
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol. , 06 September 2023
Sec. Cancer Molecular Targets and Therapeutics
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1222873
 Ying Zhang1*†
Ying Zhang1*† Xiaolu Wang2†Yuning Zhu1Chong Liang3Lijun Zhao4Qi Meng5
Xiaolu Wang2†Yuning Zhu1Chong Liang3Lijun Zhao4Qi Meng5 Jiani C. Yin5Yuqian Shi5
Jiani C. Yin5Yuqian Shi5 Fufeng Wang5Feng Qin6*Ji Xuan7*
Fufeng Wang5Feng Qin6*Ji Xuan7*Germline variations in the DNA polymerase genes, POLE and POLD1, can lead to a hereditary cancer syndrome that is characterized by frequent gastrointestinal polyposis and multiple primary malignant tumors. However, because of its rare occurrence, this disorder has not been extensively studied. In this report, we present the case of a 22-year-old female patient who had been diagnosed with gastrointestinal polyposis, breast fibroadenoma, multiple primary colorectal cancers, and glioblastoma (grade IV) within a span of 4 years. Next-generation sequencing analysis revealed a germline variant in POLD1 (c.1816C>A; p.L606M). In silico analysis using protein functional predicting software, including SIFT, Polyphen, GERP++, and CADD, further confirmed the pathogenicity of POLD1 p.L606M (classified as ACMG grade Class 4). In line with polymerase deficiency, both rectal cancer and glioblastoma tissues exhibited a high tumor mutation burden, with 16.9 muts/Mb and 347.1 muts/Mb, respectively. Interestingly, the patient has no family history of cancer, and gene examination of both parents confirms that this is a de novo germline variant. Therefore, molecular screening for POLD1 may be necessary for patients with such a cancer spectrum, regardless of their family history.
DNA replication is a fundamental process for all living organisms, and genes responsible for ensuring the accuracy of DNA replication play vital roles in maintaining health. POLD1 and POLE encode the catalytic subunits of the replicative polymerases, which are key mediators of DNA replication and repair (1–3). Germline variants in POLD1/POLE can lead to an autosomal dominant hereditary cancer syndrome that predisposes to colorectal polyposis and diverse types of cancer (4–7). Polymerase-associated polyposis syndrome is rare, but it exhibits high penetrance (5, 8–10). In addition, somatic mutations in POLD1/POLE have also been found in sporadic cancers, emphasizing the significance of DNA replication fidelity in preventing tumorigenesis (8). To date, a variety of germline and somatic POLD1/POLE alterations have been reported, with p.L424V and p.S478N being the most frequently observed germline variants for POLE and POLD1, respectively (5, 11). Here, we reported a patient who has experienced symptoms that resembled those of polymerase-associated polyposis syndrome, although with other distinct phenotypes. Genetic testing revealed a de novo germline p.L606M variant in POLD1.
In 2018, a 22-year-old female came to the hospital complaining of intermittent upper abdomen pain and occasional vomiting that had been ongoing for 8 months. An electronic gastroscope examination revealed dozens of polyps in the gastric fundus, body, and horn (Figures 1A, B). Subsequently, the patient underwent endoscopic mucosal resection (EMR) to remove all the polyps and experienced relief from her symptoms.
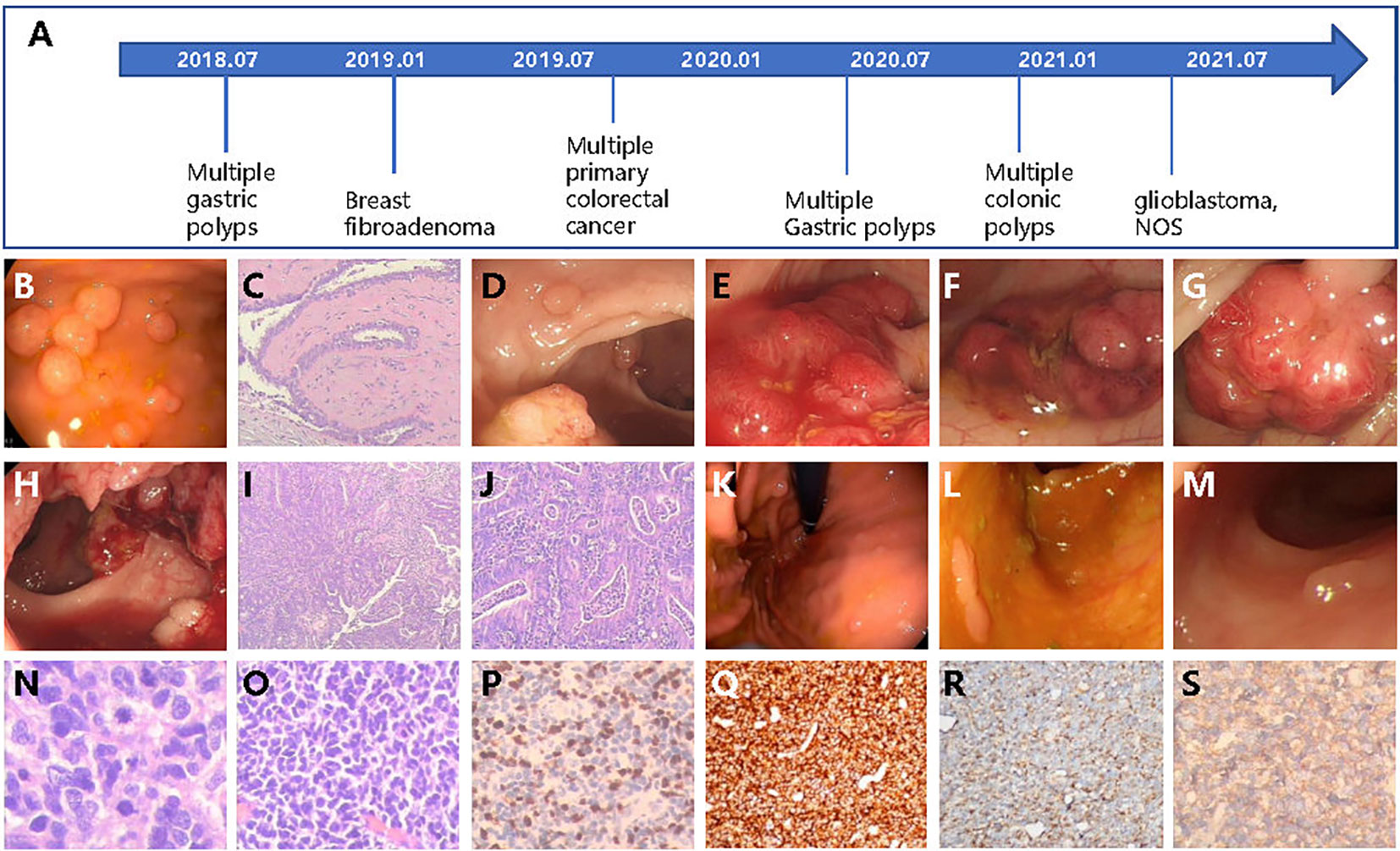
Figure 1 The patient case with multiple cancers or gastrointestinal polyps diagnosed over several years. (A) Diagnosis timeline of the patient. (B) The multiple gastric polyps found by gastroscope. (C) The HE staining of breast fibroadenoma tissue. (D–H) The colonoscopy found multiple lesions of cancer and colonic polyps. The HE stain of colonic (I) and rectal (J) cancer tissues. Newly detected gastric polyps (K) found by endoscope and colonic polyps (L, M) in 2020. (N, O) The HE staining of glioblastoma tissue. (P–S) The IHC examination of Ki-67, CD45, Nestin, and β-catenin.
In January 2019, at the age of 23, a lump was discovered in her right breast through routine health check. Subsequently, the lump was removed, and post-operative pathological testing revealed fibroadenoma (Figures 1A, C). In the same year, a routine physical examination found multiple space-occupying lesions in the rectum and colon. A further whole abdomen magnetic resonance imaging (MRI) revealed shadows in the rectosigmoid junction, mid-rectum, lower rectum, and proximal anus. Pathological biopsies from the colonoscopy indicated a diagnosis of multiple primary cancers of the colorectum with a stage of IIA (T3N0M0), accompanied by multiple colonic polyposis (Figure 1A). To be specific, the relative positions from the anus and the corresponding diagnosis are as follows: 2 cm and 27 cm, low-to-medium differentiated adenocarcinoma; 15 cm, high-grade intraepithelial neoplasia combining partial cancerization; 18 cm, tubulo-papillary structured adenocarcinoma; 30 cm, papillary adenoma; 60 cm, tubular adenoma combining partially mild-to-moderate differentiated atypical hyperplasia; and 65 cm, mixed polyposis (Figures 1D–J). The full-body positron emission tomography and computed tomography (PET-CT) as well as brain MRI examination showed no signs of distant metastasis. Therefore, the patient underwent radical surgery resection of the colorectal tumors. Owing to several high recurrent risk factors observed in this patient, such as low-to-medium differentiated cancer and the formation of cancer embolus and multiple primary cancers, the patient received adjuvant therapy (FOLFOX protocol) for 12 cycles from October 2019 to May 2020. Serum tumor markers were monitored after each cycle of adjuvant therapy. Throughout the entire treatment period, all serum markers, including CEA, AFP, CA50, and CA-199, remained within the normal ranges, except for CA125, which was abnormally high during the first three cycles of adjuvant therapy but returned to the normal range afterwards.
The patient has experienced multiple instances of primary colorectal cancers, breast fibroadenoma, and multiple gastric and colonic polyps at a young age, which suggest an inherited disease. Consequently, she was referred to genetic counseling, and an NGS test targeting tumor hot spots of 825 genes was carried out using white blood cell samples. However, no pathogenic or likely pathogenic germline variants were detected at this time.
In 2020, at the age of 24, the patient was re-admitted to the hospital twice in May and December due to the discovery of hundreds of gastric and colonic polyps ranging in size from 0.3 cm × 0.3 cm to 0.6 cm × 0.8 cm (Figure 1A). All polyps were removed through either EMR or argon plasma coagulation based on their sizes (Figures 1K–M).
In 2021, the patient returned to the hospital, reporting a headache that had persisted for the past week. An enhanced MRI examination of the head revealed an occupation in the right frontal lobe, accompanied by peritumoral brain edema and angiogenesis. Only 1 day after being admitted to the hospital, the patient experienced sudden unconsciousness and right pupil dilation. Hence, with the consent of the guardian, surgical resection of the intracranial tumor was performed. Incision of the frontal cortex showed a blood clot at 2 cm below the cortex, which was surrounded by off-white gelatinous tumor tissue that had a soft texture and clear boundary with the surrounding normal brain tissue, as well as abundant blood supply. The tumor extended to the frontal pole, midline, head of corpus callosum, and front of central anterior gyrus. Post-operative pathological diagnosis indicated glioblastoma, NOS (WHO grade IV, Figures 1A, N–S). Subsequently, the patient underwent 30 cycles of adjuvant radiotherapy, followed by oral intake of temozolomide and injection of anlotinib as intensive adjuvant therapy. Currently, the disease is under control, and the patient is in a relatively good condition.
Although the patient denied a family history of cancer and a previous genetic test failed to detect any detrimental germline variant, the presence of multiple gastrointestinal polyposis and a wide cancer spectrum still raised strong suspicions of hereditary factors. Consequently, the resected rectal tissue, glioblastoma tissue, and white blood cell samples were sent to a CLIA-certified and CAP-accredited central laboratory (Nanjing Geneseeq Technology Inc., Nanjing, China) for additional molecular testing. Next-generation sequencing (NGS) using a targeted panel covering 494 cancer-related genes (detailed gene list in Supplementary Table S1) revealed a heterozygous germline variant in the polymerase domain of POLD1 (c.1816C>A; p.L606M) (Figures 2A–C), which had been reported as somatic mutations in ultra-mutated brain, uterine, and endometrial tumors (3, 12–14), although no evidence in clinics of the germline POLD1 p.L606M has been published yet. The L606 residue in the polymerase domain is highly conserved throughout evolution (15). The yeast analog of this variant, pol3-L612M, confers a mutator phenotype with reduced DNA synthesis fidelity (16, 17), which was further confirmed by functional studies in human cells and mouse models (14, 18). Some of the most presumed somatic POLD1 driver mutations such as p.R689W also map to the polymerase domain (3, 19–21). The pathogenicity of the p.L606M variant was further confirmed by in silico analysis using protein functional predicting software, as evidenced by a SIFT score of 0 (deleterious), a PolyPhen score of 0.999 (damaging), a CADD score of 27.6, and a GERP++ RS of 3.62. A total of 304 and 13 somatic variants were detected in the brain tumor and rectal cancer tissues, respectively, with a corresponding tumor mutation burden of 347.1 and 16.9 muts/Mb. Mapping of the substitutions in the brain tumor to the mutational processes from the Catalog of Somatic Mutation in Cancer (COSMIC) database revealed high proportions of SBS15 (0.22), SBS20 (0.19), SDS 14 (0.11), and SBS6 (0.11). All these are predominantly associated with deficient mismatch repair (dMMR) signatures. Notably, SBS20 and SBS14 are also polymerase-associated signatures. Other polymerase-associated signatures, such as SBS 10a/b/c/d and SBS28, were detected at a proportion of less than 0.0001. Signature analysis of the rectal tissue showed high proportions of SBS24 (0.22) and SBS1 (0.18), which are not associated with either dMMR or polymerase signatures. Despite the signature analysis, the affected tissues were microsatellite stable (MSS) as determined by NGS. All these findings are consistent with the diagnosis of germline POLD1-induced polyposis syndrome.
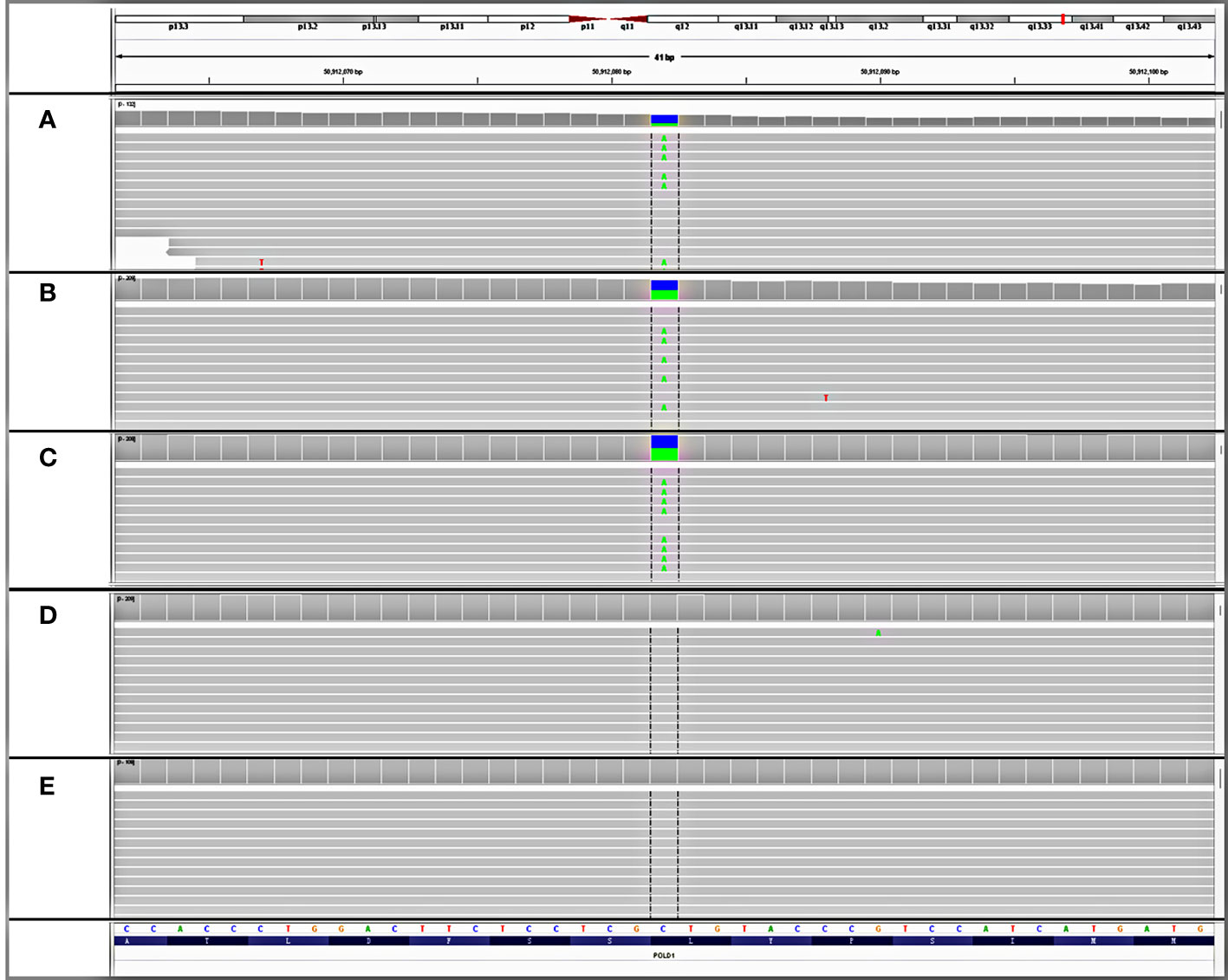
Figure 2 Detection of an unreported de novo POLD1 L606M germline variant. IGV snapshots of the heterozygous POLD1 variant tested in (A) patient’s rectal tissue, (B) patient’s glioblastoma tissue, (C) patient’s white blood cell, (D) father’s white blood cell, and (E) mother’s white blood cell.
To further investigate the origin of this germline variant, NGS tests were performed on the white blood cells of both parents. The POLD1 germline variant was not detected from either the paternal or maternal side (Figures 2D, E). Genetic fingerprint analysis confirmed the parenthood. These findings provide evidence that the POLD1 germline variant is de novo.
After the genetic diagnosis of polymerase-associated polyposis, the patient underwent surveillance and genetic counseling. Given that the disorder is associated with an increased risk of developing various types of cancers, such as colorectal breast, duodenal, ovarian, and central nervous system tumors, the patient was advised to undergo regular colonoscopies and screenings for other cancers.
Polymerase-associated polyposis syndrome is an autosomal dominant cancer syndrome caused by pathogenic germline variants in POLE and POLD1, which predisposes to multiple types of cancer, including CRC, duodenal cancer, and endometrial cancer (22). As this disease is very rare, only a limited number of clinical studies have been reported. In one study, a total of 2,813 unrelated probands were sequenced for POLE and POLD1, and it was found that polymerase-associated syndrome accounts for 0.1%–0.4% of familial cancers (7). The study also identified five POLD1 variants in the exonuclease domain, all of which were predicted to be neutral (Figure 3). In 2016, Bellido and colleagues reviewed a total number of 529 familial CRC patients and found that among germline POLD1 variant carriers, the incident rates of CRC, endometrial cancer, breast cancer, and brain tumor were 59.1%, 57.1%, 14.3%, and 4.5%, respectively. The mean ages at diagnosis for CRC and endometrial cancer were 35.9 and 51.4 years old, respectively (6). In another study, 132 patients harboring pathogenic variants in POLE/POLD1 were analyzed, including 105 with POLE variants and 27 with POLD1 variants. Among the POLD1 carriers, the incidence rates of CRC, endometrial cancer, breast cancer, and brain tumor were 44.4%, 52.9%, 23.5%, and 3.7%, respectively, with a median age at diagnosis of 41, 52, 62, and 26 years old, respectively (22) (Table 1).
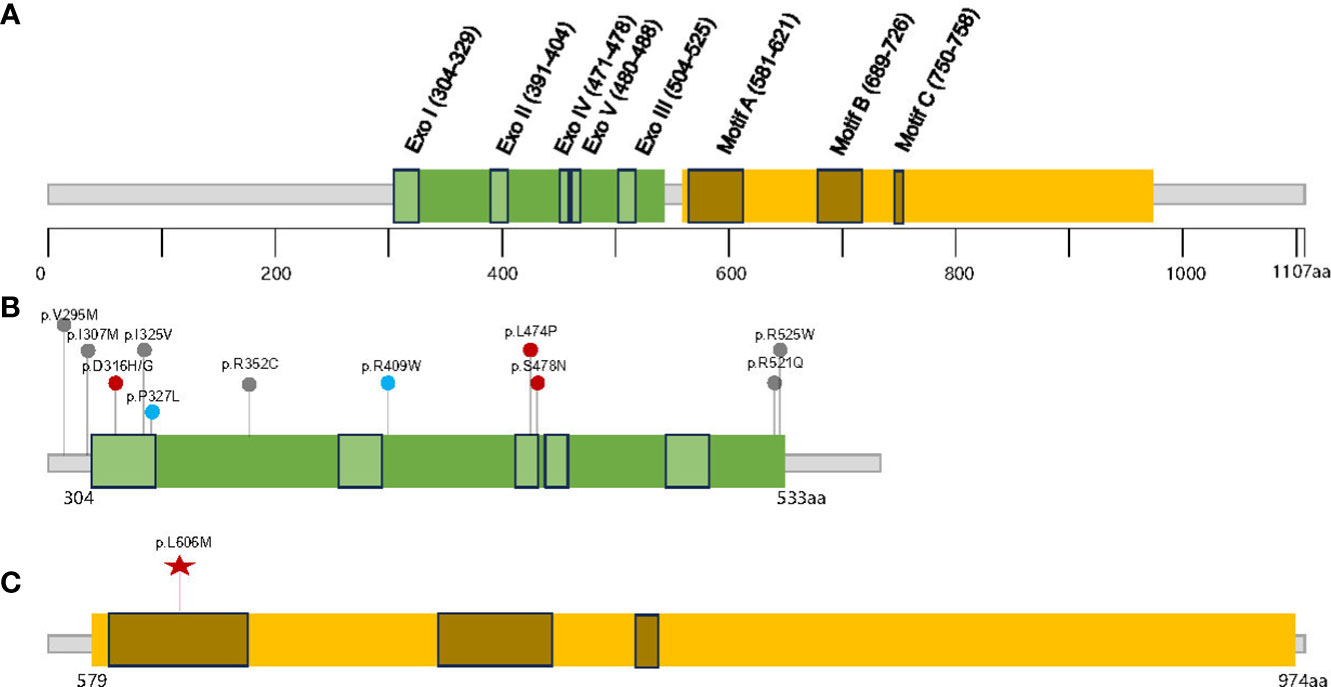
Figure 3 The distribution of the germline POLD1 variants in literature and this study. (A) Schematic domain structure and motif sites in human p125/POLD1 (14), The exonuclease and polymerase domains are shown in (B, C), respectively. Red points indicate clearly pathogenic variants, blue points indicate potentially pathogenic variants, and gray points indicate variants with unknown significance; the red star indicates variant detected in this study.
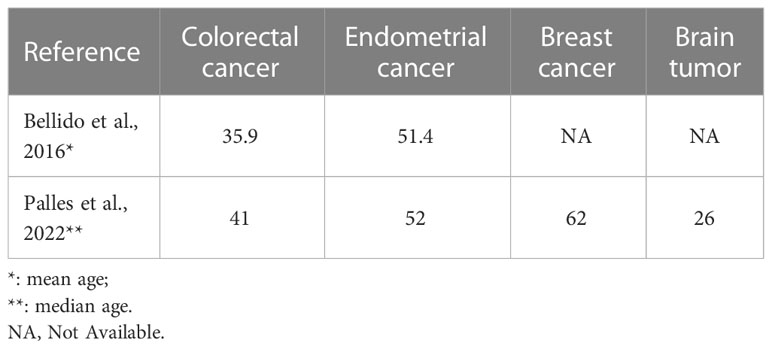
Table 1 The mean or median ages of diagnosis of multiple cancer types in germline POLD1 variant carriers.
Compared to previous reports, our case has displayed some unique characteristics. Firstly, the patient was diagnosed with breast fibroadenoma, CRC, and glioblastoma in her early twenties, which is earlier than what has been reported in the literature. Secondly, the presence of gastric polyposis, in addition to colonic polyposis, is a novel finding for the disease. Thirdly, this patient has been diagnosed with breast fibroadenoma, which is not typically associated with the cancer spectrum of polymerase-associated polyposis syndrome. We speculated that the difference in the patient’s clinical presentation may be due to the pathogenicity and penetrance of the specific POLD1 variant, as well as the occurrence of the germline variant through de novo formation.
Germline POLD1-induced polyposis also displayed a unique spectrum of somatic alterations. Thirteen somatic alterations, including a variant of uncertain significance (VUS) in APC p.S1896R, were identified in the rectal tissue. While it is uncertain whether the VUS might lead to loss of function of APC, which typically drives the oncogenesis of CRC (23, 24), we propose the APC VUS is a POLD1-induced variant rather than a driver mutation. In addition, other commonly mutated genes in CRC such as TP53 and KRAS were not detected. In the POLD1-deficient glioblastoma tissue, we observed ultra-hypermutation (tumor mutation burden of 347.1 muts/Mb), which is exceedingly rare since glioblastoma is typically classified as a TMB-low tumor type (25). Notably, the presence of the POLD1 p.L606M variant may be more prevalent in brain tumors. Among the five POLD1 p.L606M variants identified in the study by Campbell et al. (3), two were found in brain primitive neuroectodermal tumors, and two were found in brain glioblastomas, with a median TMB of 212.4 muts/Mb (range, 51.4–295 muts/Mb). In fact, upon detailed analysis of the POLD1 drivers in the Campbell et al. study, we found that POLD1-driven brain glioblastoma exhibited higher TMB than POLD1-driven CRC (median = 228.8 vs. 63.05), although the underlying cause of the tissue-specific difference is unknown. The difference in mutation burden between the rectal and brain tissue may be attributed to variations in the stage of tumor evolution, the distinct microenvironments, and exposure to additional mutational agents in the two tumor types. While both the brain and rectal tissues were MSS, we observed predominantly dMMR-associated signatures in the brain tissue and no polymerase-associated signatures in either lesion, which has also been reported in previous studies on POLE/POLD1 drivers (26). It has also been shown that SBS14 and SBS20 are associated with tumors that exhibit concurrent loss of MMR and polymerase function (27). Importantly, in all cases where the chronological sequence of events could be established, alterations in the polymerase preceded the defects in MMR (27), indicating a dynamic evolution of mutational signatures during cancer development, particularly in tumors with defects in DNA repair. This also suggests that the POLD1-altered brain tissue in our case might be at a relatively early stage in its development, transitioning towards acquiring additional MMR defects. Furthermore, other common molecular features of glioblastoma were not detected, such as 1p/19q loss, EGFR amplification, or EGFRvIII alterations (28). Thus, we hypothesized that cancers induced by germline POLD1 alterations may exhibit distinct molecular features.
It is also noteworthy that there are some other inherited syndromes that share similar clinical symptoms but with largely distinct molecular landscapes (24, 29, 30). One example is Turcot syndrome, which consistently presents a high risk of central nervous system neoplasms and gastrointestinal cancers paired with intestinal polyps. Studies have revealed that Turcot syndrome is caused by the germline alterations in APC or mismatch repair (MMR) genes and often result in microsatellite-instable (MSI) tumors (29, 31). On the other hand, hereditary colorectal cancers can manifest as both polyposis and nonpolyposis conditions (32). The most common inherited disease that predisposes individuals to colorectal and endometrial cancers is Lynch syndrome, which is a non-polyposis condition caused by pathogenic germline variants in the MMR genes that results in MSI. By contrast, tumors associated with polymerase-associated polyposis syndrome are almost always MSS. In our case, even though multiple variants were detected in MMR genes, including MLH1, MLH3, MSH2, MSH4, and MSH6, the tumor tissues were MSS. This finding is consistent with the typical manifestation of polymerase-associated polyposis syndrome. Accumulation of these MMR gene variants were likely a consequence of POLD1-induced hypermutation. Other polyposis conditions often exhibit overlapping and distinct cancer spectra. For example, familial adenomatous polyposis, caused by pathogenic germline variants in APC, is characterized by the presence of hundreds to thousands of adenomatous colonic polyps. MUTYH-associated polyposis is an autosomal recessive disorder that involves biallelic germline variants in MUTYH. Affected individuals tend to develop multiple adenomatous colonic polyps, along with an increased risk for small-bowel polyposis. Many other genetic disorders also result in an increased risk of colorectal cancer, including Li-Fraumeni syndrome (TP53), juvenile polyposis syndrome (SMAD4 and BMPR1A), Peutz–Jeghers syndrome (STK11), and Cowden syndrome (PTEN) (32, 33). Therefore, a high-throughput genetic examination can aid in accurately distinguishing these diseases, and cancer surveillance is essential for patients with these conditions after the genetic diagnosis.
In summary, our case reported a de novo germline POLD1 p.L606M variant that led to early-onset multiple types of cancer, coupling with multiple gastrointestinal polyposis. NGS targeting the whole exome of relevant genes could facilitate an accurate diagnosis of suspected hereditary diseases.
Sample processing and sequencing were performed in a CLIA-certified and CAP-accredited laboratory (Geneseeq Technology Inc., Nanjing, China) as previously described (34). In brief, genomic DNAs from FFPE sections and the whole blood control samples were extracted with the QIAamp DNA FFPE Tissue Kit and the DNeasy Blood and tissue kit (Qiagen), respectively, and quantified by Qubit 3.0 using the dsDNA HS Assay Kit (ThermoFisher Scientific). Library preparations were performed with the KAPA Hyper Prep Kit (KAPA Biosystems). Customized xGen lockdown probes (Integrated DNA Technologies) targeting whole exons of 494 cancer-relevant genes (Supplementary Table S1) were used for hybridization enrichment. The capture reaction was performed with Dynabeads M-270 (Life Technologies) and xGen Lockdown hybridization and wash kit (Integrated DNA Technologies) according to the manufacturers’ protocols. Libraries were on-beads PCR-amplified, purified, sized and quantified, and sequenced on an Illumina HiSeq4000 platform.
Trimmomatic was used for FASTQ file quality control. Leading/trailing low quality (quality reading below 20) or N bases were removed. Paired-end reads were then aligned to the reference human genome (build hg19), using the Burrows–Wheeler Aligner (BWA) with the parameters. PCR deduplication was performed using Picard and local realignment around indels, and base quality score recalibration was performed using GATK3. Furthermore, samples with a mean dedup depth <30× were removed. Cross-sample contamination was estimated using ContEst (Broad Institute). Germline and somatic mutations were called using the GATK haplotypecaller and Mutect2, respectively. Tumor mutation burden was counted by summing all base substitutions and indels in the coding region of targeted genes, including synonymous alterations to reduce sampling noise and excluding known driver mutations as they were over-represented in the panel, as previously described (34, 35).
The data presented in the study are deposited in the Genome Sequence Archive (Genomics, Proteomics & Bioinformatics 2021) in National Genomics Data Center (Nucleic Acids Res 2022), China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA-Human), publicly accessible at https://ngdc.cncb.ac.cn/gsa-human, accession number HRA005282.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
YZ, FQ, JX, and XW: Conception, design, and manuscript review. YNZ and JX: Material preparation. CL and LZ: Literature research and discussion. QM, FW, JY, and YS: Analysis of original data and bio-informatic research. All the authors agreed to the submission of the version.
We thank the patient and her relatives for participating in and cooperating with this study.
QM, FW, YS, and JY are employees of the company Nanjing Geneseeq Inc. LZ is an employee of the company Genetron Health Inc.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1222873/full#supplementary-material
Supplementary Table 1 | Detailed gene list in 494 panels.
1. Simon M, Giot L, Faye G. The 3’ to 5’ exonuclease activity located in the DNA polymerase delta subunit of Saccharomyces cerevisiae is required for accurate replication. EMBO J (1991) 10:2165–70. doi: 10.1002/j.1460-2075.1991.tb07751.x
2. Albertson TM, Ogawa M, Bugni JM, Hays LE, Chen Y, Wang Y, et al. DNA polymerase epsilon and delta proofreading suppress discrete mutator and cancer phenotypes in mice. Proc Natl Acad Sci USA (2009) 106:17101–4. doi: 10.1073/pnas.0907147106
3. Campbell BB, Light N, Fabrizio D, Zatzman M, Fuligni F, de Borja R, et al. Comprehensive analysis of hypermutation in human cancer. Cell (2017) 171:1042–1056.e10. doi: 10.1016/j.cell.2017.09.048
4. Church DN, Briggs SE, Palles C, Domingo E, Kearsey SJ, Grimes JM, et al. DNA polymerase ϵ and δ exonuclease domain mutations in endometrial cancer. Hum Mol Genet (2013) 22:2820–8. doi: 10.1093/hmg/ddt131
5. Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet (2013) 45:136–44. doi: 10.1038/ng.2503
6. Bellido F, Pineda M, Aiza G, Valdés-Mas R, Navarro M, Puente DA, et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: review of reported cases and recommendations for genetic testing and surveillance. Genet Med Off J Am Coll Med Genet (2016) 18:325–32. doi: 10.1038/gim.2015.75
7. Mur P, García-Mulero S, Del Valle J, Magraner-Pardo L, Vidal A, Pineda M, et al. Role of POLE and POLD1 in familial cancer. Genet Med Off J Am Coll Med Genet (2020) 22:2089–100. doi: 10.1038/s41436-020-0922-2
8. Briggs S, Tomlinson I. Germline and somatic polymerase ϵ and δ mutations define a new class of hypermutated colorectal and endometrial cancers. J Pathol (2013) 230:148–53. doi: 10.1002/path.4185
9. Rayner E, van Gool IC, Palles C, Kearsey SE, Bosse T, Tomlinson I, et al. A panoply of errors: polymerase proofreading domain mutations in cancer. Nat Rev Cancer (2016) 16:71–81. doi: 10.1038/nrc.2015.12
10. Mur P, Magraner-Pardo L, García-Mulero S, Díez-Villanueva A, Del Valle J, Ezquerro E, et al. Solving the enigma of POLD1 p.V295M as a potential cause of increased cancer risk. Eur J Hum Genet EJHG (2022) 30:485–9. doi: 10.1038/s41431-021-00926-6
11. Magrin L, Fanale D, Brando C, Fiorino A, Corsini LR, Sciacchitano R, et al. POLE, POLD1, and NTHL1: the last but not the least hereditary cancer-predisposing genes. Oncogene (2021) 40:5893–901. doi: 10.1038/s41388-021-01984-2
12. Shlien A, Campbell BB, de Borja R, Alexandrov LB, Merico D, Wedge D, et al. Combined hereditary and somatic mutations of replication error repair genes result in rapid onset of ultra-hypermutated cancers. Nat Genet (2015) 47:257–62. doi: 10.1038/ng.3202
13. León-Castillo A, Britton H, McConechy MK, McAlpine JN, Nout R, Kommoss S, et al. Interpretation of somatic POLE mutations in endometrial carcinoma. J Pathol (2020) 250:323–35. doi: 10.1002/path.5372
14. Nicolas E, Golemis EA, Arora S. POLD1: Central mediator of DNA replication and repair, and implication in cancer and other pathologies. Gene (2016) 590:128–41. doi: 10.1016/j.gene.2016.06.031
15. Prindle MJ, Loeb LA. DNA polymerase delta in DNA replication and genome maintenance. Environ Mol Mutagen (2012) 53:666–82. doi: 10.1002/em.21745
16. Li L, Murphy KM, Kanevets U, Reha-Krantz LJ. Sensitivity to phosphonoacetic acid: a new phenotype to probe DNA polymerase delta in Saccharomyces cerevisiae. Genetics (2005) 170:569–80. doi: 10.1534/genetics.104.040295
17. Nick McElhinny SA, Stith CM, Burgers PMJ, Kunkel TA. Inefficient proofreading and biased error rates during inaccurate DNA synthesis by a mutant derivative of Saccharomyces cerevisiae DNA polymerase delta. J Biol Chem (2007) 282:2324–32. doi: 10.1074/jbc.M609591200
18. Clausen AR, Zhang S, Burgers PM, Lee MY, Kunkel TA. Ribonucleotide incorporation, proofreading and bypass by human DNA polymerase δ. DNA Repair (2013) 12:121–7. doi: 10.1016/j.dnarep.2012.11.006
19. Daee DL, Mertz TM, Shcherbakova PV. A cancer-associated DNA polymerase delta variant modeled in yeast causes a catastrophic increase in genomic instability. Proc Natl Acad Sci USA (2010) 107:157–62. doi: 10.1073/pnas.0907526106
20. Mertz TM, Sharma S, Chabes A, Shcherbakova PV. Colon cancer-associated mutator DNA polymerase δ variant causes expansion of dNTP pools increasing its own infidelity. Proc Natl Acad Sci USA (2015) 112:E2467–2476. doi: 10.1073/pnas.1422934112
21. Mertz TM, Baranovskiy AG, Wang J, Tahirov TH, Shcherbakova PV. Nucleotide selectivity defect and mutator phenotype conferred by a colon cancer-associated DNA polymerase δ mutation in human cells. Oncogene (2017) 36:4427–33. doi: 10.1038/onc.2017.22
22. Palles C, Martin L, Domingo E, Chegwidden L, McGuire J, Cuthill V, et al. The clinical features of polymerase proof-reading associated polyposis (PPAP) and recommendations for patient management. Fam Cancer (2022) 21:197–209. doi: 10.1007/s10689-021-00256-y
23. Aghabozorgi AS, Bahreyni A, Soleimani A, Bahrami A, Khazaei M, Ferns GA, et al. Role of adenomatous polyposis coli (APC) gene mutations in the pathogenesis of colorectal cancer; current status and perspectives. Biochimie (2019) 157:64–71. doi: 10.1016/j.biochi.2018.11.003
24. Kinzler KW, Vogelstein B. Cancer-susceptibility genes. Gatekeepers and caretakers. Nature (1997) 386:761, 763. doi: 10.1038/386761a0
25. Castle JC, Uduman M, Pabla S, Stein RB, Buell JS. Mutation-derived neoantigens for cancer immunotherapy. Front Immunol (2019) 10:1856. doi: 10.3389/fimmu.2019.01856
26. Ying J, Yang L, Yin JC, Xia G, Xing M, Chen X, et al. Additive effects of variants of unknown significance in replication repair-associated DNA polymerase genes on mutational burden and prognosis across diverse cancers. J Immunother Cancer (2021) 9:e002336. doi: 10.1136/jitc-2021-002336
27. Haradhvala NJ, Kim J, Maruvka YE, Polak P, Rosebrock D, Livitz D, et al. Distinct mutational signatures characterize concurrent loss of polymerase proofreading and mismatch repair. Nat Commun (2018) 9:1746. doi: 10.1038/s41467-018-04002-4
28. Aldape K, Zadeh G, Mansouri S, Reifenberger G, von Deimling A. Glioblastoma: pathology, molecular mechanisms and markers. Acta Neuropathol (Berl.) (2015) 129:829–48. doi: 10.1007/s00401-015-1432-1
29. Dinarvand P, Davaro EP, Doan JV, Ising ME, Evans NR, Phillips NJ, et al. Familial adenomatous polyposis syndrome: an update and review of extraintestinal manifestations. Arch Pathol Lab Med (2019) 143:1382–98. doi: 10.5858/arpa.2018-0570-RA
30. Lebrun C, Olschwang S, Jeannin S, Vandenbos F, Sobol H, Frenay M. Turcot syndrome confirmed with molecular analysis. Eur J Neurol (2007) 14:470–2. doi: 10.1111/j.1468-1331.2006.01669.x
31. Hamilton SR, Liu B, Parsons RE, Papadopoulos N, Jen J, Powell SM, et al. The molecular basis of Turcot’s syndrome. N Engl J Med (1995) 332:839–47. doi: 10.1056/NEJM199503303321302
32. Hampel H, Kalady MF, Pearlman R, Stanich PP. Hereditary colorectal cancer. Hematol Oncol Clin North Am (2022) 36:429–47. doi: 10.1016/j.hoc.2022.02.002
33. Rebuzzi F, Ulivi P, Tedaldi G. Genetic predisposition to colorectal cancer: how many and which genes to test? Int J Mol Sci (2023) 24:2137. doi: 10.3390/ijms24032137
34. Fang W, Ma Y, Yin JC, Hong S, Zhou H, Wang A, et al. Comprehensive genomic profiling identifies novel genetic predictors of response to anti-PD-(L)1 therapies in non-small cell lung cancer. Clin Cancer Res Off J Am Assoc Cancer Res (2019) 25:5015–26. doi: 10.1158/1078-0432.CCR-19-0585
Keywords: NGS, POLD1, POLE, germline POLD1 L606M, polyposis syndrome
Citation: Zhang Y, Wang X, Zhu Y, Liang C, Zhao L, Meng Q, Yin JC, Shi Y, Wang F, Qin F and Xuan J (2023) Case Report: Cancer spectrum and genetic characteristics of a de novo germline POLD1 p.L606M variant-induced polyposis syndrome. Front. Oncol. 13:1222873. doi: 10.3389/fonc.2023.1222873
Received: 15 May 2023; Accepted: 20 July 2023;
Published: 06 September 2023.
Edited by:
John J. Turchi, Indiana University Bloomington, United StatesReviewed by:
Polina Shcherbakova, University of Nebraska Medical Center, United StatesCopyright © 2023 Zhang, Wang, Zhu, Liang, Zhao, Meng, Yin, Shi, Wang, Qin and Xuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ying Zhang, MTczODg3NjAzNUBxcS5jb20=; Ji Xuan, aGVsaW8wMDA5QDEyNi5jb20=; Feng Qin, cWluZmVuZzcxMTFAc2luYS5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.