 Leticia Campoverde1,2*
Leticia Campoverde1,2* Felipe Camacho1,2
Felipe Camacho1,2 Francesco Alessandrino3
Francesco Alessandrino3 Mark G. Evans4
Mark G. Evans4 Andrew Elliot4Andrew Rosenberg3Jonathan Trent3
Andrew Elliot4Andrew Rosenberg3Jonathan Trent3- 1University of Miami, Miami, FL, United States
- 2Jackson Memorial Hospital, Miami, FL, United States
- 3Sylvester Comprehensive Cancer Center, University of Miami Health System, Miami, FL, United States
- 4Caris Life Sciences Inc., Irving, TX, United States
Soft tissue sarcomas harboring EWSR1::CREM fusion are rare and challenging to treat. Pazopanib, a multi-tyrosine kinase inhibitor, is FDA-approved for advanced soft tissue sarcomas, but predictive biomarkers for its efficacy remain unidentified. We conducted a study on > 240,000 neoplasms submitted to Caris Life Sciences (Phoenix, AZ) to detect rearrangements using whole transcriptome sequencing. Two sarcoma-experienced, board-certified pathologists performed histological reviews, and treatment/outcome information was collected. Among the identified cases (n = 18), we observed a diverse range of sarcoma and other cancers, including an intracranial myxoid mesenchymal tumor, mesothelioma, hyalinizing clear cell carcinomas of the head and neck, clear cell sarcomas, and undifferentiated round cell sarcomas, as well as histologically malignant tumors with epithelioid morphology. Notably, two undifferentiated, metastatic, abdominal round cell sarcoma cases treated with pazopanib demonstrated significant sustained partial response and clinical benefit. To explore the genetic factors associated with the efficacy of pazopanib in these cases, next-generation sequencing and fluorescence in situ hybridization were analyzed for alterations in the tumors. The genomic analysis provided compelling evidence confirming the presence of EWSR1::CREM fusion in both cases, with no other pathogenic gene variants or copy number alterations detected. These cases demonstrate the potential of Pazopanib as a promising therapeutic option for patients with EWSR1::CREM fusion-positive soft tissue sarcomas, including metastatic undifferentiated round cell sarcomas. The sustained clinical benefit and partial responses observed in these cases warrant further research to validate these findings and explore the wider utility of Pazopanib in this rare and challenging subset of soft tissue sarcomas. Case studies: Case 1: A 49-year-old man presented with abdominal pain, weight loss, and chronic cough. A computed tomography (CT) of the chest, abdomen, and pelvis showed multiple lung nodules and masses and a right rectus mass that was biopsied and revealed an undifferentiated round cell sarcoma with a rare fusion EWSR1-CREM. No additional pathogenic gene variants or copy number alterations were detected. He received neoadjuvant chemotherapy with three cycles of Vincristine, Adriamycin, and Ifosfamide (VAI) and seven cycles of Vincristine/Irinotecan and Temodar (VIT). After cycle 7 of VIT, he had surgical resection of the abdominal mass and received radiation for lung metastasis. He completed 13 cycles of VIT after which he presented with progression of disease and switched to monotherapy with Pazopanib. At the time of this analysis he had stable disease for 28 months. Case 2: A 75-year-old woman presented with pelvic pain and new onset constipation. CT abdomen showed a large pelvic mass and intraperitoneal tumor spread. Exploratory laparotomy revealed a ruptured pelvic mass and a small bowel tumor. Both tumors were proved to be high-grade, poorly differentiated sarcoma. Genomic analysis demonstrated an EWSR1::CREM fusion but no other pathogenic gene variants or copy number alterations. She was treated initially for a primitive neuroectodermal tumor (PNET) with four cycles of Vincristine/Adriamycin/Cytoxan/Olaratumab but declined additional chemotherapy after progression. Two years later, she presented with recurrent abdominal mass and received one cycle of Temodar/Irinotecan, then she began Pozapanib and underwent palliative radiation to the entire pelvis. She has been on Pazopanib for 23 months with stable disease.
Introduction
Sarcomas are rare tumors with an incidence of approximately five cases per 100,000. The 2020 World Health Organization classification recognizes more than 175 soft tissue and bone tumor entities (1). Sarcomas are generally classified according to their resemblance to the tissue type from which they originate and require careful morphological assessment; however, an increasing number of new subtypes are now being defined by distinct immunohistochemical and molecular findings (2). The importance of the classification of these tumors translates into differences in treatment and outcomes. Identification of certain genes and fusions has provided insights into the mechanisms of tumorigenesis and the identification of potential targeted therapies (3).
Ewing sarcoma is a small cell sarcoma showing gene fusions involving one member of the FET protein family of genes (usually EWSR1) and a member of the TES transcription factors. Most cases are composed of uniform small round cells with scant, clear, or eosinophilic cytoplasm and, sometimes, it can present a higher grade of neuroectodermal differentiation (1). Neoplasms harboring EWSR1::CREM fusions are rare and not well characterized. This uncommon fusion includes a member of the CREB protein family of genes (ATF1, CREB1, and CREM) (4) and has been reported in tumors that include epithelioid neoplasms with a predilection for mesothelial-lined cavities (5), intracranial myxoid mesenchymal tumor (4), clear cell sarcoma of soft tissue (6), hyalinizing clear cell carcinoma (7), and myxoid angiomatoid fibrous histiocytoma (8).
Pazopanib is a multi-tyrosine kinase inhibitor that has been approved by the FDA for the treatment of advanced soft tissue sarcomas (STSs) in patients who have previously received chemotherapy (9). Pazopanib exerts its antiangiogenic effects by inhibiting the intracellular tyrosine kinase of vascular endothelial growth factor receptor (VEGFR) and platelet-derived growth factor receptor (PDGFR) (10). Although recent studies have shown complete pathological response when Pazopanib is combined with chemotherapy (11), most of these cases have been limited to synovial sarcomas, and predictive biomarkers of good response have not been well identified (12). Therefore, identifying specific genetic characteristics that predict a positive response to pazopanib may have a significant impact on therapeutic decisions and clinical outcomes. Although EWSR1 status is not currently known to be predictive of targeted therapy, there is evidence that supports the role of multi-kinase inhibitors in treating sarcomas that harbor EWSR1 fusion (13).
Methods
Detection of EWSR1::CREM rearrangements by whole transcriptome sequencing was performed for > 240,000 neoplasms submitted to Caris Life Sciences (Phoenix, AZ). Following the sequencing analysis, two board-certified pathologists (M.E. and A.R.) conducted a thorough histological review of the identified tumors. Available treatment and outcome information was obtained from the medical records.
Results
Among the cases examined, 18 exhibited an EWSR1::CREM fusion, encompassing various tumor types, including one intracranial myxoid mesenchymal tumor, one mesothelioma, four hyalinizing clear cell carcinomas of the head and neck, two clear cell sarcomas, two undifferentiated round cell sarcomas, and eight histologically malignant tumors with epithelioid morphology. Some of these cases were consistent with a recently reported disease entity in the medical literature known as “epithelioid neoplasm with a predilection for mesothelial-lined cavities.” However, some cases exhibited unexpected neuroendocrine differentiation and were located at uncommon sites, such as the kidney, uterus, and cervical spine. Additionally, three cases displayed features of squamous cell carcinoma. We identified two patients diagnosed with STSs that harbor the EWSR1::CREM fusion gene and have responded well to Pazopanib treatment. We aimed to investigate other possible EWSR1::CREM fusion alterations in STSs and their potential implications for the treatment of this type of cancer.
Case1
Case description
A 49-year-old man with a past medical history of sarcoma of the right upper extremity in his mid-30s presented with 3 months of abdominal pain and unintentional 20 lb of weight loss over 3 months, despite normal appetite and productive cough for 8 months after multiple antibiotic treatments. Exact details of his first sarcoma diagnosis are unknown, but he received neoadjuvant chemotherapy followed by resection and adjuvant radiation. He had no oncologic follow-up after completing treatment for his first tumor. Physical examination revealed a right lower quadrant abdominal mass on palpation. Computed tomography (CT) chest, abdomen, and pelvis and fluorodeoxyglucose–positron emission tomography/CT (FDG PET/CT) showed multiple FDG-avid sites of disease, including multiple lung nodules, an 8-cm mass in the left upper lung lobe with direct extension to the mediastinum, and a 2.7-cm right rectus sheath mass (Figures 1A–C).
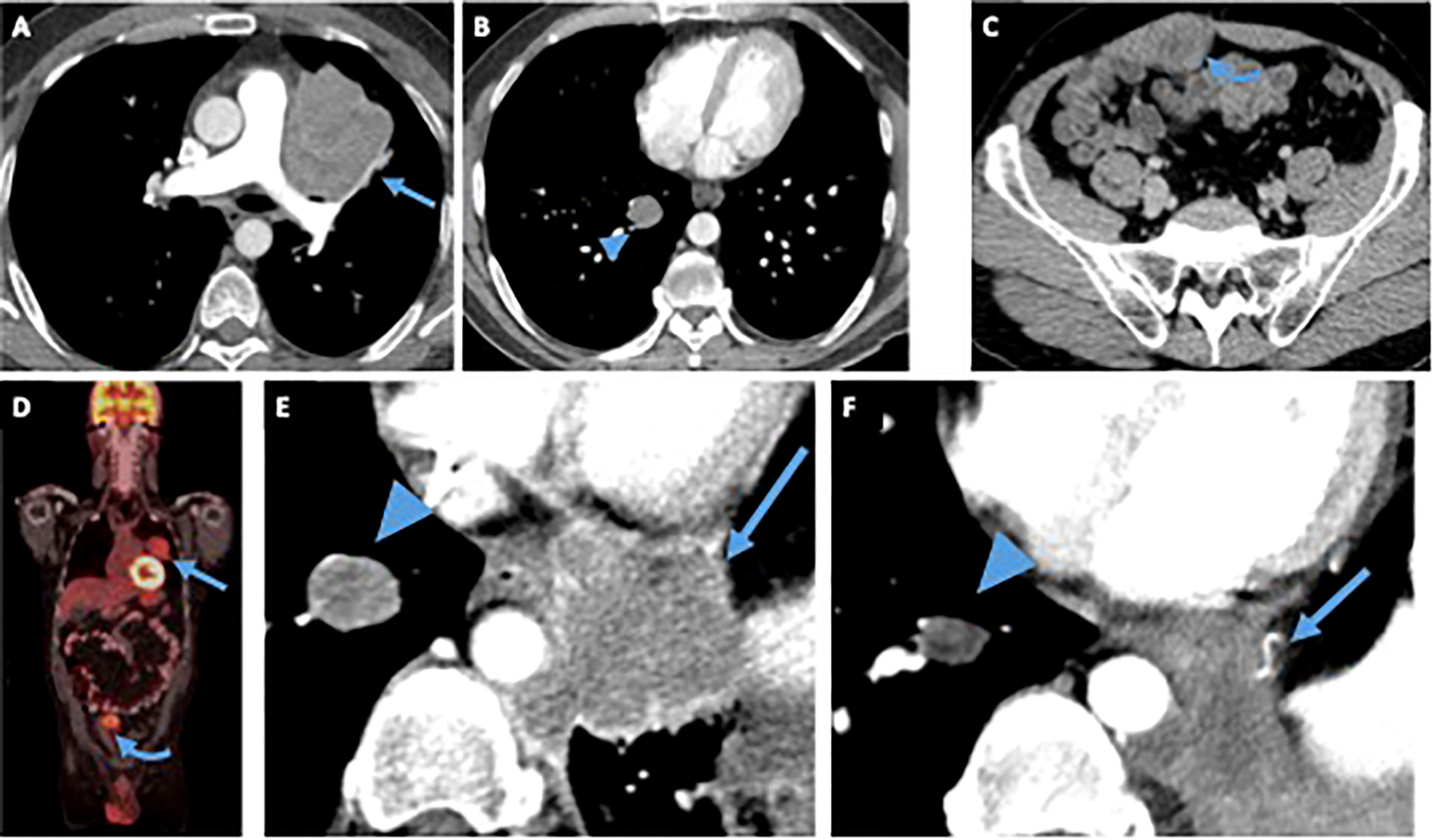
Figure 1 Patient 1. Contrast-enhanced computed tomography (CT) of the chest (A, B) abdomen and pelvis (C) at time of diagnosis demonstrates a left upper lobe paramediastinal mass (arrow), a right lower lobe lung nodule (arrowhead) and an enhancing solid right rectus sheath mass (curved arrow), fluorodeoxyglucose (FDG)-avid on a FDG positron emission tomography (PET)/CT (D) obtained at the same time. After treatment with vincristine/adriamycin/Ifosfamide, resection of the rectus sheath mass and radiation to the left lung mass, the patient started treatment with Pazopanib. Magnified view of an axial contrast-enhanced CT image of the chest (E) obtained before patient started treatment with Pazopanib shows the right lung nodule (arrowhead), and a new enhancing left paramediastinal mass (arrow), both showing mildly decreased size and enhancement on CT obtained 28 months after starting treatment (F), suggesting tumor regression.
Diagnostic assessment
A biopsy of the abdominal mass revealed a poorly differentiated malignant neoplasm, diagnosed as an unclassified round cell malignant neoplasm. The possibility of metastasis from the prior arm malignancy was considered. The lesion was composed of non-distinctive round cells with very limited amounts of eosinophilic cytoplasm and a high nuclear/cytoplasmic ratio with extensive necrosis. Immunohistochemical studies proved to be inconclusive. A month later, resection of the tumor allowed for the identification of an undifferentiated round cell sarcoma (Figure 2A). The neoplasm was characterized by sheets and nests of small to intermediate-size malignant round cells with focal eosinophilic cytoplasm. Some of the nests were surrounded by reactive fibrotic tissue (Figure 2B). Immunohistochemical studies highlighted tumor cells positive for TLE1 and EMA, and infrequent cells were positive for desmin. A poorly differentiated synovial sarcoma and a desmoplastic small round cell tumor were considered as differential diagnoses. FISH showed a deletion of the telomeric portion of EWSR1; no rearrangement of SS18 or EWSR1 was identified. Tumoral whole transcriptome sequencing identified an EWSR1::CREM fusion juxtaposing exon 10 of EWSR1 (transcript NM_005243.3) and exon 6 of CREM (transcript NM_001267562.1).
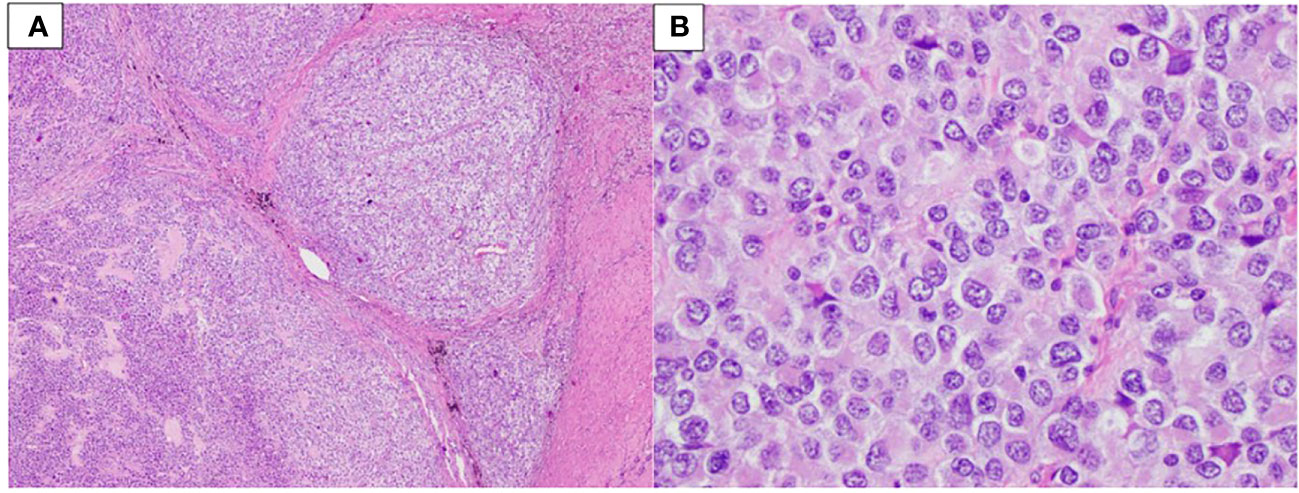
Figure 2 Patient 1. (A) Sheets and nests of neoplastic cells surrounded by fibrotic tissue (40× magnification). (B) Small to intermediate-size cells with limited amounts of eosinophilic cytoplasm and a high nuclear/cytoplasmic ratio (400× magnification).
Therapeutic intervention, follow-up, and outcome
The patient was treated with neoadjuvant chemotherapy with three cycles of VAI after which he progressed and was switched to VIT. After cycle 7 of VIT, he had surgical resection of the abdominal wall mass and received radiation for lung metastasis. Pathology showed that approximately 90% of the tumor was viable, and 10% was necrotic. Immunohistochemistry showed infrequent cells positive for desmin and negative for MYOD1, myogenin, S100, SOX10, HMB45, CD99, and TFE3. The subsequent genomic analysis confirmed an EWSR1::CREM fusion, and the lesion was diagnosed as an undifferentiated round cell sarcoma with EWSR1::CREM fusion. The patient had progression of the disease and was re-started on VIT. He completed 13 cycles of VIT after which he presented progression of the disease and switched to monotherapy with Pazopanib (Figure 1E). He has undergone imaging every 3 months consistent with stable disease of at least 28 months (Figure 1F).
Case 2:
Case description
A 75-year-old woman presented with 4 months of abdominal pain, constipation, and difficulty voiding. The patient had previously undergone a cystoscopy and was thought to have mild interstitial cystitis. Antibiotic therapy did not improve her symptoms. A CT abdomen and pelvis revealed a 12.8-cm multilobulated cystic/solid mass occupying most of the pelvis and omental and mesenteric nodularity, most likely representing intraperitoneal tumor spread. There was also evidence of ascites (Figure 3A). Surgical management was decided, and patient underwent exploratory laparotomy revealing a ruptured pelvic mass and a small bowel tumor.
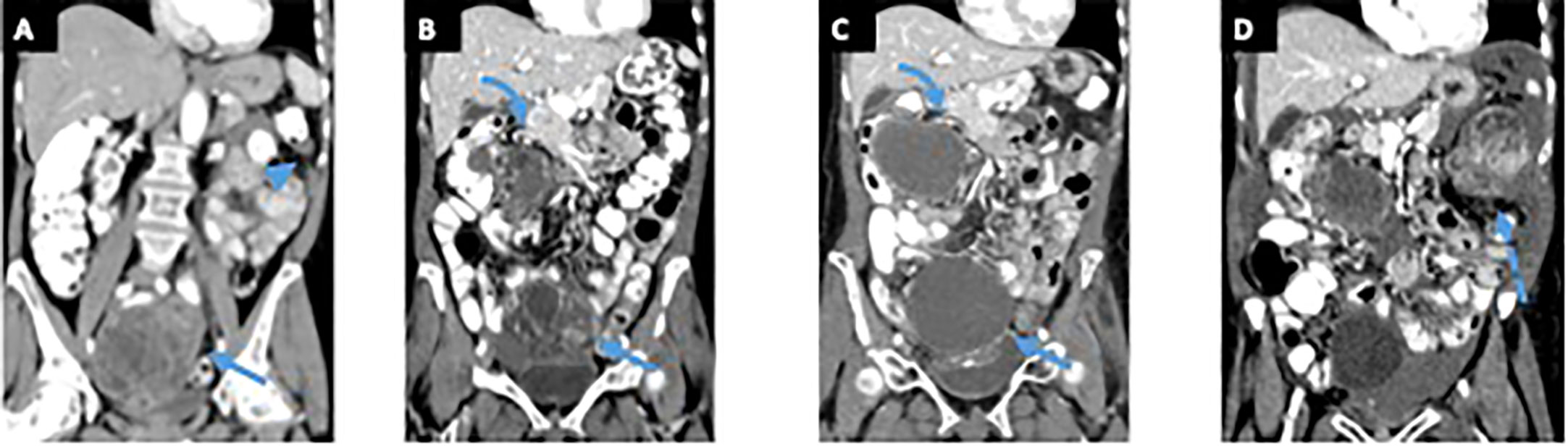
Figure 3 Patient 2. Contrast-enhanced computed tomography (CT) of the abdomen and pelvis at time of the symptoms (A) demonstrates a large pelvic mass with solid and cystic components (arrow), left upper quadrant peritoneal stranding, and peritoneal thickening (arrowhead). Patient underwent exploratory laparotomy revealing a ruptured pelvic mass and a small bowel tumor. After treatment with vincristine/adriamycin/cytoxan/olaratumab, the patient started treatment with pazopanib and underwent palliative radiation to the pelvis. Contrast-enhanced CT of the abdomen and pelvis obtained before patient started treatment with pazopanib (B) shows an enhancing solid and cystic pelvic mass (arrow) and an enhancing solid and cystic right upper quadrant mass (curved arrow), both showing decreased enhancement on contrast-enhanced CT obtained 17 months after starting treatment (C). A contrast-enhanced CT of the abdomen and pelvis obtained 23 months after starting pazopanib (D) and shows a new large enhancing solid and cystic left upper quadrant mass (dashed arrow) suggesting progression of disease. .
Diagnostic assessment
Pathologic examination of the patient’s ruptured pelvic mass and small bowel tumor revealed a high-grade, poorly differentiated sarcoma for both lesions. They were composed of malignant small to medium size round, spindled, and epithelioid cells. The cells were focally enmeshed in a myxoid stroma. Numerous mitoses were identified. Immunohistochemistry showed that the tumor cells were positive for CD117 (focal) and CD34 (a minority) and negative for DOG1, SMA, desmin, S100, HMB45, and inhibin. Full-thickness involvement of the small bowel wall was identified in the second lesion. Due to the anatomic location and CD117 expression, the possibility of a gastrointestinal stroma tumor was considered; however, morphology and lack of staining for DOG1 challenged this differential. Genomic analysis detected an EWSR1::CREM fusion identical to that observed in patient one’s tumor.
Therapeutic intervention, follow-up, and outcome
The patient was treated for a PNET and completed four cycles of Vincristine/Adriamycin/Cytoxan/Olaratumab after which, he progressed and declined additional chemotherapy. Almost 2 years later, during follow-up, the patient complained of abdominal discomfort, pain, pressure, and bloating in the upper right abdomen and pelvis. A CT scan revealed a 6 cm × 6 cm ovoid soft tissue with multiple fluid densities within the pelvis. A PET/CT showed an ill-defined soft tissue density in the right mid-abdomen with mildly increased FDG uptake; as well as heterogeneous predominantly hypoattenuating lesions in the pelvis with heterogeneous mildly increased FDG uptake. A biopsy was taken which showed malignant round, epithelioid, and small spindle cells enmeshed in a focally myxoid stroma (Figure 4A). The lesion was morphologically similar to the one previously resected. An undifferentiated round cell sarcoma was diagnosed (Figure 4B), and an EWSR1::CREM fusion was identified by genomic analysis. The patient received one cycle of Temodar/Irinotecan, then she began Pazopanib and underwent palliative radiation to the entire pelvis.
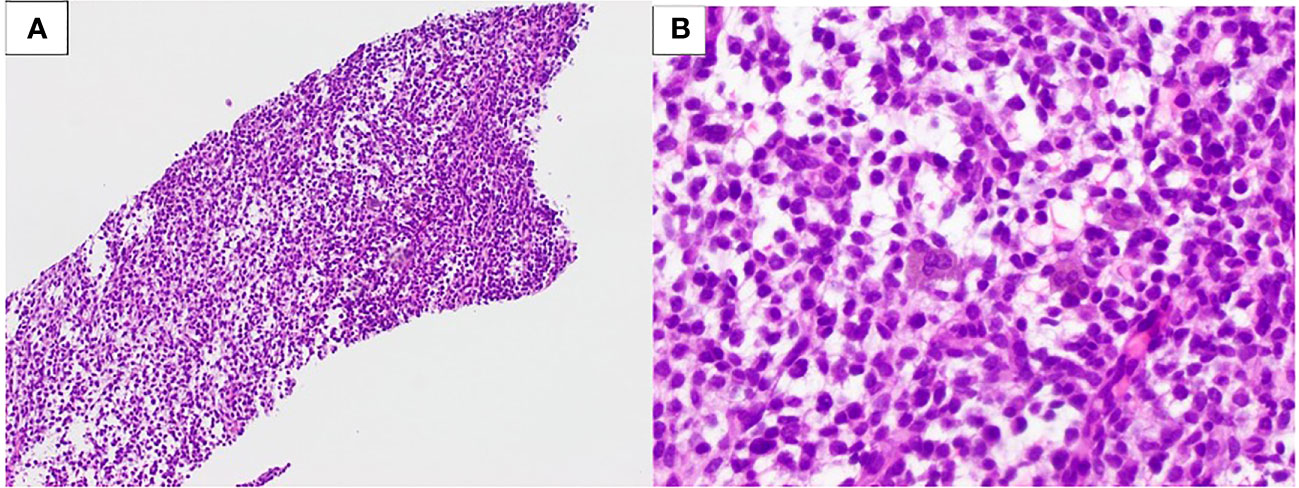
Figure 4 Patient 2. (A) Undifferentiated sarcoma with an EWSR1::CREM fusion, core biopsy (100× magnification). (B) Small- to medium-size round, spindled, and epithelioid cells in a myxoid stroma (400× magnification).
After progression of disease (Figure 3B), the patient received monotherapy with Pazopanib. She has undergone imaging every 3 months, she had stable disease for 23 months, after which she presented progression of disease by imaging (Figures 3C, D) and severe anemia.
Discussion
We present two cases of undifferentiated round cell sarcomas with EWSR1::CREM fusion, illustrating unexpectedly durable partial responses lasting nearly 2 years. Both patients presented with abdominal tumors that had metastasized at the time of diagnosis and were classified as undifferentiated round cell sarcomas of soft tissue. Treatment included neoadjuvant chemotherapy using Ewing sarcoma regimens, followed by surgery. In both cases, no other pathogenic gene variants or copy number alterations were detected. Despite EWSR1 fusions not being direct predictors of targeted therapy efficacy, emerging evidence from certain case series highlights encouraging results. Notably, there have been durable, partial responses to sunitinib in extraskeletal myxoid chondrosarcomas demonstrating EWSR1::NR4A3 fusion (13) and in c-Met/ALK inhibitor Crizotinib and Pazopanib in metastatic gastrointestinal neuroectodermal tumors harboring EWSR1::CREB1 fusion (14). Furthermore, a retrospective, multi-institutional study conducted by Frezza et al. evaluating Pazopanib in nine advanced desmoplastic small round cell tumors (characterized by 96%–97% EWSR1:WT1 fusion) reported five cases with stable disease and two with partial response (15). These findings collectively suggest that multi-kinase inhibitors with broad activity could hold promise as a potential therapeutic approach for tumors influenced by specific EWSR1 fusions and highlight the need for a comprehensive understanding of their molecular characteristics to guide targeted treatment approaches effectively.
Despite these promising results, the precise mechanism underlying the action of these inhibitors in sarcomas with ESWR1 fusions remains incompletely elucidated. EWSR1::CREM fusion is considered rare in STSs and its significance is yet to be fully evaluated (13). EWSR1::CREM fusion has been identified in rare cases of angiomatoid fibrous histiocytoma, clear cell sarcoma, hyalinizing clear cell carcinoma of the head and neck, and intracranial myxoid mesenchymal tumor (14, 15). The exact mechanism by which EWSR1::CREM fusion drives tumorigenicity is not completely understood. EWSR1 encodes a multifunctional protein involved in various cellular processes, including gene expression, cell signaling, and RNA processing, with implications for the regulation of hematopoietic stem cells (16). Pathogenic alterations in EWSR1, particularly translocation t (11;22) (q24; q12), are known to cause Ewing sarcoma and neuroectodermal tumors. EWSR1::CREM rearrangements are believed to lead to constitutive activation of CREM and subsequent dysregulation of oncogenes, such as BCL-2 family genes, EGFR1, and cyclins (17). Pazopanib is a small-molecule tyrosine kinase inhibitor. Its antiangiogenic properties can explain its main mode of action via inhibition of the intracellular tyrosine kinase of VEGFR and PDGFR (10).
STSs represent a heterogeneous and challenging group of rare cancers to treat. Molecular testing has become essential in correctly diagnosing sarcomas with unusual immunophenotypes and can help identify drugs suitable for different sarcoma histological subtypes. Although no therapies directly target EWSR1 fusion proteins, tumors harboring this fusion may ultimately be amenable to treatment with tyrosine kinase inhibitor targeted therapy. Continued research in this area holds the potential to bring about significant advancements in the management and treatment of EWSR1 fusion-associated sarcomas.
Data availability statement
The original contributions presented in the study are included in the article/Supplementary Material. Further inquiries can be directed to the corresponding author.
Ethics statement
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
LC: Conducted a comprehensive literature review, collected and meticulously analyzed the patient data, and drafted the initial version of the manuscript. FC, ME, and AR: Conducted a thorough pathological review of the patients’ pathology specimens, contributed valuable feedback to the manuscript, and ensured the accuracy of the pathological data. FA: Reviewed the radiological images, interpreted and assessed the radiological response to treatment, and provided essential input to the manuscript. ME and AE: Interpreted the results of genetic analysis of the patient specimens, and provided critical feedback on the manuscript. JT: Designed and oversaw the study, provided expert guidance and supervision throughout the research, and contributed valuable feedback to the manuscript. All authors have carefully reviewed and approved the final version of the manuscript.
Conflict of interest
Authors ME and AE were employed by the company Caris Life Sciences.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1215003/full#supplementary-material
References
1. Sbaraglia M, Bellan E, Dei Tos AP. The 2020 WHO Classification of Soft Tissue Tumours: news and perspectives. Pathologica (2020) 113:70–84. doi: 10.32074/1591-951X-213
2. Jain S, Xu R, Prieto VG, Lee P. Molecular classification of soft tissue sarcomas and its clinical applications. Int J Clin Exp Pathol (2010) 3:416–28.
3. Jo VY. EWSR1 fusions: Ewing sarcoma and beyond. Cancer Cytopathol (2020) 128:229–31. doi: 10.1002/cncy.22239
4. Kao YC, Sung YS, Zhang L, Chen CL, Vaiyapuri S, Rosenblum MK, et al. EWSR1 fusions with CREB family transcription factors define a novel myxoid mesenchymal tumor with predilection for intracranial location. Am J Surg Pathol (2017) 41:482–90. doi: 10.1097/PAS.0000000000000788
5. Argani P, Harvey I, Nielsen GP, Takano A, Suurmeijer AJH, Voltaggio L, et al. EWSR1/FUS-CREB fusions define a distinctive Malignant epithelioid neoplasm with predilection for mesothelial-lined cavities. Mod Pathol (2020) 33:2233–43. doi: 10.1038/s41379-020-0646-5
6. Segawa K, Sugita S, Aoyama T, Kubo T, Asanuma H, Sugawara T, et al. Detection of specific gene rearrangements by fluorescence in situ hybridization in 16 cases of clear cell sarcoma of soft tissue and 6 cases of clear cell sarcoma-like gastrointestinal tumor. Diagn Pathol (2018) 13:73. doi: 10.1186/s13000-018-0752-6
7. Chapman E, Skalova A, Ptakova N, Martinek P, Goytain A, Tucker T, et al. Molecular profiling of hyalinizing clear cell carcinomas revealed a subset of tumors harboring a novel EWSR1-CREM fusion: report of 3 cases. Am J Surg Pathol (2018) 42:1182–9. doi: 10.1097/PAS.0000000000001114
8. Yoshida A, Wakai S, Ryo E, Miyata K, Miyazawa M, Yoshida KI, et al. Expanding the phenotypic spectrum of mesenchymal tumors harboring the EWSR1-CREM fusion. Am J Surg Pathol (2019) 43:1622–30. doi: 10.1097/PAS.0000000000001331
9. van der Graaf WT, Blay JY, Chawla SP, Kim DW, Bui-Nguyen B, Casali PG, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet (2012) 379:1879–86. doi: 10.1016/S0140-6736(12)60651-5
10. Gotink KJ, Verheul HM. Anti-angiogenic tyrosine kinase inhibitors: what is their mechanism of action? Angiogenesis (2010) 13:1–14. doi: 10.1007/s10456-009-9160-6
11. Weiss AR, Chen YL, Scharschmidt TJ, Chi YY, Tian J, Black JO, et al. Pathological response in children and adults with large unresected intermediate-grade or high-grade soft tissue sarcoma receiving preoperative chemoradiotherapy with or without pazopanib (ARST1321): a multicentre, randomised, open-label, phase 2 trial. Lancet Oncol (2020) 21:1110–22. doi: 10.1016/S1470-2045(20)30325-9
12. Suehara Y, Kohsaka S, Yamaguchi S, Hayashi T, Kurihara T, Sano K, et al. Assessment of predictive biomarkers of the response to pazopanib based on an integrative analysis of high-grade soft-tissue sarcomas: analysis of a tumor sample from a responder and patients with other soft-tissue sarcomas. Clin Orthop Relat Res (2020) 478:2461–76. doi: 10.1097/CORR.0000000000001322
13. Stacchiotti S, Pantaleo MA, Astolfi A, Dagrada GP, Negri T, Dei Tos AP, et al. Activity of sunitinib in extraskeletal myxoid chondrosarcoma. Eur J Cancer (2014) 50:1657–64. doi: 10.1016/j.ejca.2014.03.013
14. Subbiah V, Holmes O, Gowen K, Spritz D, Amini B, Wang WL, et al. Activity of c-met/ALK inhibitor crizotinib and multi-kinase VEGF inhibitor pazopanib in metastatic gastrointestinal neuroectodermal tumor harboring EWSR1-CREB1 fusion. Oncology (2016) 91:348–53. doi: 10.1159/000449204
15. Frezza AM, Benson C, Judson IR, Litiere S, Marreaud S, Sleijfer S, et al. Pazopanib in advanced desmoplastic small round cell tumours: a multi-institutional experience. Clin Sarcoma Res (2014) 4:7. doi: 10.1186/2045-3329-4-7
16. Bale TA, Oviedo A, Kozakewich H, Giannini C, Davineni PK, Ligon K, et al. Intracranial myxoid mesenchymal tumors with EWSR1-CREB family gene fusions: myxoid variant of angiomatoid fibrous histiocytoma or novel entity? Brain Pathol (2018) 28:183–91. doi: 10.1111/bpa.12504
Keywords: Pazopanib, EWSR1::CREM fusion, sarcoma, stable response, immunophenotype
Citation: Campoverde L, Camacho F, Alessandrino F, Evans MG, Elliot A, Rosenberg A and Trent J (2023) Case report: The activity of multi-kinase VEGF inhibitor, Pazopanib, in metastatic undifferentiated round cell sarcomas harboring EWSR1::CREM fusion: clinicopathological series of two cases and literature review. Front. Oncol. 13:1215003. doi: 10.3389/fonc.2023.1215003
Received: 01 May 2023; Accepted: 14 August 2023;
Published: 27 September 2023.
Edited by:
Jill Kolesar, University of Kentucky, United StatesReviewed by:
Derek Allison, University of Kentucky, United StatesBrian Van Tine, Washington University in St. Louis, United States
Copyright © 2023 Campoverde, Camacho, Alessandrino, Evans, Elliot, Rosenberg and Trent. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Leticia Campoverde, bGVjMTYwQG1pYW1pLmVkdQ==