 Selene Cipri1
Selene Cipri1 Francesco Fabozzi1
Francesco Fabozzi1 Giada Del Baldo1,2
Giada Del Baldo1,2 Giuseppe Maria Milano1
Giuseppe Maria Milano1 Luigi Boccuto3
Luigi Boccuto3 Andrea Carai4
Andrea Carai4 Angela Mastronuzzi1*
Angela Mastronuzzi1*- 1Department of Hematology/Oncology, Cell Therapy, Gene Therapies and Hemopoietic Transplant, Bambino Gesù Children’s Hospital, Istituti di Ricovero e Cura a Carattere Scientifico (IRCCS), Rome, Italy
- 2Department of Experimental Medicine, Sapienza University of Rome, Rome, Italy
- 3Healthcare Genetics Program, School of Nursing, College of Behavioral, Social and Health Sciences, Clemson University, Clemson, SC, United States
- 4Department of Neurosciences, Neurosurgery Unit, Bambino Gesù Children’s Hospital, Istituti di Ricovero e Cura a Carattere Scientifico (IRCCS), Rome, Italy
The family of the neurotrophic tyrosine kinase receptor (NTRK) gene encodes for members of the tropomyosin receptor kinase (TRK) family. Rearrangements involving NTRK1/2/3 are rare oncogenic factors reported with variable frequencies in an extensive range of cancers in pediatrics and adult populations, although they are more common in the former than in the latter. The alterations in these genes are causative of the constitutive activation of TRKs that drive carcinogenesis. In 2017, first-generation TRK inhibitor (TRKi) larotrectinib was granted accelerated approval from the FDA, having demonstrated histologic-agnostic activity against NTRKs fusions tumors. Since this new era has begun, resistance to first-generation TRKi has been described and has opened the development of second-generation molecules, such as selitrectinib and repotrectinib. In this review, we provide a brief overview of the studies on NTRK alterations found in pediatric central nervous system tumors and first and second-generation TRKi useful in clinical practice.
1 Introduction
Central nervous system (CNS) tumors are the commonest solid neoplasm in children aged 0-14 (1).
In CNS tumors, which commonly have no effective therapies, significant frequencies of neurotrophic tyrosine receptor kinase (NTRK) fusions have been revealed and their detection has become a cornerstone in the diagnostic evaluation of these cancers and treatment through specific therapies (2, 3). NTRKs are a family of tyrosine kinases receptors of neurotrophins implicated in neuronal development, among them the development of memory and the growth and function of neuronal synapses (4). The NTRK1/2/3 genes produce three members of the tropomyosin receptor kinases (TRKs) called tropomyosin receptor kinases TRKA, TRKB and TRKC, respectively, and are characterized by an extracellular binding domain, a transmembrane region and an intracellular kinase domain (4, 5).
TRK is usually activated in tumors via fusions involving NTRK1/2/3, caused by rearrangements of chromosomes between NTRK genes, which include the kinase domain, with several partner genes. The fusion products are chimeras with a constitutively activated TRK, regardless of the ligand they bound (6, 7).
The rearrangement between tropomyosin 3 (TPM3) and NTRK1 in colorectal cancer was the first detected NTRK fusion (8). Afterward, NTRK fusions were found with several partners in a wide diversity of cancer typologies: among the fusions involving NTRK1 are known the fusions with ROS Proto-Oncogene 1, Receptor Tyrosine Kinase (ROS1) and Lamin A/C (LMNA), involved in spitzoid neoplasms and in soft tissue sarcomas (STS), respectively (9). The LMNA-NTRK1 is involved also carcinoma of lung and colorectal (10). Translocated promoter region (TPR) with NTRK1 was found in thyroid cancer, and sequestosome 1 (SQSTM1)-NTRK1 fusion in STS and non-small cell lung cancer (NSCLC) (11–15). The fusion that involved ETS variant of transcription factor 6 (ETV6) and NTRK3 was found, for example, in congenital fibrosarcoma, congenital mesoblastic nephroma, PTCs and colorectal cancer (9, 16–18). Regardless of this review, NTRK gene fusions occur in more than 2.5% of low-grade gliomas (LGGs) and 5.3% of high-grade gliomas (HGGs) in children (19), and contribute to defining infant-type hemispheric gliomas, a new type of HGG, in the 2021 WHO classification of CNS tumors (20).
In this review, we explain a brief overview of the studies on NTRK alterations found in pediatric CNS tumors and first- and second-generation TRKi targeted therapy.
2 NTRK fusions: from detection to treatment
2.1 Tropomyosin receptor kinase and cell cycle
Briefly, neurotrophin growth factors bind and activate TRKs in a specific manner: nerve growth factor neurotrophin (NGF) to TRKA; brain-derived neurotrophic factor (BDNF) and neurotrophin 4 (NT-4) that bins to TRKB; and neurotrophin 3 (NT-3) to all three TRK proteins, although it has a higher kinship for TRKC (21–26).
The RAS/MAPK, PI3K/AKT, and PLC/PKC signaling pathway is triggered by the bond between ligand to the extracellular domain that causes the homodimerization and transactivation of TRK receptors via autophosphorylation of tyrosine residues (Figure 1). Activation of the above pathways promotes cell proliferation, differentiation, and survival (5, 6, 27, 28).
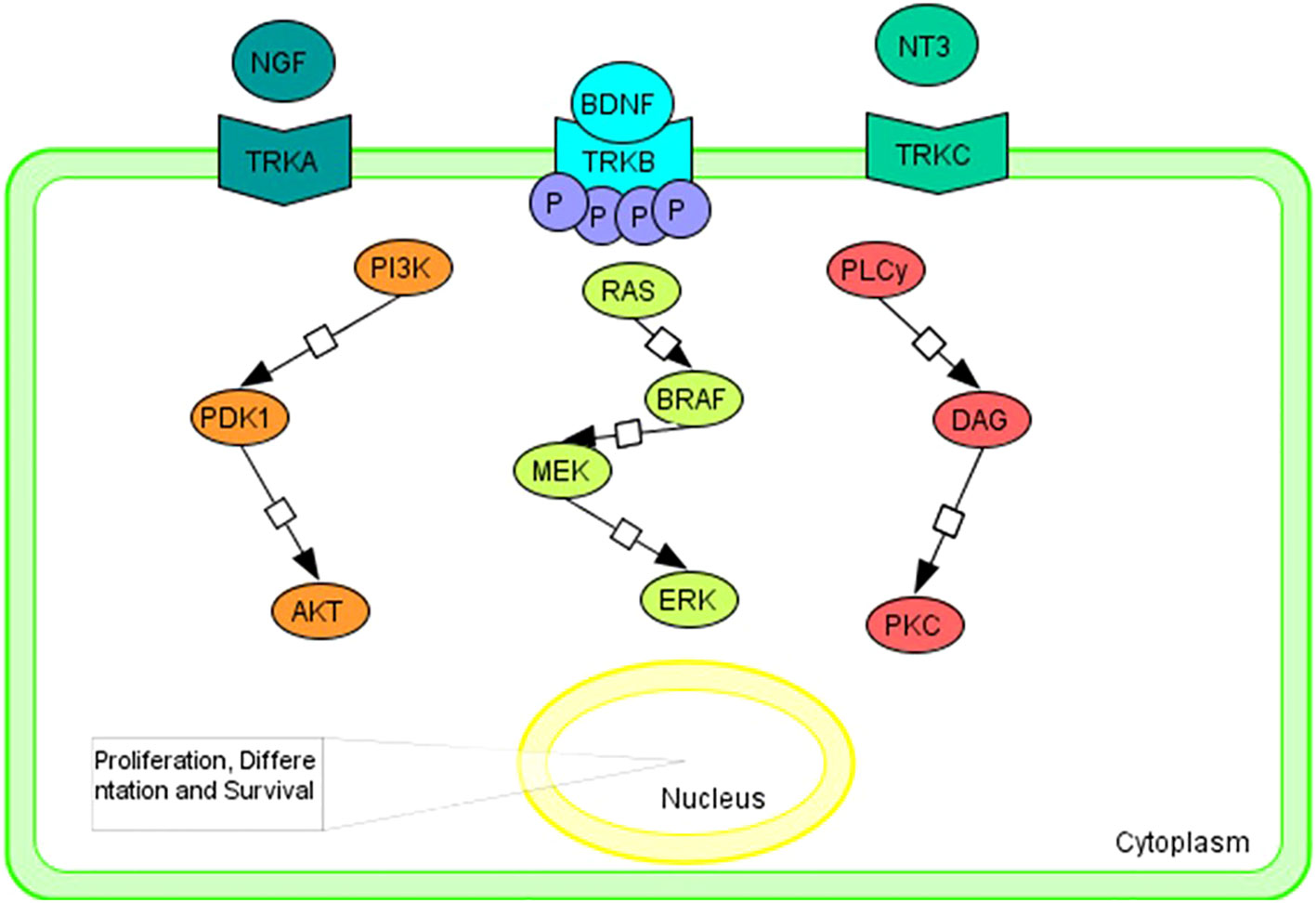
Figure 1 Graphical representation of the main intracellular signaling pathways associated with TRK family members. Tropomyosin receptor kinase A (TRKA); tropomyosin receptor kinase B (TRKB); tropomyosin receptor kinase C (TRKC); nerve growth factor neurotrophin (NGF); brain-derived neurotrophic factor (BDNF); neurotrophin 4 (NT-4); neurotrophin 3 (NT-3); PhosphatidylInositol 3-Kinase (PI3K); Pyruvate Dehydrogenase Kinase 1 (PDK1); AKT Serine/Threonine Kinase (AKT); B-Raf Proto-Oncogene, Serine/Threonine Kinase (BRAF); Mitogen-activated protein kinase kinase (MEK); extracellular signal-regulated kinase (ERK); Phospholipase C y (PLCy); diacylglycerol (DAG); Protein Kinase C (PKC).
2.2 NTRK fusions in pediatric central nervous system tumors
In CNS tumors, significant frequencies of NTRK fusions have been identified and their detection has become a cornerstone in the diagnostic evaluation of these cancers (3).
Several studies including large cohorts of pediatric CNS tumors found that NRTK1-3 alterations occur mostly in very young children and tumors localized to the hemispheric lobs (29, 30). These results converged in the 2021 WHO Classification of CNS Tumors, in which NTRK alterations contribute to defining novel entities among both HGGs and LGGs in children, namely infant-type hemispheric glioma and diffuse LGG, MAPK pathway‐altered, respectively (20). Despite the high-grade histology, the first subgroup benefits from a better outcome compared to its counterpart without tyrosine kinase fusions (29, 30).
NTRK fusions found in several studies are depicted in Figure 2.
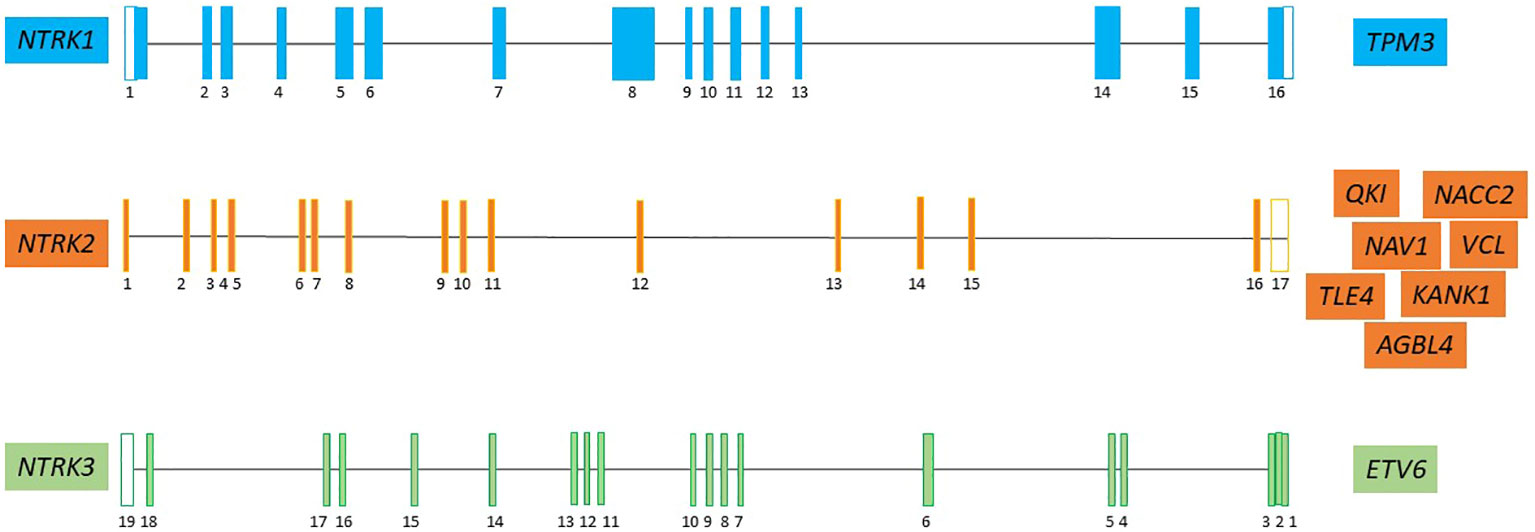
Figure 2 The major NTRK partner fusion genes in pediatric CNS tumor (7, 19, 28, 31–39). Neurotrophic tyrosine kinase receptor 1 (NTRK1), neurotrophic tyrosine kinase receptor 2 (NTRK2), neurotrophic tyrosine kinase receptor 3 (NTRK3), Tropomyosin 3 (TPM3); QKI, KH Domain Containing RNA Binding (QKI); NACC Family Member 2 (NACC2); Neuron Navigator 1 (NAV1); KN Motif And Ankyrin Repeat Domains 1 (KANK1); actin-binding protein vinculin (VCL); ATP/GTP-binding protein (AGBL4); TLE Family Member 4, Transcriptional Corepressor (TLE4); ETS variant transcription factor 6 (ETV6).
2.3 NTRK inhibitors
There have been only limited in vitro or preclinical studies of signaling performed to illuminate the effect of TRKi on downstream cascade signaling or the time span of inhibition, but meaningful clinical responsiveness to these drugs has been shown in several types of tumors such as soft tissue sarcomas, childhood fibrosarcoma, lung cancer, colon cancer, melanoma (40–45).
First-generation TRKi were developed in 2015 that included larotrectinib and entrectinib. The recruiting clinical trials of either larotrectinib or entrectinib are listed in Supplementary Table 1.
Larotrectinib, developed simultaneously for pediatric and adult cancer, is the first oral treatment with a “tumor-agnostic” indication: discovered in 2015, it obtained accelerated approval from FDA in 2017. It is a small-sized competitive inhibitor of ATP and selective pan-TRK, with a 50% inhibitory concentration (IC50) of 5-11 nm in vitro and a specificity >100 times for TRK (46, 47). Inhibition of RAF-MEK-ERK or PI3K-AKT pathways, caused by larotrectinib, inhibits the growth of some cell lines that contained targeted NTRK fusions, as well TPM3-NTRK1, TRIM24-NTRK2, and ETV6-NTRK3 (48, 49). An awesome overall response rate (ORR - the percentage of patients who experienced a complete or partial response) of 79% and a well-tolerated profile of toxicity were found in phase I/II clinical trials enrolling both adult and pediatric patients (50). In brain tumors, ORR was 30% and in particular, a retrospective study showed the results on the efficacy and safety for patients (n=33) with progressive or refractory CNS tumors enrolled in the SCOUT (NCT02637687) and NAVIGATE (NCT02576431) trials; among the 26 pediatric patients (79%), 13 pediatric HGGs and 7 pediatric LGGs were included. The observed ORR was 38% (38% in HGGs and 43% in LGGs, respectively), with three complete responses and seven partial responses. Importantly, the disease control rate at 24 weeks was 77% for pediatric HGGs and 100% for pediatric LGGs (43). In Supplementary Table 2 are reported results on patents with CNS tumor and treated with Larotectinb.
The Food and Drug Administration approved entrectinib in August 2019 to treat adult and pediatric populations with NTRK fusion tumors (51).
Robinson and colleagues published the first interim results based on 29 enrolled patients, aged 5 months to 20 years. The ORR was 100% in 11 pts [(high-grade CNS tumors (n=5) and extracranial solid tumors (n=6)] (52). In 2020, an expanded cohort of 39 patients confirmed an ORR of 77%. CNS tumors were in 14 patients, of which 11 displayed NTRK fusions. Notably, the ORR in this subgroup reached 64% (53). Desai et al. demonstrated that entrectinib had a rapid and durable responses in pediatric patients with solid tumors harboring NTRK1/2/3 or ROS1 fusions (54). In Supplementary Table 2 are reported results on patents with CNS tumor and treated with entrectinib. In addition, Liu et al. reported weight gain, dizziness and withdrawal pain in a several patients who were treated with TRKi (55).
Usually, both Larotrectinib and Entrectinib are administered until disease progression or unacceptable toxicity occurs (42, 43, 54). Treatment discontinuation is reported in extracranial tumors in which tumor size reduction has made complete resection possible; interestingly, patients who discontinued treatment following an initial response and subsequently experienced disease progression may still benefit from restart of therapy (41).
Mutations called on target and off target, respectively, on the NTRK gene or in genes associated with the MAPK pathway, are responsible for resistance to those drugs in several type of cancers (5, 56–59). In the NTRK3 gene, acquired variants p.G623E and p.G623R have been identified to confer resistance to either larotrectinib or entrectinib (48, 57, 59, 60). Additionally, acquired variants p.F617L and p.G696A specifically confer resistance to larotrectinib (50, 57, 61). In the NTRK1 gene, acquired variants p.V573M and p.G667S have been found to induce resistance to both larotrectinib and entrectinib, whereas the acquired variant p.F589L in the same gene only confers resistance to larotrectinib (50, 57, 62–64).
As a result, the need for second-generation TRKi, such as selitrectinib (loxo-195), taletrectinib (DS-6051b, AB-106), and repotrectinib (tpx-0005), has arisen (5, 30, 56). Taletrectinib works as a multi-kinase inhibitor that can overcome resistance from solvent-front replacements involving TRKA, TRKB and TRKC such as others involving ROS1 (65). Selitrectinib is a selective TRKi studied in a phase I trial involving both children and adults with tumors that have developed resistance mediated by TRK kinase mutations, in which a preliminary efficacy was found (66). Repotrectinib functions as a kinase inhibitor encoded by the NTRK, ROS1, and ALK genes. It effectively binds to the ATP-binding pocket of the kinase, preventing steric hindrance caused by various clinically resistant mutations (57). A clinical trial investigating its use in pediatric patients with solid tumors that include CNS neoplasms is currently ongoing (NCT04094610).
On the other hand, mutations that involved other RTKs or downstream pathway mediators can result in off-target resistance to TRKi. Specifically, MET amplification, BRAFV600E mutation, or KRAS alterations have been found in patients with TRK fusion and who show a progression of the tumor during the treatment of TRKi (56). Of note, the TRKi monotherapy was not effective for resistance mediated to overcome the mutational pathway, while a dual blockade of TRK and other pathways involved in the resistance mechanism could effectively control tumor growth (67). For instance, the combination of the inhibitors of TRK and MET has been found to be effective in a patient carried a TRK fusion and MET amplification that drives the resistance to the TRKi alone (56).
3 Conclusions
Tropomyosin receptor kinase inhibitors, such as larotrectinib and entrectinib, have showed high efficacy in pediatric patients, also in CNS tumors carrying alterations in NTRK genes. To date, additional research is necessary to help us to understand better the mechanism of action of these drugs and to identify biomarkers that can help identify patients who will benefit most from therapy.
Author contributions
AM, AC, and LB conceptualized the work. SC, FF, GB, and GM wrote the manuscript. AM, AC, and LB contributed to the finishing of the work and revised it critically for important intellectual content. All authors finally approved the version to be published and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All authors contributed to the article and approved the submitted version.
Acknowledgments
The authors thank Megan Eckley for revising the English language for the final version. This work was supported also by the Italian Ministry of Health with “Current Research funds”.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1235794/full#supplementary-material
References
1. Ostrom QT, Gittleman H, Truitt G, Boscia A, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: primary brain and other central nervous system tumors diagnosed in the United States in 2011-2015. Neuro-Oncol (2018) 20:iv1–iv86. doi: 10.1093/neuonc/noy131
2. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 world health organization classification of tumors of the central nervous system: a summary. Acta Neuropathol (Berl) (2016) 131:803–20. doi: 10.1007/s00401-016-1545-1
3. Gambella A, Senetta R, Collemi G, Vallero SG, Monticelli M, Cofano F, et al. NTRK fusions in central nervous system tumors: A rare, but worthy target. Int J Mol Sci (2020) 21:753. doi: 10.3390/ijms21030753
4. Solomon JP, Benayed R, Hechtman JF, Ladanyi M. Identifying patients with NTRK fusion cancer. Ann Oncol Off J Eur Soc Med Oncol (2019) 30:viii16–22. doi: 10.1093/annonc/mdz384
5. Cocco E, Scaltriti M, Drilon A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat Rev Clin Oncol (2018) 15:731–47. doi: 10.1038/s41571-018-0113-0
6. Vaishnavi A, Le AT, Doebele RC. TRKing down an old oncogene in a new era of targeted therapy. Cancer Discovery (2015) 5:25–34. doi: 10.1158/2159-8290.CD-14-0765
7. Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet (2013) 45:1141–9. doi: 10.1038/ng.2734
8. Martin-Zanca D, Hughes SH, Barbacid M. A human oncogene formed by the fusion of truncated tropomyosin and protein tyrosine kinase sequences. Nature (1986) 319:743–8. doi: 10.1038/319743a0
9. Zhao X, Kotch C, Fox E, Surrey LF, Wertheim GB, Baloch ZW, et al. NTRK fusions identified in pediatric tumors: the frequency, fusion partners, and clinical outcome. JCO Precis Oncol (2021) 5:204–14. doi: 10.1200/PO.20.00250
10. Refractory and metastatic infantile fibrosarcoma harboring LMNA–NTRK1 fusion shows complete and durable response to crizotinib . Available at: https://molecularcasestudies.cshlp.org/content/5/1/a003376.short (Accessed April 20, 2023).
11. Greco A, Pierotti MA, Bongarzone I, Pagliardini S, Lanzi C, Della Porta G. TRK-T1 is a novel oncogene formed by the fusion of TPR and TRK genes in human papillary thyroid carcinomas. Oncogene (1992) 7:237–42.
12. Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun (2014) 5:4846. doi: 10.1038/ncomms5846
13. Kachanov DY, Dobrenkov KV, Abdullaev RT, Shamanskaya TV, Varfolomeeva SR. Incidence and survival of pediatric soft tissue sarcomas in moscow region, Russian federation, 2000–2009. Sarcoma (2012) 2012:e350806. doi: 10.1155/2012/350806
14. Farago AF, Le LP, Zheng Z, Muzikansky A, Drilon A, Patel M, et al. Durable clinical response to entrectinib in NTRK1-rearranged non-small cell lung cancer. J Thorac Oncol (2015) 10:1670–4. doi: 10.1097/01.JTO.0000473485.38553.f0
15. Pavlick D, Schrock AB, Malicki D, Stephens PJ, Kuo DJ, Ahn H, et al. Identification of NTRK fusions in pediatric mesenchymal tumors. Pediatr Blood Cancer (2017) 64:e26433. doi: 10.1002/pbc.26433
16. Knezevich SR, McFadden DE, Tao W, Lim JF, Sorensen PH. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet (1998) 18:184–7. doi: 10.1038/ng0298-184
17. Leeman-Neill RJ, Kelly LM, Liu P, Brenner AV, Little MP, Bogdanova TI, et al. ETV6-NTRK3 is a common chromosomal rearrangement in radiation-associated thyroid cancer. Cancer (2014) 120:799–807. doi: 10.1002/cncr.28484
18. Wang H, Li Z, Ou Q, Wu X, Nagasaka M, Shao Y, et al. NTRK fusion positive colorectal cancer is a unique subset of CRC with high TMB and microsatellite instability. Cancer Med (2022) 11:2541–9. doi: 10.1002/cam4.4561
19. Okamura R, Boichard A, Kato S, Sicklick JK, Bazhenova L, Kurzrock R. Analysis of NTRK alterations in pan-cancer adult and pediatric Malignancies: implications for NTRK-targeted therapeutics. JCO Precis Oncol (2018) 2018:PO.18.00183. doi: 10.1200/PO.18.00183
20. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D, et al. The 2021 WHO classification of tumors of the central nervous system: a summary. Neuro-Oncol (2021) 23:1231–51. doi: 10.1093/neuonc/noab106
21. Cordon-Cardo C, Tapley P, Jing SQ, Nanduri V, O’Rourke E, Lamballe F, et al. The trk tyrosine protein kinase mediates the mitogenic properties of nerve growth factor and neurotrophin-3. Cell (1991) 66:173–83. doi: 10.1016/0092-8674(91)90149-s
22. Klein R, Jing SQ, Nanduri V, O’Rourke E, Barbacid M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell (1991) 65:189–97. doi: 10.1016/0092-8674(91)90419-y
23. Ip NY, Stitt TN, Tapley P, Klein R, Glass DJ, Fandl J, et al. Similarities and differences in the way neurotrophins interact with the Trk receptors in neuronal and nonneuronal cells. Neuron (1993) 10:137–49. doi: 10.1016/0896-6273(93)90306-c
24. Lamballe F, Klein R, Barbacid M. trkC, a new member of the trk family of tyrosine protein kinases, is a receptor for neurotrophin-3. Cell (1991) 66:967–79. doi: 10.1016/0092-8674(91)90442-2
25. Squinto SP, Stitt TN, Aldrich TH, Davis S, Bianco SM, Radziejewski C, et al. trkB encodes a functional receptor for brain-derived neurotrophic factor and neurotrophin-3 but not nerve growth factor. Cell (1991) 65:885–93. doi: 10.1016/0092-8674(91)90395-f
26. Soppet D, Escandon E, Maragos J, Middlemas DS, Reid SW, Blair J, et al. The neurotrophic factors brain-derived neurotrophic factor and neurotrophin-3 are ligands for the trkB tyrosine kinase receptor. Cell (1991) 65:895–903. doi: 10.1016/0092-8674(91)90396-g
27. Amatu A, Sartore-Bianchi A, Bencardino K, Pizzutilo EG, Tosi F, Siena S. Tropomyosin receptor kinase (TRK) biology and the role of NTRK gene fusions in cancer. Ann Oncol Off J Eur Soc Med Oncol (2019) 30:viii5–viii15. doi: 10.1093/annonc/mdz383
28. Amatu A, Sartore-Bianchi A, Siena S. NTRK gene fusions as novel targets of cancer therapy across multiple tumour types. ESMO Open (2016) 1:e000023. doi: 10.1136/esmoopen-2015-000023
29. Guerreiro Stucklin AS, Ryall S, Fukuoka K, Zapotocky M, Lassaletta A, Li C, et al. Alterations in ALK/ROS1/NTRK/MET drive a group of infantile hemispheric gliomas. Nat Commun (2019) 10:4343. doi: 10.1038/s41467-019-12187-5
30. Clarke M, Mackay A, Ismer B, Pickles JC, Tatevossian RG, Newman S, et al. Infant high-grade gliomas comprise multiple subgroups characterized by novel targetable gene fusions and favorable outcomes. Cancer Discovery (2020) 10:942–63. doi: 10.1158/2159-8290.CD-19-1030
31. Solomon JP, Linkov I, Rosado A, Mullaney K, Rosen EY, Frosina D, et al. NTRK fusion detection across multiple assays and 33,997 cases: diagnostic implications and pitfalls. Mod Pathol (2020) 33:38–46. doi: 10.1038/s41379-019-0324-7
32. Nobusawa S, Hirato J, Yokoo H. Molecular genetics of ependymomas and pediatric diffuse gliomas: a short review. Brain Tumor Pathol (2014) 31:229–33. doi: 10.1007/s10014-014-0200-6
33. Chamdine O, Gajjar A. Molecular characteristics of pediatric high-grade gliomas. CNS Oncol (2014) 3:433–43. doi: 10.2217/cns.14.43
34. Albert CM, Davis JL, Federman N, Casanova M, Laetsch TW. TRK fusion cancers in children: A clinical review and recommendations for screening. J Clin Oncol Off J Am Soc Clin Oncol (2019) 37:513–24. doi: 10.1200/JCO.18.00573
35. López GY, Perry A, Harding B, Li M, Santi M. CDKN2A/B loss is associated with anaplastic transformation in a case of NTRK2 fusion-positive pilocytic astrocytoma. Neuropathol Appl Neurobiol (2019) 45:174–8. doi: 10.1111/nan.12503
36. Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, et alThe St. Jude Children’s Research Hospital–Washington University Pediatric Cancer Genome Project. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet (2014) 46:444–50. doi: 10.1038/ng.2938
37. Prabhakaran N, Guzman MA, Navalkele P, Chow-Maneval E, Batanian JR. Novel TLE4-NTRK2 fusion in a ganglioglioma identified by array-CGH and confirmed by NGS: Potential for a gene targeted therapy: A novel gene fusion in ganglioglioma. Neuropathology (2018) 38:380–6. doi: 10.1111/neup.12458
38. Qaddoumi I, Orisme W, Wen J, Santiago T, Gupta K, Dalton JD, et al. Genetic alterations in uncommon low-grade neuroepithelial tumors: BRAF, FGFR1, and MYB mutations occur at high frequency and align with morphology. Acta Neuropathol (Berl) (2016) 131:833–45. doi: 10.1007/s00401-016-1539-z
39. Zhang J, Wu G, Miller CP, Tatevossian RG, Dalton JD, Tang B, et alWhole-genome sequencing identifies genetic alterations in pediatric low-grade gliomas. Nat Genet (2023) 45(6):602–12. doi: 10.1038/ng.2611
40. Drilon A, Laetsch TW, Kummar S, DuBois SG, Lassen UN, Demetri GD, et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N Engl J Med (2018) 378:731–9. doi: 10.1056/NEJMoa1714448
41. Laetsch TW, DuBois SG, Mascarenhas L, Turpin B, Federman N, Albert CM, et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol (2018) 19:705–14. doi: 10.1016/S1470-2045(18)30119-0
42. Drilon AE, DuBois SG, Farago AF, Geoerger B, Grilley-Olson JE, Hong DS, et al. Activity of larotrectinib in TRK fusion cancer patients with brain metastases or primary central nervous system tumors. J Clin Oncol (2019) 37:2006–6. doi: 10.1200/JCO.2019.37.15_suppl.2006
43. Doz F, van Tilburg CM, Geoerger B, Højgaard M, Øra I, Boni V, et al. Efficacy and safety of larotrectinib in TRK fusion-positive primary central nervous system tumors. Neuro-Oncol (2022) 24:997–1007. doi: 10.1093/neuonc/noab274
44. Rosen EY, Schram AM, Young RJ, Schreyer MW, Hechtman JF, Shu CA, et al. Larotrectinib demonstrates CNS efficacy in TRK fusion-positive solid tumors. JCO Precis Oncol (2019) 3:PO.19.00009. doi: 10.1200/PO.19.00009
45. Di Ruscio V, Carai A, Del Baldo G, Vinci M, Cacchione A, Miele E, et al. Molecular landscape in infant high-grade gliomas: A single center experience. Diagn Basel Switz (2022) 12:372. doi: 10.3390/diagnostics12020372
46. Shah N, Lankerovich M, Lee H, Yoon J-G, Schroeder B, Foltz G. Exploration of the gene fusion landscape of glioblastoma using transcriptome sequencing and copy number data. BMC Genomics (2013) 14:818. doi: 10.1186/1471-2164-14-818
47. Vaishnavi A, Capelletti M, Le AT, Kako S, Butaney M, Ercan D, et al. Oncogenic and drug-sensitive NTRK1 rearrangements in lung cancer. Nat Med (2013) 19:1469–72. doi: 10.1038/nm.3352
48. Drilon A. TRK inhibitors in TRK fusion-positive cancers. Ann Oncol Off J Eur Soc Med Oncol (2019) 30(Suppl 8):viii23–30. doi: 10.1093/annonc/mdz282
49. Deland L, Keane S, Olsson Bontell T, Sjögren H, Fagman H, Øra I, et al. Discovery of a rare GKAP1-NTRK2 fusion in a pediatric low-grade glioma, leading to targeted treatment with TRK-inhibitor larotrectinib. Cancer Biol Ther (2021) 22:184–95. doi: 10.1080/15384047.2021.1899573
50. Hong DS, DuBois SG, Kummar S, Farago AF, Albert CM, Rohrberg KS, et al. Larotrectinib in patients with TRK fusion-positive solid tumours: a pooled analysis of three phase 1/2 clinical trials. Lancet Oncol (2020) 21:531–40. doi: 10.1016/S1470-2045(19)30856-3
51. Marcus L, Donoghue M, Aungst S, Myers CE, Helms WS, Shen G, et al. FDA approval summary: entrectinib for the treatment of NTRK gene fusion solid tumors. Clin Cancer Res (2021) 27:928–32. doi: 10.1158/1078-0432.CCR-20-2771
52. Robinson GW, Gajjar AJ, Gauvain KM, Basu EM, Macy ME, Maese LD, et al. Phase 1/1B trial to assess the activity of entrectinib in children and adolescents with recurrent or refractory solid tumors including central nervous system (CNS) tumors. J Clin Oncol (2019) 37:10009–9. doi: 10.1200/JCO.2019.37.15_suppl.10009
53. Robinson G, Desai A, Basu E, Foster J, Gauvain K, Sabnis A, et al. Hgg-01. Entrectinib in recurrent or refractory solid tumors including primary cns tumors: updated data in children and adolescents. Neuro-Oncol (2020) 22:iii344–4. doi: 10.1093/neuonc/noaa222.293
54. Desai AV, Robinson GW, Gauvain K, Basu EM, Macy ME, Maese L, et al. Entrectinib in children and young adults with solid or primary CNS tumors harboring NTRK , ROS1 , or ALK aberrations (STARTRK-NG). Neuro-Oncol (2022) 24:1776–89. doi: 10.1093/neuonc/noac087
55. Liu D, Flory J, Lin A, Offin M, Falcon CJ, Murciano-Goroff YR, et al. Characterization of on-target adverse events caused by TRK inhibitor therapy. Ann Oncol (2020) 31:1207–15. doi: 10.1016/j.annonc.2020.05.006
56. Cocco E, Schram AM, Kulick A, Misale S, Won HH, Yaeger R, et al. Resistance to TRK inhibition mediated by convergent MAPK pathway activation. Nat Med (2019) 25:1422–7. doi: 10.1038/s41591-019-0542-z
57. Drilon A, Nagasubramanian R, Blake JF, Ku N, Tuch BB, Ebata K, et al. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion-positive solid tumors. Cancer Discov (2017) 7:963–72. doi: 10.1158/2159-8290.CD-17-0507
58. Drilon A, Li G, Dogan S, Gounder M, Shen R, Arcila M, et al. What hides behind the MASC: clinical response and acquired resistance to entrectinib after ETV6-NTRK3 identification in a mammary analogue secretory carcinoma (MASC). Ann Oncol (2016) 27:920–6. doi: 10.1093/annonc/mdw042
59. Russo M, Misale S, Wei G, Siravegna G, Crisafulli G, Lazzari L, et al. Acquired resistance to the TRK inhibitor entrectinib in colorectal cancer. Cancer Discovery (2016) 6:36–44. doi: 10.1158/2159-8290.CD-15-0940
60. Keddy C, Neff T, Huan J, Nickerson JP, Beach CZ, Akkari Y, et al. Mechanisms of targeted therapy resistance in a pediatric glioma driven by ETV6-NTRK3 fusion. Mol Case Stud (2021) 7:a006109. doi: 10.1101/mcs.a006109
61. Drilon A, Nagasubramanian R, Blake JF, Ku N, Tuch BB, Ebata K, et al. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior TRK kinase inhibition in patients with TRK fusion–positive solid tumors. Cancer Discovery (2017) 7:963–72. doi: 10.1158/2159-8290.CD-17-0507
62. Cocco E, Lee JE, Kannan S, Schram AM, Won HH, Shifman S, et al. TRK xDFG mutations trigger a sensitivity switch from type I to II kinase inhibitors. Cancer Discovery (2021) 11:126–41. doi: 10.1158/2159-8290.CD-20-0571
63. Wei G, Ardini E, Patel R, Cam N, Harris J, Vernier J-M, et al. Abstract 2136: Entrectinib is effective against the gatekeeper and other emerging resistance mutations in NTRK-, ROS1- and ALK- rearranged cancers. Cancer Res (2016) 76:2136–6. doi: 10.1158/1538-7445.AM2016-2136
64. Somwar R, Hofmann NE, Smith B, Odintsov I, Vojnic M, Linkov I, et al. NTRK kinase domain mutations in cancer variably impact sensitivity to type I and type II inhibitors. Commun Biol (2020) 3:776. doi: 10.1038/s42003-020-01508-w
65. Katayama R, Gong B, Togashi N, Miyamoto M, Kiga M, Iwasaki S, et al. The new-generation selective ROS1/NTRK inhibitor DS-6051b overcomes crizotinib resistant ROS1-G2032R mutation in preclinical models. Nat Commun (2019) 10:3604. doi: 10.1038/s41467-019-11496-z
66. Hyman D, Kummar S, Farago A, Geoerger B, Mau-Sorensen M, Taylor M, et al. Abstract CT127: Phase I and expanded access experience of LOXO-195 (BAY 2731954), a selective next-generation TRK inhibitor (TRKi). Cancer Res (2019) 79:CT127–7. doi: 10.1158/1538-7445.AM2019-CT127
Keywords: pediatric tumors, central nervous system tumors, tropomyosin receptor kinases, NTRK, gene fusions, point mutations, acquired resistance, targeted therapy
Citation: Cipri S, Fabozzi F, Del Baldo G, Milano GM, Boccuto L, Carai A and Mastronuzzi A (2023) Targeted therapy for pediatric central nervous system tumors harboring mutagenic tropomyosin receptor kinases. Front. Oncol. 13:1235794. doi: 10.3389/fonc.2023.1235794
Received: 06 June 2023; Accepted: 17 November 2023;
Published: 07 December 2023.
Edited by:
Iacopo Sardi, Meyer Children’s Hospital, ItalyReviewed by:
Joanna Stefanowicz, Medical University of Gdansk, PolandCopyright © 2023 Cipri, Fabozzi, Del Baldo, Milano, Boccuto, Carai and Mastronuzzi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Angela Mastronuzzi, angela.mastronuzzi@opbg.net