 Min Gao
Min Gao Yongwen Li
Yongwen Li Peijun Cao
Peijun Cao Hongyu Liu
Hongyu Liu Jun Chen
Jun Chen Shirong Kang
Shirong Kang- 1Department of Thoracic Surgery, The Affiliated Hospital of Inner Mongolia Medical University, Hohhot, China
- 2Inner Mongolia Medical University, First Clinical Medical College, Hohhot, China
- 3Tianjin Key Laboratory of Lung Cancer Metastasis and Tumor Microenvironment, Tianjin Lung Cancer Institute, Tianjin Medical University General Hospital, Tianjin, China
- 4Department of Lung Cancer Surgery, Tianjin Medical University General Hospital, Tianjin, China
The pathogenesis of lung cancer (LC) is a multifaceted process that is influenced by a variety of factors. Alongside genetic mutations and environmental influences, there is increasing evidence that epigenetic mechanisms play a significant role in the development and progression of LC. The Polycomb repressive complex 2 (PRC2), composed of EZH1/2, SUZ12, and EED, is an epigenetic silencer that controls the expression of target genes and is crucial for cell identity in multicellular organisms. Abnormal expression of PRC2 has been shown to contribute to the progression of LC through several pathways. Although targeted inhibition of EZH2 has demonstrated potential in delaying the progression of LC and improving chemotherapy sensitivity, the effectiveness of enzymatic inhibitors of PRC2 in LC is limited, and a more comprehensive understanding of PRC2’s role is necessary. This paper reviews the core subunits of PRC2 and their interactions, and outlines the mechanisms of aberrant PRC2 expression in cancer and its role in tumor immunity. We also summarize the important role of PRC2 in regulating biological behaviors such as epithelial mesenchymal transition, invasive metastasis, apoptosis, cell cycle regulation, autophagy, and PRC2-mediated resistance to LC chemotherapeutic agents in LC cells. Lastly, we explored the latest breakthroughs in the research and evaluation of medications that target PRC2, as well as the latest findings from clinical studies investigating the efficacy of these drugs in the treatment of various human cancers.
1 Introduction
Lung cancer (LC) is one of the most common malignancies worldwide and has taken the lead role in cancer-related deaths in recent decades (1). Although progress has been made in early detection and molecularly targeted therapies, the prognosis for patients with advanced LC remains poor due to drug resistance and tumor recurrence and metastasis (2–4). As a result, identifying new therapeutic targets to offer personalized targeted therapy in precision medicine and overcome drug resistance has become a promising approach to improve treatment outcomes.
Epigenetics is a process that alters gene expression inheritance patterns without changing the DNA sequence. Epigenetic variations, such as DNA methylation, RNA modifications, and histone modifications, are more frequent and significant than somatic mutations and contribute to cancer development (5). Polycomb repressive complex 2 (PRC2) is a chromatin-modifying complex that is essential for maintaining cell differentiation and determining cell fate via the addition of lysine 27 trimethylation to histones (H3K27me3) (6, 7). Elevated levels of H3K27me3 in LC tissues are strongly associated with the development and prognosis of LC (8). Misexpression or mutation of PRC2 can lead to aberrations in cancer signaling pathways, contributing to cancer and other diseases (6). Due to its significant role in the development and progression of LC, PRC2 is a promising target for targeted therapies. The main objective of this article is to examine the association between PRC2 and the initiation, metastasis, and drug resistance of LC. It delves into the epigenetic factors involved in the development of lung cancer and reviews the most recent research developments in PRC2-targeted therapies for various types of tumors, particularly LC.
2 PRC2
PRC2 possesses histone methyltransferase activity and is crucial for regulating cell differentiation and development. The catalytic roles of PRC2 are primarily played by the enhancer of zeste homolog 1 or 2 (EZH1/2), trimers of suppressor of zeste 12 protein homolog (SUZ12), and embryonic ectoderm development (EED) proteins, which interact interdependently to maintain the catalytic activity of PRC2 (9–13). These core subunits, along with retinoblastoma-binding protein 4 or 7 (RBBP4 and RBBP7; also known as RbAp48 and RbAp46), constitute the PRC2 complex responsible for all monomethylation, demethylation, and trimethylation (H3K27me1, H3K27me2, and H3K27me3) on histone H3, thus regulating gene expression by ensuring appropriate gene silencing (6, 14–16) (Figure 1).
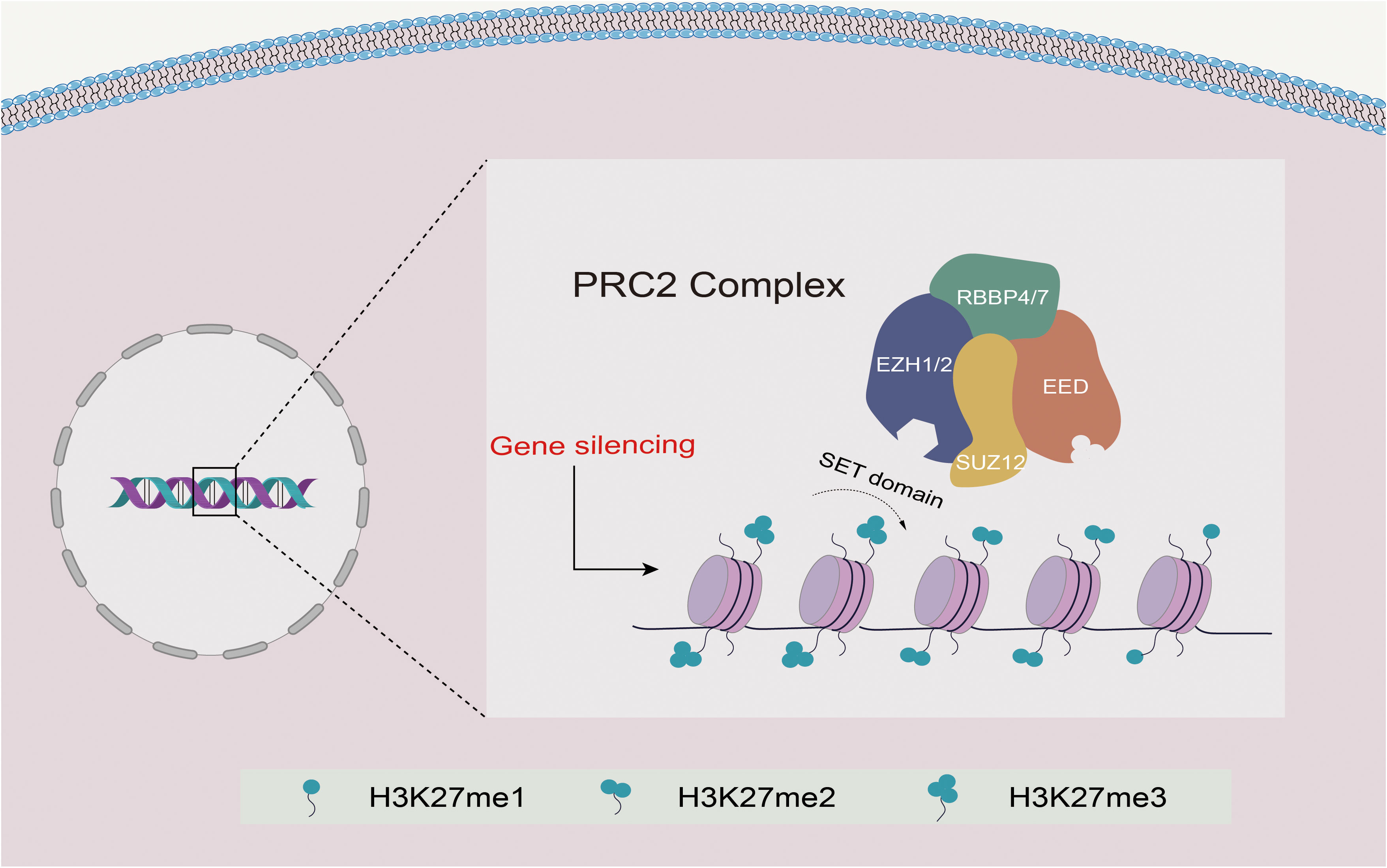
Figure 1 Schematic diagram of the structure and function of PRC2. EZH1/2 contains a SET domain with methyltransferase activity that mediates the methylation of H3K27 and promotes transcriptional silencing. EED binds to H3K27me3, resulting in allosteric activation of EZH1/2, while SUZ12 mainly stabilizes the complex. Together with RBBP4/7, they form the PRC2 complex, mediating PRC2 recruitment and promoting H3K27me1/2/3.
3 PRC2 subunits and their interactions
The core subunits of PRC2 include SUZ12, EED, two methyltransferases (EZH1 and EZH2), RBBP4, and RBBP7. Both EZH1 and EZH2 exhibit enzymatic methyltransferase activity via the C-terminal SET domain (17). While EZH2 is the primary catalytic subunit that more efficiently methylates H3K27 than EZH1, EED and SUZ12 must also contribute to the catalytic activity of PRC2 to prevent autoinhibition of EZH2 (18–20). Following EZH2 trimethylates H3K27, the EED protein recognizes and binds to H3K27me3 through its WD40 domain, resulting in the activation of EZH2 in a positive feedback mechanism. This feedback loop allows the spread of H3K27me2/me3 modifications over large stretches of DNA, leading to the formation of extended domains of repression (21, 22). SUZ12 stabilizes PRC2 through its C-terminal VEFS domain, and its delta-VEFS can be recruited to target sites independently of other core subunits (15) (Figure 1).
Negative-stain electron microscopy has preliminarily investigated the conformation of PRC2 and the physical interactions among its subunits (17). PRC2 is composed of two lobes with distinct functions: the catalytic lobe and the targeting lobe, which are connected by SUZ12 (17, 23).
Within the catalytic leaf, the VEFS domain of SUZ12 interacts with either EZH1 or EZH2. By rotating its SET-inserted domain and rearranging the post-SET domain, the SET domain of EZH2 changes from an autoinhibitory to an active conformation that promotes its methyltransferase activity (12, 20, 24–26). The targeted lobe of PRC2 is composed of SUZ12, RBBP4/7, and several accessory proteins.
4 Mechanism of abnormal expression of PRC2 in cancer
4.1 PRC2 in transcription
PRC2 is a crucial epigenetic regulator involved in transcriptional repression and cell fate regulation in cancer development. The major catalytic subunit EZH2 is repressed at the transcriptional level through its SET domain, which functions as a histone methyltransferase to regulate transcription in a PRC2-dependent manner. EZH2 can form transcriptional complexes with other factors to activate target genes downstream. For example, SETD1A can induce H3K4me3 enrichment and activate transcription of NEAT1 and EZH2 in NSCLC, leading to activation of the Wnt/β-linked protein axis and promoting tumorigenesis (27).
There are several microRNAs that are known to play a role in the transcriptional regulation of PRC2 in LC. miR-4465 can bind to the 3’-UTR of EZH2, reducing its expression and inhibiting the proliferation, migration, and invasion of NSCLC cells (28). miR-145 can significantly reduce RBBP4 promoter activity, mRNA and protein levels, and hsa_circ_0102231 can promote NSCLC proliferation and invasion by reversing the anti-tumor effects of miR-145 through the miR-145/RBBP4 axis (29). Additionally, the functional conversion of EZH2 from histone methyltransferase to non-histone methyltransferase is dependent on long-stranded non-coding RNA (lncRNA) P21. Epigenetic silencing of the lncRNA SPRY4-IT1 in NSCLC cells occurs through direct binding of EZH2 to the SPRY4-IT1 promoter region, leading to transcriptional repression. The SPRY4-IT1 plays a role in the proliferation and metastasis of NSCLC cells by modulating epithelial-mesenchymal transition (EMT) through the regulation of e-calmodulin and waveform protein expression (30). In summary, PRC2 has diverse functions in controlling target gene expression through its interactions with various transcription factors and miRNAs and lncRNAs can influence the activity of PRC2 in lung cancer.
4.2 PRC2 in translation and post-translational
Post-translational modifications (PTMs) are essential biological processes in cancer development that regulate the conformation and functionality of proteins. EZH2, the primary catalytic subunit of PRC2, is subject to various PTMs, including phosphorylation, ubiquitination, O-GlcNAcylation, acetylation, and methylation. These PTMs can modulate EZH2 histone methyltransferase activity, localization, binding partners, and stability, affecting its role in cancer development and progression.
There is a growing body of evidence supporting the critical function of PTMs of EZH2 in cancer development. For example, EZH2 can be acetylated at lysine 348 (K348) by the acetyltransferase P300/CBP-associated factor (PCAF), which increases its stability, ability to repress target genes, and enhances the migration and invasion of LC cells (31). Phosphorylation of threonine 416 by cyclin-dependent kinase 2 promotes the recruitment of EZH2 at the target gene promoter, while methylation of lysine 307 (K307) by SMYD2 enhances the stability of EZH2 and promotes breast cancer cell proliferation, epithelial mesenchymal transition, and invasion (32, 33). O-GlcNAcylation of EZH2 at multiple sites, such as S73A, S76A, S84A, T313A, and S729A, enhances its stability and methylation enzyme activity, promoting tumor progression (34). In addition, cell cycle protein-dependent kinase 1 (CDK1)-mediated phosphorylation of threonines 345 and 487 promotes ubiquitinated degradation of EZH2 in breast cancer cells, cervical cancer cells, and LC cells (35). Investigating how PRC2 is regulated by various types of PTMs and its involvement in cancer development, as well as deciphering its underlying molecular mechanisms, may present promising opportunities for LC treatment.
4.3 PRC2 in cancer immunity
The tumor microenvironment (TME) is a complex immunoregulatory network in which various immune cells and factors interact with cancer cells. The expression of EZH2, the major subunit of the PRC2 complex, in both tumor and immune cells can induce epigenetic and transcriptomic changes, which mobilize various elements of the TME and promote the development of solid tumors with immunosuppressive activity.
EZH2 mediates T-cell activity in tumors primarily through the regulation of T-cell antigen-presenting genes, such as β-2-microglobulin (β2M), CTLA-4, the HLA family, and PD-L1. EZH2 can inhibit activation of T-cell by raising PD-L1 expression (36). Additionally, the effect of EZH2 on T-cell activity is dependent on various chemokines and cytokines within the TME, such as Th1-type chemokines like CXCL9 and CXCL10, which have a remarkable effect on the transport of effector CD8+ T cells. In ovarian tumors, EZH2 expression was negatively correlated with intratumoral CD8+ T cells, and its expression inhibited the production of CXCL9 and CXCL10, thereby affecting CD8+ T cell infiltration within the tumor (37, 38). Chemokine ligand 5 (CCL5) has also been identified as one of the key chemokines recruited by macrophages in tumor cells. CCL5 plays a huge part in the proliferation and invasion of LC cells (39, 40). Overexpression of EZH2 in LC cells promotes the production of tumor-derived CCL5, leading to recruitment of M2-like macrophages and immunosuppressive Treg cells, which can facilitate tumor proliferation and metastasis while inhibiting CD8+ T cells (41). T-cell exhaustion is defined as a low T-cell response and diminished function after prolonged antigen exposure. It is usually characterized by decreased production of cytokines, such as IFN-γ, TNF-α, and granzyme b, and is associated with poor clinical prognosis in solid tumors (42, 43). Inhibition of EZH2 induces a sustained antitumor response, culminating in T-cell failure and checkpoint activation (44).
Myeloid-derived suppressor cells (MDSCs) are a heterogeneous group of myeloid cells with immunosuppressive properties, derived from myeloid progenitors and immature myeloid cells. They play a huge part in cancer progression by shaping the tumor microenvironment through immunosuppression and inflammation (45). Loss of EZH2 activity is related to the production of MDSCs. Studies have shown that circulating-associated kinases activate the EZH2/NF-κB signaling pathway. This leads to the formation of a complex between EZH2 and NF-κB, which binds to the promoter of IL-6 and promotes the accumulation of immunosuppressive MDSCs (44). This, in turn, creates a microenvironment that suppresses the immune system and promotes drug resistance. EZH2 has been demonstrated to preserve the functionality of hematopoietic progenitor cells by stabilizing chromatin structure and suppressing the transcription of genes that promote differentiation (46). The EZH2 inhibitor GSK126 promotes the differentiation of hematopoietic progenitor cells into MDSCs, which can suppress CD4+ and CD8+ T cells, contributing to antitumor immunity (47).
Regulatory T cells (Tregs) are essential for immune homeostasis as they exert their effect by suppressing immune responses. However, their presence in tumor tissue impairs antitumor immunity and promotes tumor development and metastasis. EZH2 functions as an epigenetic silencer in tumor-infiltrating Treg cells to inhibit the expression of IL-4 and IFN-γ, while enhancing Foxp3 expression and Treg activation (48). Foxp3 recruits EZH2 to repress key genes during the inflammatory response in Treg cells (49–51). On the contrary, inhibition of EZH2 may impair Foxp3 expression, reduce the suppressive activity of Tregs, reshape the tumor microenvironment, enhance CD8 and CD4 effector T cell recruitment and function, and thus enhance antitumor immunity (52, 53).
Natural killer (NK) cells are essential suppressors of cancer process and are involved in the innate immune response (54).In the tumor microenvironment, NK cell differentiation and function are inhibited. The NK activation signaling pathway relies on the presence of NK group 2 member D (NKG2D) receptors on the cell membrane, and when the NKG2D receptor binds to its ligand, NK initiates a cytotoxic immune response in TME (55). EZH2 in cancer cells downregulates NKG2D ligand expression or TME and inhibits NK cell activation (36). EZH2 deletion or inhibition improves mature NK cell function and enhances its specificity by upregulating NKG2D (56). EZH2 inhibition enhances the eradication of hepatocellular carcinoma by NK cells in hepatocellular carcinoma (57). To summarize, modifying the epigenetic landscape can potentially improve the anti-tumor immune response by altering the tumor microenvironment. Therefore, combining immunotherapy with targeted inhibition of EZH2 represents a promising approach for cancer treatment.
5 Correlation of PRC2 with pulmonic carcinogenesis and invasive metastasis
LC is a complex disease influenced by many factors, including genetics. In recent years, researchers have focused on the role of the PRC2 complex, specifically EZH2, in promoting the invasive metastasis of LC cells (Figure 2).
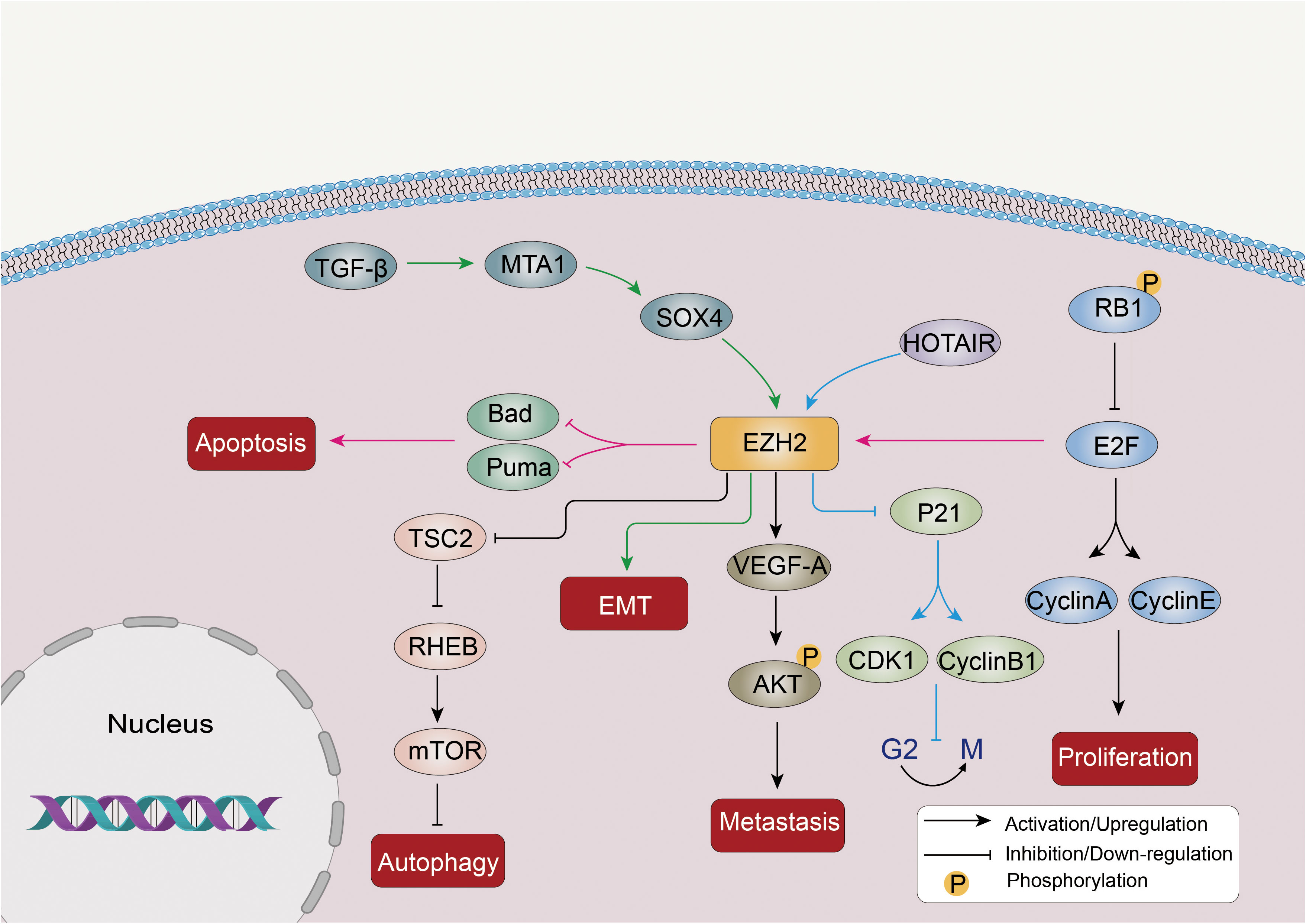
Figure 2 Abnormal expression of EZH2 promotes proliferation, invasion, metastasis and autophagy inhibition of LC cells through multiple mechanisms. EZH2 is involved in the EMT drive of the TGF-β-MTA1-SOX4-EZH2 signaling axis in cancer. In addition, EZH2 can inhibit TSC2, the negative regulator of the mTOR pathway, and further inhibit RHEB expression, thereby activating the mTOR signaling pathway and inhibiting autophagy. Other molecular pathways such as STAT3 and P21 are also affected by EZH2 in LC.
EZH2 promotes EMT, a process in which epithelial cells acquire a mesenchymal phenotype. EMT is related to many cancer features, including metastasis, immune evasion, and resistance to therapy (58). SOX4 activates EMT by directly binding to the EZH2 promoter and upregulating its expression (59). TGF-β induces the downstream targets MTA1 and SOX4, leading to further activation of EZH2 during EMT induction (60). EZH2 overexpression is linked to EMT induction and the migration of cancer cells, including those in pancreatic (61), NSCLC (62), and prostate cancer (63).
The PRC2 complex promotes LC progression by activating apoptosis and inhibiting the cell cycle. Dysregulation of the RB1/E2F pathway, in which the retinoblastoma protein (RB1) normally represses E2F transcription factors to inhibit proliferation, can lead to EZH2 overexpression in SCLC (64). Phosphorylation-mediated inactivation of RB1 can result in the release of the E2F protein, which acts as a transcription factor and triggers the activation of various factors involved in cell cycle progression, such as cyclins A and E. Overactivation of E2F can also induce p21 and activate apoptosis, leading to cell cycle inhibition (65, 66). As a downstream factor of the RB1/E2F pathway, dysregulation of this pathway can lead to the overexpression of EZH2 in SCLC (66). In addition, EZH2 exerts a negative influence on the expression of pro-apoptotic factors such as Puma and Bad, as well as cell cycle inhibitors like p21, ultimately promoting the development of SCLC tumors (66). LncRNAs, such as HOX transcript antisense intergenic RNA (HOTAIR), can affect PRC2 expression. For example, HOTAIR upregulation leads to EZH2 overexpression, which inhibits p21 and modulates CDK1 and the cell cycle at the G2/M transition (67). EZH2 overexpression also increases VEGF-A expression, activates AKT phosphorylation, and enhances proliferation, invasion, and migration of LC cells (68).
EZH2 can epigenetically suppress various negative regulators of the mTOR pathway, including TSC2, RHOA, and GPI, further inhibiting RHEB expression, thereby activating the mTOR signaling pathway and suppressing autophagy (69). A study has confirmed that enhanced protective autophagy could be one of the mechanisms leading to EGFR-TKI resistance in NSCLC, and knocking down or inhibiting EZH2 in NSCLC cells led to increased autophagy, indicating that EZH2 acts as a negative regulator of autophagy (70).
SUZ12 is an oncogene of NSCLC, and its knockdown inhibits tumor cell growth, migration, and invasion (71). Gargiulo et al. (72) discovered that EED deletion resulted in PRC2 inactivation and delayed tumor progression in KRAS-driven LC. RBBP4 is highly expressed in NSCLC tissues, and its knockout significantly reduces the proliferation and invasiveness of NSCLC cells, while its overexpression has the opposite effects (29).
6 PRC2 mediate therapy resistance in LC
Standard treatment options for LC includes surgery, platinum-based chemotherapy, radiotherapy, combination chemotherapy and targeted therapy, alone or in combination. However, treatment resistance and tumor progression remain major challenges for patients with advanced and metastatic LC. Therefore, researchers have increasingly focused on the potential of inhibitors of the PRC2 complex in tumor therapy due to the reversibility of epigenetic modifications. Studies have shown that EZH2 inhibition prevents the emergence of acquired resistance and enhances chemotherapeutic efficacy in LC.
Cancer stem cells (CSCs) are responsible for cancer progression and the induction of resistance to therapy. In LC cells, miRNA-21 promotes the expression level of EZH2 and enhances the progression of CSCs, leading to resistance to radiotherapy (73). EZH2 is also recruited through lncRNA 00665 to activate the PI3K/AKT pathway and induce acquired resistance to gefitinib in NSCLC (74). Furthermore, Riquelme et al. (75) observed that EZH2 expression can be induced by VEGF/VEGFR-2 activation through upregulation of E2F3 and hypoxia-inducible factor-1α (HIF1α), while downregulation of miR-101. They also found that depletion of EZH2 reduced the malignant potential of lung adenocarcinoma cells and increased their sensitivity to platinum-based or VEGFR-2 targeted therapies.
HOTAIR was found to be upregulated in gefitinib-resistant lung cancer cells, and its overexpression increased the resistance of LC cells to gefitinib. HOTAIR and EZH2 collaborated to silence the expression of p16 and p21 through H3K27me3 in the promoter regions, leading to accelerated gefitinib resistance. And inhibition of EZH2 sensitized LC cells to gefitinib by reversing the silencing of p16 and p21 mediated by HOTAIR and EZH2 (67).
7 Correlation between PRC2 expression and prognosis of LC patients
In a particular study, the research revealed an upregulation of EZH2 expression in NSCLC cells compared to normal human bronchial epithelial cells, as evidenced by protein blotting assay (76). Additionally, the study employed Taqman quantitative real-time RT-PCR to measure EZH2 expression in matched tumor and normal tissue samples. Notably, NSCLC patients whose tumors exhibited higher EZH2 expression experienced notably reduced overall survival rates, along with diminished disease-specific and disease-free survival rates, as compared to patients with lower EZH2 expression (P = 0.005, P = 0.001, and P = 0.003, respectively) (76). The study’s findings propose that EZH2 mRNA levels hold potential as a valuable prognostic indicator for NSCLC patients. In another study, 157 surgically resected cases of NSCLC were meticulously evaluated using immunohistochemistry. The study findings illuminated a compelling association between EZH2 expression in tumor cells and patient prognosis. Notably, patients with high EZH2 expression exhibited significantly poorer outcomes compared to those with low EZH2 expression across all pathological stages of NSCLC (P = 0.001), including the subset of patients with pathological stage I NSCLC (P = 0.006). Furthermore, a multifactorial analysis of this study underscored that high EZH2 expression independently served as a detrimental prognostic factor in patients with pathological stage I disease (P = 0.048). It’s worth noting that high EZH2 expression was notably linked with several clinicopathological characteristics, including non-adenocarcinoma histology (P = 0.001), moderate and low differentiation (P = 0.001), advanced pathological tumor classification (P = 0.02), and elevated Ki-67 and cell cycle protein E labeling index (P < 0.001) (77). They highlight the potential utility of EZH2 as an independent predictor of patient outcomes, particularly in the context of early-stage disease. Another study, confirmed through immunohistochemistry, established that EZH2 expression exhibited a significant increase in squamous lung carcinoma compared to adenocarcinoma. Within the adenocarcinoma subtype, higher EZH2 expression was notably associated with younger age, smoking history, and advanced TNM stage (78). These results shed light on the clinical significance of EZH2 in NSCLC and underscore its potential as a predictive biomarker for patient outcomes.
Furthermore, the deletion of EED has been demonstrated to promote the development of KRAS-driven NSCLCs (79). In a separate study, the expression levels of SUZ12 were assessed in 40 pairs of clinical NSCLC tissues and adjacent normal tissues using quantitative reverse transcription polymerase chain reaction (qRT-PCR). The findings from this study revealed significant dysregulation of SUZ12 expression in NSCLC tissues when compared to adjacent paracancerous tissues (P<0.05) (71). Moreover, this abnormal SUZ12 expression was closely associated with key clinical parameters, including tumor size, lymph node metastasis, and clinical stage (P<0.05) (71). Furthermore, the study conducted siRNA-mediated knockdown experiments of SUZ12, which demonstrated a noteworthy inhibition of tumor cell growth, migration, and invasion. These results strongly suggest a potential oncogenic role for SUZ12 in the initiation and progression of NSCLC. These compelling lines of evidence collectively indicate that SUZ12 functions as an oncogene in NSCLC. This implies that SUZ12 holds promise as a novel diagnostic marker for NSCLC, and it may emerge as a promising therapeutic target for interventions in NSCLC.
In summary, a compelling association emerges between the expression of the PRC2 complex and lung cancer prognosis, indicating its potential as a predictive biomarker for patient outcomes. Furthermore, techniques such as quantitative PCR and immunohistochemistry can be valuable tools in clinical studies for assessing the correlation between gene expression and various clinicopathological factors. Notably, quantitative PCR offers the advantage of simplicity and feasibility in such investigations.
8 PRC2 inhibitor
Given the pivotal role played by PRC2 in cancer development, it has become a potential therapeutic target for cancer. Different inhibitors with different mechanisms of action have been developed, including EZH2 inhibitors and EED inhibitors (Table 1).
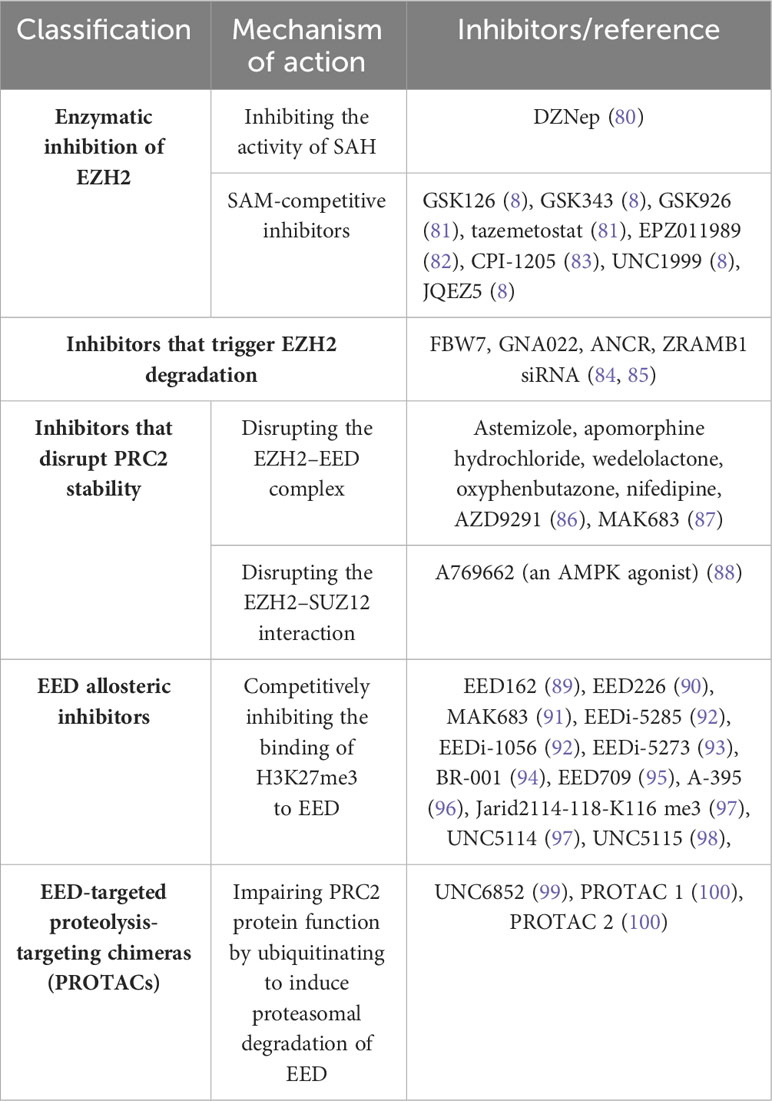
Table 1 PRC2 inhibitors and their mechanisms of action.
EZH2 inhibitors can be categorized into three groups: enzymatic inhibitors, destabilizers of the PRC2 complex, and inducers of EZH2 degradation. Enzymatic inhibitors, such as 3-Deazaneplanocin A (DZNep), indirectly inhibit EZH2 by increasing the activity of S-adenosyl-L-homocysteine hydrolase (SAH), leading to apoptosis in tumor cells (101). However, DZNep is not specific for EZH2 and can globally inhibit histone methylation. Highly selective S-adenosyl-L-methionine (SAM)-competitive inhibitors of EZH2 methyltransferase activity, including tasimetostat and GSK126, have been developed and are undergoing clinical trials (102, 103). EZH2 inhibitors such as AZD9291 (86) and MAK683 (87, 98) reduce EZH2 protein levels by disrupting the EED-EZH2 interaction. Another strategy for inhibiting EZH2 is triggering EZH2 degradation, such as Gambogenic acid (GNA) derivative GNA022 (84).
Current cancer treatment strategies for EED include protein-protein interaction inhibitors, allosteric inhibitors, and proteolytically targeted chimeras (PROTACs). Compounds that competitively occupy the H3K27me3 binding pouch in EED can combat PRC2-driven carcinogenesis by inhibiting EED-H3K27me3 interactions and disrupting allosteric activation of PRC2 (104). EED allosteric inhibitors with therapeutic potential include EED162 (89), EED226 (90), MAK683 (91), BR-001 (105), EED709 (95), A-395 (96), Jarid2114-118-K116 me3 (97), UNC5114 (97), and UNC5115 (98). In EZH2-resistant cells, EZH2 inhibitors such as GSK126 and tazemetostat fail to inhibit the development of diffuse large B-cell lymphoma (DLBCL) cells, with a significant reduction of apoptosis (106). In contrast, EED inhibitors still show efficacy in suppressing the proliferation of these cells, suggesting the potential advantage of EED inhibitors for treating SAM-competitive EZH2 inhibitor-resistant cancers (94).
9 Current status of PRC2 inhibitors in LC treatment
We review the potential of PRC2-targeted inhibition in the treatment of LC, including its use in combination with other inhibitors and chemotherapeutic agents, as well as its impact on drug sensitivity. Additionally, we summarize ongoing clinical trials investigating various PRC2 inhibitors (Table 2).
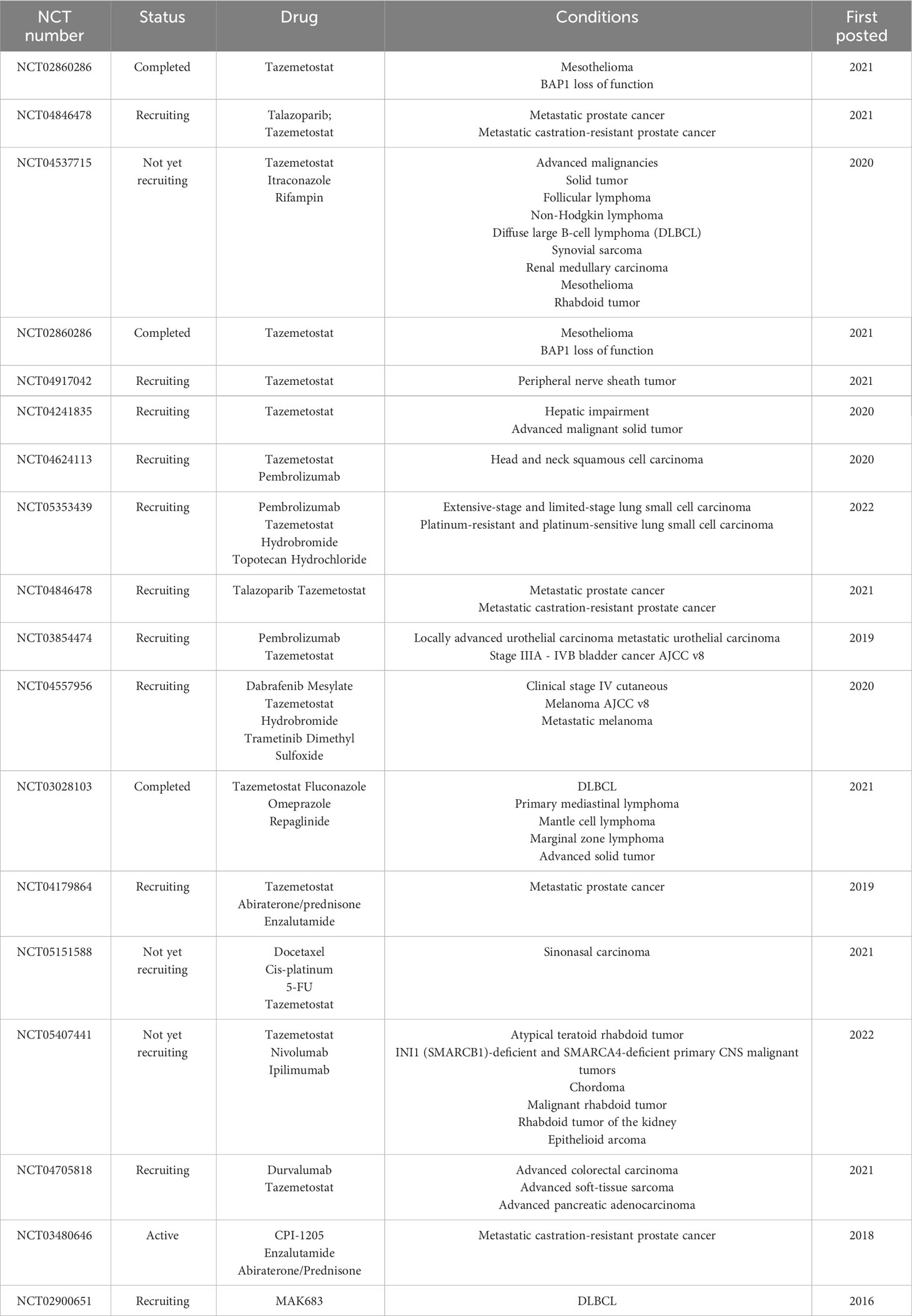
Table 2 Current clinical trials of PRC2 inhibitors.
Tazemetostat is a selective oral EZH2 inhibitor that was approved in January 2020 as the first inhibitor to treat adults and adolescents who do not meet the indications for complete resection (in metastatic or locally advanced epithelial sarcoma) (102). Recent research shows that tazemetostat has a positive antitumor effect on solid tumors such as refractory malignant pleural mesothelioma (107), PBRM1-mutated human chordoma xenograft (108), and advanced epithelioid sarcoma with the loss of INI1/SMARCB1 (109). A clinical study evaluated the EZH2 inhibitor CPI-1205, which targets SAM, in patients with metastatic castration-resistant prostate cancer and is currently in early clinical trials (NCT03480646). Another clinical study, which is currently recruiting participants, is investigating the safety and efficacy of the EED inhibitor MAK683 in patients with various advanced malignancies, including gastric cancer, DLBCL, prostate cancer, nasopharyngeal carcinoma, ovarian cancer, and sarcoma (NCT02900651). In a particular study, it was observed that the growth of SCLC cell lines was inhibited when these cells were treated with EZH2 inhibitors, specifically DZNep and GSK126 (110). Additionally, when EZH2 was specifically targeted using DZNep, it was found to enhance the sensitivity of both SCLC cells and tumors to cisplatin (111). These findings underscore the potential therapeutic benefits of EZH2 inhibitors in the context of SCLC.
Although targeting EZH2 represents an attractive strategy for treating NSCLC, including those with BRG1 or EGFR mutations, targeting EZH2 alone is sometimes not effective in inhibiting the malignant proliferation of tumors (112–116). Therefore, combination therapies of chemotherapy and PRC2 inhibitors have been explored as a better strategy for treating cancer. Recent studies have designed and synthesized double EZH2/BRD4 inhibitors, which have shown significant antiproliferative activity and resensitization to EZH2 inhibitors in several solid cancers, including LC and pancreatic cancer, in xenograft mouse models (117). Furthermore, a multifunctional nanoparticle-based delivery system has been developed for efficient and safe co-delivery of EZH2 siRNA and etoposide to in orthotopic NSCLC (113). In this study, the combination of EZH2 siRNA and etoposide showed potent inhibition of tumor proliferation and metastasis in both in vivo and in vitro experiments.
While the experimental results of DZNep imply great clinical potential, its plasma half-life is very short, and it nonspecifically inhibits histone methylation, making it toxic in animal models (118). Therefore, there is an urgent need to develop relevant combination therapies. Our previous experimental results suggest that the combination of EZH2 inhibitors (GSK343 or DZNep) and gefitinib inhibits cell proliferation, migration, and tumor activity more effectively than either drug alone (119). This suggests that the combination of EZH2 inhibitors and epidermal growth factor receptor-tyrosine kinase inhibitors (EGFR-TKIs) is effective in treating patients with EGFR wild-type NSCLC who do not respond well to conventional EGFR-TKI therapy. Additionally, the combination of a double EZH2 inhibitor and a topoisomerase II inhibitor has been shown to have a therapeutic effect on EGFR mutant NSCLC and BRG1-mutant LC, which is resistant to standard chemotherapy (112). Additionally, there is a report that DZNep used in combination with the histone deacetylase inhibitor Norville can dramatically inhibit NSCLC cell proliferation and induce apoptosis (118). These results demonstrate that epigenetic-chemotherapy combination therapies have great potential to improve current oncology treatment regimens. A list of current clinical trials of PRC2 inhibitors is provided in Table 2.
10 Conclusion
The dysregulation of PRC2-mediated epigenetics has been related to the progression of LC, driving tumor proliferation, invasion, and metastasis. In vitro and in vivo studies have demonstrated that targeting EZH2 expression is effective in slowing LC progression and enhancing chemosensitivity. Several drugs that inhibit EZH2/PRC2 are under development and being evaluated in clinical trials, although most are still in preclinical or phase 1/2 studies. Research has shown that combining PRC2 inhibitors with other therapies, such as immunotherapy, chemotherapy, and targeted therapies, may achieve complementary or synergistic antitumor effects (120), and drug delivery methods such as nanocarriers may enhance therapeutic efficacy. Overall, PRC2 is a crucial epigenetic factor in the development and progression of LC, and PRC2 inhibitors show great promise for the treatment of cancer.
Author contributions
MG, YL, and PC contributed equally to this work. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (82172569, 82072595, and 61973232), Tianjin Key Medical Discipline (Specialty) Construction Project (TJYXZDXK-061B, TJWJ2022XK005), Tianjin Health Science and Technology Project (ZC20179), and Beijing Science and Technology Innovation Medical Development Fund (KC2021-JX-0186-57).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2022. CA: Cancer J Clin (2022) 72(1):7–33. doi: 10.3322/caac.21708
2. Piperdi B, Merla A, Perez-Soler R. Targeting angiogenesis in squamous non-small cell lung cancer. Drugs (2014) 74(4):403–13. doi: 10.1007/s40265-014-0182-z
3. Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. New Engl J Med (2010) 362(25):2380–8. doi: 10.1056/NEJMoa0909530
4. Mitsudomi T, Morita S, Yatabe Y, Negoro S, Okamoto I, Tsurutani J, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): an open label, randomised phase 3 trial. Lancet Oncol (2010) 11(2):121–8. doi: 10.1016/S1470-2045(09)70364-X
5. Brzeziańska E, Dutkowska A, Antczak A. The significance of epigenetic alterations in lung carcinogenesis. Mol Biol Rep (2013) 40(1):309–25. doi: 10.1007/s11033-012-2063-4
6. Conway E, Healy E, Bracken AP. PRC2 mediated H3K27 methylations in cellular identity and cancer. Curr Opin Cell Biol (2015) 37:42–8. doi: 10.1016/j.ceb.2015.10.003
7. van Mierlo G, Veenstra GJC, Vermeulen M, Marks H. The complexity of PRC2 subcomplexes. Trends Cell Biol (2019) 29(8):660–71. doi: 10.1016/j.tcb.2019.05.004
8. Zhang H, Qi J, Reyes JM, Li L, Rao PK, Li F, et al. oncogenic deregulation of EZH2 as an opportunity for targeted therapy in lung cancer. Cancer Discovery (2016) 6(9):1006–21. doi: 10.1158/2159-8290.CD-16-0164
9. Müller J, Hart CM, Francis NJ, Vargas ML, Sengupta A, Wild B, et al. Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell (2002) 111(2):197–208. doi: 10.1016/S0092-8674(02)00976-5
10. Czermin B, Melfi R, McCabe D, Seitz V, Imhof A, Pirrotta V. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell (2002) 111(2):185–96. doi: 10.1016/S0092-8674(02)00975-3
11. Piunti A, Shilatifard A. Epigenetic balance of gene expression by Polycomb and COMPASS families. Sci (New York NY) (2016) 352(6290):aad9780. doi: 10.1126/science.aad9780
12. Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell (2004) 15(1):57–67. doi: 10.1016/j.molcel.2004.06.020
13. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Sci (New York NY) (2002) 298(5595):1039–43. doi: 10.1126/science.1076997
14. Lavarone E, Barbieri CM, Pasini D. Dissecting the role of H3K27 acetylation and methylation in PRC2 mediated control of cellular identity. Nat Commun (2019) 10(1):1679. doi: 10.1038/s41467-019-09624-w
15. Højfeldt JW, Laugesen A, Willumsen BM, Damhofer H, Hedehus L, Tvardovskiy A, et al. Accurate H3K27 methylation can be established de novo by SUZ12-directed PRC2. Nat Struct Mol Biol (2018) 25(3):225–32. doi: 10.1038/s41594-018-0036-6
16. Pengelly AR, Copur Ö, Jäckle H, Herzig A, Müller J. A histone mutant reproduces the phenotype caused by loss of histone-modifying factor Polycomb. Sci (New York NY) (2013) 339(6120):698–9. doi: 10.1126/science.1231382
17. Ciferri C, Lander GC, Maiolica A, Herzog F, Aebersold R, Nogales E. Molecular architecture of human polycomb repressive complex 2. eLife (2012) 1:e00005. doi: 10.7554/eLife.00005
18. Lee CH, Holder M, Grau D, Saldaña-Meyer R, Yu JR, Ganai RA, et al. Distinct stimulatory mechanisms regulate the catalytic activity of polycomb repressive complex 2. Mol Cell (2018) 70(3):435–448.e435. doi: 10.1016/j.molcel.2018.03.019
19. Wu H, Zeng H, Dong A, Li F, He H, Senisterra G, et al. Structure of the catalytic domain of EZH2 reveals conformational plasticity in cofactor and substrate binding sites and explains oncogenic mutations. PloS One (2013) 8(12):e83737. doi: 10.1371/journal.pone.0083737
20. Antonysamy S, Condon B, Druzina Z, Bonanno JB, Gheyi T, Zhang F, et al. Structural context of disease-associated mutations and putative mechanism of autoinhibition revealed by X-ray crystallographic analysis of the EZH2-SET domain. PloS One (2013) 8(12):e84147. doi: 10.1371/journal.pone.0084147
21. Jiao L, Liu X. Structural basis of histone H3K27 trimethylation by an active polycomb repressive complex 2. Sci (New York NY) (2015) 350(6258):aac4383. doi: 10.1126/science.aac4383
22. Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ 3rd, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature (2009) 461(7265):762–7. doi: 10.1038/nature08398
23. Kasinath V, Faini M, Poepsel S, Reif D, Feng XA, Stjepanovic G, et al. Structures of human PRC2 with its cofactors AEBP2 and JARID2. Sci (New York NY) (2018) 359(6378):940–4. doi: 10.1126/science.aar5700
24. Yu JR, Lee CH, Oksuz O, Stafford JM, Reinberg D. PRC2 is high maintenance. Genes Dev (2019) 33(15-16):903–35. doi: 10.1101/gad.325050.119
25. Brooun A, Gajiwala KS, Deng YL, Liu W, Bolaños B, Bingham P, et al. Polycomb repressive complex 2 structure with inhibitor reveals a mechanism of activation and drug resistance. Nat Commun (2016) 7:11384. doi: 10.1038/ncomms11384
26. Glancy E, Ciferri C, Bracken AP. Structural basis for PRC2 engagement with chromatin. Curr Opin Struct Biol (2021) 67:135–44. doi: 10.1016/j.sbi.2020.10.017
27. Wang R, Liu J, Li K, Yang G, Chen S, Wu J, et al. An SETD1A/Wnt/β-catenin feedback loop promotes NSCLC development. J Exp Clin Cancer research: CR (2021) 40(1):318. doi: 10.1186/s13046-021-02119-x
28. Sun J, Tian X, Lu SQ, Hu HB. MicroRNA-4465 suppresses tumor proliferation and metastasis in non-small cell lung cancer by directly targeting the oncogene EZH2. Biomed pharmacother = Biomedecine pharmacotherapie (2017) 96:1358–62. doi: 10.1016/j.biopha.2017.11.070
29. Cao X, Li F, Shao J, Lv J, Chang A, Dong W, et al. Circular RNA hsa_circ_0102231 sponges miR-145 to promote non-small cell lung cancer cell proliferation by up-regulating the expression of RBBP4. J Biochem (2021) 169(1):65–73. doi: 10.1093/jb/mvaa093
30. Sun M, Liu XH, Lu KH, Nie FQ, Xia R, Kong R, et al. EZH2-mediated epigenetic suppression of long noncoding RNA SPRY4-IT1 promotes NSCLC cell proliferation and metastasis by affecting the epithelial-mesenchymal transition. Cell Death Dis (2014) 5(6):e1298. doi: 10.1038/cddis.2014.256
31. Wan J, Zhan J, Li S, Ma J, Xu W, Liu C, et al. PCAF-primed EZH2 acetylation regulates its stability and promotes lung adenocarcinoma progression. Nucleic Acids Res (2015) 43(7):3591–604. doi: 10.1093/nar/gkv238
32. Nie L, Wei Y, Zhang F, Hsu YH, Chan LC, Xia W, et al. CDK2-mediated site-specific phosphorylation of EZH2 drives and maintains triple-negative breast cancer. Nat Commun (2019) 10(1):5114. doi: 10.1038/s41467-019-13105-5
33. Zeng Y, Qiu R, Yang Y, Gao T, Zheng Y, Huang W, et al. Regulation of EZH2 by SMYD2-mediated lysine methylation is implicated in tumorigenesis. Cell Rep (2019) 29(6):1482–1498.e1484. doi: 10.1016/j.celrep.2019.10.004
34. Lo PW, Shie JJ, Chen CH, Wu CY, Hsu TL, Wong CH. O-GlcNAcylation regulates the stability and enzymatic activity of the histone methyltransferase EZH2. Proc Natl Acad Sci United States America (2018) 115(28):7302–7. doi: 10.1073/pnas.1801850115
35. Wu SC, Zhang Y. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2) regulates its stability. J Biol Chem (2011) 286(32):28511–9. doi: 10.1074/jbc.M111.240515
36. Kang N, Eccleston M, Clermont PL, Latarani M, Male DK, Wang Y, et al. EZH2 inhibition: a promising strategy to prevent cancer immune editing. Epigenomics (2020) 12(16):1457–76. doi: 10.2217/epi-2020-0186
37. Peng D, Kryczek I, Nagarsheth N, Zhao L, Wei S, Wang W, et al. Epigenetic silencing of TH1-type chemokines shapes tumour immunity and immunotherapy. Nature (2015) 527(7577):249–53. doi: 10.1038/nature15520
38. Dangaj D, Bruand M, Grimm AJ, Ronet C, Barras D, Duttagupta PA, et al. Cooperation between constitutive and inducible chemokines enables T cell engraftment and immune attack in solid tumors. Cancer Cell (2019) 35(6):885–900.e810. doi: 10.1016/j.ccell.2019.05.004
39. Borczuk AC, Papanikolaou N, Toonkel RL, Sole M, Gorenstein LA, Ginsburg ME, et al. Lung adenocarcinoma invasion in TGFbetaRII-deficient cells is mediated by CCL5/RANTES. Oncogene (2008) 27(4):557–64. doi: 10.1038/sj.onc.1210662
40. Huang CY, Fong YC, Lee CY, Chen MY, Tsai HC, Hsu HC, et al. CCL5 increases lung cancer migration via PI3K, Akt and NF-kappaB pathways. Biochem Pharmacol (2009) 77(5):794–803. doi: 10.1016/j.bcp.2008.11.014
41. Xia L, Zhu X, Zhang L, Xu Y, Chen G, Luo J. EZH2 enhances expression of CCL5 to promote recruitment of macrophages and invasion in lung cancer. Biotechnol Appl Biochem (2020) 67(6):1011–9. doi: 10.1002/bab.1875
42. Emran AA, Chatterjee A, Rodger EJ, Tiffen JC, Gallagher SJ, Eccles MR, et al. Targeting DNA methylation and EZH2 activity to overcome melanoma resistance to immunotherapy. Trends Immunol (2019) 40(4):328–44. doi: 10.1016/j.it.2019.02.004
43. Koss B, Shields BD, Taylor EM, Storey AJ, Byrum SD, Gies AJ, et al. Epigenetic control of cdkn2a.Arf protects tumor-Infiltrating lymphocytes from metabolic exhaustion. Cancer Res (2020) 80(21):4707–19. doi: 10.1158/0008-5472.CAN-20-0524
44. Sun S, Yu F, Xu D, Zheng H, Li M. EZH2, a prominent orchestrator of genetic and epigenetic regulation of solid tumor microenvironment and immunotherapy. Biochim Biophys Acta Rev Cancer (2022) 1877(2):188700. doi: 10.1016/j.bbcan.2022.188700
45. Wang Y, Ding Y, Guo N, Wang S. MDSCs: key criminals of tumor pre-metastatic niche formation. Front Immunol (2019) 10:172. doi: 10.3389/fimmu.2019.00172
46. Herviou L, Cavalli G, Cartron G, Klein B, Moreaux J. EZH2 in normal hematopoiesis and hematological Malignancies. Oncotarget (2016) 7(3):2284–96. doi: 10.18632/oncotarget.6198
47. Huang S, Wang Z, Zhou J, Huang J, Zhou L, Luo J, et al. EZH2 inhibitor GSK126 suppresses antitumor immunity by driving production of myeloid-derived suppressor cells. Cancer Res (2019) 79(8):2009–20. doi: 10.1158/0008-5472.CAN-18-2395
48. Karantanos T, Chistofides A, Barhdan K, Li L, Boussiotis VA. Regulation of T cell differentiation and function by EZH2. Front Immunol (2016) 7:172. doi: 10.3389/fimmu.2016.00172
49. Xiong Y, Khanna S, Grzenda AL, Sarmento OF, Svingen PA, Lomberk GA, et al. Polycomb antagonizes p300/CREB-binding protein-associated factor to silence FOXP3 in a Kruppel-like factor-dependent manner. J Biol Chem (2012) 287(41):34372–85. doi: 10.1074/jbc.M111.325332
50. DuPage M, Chopra G, Quiros J, Rosenthal WL, Morar MM, Holohan D, et al. The chromatin-modifying enzyme Ezh2 is critical for the maintenance of regulatory T cell identity after activation. Immunity (2015) 42(2):227–38. doi: 10.1016/j.immuni.2015.01.007
51. Arvey A, van der Veeken J, Samstein RM, Feng Y, Stamatoyannopoulos JA, Rudensky AY. Inflammation-induced repression of chromatin bound by the transcription factor Foxp3 in regulatory T cells. Nat Immunol (2014) 15(6):580–7. doi: 10.1038/ni.2868
52. Wang D, Quiros J, Mahuron K, Pai CC, Ranzani V, Young A, et al. Targeting EZH2 reprograms intratumoral regulatory T cells to enhance cancer immunity. Cell Rep (2018) 23(11):3262–74. doi: 10.1016/j.celrep.2018.05.050
53. Goswami S, Apostolou I, Zhang J, Skepner J, Anandhan S, Zhang X, et al. Modulation of EZH2 expression in T cells improves efficacy of anti-CTLA-4 therapy. J Clin Invest (2018) 128(9):3813–8. doi: 10.1172/JCI99760
54. Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, et al. NK Cells Stimulate Recruitment of cDC1 into the Tumor Microenvironment Promoting Cancer Immune Control. Cell (2018) 172(5):1022–1037.e1014. doi: 10.1016/j.cell.2018.01.004
55. Fuertes MB, Domaica CI, Zwirner NW. Leveraging NKG2D ligands in immuno-oncology. Front Immunol (2021) 12:713158. doi: 10.3389/fimmu.2021.713158
56. Yin J, Leavenworth JW, Li Y, Luo Q, Xie H, Liu X, et al. Ezh2 regulates differentiation and function of natural killer cells through histone methyltransferase activity. Proc Natl Acad Sci United States America (2015) 112(52):15988–93. doi: 10.1073/pnas.1521740112
57. Bugide S, Green MR, Wajapeyee N. Inhibition of Enhancer of zeste homolog 2 (EZH2) induces natural killer cell-mediated eradication of hepatocellular carcinoma cells. Proc Natl Acad Sci United States America (2018) 115(15):E3509–e3518. doi: 10.1073/pnas.1802691115
58. Shenoy AK, Jin Y, Luo H, Tang M, Pampo C, Shao R, et al. Epithelial-to-mesenchymal transition confers pericyte properties on cancer cells. J Clin Invest (2016) 126(11):4174–86. doi: 10.1172/JCI86623
59. Tiwari N, Tiwari VK, Waldmeier L, Balwierz PJ, Arnold P, Pachkov M, et al. Sox4 is a master regulator of epithelial-mesenchymal transition by controlling Ezh2 expression and epigenetic reprogramming. Cancer Cell (2013) 23(6):768–83. doi: 10.1016/j.ccr.2013.04.020
60. Li L, Liu J, Xue H, Li C, Liu Q, Zhou Y, et al. A TGF-β-MTA1-SOX4-EZH2 signaling axis drives epithelial-mesenchymal transition in tumor metastasis. Oncogene (2020) 39(10):2125–39. doi: 10.1038/s41388-019-1132-8
61. Hasegawa S, Nagano H, Konno M, Eguchi H, Tomokuni A, Tomimaru Y, et al. A crucial epithelial to mesenchymal transition regulator, Sox4/Ezh2 axis is closely related to the clinical outcome in pancreatic cancer patients. Int J Oncol (2016) 48(1):145–52. doi: 10.3892/ijo.2015.3258
62. Lachat C, Bruyère D, Etcheverry A, Aubry M, Mosser J, Warda W, et al. EZH2 and KDM6B expressions are associated with specific epigenetic signatures during EMT in non small cell lung carcinomas. Cancers (2020) 12(12):3649. doi: 10.3390/cancers12123649
63. Chen H, Tu SW, Hsieh JT. Down-regulation of human DAB2IP gene expression mediated by polycomb Ezh2 complex and histone deacetylase in prostate cancer. J Biol Chem (2005) 280(23):22437–44. doi: 10.1074/jbc.M501379200
64. Cooper CS, Nicholson AG, Foster C, Dodson A, Edwards S, Fletcher A, et al. Nuclear overexpression of the E2F3 transcription factor in human lung cancer. Lung Cancer (Amsterdam Netherlands) (2006) 54(2):155–62. doi: 10.1016/j.lungcan.2006.07.005
65. Coe BP, Thu KL, Aviel-Ronen S, Vucic EA, Gazdar AF, Lam S, et al. Genomic deregulation of the E2F/Rb pathway leads to activation of the oncogene EZH2 in small cell lung cancer. PloS One (2013) 8(8):e71670. doi: 10.1371/journal.pone.0071670
66. Hubaux R, Thu KL, Coe BP, MacAulay C, Lam S, Lam WL. EZH2 promotes E2F-driven SCLC tumorigenesis through modulation of apoptosis and cell-cycle regulation. J Thorac Oncol (2013) 8(8):1102–6. doi: 10.1097/JTO.0b013e318298762f
67. Li W, Li Y, Zhang H, Liu M, Gong H, Yuan Y, et al. HOTAIR promotes gefitinib resistance through modification of EZH2 and silencing p16 and p21 in non-small cell lung cancer. J Cancer (2021) 12(18):5562–72. doi: 10.7150/jca.56093
68. Geng J, Li X, Zhou Z, Wu CL, Dai M, Bai X. EZH2 promotes tumor progression via regulating VEGF-A/AKT signaling in non-small cell lung cancer. Cancer Lett (2015) 359(2):275–87. doi: 10.1016/j.canlet.2015.01.031
69. Wei FZ, Cao Z, Wang X, Wang H, Cai MY, Li T, et al. Epigenetic regulation of autophagy by the methyltransferase EZH2 through an MTOR-dependent pathway. Autophagy (2015) 11(12):2309–22. doi: 10.1080/15548627.2015.1117734
70. Cao P, Li Y, Shi R, Yuan Y, Gong H, Zhu G, et al. Combining EGFR-TKI with SAHA overcomes EGFR-TKI-Acquired resistance by reducing the protective autophagy in non-Small cell lung cancer. Front Chem (2022) 10:837987. doi: 10.3389/fchem.2022.837987
71. Liu C, Shi X, Wang L, Wu Y, Jin F, Bai C, et al. SUZ12 is involved in progression of non-small cell lung cancer by promoting cell proliferation and metastasis. Tumour biology: J Int Soc Oncodevelopmental Biol Med (2014) 35(6):6073–82. doi: 10.1007/s13277-014-1804-5
72. Gargiulo G, Citterio E, Serresi M. Polycomb and lung cancer: When the dosage makes the (kind of) poison. Mol Cell Oncol (2016) 3(3):e1152345. doi: 10.1080/23723556.2016.1152345
73. Xia H, Zhang W, Zhang B, Zhao Y, Zhao Y, Li S, et al. miR-21 modulates the effect of EZH2 on the biological behavior of human lung cancer stem cells. vitro. Oncotarget (2017) 8(49):85442–51. doi: 10.18632/oncotarget.20006
74. Liu X, Lu X, Zhen F, Jin S, Yu T, Zhu Q, et al. LINC00665 induces acquired resistance to gefitinib through recruiting EZH2 and activating PI3K/AKT pathway in NSCLC. Mol Ther Nucleic Acids (2019) 16:155–61. doi: 10.1016/j.omtn.2019.02.010
75. Riquelme E, Suraokar M, Behrens C, Lin HY, Girard L, Nilsson MB, et al. VEGF/VEGFR-2 upregulates EZH2 expression in lung adenocarcinoma cells and EZH2 depletion enhances the response to platinum-based and VEGFR-2-targeted therapy. Clin Cancer Res (2014) 20(14):3849–61. doi: 10.1158/1078-0432.CCR-13-1916
76. Cao W, Ribeiro Rde O, Liu D, Saintigny P, Xia R, Xue Y, et al. EZH2 promotes Malignant behaviors via cell cycle dysregulation and its mRNA level associates with prognosis of patient with non-small cell lung cancer. PloS One (2012) 7(12):e52984. doi: 10.1371/journal.pone.0052984
77. Kikuchi J, Kinoshita I, Shimizu Y, Kikuchi E, Konishi J, Oizumi S, et al. Distinctive expression of the polycomb group proteins Bmi1 polycomb ring finger oncogene and enhancer of zeste homolog 2 in nonsmall cell lung cancers and their clinical and clinicopathologic significance. Cancer (2010) 116(12):3015–24. doi: 10.1002/cncr.25128
78. Behrens C, Solis LM, Lin H, Yuan P, Tang X, Kadara H, et al. et al: EZH2 protein expression associates with the early pathogenesis, tumor progression, and prognosis of non-small cell lung carcinoma. Clin Cancer Res (2013) 19(23):6556–65. doi: 10.1158/1078-0432.CCR-12-3946
79. Serresi M, Gargiulo G, Proost N, Siteur B, Cesaroni M, Koppens M, et al. Polycomb repressive complex 2 is a barrier to KRAS-Driven inflammation and epithelial-Mesenchymal transition in non-Small-Cell lung cancer. Cancer Cell (2016) 29(1):17–31. doi: 10.1016/j.ccell.2015.12.006
80. Shen X, Liu Y, Hsu YJ, Fujiwara Y, Kim J, Mao X, et al. EZH1 mediates methylation on histone H3 lysine 27 and complements EZH2 in maintaining stem cell identity and executing pluripotency. Mol Cell (2008) 32(4):491–502. doi: 10.1016/j.molcel.2008.10.016
82. Campbell JE, Kuntz KW, Knutson SK, Warholic NM, Keilhack H, Wigle TJ, et al. EPZ011989, A potent, orally-available EZH2 inhibitor with robust in vivo activity. ACS medicinal Chem Lett (2015) 6(5):491–5. doi: 10.1021/acsmedchemlett.5b00037
83. Gulati N, Béguelin W, Giulino-Roth L. Enhancer of zeste homolog 2 (EZH2) inhibitors. Leukemia lymphoma (2018) 59(7):1574–85. doi: 10.1080/10428194.2018.1430795
84. Wang X, Cao W, Zhang J, Yan M, Xu Q, Wu X, et al. A covalently bound inhibitor triggers EZH2 degradation through CHIP-mediated ubiquitination. EMBO J (2017) 36(9):1243–60. doi: 10.15252/embj.201694058
85. Gan L, Yang Y, Li Q, Feng Y, Liu T, Guo W. Epigenetic regulation of cancer progression by EZH2: from biological insights to therapeutic potential. biomark Res (2018) 6:10. doi: 10.1186/s40364-018-0122-2
86. Zhang KL, Shen QQ, Fang YF, Sun YM, Ding J, Chen Y. AZD9291 inactivates the PRC2 complex to mediate tumor growth inhibition. Acta pharmacologica Sin (2019) 40(12):1587–95. doi: 10.1038/s41401-019-0248-2
87. Huang Y, Sendzik M, Zhang J, Gao Z, Sun Y, Wang L, et al. Discovery of the clinical candidate MAK683: an EED-Directed, allosteric, and selective PRC2 inhibitor for the treatment of advanced Malignancies. J medicinal Chem (2022) 65(7):5317–33. doi: 10.1021/acs.jmedchem.1c02148
88. Han T, Yin Q, Wan L. AMPK lifts the PRC2-implemented gene repression. Mol Cell Oncol (2018) 5(3):e1441632. doi: 10.1080/23723556.2018.1441632
89. Li L, Zhang H, Zhang M, Zhao M, Feng L, Luo X, et al. Discovery and molecular basis of a diverse set of polycomb repressive complex 2 inhibitors recognition by EED. PloS One (2017) 12(1):e0169855. doi: 10.1371/journal.pone.0169855
90. Huang Y, Zhang J, Yu Z, Zhang H, Wang Y, Lingel A, et al. Discovery of first-in-Class, potent, and orally bioavailable embryonic ectoderm development (EED) inhibitor with robust anticancer efficacy. J medicinal Chem (2017) 60(6):2215–26. doi: 10.1021/acs.jmedchem.6b01576
91. Danishuddin, Subbarao N, Faheem M. Khan SN: Polycomb repressive complex 2 inhibitors: emerging epigenetic modulators. Drug Discovery Today (2019) 24(1):179–88. doi: 10.1016/j.drudis.2018.07.002
92. Rej RK, Wang C, Lu J, Wang M, Petrunak E, Zawacki KP, et al. EEDi-5285: an exceptionally potent, efficacious, and orally active small-molecule inhibitor of embryonic ectoderm development. J medicinal Chem (2020) 63(13):7252–67. doi: 10.1021/acs.jmedchem.0c00479
93. Rej RK, Wang C, Lu J, Wang M, Petrunak E, Zawacki KP, et al. Discovery of EEDi-5273 as an exceptionally potent and orally efficacious EED inhibitor capable of achieving complete and persistent tumor regression. J medicinal Chem (2021) 64(19):14540–56. doi: 10.1021/acs.jmedchem.1c01059
94. Qi W, Zhao K, Gu J, Huang Y, Wang Y, Zhang H, et al. An allosteric PRC2 inhibitor targeting the H3K27me3 binding pocket of EED. Nat Chem Biol (2017) 13(4):381–8. doi: 10.1038/nchembio.2304
95. Curtin ML, Pliushchev MA, Li HQ, Torrent M, Dietrich JD, Jakob CG, et al. SAR of amino pyrrolidines as potent and novel protein-protein interaction inhibitors of the PRC2 complex through EED binding. Bioorganic medicinal Chem Lett (2017) 27(7):1576–83. doi: 10.1016/j.bmcl.2017.02.030
96. He Y, Selvaraju S, Curtin ML, Jakob CG, Zhu H, Comess KM, et al. The EED protein-protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat Chem Biol (2017) 13(4):389–95. doi: 10.1038/nchembio.2306
97. Barnash KD, The J, Norris-Drouin JL, Cholensky SH, Worley BM, Li F, et al. Discovery of peptidomimetic ligands of EED as allosteric inhibitors of PRC2. ACS combinatorial Sci (2017) 19(3):161–72. doi: 10.1021/acscombsci.6b00174
98. Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD, et al. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat Chem Biol (2013) 9(10):643–50. doi: 10.1038/nchembio.1331
99. Potjewyd F, Turner AW, Beri J, Rectenwald JM, Norris-Drouin JL, Cholensky SH, et al. Degradation of polycomb repressive complex 2 with an EED-targeted bivalent chemical degrader. Cell Chem Biol (2020) 27(1):47–56.e15. doi: 10.1016/j.chembiol.2019.11.006
100. Hsu JH, Rasmusson T, Robinson J, Pachl F, Read J, Kawatkar S, et al. EED-targeted PROTACs degrade EED, EZH2, and SUZ12 in the PRC2 complex. Cell Chem Biol (2020) 27(1):41–46.e17. doi: 10.1016/j.chembiol.2019.11.004
101. Cheng LL, Itahana Y, Lei ZD, Chia NY, Wu Y, Yu Y, et al. TP53 genomic status regulates sensitivity of gastric cancer cells to the histone methylation inhibitor 3-deazaneplanocin A (DZNep). Clin Cancer Res (2012) 18(15):4201–12. doi: 10.1158/1078-0432.CCR-12-0036
102. Hoy SM. Tazemetostat: first approval. Drugs (2020) 80(5):513–21. doi: 10.1007/s40265-020-01288-x
103. McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature (2012) 492(7427):108–12. doi: 10.1038/nature11606
104. Zhao Y, Guan YY, Zhao F, Yu T, Zhang SJ, Zhang YZ, et al. Recent strategies targeting Embryonic Ectoderm Development (EED) for cancer therapy: Allosteric inhibitors, PPI inhibitors, and PROTACs. Eur J medicinal Chem (2022) 231:114144. doi: 10.1016/j.ejmech.2022.114144
105. Dong H, Liu S, Zhang X, Chen S, Kang L, Chen Y, et al. An allosteric PRC2 inhibitor targeting EED suppresses tumor progression by modulating the immune response. Cancer Res (2019) 79(21):5587–96. doi: 10.1158/0008-5472.CAN-19-0428
106. Bisserier M, Wajapeyee N. Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas. Blood (2018) 131(19):2125–37. doi: 10.1182/blood-2017-08-804344
107. Zauderer MG, Szlosarek PW, Le Moulec S, Popat S, Taylor P, Planchard D, et al. EZH2 inhibitor tazemetostat in patients with relapsed or refractory, BAP1-inactivated Malignant pleural mesothelioma: a multicentre, open-label, phase 2 study. Lancet Oncol (2022) 23(6):758–67. doi: 10.1016/S1470-2045(22)00277-7
108. Passeri T, Dahmani A, Masliah-Planchon J, Naguez A, Michou M, El Botty R, et al. Dramatic in vivo efficacy of the EZH2-Inhibitor tazemetostat in PBRM1-Mutated human chordoma xenograft. Cancers (2022) 14(6):1486. doi: 10.3390/cancers14061486
109. Gounder M, Schöffski P, Jones RL, Agulnik M, Cote GM, Villalobos VM, et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: an international, open-label, phase 2 basket study. Lancet Oncol (2020) 21(11):1423–32. doi: 10.1016/S1470-2045(20)30451-4
110. Sato T, Kaneda A, Tsuji S, Isagawa T, Yamamoto S, Fujita T, et al. PRC2 overexpression and PRC2-target gene repression relating to poorer prognosis in small cell lung cancer. Sci Rep (2013) 3:1911. doi: 10.1038/srep01911
111. Koyen AE, Madden MZ, Park D, Minten EV, Kapoor-Vazirani P, Werner E, et al. EZH2 has a non-catalytic and PRC2-independent role in stabilizing DDB2 to promote nucleotide excision repair. Oncogene (2020) 39(25):4798–813. doi: 10.1038/s41388-020-1332-2
112. Fillmore CM, Xu C, Desai PT, Berry JM, Rowbotham SP, Lin YJ, et al. EZH2 inhibition sensitizes BRG1 and EGFR mutant lung tumours to TopoII inhibitors. Nature (2015) 520(7546):239–42. doi: 10.1038/nature14122
113. Yuan ZQ, Chen WL, You BG, Liu Y, Yang SD, Li JZ, et al. Multifunctional nanoparticles co-delivering EZH2 siRNA and etoposide for synergistic therapy of orthotopic non-small-cell lung tumor. J Controlled release (2017) 268:198–211. doi: 10.1016/j.jconrel.2017.10.025
114. Chen AM, Zhang M, Wei D, Stueber D, Taratula O, Minko T, et al. Co-delivery of doxorubicin and Bcl-2 siRNA by mesoporous silica nanoparticles enhances the efficacy of chemotherapy in multidrug-resistant cancer cells. Small (Weinheim an der Bergstrasse Germany) (2009) 5(23):2673–7. doi: 10.1002/smll.200900621
115. Sun TM, Du JZ, Yao YD, Mao CQ, Dou S, Huang SY, et al. Simultaneous delivery of siRNA and paclitaxel via a “two-in-one” micelleplex promotes synergistic tumor suppression. ACS nano (2011) 5(2):1483–94. doi: 10.1021/nn103349h
116. Xiong XB, Lavasanifar A. Traceable multifunctional micellar nanocarriers for cancer-targeted co-delivery of MDR-1 siRNA and doxorubicin. ACS nano (2011) 5(6):5202–13. doi: 10.1021/nn2013707
117. Guo Z, Sun Y, Liang L, Lu W, Luo B, Wu Z, et al. Design and synthesis of dual EZH2/BRD4 inhibitors to target solid tumors. J medicinal Chem (2022) 65(9):6573–92. doi: 10.1021/acs.jmedchem.1c01876
118. Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, et al. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther (2009) 8(6):1579–88. doi: 10.1158/1535-7163.MCT-09-0013
119. Gong H, Li Y, Yuan Y, Li W, Zhang H, Zhang Z, et al. EZH2 inhibitors reverse resistance to gefitinib in primary EGFR wild-type lung cancer cells. BMC Cancer (2020) 20(1):1189. doi: 10.1186/s12885-020-07667-7
Keywords: polycomb repressor complex 2, lung cancer, H3K27me3, EZH2, inhibitors
Citation: Gao M, Li Y, Cao P, Liu H, Chen J and Kang S (2023) Exploring the therapeutic potential of targeting polycomb repressive complex 2 in lung cancer. Front. Oncol. 13:1216289. doi: 10.3389/fonc.2023.1216289
Received: 04 May 2023; Accepted: 02 October 2023;
Published: 16 October 2023.
Edited by:
Fayaz Malik, Indian Institute of Integrative Medicine (CSIR), IndiaReviewed by:
Terry Moody, National Cancer Institute (NIH), United StatesVera Luiza Capelozzi, University of São Paulo, Brazil
Copyright © 2023 Gao, Li, Cao, Liu, Chen and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hongyu Liu, bGl1aG9uZ3l1MTIzQGhvdG1haWwuY29t; Jun Chen, aHVudGVyY2oyMDA0QHFxLmNvbQ==; Shirong Kang, a2FuZ3NoaXJvbmcyMDEzQDE2My5jb20=
†These authors have contributed equally to this work