 Weidong Dou
Weidong Dou Yu Guan
Yu Guan Hang Zheng
Hang Zheng Xin Wang
Xin Wang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Oncol., 29 September 2023
Sec. Surgical Oncology
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1212529
Epithelioid inflammatory myofibroblastic sarcoma (EIMS) is a rare variant of the inflammatory myofibroblastic tumor, characterized by more aggressive clinical course and nuclear membrane staining of anaplastic lymphoma kinase (ALK) with ALK rearrangement. An elderly male came to the clinic because of an accidental abdominal mass. Abdominal and pelvic enhanced CT revealed a tumor apparently orginated from mesenchymal tissue. Subsequently, the abdominal mass and multiple organ resection was performed, and the mass was pathologically confirmed as EIMS. The patient developed Clavien-Dindo Grade III postoperative complications and was discharged after his condition improved. He received doxorubicin monotherapy after operation, but only one cycle was administered due to severe vomiting. The follow-up of 5 months after operation showed no evidence of recurrence. Given the rarity of EIMS, and ALk inhibitors have a long and robust effect on patients with ALK gene tumors, it is very important for clinicians to be familiar with the clinicopathological features of EIMS, which will contribute to the accurate diagnosis of EIMS and reduce misdiagnosis.
Inflammatory myofibroblastic tumor (IMT) is a distinctive mesenchymal-derived tumor characterized by predominantly myofibroblastic spindle cells with inflammatory infiltration, which may recur and rare metastasize (1). ALK rearrangement on chromosome 2p23 is found in 50% of IMT patients (2, 3). Epithelioid inflammatory myofibroblastic sarcoma (EIMS), known as a variant of IMT, which was first described by Marino-Enriquez in 2011 (4), exhibits malignant behavior, has a high likelihood of recurrence and is associated with a poor prognosis (5, 6). Most of the literatures focus on the pathological features of EIMS. Herein, we report a case of EIMS and review the previous literature to summarize its clinical features and treatment progress.
A 70-year-old man with a 10-year history of well-controlled hypertension was hospitalized due to an incidentally discovered abdominal mass. Physical examination revealed a large palpable mass with tenderness in the left upper quadrant. Abdominal and pelvic enhanced CT showed multiple soft-tissue density mass in the abdominal cavity, 14.1 cm×14 cm× 10.5 cm, lobulated, invading the stomach and pancreas. The tumor might come from the left upper abdomen and jejunum with multiple metastasis in the abdominal cavity. (Figure 1) Considering that the patient gradually developed incomplete intestinal obstruction during hospitalization, after a multi-disciplinary team (MDT) discussion, the patient accepted exploratory laparotomy, abdominal mass resection, left hemicolectomy and involved distal pancreatectomy, with splenic preservation. (Figure 2) Postoperative pathology showed that the tumor cells were short spindle and oval, and a large number of lymphocytes, plasma cells and eosinophils were infiltrated in the stroma. FISH staining showed isolated ALK signal in the nucleus. Based on the location, morphology, immunophenotype and molecular detection, the tumor was considered to be epithelioid inflammatory myofibroblastic sarcoma. The patient’s recovery was not uneventful, with a grade B pancreatic fistula and bilateral pleural effusion that improved after parenteral nutrition support, inhibition of pancreatic enzyme activity and drainage. In addition, the patient often experienced abdominal distension and occasional nausea postprandially, but no obvious signs of obstruction were seen on the postoperative plain CT scan of the abdomen and pelvis. The patient was discharged with an abdominal drainage tube while he was able to eat and move normally at 1 month after surgery.
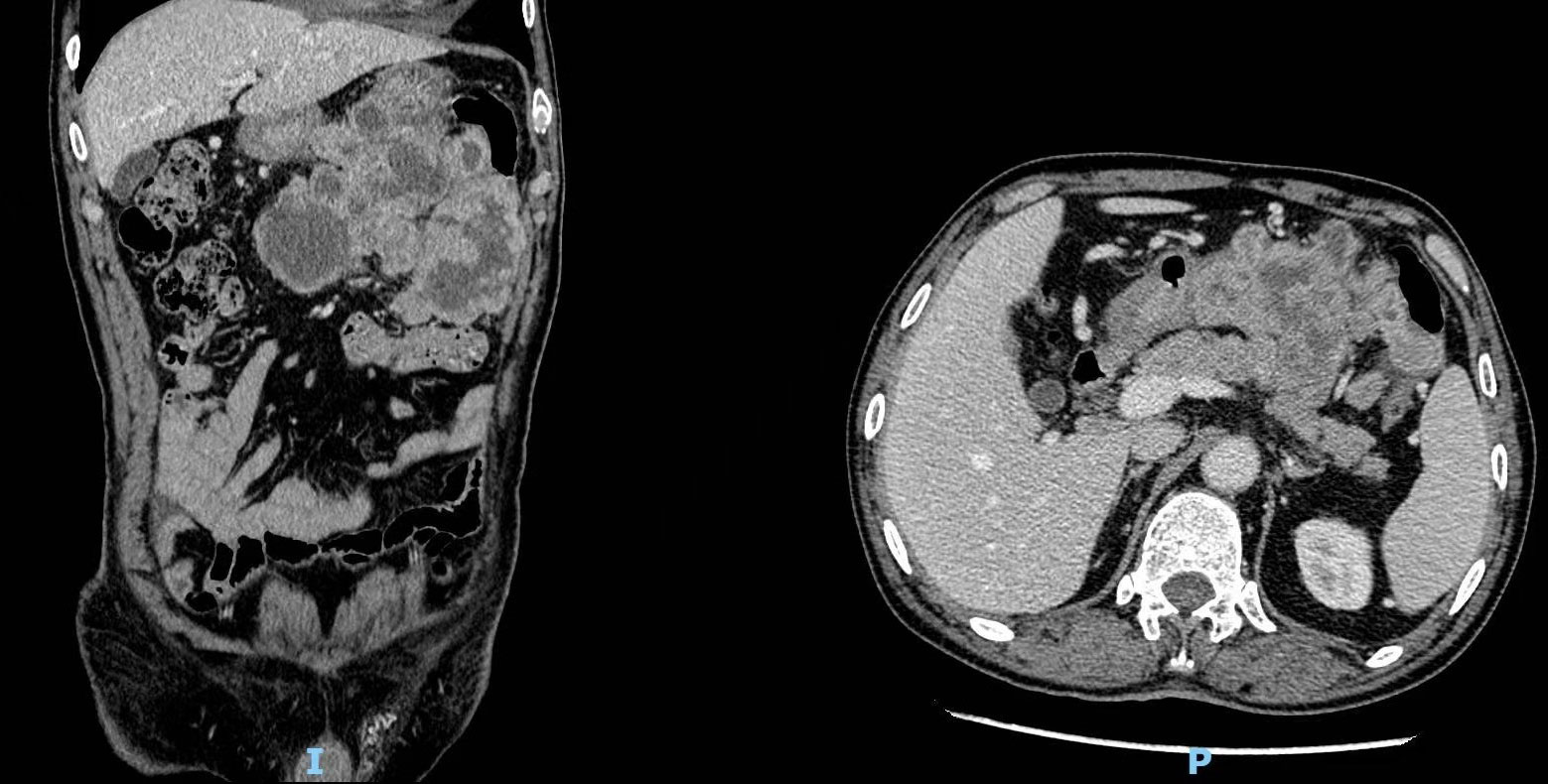
Figure 1 Computed tomography (CT) revealed multiple soft-tissue density masses in the left upper abdomen with unclear boundaries.
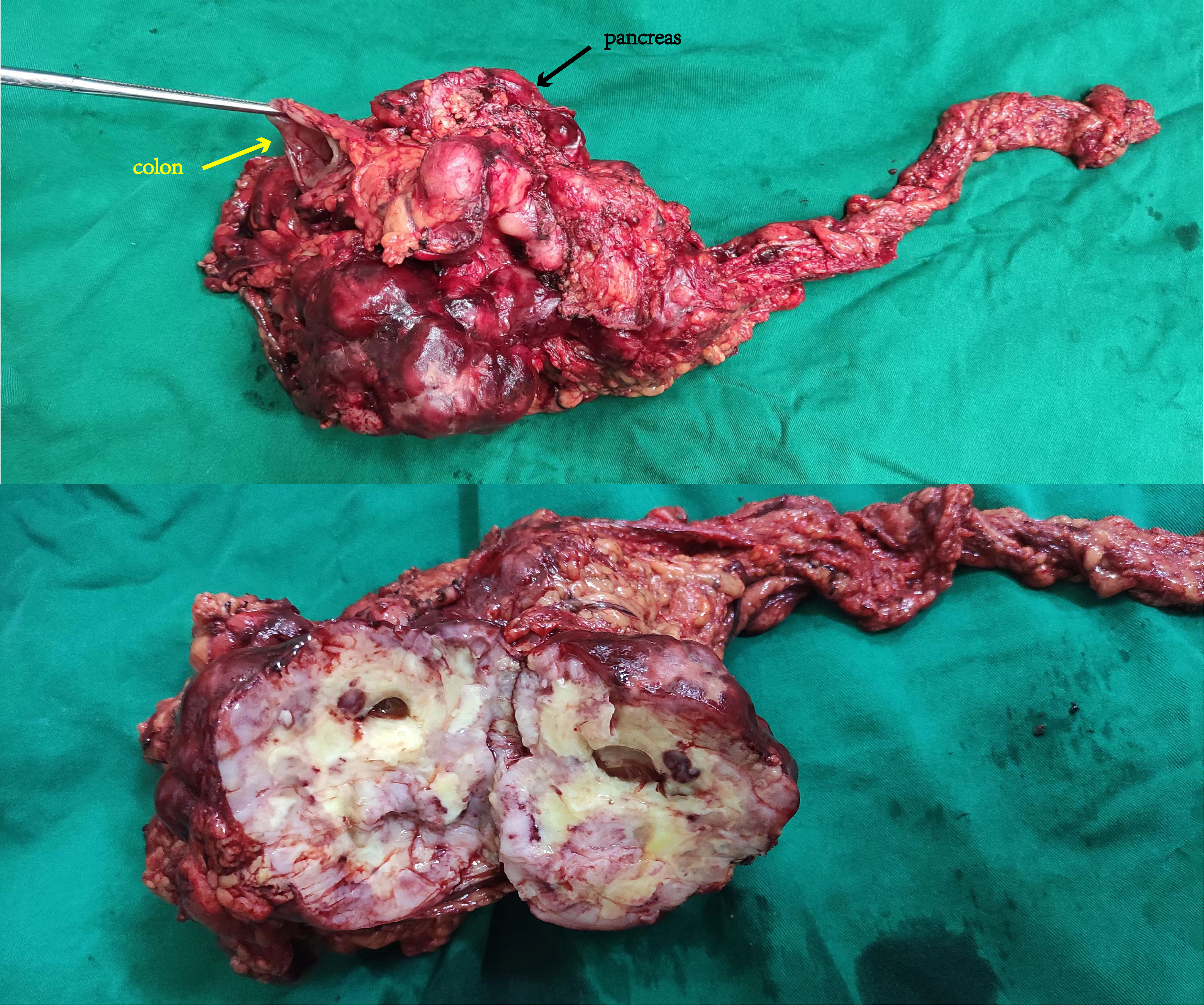
Figure 2 Grossly, the tumor is nodular, encapsulated, with gray-yellow cut surface and necrosis.
One month after discharge, the abdominal drainage tube was removed in the outpatient department, but the patient still felt abdominal distension after eating. He received doxorubicin single-drug chemotherapy, but the regime could not be continued due to severe nausea and vomiting. He and his family were relatively satisfied with the treatment and accepted the follow-up advice. The patient was still alive without disease relapse at the 5-month follow-up after operation.
Epithelioid inflammatory myofibroblastic sarcoma (EIMS) is a malignant subtype of IMT, with high invasiveness and poor clinical prognosis (4, 7). The exact incidence of EIMS is still unknown, due to the rarity of EIMS, no large-scale, convincing clinical trial is available, with only ~55 cases being reported to date (4–6, 8–33). In this study, we describe an additional case of EIMS originating from the abdomen. The clinical characteristics of these 56 EIMSs are presented in Table 1.
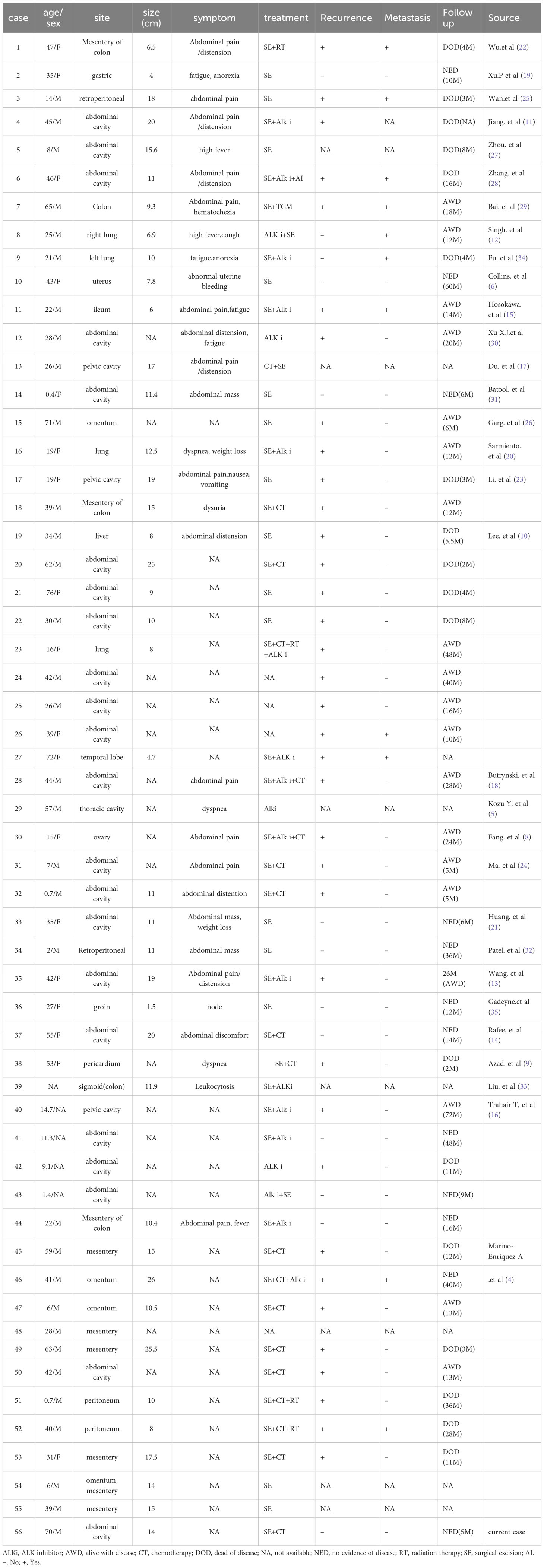
Table 1 Clinical characteristics of 56 cases of epithelioid inflammatory myofibroblastic sarcoma.
The onset age of EIMSs ranges from 4 months to 76 years (with mean age 31.6 years), and tends to occur in men and within the abdominal cavity. The reported sites of involvement include the liver (36), rectum, and transverse colon (37). The lung is also a common site of involvement (34). In addition, rare sites such as ovary (8), inguinal subcutaneous tissue (38), central nervous system (39) and pericardium (9) have also been reported. In a review of the previous literature, symptoms of EIMS have been associated with tumor location without significant specificity. Instead of abdominal pain and palpable masses, systemic fatigue and weight loss are the main clinical manifestations in some patients (34). In addition, dyspnea, cough, bleeding, repeated fever, and leukemia-like reaction have been reported as the first symptoms.
The diagnosis of EIMS is challenging because of its rarity, atypical clinical symptoms, and no characteristic findings on imaging examination. Additionally, the low positive rate of biopsy, and the atypical morphological features add to the difficulty of diagnosis. Previous studies have also proposed diagnostic criteria including: (1) round-to-epithelioid tumor cells; (2) plentiful myxoid stroma with inflammatory infiltration; (3) positive ALK immunohistochemical test (5). Nevertheless, the diagnosis of EIMS based on histology or ALK expression alone may be precarious as not all IMTs of epithelioid/round cell morphology carry the genetic changes of EIMS (2), and some mesenchymal tumors such as rhabdomyosarcoma, lipoma, leiomyosarcoma, malignant peripheral nerve sheath tumor, Ewing sarcoma/peripheral primitive neuroectodermal tumor, etc. may also have positive ALK cytoplasmic staining (40, 41). Consequently, it is recommended that further detection of ALK rearrangements by FISH, RT-PCR or next-generation sequencing(NGS) techniques be performed to confirm the diagnosis of EIMS in cases with atypical morphology or unusual immunoprofile.
Strong desmin positivity and perinuclear or cytoplasmic ALK positivity were confirmed in all described EIMS cases (39). Furthermore, ALK can be fused with multiple genes to form different chromosomal rearrangements in IMT, including TPM3 and TPM4, CLTC, RAN-BP2, CARS, ATIC, and SEC31L1. The most common form of EIMS is RANBP2-ALK fusion, which is characterized by nuclear membrane staining of ALK protein on immunohistochemistry. RRBP1-ALK (10) fusion and EML4-ALK (11) fusion have also been reported. According to the difference in fusion gene and the location of fusion protein, the displayed immunohistochemical positive pattern is different. Since RANBP2 was a macromolecular protein located in the nuclear pore, the positive position of ALK protein fusion mode by immunohistochemistry was located in the nuclear membrane. RRBP1 is a ribosomal binding protein, and the positive position by RRBP1-ALK fusion immunostaining is cytoplasmic and perinuclear. Both are associated with more aggressive biological behavior and a worse prognosis (10, 12, 37). In addition, VCL-ALK gene fusion has also been reported in the central nervous system EIMS (39).
The EMIS should be differentially diagnosed from other disease as follows: (1) Anaplastic large cell tumor (ALCL), especially the sarcomatoid variantis, the most difficult to differentiate from EIMS because they have similar histomorphology and are positive for ALK, CD30, and SMA (42). However, RANBP2-ALK fusion and typical ALK nuclear membrane expression pattern were not observed in ALCL, and most of them were expressed in the cytomembrane and Golgi apparatus. In addition, strong desmin positivity was absent in ALCL.(2) High-grade leiomyosarcoma lack of typical expression pattern of ALK in EIMS, but is generally accompanied with typical histological features in focal region, that is spindle cells arranged in bundles with red-stained cytoplasm and “cigar-shaped” nuclei (3) A solid variant of alveolar rhabdomyosarcoma, often ALK-positive (40), could usually be differentiated by histological examination. It is generally homogeneous cytologically, with inadequate cytoplasm, a lack of myxoid stroma, and prominent neutrophils. Nuclear immunopositivity for myostatin (MYFF 4) contributed to the diagnosis of rhabdomyosarcoma.(4) Dedifferentiated liposarcoma, especially those showing an “inflammatory MFH” pattern (43). Dedifferentiated liposarcoma can also mimic conventional IMT (44, 45). In such cases, overexpression of MDM2 can be misleading, as a high proportion of IMTs show nuclear staining of MDM2 (3, 46), however, the nuclear membrane staining for ALK is absent in liposarcoma.(5) Gastrointestinal stromal tumor (GIST), although epithelioid GISTs with inflammatory myxoid backgrounds are rare, GISTs should be ruled out first for mesenchymal tumors in the abdominal cavity, which can be identified by immunohistochemistry CD117, CD34, and Dog-1. Furthermore, GIST are associated with C-kit and PDGF-α mutations.
There is no consensus on the optimal treatment for EIMS, owing to its rarity and high malignancy, and surgical resection is still considered the primary treatment. However, recurrence is common after resection (13, 36). Some previous reports indicated that postoperative adjuvant chemotherapy or radiotherapy has no distinct effect in the control of aggressive progression of EIMS (4, 14–16, 37). Postoperative immunotherapies have not yet been clearly identified. Programmed cell death ligand 1 (PD-L1) is found to be diffusely positive in some cases (17). The PD-1/PD-L1 axis has also been reported to play an important role in the immune antitumor response (47, 48). Therefore, a new immunomodulatory therapy targeting the PD-1/PD-L1 pathway might be developed.
It is reported that crizotinib has strong and durable activity against ALK-positive IMT (49, 50). Since ALK gene rearrangement has been reported in all cases, ALK inhibitors such as crizotinib and ceritinib may display unexpected therapeutic prospects for EIMS. Butrynski et al. described for the first time the therapeutic effects of crizotinib in patients with RANBP2-ALK fusion EIMS (18). Trahair, T et al. suggested that crizotinib is tolerable and effective in patients with life-threatening complications (16). Subsequent reports have also shown a relatively satisfactory effect of ALK inhibitors in patients with EIMS (10, 15, 37). At the same time, ALK negative patients didn’t respond (10, 19). However, long-term reactions or responses are seldom seen and most patients with EIMS experience disease relapse, progression, or metastasis within three to six months after receiving crizotinib monotherapy (5, 20, 34). Furthermore, studies by Trahair, T et al. have shown that patients with RANBP2-ALK rearrangement EIMS still relapse despite a complete response to crizotinib initially (16). Alectinib, a highly selective ALK inhibitor, has been shown to have a high response rate and a long progression-free survival in patients with ALK-positive lung cancer in Phase I/II studies, without severe toxicity, even in patients who have failed treatment with crizotinib. In a phase III study, compared with crizotinib, alectinib showed superior efficacy and lower toxicity in primary treatment of ALK-positive NSCLC, which may suggest a more potent effect of aletinib-bick-zotinib in patients with EIMS.
CD30 positivity is another characteristic pathological feature of EIMS (4, 16, 21, 22, 35), suggesting that brentukimab vedotin (BV), a CD30-targeting antibody-drug conjugate, could be a potential therapeutic option for EIMS. Additionally, Fordham, A.M. et al. validated the potential of CD30 as a therapeutic target in EIMS, and the combination therapy targeting CD30 and ALK at the time of initial treatment was more effective than monotherapy or combination therapy at the time of relapse, which also suggests the potential of combination therapy in preventing recurrence or treating ALKi-resistant EIMS disease (51).
Regarding the biological behavior of EIMS, of the 48 patients with follow-up information, 18 (37.5%) died of the disease (15 within 1 year of diagnosis and 3 within 3 years of diagnosis), 19 (39.5%) were alive with the disease, and the remaining 11 (23%) were well without evidence of disease. The median overall survival was 12 months (mean 17.4 months). Furthermore, Only 8(16.7%) patients were followed up without recurrence, disease progression or metastasis. To date, clinicopathological factors associated with the prognosis of EIMS are unknown. Multiple studies have suggested that the aggressive course of the disease may be related to intraperitoneal origin, large tumor size, epithelioid morphology and RANBP2-ALK (2–4, 18, 23, 24).
Despite the favorable efficacy of ALK inhibitors such as crizotinib in the treatment of EIMS, there are still many unresolved issues that need to be further explored. For instance, it is currently unclear whether crizotinib should be used as first-line therapy or whether surgery should be performed before crizotinib treatment to minimize tumor size. In our opinion, ALK inhibitor therapy may be given priority to patients who need combined multiple organ resection after preoperative evaluation, because such patients usually have a poor prognosis and have many serious postoperative complications (25, 34). One of the current issues to be addressed is how to prevent or delay the development of resistance to ALK inhibitors. Further studies are needed in patients with EIMS. Moreover, further drug development is necessary to increase the life expectancy of patients and alleviate the disease.
In conclusion, we present a case of primary abdominal EIMS, a highly aggressive variant of IMT characterized by epithelioid cell morphology, often with inflammatory infiltration of neutrophils and prominent perinuclear or nuclear membrane ALK staining. It has a significant male advantage and appears in the intra-abdominal region. The diagnosis of these tumors can be difficult due to epithelioid morphological abnormalities, especially when the tumor site is atypical. In addition, we show how this can be distinguished from its histological mimicry. The detection of ALK rearrangement by FISH, RT-PCR or NGS will contribute to further clarify the diagnosis of EIMS. Furthermore, the idefication of EIMS has important implications for clinical management, as ALK inhibitors are critical and effective agents for EIMS treatment.
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding authors.
Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
WD drafted the manuscript. YG, TL, HZ, SF, YW collected data. ZL and XW designed the study and revised the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by National Project for Clinical Key Specialty Development
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Fletcher CDM, Bridge JA, Hogendoorn PCW, Mertens F eds. World Health Organization classification of tumours of soft tissueand bone. 4th edn. Lyon: IARC Press. (2013).
2. Cook JR, Dehner LP, Collins MH, Ma Z, Morris SW, Coffin CM, et al. Anaplastic lymphoma kinase (ALK) expression in the inflammatory myofibroblastic tumor: a comparative immunohistochemical study. Am J Surg Pathol (2001) 25(11):1364–71. doi: 10.1097/00000478-200111000-00003
3. Coffin CM, Hornick JL, Fletcher CD. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol (2007) 31(4):509–20. doi: 10.1097/01.pas.0000213393.57322.c7
4. Mariño-Enríquez A, Wang WL, Roy A, Lopez-Terrada D, Lazar AJ, Fletcher CD, et al. Epithelioid inflammatory myofibroblastic sarcoma: An aggressive intra-abdominal variant of inflammatory myofibroblastic tumor with nuclear membrane or perinuclear ALK. Am J Surg Pathol (2011) 35(1):135–44. doi: 10.1097/PAS.0b013e318200cfd5
5. Kozu Y, Isaka M, Ohde Y, Takeuchi K, Nakajima T. Epithelioid inflammatory myofibroblastic sarcoma arising in the pleural cavity. Gen Thorac Cardiovasc Surg (2014) 62(3):191–4. doi: 10.1007/s11748-013-0204-x
6. Collins K, Ramalingam P, Euscher ED, Reques Llanos A, García A, Malpica A. Uterine inflammatory myofibroblastic neoplasms with aggressive behavior, including an epithelioid inflammatory myofibroblastic sarcoma: A clinicopathologic study of 9 cases. Am J Surg Pathol (2022) 46(1):105–17. doi: 10.1097/PAS.0000000000001756
7. Meis JM, Enzinger FM. Inflammatory fibrosarcoma of the mesentery and retroperitoneum. A tumor closely simulating inflammatory pseudotumor. Am J Surg Pathol (1991) 15(12):1146–56. doi: 10.1097/00000478-199112000-00005
8. Fang H, Langstraat CL, Visscher DW, Folpe AL, Schoolmeester JK. Epithelioid inflammatory myofibroblastic sarcoma of the ovary with RANB2-ALK fusion: Report of a case. Int J gynecological Pathol Off J Int Soc Gynecological Pathologists (2018) 37(5):468–72. doi: 10.1097/PGP.0000000000000431
9. Azad M, Oye M, Torrente N, Mirsaeidi M. Pericardial epithelioid inflammatory myofibroblastic sarcoma: An atypical presentation. Cureus (2022) 14(7):e26827. doi: 10.7759/cureus.26827
10. Lee JC, Li CF, Huang HY, Zhu MJ, Mariño-Enríquez A, Lee CT, et al. ALK oncoproteins in atypical inflammatory myofibroblastic tumours: novel RRBP1-ALK fusions in epithelioid inflammatory myofibroblastic sarcoma. J Pathol (2017) 241(3):316–23. doi: 10.1002/path.4836
11. Jiang Q, Tong HX, Hou YY, Zhang Y, Li JL, Zhou YH, et al. Identification of EML4-ALK as an alternative fusion gene in epithelioid inflammatory myofibroblastic sarcoma. Orphanet J Rare Dis (2017) 12(1):97. doi: 10.1186/s13023-017-0647-8
12. Singh P, Nambirajan A, Gaur MK, Raj R, Kumar S, Malik PS, et al. Primary pulmonary epithelioid inflammatory myofibroblastic sarcoma: a rare entity and a literature review. J Pathol Trans Med (2022) 56(4):231–7. doi: 10.4132/jptm.2022.05.08
13. Wang Z, Geng Y, Yuan LY, Wang MM, Ye CY, Sun L, et al. Durable clinical response to ALK tyrosine kinase inhibitors in epithelioid inflammatory myofibroblastic sarcoma harboring PRRC2B-ALK rearrangement: A case report. Front Oncol (2022) 12:761558. doi: 10.3389/fonc.2022.761558
14. Rafee S, Elamin YY, Joyce E, Toner M, Flavin R, McDermott R, et al. Neoadjuvant crizotinib in advanced inflammatory myofibroblastic tumour with ALK gene rearrangement. Tumori (2015) 101(2):e35–9. doi: 10.5301/tj.5000245
15. Kurihara-Hosokawa K, Kawasaki I, Tamai A, Yoshida Y, Yakushiji Y, Ueno H, et al. Epithelioid inflammatory myofibroblastic sarcoma responsive to surgery and an ALK inhibitor in a patient with panhypopituitarism. Internal Med (Tokyo Japan) (2014) 53(19):2211–4. doi: 10.2169/internalmedicine.53.2546
16. Trahair T, Gifford AJ, Fordham A, Mayoh C, Fadia M, Lukeis R, et al. Crizotinib and surgery for long-term disease control in children and adolescents with ALK-positive inflammatory myofibroblastic tumors. JCO Precis Oncol (2019) 3:1–11. doi: 10.1200/PO.18.00297
17. Du X, Gao Y, Zhao H, Li B, Xue W, Wang D. Clinicopathological analysis of epithelioid inflammatory myofibroblastic sarcoma. Oncol Lett (2018) 15(6):9317–26. doi: 10.3892/ol.2018.8530
18. Butrynski JE, D'Adamo DR, Hornick JL, Dal Cin P, Antonescu CR, Jhanwar SC, et al. Crizotinib in ALK-rearranged inflammatory myofibroblastic tumor. N Engl J Med (2010) 363(18):1727–33. doi: 10.1056/NEJMoa1007056
19. Xu P, Shen P, Jin Y, Wang L, Wu W. Epithelioid inflammatory myofibroblastic sarcoma of stomach: diagnostic pitfalls and clinical characteristics. Int J Clin Exp Pathol (2019) 12(5):1738–44.
20. Sarmiento DE, Clevenger JA, Masters GA, Bauer TL, Nam BT. Epithelioid inflammatory myofibroblastic sarcoma: a case report. J Thorac Dis (2015) 7(10):E513–6. doi: 10.3978/j.issn.2072-1439.2015.10.55
21. Huang YH, Tian YF, Li CF. Inflammatory myofibroblastic tumor with RANBP2 and ALK gene rearrangement with bland cytological features mimicking desmoid-type fibromatosis: A case report and review of the literature. Oncol Lett (2016) 11(2):1429–34. doi: 10.3892/ol.2016.4082
22. Wu H, Meng YH, Lu P, Ning HY, Hong L, Kang XL, et al. Epithelioid inflammatory myofibroblastic sarcoma in abdominal cavity: a case report and review of literature. Int J Clin Exp Pathol (2015) 8(4):4213–9.
23. Ma Z, Hill DA, Collins MH, Morris SW, Sumegi J, Zhou M, et al. Fusion of ALK to the Ran-binding protein 2 (RANBP2) gene in inflammatory myofibroblastic tumor. Genes Chromosomes Cancer (2003) 37(1):98–105. doi: 10.1002/gcc.10177
24. Li J, Yin WH, Takeuchi K, Guan H, Huang YH, Chan JK. Inflammatory myofibroblastic tumor with RANBP2 and ALK gene rearrangement: a report of two cases and literature review. Diagn Pathol (2013) 8:147. doi: 10.1186/1746-1596-8-147
25. Wan YY, Miao CL, Liu SB, Zhang T, Luo CH. Epithelioid inflammatory myofibroblastic sarcoma with leukemoid reaction. J Coll Physicians Surg Pak (2022) 32(9):1212–5. doi: 10.29271/jcpsp.2022.09.1212
26. Garg R, Kaul S, Arora D, Kashyap V. Posttransplant epithelioid inflammatory myofibroblastic sarcoma: A case report. Indian J Pathol Microbiol (2019) 62(2):303–5. doi: 10.4103/IJPM.IJPM_284_17
27. Zhou J, Jiang G, Zhang D, Zhang L, Xu J, Li S, et al. Epithelioid inflammatory myofibroblastic sarcoma with recurrence after extensive resection: significant clinicopathologic characteristics of a rare aggressive soft tissue neoplasm. Int J Clin Exp Pathol (2015) 8(5):5803–7.
28. Zhang S, Wang Z. A case report on epithelioid inflammatory myofibroblastic sarcoma in the abdominal cavity. Int J Clin Exp Pathol (2019) 12(10):3934–9.
29. Bai Y, Jiang M, Liang W, Chen F. Incomplete intestinal obstruction caused by a rare epithelioid inflammatory myofibroblastic sarcoma of the colon: A case report. Medicine (2015) 94(51):e2342. doi: 10.1097/MD.0000000000002342
30. Xu X, Li H, Peng K, Yu Y, Chen L, Fang Y, et al. ALK-G1269A mutation in epithelioid inflammatory myofibroblastic sarcoma after progression on crizotinib: A case report. Oncol Lett (2019) 17(2):2370–6. doi: 10.3892/ol.2018.9865
31. Batool S, Ahuja A, Chauhan DS, Bhardwaj M, Meena AK. Epithelioid inflammatory myofibroblastic sarcoma: the youngest case reported. Autopsy Case Rep (2021) 11:e2021288. doi: 10.4322/acr.2021.288
32. Patel AS, Murphy KM, Hawkins AL, Cohen JS, Long PP, Perlman EJ, et al. RANBP2 and CLTC are involved in ALK rearrangements in inflammatory myofibroblastic tumors. Cancer Genet Cytogenet (2007) 176(2):107–14. doi: 10.1016/j.cancergencyto.2007.04.004
33. Liu D, Luo R, Tang H, Li T. Sigmoid epithelioid inflammatory myofibroblastic sarcoma with high white blood cell count: A case report. Asian J Surg (2020) 43(8):838–9. doi: 10.1016/j.asjsur.2020.03.010
34. Fu X, Jiang J, Tian XY, Li Z. Pulmonary epithelioid inflammatory myofibroblastic sarcoma with multiple bone metastases: case report and review of literature. Diagn Pathol (2015) 10:106. doi: 10.1186/s13000-015-0358-1
35. Gadeyne L, Creytens D, Dekeyser S, van der Meulen J, Haspeslagh M. Primary cutaneous epithelioid inflammatory myofibroblastic sarcoma harboring RANBP2-ALK fusion: Report of an exceptional case. Am J Dermatopathol (2022) 44(4):302–5. doi: 10.1097/DAD.0000000000002096
36. Chen ST, Lee JC. An inflammatory myofibroblastic tumor in liver with ALK and RANBP2 gene rearrangement: combination of distinct morphologic, immunohistochemical, and genetic features. Hum Pathol (2008) 39(12):1854–8. doi: 10.1016/j.humpath.2008.04.016
37. Yu L, Liu J, Lao IW, Luo Z, Wang J. Epithelioid inflammatory myofibroblastic sarcoma: a clinicopathological, immunohistochemical and molecular cytogenetic analysis of five additional cases and review of the literature. Diagn Pathol (2016) 11(1):67. doi: 10.1186/s13000-016-0517-z
38. Zilla ML, Khoshnoodi P, Bailey NG, Herradura A, Lee SJ, John I. Epithelioid inflammatory myofibroblastic sarcomas are not exclusive to ventral cavity sites. Histopathology (2022) 80(3):610–2. doi: 10.1111/his.14582
39. Chopra S, Maloney N, Wang WL. Epithelioid inflammatory myofibroblastic sarcoma with VCL-ALK fusion of central nervous system: case report and brief review of the literature. Brain Tumor Pathol (2022) 39(1):35–42. doi: 10.1007/s10014-021-00416-z
40. Li XQ, Hisaoka M, Shi DR, Zhu XZ, Hashimoto H. Expression of anaplastic lymphoma kinase in soft tissue tumors: an immunohistochemical and molecular study of 249 cases. Hum Pathol (2004) 35(6):711–21. doi: 10.1016/j.humpath.2003.12.004
41. Cessna MH, Zhou H, Sanger WG, Perkins SL, Tripp S, Pickering D, et al. Expression of ALK1 and p80 in inflammatory myofibroblastic tumor and its mesenchymal mimics: a study of 135 cases. Modern Pathol an Off J United States Can Acad Pathology Inc (2002) 15(9):931–8. doi: 10.1097/01.MP.0000026615.04130.1F
42. Suzuki R, Seto M, Nakamura S, Nakagawa A, Hara K, Takeuchi K. Sarcomatoid variant of anaplastic large cell lymphoma with cytoplasmic ALK and alpha-smooth muscle actin expression: a mimic of inflammatory myofibroblastic tumor. Am J Pathol (2001) 159(1):383–4. doi: 10.1016/S0002-9440(10)61706-3
43. Coindre JM, Hostein I, Maire G, Derré J, Guillou L, Leroux A, et al. Inflammatory Malignant fibrous histiocytomas and dedifferentiated liposarcomas: histological review, genomic profile, and MDM2 and CDK4 status favour a single entity. J Pathol (2004) 203(3):822–30. doi: 10.1002/path.1579
44. Gleason BC, Hornick JL. Inflammatory myofibroblastic tumours: where are we now? J Clin Pathol (2008) 61(4):428–37. doi: 10.1136/jcp.2007.049387
45. Lucas DR, Shukla A, Thomas DG, Patel RM, Kubat AJ, McHugh JB. Dedifferentiated liposarcoma with inflammatory myofibroblastic tumor-like features. Am J Surg Pathol (2010) 34(6):844–51. doi: 10.1097/PAS.0b013e3181db34d8
46. Yamamoto H, Oda Y, Saito T, Sakamoto A, Miyajima K, Tamiya S, et al. p53 Mutation and MDM2 amplification in inflammatory myofibroblastic tumours. Histopathology (2003) 42(5):431–9. doi: 10.1046/j.1365-2559.2003.01611.x
47. Stenehjem DD, Tran D, Nkrumah MA, Gupta S. PD1/PDL1 inhibitors for the treatment of advanced urothelial bladder cancer. OncoTargets Ther (2018) 11:5973–89. doi: 10.2147/OTT.S135157
48. Zandberg DP, Strome SE. The role of the PD-L1:PD-1 pathway in squamous cell carcinoma of the head and neck. Oral Oncol (2014) 50(7):627–32. doi: 10.1016/j.oraloncology.2014.04.003
49. Mossé YP, Voss SD, Lim MS, Rolland D, Minard CG, Fox E, et al. Targeting ALK with crizotinib in pediatric anaplastic large cell lymphoma and inflammatory myofibroblastic tumor: A children's oncology group study. J Clin Oncol Off J Am Soc Clin Oncol (2017) 35(28):3215–21. doi: 10.1200/JCO.2017.73.4830
50. Gambacorti-Passerini C, Orlov S, Zhang L, Braiteh F, Huang H, Esaki T, et al. Long-term effects of crizotinib in ALK-positive tumors (excluding NSCLC): A phase 1b open-label study. Am J Hematol (2018) 93(5):607–14. doi: 10.1002/ajh.25043
Keywords: EIMS, ALK-inhibitors, ALK, case report, literature review
Citation: Dou W, Guan Y, Liu T, Zheng H, Feng S, Wu Y, Wang X and Liu Z (2023) Epithelioid inflammatory myofibroblastic sarcoma: a case report and brief literature review. Front. Oncol. 13:1212529. doi: 10.3389/fonc.2023.1212529
Received: 26 April 2023; Accepted: 01 September 2023;
Published: 29 September 2023.
Edited by:
Hector Guadalajara, University Hospital Fundación Jiménez Díaz, SpainReviewed by:
Montiel Jiménez Fuertes, University Hospital Fundación Jiménez Díaz, SpainCopyright © 2023 Dou, Guan, Liu, Zheng, Feng, Wu, Wang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Wang, d2FuZ3hpbl9ndW9AMTI2LmNvbQ==; Zhanbing Liu, WmhhbmJpbmc1OEAxNjMuY29t
†Means first author
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.