 Xiao-Peng Li†
Xiao-Peng Li† Zheng-Qian Guo
Zheng-Qian Guo Bao-Feng Wang
Bao-Feng Wang Min Zhao
Min Zhao- Department of Neurosurgery, Tongji Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, China
The epidermal growth factor receptor (EGFR) is the most frequently altered gene in glioblastoma (GBM), which plays an important role in tumor development and anti-tumor immune response. While current molecular targeted therapies against the EGFR signaling pathway and its downstream key molecules have not demonstrated favorable clinical outcomes in GBM. Whereas tumor immunotherapies, especially immune checkpoint inhibitors, have shown durable antitumor responses in many cancers. However, the clinical efficacy is limited in patients carrying EGFR alterations, indicating that EGFR signaling may involve tumor immune response. Recent studies reveal that EGFR alterations not only promote GBM cell proliferation but also influence immune components in the tumor microenvironment (TME), leading to the recruitment of immunosuppressive cells (e.g., M2-like TAMs, MDSCs, and Tregs), and inhibition of T and NK cell activation. Moreover, EGFR alterations upregulate the expression of immunosuppressive molecules or cytokines (such as PD-L1, CD73, TGF-β). This review explores the role of EGFR alterations in establishing an immunosuppressive TME and hopes to provide a theoretical basis for combining targeted EGFR inhibitors with immunotherapy for GBM.
1 Introduction
Glioblastoma (GBM) is the most prevalent primary malignant tumor in the central nervous system (CNS) (1). The current standard treatment for newly diagnosed GBM involves maximal safe resection surgery, followed by temozolomide and adjuvant radiotherapy (2). Despite these interventions, only 6.9% of patients survive beyond five years post-diagnosis, based on the data from 2015 to 2019 in the United States (1). According to the 2021 World Health Organization (WHO) classification of tumors of the CNS, GBM is diagnosed based on the presence of necrosis, microvascular proliferation, and 1 or more of 3 specific genetic parameters [telomerase reverse transcriptase (TERT) promoter mutation, epidermal growth factor receptor (EGFR) gene amplification, combined gain of chromosome 7 and loss of chromosome 10 (+7/-10)] with wild-type forms of isocitrate dehydrogenase (IDH) 1/2 (3). Among these aberrances, EGFR is one of the most common oncogenic alterations (including gene amplification, mutation, rearrangement, and splicing site changes), occurring in approximately 50% of GBM samples (4, 5). After binding to ligands, EFGR forms a dimer that phosphorylates its C-terminal tail, regulating downstream physiological and pathological processes (5). Apart from this, EGFR variant III (EGFRvIII), a tumor-specific deletion of exons 2-7 which occurs in approximately 30% of GBM patients, consistently activates the EGFR signal even without ligands binding, contributing to intra-tumoral heterogeneity and resistance to targeted therapies (5–7). Aberrant activation of the EGFR and its downstream signaling pathways promotes GBM tumorigenesis and progression. Specifically, the phosphatidylinositol-3 kinase (PI3K)-AKT-mammalian target of rapamycin (mTOR) pathway and RAS-RAF-MEK-ERK pathway regulates tumor growth, survival, angiogenesis, and metabolism (7–9); the Janus kinase 2 (JAK2)-signal transducer and activator of transcription 3 (STAT3) pathway contributes to tumor growth, survival, stemness maintenance, and angiogenesis (7, 10), and the activated phospholipase C (PLC)-PKC pathway promotes tumor growth, survival, and invasiveness (11). Therefore, EGFR and its downstream signaling pathways are potential therapeutic targets for GBM. Notably, these pathways may contribute to establishing an immunosuppressive TME in EGFR-altered tumors (12–14). A bioinformatics study in glioma has revealed that EGFR mutation indicates poor prognosis and potential immune suppression within the TME (15). In terms of immunotherapy, EGFR amplification has been suggested as a biomarker of resistance to immune checkpoint inhibitors (ICIs) in GBM patients (16). In this review, we endeavor to compare the treatment targeted EGFR in GBM and discuss the mechanisms underlying the immunosuppressive TME regulated by the aberrant EGFR signaling pathway. The identified mechanisms in the EGFR pathway in GBM are illustrated in Figure 1.
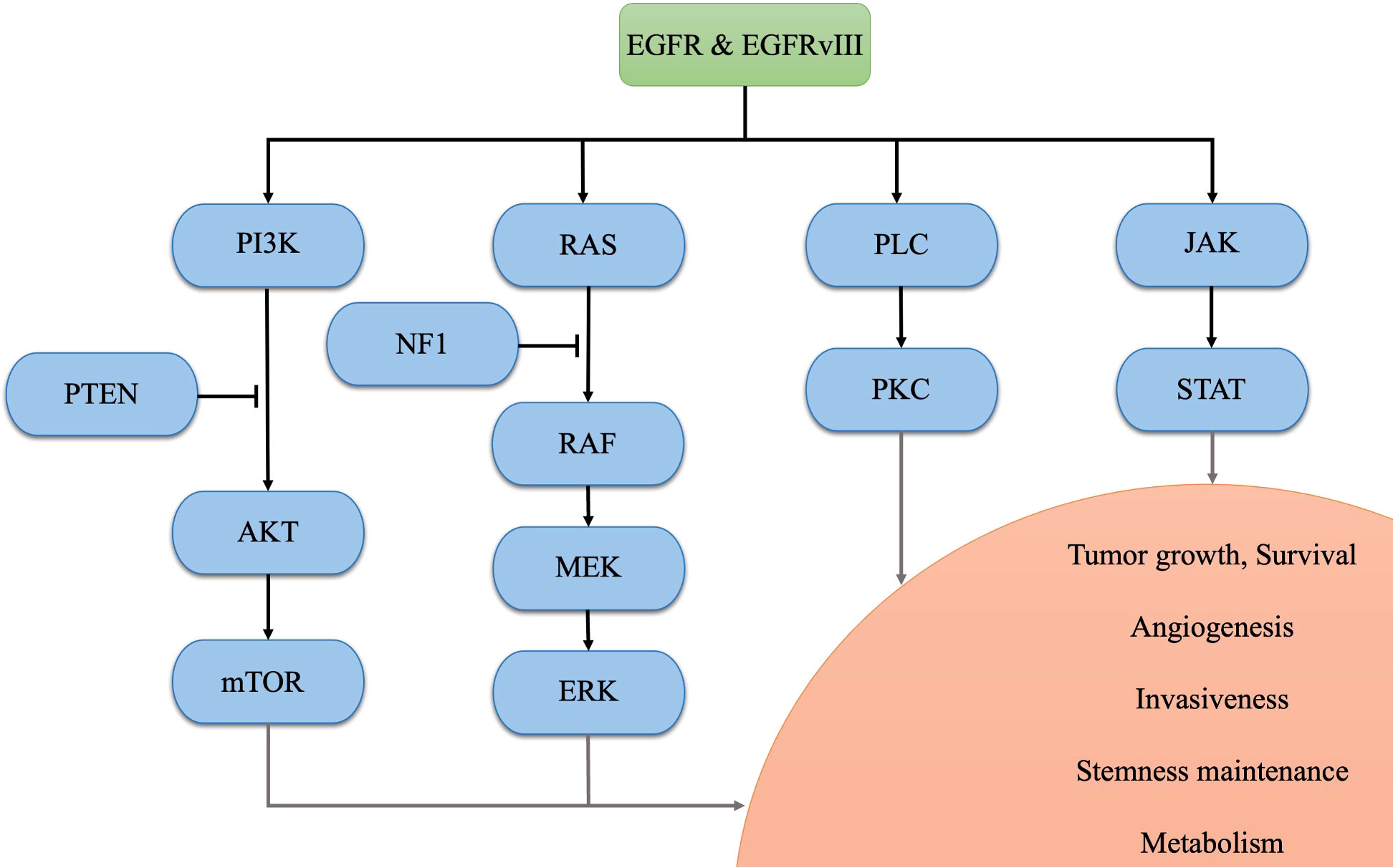
Figure 1 The identified mechanisms in the EGFR pathway in GBM.
2 Current strategies for targeting EGFR in GBM
Various treatments targeting EGFR have been explored for GBM. Three generations of EGFR tyrosine kinase inhibitors (TKIs) are approved for clinical use. First-generation TKIs (Erlotinib, Gefitinib, Lapatinib, etc.) inhibit the receptor by competitive binding with adenosine triphosphate (ATP) and second-generation TKIs (Afatinib, Dacomitinib, Tesevatinib) irreversibly inhibit all four ERBB receptors. Despite the preclinical data suggesting they affect tumor proliferation, all of them have poor responses in clinical trials on newly diagnosed and recurrent GBM due to insufficient delivery or resistance to inhibition (4–6, 17, 18). While a third-generation TKI, Osimertinib, is specifically designed to target the canonical EGFR activating mutations and the T790M resistance mutation with a good ability to cross the blood-brain barrier (BBB). It can overcome resistance to first- and second-generation TKIs by continuously blocking ERK signaling in GBM (4, 19), which may bring hope to GBM patients, but further clinical trials are needed to validate its potential. Novel methods, such as nanoparticles developed in preclinical animal models deliver and protect small interfering RNAs, allowing them to pass the BBB and knock down EGFR signaling, although their feasibility need to be verified before clinical application (20).
Immunotherapies targeting EGFR also face challenges in GBM. Rindopepimut, a vaccine against the EGFRvIII, prolonged progression-free survival (PFS) of patients with recurrent GBM but failed to increase overall survival (OS) in phase III clinical trial (21). In addition, EGFRvIII chimeric antigen receptor (CAR) T therapy has been tested in GBM. The engineered lymphocytes were detectable in both peripheral blood and tumor samples from patients after CAR T infusion, but no satisfactory results were obtained in phase I study and there was one treatment-related death due to the severe toxicity (22). ICIs in preclinical GBM mouse models confirmed the safety and efficiency of monoclonal antibodies targeting the PD-1/PD-L1 axis (23), but have little clinical benefit in GBM, especially in patients harboring EGFR amplification. A recent clinical study showed that after initiation of PD-1 inhibitor (pembrolizumab or nivolumab) for recurrent GBM patients with EGFR amplification, the median OS was 7 months compared to 18 months for those without EGFR amplification (16).
Despite achieving encouraging progress in various cancers, these treatments have not achieved the expected effectiveness in GBM patients with EGFR alterations. Factors such as complex regulatory networks, high heterogeneity of tumor cells, BBB blockage, and altered TME contribute the less therapeutic improvements against GBM (24). On the other hand, both the EGFR targeting therapy and immunotherapy demonstrate favorable results in preclinical models but fail in clinical trials, suggesting that mice still have significant limitations in modeling human cancer and immunity.
3 EGFR signaling in GBM regulates the infiltration of immune cells
The TME infiltrates various non-cancerous cells, including astrocytes, pericytes, endothelial cells, fibroblasts, and immune cells. It has long been considered that the normal brain is one of the “immune privileged” organs (25). However, the disruption of the BBB in GBM leads to immune cells infiltrating to the tumor mass from blood flow. Immunosuppressive cells include tumor-associated microglia and macrophages (TAMs), myeloid-derived suppressor cells (MDSCs), regulatory T cells (Tregs), and regulatory B cells (Bregs), while the anti-tumor immune cells include T cells (CD4+ helper T cells, CD8+ cytotoxic T cells) and natural killer (NK) cells, etc. Infiltration or dysfunction of these cells is one of the causes of the highly immunosuppressive and “cold” TME phenotype of the GBM (26).
3.1 Microglia and myeloid-derived cells
TAMs, comprising microglia and macrophages, are the dominant infiltrating immune cells in GBM and account for 30-50% of the tumor mass (27, 28). Microglia are yolk sac–derived and have limited self-renewal capacity, while macrophages are monocyte-derived from the bone marrow and peripheral circulation and constituted approximately 85% of the total TAM population (29, 30). Despite different origins, they both play a vital role in tumor progression. The activated TAMs in GBM can be simply divided into tumor-suppressing M1-like phenotype and tumor-promoting M2-like phenotype with great plasticity and heterogeneity. The main TAMs in GBM present possess an M2-like phenotype, promoting tumor invasion, proliferation, angiogenesis, and immune evasion through the expression and secretion of matrix-degrading enzymes, angiogenic factors, and immunosuppressive cytokines/chemokines (28, 31). However, it should be noted that dichotomously classified M1 and M2 phenotypes were fitted well in vitro under optimal conditions, as it does not fully reflect the complexity of TAMs activation. Additional states (such as M2a, M2b, M2c, and M2d states) have also been identified, suggesting that TAMs in vivo likely have more functions along the M1/M2 spectrum. Several studies have shown that EGFR alterations associated with TAMs infiltration in GBM, and inhibiting EGFR by pharmacy strongly decreased microglia-stimulated invasion in GL261 GBM cells (32). Another study using GBM cell lines U87 and A172 in vitro and in vivo shows that EGFR cooperates with EGFRvIII to induce macrophage infiltration by upregulation of chemokine C-C motif ligand 2 (CCL2) through the KRAS pathway, one of the major downstream pathways of the EGFR (33). In addition, dual targeting of EGFR and mTOR pathways has also been demonstrated to inhibit tumor growth and macrophage infiltration on GBM xenografts by downregulation of CCL2 (34), an immunosuppressive cytokine that shapes the macrophage polarization toward a tumor-promoting, immunosuppressive phenotype, and significantly shortened the survival of GBM-bearing mice (30, 35). Furthermore, a recent single-cell RNA sequencing study focused on the immune landscape during GBM progression found that subsets of pro-inflammatory microglia in developing GBMs, while anti-inflammatory macrophages are observed in end-stage tumors. This evolution parallels the extensive growth of EGFR+ GBM cells (36). Another single-cell level study reveals that GBM samples with EGFR and cyclin-dependent kinase 4 (CDK4) co-amplifications in the same cell exhibit higher infiltration of CD163+ immunosuppressive macrophages (37).
MDSCs are a heterogeneous population of immature bone marrow cells consisting of two cell subsets: granulocytic or polymorphonuclear MDSCs (PMN-MDSCs) and monocytic MDSCs (M-MDSCs). MDSCs exert their immunosuppressive effects in GBM by inhibiting cytotoxic T cell activity, suppressing the function of NK cells, macrophages, and dendritic cells, and augmenting the effect of Tregs (38, 39). The density of MDSCs also correlates with tumor stages, chemotherapy response, and patient prognosis in GBM (39). According to two recent studies using GBM animal models, it has been observed that, similar to TAMs, MDSCs accumulation is more pronounced in EGFR (+) GBM, when compared to the EGFR-wild type (EGFR-WT) (36, 40). These studies also found that EGFRvIII GBM highly expressed chemokine C-X-C motif ligand 1/2/3 (CXCL1/2/3) and PMN-MDSCs-expressed chemokine C-X-C motif receptor 2 (CXCR2) constitute an axis that regulates the output of PMN-MDSCs from the bone marrow, leading to increased systemic levels of these cells. Interestingly, pharmacological inhibition of this axis benefitted for response to ICIs and prolonged survival in EGFRvIII-driven GBM mice (40). In summary, the immunosuppressive TME of EGFRvIII GBM may be due to the greater infiltration by MDSCs, targeting this cell is a potential therapeutic strategy for GBM, but further research is needed in this area.
3.2 Tumor-infiltrating lymphocytes
TILs are a group of tumor-infiltrating and antigenic cell populations that exist in TME, and their amount and subtypes determine the clinical outcomes and treatment responses in glioma patients (41, 42). CD4+ helper T cells stimulate an anti-tumor response by activating CD8+ cytotoxic T cells and promoting B cell proliferation and differentiation. CD8+ cytotoxic T cells induce tumor apoptosis through T cell receptor activation or lyse tumor cells by releasing interferon-γ, perforin, and granzyme (43). However, T cells are rare in the GBM TME, accounting for less than 0.25% of total isolated cells from GBM biopsies. The majority of samples exhibit a “lymphocyte depletion” phenotype with few CD8+ cytotoxic T cells but increased infiltration of Tregs, particularly in cases with EGFR amplification (44, 45). On the other hand, dysfunction of T cells (such as senescence, tolerance, anergy, exhaustion, and ignorance) is a hallmark of GBM, leading to ineffective anti-tumor immune responses (28, 43, 46). It has been suggested that the efficacy of EGFR-TKIs depends mainly on the presence of CD4+ and CD8+ T cells, as treatment significantly enhances the immune responses against the tumor (47). In contrast to conventional CD4+ and CD8+ T cells, the FOXP3+Tregs, a subset of CD4+ T cells, play an opposite role in the GBM microenvironment. Tregs inhibit the activation of effective T cells through the interactions with cytotoxic T lymphocyte antigen-4 (CTLA-4) and CD80/86 on antigen-presenting cells (APCs). They also secrete immunosuppressive cytokines that promote tumor progression and polarization of macrophages toward an M2-like phenotype (48). How the aberrant EGFR signaling pathway regulates these T cells in the GBM microenvironment may be related to the expression of some immunosuppressive molecules or cytokines, such as programmed death-ligand 1 (PD-L1) (23), extracellular-5’-nucleotidase (CD73) (49), and transforming growth factor (TGF)-β (50), which will be discussed in detail in the next section.
NK cells are innate lymphoid cells that account for a small proportion of tumor-infiltrating cells, which exert anti-tumor effects through lytic granules secretion and recruiting other immune cells (51). However, some GBM patients show reduced NK cell activation due to decreased levels of the natural killer group 2 member D (NKG2D) receptor on the NK cell surface (28). Additionally, NKG2D ligands in cancer cells correlated positively with the activation of EGFR and its downstream MEK pathway, which is commonly hyperactivated in those tumors and reduced by EGFR inhibitors (52). However, different NKG2D ligands can function as target molecules for NK cell-mediated immunosurveillance or tumor immune escape. NKG2D ligands expressed on the cell surface of tumor cells can be recognized by NK cells through NKG2D and promote a cytotoxic response that leads to tumor cell elimination (53). However, soluble NKG2D ligands generated by a disintegrin and metalloproteinase 10 (ADAM10), ADAM17, and matrix metalloproteinase 14 (MMP14), can promote NKG2D down-regulation, impairing NK cell-effector functions and facilitating tumor immune escape (54, 55). Therefore, to determine the mechanism of EGFR signaling pathway regulation of NK cells in GBM, further studies are needed to focus on whether GBM with EGFR alterations express higher levels of NKG2D ligands or which form of the ligands are expressed in EGFR-altered GBM.
Tumor infiltrating B cells also play a role in shaping tumor development. Activated B cells display antitumor activity by producing immunoglobulins, promoting T cell responses, and killing tumor cells directly. Other B cells with tumor-promoting effects are defined as Bregs, upregulate PD-L1, CTLA-4 and secrete immunosuppressive cytokines such as IL-10 and TGF-β, attenuating the response of T and NK cells while facilitating the activation of Tregs, MDSCs, and TAMs (56). However, no relevant studies have yet shown the association between altered EGFR signaling pathways in tumor cells and the infiltration and activation of Bregs, especially in GBM. Further investigations are needed to explore this field.
4 Immunosuppressive molecules and cytokines regulated by EFGR signaling in GBM
GBM cells also express cell surface molecules or secrete various chemokines, cytokines, and growth factors that regulate immune cell infiltration or function in GBM. Some of them are modulated by EGFR and its downstream signaling.
4.1 PD-L1
The expression of PD-L1 in GBM correlates with the patient prognosis (57, 58). However, the PD-L1 positivity rate in GBM specimens ranges from 61.0% to 88% in different studies (59). Such inconsistent results might be associated with different PD-L1 detection techniques, tumor heterogeneity, and different specimen sources. PD-L1 binding to PD-1 on T cells restrains the anti-tumor response, inducing apoptosis or anergy in activated T cells and promoting the infiltration of Tregs, leading to tumor immune escape (23). Recent studies have found that PD-L1 expression in GBM cells is associated with EGFR and its downstream signaling pathways (23). Among them, EGFR-dependent PI3K activation, phosphatase and tensin homolog deleted on chromosome ten (PTEN) loss, and AKT activation induce the β-catenin binding to the CD274 gene (encode PD-L1) promoter region, resulting in increased PD-L1 expression (60, 61). Another study found that the activated EGFR-ERK signaling pathway in GBM upregulates COP9 signalosome subunit 6 (CSN6) and PD-L1 protein expression, stabilizing PD-L1 and inhibiting its degradation (62). Radiotherapy has also been shown to increase PD-L1 expression in glioma cells. In a vitro study, irradiated human GBM cell lines U87 and U251 show significant upregulation of PD-L1 expression at the protein and mRNA levels via phosphorylation of EGFR and its downstream signaling molecule JAK2 (63). A single-center retrospective study has found that GBM patients with EGFR amplification exhibit shorter median OS after receiving ICIs (16). Therefore, targeting EGFR and its downstream signaling, in combination with PD-1/PD-L1 ICIs, may hold the potential in restoring the T-lymphocyte killing capacity and improving the sensitivity to immunotherapy and targeted therapy in tumor patients (47, 64, 65).
4.2 CD73/adenosine
CD73 is anchored to cell membrane lipid rafts and highly expressed in various tumors including GBM and plays a role in immune regulation by generating adenosine in the TME. Adenosine is a component of adenine nucleotides that regulates immune cell function by the ectonucleoside triphosphate diphosphohydrolase (NTPDase1, CD39)-CD73 synergic effect in glioma TME (66). ATP and adenosine diphosphate (ADP) released from tumor cells are hydrolyzed by CD39 to adenosine monophosphate (AMP), which is then further dephosphorylated to adenosine by CD73 (67, 68). In cancer patients, alterations in adenosine deaminase (ADA) activity can lead to increased levels of adenosine as ADA normally deactivates adenosine by converting it to inosine (68). Adenosine mediates its regulatory functions by binding to adenosine receptors (A2AR and A2BR) on the tumor-infiltrating immune cells, triggering the accumulation of intracellular cyclic adenosine monophosphate (cAMP). This signaling molecule is associated with the establishment of an immunosuppressive TME characterized by the polarization of TAMs into a tumor-promoting M2-like phenotype, increased infiltration and immunosuppressive activity of Tregs and MDSCs, and suppressed activity of dendritic cells, CD8+ T cells, and NK cells (67–69). Overexpression of CD73 promotes immunosuppression and is associated with poor prognosis in multiple cancers (66, 70). A Recent in vivo study has revealed that blocking CD73 expression in the tumor cells can potentially regulate the GBM immune microenvironment and inhibit tumor growth by inducing apoptosis (71). Another study based on single-cell RNA sequencing found a correlation between high CD73 expression and EFGR amplification as well as hypoxia in GBM (49). These findings suggest that EGFR alterations signaling shape an immunosuppressive TME in GBM by promoting CD73 expression. Targeting the CD73-adenosine axis may hold promise as a therapeutic strategy in combination with EGFR-TKIs.
4.3 TGF-β
TGF-β is a pro-tumorigenic cytokine overexpressed and produced by various cells in GBM, contributes to tumor growth, angiogenesis, maintenance of glioma stem cell stemness, and the establishment of an immunosuppressive TME (72, 73). TGF-β exerts its antitumor immune response on immune cells within the TME. For innate immunity, TGF-β promotes TAMs recruitment and M2-like polarization, monocyte differentiation into MDSCs, and attenuating the tumor killing efficiency of NK cells. For adaptive immunity, it facilitates Tregs persistence, inhibiting the function of effector T cells and antigen-presenting dendritic cells (12, 50, 74). In addition, high-level TGF-β also contributes to poor response to PD-L1 immune therapy (75). Recent studies have revealed that the activation of TGF-β may be related to EGFR singling and its downstream molecules. The activation PLCγ-PKC pathway leads to intragin-αvβ activation and contributes to local TGF-β activate from its latent to its bioactive form (12, 76). Although it is well known that the correlation between EGFR signaling and TGF-β expression in many cancers. Further studies specific to GBM are needed to verify this finding.
Overall, EGFR and its downstream signaling pathways modulate immune-related molecules and cytokines in GBM, influencing the immune cell landscape and establishing an immunosuppressive TME. Targeting these interactions may hold promise for improving the effectiveness of immunotherapy and targeted therapy in GBM treatment.
5 The roles of EGFR in immune cells
A variety of immune cells express EGFR, which plays a significant role in the TME of tumors without EGFR alterations. Recently, some studies have identified the function of EGFR in macrophages and T cells. In both human and mouse tumors (hepatocellular carcinoma and colorectal carcinoma), EGFR expression in macrophages promotes tumor development (77, 78). Myeloid-specific EGFR knockout mice exhibited less carcinogenesis, possibly associated with decreased IL-6 production via the STAT3 pathway. This suggests a pro-tumorigenic role of EGFR signaling in myeloid cells (77, 78). Similarly, in a colitis-associated carcinogenesis model, EGFR signaling in macrophages plays a critical role in tumor development by activating macrophages and inducing polarization toward a tumor-promoting M2-like phenotype (79). EGFR expression has also been detected in Tregs. Amphiregulin (AREG), an EGF-like growth factor derived from mast cells, enhances the suppressive function of Tregs by activating the MAPK pathway through binding to EGFR on Tregs (80). In the central nervous system, EGFR phosphorylation and subsequent activation of ERK mediate microglia migration during inflammatory states. This process may be mediated by the lipopolysaccharide (LPS)-triggered intracellular calcium mobilization. And the calcium activity is decisive for EGFR phosphorylation initiated by its ligands (81). All these cells play key roles in the immunosuppressive microenvironment of GBM, which predicts that targeting the EGFR signaling pathway and its downstream key molecules in immune cells may be a new therapeutic strategy that should be explored in future studies.
6 Conclusion
Aberrant EGFR signaling pathways in GBM contribute to tumor progression by directly promoting tumor cell proliferation and survival, as well as establishing an immunosuppressive TME that allows for tumor immune escape. The EGFR signaling pathway recruits TAMs and MDSCs, converting M1-like TAMs to tumor-promoting M2-like phenotype, which further induce T cell and NK cell dysfunction. EGFR alterations in GBM also contribute to the upregulation of immunosuppressive molecules and cytokines, such as PD-L1, CD73, TGF-β, etc. (Figure 2). It also should be noted that the downstream signaling described in this review is very similar to other receptor tyrosine kinases (RTKs), EGFR-targeted therapies face the challenge of resistance due to the upregulation of redundant RTKs and activation of compensatory signaling pathways. Understanding how these redundant RTKs modulate the TME in GBM requires further investigation. On the other hand, to enhance the efficacy of immunotherapy in GBM patients with EGFR alteration, it is necessary to develop new therapeutic strategies targeting the immunosuppression TME associated with EGFR alterations. Future approaches could involve combining molecularly targeted therapies and immunotherapy to stimulate both the immunogenic response of GBM and the anti-tumor immune response. For example, the combination of a PD-L1 inhibitor (Atezolizumab) and EGFR-TKI (Erlotinib) has shown promising prospects in EGFR-mutant non-small cell cancer (82, 83). Implementing a similar integrated approach in GBM could improve treatment outcomes and benefit a larger number of patients.
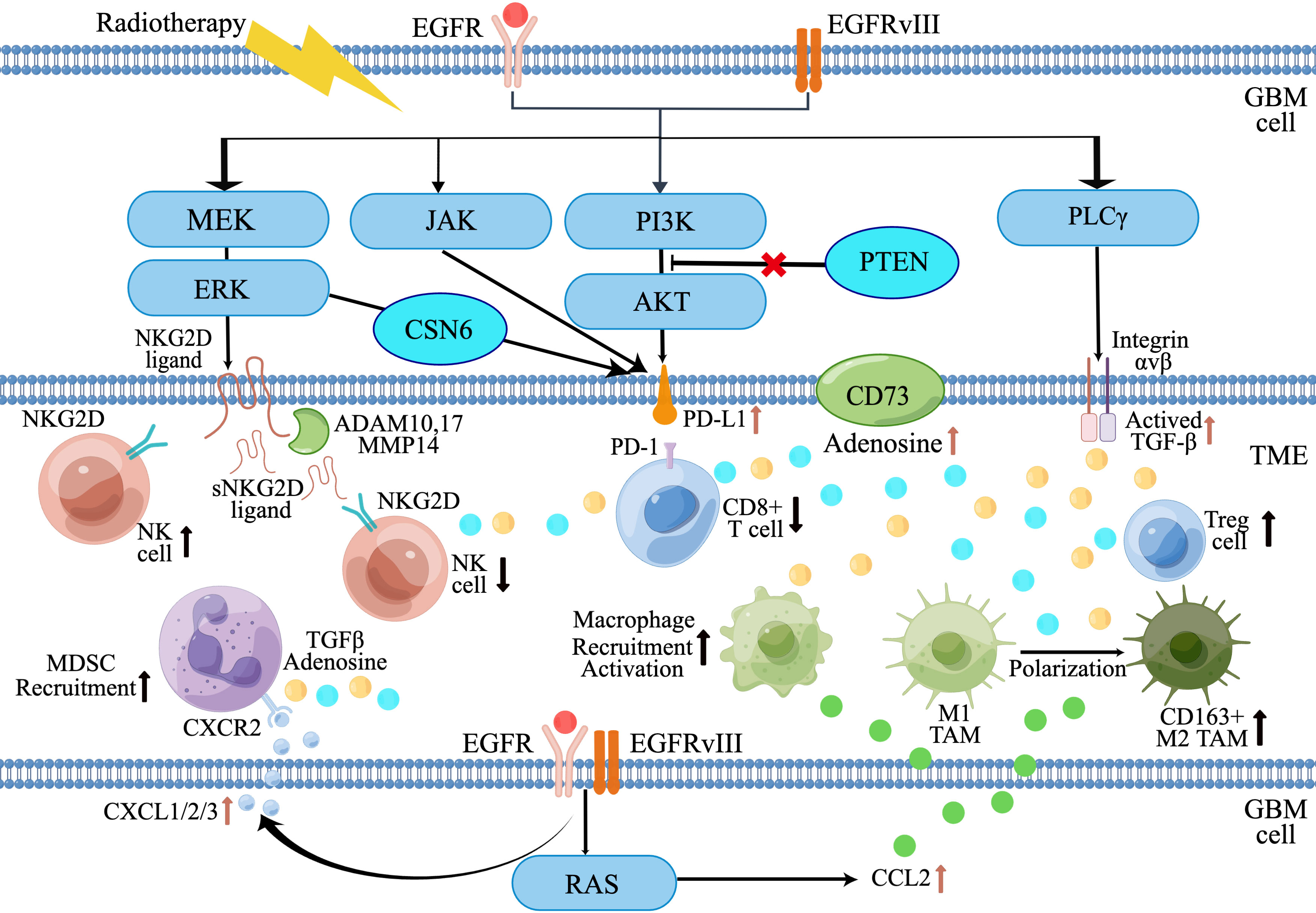
Figure 2 Diagram of EGFR alterations in GBM that promote an immunosuppressive tumor microenvironment. (By Figdraw).
Author contributions
X-PL and Z-QG wrote the manuscript and drew the figures. B-FW and MZ conceived the main outline took charge of manuscript revision in English. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ostrom QT, Price M, Neff C, Cioffi G, Waite KA, Kruchko C, et al. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2015-2019. Neuro Oncol (2022) 24(Suppl 5):v1–v95. doi: 10.1093/neuonc/noac202
2. Wen PY, Weller M, Lee EQ, Alexander BM, Barnholtz-Sloan JS, Barthel FP, et al. Glioblastoma in adults: a Society for Neuro-Oncology (SNO) and European Society of Neuro-Oncology (EANO) consensus review on current management and future directions. Neuro Oncol (2020) 22(8):1073–113. doi: 10.1093/neuonc/noaa106
3. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro Oncol (2021) 23(8):1231–51. doi: 10.1093/neuonc/noab106
4. Yang K, Wu Z, Zhang H, Zhang N, Wu W, Wang Z, et al. Glioma targeted therapy: insight into future of molecular approaches. Mol Cancer (2022) 21(1):39. doi: 10.1186/s12943-022-01513-z
5. Oprita A, Baloi SC, Staicu GA, Alexandru O, Tache DE, Danoiu S, et al. Updated insights on EGFR signaling pathways in glioma. Int J Mol Sci (2021) 22(2):587. doi: 10.3390/ijms22020587
6. Padfield E, Ellis HP, Kurian KM. Current therapeutic advances targeting EGFR and EGFRvIII in glioblastoma. Front Oncol (2015) 5:5. doi: 10.3389/fonc.2015.00005
7. An Z, Aksoy O, Zheng T, Fan QW, Weiss WA. Epidermal growth factor receptor and EGFRvIII in glioblastoma: signaling pathways and targeted therapies. Oncogene (2018) 37(12):1561–75. doi: 10.1038/s41388-017-0045-7
8. Thorpe LM, Yuzugullu H, Zhao JJ. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat Rev Cancer (2015) 15(1):7–24. doi: 10.1038/nrc3860
9. Deschênes-Simard X, Kottakis F, Meloche S, Ferbeyre G. ERKs in cancer: friends or foes? Cancer Res (2014) 74(2):412–9. doi: 10.1158/0008-5472.CAN-13-2381
10. Gong AH, Wei P, Zhang S, Yao J, Yuan Y, Zhou AD, et al. FoxM1 drives a feed-forward STAT3-activation signaling loop that promotes the self-renewal and tumorigenicity of glioblastoma stem-like cells. Cancer Res (2015) 75(11):2337–48. doi: 10.1158/0008-5472.CAN-14-2800
11. Geribaldi-Doldan N, Hervas-Corpion I, Gomez-Oliva R, Dominguez-Garcia S, Ruiz FA, Iglesias-Lozano I, et al. Targeting protein kinase C in glioblastoma treatment. Biomedicines (2021) 9(4):381. doi: 10.3390/biomedicines9040381
12. Kapoor SS, Zaiss DMW. Emerging role of EGFR mutations in creating an immune suppressive tumour microenvironment. Biomedicines (2021) 10(1):52. doi: 10.3390/biomedicines10010052
13. Madeddu C, Donisi C, Liscia N, Lai E, Scartozzi M, Maccio A. EGFR-mutated non-small cell lung cancer and resistance to immunotherapy: role of the tumor microenvironment. Int J Mol Sci (2022) 23(12):6489. doi: 10.3390/ijms23126489
14. Kumagai S, Koyama S, Nishikawa H. Antitumour immunity regulated by aberrant ERBB family signalling. Nat Rev Cancer (2021) 21(3):181–97. doi: 10.1038/s41568-020-00322-0
15. Hao Z, Guo D. EGFR mutation: novel prognostic factor associated with immune infiltration in lower-grade glioma; an exploratory study. BMC Cancer (2019) 19(1):1184. doi: 10.1186/s12885-019-6384-8
16. Friedman JS, Jun T, Rashidipour O, Huang KL, Ellis E, Kadaba P, et al. Using EGFR amplification to stratify recurrent glioblastoma treated with immune checkpoint inhibitors. Cancer Immunol Immunother (2023) 72(6):1893–901. doi: 10.1007/s00262-023-03381-y
17. Huang W, Hao Z, Mao F, Guo D. Small molecule inhibitors in adult high-grade glioma: from the past to the future. Front Oncol (2022) 12. doi: 10.3389/fonc.2022.911876
18. Sepúlveda-Sánchez JM, Vaz M, Balañá C, Gil-Gil M, Reynés G, Gallego Ó, et al. Phase II trial of dacomitinib, a pan-human EGFR tyrosine kinase inhibitor, in recurrent glioblastoma patients with EGFR amplification. Neuro Oncol (2017) 19(11):1522–31. doi: 10.1093/neuonc/nox105
19. Liu X, Chen X, Shi L, Shan Q, Cao Q, Yue C, et al. The third-generation EGFR inhibitor AZD9291 overcomes primary resistance by continuously blocking ERK signaling in glioblastoma. J Exp Clin Cancer Res (2019) 38(1):219. doi: 10.1186/s13046-019-1235-7
20. Erel-Akbaba G, Carvalho LA, Tian T, Zinter M, Akbaba H, Obeid PJ, et al. Radiation-induced targeted nanoparticle-based gene delivery for brain tumor therapy. ACS Nano (2019) 13(4):4028–40. doi: 10.1021/acsnano.8b08177
21. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H, et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol (2017) 18(10):1373–85. doi: 10.1016/S1470-2045(17)30517-X
22. Goff SL, Morgan RA, Yang JC, Sherry RM, Robbins PF, Restifo NP, et al. Pilot trial of adoptive transfer of chimeric antigen receptor-transduced T cells targeting EGFRvIII in patients with glioblastoma. J Immunother (2019) 42(4):126–35. doi: 10.1097/CJI.0000000000000260
23. Litak J, Mazurek M, Grochowski C, Kamieniak P, Rolinski J. PD-L1/PD-1 axis in glioblastoma multiforme. Int J Mol Sci (2019) 20(21):5347. doi: 10.3390/ijms20215347
24. Gatto L, Di Nunno V, Franceschi E, Tosoni A, Bartolini S, Brandes AA. Pharmacotherapeutic treatment of glioblastoma: where are we to date? Drugs (2022) 82(5):491–510. doi: 10.1007/s40265-022-01702-6
25. Engelhardt B, Vajkoczy P, Weller RO. The movers and shapers in immune privilege of the CNS. Nat Immunol (2017) 18(2):123–31. doi: 10.1038/ni.3666
26. Tomaszewski W, Sanchez-Perez L, Gajewski TF, Sampson JH. Brain tumor microenvironment and host state: implications for immunotherapy. Clin Cancer Res (2019) 25(14):4202–10. doi: 10.1158/1078-0432.CCR-18-1627
27. Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia (2012) 60(3):502–14. doi: 10.1002/glia.21264
28. Grabowski MM, Sankey EW, Ryan KJ, Chongsathidkiet P, Lorrey SJ, Wilkinson DS, et al. Immune suppression in gliomas. J Neurooncol (2021) 151(1):3–12. doi: 10.1007/s11060-020-03483-y
29. Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, et al. Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science (2010) 330(6005):841–5. doi: 10.1126/science.1194637
30. Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, et al. Cellular and molecular identity of tumor-associated macrophages in glioblastoma. Cancer Res (2017) 77(9):2266–78. doi: 10.1158/0008-5472.CAN-16-2310
31. Roesch S, Rapp C, Dettling S, Herold-Mende C. When immune cells turn bad-tumor-associated microglia/macrophages in glioma. Int J Mol Sci (2018) 19(2):436. doi: 10.3390/ijms19020436
32. Coniglio SJ, Eugenin E, Dobrenis K, Stanley ER, West BL, Symons MH, et al. Microglial stimulation of glioblastoma invasion involves epidermal growth factor receptor (EGFR) and colony stimulating factor 1 receptor (CSF-1R) signaling. Mol Med (2012) 18(1):519–27. doi: 10.2119/molmed.2011.00217
33. An Z, Knobbe-Thomsen CB, Wan X, Fan QW, Reifenberger G, Weiss WA. EGFR cooperates with EGFRvIII to recruit macrophages in glioblastoma. Cancer Res (2018) 78(24):6785–94. doi: 10.1158/0008-5472.CAN-17-3551
34. Sidorov M, Dighe P, Woo RWL, Rodriguez-Brotons A, Chen M, Ice RJ, et al. Dual targeting of EGFR and MTOR pathways inhibits glioblastoma growth by modulating the tumor microenvironment. Cells (2023) 12(4):547. doi: 10.3390/cells12040547
35. Sierra-Filardi E, Nieto C, Dominguez-Soto A, Barroso R, Sanchez-Mateos P, Puig-Kroger A, et al. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J Immunol (2014) 192(8):3858–67. doi: 10.4049/jimmunol.1302821
36. Yeo AT, Rawal S, Delcuze B, Christofides A, Atayde A, Strauss L, et al. Single-cell RNA sequencing reveals evolution of immune landscape during glioblastoma progression. Nat Immunol (2022) 23(6):971–84. doi: 10.1038/s41590-022-01215-0
37. Walentynowicz KA, Engelhardt D, Cristea S, Yadav S, Onubogu U, Salatino R, et al. Single-cell heterogeneity of EGFR and CDK4 co-amplification is linked to immune infiltration in glioblastoma. Cell Rep (2023) 42(3):112235. doi: 10.1016/j.celrep.2023.112235
38. De Leo A, Ugolini A, Veglia F. Myeloid cells in glioblastoma microenvironment. Cells (2020) 10(1):18. doi: 10.3390/cells10010018
39. Mi Y, Guo N, Luan J, Cheng J, Hu Z, Jiang P, et al. The emerging role of myeloid-derived suppressor cells in the glioma immune suppressive microenvironment. Front Immunol (2020) 11:737. doi: 10.3389/fimmu.2020.00737
40. Yeo AT, Shah R, Aliazis K, Pal R, Xu T, Zhang P, et al. Driver mutations dictate the immunological landscape and response to checkpoint immunotherapy of glioblastoma. Cancer Immunol Res (2023) 11(5):629–45. doi: 10.1158/2326-6066.c.6597334.v2
41. Wang R, Song Y, Hu T, Wang X, Jiang Y, Zhang D, et al. Decreased CD8(+) lymphocytic infiltration in multifocal and multicentric glioblastomas. Front Oncol (2021) 11:748277. doi: 10.3389/fonc.2021.748277
42. Han S, Zhang C, Li Q, Dong J, Liu Y, Huang Y, et al. Tumour-infiltrating CD4(+) and CD8(+) lymphocytes as predictors of clinical outcome in glioma. Br J Cancer (2014) 110(10):2560–8. doi: 10.1038/bjc.2014.162
43. Cordell EC, Alghamri MS, Castro MG, Gutmann DH. T lymphocytes as dynamic regulators of glioma pathobiology. Neuro Oncol (2022) 24(10):1647–57. doi: 10.1093/neuonc/noac055
44. Han S, Ma E, Wang X, Yu C, Dong T, Zhan W, et al. Rescuing defective tumor-infiltrating T-cell proliferation in glioblastoma patients. Oncol Lett (2016) 12(4):2924–9. doi: 10.3892/ol.2016.4944
45. Rutledge WC, Kong J, Gao J, Gutman DA, Cooper LA, Appin C, et al. Tumor-infiltrating lymphocytes in glioblastoma are associated with specific genomic alterations and related to transcriptional class. Clin Cancer Res (2013) 19(18):4951–60. doi: 10.1158/1078-0432.CCR-13-0551
46. Woroniecka KI, Rhodin KE, Chongsathidkiet P, Keith KA, Fecci PE. T-cell dysfunction in glioblastoma: applying a new framework. Clin Cancer Res (2018) 24(16):3792–802. doi: 10.1158/1078-0432.CCR-18-0047
47. Liu Z, Han C, Dong C, Shen A, Hsu E, Ren Z, et al. Hypofractionated EGFR tyrosine kinase inhibitor limits tumor relapse through triggering innate and adaptive immunity. Sci Immunol (2019) 4(38):eaav6473. doi: 10.1126/sciimmunol.aav6473
48. Ooi YC, Tran P, Ung N, Thill K, Trang A, Fong BM, et al. The role of regulatory T-cells in glioma immunology. Clin Neurol Neurosurg (2014) 119:125–32. doi: 10.1016/j.clineuro.2013.12.004
49. Coy S, Wang S, Stopka SA, Lin JR, Yapp C, Ritch CC, et al. Single cell spatial analysis reveals the topology of immunomodulatory purinergic signaling in glioblastoma. Nat Commun (2022) 13(1):4814. doi: 10.1038/s41467-022-32430-w
50. Crane CA, Han SJ, Barry JJ, Ahn BJ, Lanier LL, Parsa AT. TGF-beta downregulates the activating receptor NKG2D on NK cells and CD8+ T cells in glioma patients. Neuro Oncol (2010) 12(1):7–13. doi: 10.1093/neuonc/nop009
51. Liu S, Galat V, Galat Y, Lee YKA, Wainwright D, Wu J. NK cell-based cancer immunotherapy: from basic biology to clinical development. J Hematol Oncol (2021) 14(1):7. doi: 10.1186/s13045-020-01014-w
52. Vantourout P, Willcox C, Turner A, Swanson CM, Haque Y, Sobolev O, et al. Immunological visibility: posttranscriptional regulation of human NKG2D ligands by the EGF receptor pathway. Sci Transl Med (2014) 6(231):231ra49. doi: 10.1126/scitranslmed.3007579
53. Bryceson YT, Ljunggren HG. Tumor cell recognition by the NK cell activating receptor NKG2D. Eur J Immunol (2008) 38(11):2957–61. doi: 10.1002/eji.200838833
54. Fuertes MB, Domaica CI, Zwirner NW. Leveraging NKG2D ligands in immuno-oncology. Front Immunol (2021) 12:713158. doi: 10.3389/fimmu.2021.713158
55. Dhar P, Wu JD. NKG2D and its ligands in cancer. Curr Opin Immunol (2018) 51:55–61. doi: 10.1016/j.coi.2018.02.004
56. Wang SS, Liu W, Ly D, Xu H, Qu L, Zhang L. Tumor-infiltrating B cells: their role and application in anti-tumor immunity in lung cancer. Cell Mol Immunol (2019) 16(1):6–18. doi: 10.1038/s41423-018-0027-x
57. Nduom EK, Wei J, Yaghi NK, Huang N, Kong LY, Gabrusiewicz K, et al. PD-L1 expression and prognostic impact in glioblastoma. Neuro Oncol (2016) 18(2):195–205. doi: 10.1093/neuonc/nov172
58. Noronha C, Ribeiro AS, Taipa R, Leitão D, Schmitt F, Reis J, et al. PD-L1 tumor expression is associated with poor prognosis and systemic immunosuppression in glioblastoma. J Neurooncol (2022) 156(3):453–64. doi: 10.1007/s11060-021-03907-3
59. Wang X, Guo G, Guan H, Yu Y, Lu J, Yu J. Challenges and potential of PD-1/PD-L1 checkpoint blockade immunotherapy for glioblastoma. J Exp Clin Cancer Res (2019) 38(1):87. doi: 10.1186/s13046-019-1085-3
60. Du L, Lee JH, Jiang H, Wang C, Wang S, Zheng Z, et al. beta-Catenin induces transcriptional expression of PD-L1 to promote glioblastoma immune evasion. J Exp Med (2020) 217(11):e20191115. doi: 10.1084/jem.20191115
61. Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, et al. Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nat Med (2007) 13(1):84–8. doi: 10.1038/nm1517
62. Su L, Guo W, Lou L, Nie S, Zhang Q, Liu Y, et al. EGFR-ERK pathway regulates CSN6 to contribute to PD-L1 expression in glioblastoma. Mol Carcinog (2020) 59(5):520–32. doi: 10.1002/mc.23176
63. Song X, Shao Y, Jiang T, Ding Y, Xu B, Zheng X, et al. Radiotherapy upregulates programmed death ligand-1 through the pathways downstream of epidermal growth factor receptor in glioma. EBioMedicine (2018) 28:105–13. doi: 10.1016/j.ebiom.2018.01.027
64. Ebert PJR, Cheung J, Yang Y, McNamara E, Hong R, Moskalenko M, et al. MAP kinase inhibition promotes T cell and anti-tumor activity in combination with PD-L1 checkpoint blockade. Immunity (2016) 44(3):609–21. doi: 10.1016/j.immuni.2016.01.024
65. Nishii K, Ohashi K, Tomida S, Nakasuka T, Hirabae A, Okawa S, et al. CD8+ T-cell responses are boosted by dual PD-1/VEGFR2 blockade after EGFR inhibition in egfr-mutant lung cancer. Cancer Immunol Res (2022) 10(9):1111–26. doi: 10.1158/2326-6066.CIR-21-0751
66. Xu S, Shao QQ, Sun JT, Yang N, Xie Q, Wang DH, et al. Synergy between the ectoenzymes CD39 and CD73 contributes to adenosinergic immunosuppression in human Malignant gliomas. Neuro Oncol (2013) 15(9):1160–72. doi: 10.1093/neuonc/not067
67. Vijayan D, Young A, Teng MWL, Smyth MJ. Targeting immunosuppressive adenosine in cancer. Nat Rev Cancer (2017) 17(12):709–24. doi: 10.1038/nrc.2017.86
68. Zhulai G, Oleinik E, Shibaev M, Ignatev K. Adenosine-metabolizing enzymes, adenosine kinase and adenosine deaminase, in cancer. Biomolecules (2022) 12(3):418. doi: 10.3390/biom12030418
69. Sun C, Wang B, Hao S. Adenosine-A2A receptor pathway in cancer immunotherapy. Front Immunol (2022) 13:837230. doi: 10.3389/fimmu.2022.837230
70. Tang K, Zhang J, Cao H, Xiao G, Wang Z, Zhang X, et al. Identification of CD73 as a novel biomarker encompassing the tumor microenvironment, prognosis, and therapeutic responses in various cancers. Cancers (Basel) (2022) 14(22):5663. doi: 10.3390/cancers14225663
71. Azambuja JH, Schuh RS, Michels LR, Iser IC, Beckenkamp LR, Roliano GG, et al. Blockade of CD73 delays glioblastoma growth by modulating the immune environment. Cancer Immunol Immunother (2020) 69(9):1801–12. doi: 10.1007/s00262-020-02569-w
72. Gong L, Ji L, Xu D, Wang J, Zou J. TGF-beta links glycolysis and immunosuppression in glioblastoma. Histol Histopathol (2021) 36(11):1111–24. doi: 10.14670/HH-18-366
73. Kaminska B, Cyranowski S. Recent advances in understanding mechanisms of TGF beta signaling and its role in glioma pathogenesis. Adv Exp Med Biol (2020) 1202:179–201. doi: 10.1007/978-3-030-30651-9_9
74. Batlle E, Massague J. Transforming growth factor-beta signaling in immunity and cancer. Immunity (2019) 50(4):924–40. doi: 10.1016/j.immuni.2019.03.024
75. Gulley JL, Schlom J, Barcellos-Hoff MH, Wang XJ, Seoane J, Audhuy F, et al. Dual inhibition of TGF-beta and PD-L1: a novel approach to cancer treatment. Mol Oncol (2022) 16(11):2117–34. doi: 10.1002/1878-0261.13146
76. Minutti CM, Modak RV, Macdonald F, Li F, Smyth DJ, Dorward DA, et al. A macrophage-pericyte axis directs tissue restoration via amphiregulin-induced transforming growth factor beta activation. Immunity (2019) 50(3):645–54 e6. doi: 10.1016/j.immuni.2019.01.008
77. Lanaya H, Natarajan A, Komposch K, Li L, Amberg N, Chen L, et al. EGFR has a tumour-promoting role in liver macrophages during hepatocellular carcinoma formation. Nat Cell Biol (2014) 16(10):972–7. doi: 10.1038/ncb3031
78. Srivatsa S, Paul MC, Cardone C, Holcmann M, Amberg N, Pathria P, et al. EGFR in tumor-associated myeloid cells promotes development of colorectal cancer in mice and associates with outcomes of patients. Gastroenterology (2017) 153(1):178–90.e10. doi: 10.1053/j.gastro.2017.03.053
79. Hardbower DM, Coburn LA, Asim M, Singh K, Sierra JC, Barry DP, et al. EGFR-mediated macrophage activation promotes colitis-associated tumorigenesis. Oncogene (2017) 36(27):3807–19. doi: 10.1038/onc.2017.23
80. Zaiss DM, van Loosdregt J, Gorlani A, Bekker CP, Gröne A, Sibilia M, et al. Amphiregulin enhances regulatory T cell-suppressive function via the epidermal growth factor receptor. Immunity (2013) 38(2):275–84. doi: 10.1016/j.immuni.2012.09.023
81. Qu WS, Liu JL, Li CY, Li X, Xie MJ, Wang W, et al. Rapidly activated epidermal growth factor receptor mediates lipopolysaccharide-triggered migration of microglia. Neurochem Int (2015) 90:85–92. doi: 10.1016/j.neuint.2015.07.007
82. Gettinger S, Hellmann MD, Chow LQM, Borghaei H, Antonia S, Brahmer JR, et al. Nivolumab plus Erlotinib in patients with EGFR-mutant advanced NSCLC. J Thorac Oncol (2018) 13(9):1363–72. doi: 10.1016/j.jtho.2018.05.015
Keywords: glioblastoma, epidermal growth factor receptor, immunosuppression, tumor microenvironment, immune cell
Citation: Li X-P, Guo Z-Q, Wang B-F and Zhao M (2023) EGFR alterations in glioblastoma play a role in antitumor immunity regulation. Front. Oncol. 13:1236246. doi: 10.3389/fonc.2023.1236246
Received: 07 June 2023; Accepted: 20 July 2023;
Published: 04 August 2023.
Edited by:
Liang Wang, Fourth Military Medical University, ChinaReviewed by:
Po Zhang, University of Pittsburgh Medical Center, United StatesWei Yi, Renmin Hospital of Wuhan University, China
Neibla Priego, Spanish National Cancer Research Center, Spain
Paul Clark, University of Wisconsin-Madison, United States
Copyright © 2023 Li, Guo, Wang and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Bao-Feng Wang, d2JmNjIwQDE2My5jb20=; Min Zhao, NzEwMjQyNzY5QHFxLmNvbQ==
†These authors have contributed equally to this work