 Sarah M. Zimmerman
Sarah M. Zimmerman Phyo Nay Lin
Phyo Nay Lin George P. Souroullas
George P. Souroullas- 1Department of Medicine, Washington University School of Medicine in St. Louis, St. Louis, MO, United States
- 2Division of Oncology, Molecular Oncology Section, Washington University School of Medicine in St. Louis, St. Louis, MO, United States
- 3Siteman Comprehensive Cancer Center, Washington University School of Medicine in St. Louis, St. Louis, MO, United States
Mutations in chromatin modifying genes frequently occur in many kinds of cancer. Most mechanistic studies focus on their canonical functions, while therapeutic approaches target their enzymatic activity. Recent studies, however, demonstrate that non-canonical functions of chromatin modifiers may be equally important and therapeutically actionable in different types of cancer. One epigenetic regulator that demonstrates such a dual role in cancer is the histone methyltransferase EZH2. EZH2 is a core component of the polycomb repressive complex 2 (PRC2), which plays a crucial role in cell identity, differentiation, proliferation, stemness and plasticity. While much of the regulatory functions and oncogenic activity of EZH2 have been attributed to its canonical, enzymatic activity of methylating lysine 27 on histone 3 (H3K27me3), a repressive chromatin mark, recent studies suggest that non-canonical functions that are independent of H3K27me3 also contribute towards the oncogenic activity of EZH2. Contrary to PRC2’s canonical repressive activity, mediated by H3K27me3, outside of the complex EZH2 can directly interact with transcription factors and oncogenes to activate gene expression. A more focused investigation into these non-canonical interactions of EZH2 and other epigenetic/chromatin regulators may uncover new and more effective therapeutic strategies. Here, we summarize major findings on the non-canonical functions of EZH2 and how they are related to different aspects of carcinogenesis.
Introduction
Polycomb group proteins were originally described for their role in Drosophila development where they repress homeotic genes (Hox) required for proper body segmentation (1–3). The two main Polycomb group complexes are the Polycomb repressive complex 1 (PRC1) and Polycomb repressive complex 2 (PRC2), both of which are conserved from Drosophila to mammals. While PRC1 consists of highly variable subunits, PRC2 is composed of four core components: EZH1 or EZH2, EED, SUZ12 and RbAp46/48. Additionally, several secondary accessory proteins such as JARID2, PCL and AEBP2 modulate the function of PRC2 (4) (Figure 1A). Polycomb-mediated gene silencing primarily operates through the regulation of chromatin structure via post-translational modifications of histone tails (5, 6). Due to their involvement in critical biological processes like differentiation, cell identity, proliferation, stemness, and plasticity, alterations in Polycomb complexes are frequently associated with human cancers (7, 8) and developmental syndromes characterized by congenital overgrowth, dysmorphic facial features, and learning disabilities (9–13).
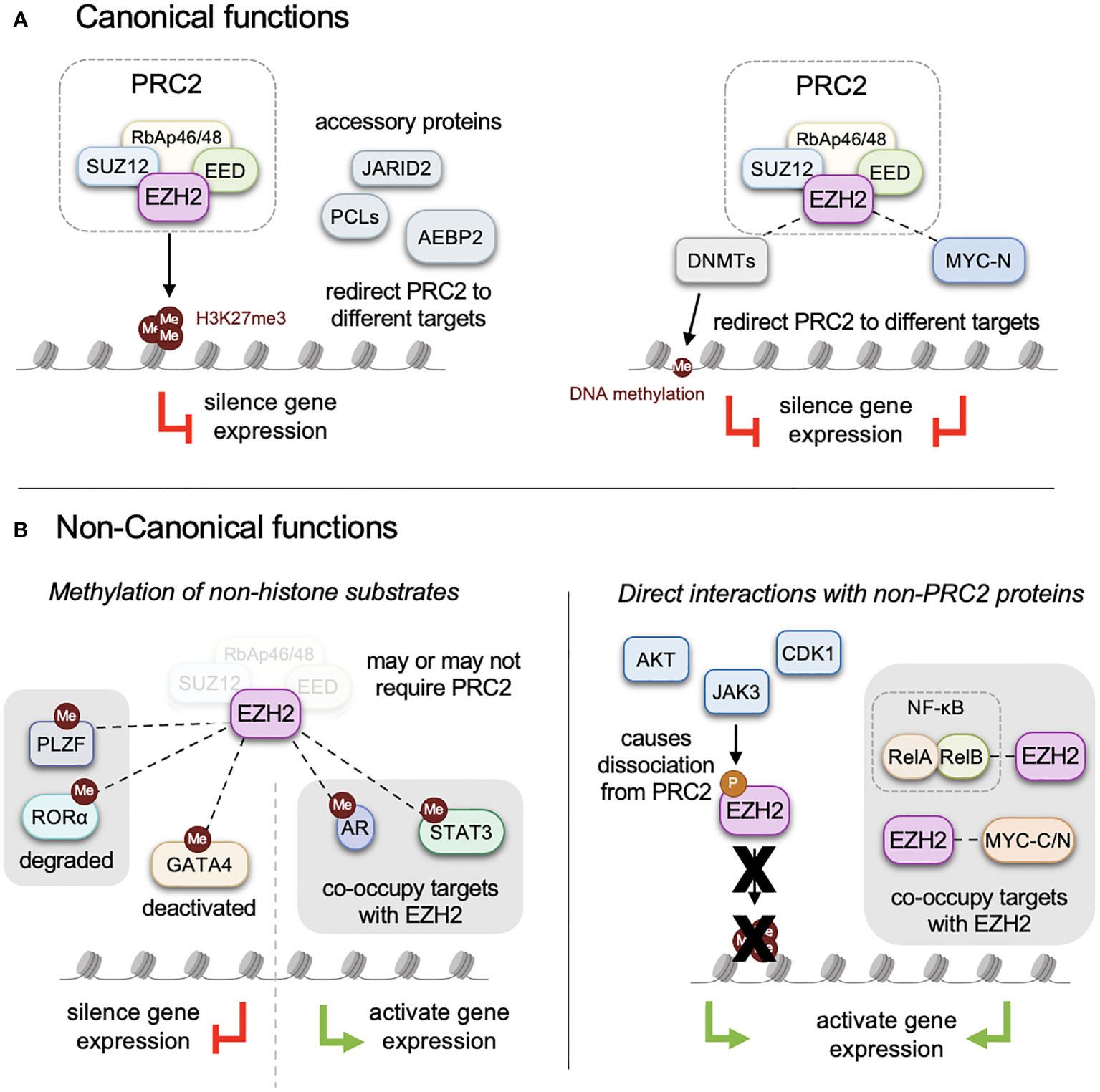
Figure 1 Canonical and non-canonical functions of EZH2. (A) EZH2 functions as the methyltransferase domain of the PRC2 complex in cooperation with core components, EED, SUZ12 and RbAp46/48, to mediate methylation of lysine 27 on histone 3 (H3K27me3). Several accessory proteins that enhance and direct PRC2 function include JARID2, PCLs, AEBP2 among others. (B) In addition to methylating histone tails EZH2 can also methylate non-histone proteins, altering their function and activity with complicated effects on downstream gene expression. These include PLZF, RORα, GATA4, AR and STAT3. Post-translational modifications of EZH2 by AKT, JAK3, CDK1, and protein interactions with non-PRC2 proteins promote non-canonical functions, which can inhibit canonical PRC2 assembly and histone methylation. PRC2-independent functions of EZH2 include interactions with transcription factors such as RelA/B, CMYC, NMYC, STAT3, where they together function as transcriptional activators.
In the context of cancer, the role of PRC2, and EZH2 specifically, is multifaceted as it can function both as a tumor-suppressor (14–18) and as an oncogene (17, 19–23) in a cell type dependent manner. This dual functionality is reflected in the observation of EZH2 amplifications and deletions in different kinds of cancer, which have been validated through functional assays and in vivo models. In addition to loss- and gain-of-function genetic events, missense mutations at tyrosine 641 (EZH2Y641) function in a neomorphic manner, cause redistribution of H3K27me3 and are strongly oncogenic (19).
While much of the oncogenic activity of PRC2, and particularly EZH2, has been attributed to its enzymatic activity of methylating lysine 27 on histone 3, recent studies revealed that this canonical enzymatic activity cannot fully explain all of its physiological functions and oncogenic effects. Considering the limited efficacy of epigenetic inhibitors in cancer patients, which primarily target enzymatic activity, a more focused investigation into EZH2’s non-canonical functions may identify more effective therapeutic strategies. In this review, we highlight and discuss the major non-canonical functions of EZH2, its interactions with non-PRC2 proteins, along with implications on therapeutic strategies.
Post-translational modifications of EZH2 may determine non-canonical functions
Several post-translational modifications of EZH2 have been identified over the years which may be responsible for switching between canonical and non-canonical EZH2 functions (Figure 1B). A study by Cha et al. was the first to find that the AKT kinase phosphorylates EZH2 at serine 21 (S21), resulting in decreased methyltransferase activity (24). Later studies found that phosphorylation at S21 not only suppressed canonical enzymatic activity, but was required for non-canonical, H3K27me3-independent functions of EZH2 in prostate cancer (25) and glioblastoma models (26). Other kinases known to modify EZH2 include JAK3 and the cyclin dependent kinases 1 and 2 (CDK1/2). JAK3 phosphorylates EZH2 at tyrosine 244, promoting the dissociation of the PRC2 complex (27), while CDKs have a more complicated effect; CDK1/2 phosphorylate EZH2 at threonine 350, enhancing recruitment of PRC2 and associations with non-coding RNAs (28), while CDK1-mediated phosphorylation of EZH2 at threonine 487 disrupts binding with PRC2 core components EED and SUZ12, inhibiting H3K27me3 (29). Different modifications, therefore, could have very different effects on the ability of EZH2 to form protein complexes with either canonical PRC2 components, or other effectors, which may be dependent on the right conformational structure. While many of these modifications are known and contribute or promote non-canonical EZH2 functions as discussed further below, many likely remain unexplored.
Direct interaction between Ezh2 and DNA methyltransferases in solid tumors
One of the earliest interaction of EZH2 outside of core PRC2 components and accessory proteins was with the DNA methyltransferases, DNMT1, 3A and 3B (30). Vire et al. found that EZH2 directly interacts and recruits DNMTs to specific target loci (Figure 1A). Repression of these genes required the presence of both H3K27me3 and DNA methylation in the promoter region. Cooperation between these two epigenetic complexes also had significant therapeutic implications with combination strategies targeting both DNMT and EZH2 activity. For example, Ning et al. showed that in gastric cancer and glioblastoma cell lines EZH2 and DNMT1 together repressed expression of tumor suppressors and that treatment with either the DNMT inhibitor 5-azadeoxycitidine and the methyltransferase inhibitor DZNep reduced tumor growth (31). These early studies, however, were limited by the fact that DZNep is not a specific EZH2 inhibitor and enhanced efficacy could be due to inhibition of other methyltransferases. Later studies with more specific EZH2 inhibitors showed that in hepatocellular carcinoma the combination of 5-azadeoxycitidine with the EZH2 inhibitor GSK126 increased expression of neoantigens, increased tumor-infiltrating lymphocytes and reduced cell growth (32, 33). In multiple myeloma, Dimopoulos et al. investigated resistance to lenalidomide and pomalidomide and found that resistance was associated with increased DNA methylation and decreased chromatin accessibility (34). Combination treatment with 5-azacytidine and the EZH2 inhibitor, EPZ-6438, reversed these epigenetic changes and re-sensitized the cells to lenalidomide and pomalidomide. Dual inhibition of DNMTs and EZH2 was also effective in prostate, breast, colon cancer and leukemia cells (35, 36), highlighting the therapeutic potential of these interactions.
Ezh2-mediated methylation of Gata4 and its role in rhabdomyosarcoma
In addition to its interactions with other epigenetic regulators, Ezh2 can directly interact with transcription factors, such as Gata4 (Figure 1B). Gata4 is a key transcription factor during heart development that recruits the acetyltransferase p300 to specific chromatin loci, activating the expression of genes critical to heart development (37). While p300 acetylates Gata4 enhancing its transcriptional activity (38), Ezh2 antagonizes this interaction by directly methylating Gata4 on lysine 299 (39). This prevents its acetylation and activation by p300, reducing its recruitment to chromatin and impeding the activation of the heart development program. This was the first report of a non-histone interaction of EZH2 and an example of a histone methyltransferase and a histone acetyltransferase that function antagonistically in a histone-independent manner. A direct interaction between EZH2 and GATA4 was also observed in rhabdomyosarcoma, a type of soft tissue sarcoma that develops from muscle or fibrous tissue, and a rare type of sarcomas of the heart. Song et al. demonstrated that GATA4 promotes normal myogenesis by binding to GRIP1 and stimulating the expression of miR-29a, which drives myogenic differentiation (40). However, in sarcoma cells, a direct interaction between GATA4 and EZH2 leads to the deposition of H3K27me3 at the miR-29a promoter suppressing its expression, inhibiting myogenic differentiation while promoting proliferation. Overall, these results suggest that interactions of GATA4 with PRC2 components are important in rhabdomyosarcoma progression.
Transcriptional activation mediated by EZH2 and the androgen receptor in prostate cancer
While many EZH2 interactions require the presence of the rest of the PRC2 complex as described above, EZH2 can engage in protein-protein interactions independently of other PRC2 components or activate downstream signaling independent of its methyltransferase activity. An early example where EZH2 functions as an activator is in androgen-dependent prostate cancer (23). Varambaly et al. initially found that EZH2 expression was highly correlative with progression of castration-resistant prostate cancer (23). Subsequent studies found that silencing of EZH2 had a profound effect on androgen-independent cell growth in vitro, with a significant number of genes directly upregulated by EZH2 (25). ChIP-seq experiments for EZH2 and H3K27me3 identified both EZH2-specific peaks, termed “solo”, as well as peaks overlapping with H3K27me3, termed “ensemble”. Additional mechanistic studies found that EZH2 directly methylated the Androgen receptor (AR), and together with AR, regulated expression of activated genes. Xu et al. further suggested that the EZH2-AR interaction was dependent on phosphorylation of EZH2 at serine 21. Interestingly, this interaction still required the intact methyltransferase activity of EZH2, although the mechanistic details remain unclear. In contrast to Xu et al., Kim et al. proposed that EZH2 directly binds to the promoter of the AR, controlling its expression independent of its methyltransferase activity (41). Differences in methodology and models could account for these disparate results and both mechanisms could be true.
Non-canonical functions of EZH2 in breast cancer involve transcriptional activation and protein degradation mechanisms
In breast cancer, EZH2 plays a significant role via both canonical and non-canonical functions, which are summarized by Anwar et al. (42). Interestingly, the non-canonical function of EZH2 in breast cancer is not the same across different subtypes. In triple-negative breast cancer, EZH2 acts as a transcriptional activator of the NF-KB subunit RelB to drive self-renewal and promote tumor initiation (43, 44). In Estrogen Receptor (ER)-negative breast cancer, EZH2 physically interacts with RelA/RelB and promotes the transcription of NF-KB targets such as IL6 and TNF, independent of its histone methyltransferase activity. However, in ER-positive breast cancer EZH2 assumes its canonical role, interacting with the ER in a PRC2-dependent manner mediating H3K27me3 at NF-KB target genes and repressing their expression (44).
EZH2 also non-canonically regulates gene expression by targeting proteins for degradation via E3 ubiquitin ligases. Lee et al. discovered that in breast cancer cells EZH2 recognizes and monomethylates the lysine of an Arginine(R)-Serine(S)-Lysine(K) histone-like sequence on RORα, leading to its subsequent ubiquitination and degradation by DCAF1 (45). These data may be biologically significant in cancer as well, as expression of RORα and EZH2 is inversely correlated in breast cancer tissues (45).
Ezh2 association with protein degradation machinery in nature killer T cell lymphoma
Interactions of EZH2 with protein degradation machinery are not unique to breast cancer. While studying the role of EZH2 during T cell development, Vasanthakumar et al. found diametrically different results when deleting different core components of the PRC2 complex; depletion of Ezh2 resulted in expansion of NKT cells, while depletion of Eed and Suz12 resulted in a dramatic loss of NKT cells (46). They subsequently found that PLZF, a master regulator of hematopoiesis and NKT cell expansion (47) was directly methylated by EZH2 at lysine 430, which lead to its ubiquitination and degradation, providing an explanation for the observed phenotypes. EZH2 and PLZF also interact directly on chromatin in myeloblastic cells, co-occupying loci that are not associated with SUZ12, EED, or H3K27me3, but are instead associated with H3K4me3 and active transcription (48), highlighting the multilevel complexity of EZH2’s functions.
Cell-intrinsic and cell-extrinsic effects of an EZH2-STAT3 interaction in multiple solid tumors
A direct interaction between EZH2 and the immune-regulator, STAT3, has been observed in multiple solid tumors. In melanoma, EZH2 is either amplified, overexpressed or mutated at Tyrosine 641 (Y641), a hotspot mutation within the methyltransferase domain which creates a hyperactive and neomorphic protein (19). To understand the mechanisms of the oncogenic activity of this mutant, we recently investigated potential non-canonical interactions in melanoma and found that in mouse melanoma cells, mutant Ezh2Y641F protein interacts directly with and methylates Stat3 protein (49). We found that the interaction between Ezh2 and Stat3 was highly enriched in the presence of mutant Ezh2Y641F protein, which exhibits increased methyltransferase activity, suggesting that this interaction is dependent on Ezh2’s activity. Using chromatin-immunoprecipitation followed by sequencing (ChIP-seq) and expression profile analysis, we found that Ezh2Y641F and Stat3 cooperate as transcriptional activators, binding to new regions on chromatin to increase expression of several target genes. These include MHC Class 1b antigen processing genes and autophagy regulators. Functionally, the Ezh2Y641F-Stat3 interaction mediated the recruitment of cytotoxic CD8+ T cells to the melanoma tumor microenvironment, which slowed down in vivo tumor growth (49). This was the first report of an enhanced non-canonical interaction between the Ezh2Y641F mutant and a non-histone protein, suggesting that perhaps this is an important aspect of the oncogenic activity of EZH2 in cancers with Y641 mutations (7, 8).
The interaction between EZH2 and STAT3 has also been reported in glioblastoma and colon carcinomas. In glioblastoma, EZH2 binds to and methylates STAT3 at lysine 180, resulting in enhanced STAT3 activity (26), while in colon carcinoma Dasgupta et al. found that dimethylation of STAT3 at lysine 49 was crucial for activating the IL6-STAT3 transcriptional program (50). EZH2 can also methylate STAT3 in breast cancer cells, which was necessary for tumor growth (51). Overall, the function and downstream mechanisms of the EZH2-STAT3 interaction appears to be determined by cellular context and may depend on availability of other co-factors or post-translational modifications.
Multifaceted interactions between EZH2 and MYC in both solid and blood cancers
MYC is a well-studied oncogene that is implicated in many types of cancers. The relationship between MYC and EZH2 was established more than a decade ago with studies demonstrating their co-regulation. For example, EZH2 directly regulates CMYC expression via association with the estrogen receptor alpha (ERα) and β-catenin (52), and conversely, MYC binds the promoter of EZH2 and regulates its expression in prostate cancer cells (53) and B cell lymphomas (19). The latter raised the intriguing possibility that EZH2 may be mediating some of the oncogenic activity of MYC and account for the repressed genes in cancers with MYC overexpression. This was demonstrated by Kaur and Cole in immortalized mammary epithelial cells. Specifically, they showed that MYC transcriptionally activates PTEN, leading to suppression of AKT kinase activity, which in turn results in increased EZH2 protein stability and enzymatic activity. They further showed with rescue assays and EZH2 mutants that EZH2 was responsible for the transcriptional repression of nearly half of all the MYC-repressed genes, consistent with EZH2 binding and accumulation of H3K27me3 at MYC-repressed gene loci (54).
While the above studies suggested EZH2/PRC2 may be mediating some of the repressive properties of MYC through indirect signaling, they did not exclude the possibility of a more direct interaction. More recent studies proposed that possibility, but it remained unclear how EZH2 and MYC interacted at the chromatin interface. Two recent studies shed more light into these questions by investigating the interaction between EZH2 and CMYC in acute leukemia and multiple myeloma models (55, 56). Specifically, Wang J et al. discovered that EZH2 not only binds to regions characterized by repressive H3K27me3 marks, as is typical of canonical EZH2 activity, but also to regions with no H3K27me3 (termed “EZH2-solo” sites). In fact, significant EZH2 binding overlaps with multiple gene-active chromatin marks, such as H3K4me3, H3K9ac and H3K27ac. Interestingly, they found that these EZH2-solo sites were also bound by CMYC. They further demonstrated that EZH2 and CMYC directly interact through EZH2’s transactivation domain (TAD) and that this interaction is essential for malignant growth of leukemia cells. This result was consistent with a previous report that identified a hidden TAD within EZH2 that can be unlocked by cancer-specific EZH2-phosphorylation, resulting in structural conformation changes that mediate interactions with active transcription machinery (57).
In addition to interacting with CMYC, EZH2 also interacts with NMYC, both in a canonical and non-canonical way. In neuroblastoma cells, Corvetta et al. demonstrated that NMYC directly interacts with EZH2 and recruits the PRC2 complex to the promoter region of the tumor suppressor, clusterin (CLU). This resulted in increased deposition of H3K27me3, suppressing expression of CLU, and causing significant phenotypic effects such as increased invasion and cell cycle progression (58). In prostate cancer cells, Dardenne et al. showed that NMYC forms a complex with EZH2 and the Androgen Receptor (AR), which enhances PRC2 target gene repression, regulating gene expression programs critical for prostate cancer progression (59). Contrary to these canonical functions of EZH2 in cooperation with NMYC, Vanden Bempt et al. recently found that EZH2 exhibits non-canonical functions in coordination with NMYC in peripheral T-cell lymphoma (PTCL). Specifically, using a mouse model of NMYC-driven PTCL, they found that EZH2 acts as a cofactor of NMYC, activating expression of EZH2-NMYC bound sites without association with other canonical PRC2 components or activity (60).
A noteworthy observation from these studies was that the interaction between EZH2 and MYC was unique to transformed leukemia cell lines and primary leukemia patient samples, and was not detected in healthy, non-leukemic cells. This suggests that the EZH2-MYC interactions may depend on the presence of specific co-factors or post-translational modifications that facilitate that interaction and are not present in healthy cells or in all types of cancers.
Therapeutic implications of EZH2 non-canonical functions
To investigate the importance of EZH2’s canonical and non-canonical activities in cancer and therapeutics, several studies have explored the concept of synthetic lethality between EZH2 inhibition and mutations in other pathways. A notable example is the relationship between PRC2/EZH2 and the SWI/SNF complex in cancer. Several cancers with mutations in SWI/SNF subunits exhibit increased sensitivity to PRC2 inhibition (61, 62). However, Kim et al. demonstrated that SWI/SNF-mutant lung and ovarian cancer cells are primarily dependent on the non-catalytic functions of EZH2 and only partially dependent on its methyltransferase activity (61). This strongly suggests that non-enzymatic functions of EZH2 are not only relevant in cancer progression, but also to therapeutic approaches.
The non-canonical functions of EZH2 may also explain the relatively weak responses of EZH2 inhibitors in some clinical trials and must be taken into serious consideration when designing therapeutic strategies. Several recent studies have utilized proteolysis targeting chimeras (PROTACs) technology to hijack the ubiquitin-proteasome pathway to degrade targeted proteins for loss-of-function studies. Degradation of EZH2 by the PROTAC-degrader MS-177 effectively inhibited on-target EZH2-PRC2 and MYC-related oncogenic nodes and inhibited leukemia growth in vivo more effectively than enzymatic inhibition (55, 56). In a separate study, using the EZH2 PROTAC-degrader MS1973, EZH2 degradation was necessary and more effective than enzymatic inhibition in eliminating relapse-initiating cells in a retinoic acid-dependent model of acute promyelocytic leukemia (63). These studies highlight the potential clinical significance of targeting non-canonical functions of EZH2, and may even be more broadly applicable to other chromatin-modifying genes.
Discussion
The non-canonical interactions of EZH2 encompass a wide range of functions. Recent studies provided significant insights into the intricate roles of epigenetic regulators and their interactions with oncogenes such as MYC. It is important to acknowledge that while these interactions are intriguing, many of them have yet to be validated in vivo. Considering that the proper biological context greatly influences protein functionality, it is possible that these interactions may be different in vivo. Nonetheless, these studies underscore the notion that proteins often have additional roles beyond their established functions as epigenetic regulators or chromatin modifiers. These additional roles can arise from association with other protein complexes aberrantly expressed in cancer cells, or by post-translational modifications, which can cause structural conformational changes, facilitating novel interactions not typically present in healthy cells. Considering these discoveries, targeting the degradation of EZH2, or its post-translational modifiers could be a promising therapeutic approach for cancers that rely on these non-canonical interactions. However, the specificities of these interactions to cancer cells, their occurrence in different contexts, and the involvement of other epigenetic regulators in similar functionalities are still not fully understood. Ongoing and future research will undoubtedly uncover more of these interactions, providing a deeper understanding of disease mechanisms and guiding the development of effective therapeutic strategies.
Author contributions
All authors researched the literature and wrote the manuscript with GPS’s supervision. All authors contributed to the article and approved the submitted version.
Funding
GPS was supported by NCI K22CA229612, Leukemia Research Foundation, Cancer Research Foundation and Harry J Lloyd Young Investigator Awards. SMZ was supported by an NCI training grant 5T32CA113275.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Lewis EB. A gene complex controlling segmentation in Drosophila. Nature (1978) 276:565–70. doi: 10.1038/276565a0
2. Struhl G. A gene product required for correct initiation of segmental determination in Drosophila. Nature (1981) 293:36–41. doi: 10.1038/293036a0
3. Crick FHC, Lawrence PA. Compartments and polyclones in insect development. Science (1975) 189(4200):340–7. doi: 10.1126/science.806966
4. Aranda S, Mas G, Di Croce L. Regulation of gene transcription by Polycomb proteins. Sci Adv (2015) 1:e1500737. doi: 10.1126/sciadv.1500737
5. Laugesen A, Højfeldt JW, Helin K. Molecular mechanisms directing PRC2 recruitment and H3K27 methylation. Mol Cell (2019) 74:8–18. doi: 10.1016/j.molcel.2019.03.011
6. Laugesen A, Højfeldt JW, Helin K. Role of the polycomb repressive complex 2 (PRC2) in transcriptional regulation and cancer. Cold Spring Harb Perspect Med (2016) 6:a026575. doi: 10.1101/cshperspect.a026575
7. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data: Figure 1. Cancer Discov (2012) 2:401–4. doi: 10.1158/2159-8290.CD-12-0095
8. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal (2013) 6:pl1–1. doi: 10.1126/scisignal.2004088
9. Tatton-Brown K, Murray A, Hanks S, Douglas J, Armstrong R, Banka S, et al. Weaver syndrome and EZH2mutations: Clarifying the clinical phenotype. Am J Med Genet A (2013) 161:2972–80. doi: 10.1002/ajmg.a.36229
10. Tatton-Brown K, Rahman N. The NSD1 and EZH2 overgrowth genes, similarities and differences. Am J Med Genet C Semin Med Genet (2013) 163C:86–91. doi: 10.1002/ajmg.c.31359
11. Cohen AS, Gibson WT. EED-associated overgrowth in a second male patient. J Hum Genet (2016) 61:831–4. doi: 10.1038/jhg.2016.51
12. Cooney E, Bi W, Schlesinger AE, Vinson S, Potocki L. Novel EED mutation in patient with Weaver syndrome. Am J Med Genet A (2017) 173:541–5. doi: 10.1002/ajmg.a.38055
13. Cohen ASA, Yap DB, Lewis MES, Chijiwa C, Ramos-Arroyo MA, Tkachenko N, et al. Weaver syndrome-associated EZH2 protein variants show impaired histone methyltransferase function in vitro. Hum Mutat (2016) 37:301–7. doi: 10.1002/humu.22946
14. Ntziachristos P, Tsirigos A, Vlierberghe PV, Nedjic J, Trimarchi T, Flaherty MS, et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med (2012) 18:298–302. doi: 10.1038/nm.2651
15. Mochizuki-Kashio M, Aoyama K, Sashida G, Oshima M, Tomioka T, Muto T, et al. Ezh2 loss in hematopoietic stem cells predisposes mice to develop heterogeneous Malignancies in an Ezh1-dependent manner. Blood (2015) 126:1172–83. doi: 10.1182/blood-2015-03-634428
16. Muto T, Sashida G, Oshima M, Wendt GR, Mochizuki-Kashio M, Nagata Y, et al. Concurrent loss of Ezh2and Tet2cooperates in the pathogenesis of myelodysplastic disorders. J Exp Med (2013) 210:2627–39. doi: 10.1084/jem.20131144
17. Wang Y, Hou N, Cheng X, Zhang J, Tan X, Zhang C, et al. Ezh2 acts as a tumor suppressor in kras-driven lung adenocarcinoma. Int J Biol Sci (2017) 13:652. doi: 10.7150/ijbs.19108
18. Clair JM-S, Soydaner-Azeloglu R, Lee KE, Taylor L, Livanos A, Pylayeva-Gupta Y, et al. EZH2 couples pancreatic regeneration to neoplastic progression. Genes Dev (2012) 26:439–44. doi: 10.1101/gad.181800.111
19. Souroullas GP, Jeck WR, Parker JS, Simon JM, Liu J-Y, Paulk J, et al. An oncogenic Ezh2 mutation induces tumors through global redistribution of histone 3 lysine 27 trimethylation. Nat Med (2016) 22:632–40. doi: 10.1038/nm.4092
20. Zingg D, Debbache J, Schaefer SM, Tuncer E, Frommel SC, Cheng P, et al. The epigenetic modifier EZH2 controls melanoma growth and metastasis through silencing of distinct tumour suppressors. Nat Commun (2015) 6:6051. doi: 10.1038/ncomms7051
21. Zhang H, Qi J, Reyes JM, Li L, Rao PK, Li F, et al. Oncogenic deregulation of EZH2 as an opportunity for targeted therapy in lung cancer. Cancer Discov (2016) 6:1006–21. doi: 10.1158/2159-8290.CD-16-0164
22. Béguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell (2013) 23:677–92. doi: 10.1016/j.ccr.2013.04.011
23. Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature (2002) 419:624–9. doi: 10.1038/nature01075
24. Cha T-L, Zhou BP, Xia W, Wu Y, Yang C-C, Chen C-T, et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science (2005) 310:306–10. doi: 10.1126/science.1118947
25. Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is polycomb-independent. Science (2012) 338:1465–9. doi: 10.1126/science.1227604
26. Kim E, Kim M, Woo D-H, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell (2013) 23:839–52. doi: 10.1016/j.ccr.2013.04.008
27. Yan J, Li B, Lin B, Lee PT, Chung T-H, Tan J, et al. EZH2 phosphorylation by JAK3 mediates a switch to noncanonical function in natural killer/T-cell lymphoma. Blood J Am Soc Hematol (2016) 128:948–58. doi: 10.1182/blood-2016-01-690701
28. Zeng X, Chen S, Huang H. Phosphorylation of EZH2 by CDK1 and CDK2. Cell Cycle (2011) 10:579–83. doi: 10.4161/cc.10.4.14722
29. Wei Y, Chen Y-H, Li L-Y, Lang J, Yeh S-P, Shi B, et al. CDK1-dependent phosphorylation of EZH2 suppresses methylation of H3K27 and promotes osteogenic differentiation of human mesenchymal stem cells. Nat Cell Biol (2011) 13:87–94. doi: 10.1038/ncb2139
30. Viré E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature (2005) 439:871–4. doi: 10.1038/nature04431
31. Ning X, Shi Z, Liu X, Zhang A, Han L, Jiang K, et al. DNMT1 and EZH2 mediated methylation silences the microRNA-200b/a/429 gene and promotes tumor progression. Cancer Lett (2015) 359:198–205. doi: 10.1016/j.canlet.2015.01.005
32. Zhang L, Li H-T, Shereda R, Lu Q, Weisenberger DJ, O’Connell C, et al. DNMT and EZH2 inhibitors synergize to activate therapeutic targets in hepatocellular carcinoma. Cancer Lett (2022) 548:215899. doi: 10.1016/j.canlet.2022.215899
33. Hong YK, Li Y, Pandit H, Li S, Pulliam Z, Zheng Q, et al. Epigenetic modulation enhances immunotherapy for hepatocellular carcinoma. Cell Immunol (2019) 336:66–74. doi: 10.1016/j.cellimm.2018.12.010
34. Dimopoulos K, Søgaard Helbo A, Fibiger Munch-Petersen H, Sjö L, Christensen J, Sommer Kristensen L, et al. Dual inhibition of DNMTs and EZH2 can overcome both intrinsic and acquired resistance of myeloma cells to IMiDs in a cereblon-independent manner. Mol Oncol (2018) 12:180–95. doi: 10.1002/1878-0261.12157
35. Takeshima H, Wakabayashi M, Hattori N, Yamashita S, Ushijima T. Identification of coexistence of DNA methylation and H3K27me3 specifically in cancer cells as a promising target for epigenetic therapy. Carcinogenesis (2015) 36:192–201. doi: 10.1093/carcin/bgu238
36. Yang Y, Dai Y, Yang X, Wu S, Wang Y. DNMT3A mutation-induced CDK1 overexpression promotes leukemogenesis by modulating the interaction between EZH2 and DNMT3A. Biomolecules (2021) 11:781. doi: 10.3390/biom11060781
37. Dai Y-S, Markham BE. p300 functions as a coactivator of transcription factor GATA-4 *. J Biol Chem (2001) 276:37178–85. doi: 10.1074/jbc.M103731200
38. Takaya T, Kawamura T, Morimoto T, Ono K, Kita T, Shimatsu A, et al. Identification of p300-targeted Acetylated Residues in GATA4 during Hypertrophic Responses in Cardiac Myocytes*♦. J Biol Chem (2008) 283:9828–35. doi: 10.1074/jbc.M707391200
39. He A, Shen X, Ma Q, Cao J, von Gise A, Zhou P, et al. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev (2012) 26:37–42. doi: 10.1101/gad.173930.111
40. Song Y-L, Yang M-H, Zhang S, Wang H, Kai K-L, Yao C-X, et al. A GRIP-1–EZH2 switch binding to GATA-4 is linked to the genesis of rhabdomyosarcoma through miR-29a. Oncogene (2022) 41:5223–37. doi: 10.1038/s41388-022-02521-5
41. Kim J, Lee Y, Lu X, Song B, Fong K-W, Cao Q, et al. Polycomb- and methylation-independent roles of EZH2 as a transcription activator. Cell Rep (2018) 25:2808–2820.e4. doi: 10.1016/j.celrep.2018.11.035
42. Anwar T, Gonzalez ME, Kleer CG. Noncanonical functions of the polycomb group protein EZH2 in breast cancer. Am J Pathol (2021) 191:774–83. doi: 10.1016/j.ajpath.2021.01.013
43. Lawrence CL, Baldwin AS. Non-canonical EZH2 transcriptionally activates RelB in triple negative breast cancer. PloS One (2016) 11:e0165005. doi: 10.1371/journal.pone.0165005
44. Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RM, et al. Context-specific regulation of NF-κB target gene expression by EZH2 in breast cancers. Mol Cell (2011) 43:798–810. doi: 10.1016/j.molcel.2011.08.011
45. Lee JM, Lee JS, Kim H, Kim K, Park H, Kim J-Y, et al. EZH2 generates a methyl degron that is recognized by the DCAF1/DDB1/CUL4 E3 ubiquitin ligase complex. Mol Cell (2012) 48:572–86. doi: 10.1016/j.molcel.2012.09.004
46. Vasanthakumar A, Xu D, Lun AT, Kueh AJ, Gisbergen KP, Iannarella N, et al. A non-canonical function of Ezh2 preserves immune homeostasis. EMBO Rep (2017) 18:619–31. doi: 10.15252/embr.201643237
47. Dick JE, Doulatov S. The role of PLZF in human myeloid development. Ann N Y Acad Sci (2009) 1176:150–3. doi: 10.1111/j.1749-6632.2009.04965.x
48. Koubi M, Poplineau M, Vernerey J, N’Guyen L, Tiberi G, Garciaz S, et al. Regulation of the positive transcriptional effect of PLZF through a non-canonical EZH2 activity. Nucleic Acids Res (2018) 46:3339–50. doi: 10.1093/nar/gky080
49. Zimmerman SM, Nixon SJ, Chen PY, Raj L, Smith SR, Paolini RL, et al. Ezh2Y641F mutations co-operate with Stat3 to regulate MHC class I antigen processing and alter the tumor immune response in melanoma. Oncogene (2022) 41:4983–93. doi: 10.1038/s41388-022-02492-7
50. Dasgupta M, Dermawan JKT, Willard B, Stark GR. STAT3-driven transcription depends upon the dimethylation of K49 by EZH2. Proc Natl Acad Sci (2015) 112:3985–90. doi: 10.1073/pnas.1503152112
51. Zhao Y, Hu Z, Li J, Hu T. EZH2 exacerbates breast cancer by methylating and activating STAT3 directly. J Cancer (2021) 12:5220–30. doi: 10.7150/jca.50675
52. Shi B, Liang J, Yang X, Wang Y, Zhao Y, Wu H, et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol (2007) 27:5105–19. doi: 10.1128/MCB.00162-07
53. Koh CM, Iwata T, Zheng Q, Bethel C, Yegnasubramanian S, De Marzo AM. Myc enforces overexpression of EZH2 in early prostatic neoplasia via transcriptional and post-transcriptional mechanisms. Oncotarget (2011) 2:669–83. doi: 10.18632/oncotarget.327
54. Kaur M, Cole MD. MYC acts via the PTEN tumor suppressor to elicit autoregulation and genome-wide gene repression by activation of the Ezh2 methyltransferase. Cancer Res (2013) 73:695–705. doi: 10.1158/0008-5472.CAN-12-2522
55. Wang J, Yu X, Gong W, Liu X, Park K-S, Ma A, et al. EZH2 noncanonically binds cMyc and p300 through a cryptic transactivation domain to mediate gene activation and promote oncogenesis. Nat Cell Biol (2022) 24:384–99. doi: 10.1038/s41556-022-00850-x
56. Yu X, Wang J, Gong W, Ma A, Shen Y, Zhang C, et al. Dissecting and targeting noncanonical functions of EZH2 in multiple myeloma via an EZH2 degrader. Oncogene (2023), 42:994–1009. doi: 10.1038/s41388-023-02618-5
57. Jiao L, Shubbar M, Yang X, Zhang Q, Chen S, Wu Q, et al. A partially disordered region connects gene repression and activation functions of EZH2. Proc Natl Acad Sci (2020) 117:16992–7002. doi: 10.1073/pnas.1914866117
58. Corvetta D, Chayka O, Gherardi S, D’Acunto CW, Cantilena S, Valli E, et al. Physical interaction between MYCN oncogene and polycomb repressive complex 2 (PRC2) in neuroblastoma. J Biol Chem (2013) 288:8332–41. doi: 10.1074/jbc.M113.454280
59. Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell (2016) 30:563–77. doi: 10.1016/j.ccell.2016.09.005
60. Vanden Bempt M, Debackere K, Demeyer S, Van Thillo Q, Meeuws N, Prieto C, et al. Aberrant MYCN expression drives oncogenic hijacking of EZH2 as a transcriptional activator in peripheral T-cell lymphoma. Blood (2022) 140:2463–76. doi: 10.1182/blood.2022016428
61. Kim KH, Kim W, Howard TP, Vazquez F, Tsherniak A, Wu JN, et al. SWI/SNF-mutant cancers depend on catalytic and non-catalytic activity of EZH2. Nat Med (2015) 21:1491–6. doi: 10.1038/nm.3968
62. Bitler BG, Aird KM, Garipov A, Li H, Amatangelo M, Kossenkov AV, et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat Med (2015) 21:231–8. doi: 10.1038/nm.3799
Keywords: EZH2, H3K27me3, non-canonical, cancer, oncogene, tumor suppressor
Citation: Zimmerman SM, Lin PN and Souroullas GP (2023) Non-canonical functions of EZH2 in cancer. Front. Oncol. 13:1233953. doi: 10.3389/fonc.2023.1233953
Received: 03 June 2023; Accepted: 21 July 2023;
Published: 18 August 2023.
Edited by:
Alessandra Montecucco, National Research Council (CNR), ItalyReviewed by:
Estelle Duprez, INSERM U1068 Centre de Recherche en Cancérologie de Marseille (CRCM), FranceMarlies Vanden Bempt, KU Leuven, Belgium
Copyright © 2023 Zimmerman, Lin and Souroullas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George P. Souroullas, Z2VvcmdlLnNvdXJvdWxsYXNAd3VzdGwuZWR1