 Takeshi Hori
Takeshi Hori Kosuke Yokobori
Kosuke Yokobori Rick Moore1
Rick Moore1 Tatsuya Sueyoshi
Tatsuya Sueyoshi- 1Pharmacogenetics Section, Reproductive and Developmental Biology Laboratory, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC, United States
- 2Department of Biomechanics, Institute of Biomaterials and Bioengineering (IBB), Tokyo Medical and Dental University (TMDU), Tokyo, Japan
Phenobarbital (PB) is an archetypal substance used as a mouse hepatocellular carcinoma (HCC) promotor in established experimental protocols. Our previous results showed CAR is the essential factor for PB induced HCC promotion. Subsequent studies suggested Gadd45β, which is induced by PB through CAR activation, is collaborating with CAR to repress TNF-α induced cell death. Here, we used Gadd45β null mice (Gadd45β KO) treated with N-diethylnitrosamine (DEN) at 5 weeks of age and kept the mice with PB supplemented drinking water from 7 to 57 weeks old. Compared with wild type mice, Gadd45β KO mice developed no HCC in the PB treated group. Increases in liver weight were more prominent in wild type mice than KO mice. Microarray analysis of mRNA derived from mouse livers found multiple genes specifically up or down regulated in wild type mice but not null mice in DEN + PB groups. Further qPCR analysis confirmed two genes, Tgfbr2 and irisin/Fndc5, were up-regulated in PB treated wild type mice but no significant increase was observed in Gadd45β KO mice. We focused on these two genes because previous reports showed that hepatic Irisin/Fndc5 expression was significantly higher in HCC patients and that irisin binds to TGF-β receptor complex that includes TGFBR2 subunit. Our results revealed irisin peptide in cell culture media increased the growth rate of mouse hepatocyte-derived AML12 cells. Microarray analysis revealed that irisin-regulated genes in AML12 cells showed a significant association with the genes in the TGF-β pathway. Expression of irisin/Fndc5 and Tgfbr2 induced growth of human HCC cell line HepG2. Thus, Gadd45β plays an indispensable role in mouse HCC development regulating the irisin/Fndc5 and Tgfbr2 genes.
Introduction
According to current information from the American Cancer Society, liver cancer is a common neoplasm with more than 80000 diagnoses and causing more than 70000 deaths each year worldwide (1). It is one of the most common cancers and the third leading cause of cancer-related death in 2020 (2). Hepatocellular carcinoma (HCC) is the most frequent histologic type of liver cancer (3). Many rodent model systems were developed for studying HCC mechanisms and testing drug candidates. Among them, a two stage chemically induced HCC that employs N-diethylnitrosamine (DEN) as the genotoxic initiator and phenobarbital (PB) as the non genotoxic promoter has been used in numerous studies as an archetypal system (4).
PB is a barbiturate anticonvulsant and activates constitutive active/androstane receptor (CAR, NR1i3) (5). CAR, originally cloned as a homologue of orphan nuclear receptors, was found to be a transcription factor that activates drug/chemical metabolizing enzymes in the liver for detoxification in late ‘90 (6). Our group has revealed that CAR is an essential factor for liver tumor promotion by PB in the two stage experimental system (7). Although a substantial number of publications about molecular mechanisms about PB tumor promotion mediated by CAR were published since then, the details of that were poorly understood (8–16). One such study found that the expression of growth arrest and DNA-damage inducible 45 beta (Gadd45β), a molecule known to be involved in cell stress responses, was strongly induced in a CAR dependent manner by PB (13). Gadd45β, a member of Gadd45 family comprised of α, β, and γ, is induced by stress signals and mediates various protein interplay among protein factors that causes a wide variety of cellular responses including DNA repair, cell cycle control, senescence, and apoptosis (17, 18). For example, Gadd45β mediates its anti-apoptotic effect by promoting degradation of p53 via Src/PP2A/MDM2 pathway following arsenite treatment (19). Moreover, Gadd45β downregulates pro apoptotic JNK signaling by interacting with MAPK kinase 7 (MKK7) (20). In addition to the anti-tumorigenesis functions of Gadd45β, it also has pro-apoptotic activities suggesting that Gadd45β functions are contingent on the context of cell circumstances and interplay with other factors (17, 18, 21).
Among the three GADD45 molecules, only Gadd45β was strongly induced by PB, in a CAR dependent manner. Provocatively, Gadd45β protein interacts with CAR and serves as a scaffold protein for regulating the activity of mitogen-activated protein kinase (MAPK). Our recent results suggested that the CAR-Gadd45β complex reduces p38 MAPK phosphorylation and acts as a tumor suppressor (22). Moreover, p38 MAPK activation in mouse liver dramatically reduced tumor development (23) thus suggesting p38 MAPK activity mitigation by CAR-Gadd45β complex may be causing the observed HCC development. Furthermore, CAR-Gadd45β complex was found to suppress phosphorylation of JNK1 by MAPK kinase 7 (MKK7) and repress TNFα induced cell death. (13). Fittingly, in the absence of PB, hepatocyte-specific knock-out (KO) of either p38 MAPK or JNK1 signaling promotes DEN-initiated HCC in mice (24, 25). In other words, suppressing these kinase activities by PB or gene knockout may promote DEN-induced HCC in a similar mechanism. Thus, we hypothesized that MAPK suppression by CAR-Gadd45β complex is responsible for PB-induced HCC promotion. In a previous report, as a first step for evaluating Gadd45β in HCC development, we analyzed Gadd45β KO mice with short term treatment with PB for hepatocyte growth. Gadd45β KO mice treated with PB for 48 hr showed suppressed BrdUrd intake into hepatocytes (22). Consistent with our findings, Tian et al. showed that potent CAR activator 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP), induced liver growth and that Gadd45β slowed this growth (26).
Our new finding through a cancer patient database analysis which suggests low survival rates associated with low GADD45β expression levels in the liver further prompted us to focus on Gadd45β. Although nongenotoxic chemicals including PB have been well established to activate CAR and eventually induce HCC in rodents, the same mode of action involving nongenotoxic chemicals and CAR was not observed in human HCC. However molecular pathways found in HCC development in rodents may partially overlap with that of humans despite CAR activation may not be involved. Hence, in this report, we compared tumorigenesis in WT and Gadd45β null mice from a C57BL/6J background for analyzing roles of Gadd45β in HCC development. Mice were treated with DEN at 5 weeks old and PB was administered from 7 weeks to 57 weeks of age. Obtained results showed the disappearance of PB-induced adenoma in Gadd45β null mice. HCC was observed in only WT mice; thus, these results suggest that Gadd45β plays critical roles in PB-induced tumor promotion. Among numerous PB-modulated liver genes differentially affected by the Gadd45β null mutation in microarray analysis, we focused on two genes, Tgfbr2 and Irisin/Fndc5, for this report. Both genes were induced by PB in wild type but not in Gadd45β null mouse liver. Irisin, a peptide produced by the cleavage of Fndc5 protein, was originally found as a myokine induced by physical exercise (27) and now this peptide is known to be expressed in multiple organs including the liver (28). Interestingly, a recent report showed increased irisin expression in HCC patients. These genes were found to be involved in the regulation of cell growth and TGFBR2 expression levels showed a strong association with the survival of liver cancer patients. Further studies about the genes affected by Gadd45β including Tgfbr2 and Fndc5 and their roles in the MAPK activity modulation by Gadd45β will contribute to mechanistic understandings of CAR-dependent HCC promotion in mice.
Materials and methods
Materials
Phenobarbital sodium salt, 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP), anisomycin, and DEN were purchased from Sigma-Aldrich (St. Louis, MO, USA). Fugene 6 from Promega (Ann Arbor, MI) and Lipofectamine 3000 from Life Technologies (Grand Island, NY, USA) were used for cell transfection of plasmid DNA; TaqMan Gene Expression Assays (Thermo Fisher, Waltham, MA) for gene expression analyses used in this study were as follows: Akr1b7, Mm00477605_m1; Casp1, Mm00438023_m1; Cdh1, Mm01247357_m1; Cyp2b9, Mm00657910_m1; Cyp2b10, Mm00456591_m1; Cyp2b13, Mm00771172_g1; Gadd45β, Mm00435123_m1; Fndc5, Mm01181543_m1; Krt19, Mm00491980_m1; Nedd9, Mm01324843_m1; Sult1e1, Mm00499178_m1; Tgfbr2, Mm03024091_m1; and Mouse GAPDH Endogenous Control. Irisin peptide was a product of Raybiotech (Peachtree Corners, GA, USA). Expression plasmid for FNDC5/Irisin was constructed by amplifying cDNA fragment using mouse liver cDNA and cloned into pcDNA3.1 V5-His TOPO (Thermo Fisher). Human TGFBR2 expression vector (pCMV5B-TGFbeta receptor II) was a gift from Joan Massague and Jeff Wrana (Addgene plasmid #11766) (29),
Animals and drug treatments
C57BL/6J (Catalog# JAX 000664) and Gadd45β KO (B6;129S6-Gadd45βtm1Flv/J) (Catalog# JAX 013101) were obtained from Jackson Laboratories (Bar Harbor, ME, USA) and were maintained at the National Institute of Environmental Health Sciences (NIEHS, USA). Mice received a single i.p. treatment with DEN (90mg/kg body weight) at 5 weeks of age as in previous reports (7, 30, 31). Two weeks after the DEN treatment, mice were placed on PB (0.5 g/l) via drinking water or normal water as a control for 50 weeks. The water bottle with or without PB was changed every week. Based on 5 ml water consumption per mouse for one day, we expected 0.875 g of PB/mouse for the study. At the end of the 50-week period, mice were euthanized by CO2 and livers were excised for mRNA analyses. Histopathological evaluation of livers for tumor induction in Gadd45β KO mice and wild-type (WT) C57BL/6J mice was performed to see changes in hepatocytes including hypertrophy, foci and tumors. All animal procedures were approved by the Animal Care and Use Committee at NIEHS, NIH, and performed humanely in accordance with Public Health Service Policy.
Cell cultures and transient transfection
HepG2 (human hepatocellular carcinoma cell line) and AML12 (mouse hepatocyte cell line) were obtained from the American Type Culture Collection (ATCC). HepG2 and AML12 cells were cultured in Eagle’s minimum essential media and DMEM/F12 media, respectively, supplemented with 10% fetal bovine serum (FBS) and penicillin/streptomycin at 37°C with 5% CO2. HepG2 cells were transfected by Fndc5/Irisin and Tgfbr2 expression plasmids using Fugene 6 reagent according to the manufacturer’s instructions. FNDC5/Irisin and Tgfbr2 protein expressions were detected by western blotting analyses using 10% SDS page gels employing anti V5 tag antibody (Thermo Fisher) and anti Tgfbr2 monoclonal antibody (Santa Cruz Biotechnology), respectively. AML12 cells in 96 well plates (seeded 5000 cells per well) were treated with irisin (1 μg/ml or 2 μg/ml) at 24 hr. Seventy-two hr after transfection (HepG2) or irisin treatment (AML12), cell viability was determined using Cell Counting Kit-8 (Dojindo).
Gene arrays
All tissue samples for gene expression analysis were obtained from non-tumorous regions of liver tissues. RNAs were purified from mouse livers using Trizol reagent (Thermo Fisher Scientific) and the RNeasy mini kit (QIAGEN, Valencia, CA, USA). Gene expression analysis was conducted using Agilent Whole Mouse Genome 4 × 44 multiplex format oligo arrays (014868) (Agilent Technologies, Palo Alto, CA, USA) following the Agilent 1-color microarray-based gene expression analysis protocol. Cy3 labeled cRNA was produced following the manufacturer’s protocol. Cy3 labeled cRNAs were fragmented and hybridized for 17 h in a rotating hybridization oven. Slides were scanned with an Agilent Scanner. Data were obtained using the Agilent Feature Extraction software (v12). The Agilent Feature Extraction Software performed error modeling, adjusting for additive and multiplicative noise. Genes were considered differentially expressed if they showed a fold-change of at least 1.5 with p-value < 0.05 tested by an ANOVA and Benjamini-Hochberg multiple test correction performed using OmicSoft Array Studio (Version 10) software. Ingenuity Pathway Analysis software (IPA, Qiagen) was utilized to search the canonical pathway, toxicity function, or upstream regulator. The pathways with Z-score >2 and <-2 were considered significantly activated and repressed pathways, respectively. Gene set enrichment analysis (GSEA, version 4.0.3) was performed with the oncogenic gene sets (H) from Molecular Signature Database (MSigDB, v7.1). Gene set with NOM p-value < 0.05 was considered as significantly enriched.
Bioinformatics analysis
The transcriptome data as fragments per kilobase of transcript per million mapped reads upper quartile (FMPK-UQ) and clinical information of hepatocarcinoma patients in TCGA-LIHC was downloaded from the GDC data portal (https://portal.gdc.cancer.gov/). The hepatocarcinoma samples of 377 patients were downloaded. The patients were grouped as low and high GADD45B by FMPK score of GADD45B as lower than 40 (n=121) and greater than 85 (n=133), respectively. Subsequently, the patients in high GADD45B were grouped as low and high FNDC5 by FMPK score of FNDC5 as lower than 4 (n=55) and greater than 10 (n=57), respectively. The hepatocarcinoma patients were grouped as low and high FNDC5 by FMPK score of FNDC5 as lower than 1 (n=133) and greater than 6 (n=137), respectively. Kaplan-Meier survival curve with log-rank test was performed to compare differences in survival distributions by using Python lifelines (https://lifelines.readthedocs.io/en/latest/, version 0.27.1).
Statistical analysis
Statistical analyses were performed using Graphpad Prism (version 8.3.0, Graphpad Software, La Jolla, CA). A Grubbs test was performed for the statistical outlier evaluation for the assessment of liver weight/body weight.
Results
Low GADD45B expression is associated with poorer overall survival in hepatocarcinoma patients in public database
In previous studies using the DEN/PB mouse HCC experimental system, our group found Gadd45β is induced in mouse liver through CAR activation by PB. Since Gadd45β has been considered to play an anti-tumor role in some cancers (18), our logical hypothesis was that this molecule is playing a key role in HCC tumorigenesis. Here we compared HCC patient survival rates between groups with low and high GADD45B expression. For this analysis, human HCC patients from the public domain (TCGA-LIHC) were grouped based on liver cancer stages. The data shows that GADD45B expression levels decreased according to liver cancer stage progression (Figure 1A). Strikingly, the Kaplan-Meier survival curve with log-rank test showed that the group with lower GADD45B expression has a poorer prognosis than the group with higher GADD45B expression, suggesting that GADD45B has antitumor functions in human HCC.
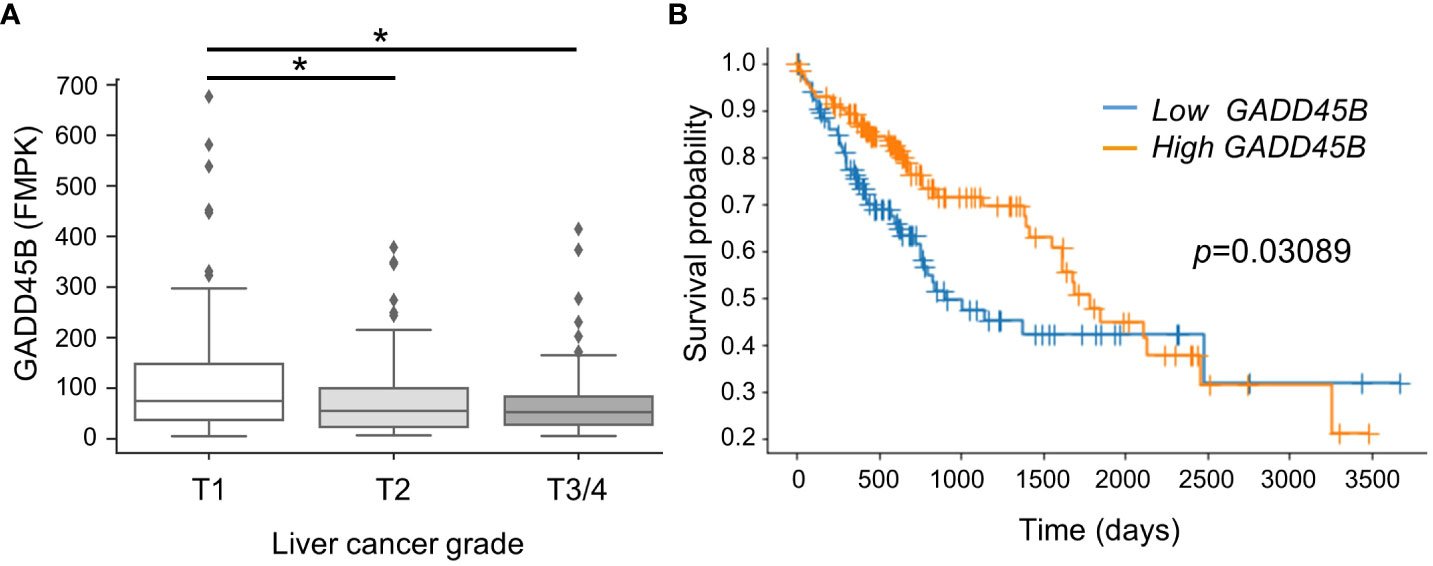
Figure 1 Lower expression of Gadd45β associated with poorer prognosis in human hepatocarcinoma. (A) Gadd45β expression levels in HCC patients. The box plot presented the median, first quartile, third quartile, and the vertical bars represent the 1.5*interquartile range (IQR). *P < 0.05 by one-way ANOVA followed by Sidak’s multiple comparisons. (B) Kaplan-Meier survival curves of patients with the high- (orange) or low- (blue) Gadd45β expression are displayed. Differences between survival curves were statistically analyzed by using the log-rank test.
Gadd45β is essential for PB promotion of HCC
To evaluate the effect of Gadd45β null mutation for PB-promoted liver tumors in mice, approximately 40 C57BL/6 WT (Gadd45β WT) and 40 Gadd45β null mutant mice (Gadd45β KO) were injected with DEN, and half of the mice in each group were treated with PB in drinking water. As shown in Figure 2A, fifty weeks after administration of PB, necropsies were performed, livers were removed, and mRNA expression levels were analyzed (N=6). Real-time PCR confirmed that Gadd45β was not expressed in Gadd45β KO mice (Figure 2B). Consistent with our previous results, PB treatment significantly increased the expression of Gadd45β. The numbers of basophilic foci, eosinophilic foci, adenoma, and carcinoma were analyzed in all livers excised from each group. The numbers of foci and tumors in each group are shown in Table 1. Five cases of adenoma were observed in Gadd45β WT and only one in Gadd45β KO in the groups with PB treatment (Table 1). In addition, carcinoma was observed in only one case in Gadd45β WT, which was not observed in Gadd45β KO (Table 1, Figure 2C). These results strongly suggested that Gadd45β plays a promoting role in PB-mediated tumorigenesis.
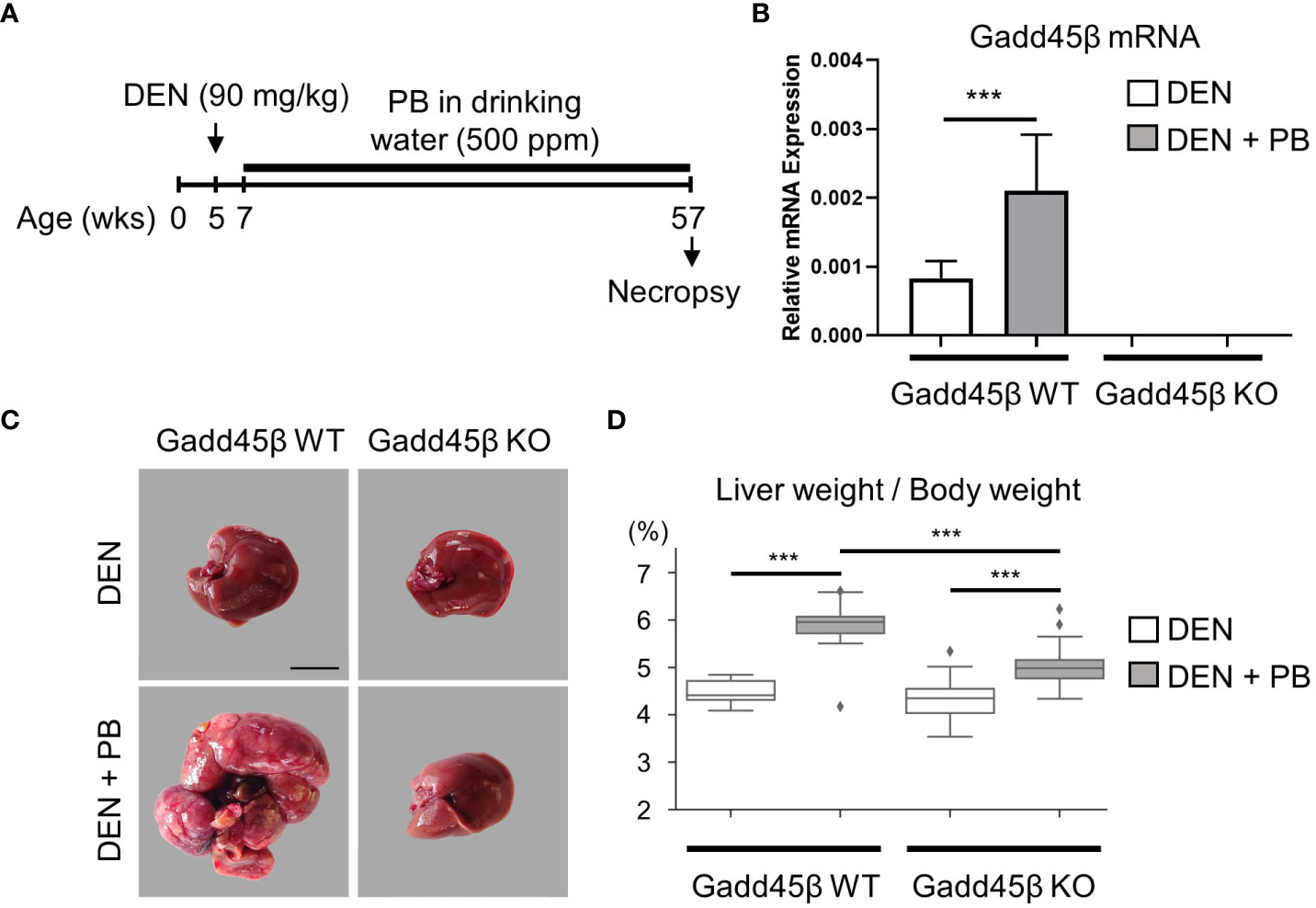
Figure 2 Gadd45β-null mutation compromised HCC promotion by PB. (A) Schematic representation of animal treatment. Numbers indicate age of the animals. DEN, diethylnitrosamine; PB, phenobarbital. (B) Induction of Gadd45β mRNA by PB in mouse livers. Each value is shown as the mean ± S.D. (n=6). ***, P < 0.01 by Student’s t-test. (C) Carcinoma was developed in DEN/PB-treated wild type mice but not in Gadd45β KO mice. Scale bar = 1 cm. Adenoma and carcinoma development in these mice were summarized in Table 1 and other liver images of DEN/PB-treated wild type mice were shown in Supplement Figure 1A. Representative livers from mice at 50 weeks after PB promotion. The liver from a DEN/PB-treated Gadd45β KO mouse has multiple tumors. Scale bar = 1 cm. (D) The liver weight per body weight values in DEN or DEN/PB-treated Gadd45β WT or Gadd45β KO were evaluated. The box plot presents the median, first quartile, and third quartile (n = 16). The vertical bars represent the 1.5*interquartile range (IQR). ***, P < 0.001 by one-way ANOVA followed by Sidak’s multiple comparisons.
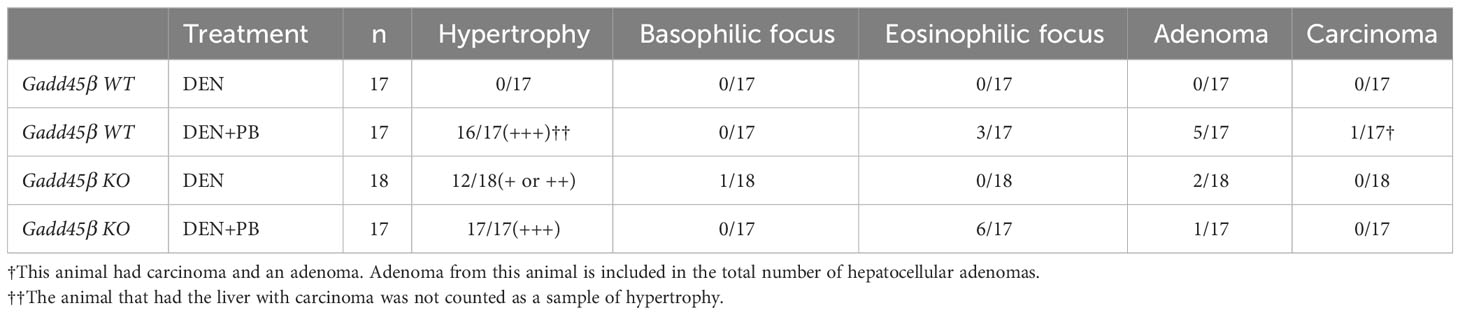
Table 1 Incidences of proliferative liver lesions after 50 weeks of PB treatment.
Subsequently, liver weight was analyzed for the assessment of hepatocyte proliferation. The Grubbs test detected the values of liver weight per body weight of two mice, one with carcinoma and the other with adenoma, of PB/DEN treated Gadd45β WT group as outliers. Therefore, these two samples were excluded in the following analysis (data including those two are shown in Supplemental Figure 1). Although liver weight per body weight was increased by PB in both Gadd45β WT and Gadd45β KO mice, that of livers from PB-treated Gadd45β KO mice was significantly smaller compared to that of livers from Gadd45β WT mice (Figure 2D). These results support that Gadd45β promotes hepatocyte proliferation in the presence of PB. In contrast, without PB, Gadd45β WT mice developed no adenoma, whereas Gadd45β KO had two adenomas (Table 1). This result, which may indicate that Gadd45β functions as a tumor suppressor in the absence of PB, is consistent with previously known Gadd45β function in tumorigenesis.
Eosinophilic foci and adenoma development in rodents are characteristic of liver tumor promotion by PB (32). Eosinophilic foci typically consist of hepatocytes which are stained more eosinophilic and are often larger than surrounding hepatocytes (33.). This is consistent with our previous tumorigenesis study with Car KO mice, which were changed to a liver tumor-susceptible mouse strain by repeated backcrossing to C3H/HeNCrlBR mice. In the present study we also observed PB administration increased eosinophilic foci. However, even though Gadd45β KO mice exhibited lower incidences of adenoma and carcinoma than Gadd45β WT mice in the presence of PB, the same mice showed higher incidences of eosinophilic foci. This histological result indicates that hepatic conditions of eosinophilic foci may not be enough to promote tumorigenesis.
Gadd45β null mutation causes significant changes in DEN + PB-induced liver gene expressions
Results obtained in Figure 2 suggest that the Gadd45β null mutation provoked fundamental changes in mouse liver responses against DEN/PB treatment. Therefore, we analyzed PB-induced transcriptome responses of wild type and Gadd45β null mutant mice livers expecting to find the key gene expression changes derived from the null mutation resulted in altered tumor development. For this purpose, RNAs isolated from the livers in Figure 2 were subjected to cDNA microarray analyses as described in the Methods section. We found differentially expressed genes by PB treatment in Gadd45β WT (134 genes) and Gadd45β KO (262 genes) and applied Ingenuity Pathway Analysis (IPA) for the results (Figures 3A, B). Canonical pathway analysis results shown in Figure 3A suggested Gadd45β gene null mutation provoked widespread effects on PB-induced gene expression in numerous pathways including those involved in endo- and exogenous metabolism. For example, genes in nicotine degradation, estrogen biosynthesis, and melatonin degradation were strongly affected by the null mutation. Furthermore, provocative differences were found by IPA Toxicity functional analysis (Figure 3B) in which clear differences were observed in cancer-related genes of multiple tissues including those of the liver. Subsequently, the Venn diagram visualized the unique differentially expressed genes in Gadd45β WT and KO mice (Figure 3C, Supplementary Table 1). Among those genes, we focused on two genes, Fndc5 and Tgfbr2. Increased FNDC5 expression in human HCC was observed in a previous report (28) and this gene was known to be a CAR target gene (34). On the other hand, Tgfbr2 was reported to have putative CAR response elements and to be CAR dependently induced in DEN/PB-treated mice (11). Our qPCR confirmed these two genes were induced by DEN/PB compared with DEN alone in WT mice and the inductions were much less prominent in Gadd45β KO mice (Figures 3D, E).
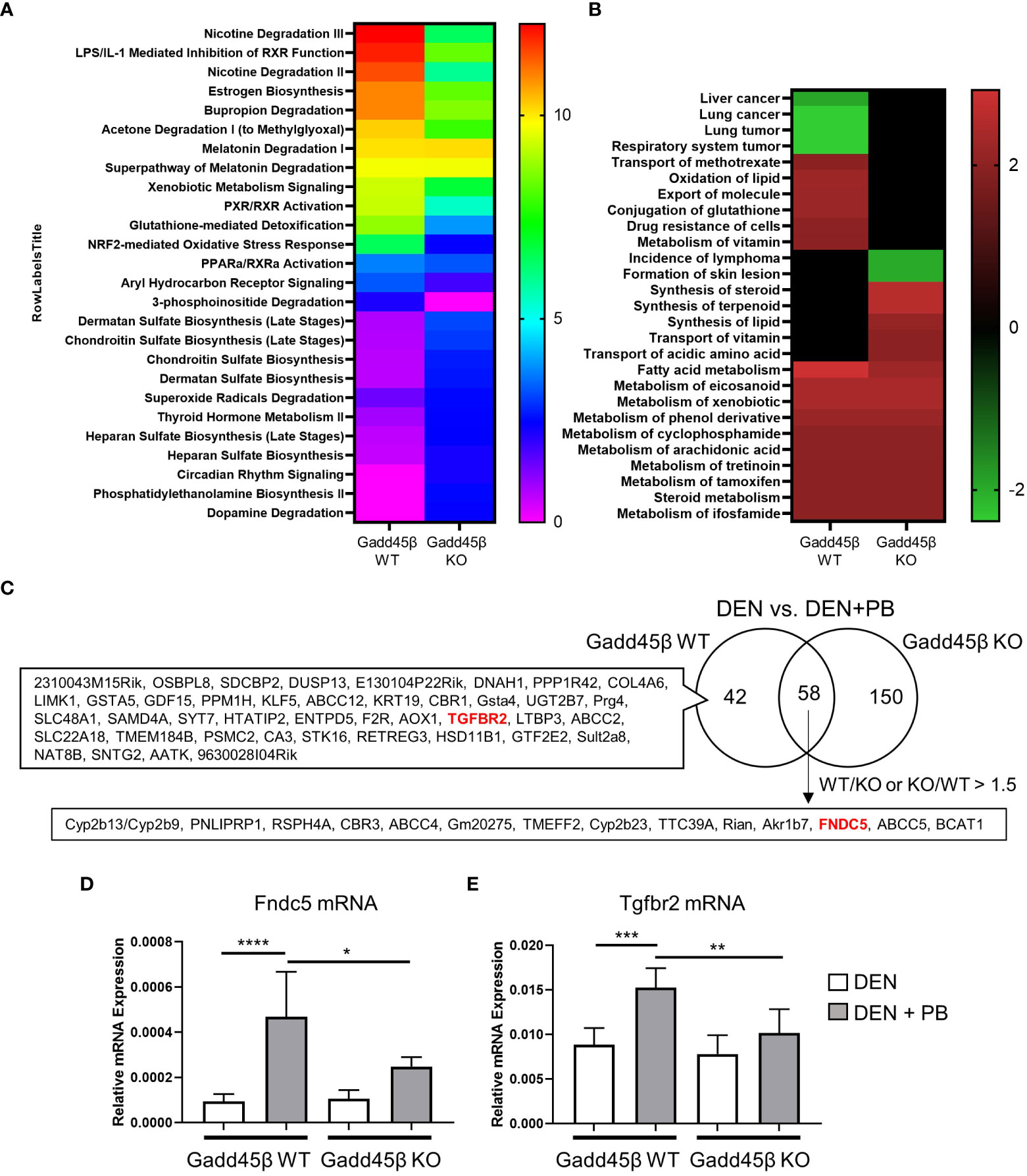
Figure 3 Gadd45β null mutation alters liver transcriptome in DEN + PB treated mice. (A, B) Differentially expressed genes of DEN/PB vs DEN treated Gadd45β WT or Gadd45β KO mice were analyzed by IPA for Canonical pathways (A, -log p value) or Toxicity function (B, Z score). (C) Venn diagram showing differentially expressed genes in DEN/PB treated vs DEN treated Gadd45β WT or Gadd45β KO mice. (D, E) Expression levels of Fndc5 mRNA (D) or Tgfbr2 mRNA (E) in DEN or DEN/PB treated Gadd45β WT or Gadd45β KO mice (n = 6). Data represent the means ± S.D. *P < 0.05, **P < 0.01, ***P < 0.001, or ****P < 0.0001 by one-way ANOVA followed by Sidak’s multiple comparisons.
Direct activation of CAR by PB is involved for Fndc5 but not for Tgfbr2 gene inductions
Next, we assessed the effect of single-dose PB treatment on gene expression levels of Fndc5 and Tgfbr2 in CAR WT and KO mice (Figure 4). Fndc5 mRNA was induced by PB treatment in CAR WT mice whereas no change was observed in CAR KO mice, indicating Fndc5 is a CAR target gene. In contrast, Tgfbr2 was induced by PB treatment in neither CAR WT mice nor CAR KO mice, suggesting the expression change of Tgfbr2 in DEN/PB-treated Gadd45β WT mice was caused by in-direct functions of CAR activated by long-term treatment with PB.
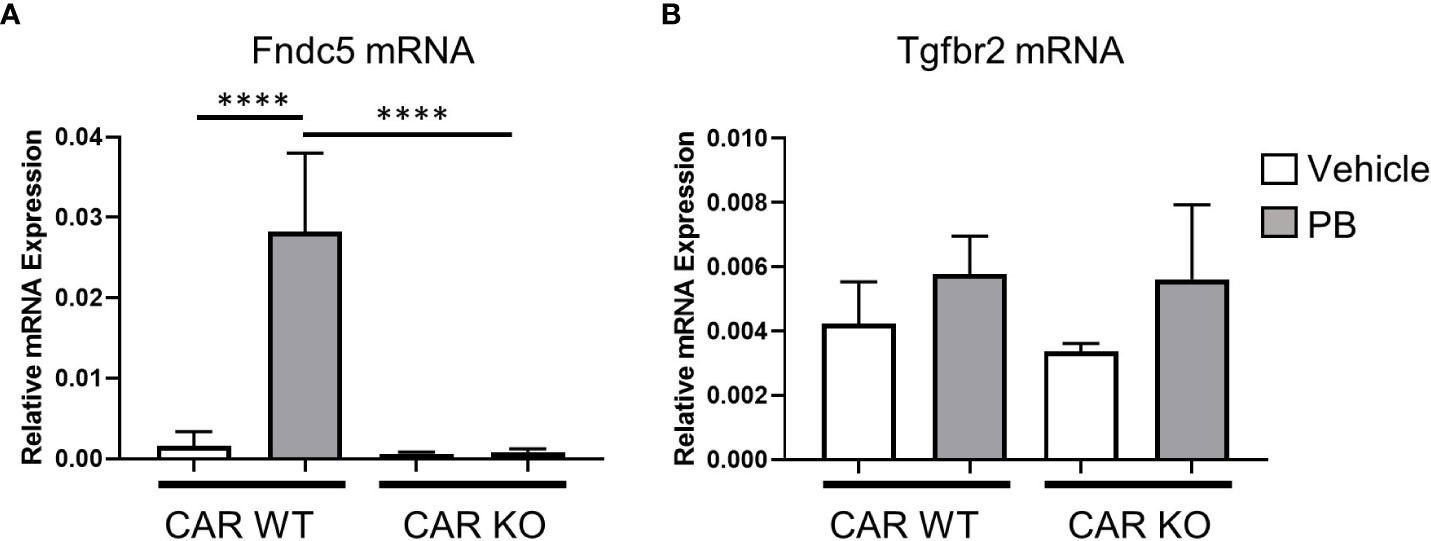
Figure 4 CAR is indispensable for Fndc5 gene induction by PB. Expression levels of Fndc5 mRNA (A) or Tgfbr2 mRNA (B) in vehicle or PB-treated Car WT or Car KO mice. Data represent means ± S.D. ****, P < 0.0001 by one-way ANOVA followed by Sidak’s multiple comparisons.
Irisin treatment induces proliferation of mouse hepatocyte cell line cells
Irisin has been reported to induce cell proliferations for many cell types including HepG2 cells, one of human hepatocellular carcinoma derived cell lines. Here we analyzed the effects of irisin treatment on the proliferation of mouse hepatocyte AML12. The commercial Irisin we used here has 100% identical peptide sequences between humans and mice. Cell numbers of AML12 cells were increased by irisin treatment after 72 hr, indicating proliferation was promoted by irisin (Figure 5A). Irisin induced gene expression changes in AML12 cells were analyzed by cDNA microarray followed by GSEA with C6 gene sets and IPA with upstream analysis (Figure 5B, Table 2). In this GSEA analysis, we compared Fndc5/Irisin-regulated genes found in our microarray with previously found genes regulated by TGF-β. The comparison revealed genes down-regulated by transiently expression of TGF-β, were enriched in up-regulated genes by irisin treatment (Figure 5B). Moreover, the IPA analysis found 10 upstream regulator candidates that are affected by irisin treatment. Among them, irisin has a potential function for suppression of the Tgfbr1 signaling pathway (Table 2), suggesting that these factors, Fndc5/Irisin, TGF-β and Tgfbr1, have functional interactions.
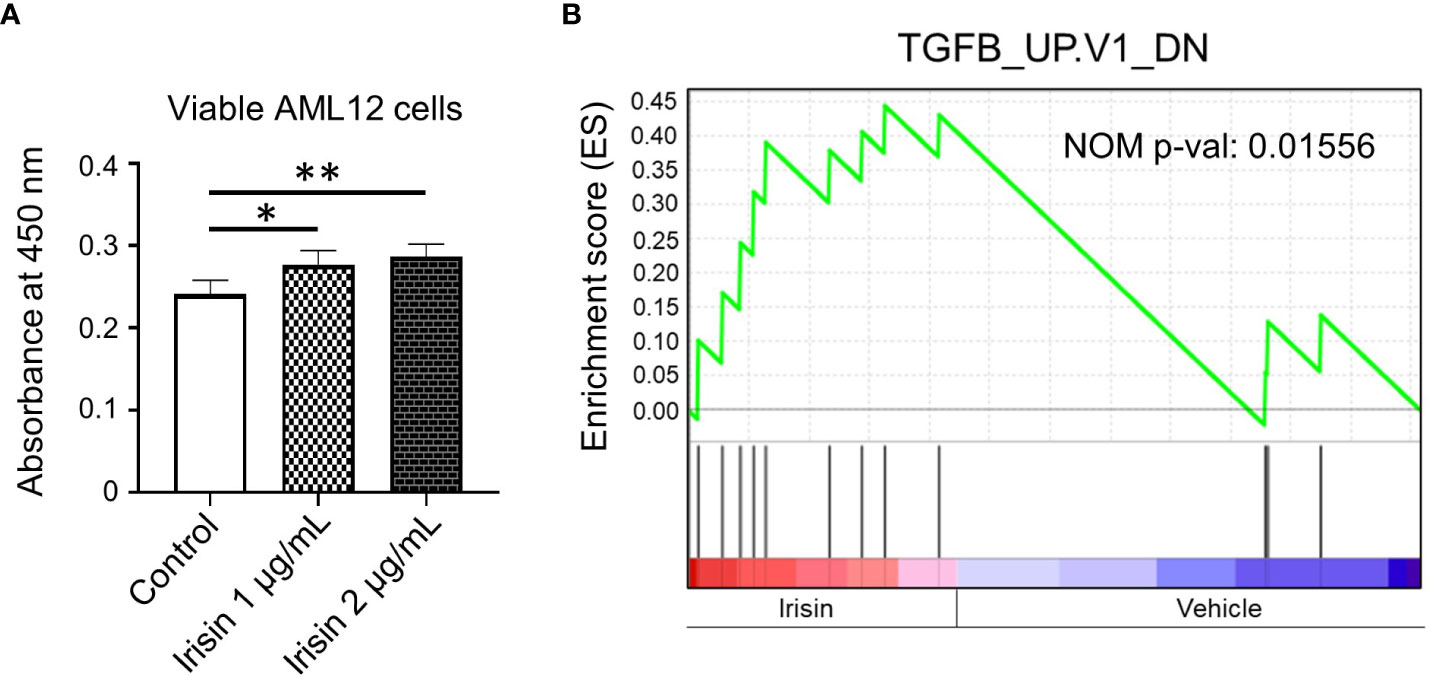
Figure 5 Irisin enhances growth of AML12 mouse liver cells. (A) AML12 cells were treated with 1 µg/mL or 2 µg/mL of irisin or vehicle for 72 hr, followed by cell viability determination. Data represent means ± S.D. (n=3). *P < 0.05 and **P < 0.01 by one-way ANOVA followed by Sidak’s multiple comparisons. (B) Enrichment plots of genes, downregulated by TGF-β overexpression, in differentially expressed genes by irisin treatment in AML12 cells were presented by GSEA. NOM p-val, nominal p-value. The p-value was calculated by GSEA.
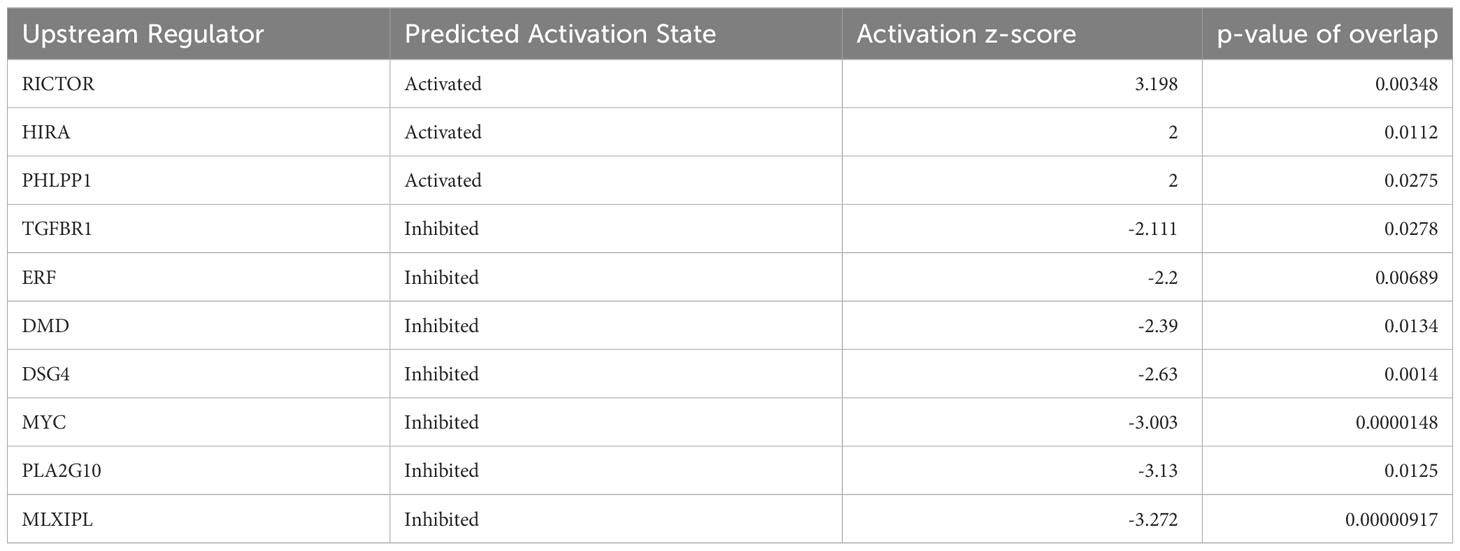
Table 2 Up-stream analysis for FNDC5 responsive genes in AML12 cells.
Irisin/Fndc5 has functional interaction with TGRBR2 for cell proliferation
Next, we assessed the effect of irisin treatment on human hepatocarcinoma HepG2 cell growth. Irisin has been reported to bind with Tgfbr2 resulting in interrupted TGF-β1 signaling pathway (35). We assessed the effect of the combination of transiently expressing irisin and TGFBR2 on the proliferation of HepG2 cells. FNDC5/Irisin and Tgfbr2 protein expressions were confirmed by western blotting analyses in Figure 6A. The expression of either irisin or TGFBR2 promoted cell proliferation and simultaneous expression of both factors yielded a greater promotion (Figure 6B). These results suggested that the two factors have a functional interaction for HepG2 cell proliferation. Next, we analyzed the relation of FNDC5 expression levels with HCC patients’ survival rates (Figures 6C, D). Whereas a higher FNDC5 expression group does not show a poorer prognosis (Figure 6C), higher FNDC5 expression in higher GADD45B expression groups tends to have a poorer prognosis (Figure 6D). Thus, FNDC5 has cooperative effects with GADD45B on prognosis of cancer patients.
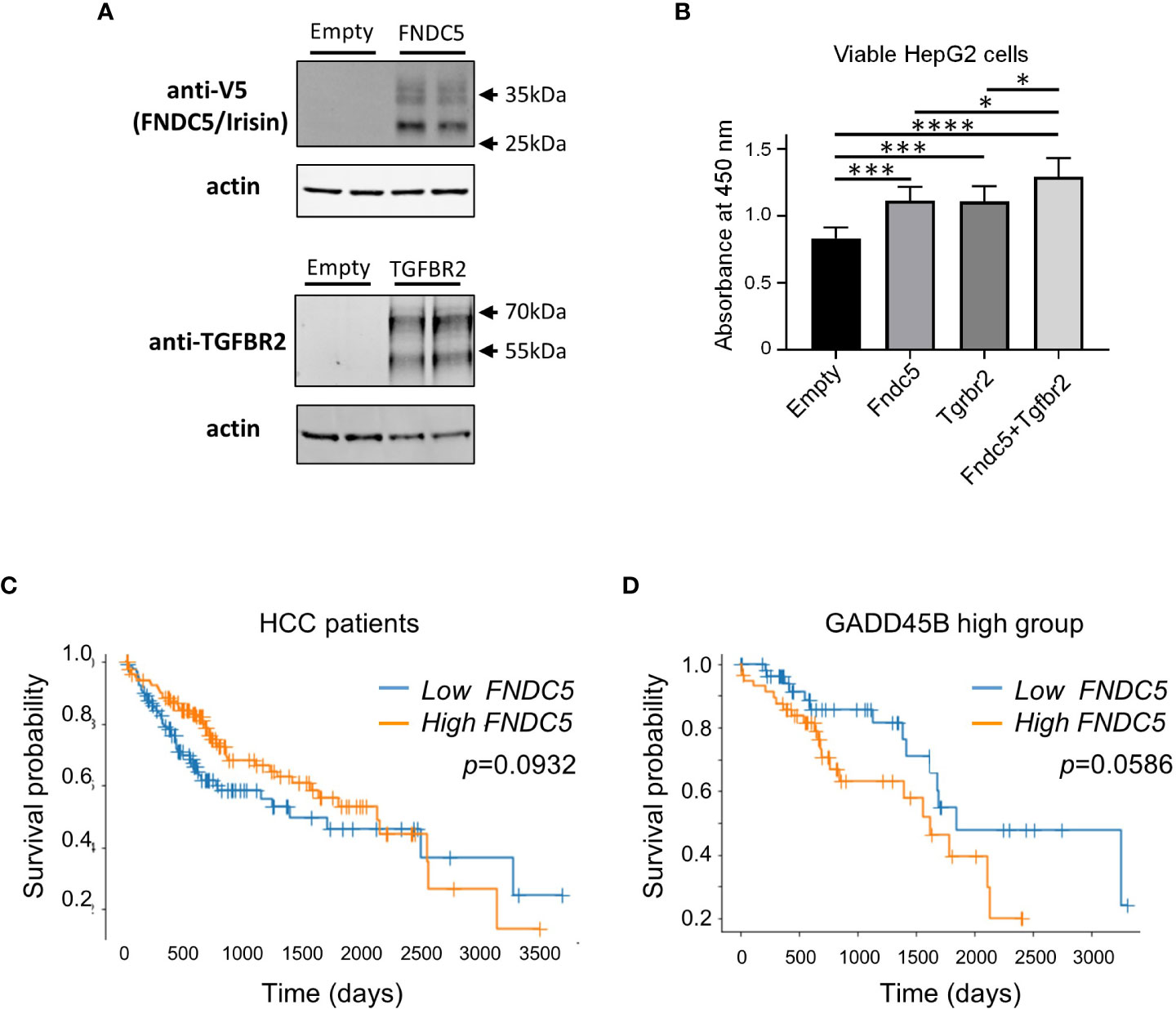
Figure 6 Ectopic expression of Fndc5 and Tgfbr2 enhances HepG2 cell growth. (A) FNDC5/Irisin and Tgfbr2 protein expression in HepG2 cells. HepG2 cells were transfected by each expression plasmid and the proteins were detected 72 hr after the transfection by western blotting analyses as described in Methods section. (B) HepG2 cells were transfected with mock, Tgfbr2, or Fndc5 expression plasmids and then cultured for 72 hr, followed by cell viability determination. Data represent means ± S.D. (n=3). *P < 0.05, ***P < 0.001, and ****P < 0.0001 by one-way ANOVA followed by Sidak’s multiple comparisons. (C) Kaplan-Meier survival curves of patients in the high- (orange) or low- (blue) FNDC5 expression in the patients are displayed. Differences between survival curves were statistically analyzed by using the log-rank test. (D) Kaplan-Meier survival curves of patients in the high- (orange) or low- (blue) FNDC5 expression in the patients with higher Gadd45β expression are displayed. Differences between survival curves were statistically analyzed by using the log-rank test.
Discussion
Our primary target of this study is defining the molecular mechanism of HCC development in rodent livers in the widely adapted HCC animal model system using DEN as the initiator and PB as the promoter. Here we focused on the roles of Gadd45β in the molecular mechanisms since several lines of evidence suggest possible involvement of this molecule in HCC development. Beyond our expectations, the results in this paper showed that Gadd45β null mutation strongly attenuated HCC development in DEN/PB treated mice (Figure 2, Table 1). Moreover, in a database analysis we found human survival rates have a strong association with GADD45β expression levels (Figure 1), suggesting that Gadd45β is involved in not only HCC of rodents but also that of humans. Although we employed C57BL/6, not a carcinogen-susceptible strain such as C3H, and the number of adenomas and carcinomas observed was not large in this study, the results obtained will be valuable to understand tumorigenesis molecular mechanism in rodent HCC. Nonetheless, the validity of our findings needs to be evaluated in different mouse systems using susceptible strains like C3H. Our genomics analysis using microarray revealed that many genes are affected differently by DEN/PB treatment in wild type compared with Gadd45β null mice. We focused on Fndc5 and Tgfbr2 genes among several genes reported to be involved in cell proliferation and apoptosis (Figure 3) (27, 36, 37).
Irisin was originally identified as a myokine hormone cleaved from Fndc5 and secreted from muscle cells regulating the browning of white fat tissue (38). Although CAR activation was shown to induce Fndc5 mRNA expression in the liver (34), the molecular and physiological roles of Fndc5 in this organ have not been well understood (27, 37). Recent studies have shown that irisin promotes the proliferation of osteoblasts (39), endothelial cells (40), neural cells (41), and pancreatic β cells (42) but suppresses the proliferation of lung cancer cells (43). In this study, we demonstrated that the growth of mouse AML12 cells and human HepG2 cells are induced by irisin treatment. Irisin binds to the TGF receptor complex that is composed of the TGFBR2 dimer (35) and may disrupt the TGF signaling pathway and affect cell growth (44, 45) (Figure 7). Indeed, cDNA microarray analysis of AML12 cells suggested an intimate relation of irisin and TGFBR2. Gene expression changes induced by irisin in AML12 cells are negatively associated with genes under the regulation of TGFBR1 in IPA analysis (Table 2). TGFBR2 is playing a major role, forming a heterotetramer with TGFBR1 or forming homotetramer, in the TGF-β signaling pathway (36, 46). This means a substantial part of the genes under TGFBR1 regulation found in Table 2 are also regulated as downstream genes of TGFBR2. Therefore, the results in Table 2 are implying that irisin and TGFBR2 are functionally interacting in AML12 cells treated with irisin.
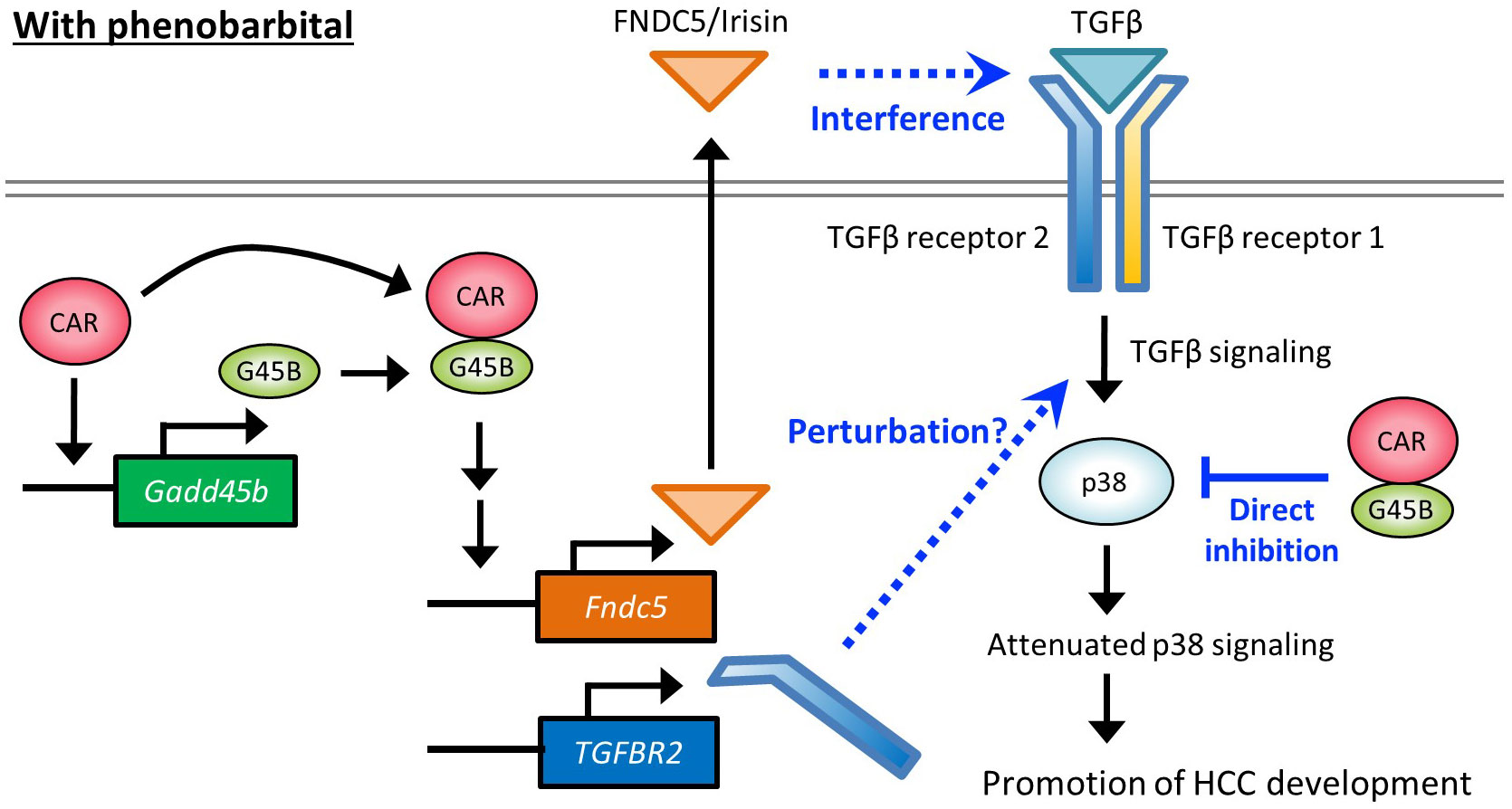
Figure 7 Hypothetical molecular pathways depicting HCC promotion by PB.
Figure 7 proposes hypothetical molecular pathways involved in HCC promotion by PB. Our previous reports using mice treated with PB for long or short periods and cell-based assays have indicated that PB increased CAR-Gadd45β complex that suppresses anti-tumor p38 MAPK activity. The present study with Gadd45β KO mice demonstrated that Gadd45β is essential to the PB-mediated full development of hepatic adenoma and HCC as well as irisin/Fndc5 and Tgfbr2 up-regulation by PB. Irisin can interfere with the TGF-β signaling pathway through the direct interaction with the receptor complex and the induction of Tgfbr2 may perturb this pathway by changing the receptor complex stoichiometry. Overall, our data implies that repression of the p38 MAPK through interference with the TGF-β pathway by the two molecules contributes to HCC development promotion. However, evidence at this point is not enough to describe the details of the molecular functions of these proteins in this pathway, and further studies are necessary for deciphering molecular-level mechanisms that depict how these molecules are involved in PB promoted HCC development in rodents.
In rodents, various non-genotoxic substances, including PB, induce liver carcinogenesis through the mode of action involving CAR activation. However, this phenomenon has not been established in humans (47), and the molecular mechanism depicted in Figure 7 may not have full legitimacy in humans. Nevertheless, even without the direct involvement of CAR, partially overlapped signaling pathways may be encompassed in human HCC development. In fact, there are reports of elevated expression of FNDC5/irisin in human hepatocellular carcinoma (HCC) (28) and our bioinformatic analysis also indicates that high expression of both GADD45β and FNDC5 negatively impacts the prognosis of cancer patients (Figure 6D). Thus, the elucidation of the detailed molecular mechanistic roles played by GADD45β and FNDC5 in liver cancer will contribute to finding new therapeutic targets. On the other hand, multiple studies suggest associations of human liver carcinogenesis with decreased expression of TGFBR2, which appears to contradict our findings in mice (48, 49) (Figure 2C, Figure 3E). Therefore, TGF-β pathways disruption in rodents arises from an imbalance of receptor subunits caused by augmented TGFBR2 expression may not happen in humans for HCC development in the same fashion. This difference may be related to the difference between humans and rodents regarding PB-induced HCC mode of action.
Recently, the Yes-associated protein (YAP) signaling in the Hippo pathway is reported to be an accelerator of hepatocyte proliferation by CAR activation. The Hippo pathway regulates organ size in mammals (50), and functioning as a downstream effector, YAP is involved in the development of HCC (50–53). YAP has been shown to facilitate CAR-mediated hepatocyte proliferation (54–56). TCPOBOP-induced activation of CAR increased the accumulation of YAP in the nucleus and enhanced cell proliferation (57). The functional interaction between TGFBR2 and YAP has been reported in both humans and mice (58, 59), implying that the CAR-YAP pathway could affect TGFBR2 molecular roles proposed in Figure 7 for the CAR-dependent proliferation of hepatocytes. In addition, recent findings suggest that CAR suppresses hepatocellular carcinoma through the erythropoietin signaling pathway in humans (60). Exploring the relationship between YAP pathway or erythropoietin pathway and the pathway depicted in Figure 7 would be a crucial research objective in the future.
In conclusion, the results of this study demonstrate for the first time, to our knowledge, that Gadd45β is a factor in promoting PB-mediated tumorigenesis in mice. In addition, we identified irisin as a possible curial player for HCC development in mice and humans. These findings shed more light on the biological process underlying HCC development.
Data availability statement
GEO accession numbers for microarray study results in this report are GSE232188 and GSE231793. The data will be released to the public on May 10, 2024.
Ethics statement
The animal study was approved by Animal Care and Use Committee, NIEHS, NIH. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
TH, MN, KY, and TS contributed to conception and design of the study. TH and RM collected animal study data. KY and TS performed genomic data analyses. TH and KY performed the statistical analysis. TH, KY, MN, and TS wrote the first draft of the manuscript. All authors contributed to manuscript revision, read, and approved the submitted version.
Funding
This work was supported by the Intramural Research Program of the National Institutes of Health and the National Institute of Environmental Health Sciences [Grant numbers Z01ES71005-01].
Acknowledgments
We thank the molecular genomics core laboratory and histology core laboratory (NIEHS/NIH), Dr. Kyathanahalli Janardhan (Integrated Laboratory Systems/CMPB, DNTP, NIEHS), and Dr. Gordon P. Flake (Cellular and Molecular Pathology Branch, NTP, NIEHS). We also thank Mack Sobhany for his carful editing of this manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1217847/full#supplementary-material
Supplementary Figure 1 | Effects of DEN/PB on liver and body weight in Gadd45β WT and KO. (A) Liver images of WT DEN/PB treated mice. Adenomas were found in livers shown in panels 1 – 3. No tumor was observed in the livers in panels 4 – 8. The liver weight (B), body weight (C), and the liver weight per body weight values (D) in DEN or DEN/PB-treated Gadd45β WT or Gadd45β KO were evaluated. The box plot presents the median, first quartile, and third quartile. The vertical bars represent the 1.5*interquartile range (IQR). *P < 0.05 and **P < 0.01 by one-way ANOVA followed by Sidak’s multiple comparisons.
Supplementary Figure 2 | Effects of Gadd45β on the expression of Cyp2b10 mRNA, Cyp2b9 mRNA, and Cyp2b13 mRNA. Expression of Cyp1b10 mRNA (A), Cyp2b9 mRNA (B), and Cyp2b13 mRNA (C) were analyzed in Cadd45β WT and KO mice. (D) Expression of Cyp2b10 mRNA was analyzed in CAR WT and KO mice. *P < 0.05, **P < 0.01, and ***P < 0.001 by one-way ANOVA followed by Sidak’s multiple comparisons.
Supplementary Figure 3 | Effects of Gadd45β on the expression of multiple genes. Relative mRNA expression was analyzed by real-time PCR for liver samples obtained from Gadd45β WT or KO mice. *P < 0.05, **P < 0.01, and ***P < 0.001 by one-way ANOVA followed by Sidak’s multiple comparisons.
Supplementary Table 1 | Differentially expressed genes in DEN/PB treated vs DEN treated Gadd45β WT or Gadd45β KO mice. (A) Differentially expressed genes by PB/DEN treatment compared with DEN treatment in Gadd45β WT mice. (B) Genes grouped in the common group in Figure 3C, differentially expressed at least 1.5-fold between Gaddd45β WT and Gadd45β KO mice.
References
1. American Cancer Society. Global Cancer Facts & Figures. 4th Edition. Atlanta: American Cancer Society (2018).
2. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
3. Stuver S, Trichopoulos D. 308Cancer of the liver and biliary tract. In: Adami H-O, Hunter D, Trichopoulos D, editors. Textbook of Cancer Epidemiology. Oxford University Press (2008). p. 308–332.
4. Heindryckx F, Colle I, Van Vlierberghe H. Experimental mouse models for hepatocellular carcinoma research. Int J Exp Pathol (2009) 90:367–86. doi: 10.1111/j.1365-2613.2009.00656.x
5. Sueyoshi T, Kawamoto T, Zelko I, Honkakoski P, Negishi M. The repressed nuclear receptor CAR responds to phenobarbital in activating the human CYP2B6 gene. J Biol Chem (1999) 274:6043–6. doi: 10.1074/jbc.274.10.6043
6. Honkakoski P, Zelko I, Sueyoshi T, Negishi M. The nuclear orphan receptor CAR-retinoid X receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol Cell Biol (1998) 18:5652–8. doi: 10.1128/MCB.18.10.5652
7. Yamamoto Y, Moore R, Goldsworthy TL, Negishi M, Maronpot RR. The orphan nuclear receptor constitutive active/androstane receptor is essential for liver tumor promotion by phenobarbital in mice. Cancer Res (2004) 64:7197–200. doi: 10.1158/0008-5472.CAN-04-1459
8. Phillips JM, Yamamoto Y, Negishi M, Maronpot RR, Goodman JI. Orphan nuclear receptor constitutive active/androstane receptor-mediated alterations in DNA methylation during phenobarbital promotion of liver tumorigenesis. Toxicol Sci (2007) 96:72–82. doi: 10.1093/toxsci/kfl188
9. Phillips JM, Goodman JI. Identification of genes that may play critical roles in phenobarbital (PB)-induced liver tumorigenesis due to altered DNA methylation. Toxicol Sci (2008) 104:86–99. doi: 10.1093/toxsci/kfn063
10. Phillips JM, Goodman JI. Multiple genes exhibit phenobarbital-induced constitutive active/androstane receptor-mediated DNA methylation changes during liver tumorigenesis and in liver tumors. Toxicol Sci (2009) 108:273–89. doi: 10.1093/toxsci/kfp031
11. Phillips JM, Burgoon LD, Goodman JI. The constitutive active/androstane receptor facilitates unique phenobarbital-induced expression changes of genes involved in key pathways in precancerous liver and liver tumors. Toxicol Sci (2009) 110:319–33. doi: 10.1093/toxsci/kfp108
12. Phillips JM, Burgoon LD, Goodman JI. Phenobarbital elicits unique, early changes in the expression of hepatic genes that affect critical pathways in tumor-prone B6C3F1 mice. Toxicol Sci (2009) 109:193–205. doi: 10.1093/toxsci/kfp050
13. Yamamoto Y, Moore R, Flavell RA, Lu B, Negishi M. Nuclear receptor CAR represses TNFalpha-induced cell death by interacting with the anti-apoptotic GADD45B. PLoS One (2010) 5:e10121. doi: 10.1371/journal.pone.001012
14. Ohara A, Takahashi Y, Kondo M, Okuda Y, Takeda S, Kushida M, et al. Candidate genes responsible for early key events of phenobarbital-promoted mouse hepatocellular tumorigenesis based on differentiation of regulating genes between wild type mice and humanized chimeric mice. Toxicol Res (Camb) (2017) 6:795–813. doi: 10.1039/C7TX00163K
15. Oomman A, Bansal M. Adding ivabradine to beta-blockers in chronic heart failure: Do not rest without lowering the resting heart rate sufficiently. Indian Heart J (2018) 70:201–3. doi: 10.1016/j.ihj.2018.03.009
16. Yamamoto Y, Negishi M. The antiapoptotic factor growth arrest and DNA-damage-inducible 45 beta regulates the nuclear receptor constitutive active/androstane receptor-mediated transcription. Drug Metab Dispos (2008) 36:1189–93. doi: 10.1124/dmd.108.020628
17. Liebermann DA, Hoffman B. Gadd45 in stress signaling. J Mol Signaling (2008) 3:15. doi: 10.1186/1750-2187-3-15
18. Tamura RE, de Vasconcellos JF, Sarkar D, Libermann TA, Fisher PB, Zerbini LF. GADD45 proteins: central players in tumorigenesis. Curr Mol Med (2012) 12:634–51. doi: 10.2174/156652412800619978
19. Yu Y, Huang H, Li J, Zhang J, Gao J, Lu B, et al. GADD45beta mediates p53 protein degradation via Src/PP2A/MDM2 pathway upon arsenite treatment. Cell Death Dis (2013) 4:e637. doi: 10.1038/cddis.2013.162
20. De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, et al. Induction of gadd45beta by NF-kappaB downregulates pro-apoptotic JNK signalling. Nature (2001) 414:308–13. doi: 10.1038/35104560
21. Cho HJ, Park SM, Hwang EM, Baek KE, Kim IK, Nam IK, et al. Gadd45b mediates Fas-induced apoptosis by enhancing the interaction between p38 and retinoblastoma tumor suppressor. J Biol Chem (2010) 285:25500–5. doi: 10.1074/jbc.M109.091413
22. Hori T, Saito K, Moore R, Flake GP, Negishi M. Nuclear receptor CAR suppresses GADD45B-p38 MAPK signaling to promote phenobarbital-induced proliferation in mouse liver. Mol Cancer Res (2018) 16:1309–18. doi: 10.1158/1541-7786.MCR-18-0118
23. Orlik J, Schungel S, Buitrago-Molina LE, Marhenke S, Geffers R, Endig J, et al. The BH3-only protein BID impairs the p38-mediated stress response and promotes hepatocarcinogenesis during chronic liver injury in mice. Hepatology (2015) 62:816–28. doi: 10.1002/hep.27888
24. Sakurai T, He G, Matsuzawa A, Yu GY, Maeda S, Hardiman G, et al. Hepatocyte necrosis induced by oxidative stress and IL-1 alpha release mediate carcinogen-induced compensatory proliferation and liver tumorigenesis. Cancer Cell (2008) 14:156–65. doi: 10.1016/j.ccr.2008.06.016
25. Das M, Garlick DS, Greiner DL, Davis RJ. The role of JNK in the development of hepatocellular carcinoma. Genes Dev (2011) 25:634–45. doi: 10.1101/gad.1989311
26. Tian J, Huang H, Hoffman B, Liebermann DA, Ledda-Columbano GM, Columbano A, et al. Gadd45beta is an inducible coactivator of transcription that facilitates rapid liver growth in mice. J Clin Invest (2011) 121:4491–502. doi: 10.1172/JCI38760
27. Waseem R, Shamsi A, Mohammad T, Hassan MI, Kazim SN, Chaudhary AA, et al. FNDC5/irisin: physiology and pathophysiology. Molecules (2022) 27. doi: 10.3390/molecules27031118
28. Gaggini M, Cabiati M, Del Turco S, Navarra T, De Simone P, Filipponi F, et al. Increased FNDC5/Irisin expression in human hepatocellular carcinoma. Peptides (2017) 88:62–6. doi: 10.1016/j.peptides.2016.12.014
29. Wrana JL, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, et al. TGF beta signals through a heteromeric protein kinase receptor complex. Cell (1992) 71:1003–14. doi: 10.1016/0092-8674(92)90395-S
30. Bursch W, Grasl-Kraupp B, Wastl U, Hufnagl K, Chabicovsky M, Taper H, et al. Role of apoptosis for mouse liver growth regulation and tumor promotion: comparative analysis of mice with high (C3H/He) and low (C57Bl/6J) cancer susceptibility. Toxicol Lett (2004) 149:25–35. doi: 10.1016/j.toxlet.2003.12.018
31. Huang W, Zhang J, Washington M, Liu J, Parant JM, Lozano G, et al. Xenobiotic stress induces hepatomegaly and liver tumors via the nuclear receptor constitutive androstane receptor. Mol Endocrinol (2005) 19:1646–53. doi: 10.1210/me.2004-0520
32. Evans JG, Collins MA, Savage SA, Lake BG, Butler WH. The histology and development of hepatic nodules in C3H/He mice following chronic administration of phenobarbitone. Carcinogenesis (1986) 7:627–31. doi: 10.1093/carcin/7.4.627
33. Maronpot RR. NTP Nonneoplastic lesion atlas. Available at: https://ntp.niehs.nih.gov/nnl/hepatobiliary/liver/foci/liver-foci_508.pdf.
34. Mo L, Shen J, Liu Q, Zhang Y, Kuang J, Pu S, et al. Irisin is regulated by CAR in liver and is a mediator of hepatic glucose and lipid metabolism. Mol Endocrinol (2016) 30:533–42. doi: 10.1210/me.2015-1292
35. Peng H, Wang Q, Lou T, Qin J, Jung S, Shetty V, et al. Myokine mediated muscle-kidney crosstalk suppresses metabolic reprogramming and fibrosis in damaged kidneys. Nat Commun (2017) 8:1493. doi: 10.1038/s41467-017-01646-6
36. Colak S, Ten Dijke P. Targeting TGF-beta signaling in cancer. Trends Cancer (2017) 3:56–71. doi: 10.1016/j.trecan.2016.11.008
37. Ma J, Chen K. The role of Irisin in multiorgan protection. Mol Biol Rep (2021) 48:763–72. doi: 10.1007/s11033-020-06067-1
38. Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature (2012) 481:463–8. doi: 10.1038/nature10777
39. Qiao X, Nie Y, Ma Y, Chen Y, Cheng R, Yin W, et al. Irisin promotes osteoblast proliferation and differentiation via activating the MAP kinase signaling pathways. Sci Rep (2016) 6:18732. doi: 10.1038/srep18732
40. Song H, Wu F, Zhang Y, Zhang Y, Wang F, Jiang M, et al. Irisin promotes human umbilical vein endothelial cell proliferation through the ERK signaling pathway and partly suppresses high glucose-induced apoptosis. PloS One (2014) 9:e110273. doi: 10.1371/journal.pone.0110273
41. Moon HS, Dincer F, Mantzoros CS. Pharmacological concentrations of irisin increase cell proliferation without influencing markers of neurite outgrowth and synaptogenesis in mouse H19-7 hippocampal cell lines. Metabolism (2013) 62:1131–6. doi: 10.1016/j.metabol.2013.04.007
42. Liu S, Du F, Li X, Wang M, Duan R, Zhang J, et al. Effects and underlying mechanisms of irisin on the proliferation and apoptosis of pancreatic beta cells. PLoS One (2017) 12:e0175498. doi: 10.1371/journal.pone.0175498
43. Shao L, Li H, Chen J, Song H, Zhang Y, Wu F, et al. Irisin suppresses the migration, proliferation, and invasion of lung cancer cells via inhibition of epithelial-to-mesenchymal transition. Biochem Biophys Res Commun (2017) 485:598–605. doi: 10.1016/j.bbrc.2016.12.084
44. Fabregat I, Caballero-Diaz D. Transforming growth factor-beta-induced cell plasticity in liver fibrosis and hepatocarcinogenesis. Front Oncol (2018) 8:357. doi: 10.3389/fonc.2018.00357
45. Tu S, Huang W, Huang C, Luo Z, Yan X. Contextual regulation of TGF-beta signaling in liver cancer. Cells (2019) 8. doi: 10.3390/cells8101235
47. Lake BG. Human relevance of rodent liver tumour formation by constitutive androstane receptor (CAR) activators. Toxicol Res (Camb) (2018) 7:697–717. doi: 10.1039/c8tx00008e
48. Mamiya T, Yamazaki K, Masugi Y, Mori T, Effendi K, Du W, et al. Reduced transforming growth factor-beta receptor II expression in hepatocellular carcinoma correlates with intrahepatic metastasis. Lab Invest (2010) 90:1339–45. doi: 10.1038/labinvest.2010.105
49. Zaidi S, Amdur R, Xiang X, Yu H, Wong LL, Rao S, et al. Using quantitative immunohistochemistry in patients at high risk for hepatocellular cancer. Genes Cancer (2022) 13:9–20. doi: 10.18632/genesandcancer.220
50. Harvey KF, Zhang X, Thomas DM. The Hippo pathway and human cancer. Nat Rev Cancer (2013) 13:246–57. doi: 10.1038/nrc3458
51. Zheng T, Wang J, Jiang H, Liu L. Hippo signaling in oval cells and hepatocarcinogenesis. Cancer Lett (2011) 302:91–9. doi: 10.1016/j.canlet.2010.12.008
52. Yu FX, Zhao B, Guan KL. Hippo pathway in organ size control, tissue homeostasis, and cancer. Cell (2015) 163:811–28. doi: 10.1016/j.cell.2015.10.044
53. Michalopoulos GK. Hepatostat: Liver regeneration and normal liver tissue maintenance. Hepatology (2017) 65:1384–92. doi: 10.1002/hep.28988
54. Kowalik MA, Saliba C, Pibiri M, Perra A, Ledda-Columbano GM, Sarotto I, et al. Yes-associated protein regulation of adaptive liver enlargement and hepatocellular carcinoma development in mice. Hepatology (2011) 53:2086–96. doi: 10.1002/hep.24289
55. Bhushan B, Molina L, Koral K, Stoops JW, Mars WM, Banerjee S, et al. Yes-Associated protein is crucial for constitutive androstane receptor-driven hepatocyte proliferation but not for induction of drug metabolism genes in mice. Hepatology (2021) 73:2005–22. doi: 10.1002/hep.31521
56. Gao Y, Fan S, Li H, Jiang Y, Yao X, Zhu S, et al. Constitutive androstane receptor induced-hepatomegaly and liver regeneration is partially via yes-associated protein activation. Acta Pharm Sin B (2021) 11:727–37. doi: 10.1016/j.apsb.2020.11.021
57. Abe T, Amaike Y, Shizu R, Takahashi M, Kano M, Hosaka T, et al. Role of YAP activation in nuclear receptor CAR-mediated proliferation of mouse hepatocytes. Toxicol Sci (2018) 165:408–19. doi: 10.1093/toxsci/kfy149
58. Nishio M, Sugimachi K, Goto H, Wang J, Morikawa T, Miyachi Y, et al. Dysregulated YAP1/TAZ and TGF-beta signaling mediate hepatocarcinogenesis in Mob1a/1b-deficient mice. Proc Natl Acad Sci U S A (2016) 113:E71–80. doi: 10.1073/pnas.1517188113
59. Fan Y, Gao Y, Rao J, Wang K, Zhang F, Zhang C. YAP-1 promotes tregs differentiation in hepatocellular carcinoma by enhancing TGFBR2 transcription. Cell Physiol Biochem (2017) 41:1189–98. doi: 10.1159/000464380
Keywords: phenobarbital, chemical carcinogenesis, growth arrest and DNA-damageinducible 45 beta, TGFbeta signaling, irisin
Citation: Hori T, Yokobori K, Moore R, Negishi M and Sueyoshi T (2023) CAR requires Gadd45β to promote phenobarbital-induced mouse liver tumors in early stage. Front. Oncol. 13:1217847. doi: 10.3389/fonc.2023.1217847
Received: 19 May 2023; Accepted: 17 August 2023;
Published: 07 September 2023.
Edited by:
Yusuke Kanno, Tokyo Institute of Technology, JapanReviewed by:
Kouichi Yoshinari, University of Shizuoka, JapanToshiyuki Sakaki, Toyama Prefectural University, Japan
Copyright © 2023 Hori, Yokobori, Moore, Negishi and Sueyoshi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tatsuya Sueyoshi, c3VleW9zaGlAbmllaHMubmloLmdvdg==