bioRxiv (2020). doi: 10.1101/2020.01.20.910752
43. Kim JY, Kim H, Suk K, Lee WH. Activation of CD147 with cyclophilin a induces the expression of IFITM1 through ERK and PI3K in THP-1 cells. Mediators Inflammation (2010) 2010:821940. doi: 10.1155/2010/821940
44. Garcia-Bermudez J, Baudrier L, Bayraktar EC, Shen Y, La K, Guarecuco R, et al. Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature (2019) 567:7746. doi: 10.1038/s41586-019-0945-5
45. Salaverria I, Beà S, Lopez-Guillermo A, Lespinet V, Pinyol M, Burkhardt B, et al. Genomic profiling reveals different genetic aberrations in systemic ALK-positive and ALK-negative anaplastic large cell lymphomas. Br J Haematol (2008) 140(5):516–26. doi: 10.1111/j.1365-2141.2007.06924.x
46. Lobello C, Tichy B, Bystry V, Radova L, Filip D, Mraz M, et al. STAT3 and TP53 mutations associate with poor prognosis in anaplastic large cell lymphoma. Leukemia (2021) 35(5):1500–5. doi: 10.1038/s41375-020-01093-1
47. Youssif C, Goldenbogen J, Hamoudi R, Carreras J, Viskaduraki M, Cui YX, et al. Genomic profiling of pediatric ALK-positive anaplastic large cell lymphoma: a Children’s Cancer and Leukaemia Group Study. Genes Chromosomes Cancer (2009) 48(11):1018–26. doi: 10.1002/gcc.20701
48. Larose H, Prokoph N, Matthews JD, Schlederer M, Högler S, Alsulami AF, et al. Whole Exome Sequencing reveals NOTCH1 mutations in anaplastic large cell lymphoma and points to Notch both as a key pathway and a potential therapeutic target. Haematologica (2021) 106(6):1693–704. doi: 10.3324/haematol.2019.238766
49. Lyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem Rev (2006) 106(2):302–23. doi: 10.1021/cr0404794
50. Lobello C, Bikos V, Janikova A, Pospisilova S. The role of oncogenic tyrosine kinase NPM-ALK in genomic instability. Cancers (Basel) (2018) 10(3):64. doi: 10.3390/cancers10030064
51. Pawlicki JM, Cookmeyer DL, Maseda D, Everett JK, Wei F, Kong H, et al. NPM-ALK-induced reprogramming of mature TCR-stimulated T cells results in dedifferentiation and Malignant transformation. Cancer Res (2021) 81(12):3241–54. doi: 10.1158/0008-5472.CAN-20-2297
52. Jin B, Li Y, Robertson KD. DNA methylation: superior or subordinate in the epigenetic hierarchy? Genes Cancer (2011) 2(6):607. doi: 10.1177/1947601910393957
53. Millán-Zambrano G, Burton A, Bannister AJ, Schneider R. Histone post-translational modifications — cause and consequence of genome function. Nat Rev Genet (2022) 23(9):563–80. doi: 10.1038/s41576-022-00468-7
54. Cedar H, Bergman Y. Linking DNA methylation and histone modification: patterns and paradigms. Nat Rev Genet (2009) 10(5):295–304. doi: 10.1038/nrg2540
55. Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell (1997) 88(4):471–81. doi: 10.1016/S0092-8674(00)81887-5
56. Hassler MR, Pulverer W, Lakshminarasimhan R, Redl E, Hacker J, Garland GD, et al. Insights into the Pathogenesis of Anaplastic Large-Cell Lymphoma through Genome-wide DNA Methylation Profiling. Cell Rep (2016) 17(2):596–608. doi: 10.1016/j.celrep.2016.09.018
57. Zhang Q, Wang HY, Woetmann A, Raghunath PN, Odum N, Wasik MA. STAT3 induces transcription of the DNA methyltransferase 1 gene (DNMT1) in Malignant T lymphocytes. Blood (2006) 108(3):1058–64. doi: 10.1182/blood-2005-08-007377
58. Zhang Q, Wang HY, Liu X, Bhutani G, Kantekure K, Wasik M. IL-2R common gamma-chain is epigenetically silenced by nucleophosphin-anaplastic lymphoma kinase (NPM-ALK) and acts as a tumor suppressor by targeting NPM-ALK. Proc Natl Acad Sci USA (2011) 108(29):11977–82. doi: 10.1073/pnas.1100319108
59. Redl E, Sheibani-Tezerji R, Cardona CDJ, Hamminger P, Timelthaler G, Hassler MR, et al. Requirement of DNMT1 to orchestrate epigenomic reprogramming for NPM-ALK-driven lymphomagenesis. Life Sci Alliance (2020) 4(2):e202000794. doi: 10.26508/lsa.202000794
60. Zhang Q, Wang HY, Liu X, Wasik MA. STAT5A is epigenetically silenced by the tyrosine kinase NPM1-ALK and acts as a tumor suppressor by reciprocally inhibiting NPM1-ALK expression. Nat Med (2007) 13(11):1341–8. doi: 10.1038/nm1659
61. Wu R. Untangling the epigenetic imbalance in B cell lymphoma. Curr Pharmacol Rep (2020) 6(3):110–20. doi: 10.1007/s40495-020-00214-9
62. Nair SS, Kumar R. Chromatin remodeling in Cancer: A Gateway to regulate gene Transcription. Mol Oncol (2012) 6(6):611. doi: 10.1016/j.molonc.2012.09.005
63. Mittal P, Roberts CWM. The SWI/SNF complex in cancer — biology, biomarkers and therapy. Nat Rev Clin Oncol (2020) 17(7):435–48. doi: 10.1038/s41571-020-0357-3
64. Mardinian K, Adashek JJ, Botta GP, Kato S, Kurzrock R. SMARCA4: Implications of an altered chromatin-remodeling gene for cancer development and therapy. Mol Cancer Ther (2021) 20(12):2341–51. doi: 10.1158/1535-7163.MCT-21-0433
65. Garland GD, Ducray SP, Jahangiri L, Pucci P, Burke GAA, Monahan J, et al. Brg1 and npm-alk are co-regulated in anaplastic large-cell lymphoma; brg1 is a potential therapeutic target in alcl. Cancers (Basel) (2022) 14(1):151. doi: 10.3390/cancers14010151
66. Joosten M, Seitz V, Zimmermann K, Sommerfeld A, Berg E, Lenze D, et al. Histone acetylation and DNA demethylation of T cells result in an anaplastic large cell lymphoma-like phenotype. Haematologica (2013) 98(2):247–54. doi: 10.3324/haematol.2011.054619
67. Shilatifard A. The COMPASS family of histone H3K4 methylases: Mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem (2012) 81(1):65–95. doi: 10.1146/annurev-biochem-051710-134100
68. Cloos PAC, Christensen J, Agger K, Helin K. Erasing the methyl mark: histone demethylases at the center of cellular differentiation and disease. Genes Dev (2008) 22(9):1115–40. doi: 10.1101/gad.1652908
69. D’Oto A, Tian Q, Davidoff AM, Yang J. Histone demethylases and their roles in cancer epigenetics. J Med Oncol Ther (2016) 01(02):34–40. doi: 10.35841/medical-oncology.1.2.34-40
70. Haery L, Thompson RC, Gilmore TD. Histone acetyltransferases and histone deacetylases in B- and T-cell development, physiology and Malignancy. Genes Cancer Impact Journals LLC; (2015) 6:184–213. doi: 10.18632/genesandcancer.65
71. Piazza R, Magistroni V, Mogavero A, Andreoni F, Ambrogio C, Chiarle R, et al. Epigenetic silencing of the proapoptotic gene BIM in anaplastic large cell lymphoma through an meCP2/SIN3a deacetylating complex. Neoplasia (2013) 15(5):511–IN17. doi: 10.1593/neo.121784
72. Onder O, Wu R, Wysocka M, Haun PL, Kim E, Garcia BA, et al. Epiproteomic landscape and histone code of cutaneous T-cell lymphoma/sézary syndrome. Blood (2018) 132(Supplement 1):780. doi: 10.1182/blood-2018-99-115347
73. Suraweera A, O’Byrne KJ, Richard DJ. Combination therapy with histone deacetylase inhibitors (HDACi) for the treatment of cancer: Achieving the full therapeutic potential of HDACi. Front Oncol (2018) 8(MAR). doi: 10.3389/fonc.2018.00092
74. Jones PA, Issa JPJ, Baylin S. Targeting the cancer epigenome for therapy. Nat Rev Genet (2016) 17(10):630–41. doi: 10.1038/nrg.2016.93
75. Guha M. HDAC inhibitors still need a home run, despite recent approval. Nat Rev Drug Discovery (2015) 14(4):225–6. doi: 10.1038/nrd4583
76. Zhang P, Zhang M. Epigenetic alterations and advancement of treatment in peripheral T-cell lymphoma. Clin Epigenet (2020) 12(1):169. doi: 10.1186/s13148-020-00962-x
77. Pérez-Cadahía B, Drobic B, Davie JR. H3 phosphorylation: dual role in mitosis and interphase. Biochem Cell Biol (2009) 87(5):695–709. doi: 10.1139/O09-053
78. Weake VM, Workman JL. Histone ubiquitination: triggering gene activity. Mol Cell (2008) 29(6):653–63. doi: 10.1016/j.molcel.2008.02.014
79. Shiio Y, Eisenman RN. Histone sumoylation is associated with transcriptional repression. Proc Natl Acad Sci USA (2003) 100(23):13225–30. doi: 10.1073/pnas.1735528100
80. Li W, Sun Z. Mechanism of action for HDAC inhibitors-insights from omics approaches. Int J Mol Sci (2019) 20(7):1616. doi: 10.3390/ijms20071616
82. Merkel O, Hamacher F, Laimer D, Sifft E, Trajanoski Z, Scheideler M, et al. Identification of differential and functionally active miRNAs in both anaplastic lymphoma kinase (ALK)+ and ALK- anaplastic large-cell lymphoma. Proc Natl Acad Sci USA (2010) 107(37):16228–33. doi: 10.1073/pnas.1009719107
83. Fuchs S, Naderi J, Meggetto F. Non-coding RNA networks in ALK-positive anaplastic-large cell lymphoma. Int J Mol Sci (2019) 20:2150. doi: 10.3390/ijms20092150
84. Gregory RI, Chendrimada TP, Cooch N, Shiekhattar R. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell (2005) 123(4):631–40. doi: 10.1016/j.cell.2005.10.022
85. Liu C, Iqbal J, Teruya-Feldstein J, Shen Y, Dabrowska MJ, Dybkaer K, et al. MicroRNA expression profiling identifies molecular signatures associated with anaplastic large cell lymphoma. Blood (2013) 122(12):2083–92. doi: 10.1182/blood-2012-08-447375
86. Steinhilber J, Bonin M, Walter M, Fend F, Bonzheim I, Quintanilla-Martinez L. Next-generation sequencing identifies deregulation of microRNAs involved in both innate and adaptive immune response in ALK+ ALCL. PloS One (2015) 10(2):e0117780. doi: 10.1371/journal.pone.0117780
87. Damanti CC, Ferrone L, Gaffo E, Garbin A, Tosato A, Contarini G, et al. Plasma small-extracellular vesicles enriched in miR-122-5p promote disease aggressiveness in pediatric anaplastic large-cell lymphoma. Cancer Commun (Lond) (2023) 43(5):630–4. doi: 10.1002/cac2.12415
88. Kasprzycka M, Marzec M, Liu X, Zhang Q, Wasik MA. Nucleophosmin/anaplastic lymphoma kinase (NPM/ALK) oncoprotein induces the T regulatory cell phenotype by activating STAT3. Proc Natl Acad Sci (2006) 103(26):9964–9. doi: 10.1073/pnas.0603507103
89. Wu R, Ivan E, Sahasrabuddhe AA, Shaw T, Mullighan CG, Leventaki V, et al. Epigenetic modulation of CD48 by NPM-ALK promotes immune evasion in ALK+ ALCL. Blood (2019) 134(Supplement_1):1510. doi: 10.1182/blood-2019-127453
90. Zhang Q, Wang H, Kantekure K, Paterson JC, Liu X, Schaffer A, et al. Oncogenic tyrosine kinase NPM-ALK induces expression of the growth-promoting receptor ICOS. Blood (2011) 118(11):3062–71. doi: 10.1182/blood-2011-01-332916
91. Zhang Q, Wang HY, Bhutani G, Liu X, Paessler M, Tobias JW, et al. Lack of TNFalpha expression protects anaplastic lymphoma kinase-positive T-cell lymphoma (ALK+ TCL) cells from apoptosis. Proc Natl Acad Sci USA (2009) 106(37):15843–8. doi: 10.1073/pnas.0907070106
92. Mussolin L, Damm-Welk C, Pillon M, Zimmermann M, Franceschetto G, Pulford K, et al. Use of minimal disseminated disease and immunity to NPM-ALK antigen to stratify ALK-positive ALCL patients with different prognosis. Leukemia (2013) 27(2):416–22. doi: 10.1038/leu.2012.205
93. Krenacs L, Wellmann A, Sorbara L, Himmelmann AW, Bagdi E, Jaffe ES, et al. Cytotoxic cell antigen expression in anaplastic large cell lymphomas of T- and null-cell type and hodgkin’s disease: Evidence for distinct cellular origin. Blood (1997) 89(3):980–9. doi: 10.1182/blood.V89.3.980
94. Schleussner N, Merkel O, Costanza M, Liang HC, Hummel F, Romagnani C, et al. The AP-1-BATF and -BATF3 module is essential for growth, survival and TH17/ILC3 skewing of anaplastic large cell lymphoma. Leukemia (2018) 32(9):1994–2007. doi: 10.1038/s41375-018-0045-9
95. Congras A, Hoareau-Aveilla C, Caillet N, Tosolini M, Villarese P, Cieslak A, et al. ALK-transformed mature T lymphocytes restore early thymus progenitor features. J Clin Invest (2020) 130(12):6395–408. doi: 10.1172/JCI134990
96. Shinkai Y, Rathbun G, Lam KP, Oltz EM, Stewart V, Mendelsohn M, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell (1992) 68(5):855–67. doi: 10.1016/0092-8674(92)90029-C
97. Malcolm TIM, Villarese P, Fairbairn CJ, Lamant L, Trinquand A, Hook CE, et al. Anaplastic large cell lymphoma arises in thymocytes and requires transient TCR expression for thymic egress. Nat Commun (2016) 7:10087. doi: 10.1038/ncomms10087
98. Bromberg J, Darnell JE. The role of STATs in transcriptional control and their impact on cellular function. Oncogene (2000) 19(21):2468–73. doi: 10.1038/sj.onc.1203476
99. Crockett DK, Lin Z, Elenitoba-Johnson KSJ, Lim MS. Identification of NPM-ALK interacting proteins by tandem mass spectrometry. Oncogene (2004) 23(15):2617–29. doi: 10.1038/sj.onc.1207398
100. Amin HM, Medeiros LJ, Ma Y, Feretzaki M, Das P, Leventaki V, et al. Inhibition of JAK3 induces apoptosis and decreases anaplastic lymphoma kinase activity in anaplastic large cell lymphoma. Oncogene (2003) 22(35):5399–407. doi: 10.1038/sj.onc.1206849
101. Khoury JD, Rassidakis GZ, Medeiros LJ, Amin HM, Lai R. Methylation of SHP1 gene and loss of SHP1 protein expression are frequent in systemic anaplastic large cell lymphoma. Blood (2004) 104(5):1580–1. doi: 10.1182/blood-2004-03-1151
102. Honorat JF, Ragab A, Lamant L, Delsol G, Ragab-Thomas J. SHP1 tyrosine phosphatase negatively regulates NPM-ALK tyrosine kinase signaling. Blood (2006) 107(10):4130–8. doi: 10.1182/blood-2005-06-2421
103. Klingmüller U, Lorenz U, Cantley LC, Neel BG, Lodish HF. Specific recruitment of SH-PTP1 to the erythropoietin receptor causes inactivation of JAK2 and termination of proliferative signals. Cell (1995) 80(5):729–38. doi: 10.1016/0092-8674(95)90351-8
104. Mathas S, Hinz M, Anagnostopoulos I, Krappmann D, Lietz A, Jundt F, et al. Aberrantly expressed c-Jun and JunB are a hallmark of Hodgkin lymphoma cells, stimulate proliferation and synergize with NF-kappa B. EMBO J (2002) 21(15):4104–13. doi: 10.1093/emboj/cdf389
105. Schiefer AI, Vesely P, Hassler MR, Egger G, Kenner L. The role of AP-1 and epigenetics in ALCL. Front Biosci (Schol Ed) (2015) 7:226–35. doi: 10.2741/s436
106. Wu Z, Nicoll M, Ingham RJ. AP-1 family transcription factors: a diverse family of proteins that regulate varied cellular activities in classical hodgkin lymphoma and ALK+ ALCL. Exp Hematol Oncol (2021) 10(1):4. doi: 10.1186/s40164-020-00197-9
107. Turner SD, Yeung D, Hadfield K, Cook SJ, Alexander DR. The NPM-ALK tyrosine kinase mimics TCR signalling pathways, inducing NFAT and AP-1 by RAS-dependent mechanisms. Cell Signal (2007) 19(4):740–7. doi: 10.1016/j.cellsig.2006.09.007
108. Quintanilla-Martinez L, Pittaluga S, Miething C, Klier M, Rudelius M, Davies-Hill T, et al. NPM-ALK-dependent expression of the transcription factor CCAAT/enhancer binding protein beta in ALK-positive anaplastic large cell lymphoma. Blood (2006) 108(6):2029–36. doi: 10.1182/blood-2005-10-014258
109. Anastasov N, Bonzheim I, Rudelius M, Klier M, Dau T, Angermeier D, et al. C/EBPβ expression in ALK-positive anaplastic large cell lymphomas is required for cell proliferation and is induced by the STAT3 signaling pathway. Haematologica (2010) 95(5):760. doi: 10.3324/haematol.2009.014050
110. Bergalet J, Fawal M, Lopez C, Desjobert C, Lamant L, Delsol G, et al. HuR-mediated control of C/EBPbeta mRNA stability and translation in ALK-positive anaplastic large cell lymphomas. Mol Cancer Res (2011) 9(4):485–96. doi: 10.1158/1541-7786.MCR-10-0351
111. Bonzheim I, Irmler M, Klier-Richter M, Steinhilber J, Anastasov N, Schäfer S, et al. Identification of C/EBPβ target genes in ALK+ anaplastic large cell lymphoma (ALCL) by gene expression profiling and chromatin immunoprecipitation. PloS One (2013) 8(5):e64544. doi: 10.1371/journal.pone.0064544
112. Piva R, Pellegrino E, Mattioli M, Agnelli L, Lombardi L, Boccalatte F, et al. Functional validation of the anaplastic lymphoma kinase signature identifies CEBPB and BCl2A1 as critical target genes. J Clin Invest (2006) 116(12):3171–82. doi: 10.1172/JCI29401
113. O’Shea JJ, Gadina M, Siegel RM. Cytokines and cytokine receptors. Clinical immunology: principles and practice. (Fifth Edition) (2019) 127–155.e1. doi: 10.1016/B978-0-7020-6896-6.00009-0
114. Knörr F, Damm-Welk C, Ruf S, Singh VK, Zimmermann M, Reiter A, et al. Blood cytokine concentrations in pediatric patients with anaplastic lymphoma kinase-positive anaplastic large cell lymphoma. Haematologica (2018) 103(3):477–85. doi: 10.3324/haematol.2017.177972
115. Sueki Y, Nozaki Y, Kawashima I, Yamamoto T, Nakajima K, Mitumori T, et al. Anaplastic large cell lymphoma with paraneoplastic neutrophilia: an association between IL-17 elevation and aggressive disease progression. Int J Hematol (2014) 99(6):773–6. doi: 10.1007/s12185-014-1557-3
116. Siebert S, Amos N, Williams BD, Lawson TM. Cytokine production by hepatic anaplastic large-cell lymphoma presenting as a rheumatic syndrome. Semin Arthritis Rheum (2007) 37(1):63–7. doi: 10.1016/j.semarthrit.2006.12.007
117. Zhang Q, Raghunath PN, Xue L, Majewski M, Carpentieri DF, Odum N, et al. Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J Immunol (2002) 168(1):466–74. doi: 10.4049/jimmunol.168.1.466
118. Yoshimura A, Ichihara M, Kinjyo I, Moriyama M, Copeland NG, Gilbert DJ, et al. Mouse oncostatin M: an immediate early gene induced by multiple cytokines through the JAK-STAT5 pathway. EMBO J (1996) 15(5):1055–63. doi: 10.1002/j.1460-2075.1996.tb00443.x
119. Wu C, Molavi O, Zhang H, Gupta N, Alshareef A, Bone KM, et al. STAT1 is phosphorylated and downregulated by the oncogenic tyrosine kinase NPM-ALK in ALK-positive anaplastic large-cell lymphoma. Blood (2015) 126(3):336–45. doi: 10.1182/blood-2014-10-603738
120. Bluyssen HAR, Rastmanesh MM, Tilburgs C, Jie K, Wesseling S, Goumans MJ, et al. IFN gamma-dependent SOCS3 expression inhibits IL-6-induced STAT3 phosphorylation and differentially affects IL-6 mediated transcriptional responses in endothelial cells. Am J Physiol Cell Physiol (2010) 299(2):C354–62. doi: 10.1152/ajpcell.00513.2009
121. Delgoffe GM, Vignali DAA. STAT heterodimers in immunity: A mixed message or a unique signal? JAKSTAT (2013) 2(1):e23060. doi: 10.4161/jkst.23060
122. Gopal K, Kneteman NM, Zhang H, Lai R, Bone KM, Wu F, et al. STAT1 is phosphorylated and downregulated by the oncogenic tyrosine kinase NPM-ALK in ALK-positive anaplastic large-cell lymphoma. Blood (2015) 126(3):336–45. doi: 10.1182/blood-2014-10-603738
123. Haque SJ, Harbor P, Tabrizi M, Yi T, Williams BRG. Protein-tyrosine phosphatase Shp-1 is a negative regulator of IL-4- and IL-13-dependent signal transduction. J Biol Chem (1998) 273(51):33893–6. doi: 10.1074/jbc.273.51.33893
124. Han Y, Amin HM, Frantz C, Franko B, Lee J, Lin Q, et al. Restoration of shp1 expression by 5-AZA-2’-deoxycytidine is associated with downregulation of JAK3/STAT3 signaling in ALK-positive anaplastic large cell lymphoma. Leukemia (2006) 20(9):1602–9. doi: 10.1038/sj.leu.2404323
125. Liu Y, Sattarzadeh A, Diepstra A, Visser L, Van Den Berg A. The microenvironment in classical Hodgkin lymphoma: An actively shaped and essential tumor component. Semin Cancer Biol (2014) 24:15–22. doi: 10.1016/j.semcancer.2013.07.002
126. Wu R, Sattarzadeh A, Rutgers B, Diepstra A, Van Den Berg A, Visser L. The microenvironment of classical Hodgkin lymphoma: heterogeneity by Epstein–Barr virus presence and location within the tumor. Blood Cancer J (2016) 6(5):e417–7. doi: 10.1038/bcj.2016.26
127. Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med (2014) 20(11):1242–53. doi: 10.1038/nm.3739
128. Lee JS, Choi HJ, Baek SH. Sumoylation and its contribution to cancer. Adv Exp Med Biol (2017) 963:283–98. doi: 10.1007/978-3-319-50044-7_17
129. Vishwamitra D, Curry C v, Shi P, Alkan S, Amin HM. SUMOylation confers posttranslational stability on NPM-ALK oncogenic protein. Neoplasia (2015) 17(9):742–54. doi: 10.1016/j.neo.2015.09.005
130. Zhang C, Han XR, Yang X, Jiang B, Liu J, Xiong Y, et al. Proteolysis targeting chimeras (PROTACs) of anaplastic lymphoma kinase (ALK). Eur J Med Chem (2018) 151:304–14. doi: 10.1016/j.ejmech.2018.03.071
131. Wang Y, Han L, Liu F, Yang F, Jiang X, Sun H, et al. Targeted degradation of anaplastic lymphoma kinase by gold nanoparticle-based multi-headed proteolysis targeting chimeras. Colloids Surf B Biointerfaces (2020) 188:110795. doi: 10.1016/j.colsurfb.2020.110795
132. Yan G, Zhong X, Yue L, Pu C, Shan H, Lan S, et al. Discovery of a PROTAC targeting ALK with in vivo activity. Eur J Med Chem (2021) 212:113150. doi: 10.1016/j.ejmech.2020.113150
133. Kang CH, Lee DH, Lee CO, du Ha J, Park CH, Hwang JY. Induced protein degradation of anaplastic lymphoma kinase (ALK) by proteolysis targeting chimera (PROTAC). Biochem Biophys Res Commun (2018) 505(2):542–7. doi: 10.1016/j.bbrc.2018.09.169
134. Pileri SA, Agostinelli C, Bacci F, Sabattini E, Sagramoso C, Falini B, et al. Pathobiology of ALK-negative anaplastic large cell lymphoma. Pediatr Rep (2011) 3 Suppl 2(Suppl 2):8–10. doi: 10.4081/pr.2011.s2.e5
135. Savage KJ, Harris NL, Vose JM, Ullrich F, Jaffe ES, Connors JM, et al. ALK- anaplastic large-cell lymphoma is clinically and immunophenotypically different from both ALK+ ALCL and peripheral T-cell lymphoma, not otherwise specified: report from the International Peripheral T-Cell Lymphoma Project. Blood (2008) 111(12):5496–504. doi: 10.1182/blood-2008-01-134270
136. Hapgood G, Savage KJ. The biology and management of systemic anaplastic large cell lymphoma. Blood (2015) 126(1):17–25. doi: 10.1182/blood-2014-10-567461
137. Luchtel RA, Dasari S, Oishi N, Pedersen MB, Hu G, Rech KL, et al. Molecular profiling reveals immunogenic cues in anaplastic large cell lymphomas with DUSP22 rearrangements. Blood (2018) 132(13):1386–98. doi: 10.1182/blood-2018-03-838524
138. Luchtel RA, Zimmermann MT, Hu G, Dasari S, Jiang M, Oishi N, et al. Recurrent MSC E116K mutations in ALK-negative anaplastic large cell lymphoma. Blood (2019) 133(26):2776–89. doi: 10.1182/blood.2019000626
139. Sekine Y, Tsuji S, Ikeda O, Sato N, Aoki N, Aoyama K, et al. Regulation of STAT3-mediated signaling by LMW-DSP2. Oncogene (2006) 25(42):5801–6. doi: 10.1038/sj.onc.1209578
140. Parkhi M, Bal A, Das A, Kashyap D, Bhardwaj S, Prakash G, et al. ALK-negative anaplastic large cell lymphoma (ALCL): prognostic implications of molecular subtyping and JAK-STAT pathway. Appl Immunohistochem Mol Morphol (2021) 29(9):648–56. doi: 10.1097/PAI.0000000000000936
141. Candi E, Dinsdale D, Rufini A, Salomoni P, Knight RA, Mueller M, et al. TAp63 and DeltaNp63 in cancer and epidermal development. Cell Cycle (2007) 6(3):274–84. doi: 10.4161/cc.6.3.3797
142. Melino G. p63 is a suppressor of tumorigenesis and metastasis interacting with mutant p53. Cell Death Differ (2011) 18(9):1487–99. doi: 10.1038/cdd.2011.81
143. Parrilla Castellar ER, Jaffe ES, Said JW, Swerdlow SH, Ketterling RP, Knudson RA, et al. ALK-negative anaplastic large cell lymphoma is a genetically heterogeneous disease with widely disparate clinical outcomes. Blood (2014) 124(9):1473–80. doi: 10.1182/blood-2014-04-571091
144. Crescenzo R, Abate F, Lasorsa E, Tabbo’ F, Gaudiano M, Chiesa N, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell (2015) 27(4):516–32. doi: 10.1016/j.ccell.2015.03.006
145. Scarfò I, Pellegrino E, Mereu E, Kwee I, Agnelli L, Bergaggio E, et al. Identification of a new subclass of ALK-negative ALCL expressing aberrant levels of ERBB4 transcripts. Blood (2016) 127(2):221–32. doi: 10.1182/blood-2014-12-614503
146. Turner SD, Inghirami G, Miranda RN, Kadin ME. Cell of origin and immunologic events in the pathogenesis of breast implant–associated anaplastic large-cell lymphoma. Am J Pathol (2020) 190(1):2–10. doi: 10.1016/j.ajpath.2019.09.005
147. Cappellano G, Ploner C, Lobenwein S, Sopper S, Hoertnagl P, Mayerl C, et al. Immunophenotypic characterization of human T cells after in vitro exposure to different silicone breast implant surfaces. PloS One (2018) 13(2):e0192108. doi: 10.1371/journal.pone.0192108
148. Kadin ME, Morgan J, Xu H, Epstein AL, Sieber D, Hubbard BA, et al. IL-13 is produced by tumor cells in breast implant-associated anaplastic large cell lymphoma: implications for pathogenesis. Hum Pathol (2018) 78:54–62. doi: 10.1016/j.humpath.2018.04.007
149. Quesada AE, Medeiros LJ, Clemens MW, Ferrufino-Schmidt MC, Pina-Oviedo S, Miranda RN. Breast implant-associated anaplastic large cell lymphoma: a review. Mod Pathol (2019) 32(2):166–88. doi: 10.1038/s41379-018-0134-3
150. Laurent C, Nicolae A, Laurent C, le Bras F, Haioun C, Fataccioli V, et al. Gene alterations in epigenetic modifiers and JAK-STAT signaling are frequent in breast implant–associated ALCL. Blood (2020) 135(5):360–70. doi: 10.1182/blood.2019001904
151. Blombery P, Thompson E, Ryland GL, Joyce R, Byrne DJ, Khoo C, et al. Frequent activating STAT3 mutations and novel recurrent genomic abnormalities detected in breast implant-associated anaplastic large cell lymphoma. Oncotarget (2018) 9(90):36126–36. doi: 10.18632/oncotarget.26308
152. Oishi N, Brody GS, Ketterling RP, Viswanatha DS, He R, Dasari S, et al. Genetic subtyping of breast implant-associated anaplastic large cell lymphoma. Blood (2018) 132(5):544–7. doi: 10.1182/blood-2017-12-821868
153. di Napoli A, Jain P, Duranti E, Margolskee E, Arancio W, Facchetti F, et al. Targeted next generation sequencing of breast implant-associated anaplastic large cell lymphoma reveals mutations in JAK/STAT signalling pathway genes, TP53 and DNMT3A. Br J Haematol (2018) 180(5):741–4. doi: 10.1111/bjh.14431
154. Quesada AE, Zhang Y, Ptashkin R, Ho C, Horwitz S, Benayed R, et al. Next generation sequencing of breast implant-associated anaplastic large cell lymphomas reveals a novel STAT3-JAK2 fusion among other activating genetic alterations within the JAK-STAT pathway. Breast J (2021) 27(4):314–21. doi: 10.1111/tbj.14205
155. Oishi N, Miranda RN, Feldman AL. Genetics of breast implant-associated anaplastic large cell lymphoma (BIA-ALCL). Aesthet Surg J (2019) 39(Suppl_1):S14–20. doi: 10.1093/asj/sjy311
156. Melchers RC, Willemze R, Van De Loo M, Van Doorn R, Jansen PM, Cleven AHG, et al. Clinical, histologic, and molecular characteristics of anaplastic lymphoma kinase-positive primary cutaneous anaplastic large cell lymphoma. Am J Surg Pathol (2020) 44(6):776–781. doi: 10.1097/PAS.0000000000001449
157. Quintanilla-Martinez L, Jansen PM, Kinney MC, Swerdlow SH, Willemze R. Non-mycosis fungoides cutaneous T-cell lymphomas: report of the 2011 Society for Hematopathology/European Association for Haematopathology workshop. Am J Clin Pathol (2013) 139(4):491–514. doi: 10.1309/AJCP83AOQTMLOJTM
158. Attygalle AD, Cabeçadas J, Gaulard P, Jaffe ES, de Jong D, Ko YH, et al. Peripheral T-cell and NK-cell lymphomas and their mimics; taking a step forward - report on the lymphoma workshop of the XVIth meeting of the European Association for Haematopathology and the Society for Hematopathology. Histopathology (2014) 64(2):171–99. doi: 10.1111/his.12251
159. Gallardo F, Barranco C, Toll A, Pujol RM. CD30 antigen expression in cutaneous inflammatory infiltrates of scabies: a dynamic immunophenotypic pattern that should be distinguished from lymphomatoid papulosis. J Cutan Pathol (2002) 29(6):368–73. doi: 10.1034/j.1600-0560.2002.290608.x
160. Oschlies I, Lisfeld J, Lamant L, Nakazawa A, d’Amore ESG, Hansson U, et al. ALK-positive anaplastic large cell lymphoma limited to the skin: clinical, histopathological and molecular analysis of 6 pediatric cases. A report from the ALCL99 study. Haematologica (2013) 98(1):50–6. doi: 10.3324/haematol.2012.065664
161. Kadin ME, Pinkus JL, Pinkus GS, Duran IH, Fuller CE, Onciu M, et al. Primary cutaneous ALCL with phosphorylated/activated cytoplasmic ALK and novel phenotype: EMA/MUC1+, cutaneous lymphocyte antigen negative. Am J Surg Pathol (2008) 32(9):1421–6. doi: 10.1097/PAS.0b013e3181648d6d
162. Mao X, Orchard G, Lillington DM, Russell-Jones R, Young BD, Whittaker S. Genetic alterations in primary cutaneous CD30+ anaplastic large cell lymphoma. Genes Chromosomes Cancer (2003) 37(2):176–85. doi: 10.1002/gcc.10184
163. Szuhai K, van Doorn R, Tensen CP, Kester V. Array-CGH analysis of cutaneous anaplastic large cell lymphoma. Methods Mol Biol (2013) 973:197–212. doi: 10.1007/978-1-62703-281-0_13
164. Velusamy T, Kiel MJ, Sahasrabuddhe AA, Rolland D, Dixon CA, Bailey NG, et al. A novel recurrent NPM1-TYK2 gene fusion in cutaneous CD30-positive lymphoproliferative disorders. Blood (2014) 124(25):3768–71. doi: 10.1182/blood-2014-07-588434
165. Liang HC, Kapoor R, Onder O, Dennis K, Lim MS, Elenitoba-Johnson KSJ. The NPM1-TYK2 chimeric fusion promotes activation of STAT family signaling, skewing towards tfh functional subset differentiation and mature T-cell lymphomagenesis. Blood (2022) 140(Supplement 1):9281–2. doi: 10.1182/blood-2022-169494
Keywords: pathogenesis, ALCL, lymphoma biology, genetic abnormalities, proteomics
Citation: Wu R and Lim MS (2023) Updates in pathobiological aspects of anaplastic large cell lymphoma. Front. Oncol. 13:1241532. doi: 10.3389/fonc.2023.1241532
Received: 16 June 2023; Accepted: 04 September 2023;
Published: 22 September 2023.
Edited by:
Juliana Pereira, University of São Paulo, BrazilReviewed by:
Shih-Sung Chuang, Chi Mei Medical Center, TaiwanSuzanne Dawn Turner, University of Cambridge, United Kingdom
Copyright © 2023 Wu and Lim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Megan S. Lim, TGltbTRAbXNrY2Mub3Jn