
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Oncol. , 16 August 2023
Sec. Hematologic Malignancies
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1231601
 Julia Böck1
Julia Böck1 Katja Maurus1Elena Gerhard-Hartmann1Stephanie Brändlein1Katrin S. Kurz2German Ott2Ioannis Anagnostopoulos1Andreas Rosenwald1
Katja Maurus1Elena Gerhard-Hartmann1Stephanie Brändlein1Katrin S. Kurz2German Ott2Ioannis Anagnostopoulos1Andreas Rosenwald1 Alberto Zamò1*
Alberto Zamò1*Diagnosing any of the more than 30 types of T-cell lymphomas is considered a challenging task for many pathologists and currently requires morphological expertise as well as the integration of clinical data, immunophenotype, flow cytometry and clonality analyses. Even considering all available information, some margin of doubt might remain using the current diagnostic procedures. In recent times, the genetic landscape of most T-cell lymphomas has been elucidated, showing a number of diagnostically relevant mutations. In addition, recent data indicate that some of these genetic alterations might bear prognostic and predictive value. Extensive genetic analyses, such as whole exome or large panel sequencing are still expensive and time consuming, therefore limiting their application in routine diagnostic. We therefore devoted our effort to develop a lean approach for genetic analysis of T-cell lymphomas, focusing on maximum efficiency rather than exhaustively covering all possible targets. Here we report the results generated with our small amplicon-based panel that could be used routinely on paraffin-embedded and even decalcified samples, on a single sample basis in parallel with other NGS-panels used in our routine diagnostic lab, in a relatively short time and with limited costs. We tested 128 available samples from two German reference centers as part of our routine work up (among which 116 T-cell lymphomas), which is the largest routine diagnostic series reported to date. Our results showed that this assay had a very high rate of technical success (97%) and could detect mutations in the majority (79%) of tested T-cell lymphoma samples.
T- and NK-cell lymphomas are relatively rare and usually aggressive neoplastic diseases, which notoriously pose diagnostic difficulties due to their rarity, but also due to their intrinsic morphological subtlety and partial lack of strongly specific diagnostic markers. A diagnosis of mature T- or NK-cell lymphoma (actually comprising more than 30 diagnostic categories in the 5th edition of WHO classification as well as in the International Consensus Classification - ICC) (1, 2) requires a combination of clinical data, morphological alterations, immunophenotypical features and clonality testing, none of which is singularly diagnostic. The molecular detection of clonal T-cell populations by means of consensus primer PCR amplification and capillary electrophoresis of the TCR gamma (TCRG) or beta (TCRB) rearranged gene segments has become a determining factor for the diagnosis of difficult cases (3). However, it is commonly negative in NK-derived lymphomas and can be positive in a number of reactive conditions as well as in many normal individuals.
Due to the recently discovered molecular alterations in T-cell lymphomas, targeted next-generation sequencing (NGS) of commonly mutated genes seems an appealing complementary approach for the detection of abnormal T-cell populations (4–6), which might also help in the precise subclassification and provide therapeutically relevant information.
Only few reports describe real-life experience with targeted NGS panels (7–10), and only one focuses specifically on an approach for T-cell lymphomas (10). Focusing on a purely diagnostic approach, we have designed and tested a custom-made targeted NGS panel for T-/NK-cell neoplasias with the following features in mind: a) the mutational pattern should provide support for differential diagnosis within a reference setting as well as answer clinical questions; b) the test should be easily and rapidly implementable within our common NGS routine diagnostic pipeline; c) the panel should be relatively small (≤ 30kb), so that it could be run on a single case basis (i.e. not requiring batch runs), and might allow sequencing in a pool with different NGS panels of other diagnostic cases of the day-to-day routine; d) the test should work on difficult samples (formalin-fixed paraffin-embedded (FFPE) material, low input, decalcified bone marrow samples, poor DNA quality). Here we report on the design and performance of our panel, tested on 128 prospective cases from the clinical routine of two major German reference centers for hematopathology (Würzburg and Stuttgart).
The majority of cases was tested immediately after completion of standard diagnostic procedures (morphology, immunohistochemistry and in a subset of cases clonality studies), in 10 cases the sequencing was performed following a specific clinical request (mutation status of TET2, STAT3, STAT5B, IDH2, DNMT3A or RHOA). DNA was either previously available from clonality testing or newly extracted. Genomic DNA was isolated from FFPE tissue using the Maxwell RSC Blood DNA Kit (Promega GmbH, Walldorf, Germany). Briefly, samples were pre-treated with a THG1-Thioglycerol/incubation buffer mix for 10 minutes at 80°C followed by an incubation-step overnight with proteinase K at 65°C. Extraction was performed on the Maxwell® RSC 48 instrument. DNA was quantified by quantitative PCR using the TaqMan RNase P Detection Reagents Kit (Life Technologies, Darmstadt, Germany).
T-cell receptor rearrangement assays were performed according to the Euroclonality/BIOMED-2 protocol (3). Ethical approval was granted from the local ethical committee for the use of leftover samples. Samples were reviewed from expert hematopathologists to determine tumor-cell content/cell population of interest. Criteria for inclusion were: a) diagnosis of T-cell lymphoma according to the WHO classification (revised 4th edition) or T-cell proliferation suspicious for T-cell lymphoma; b) tumor cell content/cell population of interest ≥ 10% and; c) DNA concentration of ≥ 1ng/µl (with total DNA quantity ≥ 10ng).
In total, we included 128 cases consisting of 60 T-cell lymphomas with T follicular helper (T)FH-cell phenotype (TCL-TFH; including both angioimmunoblastic T-cell lymphomas (AITL) and nodal peripheral T-cell lymphoma with TFH-phenotype (NPTCL-TFH)); 21 peripheral T-cell lymphomas, not otherwise specified (PTCL, NOS); a group of 7 intestinal T-cell neoplasias (ITCN), including 2 enteropathy-associated T-cell lymphomas (EATL) and 3 monomorphic epitheliotropic intestinal T-cell lymphomas (MEITL) as well as 1 intestinal T-cell lymphoma, NOS (ITCL, NOS) and 1 indolent T-cell lymphoma of the gastrointestinal tract (iTCLGIT); a group of 12 anaplastic large-cell lymphomas (ALCL, of those one breast-implant associated (BIA-ALCL), 2 ALK-positive, the remaining ALK-negative); a group of 8 cutaneous T-cell neoplasias (CTCN), including 3 Mycosis fungoides samples (MF), 3 Sezary syndrome samples (SS) and 2 subcutaneous panniculitis-like T-cell lymphomas (SCPL-TCL); 3 extranodal NK/T-cell lymphomas (ENNK/TCL); and a group of 5 other T-cell lymphoma cases (other T); we also included a group of 8 lymphoma cases which were finally classified as B-cell lymphoma (other non-T, thereof 2 classical Hodgkin lymphomas (C-HL), 1 nodular lymphocyte predominant Hodgkin lymphoma (NLPHL), 1 diffuse large B-cell lymphoma, not otherwise specified (DLBCL, NOS), 1 EBV-positive DLBCL and 2 marginal zone lymphomas (MZL)); and a group of 5 cases, which were eventually classified as reactive (including 1 infectious mononucleosis case (IM)).
Panel design was conducted with the Ion AmpliSeq Designer software (v7.48). A full list of covered genes with the respective genomic coordinates is shown in Supplementary Table S1. Libraries were prepared using the Ion AmpliSeq Library Kit 2.0 according to the manufacturer’s recommendations and quantified via qPCR using the Ion Library TaqMan Quantitation Kit (Life Technologies). Afterwards, templating and enrichment was performed with the Ion OneTouch 2 and Ion OneTouch ES automated systems or the Ion Chef System, followed by sequencing on the Ion GeneStudio S5 Plus System. Data were analyzed using Torrent Suite Software (v5.16) and Ion Reporter Software (v5.18) (Thermo Fisher Scientific, Darmstadt, Germany).
Analyzed data were filtered for somatic exonic non-synonymous variants, small insertions/deletions and splice site variants in the flanking regions with an allele frequency ≥ 2%. To remove technical artefacts (e.g., PCR artefacts, fixation artefacts), all variants with an allele frequency lower than 10% and a coverage lower than 200x were visualized by Integrative Genomics Viewer (v2.15.4) (11). Variants with a minor allele frequency of ≥ 0.2% in the general population listed in dbSNP database, ExAC database and gnomAD database were also excluded from further analysis. Variants were classified in likely oncogenic and oncogenic variants or variants of unknown significance (VUS) using the current literature and various databases including cBioPortal, ClinVar, OncoKB, CKB and COSMIC. Nonsense and frameshift mutations in tumor suppressor genes, that were not molecularly characterized yet, were classified as loss of function mutations and therefore as likely oncogenic variants. Results are shown in Supplementary Table S2.
We have analyzed a cohort of 128 cases from two reference centers, comprising 116 T-cell lymphoma samples and 12 non T-cell lymphoma samples. After DNA quantification, all 128 samples were deemed appropriate for further processing. Quantity of all prepared libraries was sufficient for sequencing. Over all samples mean coverage was 4856x, mean of bases above 500 reads was 94% and mean of reads on target was 99% (Supplementary Table S4). Defining a threshold for quality control after sequencing, 99% (127/128) of cases showed ≥ 95% reads on target sequence, 91% (116/128) of cases ≥ 90% bases with a coverage above 500 reads and 97% (124/128) of cases a mean coverage of ≥ 1000 reads (Figure 1A). Only four samples (3%) failed two of these three quality control criteria. However, in three of those cases mutations with a limit of detection of ≥ 2% allele frequency were reliably detectable, so that the overall quality was individually and finally classified as sufficient.
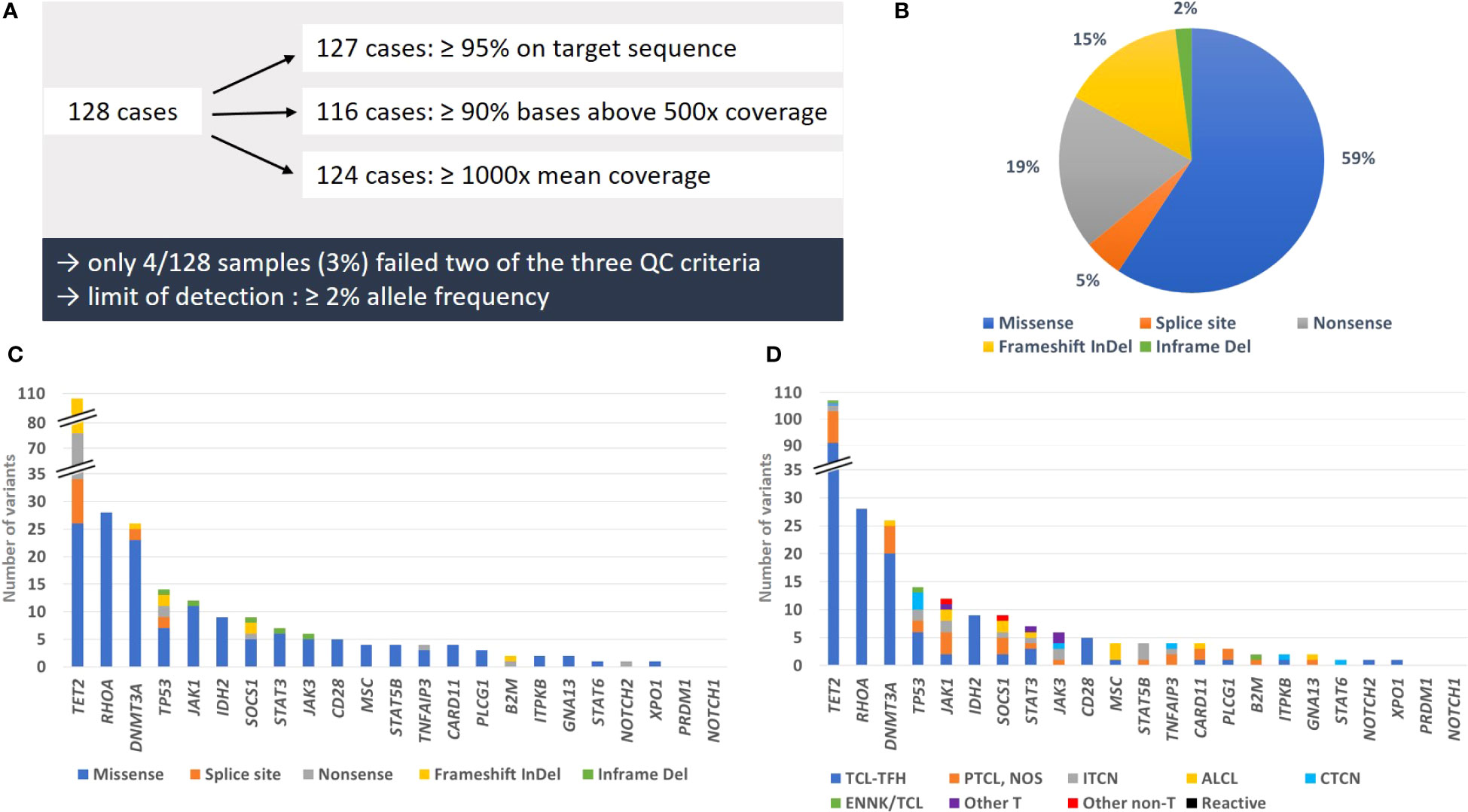
Figure 1 Summary of quality parameters, types of mutations and mutation frequency. (A) Overview of cases matching the different QC criteria. (B) Proportion of mutation types detected in the studied cohort. (C) Number of genetic alterations per mutation type for each analyzed gene. (D) Number of variants per T-cell lymphoma subtype and control groups for each analyzed gene. All likely oncogenic/oncogenic variants and variants of unknown significance we detected in this study were counted for illustrations (B–D).
Considering the whole collective, we detected 249 somatic mutations, which could be classified as likely oncogenic and oncogenic variants or VUS. Counting all variants per gene as one alteration, we found in 40 samples (31%) only one altered gene, whereas 29 (23%) showed two, 16 (13%) three, 5 (4%) four, 2 (2%) five, 2 (2%) six and 1 (1%) seven altered genes. Overall, 95/128 of the total tested samples (74%) and 92/116 T-cell lymphoma samples (79%) had at least one relevant mutation (Figure 2). In 23 out of 116 T-cell lymphoma samples (20%), we could not detect any genetic alteration.
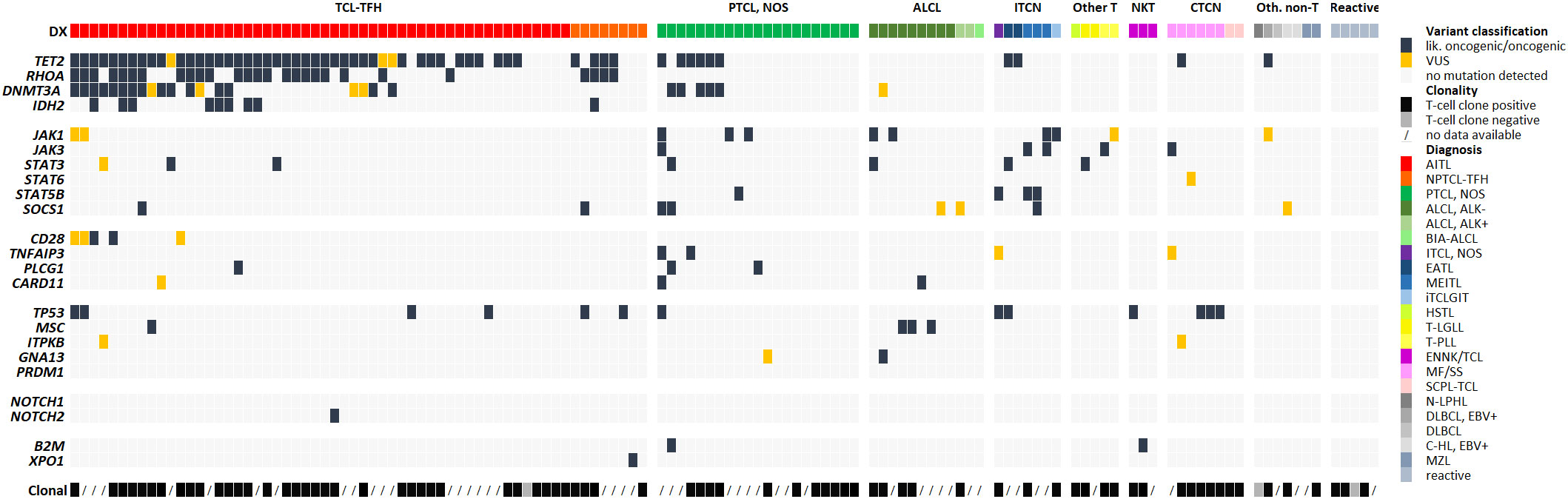
Figure 2 Genetic alterations detected via NGS-based panel sequencing and clonality results. Each row represents one of the genes included in the panel and each column one analyzed sample. If several variants were present in a gene, the most damaging mutation is illustrated and counted as one alteration. Cases are sorted by diagnosis and within the group per frequency of altered genes. Available clonality results are shown in the last row.
The most frequently mutated genes were TET2 (58/128 total cases, 45%; and 57/116 T-cell lymphoma cases, 49%), RHOA (24/116, 21%), DNMT3A (20/116, 17%), TP53 (13/116, 11%) and JAK1 (11/128 total cases, 9%; and 10/116 T-cell lymphoma cases, 9%) (Figure 1C). All affected genes were exclusively mutated in T-cell lymphoma, except for TET2 and JAK1. The largest proportion of mutations were missense mutations (59%), followed by nonsense mutations (19%), frameshift mutations (15%), splice site mutations (5%) and inframe deletions (2%) (Figure 1B). Excluding VUS, the overall picture changed only slightly, with missense mutations now constituting 54% of all variants, followed by nonsense mutations (22%), frameshift mutations (17%), splice site mutations (6%) and inframe deletions (1%) (Supplementary Tables S2, S3). Solely missense mutations were detected in RHOA, hotspot p.G17V and in IDH2, hotspot p.R172K/G/S, typical for AITL, and in MSC, hotspot p.E116K, typical for ALCL with DUSP22-translocation (Supplementary Table S2). The tumor suppressor genes TET2, TP53 and SOCS1 showed in addition to missense mutations also different types of truncating mutations, including frameshift, nonsense and splice site mutations (Figure 1C). All other genes displayed mostly missense mutations.
Of all detected mutations, 85% (212/249) were classified as likely oncogenic and oncogenic variants and only 15% (37/249) as VUS. About one third of all functionally relevant mutations were truncating mutations in tumor suppressor genes (95/212, 45%) or classical hotspot mutations (40/212, 19%). VUS were detected in 12/23 genes, most frequently in TET2 (11/107 variants), DNMTA3 (6/25 variants) and JAK1 (5/12 variants). They were almost exclusively missense mutations, with four exceptions in JAK1, JAK3, DNMT3A and SOCS1, which had inframe deletions and one frameshift mutation (Supplementary Table S2).
Data evaluation showed that the panel is very well suited for diagnostics of T-cell lymphomas with TFH phenotype (AITL and nodal PTCL with TFH-phenotype, currently under the common umbrella of TFH-cell lymphomas both in the 5th edition of the WHO classification and in the ICC) (1, 2), with 48/53 (91%) cases showing at least one mutation, most frequently of TET2 (45/53, 85%), RHOA (23/53, 47%), DNMT3A (19/53, 36%) and IDH2 (8/53, 15%) (Figure 1D). In PTCL, NOS the detection rate was only moderate, with 12/21 cases (57%) showing at least one mutation, whereas performance of the panel was high for ALCL (overall 9/12 positive; 75%). Additionally, we obtained a high detection rate for MEITL (3/3 cases), EATL (2/2 cases) and MF/SS (6/6 cases), but the number of tested patients was too low to draw a definitive conclusion (Figure 2).
Data about clonally rearranged T-cell receptor genes was available in 78 of 128 (61%) cases from routine testing. We also correlated our sequencing results with standard clonality detection (Figure 2; Supplementary Table S5). Clonally rearranged T-cell receptor genes were detected in 75 out of 78 (96%) tested samples. Of these positive samples, 57 (76%) had at least one mutation, while no genetic alteration was detected in the negative cases. However, 18 cases (24%) with no mutation showed rearranged T-cell-receptor gene fragments. 38 of the 50 non-tested samples (75%) comprised at least one mutation whereas 12 cases (24%) showed no genetic alteration at all. However, the mutation detection rate in rearranged positive samples and non-tested cases was nearly identical (76% compared to 75%).
Mature T-cell lymphomas include more than 30 entities in the 5th edition of the WHO classification as well as in the ICC (1, 2), with the most frequent group AITL (now denominated nodal TFH-cell lymphoma, angioimmunoblastic type or TFH- lymphoma, angioimmunoblastic type) comprising about 1-2% of total lymphoma diagnoses. Practically all T-/NK-cell lymphomas are therefore rare entities, a fact that, in addition to a sometimes challenging morphology, poses diagnostic difficulties in the differential diagnosis with reactive/benign processes as well as with B-cell lymphomas or lymphoproliferations, and occasionally also with Hodgkin lymphomas. A diagnosis of T-/NK-cell lymphoma requires the integration of clinical data, morphology, immunophenotype and molecular information, with no single element per se being completely diagnostic. T-cell clonality detection through multiplex PCR of the rearranged T-cell-receptor gene fragments has proven a valuable tool to facilitate a diagnostic decision that usually bears heavy therapeutic consequences. The availability of a nearly complete genetic landscape of mature T-cell lymphomas (12–18) has recently allowed a mutation-based approach to diagnosing these malignancies. The first two notable reports in this sense are the results of a Swiss group (9) and an American group (8), which independently reported their routine experience with the application of targeted panel sequencing in lymphoma diagnostics. Pillonel et al. reported their experience with 80 highly selected, mixed lymphoma cases (including 17 T- and NK-cell neoplasias) collected over three years. Panel sequencing was requested either by the treating physician or by the reference pathologist. Their custom panel included 68 genes and they used Thermo Fisher technology (S5 system) with a sequencing success rate of 98% (78/80 samples with complete panel coverage) (9). The approach by Davis et al. was diametrically opposite, with a large-scale approach sequencing of 518 patients/598 samples (corresponding to 55% of all lymphoma diagnoses during one year) using a commercially available targeted panel (Illumina TruSight Lymphoma). The panel contained 40 lymphoma-related genes and was run on an Illumina MiSeq instrument; the authors report a sequencing success (defined as the detection of a disease-associated variant, of a variant of unknown significance or no variants) of 95% for intramural cases and 77% for extramural cases. Their series included 40 T-cell neoplasms (miscellaneous) where they identified 18 disease-associated variants and 8 variants of unknown significance (8).
More recently, a French group reported on their experience in routine use of a custom capture-based T-cell-oriented panel including 69 genes, which was sequenced using Illumina technology (MiseqDX or NextSeq 550) (10). In contrast to the first two papers mentioned, the authors focused on T-cell lymphomas (82 cases) and reactive T-cell infiltrates (25 cases), comparing the results of panel sequencing with conventional clonality analysis. They detected mutations in 79/82 (96%) T-cell lymphomas, including four cases, which were non-amplifiable by conventional PCR, but none in “reactive T-cell infiltrates” (not further specified). However, the latter showed a clonal population in 13/25 (52%) cases, which argues for a higher specificity of panel sequencing in comparison to T-cell clonality studies.
With special focus on T-cell lymphomas, our approach is more comparable with the work of the French group, but we analyzed a larger series of samples in a real-world environment; to our knowledge, we tested the largest diagnostic series of T-cell lymphomas so far (116 cases). Compared to previous publications, we have focused on developing a NGS-based test providing diagnostically relevant and robust information supporting a diagnosis of T-cell neoplasia within a clinically acceptable time-frame and at a relatively low cost. In our opinion, the panel could be easily implemented in most molecular pathology laboratories that are using a similar NGS pipeline. Our panel design proposes an innovative approach, because: a) it assists in answering diagnostically, prognostically or therapeutically relevant questions, based on our experience within the two reference centers; b) it comprises a small number of genes (23, about 29kb), mostly focusing on hotspots and genes mutated with high frequency and non-redundantly in T-/NK-cell lymphomas; c) it follows a mix-and-match approach, with the intent of maximum flexibility, because it can be combined with other diagnostic panels on an everyday basis, also single samples can be run; d) our protocol has a short turn-around-time (ten working days to final report) thanks to the use of a PCR-based protocol and implementation on an Ion S5 system, as well as relatively low costs. In order to optimize the size and efficiency of our panel, we did not take copy number variations (CNV) into consideration.
As detailed in the results section, we had a very high sequencing success rate (97%), despite using samples from different external sources and decalcified samples. This high rate is probably due to the small size of the panel, which provides a high level of coverage for each region and the power of amplicon-based library preparation of samples with poor DNA quality and quantity. In fact, only four samples (3%) failed two of the three stringent quality control criteria, but in three cases relevant mutations were reliably detectable and were therefore deemed as valid.
We found one or more likely oncogenic or oncogenic variants in 89 samples and 6 samples with only VUS, which is higher than the number reported by Pillonel et al. and Davis et al., but lower than the mutation rate declared by Syrykh et al. However, the French group has used capture-based library generation and a different bioinformatics pipeline for variant calling, which might explain the differences with both our results and those of previous studies. A major source of variance is of course panel design itself, which in our case was focused on the inclusion of genes known to be mutated mostly in T-cell lymphomas, whereas the panels reported in the other publications contained also genes known to be mutated in both B- and T-cell neoplasias. Figure 3 visualizes the overlap between our panel and the one designed by the French group. The comparison of both panels shows that most genes missing in our panel are indeed B-cell lymphoma associated genes, being diagnostically and biologically less relevant in T-cell lymphomas. We deliberately chose not to include large tumor-suppressor genes (except for TET2), especially if they rarely occurred as isolated mutations, since they would dramatically increase the panel size. On the other hand, we included part of genes which are frequently mutated in Hodgkin lymphomas (STAT6, B2M), since in our experience the differential diagnosis between a T-cell lymphoma with Hodgkin- and Reed-Sternberg-like cells and true classical Hodgkin lymphoma can be challenging and therapeutically relevant. The efficiency of our panel is shown in the percentage of mutated genes (21/23; 91%), compared with 39/69 (57%) in the panel designed by the French authors. This is also reflected in the high number of detected variants that could be classified as biologically relevant mutations (85%, 212/249).
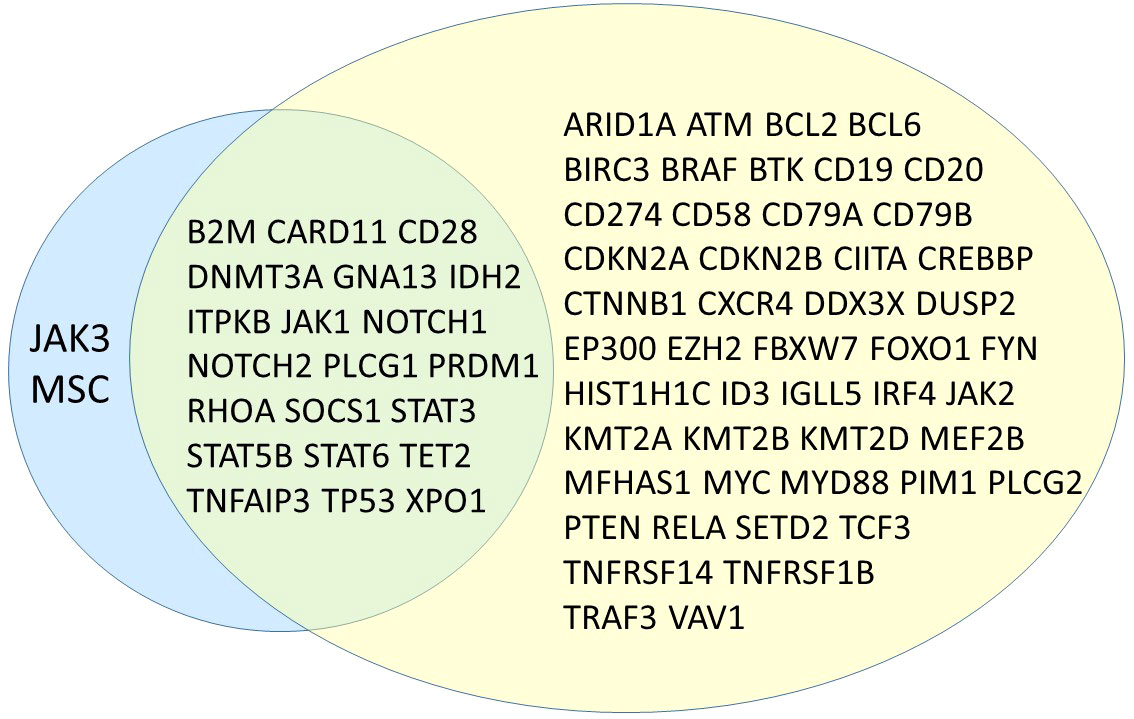
Figure 3 Venn diagram showing overlapping genes between the proposed panel in this study (left, blue) and the one presented by Syrykh et al. (right, yellow) (10).
As already shown in the results section, our success in detecting mutations was entity-dependent, with a very high efficiency for the TFH-cell lymphoma group (nearly 91% detection rate). Otherwise, we had only a limited success with PTCL, NOS cases (about 57% detection rate), due to the more complex genetic landscape that is not reflected in our simple design. Our finding confirms previously described data in the literature, including that RHOA and IDH2 mutations appear to be highly specific for TFH-cell lymphomas, since we found no mutations in any of the 21 tested PTCL, NOS or in any other entity).
Interestingly, sometimes the panel results suggested new approaches to old problems. For instance, ALCL showed no mutations in TET2 and only one (1/12, 8%) mutation in DNMT3A, a finding that might help in the differential diagnosis between this entity and cases of PTCL, NOS, where TET2 mutations were relatively frequent (6/21, 29%). In some cases, genetic data might be a surrogate for other analyses. As an example, it was previously published, that mutations in MSC are specific for ALCL cases bearing a DUSP22 translocation (19); although not perfectly sensitive (only 2/3 cases with the translocation showed a MSC mutation) the detection of this mutation might warrant the performance of an appropriate FISH test.
The correlation of our sequencing data with standard clonality detection showed that the latter still has a superior sensitivity (as expected from the biology of T-cells). We detected clonally rearranged T-cell receptor genes in 75/78 (96%) tested samples, among positive cases, 57 (73%) had at least one mutation, while 18 cases with no genetic alteration showed rearranged T-cell-receptor gene fragments. GeneScan analysis confirms itself as a very sensitive technique, which will probably continue to be performed; however, its relative lack of specificity (being positive also in some non-neoplastic conditions) supports the use of a more specific molecular technology to corroborate a lymphoma diagnosis.
We analyzed our cases without selecting for specific entities, but we might have a selection bias since we receive external cases for a second opinion. In most cases, the panel results were only of confirmatory nature, adding further objective data to the results of morphology and immunophenotyping. In some cases, however, the results helped in the differential diagnosis or confirmed the neoplastic nature of a minor component, which was suspected to be neoplastic. Emblematic cases, for example, were cases of T-cell lymphomas in which the associated EBV-positive B-cell proliferation was exceptionally dominant, obscuring the neoplastic T-cell component and making it difficult to assess both morphologically and immunophenotypically; in this case the combination of clonality studies and panel sequencing eventually provided evidence of T-cell neoplasia (also confirmed in a later biopsy, not included in this series). In one case of NLPHL with slightly atypical and expanded intranodular T-cells, the detection of a classic RHOA and TET2 mutation provided definitive evidence of an associated T-cell lymphoma (which could be classified as nodal T-cell lymphoma with TFH-phenotype). In other cases with only minimal lymphomatous infiltration or an atypical clinical presentation (e.g., isolated lymphadenopathy, young age) and considering the clinical consequence, we felt more comfortable having additional objective data to support the diagnosis of malignant lymphoma. In addition, we had several clinical requests for detection of mutations, particularly for TET2, STAT3, STAT5B, IDH2, DNMT3A, RHOA (10 cases), as cases with TET2 mutations may correlate with response to therapy with azacitidine (20–23).
In the context of low variant allele frequency in genes which are strongly correlated to clonal hematopoiesis (CH) like TET2 or DNMT3A it is difficult (if not impossible) to certainly attribute the mutation to the lymphoma cells; in this case, we normally add a comment highlighting this difficulty and recommending further studies (on peripheral blood or bone marrow). In our opinion, it is important to highlight that no single element is per se completely diagnostic, and in case of doubt a re-biopsy might be discussed. The relationship between CH and T-cell lymphomas is biologically relevant and warrants further investigation, since both conditions probably derive from a common mutated progenitor and significant clinical correlations start to emerge (24).
In conclusion, we report our experience with a small and efficient targeted NGS panel that could be a powerful tool for the diagnosis of selected cases or a useful diagnostic aid for specific predictive purposes. Our panel could be easily reproduced and applied in routine molecular diagnostics at a single samples level and provide reliable results in a relatively short time. Moreover, its application could be a useful complement to classical clonality detection.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: NCBI, PRJNA992223.
The studies involving humans were approved by Local ethics committee University of Würzburg. The studies were conducted in accordance with the local legislation and institutional requirements. The ethics committee/institutional review board waived the requirement of written informed consent for participation from the participants or the participants’ legal guardians/next of kin because use of left-over and anonymized material.
AZ, AR and JB contributed to conception and design of the study. AZ, AR, EG-H, IA, GO, and KSK reviewed the samples. JB and AZ designed the panel, interpreted the data and wrote the first draft of the manuscript. JB and KM supervised the panel implementation and the lab work. JB analyzed the mutational data and classified the variants. JB, AZ, SB, and KM analyzed the clonality data. JB submitted the final draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version. All authors contributed to the article and approved the submitted version.
This publication was supported by the Open Access Publication Fund of the University of Würzburg.
We thank the entire team of the molecular pathology lab for their support in the realization of this project, especially Dr. Julia Doll and Dr. Qunpei Yang.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1231601/full#supplementary-material
1. Alaggio R, Amador C, Anagnostopoulos I, Attygalle AD, Araujo IBO, Berti E, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: lymphoid neoplasms. Leukemia (2022) 36(7):1720–48. doi: 10.1038/s41375-022-01620-2
2. Campo E, Jaffe ES, Cook JR, Quintanilla-Martinez L, Swerdlow SH, Anderson KC, et al. The international consensus classification of mature lymphoid neoplasms: a report from the clinical advisory committee. Blood (2022) 140(11):1229–53. doi: 10.1182/blood.2022015851
3. van Dongen JJ, Langerak AW, Bruggemann M, Evans PA, Hummel M, Lavender FL, et al. Design and standardization of PCR primers and protocols for detection of clonal immunoglobulin and T-cell receptor gene recombinations in suspect lymphoproliferations: report of the BIOMED-2 Concerted Action BMH4-CT98-3936. Leukemia (2003) 17(12):2257–317. doi: 10.1038/sj.leu.2403202
4. Heimann P, Dewispelaere L. Indications of next-generation sequencing in non-Hodgkin’s lymphoma. Curr Opin Oncol (2020) 32(5):391–7. doi: 10.1097/CCO.0000000000000666
5. van Dongen JJ, Langerak AW, Bruggemann M, Evans PA, Hummel M, Lavender FL, et al. The need for a consensus next-generation sequencing panel for mature lymphoid Malignancies. Hemasphere (2019) 3(1):e169. doi: 10.1097/HS9.0000000000000169
6. Vega F, Amador C, Chadburn A, Hsi ED, Slack G, Medeiros LJ, et al. Genetic profiling and biomarkers in peripheral T-cell lymphomas: current role in the diagnostic work-up. Mod Pathol (2022) 35(3):306–18. doi: 10.1038/s41379-021-00937-0
7. Bommier C, Mauduit C, Fontaine J, Bourbon E, Sujobert P, Huet S, et al. Real-life targeted next-generation sequencing for lymphoma diagnosis over 1 year from the French Lymphoma Network. Br J Haematol (2021) 193(6):1110–22. doi: 10.1111/bjh.17395
8. Davis AR, Stone SL, Oran AR, Sussman RT, Bhattacharyya S, Morrissette JJD, et al. Targeted massively parallel sequencing of mature lymphoid neoplasms: assessment of empirical application and diagnostic utility in routine clinical practice. Mod Pathol (2021) 34(5):904–21. doi: 10.1038/s41379-020-00720-7
9. Pillonel V, Juskevicius D, Bihl M, Stenner F, Halter JP, Dirnhofer S, et al. Routine next generation sequencing of lymphoid Malignancies: clinical utility and challenges from a 3-Year practical experience. Leuk Lymphoma (2020) 61(11):2568–83. doi: 10.1080/10428194.2020.1786560
10. Syrykh C, Gorez P, Pericart S, Grand D, Escudie F, Cabarrou B, et al. Molecular diagnosis of T-cell lymphoma: a correlative study of PCR-based T-cell clonality assessment and targeted NGS. Blood Adv (2021) 5(22):4590–3. doi: 10.1182/bloodadvances.2021005249
11. Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform (2013) 14(2):178–92. doi: 10.1093/bib/bbs017
12. Watatani Y, Sato Y, Miyoshi H, Sakamoto K, Nishida K, Gion Y, et al. Molecular heterogeneity in peripheral T-cell lymphoma, not otherwise specified revealed by comprehensive genetic profiling. Leukemia (2019) 33(12):2867–83. doi: 10.1038/s41375-019-0473-1
13. Couronne L, Bastard C, Bernard OA. TET2 and DNMT3A mutations in human T-cell lymphoma. N Engl J Med (2012) 366(1):95–6. doi: 10.1056/NEJMc1111708
14. Palomero T, Couronne L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet (2014) 46(2):166–70. doi: 10.1038/ng.2873
15. Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet (2014) 46(2):171–5. doi: 10.1038/ng.2872
16. Crescenzo R, Abate F, Lasorsa E, Tabbo F, Gaudiano M, Chiesa N, et al. Convergent mutations and kinase fusions lead to oncogenic STAT3 activation in anaplastic large cell lymphoma. Cancer Cell (2015) 27(4):516–32. doi: 10.1016/j.ccell.2015.03.006
17. Schatz JH, Horwitz SM, Teruya-Feldstein J, Lunning MA, Viale A, Huberman K, et al. Targeted mutational profiling of peripheral T-cell lymphoma not otherwise specified highlights new mechanisms in a heterogeneous pathogenesis. Leukemia (2015) 29(1):237–41. doi: 10.1038/leu.2014.261
18. Vallois D, Dobay MP, Morin RD, Lemonnier F, Missiaglia E, Juilland M, et al. Activating mutations in genes related to TCR signaling in angioimmunoblastic and other follicular helper T-cell-derived lymphomas. Blood (2016) 128(11):1490–502. doi: 10.1182/blood-2016-02-698977
19. Luchtel RA, Zimmermann MT, Hu G, Dasari S, Jiang M, Oishi N, et al. Recurrent MSC (E116K) mutations in ALK-negative anaplastic large cell lymphoma. Blood (2019) 133(26):2776–89. doi: 10.1182/blood.2019000626
20. Ruan J, Moskowitz A, Mehta-Shah N, Sokol L, Chen Z, Kotlov N, et al. Multicenter Phase 2 Study of Oral azacitidine (CC-486) plus CHOP as initial treatment for peripheral T-cell lymphoma. Blood (2023) 141(18):2194–205. doi: 10.1182/blood.2022018254
21. Falchi L, Ma H, Klein S, Lue JK, Montanari F, Marchi E, et al. Combined oral 5-azacytidine and romidepsin are highly effective in patients with PTCL: a multicenter phase 2 study. Blood (2021) 137(16):2161–70. doi: 10.1182/blood.2020009004
22. O'Connor OA, Falchi L, Lue JK, Marchi E, Kinahan C, Sawas A, et al. Oral 5-azacytidine and romidepsin exhibit marked activity in patients with PTCL: a multicenter phase 1 study. Blood (2019) 134(17):1395–405. doi: 10.1182/blood.2019001285
23. Lemonnier F, Dupuis J, Sujobert P, Tournillhac O, Cheminant M, Sarkozy C, et al. Treatment with 5-azacytidine induces a sustained response in patients with angioimmunoblastic T-cell lymphoma. Blood (2018) 132(21):2305–9. doi: 10.1182/blood-2018-04-840538
Keywords: T-cell lymphoma, panel-sequencing, NGS, diagnostics, mutation, FFPE
Citation: Böck J, Maurus K, Gerhard-Hartmann E, Brändlein S, Kurz KS, Ott G, Anagnostopoulos I, Rosenwald A and Zamò A (2023) Targeted panel sequencing in the routine diagnosis of mature T- and NK-cell lymphomas: report of 128 cases from two German reference centers. Front. Oncol. 13:1231601. doi: 10.3389/fonc.2023.1231601
Received: 30 May 2023; Accepted: 31 July 2023;
Published: 16 August 2023.
Edited by:
Lorenzo Leoncini, University of Siena, ItalyReviewed by:
Giorgio Alberto Croci, University of Milan, ItalyCopyright © 2023 Böck, Maurus, Gerhard-Hartmann, Brändlein, Kurz, Ott, Anagnostopoulos, Rosenwald and Zamò. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alberto Zamò, YWxiZXJ0by56YW1vQHVuaS13dWVyemJ1cmcuZGU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.