 Junghwa Lee
Junghwa Lee Eui Ho Kim
Eui Ho Kim
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Oncol., 28 July 2023
Sec. Molecular and Cellular Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1233376
This article is part of the Research TopicPromising Biomarkers and Molecular Mechanisms for Predicting Response to Immunotherapy in Different Types of CancerView all 8 articles
Cancer immunotherapies targeting immune checkpoint pathways, such as programmed cell death-1 (PD-1)/programmed cell death ligand-1 (PD-L1) and cytotoxic T-lymphocyte-associated antigen-4 (CTLA-4), have achieved unprecedented therapeutic success in treating various types of cancer. The prominent and persistent clinical responses to immune checkpoint blockade (ICB) therapy are currently constrained to a subset of patients. Owing to discrete individual tumor and immune heterogeneity, most patients fail to benefit from ICB treatment, demonstrating either primary or acquired resistance. A thorough comprehension of the mechanisms restricting the efficacy of immune checkpoint inhibitors (ICIs) is required to extend their clinical applicability to a broader spectrum of patients and cancer types. Numerous studies are presently investigating potential prognostic markers of responsiveness, the complex dynamics underlying the therapeutic and adverse effects of ICB, and tumor immune evasion throughout the course of immunotherapy. In this article, we have reviewed the extant literature elucidating the mechanisms underlying the response and resistance to ICB, with a particular emphasis on PD-1 and CTLA-4 pathway blockade in the context of anti-tumor immunity. Furthermore, we aimed to explore potential approaches to overcome cancer therapeutic resistance and develop a rational design for more personalized ICB-based combinational regimens.
Prolonged antigenic stimulation and the ensuing inflammation in tumor microenvironments (TME) lead to immunosuppression, which results in different states of T cell exhaustion. Continuous exposure to antigens causes antigen-specific T cells to lose their effector functions and proliferative capacity and acquire multiple inhibitory receptors. Therefore, restoring the functionality of tumor-specific T cells is a crucial objective of cancer immunotherapy. Immune checkpoint-targeting interventions have demonstrated significant anti-tumor efficacy; nonetheless, ICB has elicited therapeutic responses only in a subset of patients (1). The molecular mechanisms associated with the PD-1 or CTLA-4 pathway-mediated negative regulation of T cell responses and the alterations in PD-1 or CTLA-4 signaling upon blocking their interactions to revive exhausted T cells warrant further comprehensive elucidation. Moreover, it is essential to identify the predictive biomarkers of ICB-mediated responsiveness for designing personalized therapies.
CTLA-4 is a co-inhibitory receptor primarily expressed by T cells. Intracellular CTLA-4 is translocated to the cell surface following the recognition of cognate antigens early after T cell activation where they compete with CD28 for interacting with B7 molecules on antigen-presenting cells (APCs), exhibiting a higher affinity (2). Subsequently, this interaction delivers negative signaling to T cells, leading to the attenuation of T cell proliferation, activation, and overall function (3, 4) (Figure 1). CTLA-4 is also constitutively expressed in regulatory T cells (Tregs) (6–8). A close association between CTLA-4 and the maintenance and function of Tregs has been demonstrated. CTLA-4-knockout mice suffer from lymphoproliferative disorders and early fatality (9, 10). Treatment with an anti-CTLA-4 antibody effected a pronounced and persistent regression of established tumors in syngeneic mouse models (11). Subsequently, clinical trials evaluated fully human anti-CTLA-4 blocking antibodies in patients with advanced cancers, revealing a durable response in a certain proportion of patients, irrespective of the relatively high frequency of associated toxicity. In an initial clinical trial where an anti-CTLA-4 blocking antibody was administered to patients with metastatic melanoma in combination with peptide vaccines containing melanoma-associated antigens, 43% of patients (6 out of 14) experienced grade III/IV autoimmunity affecting multiple organs (12). In 2011, ipilimumab was licensed as the first ICI for the treatment of metastatic melanoma (13, 14).
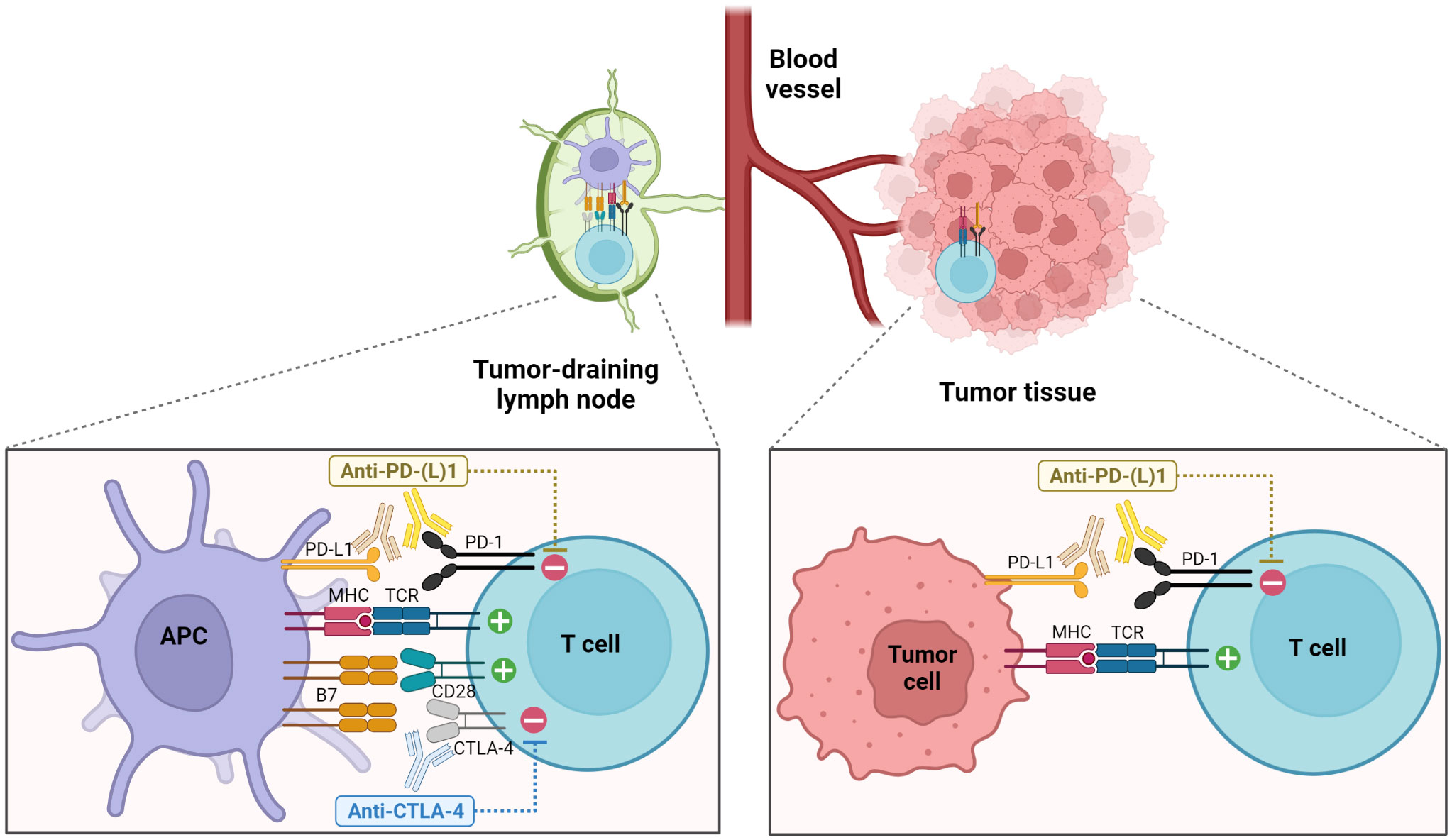
Figure 1 Enhancement of anti-tumor T cell responses through the blockade of PD-1 and CTLA-4 pathways. Blockade of CTLA-4 primarily acts during the priming phase in secondary lymphoid organs, promoting the interactions between T cells and APCs and leading to increased T cell proliferation and activation. On the other hand, PD-1 pathway blockade elicits the functional restoration of exhausted T cells in the TME as well as the enhanced priming of tumor-specific T cells in the TDLNs (1, 5).
The anti-tumor effects of CTLA-4 blockade have been shown to be accompanied by an increase in effector T cells and a decrease in Tregs, thereby leading to an elevated ratio of intratumoral effector T cells to Tregs in mouse tumor models (15, 16). These findings have also been observed in studies involving patients with melanoma who received ipilimumab, both in peripheral blood and tumor tissues (17, 18). Additionally, a recent study demonstrated that local administration of tremelimumab resulted in reduced Tregs in the sentinel lymph nodes and peripheral blood of melanoma patients (19). However, it is noteworthy to mention that contrasting results have been reported in multiple studies where increased or maintained levels of Tregs in peripheral blood and tumor-infiltrating lymphocytes (TILs) of cancer patients were indicated following anti-CTLA-4 therapy (20), suggesting variable outcomes regarding the impact of CTLA-4 blockade on Tregs in relation to human tumors.
PD-1, an inhibitory receptor of the CD28 family, predominantly regulates T cell exhaustion and suppresses T cell anti-tumor effector functions under persistent antigenic stimulation. PD-1 is expressed in various hematopoietic cells and binds to two members of the B7 family of ligands— PD-L1 and PD-L2 (21–24). PD-L1 is widely expressed in both hematopoietic and non-hematopoietic cells. Contrarily, PD-L2 expression is more restricted to professional APCs and a specific subset of B cells. PD-L1 and PD-L2 expression is induced by inflammatory cytokines such as interferons (25). Activation-driven T-cell receptor (TCR) signaling induces the expression of PD-1, which is further upregulated by cytokines. Engagement of PD-1 with its ligands recruits the tyrosine phosphatase 2-containing SH2-domain (SHP-2) that inactivates proximal effector molecules, namely zeta-chain-associated protein kinase-70 and phosphoinositide 3-kinase (PI3K), thereby attenuating TCR- and CD28-mediated signaling (26–28). The immune-regulatory function of PD-1 is evidenced through the development of an autoimmune-like phenotype in mice lacking PD-1 with a delayed onset compared with CTLA-4-knockout mice (29, 30). In the TME, continuous antigenic stimulation leads to sustained high-levels of PD-1 expression on T cells. Additionally, inflammatory cytokines such as interferon-γ (IFN-γ) induce upregulation of PD-L1 on tumor cells, facilitating the PD-1/PD-L1 interaction and ultimately contributing to T cell exhaustion. The PD-1 pathway restrains the interactions between tumor-infiltrating PD-1+ T cells and other immune cells or tumor cells expressing PD-1 ligands in the TME as well as tumor antigen presentation by APCs to tumor-specific T cells during priming in the tumor-draining lymph nodes (TDLNs) (Figure 1). In vivo studies investigating the treatment with anti-PD-1 or PD-L1 antibodies have reported delayed tumor progression and enhanced viral control accompanied by the reinvigoration of virus-specific T cells in syngeneic murine tumor models and chronic viral infection models, respectively (31, 32). These preclinical studies have prompted the development of human PD-1- and PD-L1-targeting antibodies and the initiation of clinical trials involving patients presenting with advanced malignancies refractory to conventional treatment (33–36). In 2014, the Food and Drug Administration (FDA) authorized a PD-1-targeting ICI for treating patients with advanced melanoma. Currently, multiple FDA-approved PD-1-targeting ICIs (pembrolizumab, nivolumab, cemiplimab, dostarlimab, and retifanlimab) or PD-L1-targeting ICIs (atezolizumab, durvalumab, and avelumab) are available for remedying various types of cancer. Additionally, PD-1 blockade therapy exhibits a relatively favorable toxicity profile in comparison to CTLA-4 blockade therapy. Furthermore, the distinct mechanisms of action of PD-1 and CTLA-4 pathway blockade, along with the observed synergistic anti-tumor effects of combined treatment in preclinical models (37–39), led to the clinical evaluation of these two ICIs for cancer patients, demonstrating an enhanced clinical response (40). Subsequently, the combination therapy of nivolumab and ipilimumab was approved in 2016 for the treatment of metastatic melanoma patients.
Recent findings on heterogeneous exhausted CD8 T cell subsets with varying qualities have revealed that PD-1 blockade therapy acts primarily on progenitor exhausted CD8 T cells, promoting their proliferation and differentiation into more terminally exhausted CD8 T cells with effector functions (41–44). Considering the significance of progenitor exhausted CD8 T cells in sustaining the exhausted CD8 T-cell pool, it is crucial to identify the mechanisms regulating their maintenance during chronic antigenic stimulation. Moreover, recent discoveries have explicitly demonstrated the prominence of anatomical effector sites in PD-1 pathway blockade. FTY720 treatment-mediated prevention of T cell egress via secondary lymphoid organs in murine tumor models abolished the therapeutic effect of anti-PD-L1 antibodies (45, 46). These findings suggest that the restoration of exhausted T cell function by anti-PD-L1 antibodies could be more closely associated with immune cells located in TDLNs than with those in the TME. Compared with systemic treatments, local administration of anti-PD-L1 antibodies targeting TDLNs elicited superior anti-tumor therapeutic effects, and PD-L1+ conventional dendritic cells (DCs), rather than other myeloid cells, were the primary targets of PD-L1 inhibition (47–49). Studies conducted on cancer patients have provided that anti-PD-L1 antibodies exert a therapeutic effect through the expansion of pre-existing tumor-specific T cells and the infiltration of new clonotype T cells, thereby effecting clonal replacement (1, 50, 51). PD-1 is also highly expressed in tumor-infiltrating Tregs (52). Whether cell-intrinsic PD-1 exerts a stimulatory or inhibitory effect in Tregs remains controversial; nonetheless, a recent study demonstrated that PD-1 deletion in Tregs impairs their proliferation and inhibitory function in the TME (53).
Despite the unprecedented durable outcomes achieved with cancer immunotherapies, many patients may present with either primary resistance, which indicates irresponsiveness to ICB, or acquired resistance, which develops in patients who initially respond to treatment, and eventually deems the tumor refractory. Various mechanisms regulating the therapeutic resistance to ICBs have been elucidated and implicated in multiple cancer indications (Table 1).

Table 1 Mechanisms of primary and acquired resistance to ICB.
To generate anti-tumor T cell responses, the first step is the recognition of tumor antigens that are distinct from self-antigens by T cells. Although tumor mutational burden (TMB) alone is not sufficient to prognosticate the benefit of treatment, prior research has depicted a relationship between the neoantigen load arising from tumor-specific mutations and the clinical activity of ICB. A significant correlation has been established between the high TMB and improved clinical outcomes from anti-PD-1/PD-L1 therapy across diverse types of cancer (54–56). Similarly, a higher mutational burden and neoantigen load were associated with enhanced clinical responses and survival after anti-CTLA-4 therapy in patients with melanoma (57, 58). Furthermore, patients with mismatch repair-deficient/high microsatellite instability tumors, which generate a high level of somatic mutations, are more likely to benefit from anti-PD-1 therapy in multiple cancer types (59, 60). Therefore, these data indicate that low levels of available neoantigens, resulting from a lack of sufficient tumor mutational load, can influence primary resistance to ICB. Besides insufficient tumor antigenicity, defective antigen presentation may be associated with tumor resistance to ICB by rendering tumor cells incapable of being recognized and killed by T cells. Consequently, the expression of MEX3B mRNA in the pretreated melanoma tumors, which downregulated human leukocyte antigen (HLA)-A expression on tumor cells by binding to the 3’ untranslated region, was noted to be higher in non-responders than responders to anti-PD-1 therapy (63).
Tumor-intrinsic oncogenic signaling plays a role in ICB resistance. Activation of the tumor-intrinsic PI3K pathway via phosphatase and tensin homolog (PTEN) loss is inversely associated with the efficacy of anti-PD-1 therapy. Patients with metastatic melanoma who received anti-PD-1 therapy have shown that PTEN-expressing tumors were associated with an improved objective response compared with tumors with PTEN deficiency (67). The Cancer Genome Atlas (TCGA) analysis of melanoma demonstrated that a higher frequency of PTEN deletion and loss of function was observed in non-T cell-inflamed tissues than in T cell-inflamed tissues. A mouse melanoma model has shown that tumor-intrinsic PTEN loss results in reduced T-cell tumor infiltration accompanied by elevated expression of inhibitory cytokines and inhibition of autophagy, with subsequent escape from T-cell killing. Furthermore, PI3K inhibition combined with anti-PD-1 therapy enhanced the therapeutic effects of ICB, leading to further tumor regression and improved survival in tumor-bearing mice. In another study on metastatic cutaneous melanoma patients, the activation of the β-catenin signaling was one of the primary features of non-T cell-inflamed tumors compared with T cell-inflamed tumors (68). In a preclinical melanoma model, tumor-intrinsic β-catenin activation was shown to impair the infiltration of T cells and CD103+ DCs, which is related to reduced CCL4 secretion from tumors and leads to therapeutic resistance to the combined PD-1 and CTLA-4 blockade. These data indicate that specific oncogenic signals in tumors can cause immune exclusion, contributing to primary resistance to ICB.
In the TME, T cells produce cytokines, such as IFN-γ, upon recognizing specific tumor antigens. IFN-γ serves as a strong inducer of PD-L1 expression, enabling the tumor to evade the anti-tumor immune response. The therapeutic effects of PD-1 pathway blockade involve the disruption of adaptive immune resistance that relies on pre-existing anti-tumor immunity (90). The presence of an IFN-γ signature from the tumor has been proposed as an indicator of responsiveness to anti-PD-1 therapy (91, 92). IFN-γ has both stimulatory and inhibitory effects on the anti-tumor immune response (93). In the TME, IFN-γ promotes anti-tumor immunity not only by activating and recruiting immune cells but also by increasing the expression of major histocompatibility complex (MHC) molecules by tumor cells and directly exerting an anti-proliferative and pro-apoptotic effect on tumor cells. Considering the critical role of IFN-γ in anti-tumor immunity, tumor-intrinsic alterations that disrupt the IFN-γ pathway could interfere with the efficacy of ICB therapy. Melanoma tumors from non-responders to ipilimumab have displayed a higher frequency of mutations such as copy-number alterations in IFN-γ pathway genes, including IFNGR1, IRF1, and JAK2, than that from the responders (70). Consistently, tumor-intrinsic IFNGR1 deficiency was associated with reduced therapeutic effects of CTLA-4 blockade, resulting in accelerated tumor growth and reduced survival of B16/BL6-bearing mice, indicating an association between the lack of tumor-intrinsic IFN-γ signaling and primary resistance to CTLA-4 blockade. Another study has demonstrated that human melanoma cell lines harboring loss-of-function mutations of JAK2 failed to upregulate PD-L1 in response to IFN-γ exposure (71), implying that patients with defective JAK2-harboring tumors would result in the absence of inducible PD-L1 expression and be unlikely to benefit from anti-PD-1 therapy, representing primary resistance.
Epigenetic modifications occurring in tumor cells may contribute to immune evasion and resistance to ICIs. In an ovarian cancer model, mice harboring tumors deficient in ARID1A, a component of the switch/sucrose non-fermentable (SWI/SNF) chromatin remodeling complex, exhibited increased TMB and impaired mismatch repair capacity, and notably demonstrated reduced tumor growth and improved survival in response to anti-PD-1 therapy compared to those with control tumors (72). Similarly, mouse melanoma lacking Pbrm1, another subunit of the SWI/SNF complex, displayed increased susceptibility to combined PD-1 and CTLA-4 blockade than control tumors (73). In line with these findings, a study involving renal cell carcinoma patients receiving PD-1 pathway blockade monotherapy or combination therapy with CTLA-4 blockade showed that tumors with loss of function mutations in Pbrm1 correlated with clinical benefits from the treatment (74). Additionally, epigenetic modifying agents, such as DNA methyltransferase inhibitors and histone deacetylases inhibitors, have the potential to significantly impact genes associated with tumor antigenicity, cytokine and chemokine production, as well as oncogenic or tumor-suppressive signaling (75, 94). These agents have been proposed as potential therapeutic strategies for restoring anti-tumor immunity and the clinical efficacy of combing these inhibitors with ICB has been evaluated.
Suppressive immune cells have been implicated as mediating ICB resistance. Myeloid-derived suppressor cells (MDSCs) play a role in promoting tumor growth, angiogenesis, and metastasis, as well as inhibiting anti-tumor immunity (95). A study on patients with metastatic melanoma receiving ipilimumab showed that responders had a significantly lower frequency of circulating monocytic MDSCs than non-responders (76). In addition, treatment with the selective PI3K inhibitor to inhibit MDSCs, in combination with ICIs enhanced tumor regression and survival than ICB blockade alone in multiple tumor models, indicating that reprograming MDSCs could stimulate the response to ICB (77, 78). Tumor-associated macrophages (TAMs) exert immunosuppressive effects by upregulating the molecules involved in the inhibitory pathway, secreting regulatory cytokines, and promoting tumor invasion and metastasis (96). An association between a higher frequency of TAMs and a poor prognosis in human cancer has been cataloged for several types of cancer. Macrophage depletion has demonstrated reduced tumor growth in various preclinical models. In a study conducted on murine pancreatic cancer, blocking the colony-stimulating factor-1 receptor to deplete macrophages led to substantial tumor regression when combined with PD-1 and CTLA-4 blockade, which otherwise had no significant anti-tumor effects, indicating the role of TAMs in the therapeutic effects of ICB (79). Numerous studies have reported Treg infiltration in human tumors (20). Preclinical models have demonstrated that depletion of Tregs in the TME can enhance anti-tumor immunity. In patients receiving nivolumab, a reduced frequency of peripheral blood Tregs was observed in responders and patients with stable diseases whereas an increased Treg frequency was noted in non-responders (80). These results indicate the potential of reprogramming suppressive immune cells to enhance the response to ICB.
The gut microbiome has been reported to influence and modulate responses to ICB therapy. Considerable variations in the diversity and composition of the gut microbiome have been observed between responders and non-responder melanoma patients receiving anti-PD-1 therapy (81). Responders had a favorable gut microbiome that promotes anti-tumor immunity. Another study showed correlations between clinical responses to anti-PD-1 therapy and Akkermansia muciniphila in the gut microbiome. Oral supplementation with A. muciniphila following fecal microbiota transfer from non-responders resulted in the restoration of anti-PD-1 efficacy (82). In addition, antibiotic treatment reduced the clinical benefits of anti-PD-1 therapy in patients with advanced cancer (83). Similarly, the efficacy of anti-CTLA-4 therapy was associated with specific bacterial species, and antibiotic-treated or germ-free mice did not respond to CTLA-4 blockade (84). These data suggest that the microbiome could be a critical factor in causing resistance and implicate the manipulation of the microbiome to augment the efficacy of ICB.
Immunosuppressive cytokines such as transforming growth factor-β (TGF-β) contribute to ICB resistance. TGF-β signaling in fibroblasts was associated with CD8 T cell exclusion from the tumor parenchyma and resistance to anti-PD-1 therapy in patients with metastatic urothelial cancer (87). A preclinical study has shown that the combination of PD-L1 and TGF-β blocking antibodies resulted in increased infiltration of T cells into the tumor and a significant reduction in tumor growth compared with either blockade alone. Additionally, growth factors such as vascular endothelial growth factor (VEGF) have been found to correlate with resistance to anti-PD-1 therapy in melanoma patients (88). The combined blockade targeting PD-1 and VEGF induced an improved anti-tumor effect in patients with renal cell carcinoma (89). Other inhibitory molecules and pathways have been implicated in limiting the efficacy of ICB, and further study will be required to verify the relationship with ICI resistance.
Cancer immunoediting involves three distinct phases: elimination, equilibrium, and escape. It reflects the intricate interplay between the tumor and the immune system during tumor progression, and these dynamics can also occur in response to immunotherapy (97). When the tumor reaches a state where it can evade the anti-tumor immune response and the immunotherapy fails to adequately counteract tumor-mediated immune suppression, the tumor acquires resistance to the treatment.
It has been reported that loss of neoantigens could be associated with acquired resistance to ICB. Examination of the emergence of acquired resistance in patients with non-small cell lung cancer who initially responded to PD-1 blockade or combined PD-1 and CTLA-4 blockade therapy revealed a decrease in mutation-associated neoantigens in resistant tumors compared with pretreatment tumor samples (61). This loss of neoantigens occurred due to subclonal elimination and chromosomal deletion and it was associated with decreased T-cell clonotypes. Additionally, another study reported a reduction in neoantigen expression from patients with uterine leiomyosarcoma who developed acquired resistance to anti-PD-1 therapy (62).
β-2-microglobulin (B2M), a subunit of the MHC class I complex, plays an essential role in the assembly and trafficking of MHC class I molecules and antigen presentation. Accordingly, B2M deficiency can lead to immune evasion by tumors. Genetic alterations in B2M have been reported in several types of cancer that have developed acquired resistance to ICB. Whole-exosome sequencing assessment of baseline and relapsed tumors from four patients with melanoma who developed resistance to anti-PD-1 therapy after the initial response identified an acquired homozygous truncating mutation in the gene encoding B2M at relapse in one patient (64). Another longitudinal study examining five melanoma patients with acquired resistance to ICB reported a patient with two frameshift mutations in B2M and a second patient harboring a single B2M loss with two frameshift mutations during progression, which resulted in a significant reduction in tumor expression of B2M and a lack of HLA class I protein expression on the outer membrane (65). In addition, a newly generated truncating B2M mutation was reported in two patients with brain metastasis among five patients with advanced mismatch repair-deficient cancers who demonstrated acquired resistance to anti-PD-1 therapy (59). A study of lung cancer with 14 ICB-resistant tumor samples also revealed the homozygous loss of B2M in a patient who acquired resistance to combined anti-PD-L1 and anti-CTLA-4 therapy (66). Therefore, these data indicate that impaired tumor antigen presentation could also be a mechanism of acquired resistance that develops among responders to ICB.
Tumors exhibiting PTEN loss have also been observed in patients who experienced relapse after an initial clinical response to ICB. In the case of uterine leiomyosarcoma patients who developed acquired resistance following an initial response to anti-PD-1 therapy, biallelic PTEN loss was detected (62). Similarly, a separate study identified a patient with metastatic melanoma who demonstrated a partial response to the combination of anti-PD-1 and anti-CTLA-4 therapy but later developed treatment-resistant metastasis with acquired PTEN loss (69).
Defective IFN-γ signaling has also been documented in ICB-resistant tumors upon relapse. A study has depicted that loss-of-function mutations in JAK1 or JAK2 were found in two of four melanoma patients presenting with acquired resistance to anti-PD-1 therapy (64). The cell lines established from relapsed tumors of the patient harboring the JAK2 mutation exhibited an absence of JAK2 protein and loss of sensitivity to IFN-γ signaling, concurrently with reduced expression of IFN-γ-responsive PD-L1 and MHC I, in contrast to those derived from the baseline tumors.
It has been documented that in the TME at the time of acquired resistance, there is an upregulation of other immune checkpoint molecules, which may have non-redundant roles. This suggests the existence of potential resistance mechanisms and implies that combined blockade targeting multiple immune checkpoints could result in synergistic therapeutic efficacy. Notably, an analysis of two lung cancer patients who experienced a partial response to anti-PD-1 therapy but eventually progressed revealed upregulated expression of T cell immunoglobulin and mucin domain-containing protein 3 (TIM-3) on TILs, to which anti-PD-1 antibodies were bound, at the time of resistance (85). In a mouse model of lung cancer, elevated expression of TIM-3 was similarly observed on anti-PD-1 antibody-bound T cells, and subsequent blockade of TIM-3 improved overall survival than PD-1 blockade alone. Additionally, a separate report found that 67% of melanoma patients who developed resistance following the initial response to anti-PD-1 monotherapy or a combination of anti-PD-1 and anti-CTLA-4 therapy exhibited increased numbers of intratumoral V-domain Ig suppressor of T cell activation (VISTA)+ lymphocytes upon disease progression compared with pretreatment levels (86). Additionally, an upregulation of the expression of multiple inhibitory receptor genes, including PD-1, CTLA-4, TIM-3, lymphocyte activation gene-3 (LAG-3), T cell immunoreceptor with Ig and ITIM domains (TIGIT), and 2B4, was observed in lung cancer samples that developed resistance to PD-1 pathway blockade therapy compared with their corresponding pretreatment samples (66).
Numerous factors and mechanisms contribute to primary and acquired ICB resistance. Specific immunomodulatory strategies are envisaged to induce cancer immunoediting and consequent immune escape. Clinical trials are actively assessing the efficacy of combination cancer therapies involving multiple ICIs and other therapeutic interventions. Therefore, endeavors directed toward comprehensively elucidating the response, limitations, and irresponsiveness to ICB can provide insight into identifying prognostic biomarkers of ICB-mediated response to formulate personalized combinational therapeutic regimens. Consequently, this would enhance clinical outcomes for a broader spectrum of cancer patients.
JL researched and drafted the manuscript. EHK supervised the content. All authors contributed to the article and approved the submitted version.
This work was supported by National Research Foundation of Korea (NRF) grants funded by the Korean government (MSIT), grant number NRF-2021R1C1C1012962 and NRF-2023M3A9G6057281.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Chow A, Perica K, Klebanoff CA, Wolchok JD. Clinical implications of T cell exhaustion for cancer immunotherapy. Nat Rev Clin Oncol (2022) 19(12):775–90. doi: 10.1038/s41571-022-00689-z
2. Krummel MF, Allison JP. Cd28 and ctla-4 have opposing effects on the response of T cells to stimulation. J Exp Med (1995) 182(2):459–65. doi: 10.1084/jem.182.2.459
3. Linsley PS, Brady W, Urnes M, Grosmaire LS, Damle NK, Ledbetter JA. Ctla-4 is a second receptor for the B cell activation antigen B7. J Exp Med (1991) 174(3):561–9. doi: 10.1084/jem.174.3.561
4. Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. Ctla-4 can function as a negative regulator of T cell activation. Immunity (1994) 1(5):405–13. doi: 10.1016/1074-7613(94)90071-x
5. Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer (2016) 16(5):275–87. doi: 10.1038/nrc.2016.36
6. Read S, Greenwald R, Izcue A, Robinson N, Mandelbrot D, Francisco L, et al. Blockade of ctla-4 on cd4+Cd25+ Regulatory T cells abrogates their function in vivo. J Immunol (2006) 177(7):4376–83. doi: 10.4049/jimmunol.177.7.4376
7. Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, et al. Ctla-4 control over foxp3+ Regulatory T cell function. Science (2008) 322(5899):271–5. doi: 10.1126/science.1160062
8. Friedline RH, Brown DS, Nguyen H, Kornfeld H, Lee J, Zhang Y, et al. Cd4+ Regulatory T cells require ctla-4 for the maintenance of systemic tolerance. J Exp Med (2009) 206(2):421–34. doi: 10.1084/jem.20081811
9. Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in ctla-4. Science (1995) 270(5238):985–8. doi: 10.1126/science.270.5238.985
10. Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of ctla-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of ctla-4. Immunity (1995) 3(5):541–7. doi: 10.1016/1074-7613(95)90125-6
11. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by ctla-4 blockade. Science (1996) 271(5256):1734–6. doi: 10.1126/science.271.5256.1734
12. Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ, et al. Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci U.S.A. (2003) 100(14):8372–7. doi: 10.1073/pnas.1533209100
13. Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med (2010) 363(8):711–23. doi: 10.1056/NEJMoa1003466
14. Robert C, Thomas L, Bondarenko I, O'Day S, Weber J, Garbe C, et al. Ipilimumab plus dacarbazine for previously untreated metastatic melanoma. N Engl J Med (2011) 364(26):2517–26. doi: 10.1056/NEJMoa1104621
15. Quezada SA, Peggs KS, Curran MA, Allison JP. Ctla4 blockade and gm-csf combination immunotherapy alters the intratumor balance of effector and regulatory T cells. J Clin Invest (2006) 116(7):1935–45. doi: 10.1172/JCI27745
16. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-ctla-4 therapy against melanoma. J Exp Med (2013) 210(9):1695–710. doi: 10.1084/jem.20130579
17. Hodi FS, Butler M, Oble DA, Seiden MV, Haluska FG, Kruse A, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U.S.A. (2008) 105(8):3005–10. doi: 10.1073/pnas.0712237105
18. Liakou CI, Kamat A, Tang DN, Chen H, Sun J, Troncoso P, et al. Ctla-4 blockade increases ifngamma-producing cd4+Icoshi cells to shift the ratio of effector to regulatory T cells in cancer patients. Proc Natl Acad Sci U.S.A. (2008) 105(39):14987–92. doi: 10.1073/pnas.0806075105
19. van Pul KM, Notohardjo JCL, Fransen MF, Koster BD, Stam AGM, Chondronasiou D, et al. Local delivery of low-dose anti-ctla-4 to the melanoma lymphatic basin leads to systemic T(Reg) reduction and effector T cell activation. Sci Immunol (2022) 7(73):eabn8097. doi: 10.1126/sciimmunol.abn8097
20. Chaudhary B, Elkord E. Regulatory T cells in the tumor microenvironment and cancer progression: role and therapeutic targeting. Vaccines (Basel) (2016) 4(3). doi: 10.3390/vaccines4030028
21. Dong H, Zhu G, Tamada K, Chen L. B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat Med (1999) 5(12):1365–9. doi: 10.1038/70932
22. Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the pd-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med (2000) 192(7):1027–34. doi: 10.1084/jem.192.7.1027
23. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. Pd-L2 is a second ligand for pd-1 and inhibits T cell activation. Nat Immunol (2001) 2(3):261–8. doi: 10.1038/85330
24. Tseng SY, Otsuji M, Gorski K, Huang X, Slansky JE, Pai SI, et al. B7-dc, a new dendritic cell molecule with potent costimulatory properties for T cells. J Exp Med (2001) 193(7):839–46. doi: 10.1084/jem.193.7.839
25. Kinter AL, Godbout EJ, McNally JP, Sereti I, Roby GA, O'Shea MA, et al. The common gamma-chain cytokines il-2, il-7, il-15, and il-21 induce the expression of programmed death-1 and its ligands. J Immunol (2008) 181(10):6738–46. doi: 10.4049/jimmunol.181.10.6738
26. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. Pd-1 inhibits T-cell receptor induced phosphorylation of the zap70/cd3zeta signalosome and downstream signaling to pkctheta. FEBS Lett (2004) 574(1-3):37–41. doi: 10.1016/j.febslet.2004.07.083
27. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by recruiting phosphatase shp2. J Exp Med (2012) 209(6):1201–17. doi: 10.1084/jem.20112741
28. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor cd28 is a primary target for pd-1-mediated inhibition. Science (2017) 355(6332):1428–33. doi: 10.1126/science.aaf1292
29. Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus-like autoimmune diseases by disruption of the pd-1 gene encoding an itim motif-carrying immunoreceptor. Immunity (1999) 11(2):141–51. doi: 10.1016/s1074-7613(00)80089-8
30. Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in pd-1 receptor-deficient mice. Science (2001) 291(5502):319–22. doi: 10.1126/science.291.5502.319
31. Hirano F, Kaneko K, Tamura H, Dong H, Wang S, Ichikawa M, et al. Blockade of B7-H1 and pd-1 by monoclonal antibodies potentiates cancer therapeutic immunity. Cancer Res (2005) 65(3):1089–96. doi: 10.1158/0008-5472.1089.65.3
32. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted cd8 T cells during chronic viral infection. Nature (2006) 439(7077):682–7. doi: 10.1038/nature04444
33. Brahmer JR, Drake CG, Wollner I, Powderly JD, Picus J, Sharfman WH, et al. Phase I study of single-agent anti-programmed death-1 (Mdx-1106) in refractory solid tumors: safety, clinical activity, pharmacodynamics, and immunologic correlates. J Clin Oncol (2010) 28(19):3167–75. doi: 10.1200/JCO.2009.26.7609
34. Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-pd-1 antibody in cancer. N Engl J Med (2012) 366(26):2443–54. doi: 10.1056/NEJMoa1200690
35. Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-pd-L1 antibody in patients with advanced cancer. N Engl J Med (2012) 366(26):2455–65. doi: 10.1056/NEJMoa1200694
36. Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (Anti-pd-1) in melanoma. N Engl J Med (2013) 369(2):134–44. doi: 10.1056/NEJMoa1305133
37. Das R, Verma R, Sznol M, Boddupalli CS, Gettinger SN, Kluger H, et al. Combination therapy with anti-ctla-4 and anti-pd-1 leads to distinct immunologic changes in vivo. J Immunol (2015) 194(3):950–9. doi: 10.4049/jimmunol.1401686
38. Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature (2014) 515(7528):577–81. doi: 10.1038/nature13988
39. Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discovery (2018) 8(9):1069–86. doi: 10.1158/2159-8290.CD-18-0367
40. Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med (2015) 373(1):23–34. doi: 10.1056/NEJMoa1504030
41. Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, et al. Defining cd8+ T cells that provide the proliferative burst after pd-1 therapy. Nature (2016) 537(7620):417–21. doi: 10.1038/nature19330
42. Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, et al. T cell factor 1-expressing memory-like cd8(+) T cells sustain the immune response to chronic viral infections. Immunity (2016) 45(2):415–27. doi: 10.1016/j.immuni.2016.07.021
43. He R, Hou S, Liu C, Zhang A, Bai Q, Han M, et al. Follicular cxcr5- expressing cd8(+) T cells curtail chronic viral infection. Nature (2016) 537(7620):412–28. doi: 10.1038/nature19317
44. Wu T, Ji Y, Moseman EA, Xu HC, Manglani M, Kirby M, et al. The tcf1-bcl6 axis counteracts type I interferon to repress exhaustion and maintain T cell stemness. Sci Immunol (2016) 1(6). doi: 10.1126/sciimmunol.aai8593
45. Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, et al. Systemic immunity is required for effective cancer immunotherapy. Cell (2017) 168(3):487–502 e15. doi: 10.1016/j.cell.2016.12.022
46. Fransen MF, Schoonderwoerd M, Knopf P, Camps MG, Hawinkels LJ, Kneilling M, et al. Tumor-draining lymph nodes are pivotal in pd-1/pd-L1 checkpoint therapy. JCI Insight (2018) 3(23). doi: 10.1172/jci.insight.124507
47. Dammeijer F, van Gulijk M, Mulder EE, Lukkes M, Klaase L, van den Bosch T, et al. The pd-1/pd-L1-checkpoint restrains T cell immunity in tumor-draining lymph nodes. Cancer Cell (2020) 38(5):685–700 e8. doi: 10.1016/j.ccell.2020.09.001
48. Francis DM, Manspeaker MP, Schudel A, Sestito LF, O'Melia MJ, Kissick HT, et al. Blockade of immune checkpoints in lymph nodes through locoregional delivery augments cancer immunotherapy. Sci Transl Med (2020) 12(563). doi: 10.1126/scitranslmed.aay3575
49. Oh SA, Wu DC, Cheung J, Navarro A, Xiong H, Cubas R, et al. Pd-L1 expression by dendritic cells is a key regulator of T-cell immunity in cancer. Nat Cancer (2020) 1(7):681–91. doi: 10.1038/s43018-020-0075-x
50. Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following pd-1 blockade. Nat Med (2019) 25(8):1251–9. doi: 10.1038/s41591-019-0522-3
51. Liu B, Hu X, Feng K, Gao R, Xue Z, Zhang S, et al. Temporal single-cell tracing reveals clonal revival and expansion of precursor exhausted T cells during anti-pd-1 therapy in lung cancer. Nat Cancer (2022) 3(1):108–21. doi: 10.1038/s43018-021-00292-8
52. Kim MJ, Ha SJ. Differential role of pd-1 expressed by various immune and tumor cells in the tumor immune microenvironment: expression, function, therapeutic efficacy, and resistance to cancer immunotherapy. Front Cell Dev Biol (2021) 9:767466. doi: 10.3389/fcell.2021.767466
53. Kim MJ, Kim K, Park HJ, Kim GR, Hong KH, Oh JH, et al. Deletion of pd-1 destabilizes the lineage identity and metabolic fitness of tumor-infiltrating regulatory T cells. Nat Immunol (2023) 24(1):148–61. doi: 10.1038/s41590-022-01373-1
54. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to pd-1 inhibition. N Engl J Med (2017) 377(25):2500–1. doi: 10.1056/NEJMc1713444
55. Goodman AM, Kato S, Bazhenova L, Patel SP, Frampton GM, Miller V, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther (2017) 16(11):2598–608. doi: 10.1158/1535-7163.MCT-17-0386
56. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to pd-1 blockade in non-small cell lung cancer. Science (2015) 348(6230):124–8. doi: 10.1126/science.aaa1348
57. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to ctla-4 blockade in melanoma. N Engl J Med (2014) 371(23):2189–99. doi: 10.1056/NEJMoa1406498
58. Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to ctla-4 blockade in metastatic melanoma. Science (2015) 350(6257):207–11. doi: 10.1126/science.aad0095
59. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to pd-1 blockade. Science (2017) 357(6349):409–13. doi: 10.1126/science.aan6733
60. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. Pd-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372(26):2509–20. doi: 10.1056/NEJMoa1500596
61. Anagnostou V, Smith KN, Forde PM, Niknafs N, Bhattacharya R, White J, et al. Evolution of neoantigen landscape during immune checkpoint blockade in non-small cell lung cancer. Cancer Discovery (2017) 7(3):264–76. doi: 10.1158/2159-8290.CD-16-0828
62. George S, Miao D, Demetri GD, Adeegbe D, Rodig SJ, Shukla S, et al. Loss of pten is associated with resistance to anti-pd-1 checkpoint blockade therapy in metastatic uterine leiomyosarcoma. Immunity (2017) 46(2):197–204. doi: 10.1016/j.immuni.2017.02.001
63. Huang L, Malu S, McKenzie JA, Andrews MC, Talukder AH, Tieu T, et al. The rna-binding protein mex3b mediates resistance to cancer immunotherapy by downregulating hla-a expression. Clin Cancer Res (2018) 24(14):3366–76. doi: 10.1158/1078-0432.CCR-17-2483
64. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to pd-1 blockade in melanoma. N Engl J Med (2016) 375(9):819–29. doi: 10.1056/NEJMoa1604958
65. Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, et al. Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun (2017) 8(1):1136. doi: 10.1038/s41467-017-01062-w
66. Gettinger S, Choi J, Hastings K, Truini A, Datar I, Sowell R, et al. Impaired hla class I antigen processing and presentation as a mechanism of acquired resistance to immune checkpoint inhibitors in lung cancer. Cancer Discovery (2017) 7(12):1420–35. doi: 10.1158/2159-8290.CD-17-0593
67. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of pten promotes resistance to T cell-mediated immunotherapy. Cancer Discovery (2016) 6(2):202–16. doi: 10.1158/2159-8290.CD-15-0283
68. Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature (2015) 523(7559):231–5. doi: 10.1038/nature14404
69. Trujillo JA, Luke JJ, Zha Y, Segal JP, Ritterhouse LL, Spranger S, et al. Secondary resistance to immunotherapy associated with beta-catenin pathway activation or pten loss in metastatic melanoma. J Immunother Cancer (2019) 7(1):295. doi: 10.1186/s40425-019-0780-0
70. Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, et al. Primary resistance to pd-1 blockade mediated by jak1/2 mutations. Cancer Discovery (2017) 7(2):188–201. doi: 10.1158/2159-8290.CD-16-1223
71. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of ifn-gamma pathway genes in tumor cells as a mechanism of resistance to anti-ctla-4 therapy. Cell (2016) 167(2):397–404 e9. doi: 10.1016/j.cell.2016.08.069
72. Shen J, Ju Z, Zhao W, Wang L, Peng Y, Ge Z, et al. Arid1a deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat Med (2018) 24(5):556–62. doi: 10.1038/s41591-018-0012-z
73. Pan D, Kobayashi A, Jiang P, Ferrari de Andrade L, Tay RE, Luoma AM, et al. A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science (2018) 359(6377):770–5. doi: 10.1126/science.aao1710
74. Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science (2018) 359(6377):801–6. doi: 10.1126/science.aan5951
75. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of th1-type chemokines shapes tumour immunity and immunotherapy. Nature (2015) 527(7577):249–53. doi: 10.1038/nature15520
76. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating mdsc correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother (2014) 63(3):247–57. doi: 10.1007/s00262-013-1508-5
77. De Henau O, Rausch M, Winkler D, Campesato LF, Liu C, Cymerman DH, et al. Overcoming resistance to checkpoint blockade therapy by targeting pi3kgamma in myeloid cells. Nature (2016) 539(7629):443–7. doi: 10.1038/nature20554
78. Davis RJ, Moore EC, Clavijo PE, Friedman J, Cash H, Chen Z, et al. Anti-pd-L1 efficacy can be enhanced by inhibition of myeloid-derived suppressor cells with a selective inhibitor of pi3kdelta/gamma. Cancer Res (2017) 77(10):2607–19. doi: 10.1158/0008-5472.CAN-16-2534
79. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. Csf1/csf1r blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res (2014) 74(18):5057–69. doi: 10.1158/0008-5472.CAN-13-3723
80. Weber JS, Kudchadkar RR, Yu B, Gallenstein D, Horak CE, Inzunza HD, et al. Safety, efficacy, and biomarkers of nivolumab with vaccine in ipilimumab-refractory or -naive melanoma. J Clin Oncol (2013) 31(34):4311–8. doi: 10.1200/JCO.2013.51.4802
81. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-pd-1 immunotherapy in melanoma patients. Science (2018) 359(6371):97–103. doi: 10.1126/science.aan4236
82. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of pd-1-based immunotherapy against epithelial tumors. Science (2018) 359(6371):91–7. doi: 10.1126/science.aan3706
83. Derosa L, Routy B, Fidelle M, Iebba V, Alla L, Pasolli E, et al. Gut bacteria composition drives primary resistance to cancer immunotherapy in renal cell carcinoma patients. Eur Urol (2020) 78(2):195–206. doi: 10.1016/j.eururo.2020.04.044
84. Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by ctla-4 blockade relies on the gut microbiota. Science (2015) 350(6264):1079–84. doi: 10.1126/science.aad1329
85. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG, et al. Adaptive resistance to therapeutic pd-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun (2016) 7:10501. doi: 10.1038/ncomms10501
86. Kakavand H, Jackett LA, Menzies AM, Gide TN, Carlino MS, Saw RPM, et al. Negative immune checkpoint regulation by vista: A mechanism of acquired resistance to anti-pd-1 therapy in metastatic melanoma patients. Mod Pathol (2017) 30(12):1666–76. doi: 10.1038/modpathol.2017.89
87. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. Tgfbeta attenuates tumour response to pd-L1 blockade by contributing to exclusion of T cells. Nature (2018) 554(7693):544–8. doi: 10.1038/nature25501
88. Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discovery (2016) 6(8):827–37. doi: 10.1158/2159-8290.CD-15-1545
89. Wallin JJ, Bendell JC, Funke R, Sznol M, Korski K, Jones S, et al. Atezolizumab in combination with bevacizumab enhances antigen-specific T-cell migration in metastatic renal cell carcinoma. Nat Commun (2016) 7:12624. doi: 10.1038/ncomms12624
90. Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-H1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med (2012) 4(127):127ra37. doi: 10.1126/scitranslmed.3003689
91. Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. Ifn-gamma-related mrna profile predicts clinical response to pd-1 blockade. J Clin Invest (2017) 127(8):2930–40. doi: 10.1172/JCI91190
92. Karachaliou N, Gonzalez-Cao M, Crespo G, Drozdowskyj A, Aldeguer E, Gimenez-Capitan A, et al. Interferon gamma, an important marker of response to immune checkpoint blockade in non-small cell lung cancer and melanoma patients. Ther Adv Med Oncol (2018) 10:1758834017749748. doi: 10.1177/1758834017749748
93. Jorgovanovic D, Song M, Wang L, Zhang Y. Roles of ifn-gamma in tumor progression and regression: A review. biomark Res (2020) 8:49. doi: 10.1186/s40364-020-00228-x
94. Villanueva L, Alvarez-Errico D, Esteller M. The contribution of epigenetics to cancer immunotherapy. Trends Immunol (2020) 41(8):676–91. doi: 10.1016/j.it.2020.06.002
95. Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther (2021) 6(1):362. doi: 10.1038/s41392-021-00670-9
96. Xiang X, Wang J, Lu D, Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct Target Ther (2021) 6(1):75. doi: 10.1038/s41392-021-00484-9
Keywords: cancer, immune checkpoint blockade, PD-1, CTLA-4, response, resistance
Citation: Lee J and Kim EH (2023) Mechanisms underlying response and resistance to immune checkpoint blockade in cancer immunotherapy. Front. Oncol. 13:1233376. doi: 10.3389/fonc.2023.1233376
Received: 02 June 2023; Accepted: 10 July 2023;
Published: 28 July 2023.
Edited by:
Huiyan Zeng, Beth Israel Deaconess Medical Center and Harvard Medical School, United StatesReviewed by:
Hiroshi Yano, Cornell University, United StatesCopyright © 2023 Lee and Kim. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eui Ho Kim, ZXVpaG8ua2ltQGlwLWtvcmVhLm9yZw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.