 Jiangbo Shao
Jiangbo Shao Ying Jin
Ying Jin Chunxiang Jin
Chunxiang Jin
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 10 August 2023
Sec. Pharmacology of Anti-Cancer Drugs
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1210245
This article is part of the Research Topic Overcoming Obstacles of Cancer Immunotherapy: The Important Role of Emerging Nanomedicine View all 6 articles
Immunotherapy for immune response has ushered in a new era of cancer treatment. In recent years, new immunotherapeutic agents have been introduced into clinical trials and even approved for marketing. However, the widespread use of immunotherapeutic agents faces an unavoidable challenge: immunotherapy does not work at all for some patients, or has good efficacy in the initial phase, but immunotherapy resistance develops within a short period of time, and immunotherapy can also cause serious adverse effects such as autoimmune inflammation and non-specific inflammation. How to enable patients to overcome drug resistance, reduce the toxic side effects of drugs, enhance patient compliance and improve patient survival has become a problem that clinicians have to face. The advent of nanotechnology provides an encouraging platform for immunotherapy. It can not only improve the bioavailability and stability of drugs and reduce toxic side effects, but also reduce resistance to immunotherapy. Here, we discuss these research advances and discuss potential challenges and future directions.
In 2013, tumor immunotherapy was named the most important scientific breakthrough of the year by the journal Science for its outstanding efficacy and innovation (1). Immunotherapy has now become an important treatment for many malignant tumors, ushering in a new era of tumor treatment and bringing light to the fight against tumors. However, as the use of immunotherapy has become more popular, the problem of drug resistance to immunotherapy has gradually emerged. Due to its special mechanism of action, the mechanism of resistance to immunotherapy is different from that of resistance to conventional chemotherapy (2–6), so how to overcome resistance to immunotherapy is currently an urgent challenge to be solved.
Recent advances in nanomedicine have led to a plethora of new nanomedicines targeting immunotherapeutic targets (7–11). Furthermore, as research has progressed, it has been found that nanomedicines not only have the ability to target drug delivery, but also have the ability to remodel the immune microenvironment, thereby enhancing the efficacy of anti-tumor immunotherapy (12). Therefore, we believe that nanomedicines provide a relevant reference for basic research and the development of improved clinical treatment regimens, and are expected to be an important weapon in overcoming drug resistance in immunotherapy. In this article, we review the mechanisms of immunotherapy resistance and summarize the recent research advances in nanomedicine with respect to the mechanisms of resistance, with the aim of providing clues for overcoming immunotherapy resistance.
Tumors are inherently complex and heterogeneous, and resistance involves not only the tumor but also complex metabolic, inflammatory and neovascular mechanisms, many of which are still unknown. At present, the mechanisms of resistance to immunotherapy can be divided into intrinsic and extrinsic mechanisms. Intrinsic mechanisms of tumor immune resistance include alterations in anti-tumor immune response pathways and alterations in tumor cell signaling leading to a suppressive immunosuppressive microenvironment. External factors mainly include those associated with the local tumor microenvironment. The tumor immune microenvironment is a complex network of interactions involving tumor and various non-tumor cells, including fibroblasts, macrophages, B/T lymphocytes and antigen presenting cells (APCs), which in turn affect the whole body.
The immune response pathway involves the processing of tumor-associated antigenic peptides by antigen-presenting cells (APCs), presentation to CD8+ T cells, stimulation of T cell proliferation and activation, and activation of T cells to kill tumor cells in the TME (13). Any alteration in the antitumor immune response pathway has the potential to lead to resistance to immunotherapy.
High tumor mutational burden (TMB) (non-synonymous mutations), microsatellite instability (MSI) and defective mismatch repair (dMMR) are intrinsic tumor features associated with anti-tumor immune responses and responses to immune checkpoint inhibitors (ICIs) (14, 15). These responses are closely associated with increased generation of tumor-associated antigens (TAA) and tumor-specific antigens (TSA), with neoantigens conferring greater immunogenicity and increased T-cell infiltration, as has been demonstrated in a variety of tumors (16–19). However, tumor cells tend to suppress T-cell activation by reducing or losing antigen expression (20) and regulate autoantigenicity by endocytosis of antigens or antigen shedding to mediate immune escape. In addition, the host can selectively eliminate TSA-expressing cells and, to some extent, promote the production of tumor antigen-losing variants (21). Tumor cells, like viruses, can also undergo ‘antigenic drift’, resulting in epitope mutations that alter tumor antigenicity and thus evade T cell-mediated attack (22). Endoplasmic reticulum (ER) stress and autophagy determine the immunogenicity of tumor cell death (23, 24). The high presence of LC3b puncta in the cytoplasm of tumor cells represents an active autophagic mechanism within tumor cells and is associated with the infiltration of tumor-infiltrating lymphocytes (TILs) and a favorable clinical outcome, whereas tumor cells do not respond to ER stress or autophagy induction may lead to resistance to ICIS (25, 26).
Deletion of the phosphatase and tensin homologue (PTEN) gene on chromosome 10 has been shown to be directly involved in the regulation of antitumor immunity. First, there is a significant correlation between loss of PTEN and reduced T-cell infiltration, ultimately leading to resistance to PD-1 monoclonal antibodies (27–29). In addition, dysfunctional PTEN also promotes the aggregation of suppressor immune cells, such as myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) (30–33). More importantly, deletion of PTEN expression has been shown to down-regulate autophagy (34, 35), which can effectively support tumor progression.
In addition, alterations associated with the IFN-γ pathway can also affect immune resistance. In patients receiving immunotherapy, tumor cells can downregulate or alter the IFN-γ pathway, such as loss-of-function alleles in the gene encoding JAK1/2 and alterations in STAT1, to evade the effects of IFN-γ and thus develop resistance (36, 37). Zaretsky et al (38) showed that melanoma patients resistant to PD-1 treatment acquired loss-of-function mutations in JAK1/2. Although tumor cells were still recognized by T cells, their JAK1/2 mutation rendered them insensitive to the anti-proliferative effects of IFN-γ and they lacked IFN-γ-induced PD-L1 and MHC class I surface expression. Similarly, analysis of tumors from ipilimumab-refractory patients showed that mutations in the IFN-γ pathway genes IFNGR1/2, JAK1/2 and IRF1 inhibit the response of tumor cells to IFN-γ signaling (39). This facilitates tumor escape from T-cell immunity, thereby conferring resistance to anti-CTLA4 therapy.
Checkpoint blockade with specific mAbs helps to inhibit pathways that maintain the duration and strength of the immune system. Inhibition of these checkpoint molecules works by reinvigorating the adaptive immune system and selectively eliminating primary and metastatic tumors. Currently, cytotoxic T lymphocyte-associated antigen-4(CTLA4), programmed death receptor-1(PD-1) and its ligand(PD-L1) are the main targets of immunotherapy and are the most widely used in clinical practice (40). In one study, RNA sequencing analysis of patients with NSCLC resistant to PD-L1 inhibitors demonstrated the presence of PD-L1 variant fragments in patients and confirmed their inhibitory effect on T-cell activity (37). TIM-3 (T cell immunoglobulin domain and mucin domain-3) is a negatively regulated immune checkpoint expressed on the surface of T cells, Treg cells and other innate immune cells and is capable of causing T cell depletion in cancer and chronic viral infections. It has been shown that T cells co-expressing TIM-3/PD-1 are more likely to fail, and that patients who fail to respond to anti-PD-1 therapy are often highly correlated with TIM-3 expression, while patients who are resistant to PD-1 therapy are also due to selective overexpression of TIM-3, resulting in tumor immune escape (41). Drugs and drug combinations targeting TIM-3 are therefore being developed to overcome immunotherapy resistance.
Tumor-associated macrophages (TAMs) have also been implicated in patient response to immunotherapy (42, 43).TAMs are classified into M1 and M2 macrophages based on their different activation pathways and expression of surface molecules: M1 type macrophages promote anti-tumor immunity and M2 type macrophages promote tumor progression by supporting angiogenesis, tumor cell metastasis and suppression of effector CD8+ T cells and NK cells due to reduced efficiency of antigen presentation (44–46). Recruitment of TAMs to tumor sites is mediated by tumor-derived proteins (e.g. CSF-1, VEGF and chemokines). The recruitment of TAMs to tumor sites is mediated by tumor-derived effector proteins such as CSF-1, VEGF and chemokines (47). tumors (48–51). A higher density of TAMs is associated with a poor clinical prognosis in cancer patients (52, 53). Fritz et al (54) found that depletion of TAMs can reduce tumor growth in a mouse model of lung adenocarcinoma through downregulation of M2 macrophages. It has been suggested that inactivation of CCL2 and/or CCR2 signaling is responsible for this phenomenon. Similar findings have been reported in other cancer types (e.g. T-cell lymphoma (55), colorectal cancer (56), lung cancer and breast cancer (56–58), where suppression or elimination of these macrophages in the TME may improve patient prognosis.
MDSCs contain a panel of neutrophils and monocytes with potent immunosuppressive properties that mediate immune responses induced by T cells, B cells and NK cells (59). Higher levels of neutrophils within the tumor are negatively associated with clinical outcome in cancer patients (60). Indeed, using multidimensional imaging, Si et al (61) provided direct evidence that MDSCs inhibit the expression of Teff-secreted granzyme B and Ki67 (markers of Teff cytotoxicity and proliferation, respectively). The presence of MDSCs in TME is strongly correlated with the efficacy of immunotherapy, as blockade of these cells leads to improved preclinical (62) and clinical outcomes (63). Due to the important role of MDSCs in promoting angiogenesis, tumor invasion and metastasis, these cells have become therapeutic targets for cancer (59, 64–68).
Cancer-associated fibroblasts (CAFs) are diverse stromal cell populations with multiple functions, including stromal deposition and remodeling, crosstalk with infiltrating leukocytes, and interaction with cancer cells (69). The presence and role of activated CAFs in the microenvironment has been associated with poor prognosis in several cancers (70). CAFs suppress the immune system by promoting physical and chemical barriers. CAFs induce a TH2 phenotype in neighboring cells, and cytokines produced by TH2 cells induce myeloid differentiation into MDSCs (71). CAFs also induce monocytes to differentiate into M2-type TAMs (72, 73), produce fibronectin and secrete TGF-β and IDO (74), further indicating the expansion of Treg cells. In addition, CAFs inhibit the activity of CD8+ T cells by expressing PD-L1 (75, 76).
NK cells can facilitate the recruitment of DC to solid tumors by releasing CC-chemokine ligand 5 (CCL5), XC-chemokine ligand 1 (XCL1) and XCL2. In addition, NK cells enter lymph nodes and influence the T cell response; and through the regulation of antigen-presenting cells, thereby regulating T cells. Activated NK cells can kill immature DCs while retaining mature DCs, thus ensuring successful T cell priming. Thus, the manipulation of NK cells in cancer aims to initiate a multilayered immune response, ultimately leading to protective and long-lasting immunity against the tumor (77).
In recent years, NK cell-associated immunotherapy has emerged as an alternative to ICB-based or vaccine-based immunotherapy (78–81). However, its therapeutic effects are largely limited by the downregulation of recognition ligands, and its immune effects can be further blocked by the secretion of tumor microenvironment such as transforming growth factor-β (TGF-β) (82–84). Researchers have demonstrated the benefits of combining immunotherapy with chemotherapy in cancer treatment (85–87). Therefore, there is great interest in developing combined strategies to enhance NK cell immunity.
Interferon gamma is a cytokine produced and secreted by effector T cells (TEFFs) and APCs. It acts through the JAK-STAT pathway (36) and has a dual role in anti-tumor immunity. Interferon γ can induce the production of the chemokines CXCL9 and CXCL10 and promote the recruitment of CXCR3+ lymphocytes and other immune cells around tumor cells, thereby exerting an anti-tumor immune effect (88); it can also exert direct anti-tumor cell proliferation and pro-apoptotic effects by binding to cell surface receptors and triggering a series of events that inhibit tumor cell growth and promote tumor cell death (89); In addition, IFN-γ can play a role in immune escape by increasing the expression of PD-L1 on the surface of tumor cells (89), and therefore altered IFN-γ secretion is thought to be closely associated with immune drug resistance.
Tregs are an important subset of T cells that help prevent excessive immune responses and autoimmunity, and can infiltrate human tumors and promote tumor growth (90). These FoxP3-expressing cells inhibit the Teff response either directly through physical contact or indirectly by suppressing the secretion of inhibitory cytokines including IL-10, IL-35 and TGF-β (91–94). After anti-CTLA-4 mAb treatment, the ratio of Teffs to Tregs was positively correlated with treatment response depending on the presence of macrophages expressing the Fcγ receptor (95), and the use of anti-CTLA4 antibodies with an enhanced Fcγ R binding profile is recommended to achieve robust anti-tumour responses and improved survival (96). In a clinical trial using ipilimumab to treat patients with advanced melanoma, increased TIL was found to be associated with better outcomes (97). A clinical follow-up study showed that although anti-CTLA-4 immunotherapy promoted intra-tumoral Teff infiltration, it did not lead to FoxP3 T-cell depletion in human cancers (98). These seminal studies on the balance between Teffs and Tregs suggest that increased numbers of tumour-infiltrating Teffs, rather than depletion of Tregs, can be used to predict sensitivity to anti-CTLA-4 immunotherapy. If the ratio of these two T cell subpopulations responding to immunotherapy in TME is unfavorable to Teffs, resistance may emerge throughout the course of treatment.
Over the past few decades, tremendous progress has been made in the clinical application of peripatetic immunotherapy in the fight against tumors. However, given the cost and complexity of generating tumor-specific T cells, there are many limitations to the practical clinical application of secondary immunotherapy. Nanomedicine can provide additional technical support to overcome these limitations (Figure 1). Using artificial antigen-presenting cells (aAPCs), scientists have loaded specific MHC peptides and co-stimulatory molecules onto nanoparticles to activate and promote the expansion of antigen-specific CD8+ T cells (99). In some aAPCs, cytokines can also be used to promote activation and expansion of T cells in lymph nodes (100, 101). Iron-dextran nanoparticles and anti-biotin protein-coated quantum dot crystals have been synthesized using surface-coated MHC-I peptide complexes and biotin-CD28. These nanoparticles(NPs) are magnetic and can promote T-cell enrichment and activation using magnetic fields to promote TCR aggregation (102). IL-2 can activate T cells and with this in mind, Steenblock et al (100) constructed carbon nanotube polymers as aAPCs in which IL-2 was encapsulated and modified with MHC-I and αCD28. These aAPCs can induce activation and expansion of CD8+T cells with very low concentrations of IL-2 compared to conventional methods. In addition, Kelly et al (103) synthesized PLGA/PBAE aAPCs from poly(lactic acid-glycolic acid) (PLGA) and cationic polybasic amino esters (PBAE), nanoparticles that can expand antigen-specific cytotoxic CD8+ T cells in vivo.
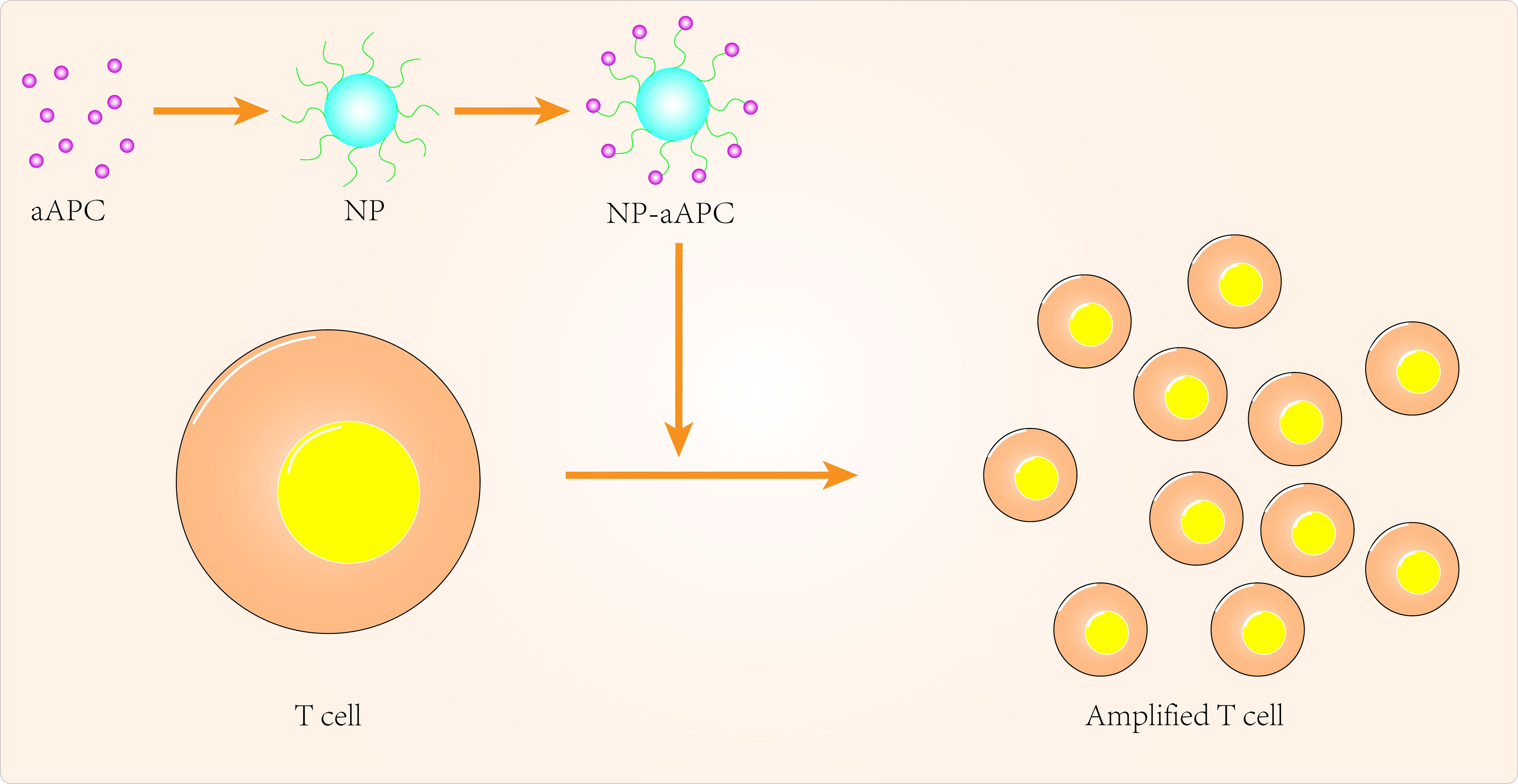
Figure 1 Targeted APCs to improve cancer immunotherapy.
As current T cell expansion rates are not ideal, research should also focus on biomaterials to improve in vitro T cell expansion (101, 102, 104). Cheung et al (105) designed an APC mimetic scaffold (APC-ms) that mimics natural APCs and compared this APC-ms to commercial expansion beads. This APC-ms promoted polyclonal expansion of mouse and human T cell generations by 2-10 times compared to commercial expansion beads (Dynabeads). The APC-ms consist of a liquid lipid bilayer supported by mesoporous silica microrods. After a single stimulation, the APC-ms resulted in antigen-specific expansion of a subpopulation of cytotoxic T cells in much larger numbers than autologous monocyte-derived dendritic cells. aAPCs may become the next focus of research in cancer nanomedicine, so more attention needs to be invested in optimizing the physicochemical properties of aAPCs, for example.
Given the important role of PTEN in immunotherapy resistance, the design of PTEN-targeted nanomaterials has been the focus of many scientists. Kinoh et al (106) used pH-sensitive epirubicin-loaded micellar nanodrugs to synergize the efficacy of anti-PD-1 antibody (aPD-1) against PTEN-positive and PTEN-negative glioblastoma in situ (GBM). The combination of epirubicin-loaded micelles (Epi/m) with aPD-1 overcame the resistance of GBM to ICIs and reduced PD-L1 expression on tumor cells by inducing immunogenic cell death (ICD), eliminating MDSC and transforming the otherwise immune insensitive GBM into a hot tumor with high infiltration of anti-tumor immune cells. Catania et al (107) designed a combined local treatment based on Adriamycin (DOX) as an inducer of ICD and CpG (toll-like receptor-9 agonist, TLR-9 agonist) to synergistically eliminate GBM and found that a single intratumoral administration of HA-DOX + HA-CpG was effective in prolonging the survival of GBM animals. Teo et al (108) combined siRNA with folic acid-modified PEI, resulting in significant PD-1 silencing and enhanced T-cell activation. Yang et al (109) combined anti-PD-L1(aPD-L1) with multiple polyethylene glycol (PEG) chains to improve the efficacy and safety of checkpoint blockade treatment in GBM. In mice with GBM in situ, single doses of glycosylated and polyethylene glycol-linked antibodies reactivated anti-tumor immune responses and induced immune memory to protect the animals from recurrent tumor cell attack and to suppress autoimmune responses in healthy tissues of the animals.
In addition to GBM, PTEN-targeted nanomaterials have been investigated for other tumor applications. Lin et al (110) delivered mRNA via polymeric nanoparticles to effectively induce PTEN expression in melanoma and prostate cancer cells, which in turn induced autophagy and triggered cell death-related immune activation by releasing damage-related molecular patterns. In vivo experiments showed that PTEN mRNA nanoparticles reversed the immunosuppressive TME by promoting CD8+ T cell infiltration into tumor tissue, increasing pro-inflammatory cytokine expression and reducing Treg cells and MDSCs. The combination of PTEN mRNA nanoparticles with immune checkpoint inhibitors, aPD-1 antibodies, in a subcutaneous model of PTEN-mutant melanoma and PTEN-negative prostate cancer in situ models, produced potent anti-tumor effects (Figure 2).
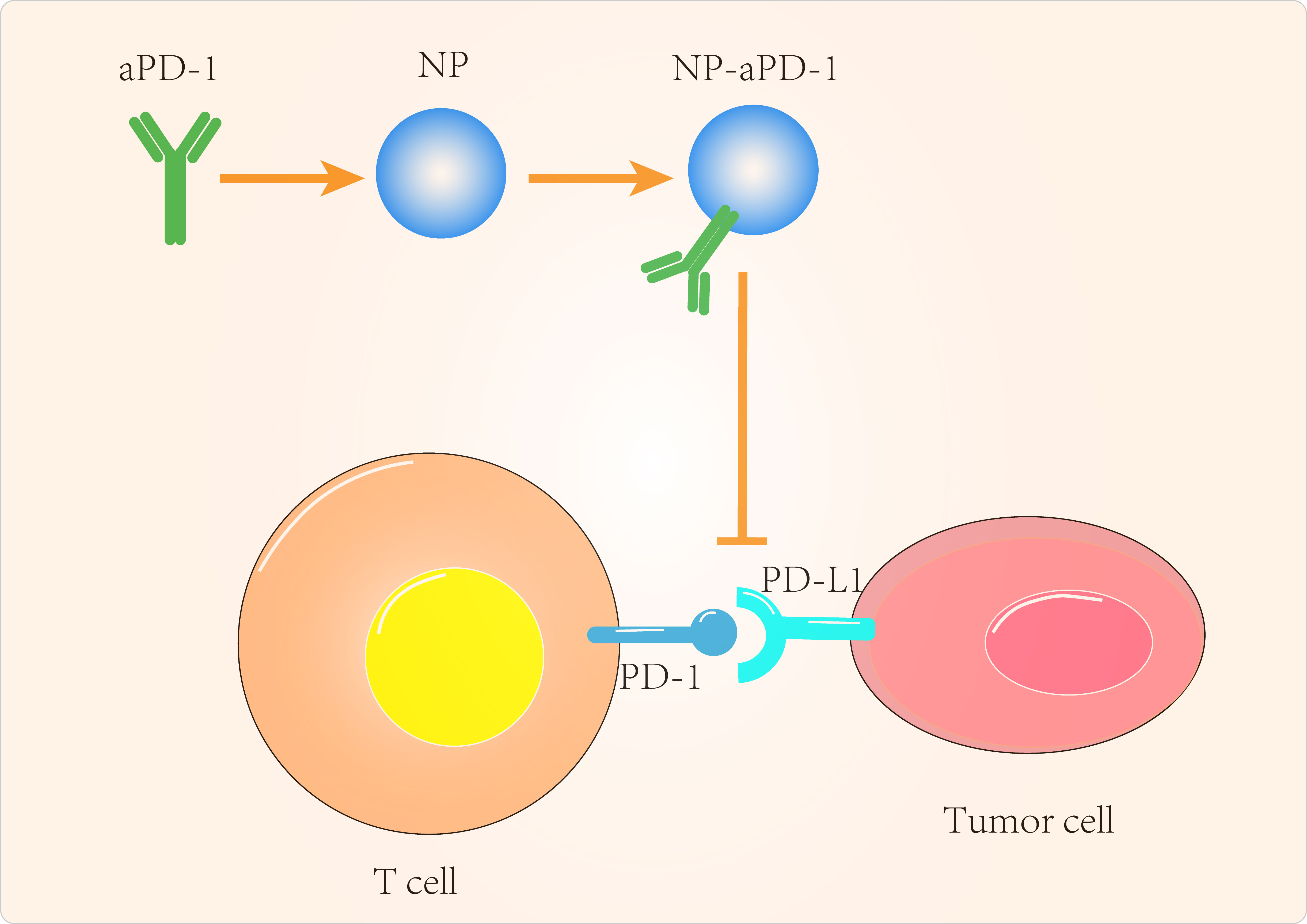
Figure 2 Targeted tumor cell-Associated signaling pathway to improve cancer immunotherapy.
When anti-CTLA4 antibodies are loaded into functionalized mesoporous silica (FMS) and administered intratumorally, they prolong local release under physiological conditions compared to free antibodies. FMS can be non-covalently linked to antibodies to promote sustained prolonged release (111, 112). Blockade of PD-1 with siRNA loaded into cationic lipids and polymeric NPs has been investigated (112). Wang et al (113) combined the synergistic delivery of anti-PD-1 antibodies and CpG oligodeoxynucleotides to prevent cancer recurrence. ye et al (114) demonstrated dual targeting of IDO and PD-1 by a microneedle-based transdermal delivery approach. Xiao et al (115) designed aPD-L1 functionalized mimetic polydopamine-modified gold nanostellar nanoparticles (PDA/GNS@aPD-L1 NPs) which, in addition to disrupting PD-1/PD-L1 immunosuppressive signaling, aPD-L1 scFv on the membranes of PDA/GNS@aPD-L1 NPs contributed to the accumulation of PDA-GNS at tumor sites. Importantly, PDA-GNS-induced photothermal ablation of tumors reverses the immunosuppressive tumor microenvironment, further enhancing the efficacy of PD-1/PD-L1 blockade therapy.
Combination immunotherapy with PD-L1 antibodies and CXCL12 inhibitors has better anti-tumor efficacy than single immunotherapy (116, 117). However, combination therapy has some drawbacks, including unpredictable PK/PD and overlapping toxicity. Therefore, targeting PD-L1 while reducing the duration of drug administration in vivo may help to reduce the cytotoxicity caused by free aPD-L1. Based on this concept, Miao et al (118) designed a chimeric PD-L1 trap protein. When a plasmid encoding the PD-L1 trap protein was encapsulated in lipid-fisetin DNA NPs and delivered to CT26 colon tumors and KPC pancreatic tumors, the highest expression was observed from day 2 to day 4 and finally declined by day 6. This transient expression and high affinity for PD-L1 molecules could serve as a promising therapeutic approach with reduced side effects.
NP can also regulate TAMs by inhibiting macrophage recruitment, by inhibiting TAMs survival through the use of chemicals, by enhancing the activity of M1-type TAMs and by blocking the activity of M2-type TAMs (Figure 3) (119). Qian et al (120) used a novel bi-peptide in which scavenger receptor B type 1 (SRB1) was linked to a specific TAMs binding peptide which specifically blocked the M2-TAMs survival signals. Alternatively, we could try to reprogramme M2-type TAMs back to M1-type (121–123). When IL-12 in poly(β-amino ester) NPs was administered intravenously in a B16F10 melanoma model, M2-type macrophages were reduced and M1-type macrophages were increased (121). Specific miRNA-125b increased expression of MHCII, CD40, CD80, CD86 and increased responsiveness to IFN-γ in macrophages (124). Parayath et al (125) intraperitoneal injection of miRNA 125-b complexed with hyaluronic acid-fixed poly(ethyleneimine) NPs into the TME of G12/P53 mice resulted in increased M1-type macrophages and decreased M2-type macrophages. Zanganeh et al (126) repolarized M2-type TAMs back to M1-type via hydroxyl radicals for breast cancer treatment. Macrophages produce H2O2, which can be converted to cytotoxic hydroxyl radicals via iron oxide NPs. When co-cultures of macrophages and MMTV-PyMT cancer cells were treated with iron oxide NPs, there was an increase in the number of M1-type cells and a decrease in the number of M2-type cells. The effect of M1-type TAMs in the TME could be maximized by the use of immune checkpoint blockers or combination therapy with NPs carrying these modulators. Parayath et al (127) found that hyaluronic acid-based nanoparticles encapsulated with miR-125b (HA-PEI-miR-125b) could specifically target homozygous ID8-VEGF ovarian cancer mouse The imbalance in the ratio of M1 to M2 TAMs populations and the uncontrolled increase of M1 TAMs stimulate the inflammatory response. In contrast, TAMs can sometimes express both M1- and M2-type markers (128), which limits the application of specific targeting of M2-type TAMs.
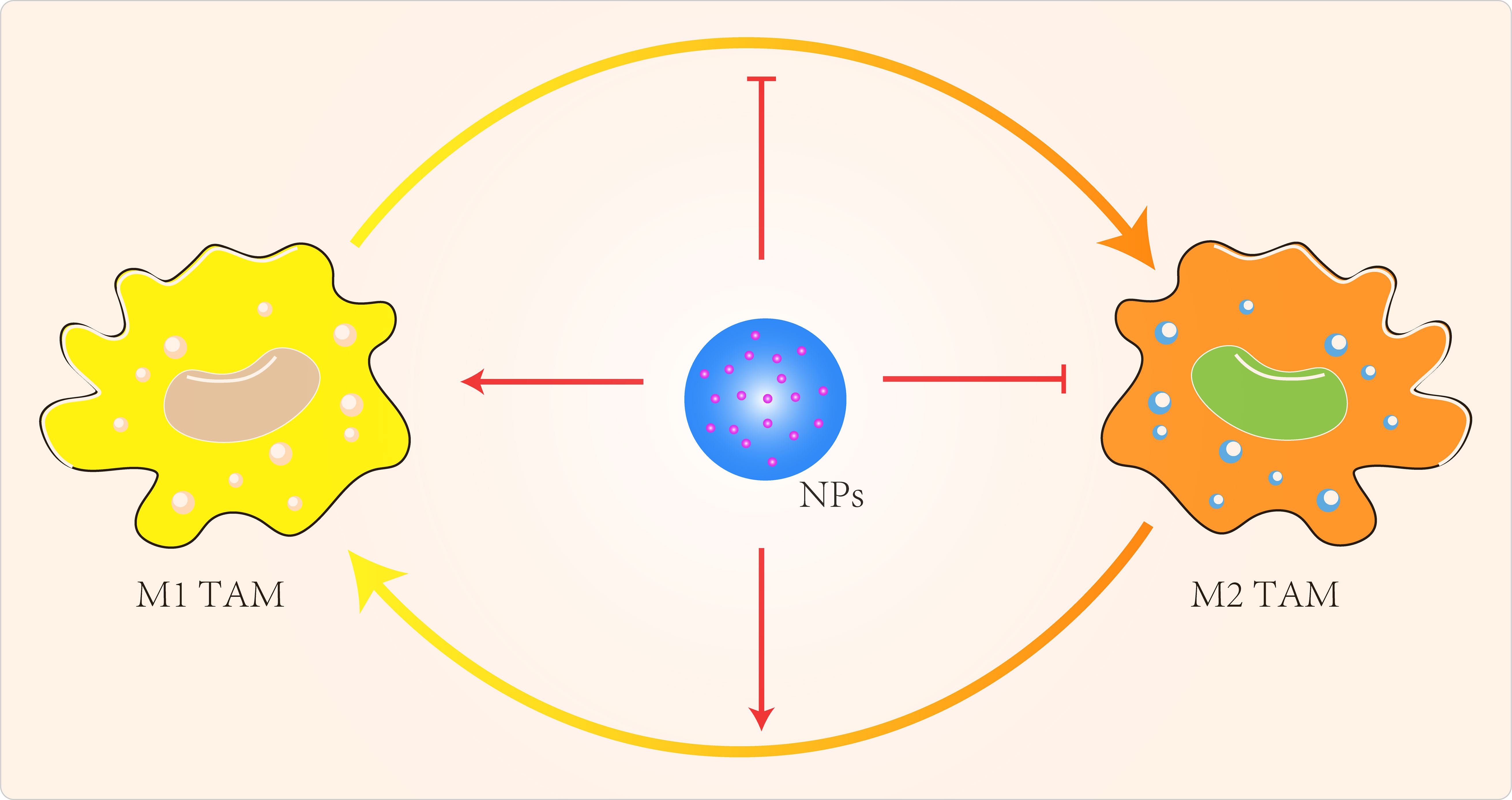
Figure 3 Targeted TAMs to improve cancer immunotherapy.
Strategies to regulate MDSCs primarily require specific blockade of MDSCs development, differentiation of MDSCs into mature cells, and depletion and inhibition of MDSC function by small molecule drugs (129–133). Gold NPs based on high-density lipoprotein (HDL) loaded with apoA-1 have been used to inhibit MDSCs, and in a study in B16F10 melanoma mice, MDSCs were depleted by lipid-encapsulated calcium phosphate NPs loaded with gemcitabine (134, 135). Plebanek et al (136) synthesized a HDL receptor with high affinity for HDL that inhibited MDSCs activity by specifically binding SCARB1. In another study, Kong et al (137) used mesoporous silica NPs loaded with all-trans retinoic acid and Dox, coated with IL-2 and subsequently modified with dipalmitoylphosphatidylcholine cholesterol and DSPE-PEG 2000. Intravenous administration of these NPs reduced the MDSCs population by 2.7-fold and increased the population of NK cells, mature DCs and cytotoxic T cells in the TME. Similarly, IL-2 encapsulated NPs were designed to remodel the TME cell population, but were associated with poor secondary cytokine responses (138).
Blocking the pathway activated by CAFs can reverse mesenchymal-mediated multidrug resistance. In one study, targeting CAFs with anti-FAP SCFV-modified ferritin nanomaterials containing the photosensitizer zinc hexadecafluorophthalocyanine led to CAF ablation and increased the number of CD8+ T cells in the TME when irradiated with 671nm light (139). Intraperitoneal injection of homogeneous gold nanoparticles in the ASPC1 human pancreatic cancer cell and CAF19 pancreatic stellate tumor mouse models led to a reduction in fibronectin, collagen and α-SMA (140). These NPs specifically target α-SMA-positive CAFs through the interaction of serum albumin encapsulated on NPs during circulation with cysteine-rich acidic protein (SPARC) secreted on the surface of CAFs (141).
NK cells play a pivotal role in tumor immunotherapy, and more and more scholars have started to design nanomedicines for NK cell-related immune resistance. Liu et al (142) designed a nanoemulsion system (SSB-NMs) to load TGF-β inhibitors and selenocysteine (Se-C), and the experimental results showed that the nanoparticles significantly enhanced the efficacy of NK cell-mediated immunotherapy against triple-negative breast cancer. Lai et al. (143) used a selenium-containing ruthenium complex (Ru-Se) to synergistically enhance NK cell-mediated killing of prostate cancer cells. The complex was found to effectively enhance NK cell lysis of PC3 cells and was demonstrated in 10 clinical patients. NK cells are superior to T cells in their ability to fight tumors in the absence of specific antigens, making them a potential target for tumor immunotherapy.
Zaretsky et al (38) melanoma patients resistant to PD-1 treatment acquired loss-of-function mutations in JAK1/2. Although tumor cells were still recognized by T cells, their JAK1/2 mutation rendered them insensitive to the anti-proliferative effects of IFN-γ and they lacked IFN-γ induced PD-L1 and MHC class I surface expression. Similarly, analysis of tumors from patients resistant to treatment with the anti-CTLA4 drug Ipilimumab showed that mutations in the IFN-γ pathway genes IFNGR1/2, JAK1/2 and IRF1 inhibit the response of tumor cells to IFN-γ signaling (39). This facilitates tumor escape from T-cell immunity, thereby conferring resistance to anti-CTLA4 therapy.
After treatment with Frax NEs, the T cofactor 1 (Th1) cytokine of IFN-γ was effective in inducing anti-tumor immunity (144). Transforming growth factor-β (TGF-β), chemokine (C-C motif) ligand 2 (CCL2) and interleukin 6 (IL6), which inhibit the development of anti-tumor immunity, were reduced. Although Frax NEs have shown an inhibitory effect on tumor growth, this monotherapy is only partially effective against tumors and does not maintain the tumor growth effect long after administration has stopped.
Lin et al. (110) constructed PTEN mRNA nanoparticles (mPTEN @ NPs) delivered to tumour cells with PTEN deletion or mutation to restore the TME, stimulate immune response and enhance the efficacy of immune checkpoint blockade (ICB) therapy by inducing autophagy activation and damage-associated molecular patterns (DAMPs) release. In their study, mPTEN @ NPs restored tumour sensitivity to immunotherapy and triggered the release of DAMPs and autophagy, thereby promoting the formation of autophagosomes. In vivo studies showed that PTEN repair induced a strong CD8+ T cell response and restored TME by inhibiting the production of Tregs and monocyte MDSCs and promoting the production of pro-inflammatory cytokines. In addition, they evaluated the anti-tumour effects of mPTEN @ NPs in combination with anti-PD-1 immunotherapy in a PTEN-deficient or mutated tumour model, showing that this combination therapy strategy has significant therapeutic efficacy and immunological memory. These results suggest that nanomedicine mRNA repair of tumour suppressors can enhance the sensitivity of tumors to ICB therapy and provide an effective cooperative treatment strategy for a variety of malignancies.
Despite the remarkable achievements of immunotherapy in cancer treatment, all current treatment strategies have serious limitations and often encounter difficulties in clinical treatment. There are many reasons for treatment failure, including poor oral bioavailability of some drugs and serious drug-related side effects. The most challenging problem is that patients receiving immunotherapy almost always develop drug resistance. While much progress has been made in recent years in understanding the mechanisms of resistance to cancer immunotherapy, overcoming drug resistance remains an important unmet clinical need. Thus, immune resistance in tumors has become a major obstacle to cancer immunotherapy, and overcoming resistance is a goal actively pursued by current and future oncologists (145).
The emergence of nanotechnology offers a novel solution to immunotherapy resistance. Nanomedicines can improve the bioavailability of insoluble drugs, prolong drug circulation, cross the biological barrier, achieve tumor-targeted therapy through passive or active targeting, improve anti-tumor effects and reduce tumor resistance to immunotherapy. A number of nanomaterials have now completed Phase III clinical trials (Table 1).The convergence of these two disciplines will certainly provide a tremendous impetus to improve cancer treatment. The advantages of nanomedicines over current therapeutic strategies will continue to be exploited as more nanomedicines are developed and optimized. It is therefore believed that nanomedicines will be an attractive strategy for reversing or overcoming resistance to cancer immunotherapy.
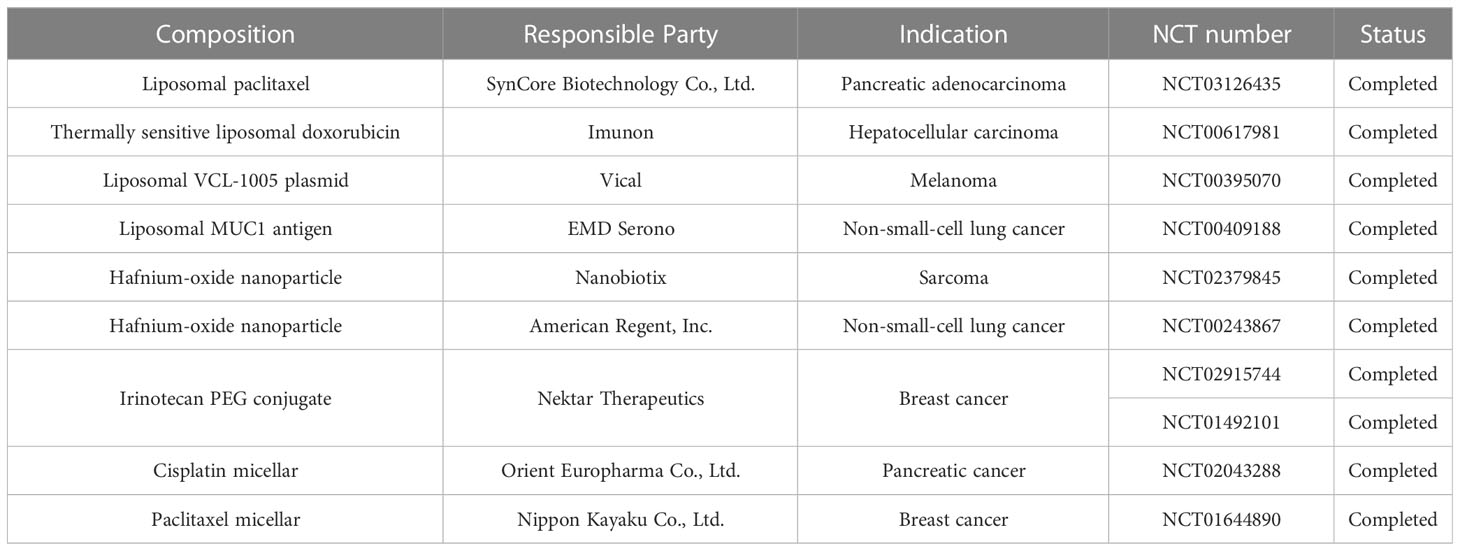
Table 1 Summary of Phase 3 Clinical Trials for Cancer Nanomedicine Products.
Drug resistance in tumor immunotherapy involves multiple mechanisms working together, so here we have only reviewed the more mature aspects of nanomaterial development, as for other aspects of drug resistance mechanisms such as hypoxic environment (146, 147), lactate metabolism (148), glycolytic metabolism (149), tryptophan metabolism (150), cholesterol metabolism (151–154), which also play an important role in drug resistance. These mechanisms of drug resistance may also provide additional clues for the application of nanomedicine.
Currently, a lot of researchers are concentrating on creating organically produced NPs, which are safer as well as more biocompatible and able to activate the body’s own immune cells and use that immune response to help eradicate cancers (155). But in this case, there is a more subtle biological element and a well-defined treatment process. In conclusion, it is encouraging to see how NPs might increase the body’s immunological resilience. It is thought that through in-depth research in this area and the design of more effective nanomaterials, it is possible to overcome immune resistance, reduce toxic side effects on patients, and achieve better therapeutic effects for the benefit of cancer patients, despite some limitations in its clinical application.
JS carried out the primary literature search, drafted and revised the manuscript. YJ helped modify the manuscript and participated in discussions. CJ conceived and approved the final manuscript. All authors contributed to the article and approved the submitted version.
This research was supported by Natural Science Foundation of Jilin Provincial (Grant ID: YDZJ202201ZYTS643) and National Natural Science Foundation of China (Grant ID: 82003173).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Breakthrough of the year 2013. Notable developments. Science (2013) 342:1435–41. doi: 10.1126/science.342.6165.1444
2. Vesely MD, Zhang T, Chen L. Resistance mechanisms to anti-PD cancer immunotherapy. Annu Rev Immunol (2022) 40:45–74. doi: 10.1146/annurev-immunol-070621-030155
3. Jerby-Arnon L, Shah P, Cuoco MS, Rodman C, Su M-J, Melms JC, et al. A cancer cell program promotes T cell exclusion and resistance to checkpoint blockade. Cell (2018) 175:984–997.e24. doi: 10.1016/j.cell.2018.09.006
4. Baldominos P, Barbera-Mourelle A, Barreiro O, Huang Y, Wight A, Cho J-W, et al. Quiescent cancer cells resist T cell attack by forming an immunosuppressive niche. Cell (2022) 185:1694–1708.e19. doi: 10.1016/j.cell.2022.03.033
5. Yu J, Green MD, Li S, Sun Y, Journey SN, Choi JE, et al. Liver metastasis restrains immunotherapy efficacy via macrophage-mediated T cell elimination. Nat Med (2021) 27:152–64. doi: 10.1038/s41591-020-1131-x
6. Goc J, Lv M, Bessman NJ, Flamar A-L, Sahota S, Suzuki H, et al. Dysregulation of ILC3s unleashes progression and immunotherapy resistance in colon cancer. Cell (2021) 184:5015–5030.e16. doi: 10.1016/j.cell.2021.07.029
7. Wu P, Han J, Gong Y, Liu C, Yu H, Xie N. Nanoparticle-based drug delivery systems targeting tumor microenvironment for cancer immunotherapy resistance: current advances and applications. Pharmaceutics (2022) 14:1990. doi: 10.3390/pharmaceutics14101990
8. Shen L, Li J, Liu Q, Song W, Zhang X, Tiruthani K, et al. Local blockade of interleukin 10 and C-X-C motif chemokine ligand 12 with nano-delivery promotes antitumor response in murine cancers. ACS Nano (2018) 12:9830–41. doi: 10.1021/acsnano.8b00967
9. Gao S, Yang D, Fang Y, Lin X, Jin X, Wang Q, et al. Engineering nanoparticles for targeted remodeling of the tumor microenvironment to improve cancer immunotherapy. Theranostics (2019) 9:126–51. doi: 10.7150/thno.29431
10. Chen Y. Nanotechnology for next-generation cancer immunotherapy: State of the art and future perspectives. J Control Release (2023) 356:14–25. doi: 10.1016/j.jconrel.2023.02.016
11. Riley RS, June CH, Langer R, Mitchell MJ. Delivery technologies for cancer immunotherapy. Nat Rev Drug Discovery (2019) 18:175–96. doi: 10.1038/s41573-018-0006-z
12. Xu Y, Xiong J, Sun X, Gao H. Targeted nanomedicines remodeling immunosuppressive tumor microenvironment for enhanced cancer immunotherapy. Acta Pharm Sin B (2022) 12:4327–47. doi: 10.1016/j.apsb.2022.11.001
13. Reeves E, James E. Antigen processing and immune regulation in the response to tumours. Immunology (2017) 150:16–24. doi: 10.1111/imm.12675
14. Łuksza M, Riaz N, Makarov V, Balachandran VP, Hellmann MD, Solovyov A, et al. A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature (2017) 551:517–20. doi: 10.1038/nature24473
15. Carbone DP, Reck M, Paz-Ares L, Creelan B, Horn L, Steins M, et al. First-line nivolumab in stage IV or recurrent non-small-cell lung cancer. N Engl J Med (2017) 376:2415–26. doi: 10.1056/NEJMoa1613493
16. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science (2015) 348:69–74. doi: 10.1126/science.aaa4971
17. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
18. Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science (2017) 357:409–13. doi: 10.1126/science.aan6733
19. Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature (2013) 499:214–8. doi: 10.1038/nature12213
20. de Vries TJ, Fourkour A, Wobbes T, Verkroost G, Ruiter DJ, van Muijen GN. Heterogeneous expression of immunotherapy candidate proteins gp100, MART-1, and tyrosinase in human melanoma cell lines and in human melanocytic lesions. Cancer Res (1997) 57:3223–9.
21. DuPage M, Mazumdar C, Schmidt LM, Cheung AF, Jacks T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature (2012) 482:405–9. doi: 10.1038/nature10803
22. Bai X-F, Liu J, Li O, Zheng P, Liu Y. Antigenic drift as a mechanism for tumor evasion of destruction by cytolytic T lymphocytes. J Clin Invest (2003) 111:1487–96. doi: 10.1172/JCI17656
23. Galluzzi L, Buqué A, Kepp O, Zitvogel L, Kroemer G. Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell (2015) 28:690–714. doi: 10.1016/j.ccell.2015.10.012
24. Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol (2013) 31:51–72. doi: 10.1146/annurev-immunol-032712-100008
25. Fucikova J, Becht E, Iribarren K, Goc J, Remark R, Damotte D, et al. Calreticulin expression in human non-small cell lung cancers correlates with increased accumulation of antitumor immune cells and favorable prognosis. Cancer Res (2016) 76:1746–56. doi: 10.1158/0008-5472.CAN-15-1142
26. Ladoire S, Penault-Llorca F, Senovilla L, Dalban C, Enot D, Locher C, et al. Combined evaluation of LC3B puncta and HMGB1 expression predicts residual risk of relapse after adjuvant chemotherapy in breast cancer. Autophagy (2015) 11:1878–90. doi: 10.1080/15548627.2015.1082022
27. Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN promotes resistance to T cell-mediated immunotherapy. Cancer Discovery (2016) 6:202–16. doi: 10.1158/2159-8290.CD-15-0283
28. Cretella D, Digiacomo G, Giovannetti E, Cavazzoni A. PTEN alterations as a potential mechanism for tumor cell escape from PD-1/PD-L1 inhibition. Cancers (Basel) (2019) 11:1318. doi: 10.3390/cancers11091318
29. Cetintas VB, Batada NN. Is there a causal link between PTEN deficient tumors and immunosuppressive tumor microenvironment? J Transl Med (2020) 18:45. doi: 10.1186/s12967-020-02219-w
30. Feng S, Cheng X, Zhang L, Lu X, Chaudhary S, Teng R, et al. Myeloid-derived suppressor cells inhibit T cell activation through nitrating LCK in mouse cancers. Proc Natl Acad Sci U.S.A. (2018) 115:10094–9. doi: 10.1073/pnas.1800695115
31. Sharma MD, Shinde R, McGaha TL, Huang L, Holmgaard RB, Wolchok JD, et al. The PTEN pathway in Tregs is a critical driver of the suppressive tumor microenvironment. Sci Adv (2015) 1:e1500845. doi: 10.1126/sciadv.1500845
32. Piro G, Carbone C, Carbognin L, Pilotto S, Ciccarese C, Iacovelli R, et al. Revising PTEN in the era of immunotherapy: new perspectives for an old story. Cancers (Basel) (2019) 11:1525. doi: 10.3390/cancers11101525
33. Vidotto T, Melo CM, Castelli E, Koti M, Dos Reis RB, Squire JA. Emerging role of PTEN loss in evasion of the immune response to tumours. Br J Cancer (2020) 122:1732–43. doi: 10.1038/s41416-020-0834-6
34. Conciatori F, Bazzichetto C, Falcone I, Ciuffreda L, Ferretti G, Vari S, et al. PTEN Function at the Interface between Cancer and Tumor Microenvironment: Implications for Response to Immunotherapy. Int J Mol Sci (2020) 21:5337. doi: 10.3390/ijms21155337
35. Arico S, Petiot A, Bauvy C, Dubbelhuis PF, Meijer AJ, Codogno P, et al. The tumor suppressor PTEN positively regulates macroautophagy by inhibiting the phosphatidylinositol 3-kinase/protein kinase B pathway. J Biol Chem (2001) 276:35243–6. doi: 10.1074/jbc.C100319200
36. Darnell JE, Kerr IM, Stark GR. Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science (1994) 264:1415–21. doi: 10.1126/science.8197455
37. Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, et al. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature (2017) 547:413–8. doi: 10.1038/nature23270
38. Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N Engl J Med (2016) 375:819–29. doi: 10.1056/NEJMoa1604958
39. Possick JD. Pulmonary toxicities from checkpoint immunotherapy for Malignancy. Clin Chest Med (2017) 38:223–32. doi: 10.1016/j.ccm.2016.12.012
40. Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med (2015) 372:2521–32. doi: 10.1056/NEJMoa1503093
41. Kandel S, Adhikary P, Li G, Cheng K. The TIM3/Gal9 signaling pathway: An emerging target for cancer immunotherapy. Cancer Lett (2021) 510:67–78. doi: 10.1016/j.canlet.2021.04.011
42. Pathria P, Louis TL, Varner JA. Targeting tumor-associated macrophages in cancer. Trends Immunol (2019) 40:310–27. doi: 10.1016/j.it.2019.02.003
43. Xiang X, Wang J, Lu D, Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct Target Ther (2021) 6:75. doi: 10.1038/s41392-021-00484-9
44. Chanmee T, Ontong P, Konno K, Itano N. Tumor-associated macrophages as major players in the tumor microenvironment. Cancers (Basel) (2014) 6:1670–90. doi: 10.3390/cancers6031670
45. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity (2014) 41:49–61. doi: 10.1016/j.immuni.2014.06.010
46. Ramanathan S, Jagannathan N. Tumor associated macrophage: a review on the phenotypes, traits and functions. Iran J Cancer Prev (2014) 7:1–8.
47. Lin Y, Xu J, Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J Hematol Oncol (2019) 12:76. doi: 10.1186/s13045-019-0760-3
48. Neubert NJ, Schmittnaegel M, Bordry N, Nassiri S, Wald N, Martignier C, et al. T cell-induced CSF1 promotes melanoma resistance to PD1 blockade. Sci Transl Med (2018) 10:eaan3311. doi: 10.1126/scitranslmed.aan3311
49. Mok S, Koya RC, Tsui C, Xu J, Robert L, Wu L, et al. Inhibition of CSF-1 receptor improves the antitumor efficacy of adoptive cell transfer immunotherapy. Cancer Res (2014) 74:153–61. doi: 10.1158/0008-5472.CAN-13-1816
50. Zhu Y, Yang J, Xu D, Gao X-M, Zhang Z, Hsu JL, et al. Disruption of tumour-associated macrophage trafficking by the osteopontin-induced colony-stimulating factor-1 signalling sensitises hepatocellular carcinoma to anti-PD-L1 blockade. Gut (2019) 68:1653–66. doi: 10.1136/gutjnl-2019-318419
51. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in pancreatic cancer models. Cancer Res (2014) 74:5057–69. doi: 10.1158/0008-5472.CAN-13-3723
52. Hu W, Li X, Zhang C, Yang Y, Jiang J, Wu C. Tumor-associated macrophages in cancers. Clin Transl Oncol (2016) 18:251–8. doi: 10.1007/s12094-015-1373-0
53. Jung KY, Cho SW, Kim YA, Kim D, Oh B-C, Park DJ, et al. Cancers with higher density of tumor-associated macrophages were associated with poor survival rates. J Pathol Transl Med (2015) 49:318–24. doi: 10.4132/jptm.2015.06.01
54. Fritz JM, Tennis MA, Orlicky DJ, Lin H, Ju C, Redente EF, et al. Depletion of tumor-associated macrophages slows the growth of chemically induced mouse lung adenocarcinomas. Front Immunol (2014) 5:587. doi: 10.3389/fimmu.2014.00587
55. Wu X, Schulte BC, Zhou Y, Haribhai D, Mackinnon AC, Plaza JA, et al. Depletion of M2-like tumor-associated macrophages delays cutaneous T-cell lymphoma development in vivo. J Invest Dermatol (2014) 134:2814–22. doi: 10.1038/jid.2014.206
56. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell (2014) 25:846–59. doi: 10.1016/j.ccr.2014.05.016
57. Luo Y, Zhou H, Krueger J, Kaplan C, Lee S-H, Dolman C, et al. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J Clin Invest (2006) 116:2132–41. doi: 10.1172/JCI27648
58. Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CMT, Pryer N, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell (2014) 26:623–37. doi: 10.1016/j.ccell.2014.09.006
59. Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol (2021) 21:485–98. doi: 10.1038/s41577-020-00490-y
60. Solito S, Falisi E, Diaz-Montero CM, Doni A, Pinton L, Rosato A, et al. A human promyelocytic-like population is responsible for the immune suppression mediated by myeloid-derived suppressor cells. Blood (2011) 118:2254–65. doi: 10.1182/blood-2010-12-325753
61. Si Y, Merz SF, Jansen P, Wang B, Bruderek K, Altenhoff P, et al. Multidimensional imaging provides evidence for down-regulation of T cell effector function by MDSC in human cancer tissue. Sci Immunol (2019) 4:eaaw9159. doi: 10.1126/sciimmunol.aaw9159
62. Kodumudi KN, Weber A, Sarnaik AA, Pilon-Thomas S. Blockade of myeloid-derived suppressor cells after induction of lymphopenia improves adoptive T cell therapy in a murine model of melanoma. J Immunol (2012) 189:5147–54. doi: 10.4049/jimmunol.1200274
63. Meyer C, Cagnon L, Costa-Nunes CM, Baumgaertner P, Montandon N, Leyvraz L, et al. Frequencies of circulating MDSC correlate with clinical outcome of melanoma patients treated with ipilimumab. Cancer Immunol Immunother (2014) 63:247–57. doi: 10.1007/s00262-013-1508-5
64. Yang L, DeBusk LM, Fukuda K, Fingleton B, Green-Jarvis B, Shyr Y, et al. Expansion of myeloid immune suppressor Gr+CD11b+ cells in tumor-bearing host directly promotes tumor angiogenesis. Cancer Cell (2004) 6:409–21. doi: 10.1016/j.ccr.2004.08.031
65. Yang L, Huang J, Ren X, Gorska AE, Chytil A, Aakre M, et al. Abrogation of TGF beta signaling in mammary carcinomas recruits Gr-1+CD11b+ myeloid cells that promote metastasis. Cancer Cell (2008) 13:23–35. doi: 10.1016/j.ccr.2007.12.004
66. Li K, Shi H, Zhang B, Ou X, Ma Q, Chen Y, et al. Myeloid-derived suppressor cells as immunosuppressive regulators and therapeutic targets in cancer. Signal Transduct Target Ther (2021) 6:362. doi: 10.1038/s41392-021-00670-9
67. Fujimura T, Kambayashi Y, Aiba S. Crosstalk between regulatory T cells (Tregs) and myeloid derived suppressor cells (MDSCs) during melanoma growth. Oncoimmunology (2012) 1:1433–4. doi: 10.4161/onci.21176
68. Wesolowski R, Markowitz J, Carson WE. Myeloid derived suppressor cells - a new therapeutic target in the treatment of cancer. J Immunother Cancer (2013) 1:10. doi: 10.1186/2051-1426-1-10
69. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM, et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer (2020) 20:174–86. doi: 10.1038/s41568-019-0238-1
70. Servais C, Erez N. From sentinel cells to inflammatory culprits: cancer-associated fibroblasts in tumour-related inflammation. J Pathol (2013) 229:198–207. doi: 10.1002/path.4103
71. Deng Y, Cheng J, Fu B, Liu W, Chen G, Zhang Q, et al. Hepatic carcinoma-associated fibroblasts enhance immune suppression by facilitating the generation of myeloid-derived suppressor cells. Oncogene (2017) 36:1090–101. doi: 10.1038/onc.2016.273
72. Takahashi H, Sakakura K, Kudo T, Toyoda M, Kaira K, Oyama T, et al. Cancer-associated fibroblasts promote an immunosuppressive microenvironment through the induction and accumulation of protumoral macrophages. Oncotarget (2017) 8:8633–47. doi: 10.18632/oncotarget.14374
73. Komohara Y, Takeya M. CAFs and TAMs: maestros of the tumour microenvironment. J Pathol (2017) 241:313–5. doi: 10.1002/path.4824
74. Cheng J-T, Deng Y-N, Yi H-M, Wang G-Y, Fu B-S, Chen W-J, et al. Hepatic carcinoma-associated fibroblasts induce IDO-producing regulatory dendritic cells through IL-6-mediated STAT3 activation. Oncogenesis (2016) 5:e198. doi: 10.1038/oncsis.2016.7
75. Pinchuk IV, Saada JI, Beswick EJ, Boya G, Qiu SM, Mifflin RC, et al. PD-1 ligand expression by human colonic myofibroblasts/fibroblasts regulates CD4+ T-cell activity. Gastroenterology (2008) 135:1228–1237, 1237.e1–2. doi: 10.1053/j.gastro.2008.07.016
76. Ghebeh H, Dermime S. Comment on “Characterization of human lung tumor-associated fibroblasts and their ability to modulate the activation of tumor-associated T cells”. J Immunol (2007) 179:732. doi: 10.4049/jimmunol.179.2.732
77. Chiossone L, Dumas P-Y, Vienne M, Vivier E. Natural killer cells and other innate lymphoid cells in cancer. Nat Rev Immunol (2018) 18:671–88. doi: 10.1038/s41577-018-0061-z
78. Cifaldi L, Di Santo J, Olive D. Editorial: molecular strategies aimed to boost NK cell-based immunotherapy of cancer. Front Immunol (2020) 11:1132. doi: 10.3389/fimmu.2020.01132
79. Tarannum M, Romee R, Shapiro RM. Innovative strategies to improve the clinical application of NK cell-based immunotherapy. Front Immunol (2022) 13:859177. doi: 10.3389/fimmu.2022.859177
80. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nat Immunol (2016) 17:1025–36. doi: 10.1038/ni.3518
81. Mikelez-Alonso I, Magadán S, González-Fernández Á, Borrego F. Natural killer (NK) cell-based immunotherapies and the many faces of NK cell memory: A look into how nanoparticles enhance NK cell activity. Adv Drug Delivery Rev (2021) 176:113860. doi: 10.1016/j.addr.2021.113860
82. Otegbeye F, Ojo E, Moreton S, Mackowski N, Lee DA, de Lima M, et al. Inhibiting TGF-beta signaling preserves the function of highly activated, in vitro expanded natural killer cells in AML and colon cancer models. PloS One (2018) 13:e0191358. doi: 10.1371/journal.pone.0191358
83. Regis S, Dondero A, Caliendo F, Bottino C, Castriconi R. NK cell function regulation by TGF-β-induced epigenetic mechanisms. Front Immunol (2020) 11:311. doi: 10.3389/fimmu.2020.00311
84. Huang C-H, Liao Y-J, Chiou T-J, Huang H-T, Lin Y-H, Twu Y-C. TGF-β regulated leukemia cell susceptibility against NK targeting through the down-regulation of the CD48 expression. Immunobiology (2019) 224:649–58. doi: 10.1016/j.imbio.2019.07.002
85. Shi L, Lin H, Li G, Sun Y, Shen J, Xu J, et al. Cisplatin enhances NK cells immunotherapy efficacy to suppress HCC progression via altering the androgen receptor (AR)-ULBP2 signals. Cancer Lett (2016) 373:45–56. doi: 10.1016/j.canlet.2016.01.017
86. Yoo JY, Jaime-Ramirez AC, Bolyard C, Dai H, Nallanagulagari T, Wojton J, et al. Bortezomib treatment sensitizes oncolytic HSV-1-treated tumors to NK cell immunotherapy. Clin Cancer Res (2016) 22:5265–76. doi: 10.1158/1078-0432.CCR-16-1003
87. Padrón LJ, Maurer DM, O’Hara MH, O’Reilly EM, Wolff RA, Wainberg ZA, et al. Sotigalimab and/or nivolumab with chemotherapy in first-line metastatic pancreatic cancer: clinical and immunologic analyses from the randomized phase 2 PRINCE trial. Nat Med (2022) 28:1167–77. doi: 10.1038/s41591-022-01829-9
88. Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol (2005) 5:375–86. doi: 10.1038/nri1604
89. Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ Pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell (2016) 167:397–404.e9. doi: 10.1016/j.cell.2016.08.069
90. Ohue Y, Nishikawa H. Regulatory T (Treg) cells in cancer: Can Treg cells be a new therapeutic target? Cancer Sci (2019) 110:2080–9. doi: 10.1111/cas.14069
91. Sundstedt A, O’Neill EJ, Nicolson KS, Wraith DC. Role for IL-10 in suppression mediated by peptide-induced regulatory T cells in vivo. J Immunol (2003) 170:1240–8. doi: 10.4049/jimmunol.170.3.1240
92. Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell (2008) 133:775–87. doi: 10.1016/j.cell.2008.05.009
93. Oida T, Zhang X, Goto M, Hachimura S, Totsuka M, Kaminogawa S, et al. CD4+CD25- T cells that express latency-associated peptide on the surface suppress CD4+CD45RBhigh-induced colitis by a TGF-beta-dependent mechanism. J Immunol (2003) 170:2516–22. doi: 10.4049/jimmunol.170.5.2516
94. Nishikawa H, Koyama S. Mechanisms of regulatory T cell infiltration in tumors: implications for innovative immune precision therapies. J Immunother Cancer (2021) 9:e002591. doi: 10.1136/jitc-2021-002591
95. Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med (2013) 210:1695–710. doi: 10.1084/jem.20130579
96. Arce Vargas F, Furness AJS, Litchfield K, Joshi K, Rosenthal R, Ghorani E, et al. Fc effector function contributes to the activity of human anti-CTLA-4 antibodies. Cancer Cell (2018) 33:649–663.e4. doi: 10.1016/j.ccell.2018.02.010
97. Hamid O, Schmidt H, Nissan A, Ridolfi L, Aamdal S, Hansson J, et al. A prospective phase II trial exploring the association between tumor microenvironment biomarkers and clinical activity of ipilimumab in advanced melanoma. J Transl Med (2011) 9:204. doi: 10.1186/1479-5876-9-204
98. Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, et al. Anti-CTLA-4 immunotherapy does not deplete FOXP3+ Regulatory T cells (Tregs) in human cancers. Clin Cancer Res (2019) 25:1233–8. doi: 10.1158/1078-0432.CCR-18-0762
99. Rhodes KR, Green JJ. Nanoscale artificial antigen presenting cells for cancer immunotherapy. Mol Immunol (2018) 98:13–8. doi: 10.1016/j.molimm.2018.02.016
100. Steenblock ER, Fadel T, Labowsky M, Pober JS, Fahmy TM. An artificial antigen-presenting cell with paracrine delivery of IL-2 impacts the magnitude and direction of the T cell response. J Biol Chem (2011) 286:34883–92. doi: 10.1074/jbc.M111.276329
101. Fadel TR, Sharp FA, Vudattu N, Ragheb R, Garyu J, Kim D, et al. A carbon nanotube-polymer composite for T-cell therapy. Nat Nanotechnol (2014) 9:639–47. doi: 10.1038/nnano.2014.154
102. Perica K, Tu A, Richter A, Bieler JG, Edidin M, Schneck JP. Magnetic field-induced T cell receptor clustering by nanoparticles enhances T cell activation and stimulates antitumor activity. ACS Nano (2014) 8:2252–60. doi: 10.1021/nn405520d
103. Rhodes KR, Isser A, Hickey JW, Ben-Akiva E, Meyer RA, Kosmides AK, et al. Biodegradable cationic polymer blends for fabrication of enhanced artificial antigen presenting cells to treat melanoma. ACS Appl Mater Interfaces (2021) 13:7913–23. doi: 10.1021/acsami.0c19955
104. Sunshine JC, Perica K, Schneck JP. Green JJ. Particle shape dependence of CD8+ T cell activation by artificial antigen presenting cells. Biomaterials (2014) 35:269–77. doi: 10.1016/j.biomaterials.2013.09.050
105. Cheung AS, Zhang DKY, Koshy ST, Mooney DJ. Scaffolds that mimic antigen-presenting cells enable ex vivo expansion of primary T cells. Nat Biotechnol (2018) 36:160–9. doi: 10.1038/nbt.4047
106. Kinoh H, Quader S, Shibasaki H, Liu X, Maity A, Yamasoba T, et al. Translational nanomedicine boosts anti-PD1 therapy to eradicate orthotopic PTEN-negative glioblastoma. ACS Nano (2020) 14:10127–40. doi: 10.1021/acsnano.0c03386
107. Catania G, Rodella G, Vanvarenberg K, Préat V, Malfanti A. Combination of hyaluronic acid conjugates with immunogenic cell death inducer and CpG for glioblastoma local chemo-immunotherapy elicits an immune response and induces long-term survival. Biomaterials (2023) 294:122006. doi: 10.1016/j.biomaterials.2023.122006
108. Teo PY, Yang C, Whilding LM, Parente-Pereira AC, Maher J, George AJT, et al. Ovarian cancer immunotherapy using PD-L1 siRNA targeted delivery from folic acid-functionalized polyethylenimine: strategies to enhance T cell killing. Adv Healthc Mater (2015) 4:1180–9. doi: 10.1002/adhm.201500089
109. Yang T, Mochida Y, Liu X, Zhou H, Xie J, Anraku Y, et al. Conjugation of glucosylated polymer chains to checkpoint blockade antibodies augments their efficacy and specificity for glioblastoma. Nat BioMed Eng (2021) 5:1274–87. doi: 10.1038/s41551-021-00803-z
110. Lin Y-X, Wang Y, Ding J, Jiang A, Wang J, Yu M, et al. Reactivation of the tumor suppressor PTEN by mRNA nanoparticles enhances antitumor immunity in preclinical models. Sci Transl Med (2021) 13:eaba9772. doi: 10.1126/scitranslmed.aba9772
111. Lei C, Liu P, Chen B, Mao Y, Engelmann H, Shin Y, et al. Local release of highly loaded antibodies from functionalized nanoporous support for cancer immunotherapy. J Am Chem Soc (2010) 132:6906–7. doi: 10.1021/ja102414t
112. Roeven MWH, Hobo W, van der Voort R, Fredrix H, Norde WJ, Teijgeler K, et al. Efficient nontoxic delivery of PD-L1 and PD-L2 siRNA into dendritic cell vaccines using the cationic lipid SAINT-18. J Immunother (2015) 38:145–54. doi: 10.1097/CJI.0000000000000071
113. Wang X, Li X, Ito A, Watanabe Y, Sogo Y, Tsuji NM, et al. Stimulation of in vivo antitumor immunity with hollow mesoporous silica nanospheres. Angew Chem Int Ed Engl (2016) 55:1899–903. doi: 10.1002/anie.201506179
114. Ye Y, Wang J, Hu Q, Hochu GM, Xin H, Wang C, et al. Synergistic transcutaneous immunotherapy enhances antitumor immune responses through delivery of checkpoint inhibitors. ACS Nano (2016) 10:8956–63. doi: 10.1021/acsnano.6b04989
115. Xiao Y, Zhu T, Zeng Q, Tan Q, Jiang G, Huang X. Functionalized biomimetic nanoparticles combining programmed death-1/programmed death-ligand 1 blockade with photothermal ablation for enhanced colorectal cancer immunotherapy. Acta Biomater (2023) 157:451–66. doi: 10.1016/j.actbio.2022.11.043
116. Feig C, Jones JO, Kraman M, Wells RJB, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U.S.A. (2013) 110:20212–7. doi: 10.1073/pnas.1320318110
117. Tang T, Huang X, Zhang G, Hong Z, Bai X, Liang T. Advantages of targeting the tumor immune microenvironment over blocking immune checkpoint in cancer immunotherapy. Signal Transduct Target Ther (2021) 6:72. doi: 10.1038/s41392-020-00449-4
118. Miao L, Li J, Liu Q, Feng R, Das M, Lin CM, et al. Transient and local expression of chemokine and immune checkpoint traps to treat pancreatic cancer. ACS Nano (2017) 11:8690–706. doi: 10.1021/acsnano.7b01786
119. Reichel D, Tripathi M, Perez JM. Biological effects of nanoparticles on macrophage polarization in the tumor microenvironment. Nanotheranostics (2019) 3:66–88. doi: 10.7150/ntno.30052
120. Qian Y, Qiao S, Dai Y, Xu G, Dai B, Lu L, et al. Molecular-targeted immunotherapeutic strategy for melanoma via dual-targeting nanoparticles delivering small interfering RNA to tumor-associated macrophages. ACS Nano (2017) 11:9536–49. doi: 10.1021/acsnano.7b05465
121. Wang Y, Lin Y-X, Qiao S-L, An H-W, Ma Y, Qiao Z-Y, et al. Polymeric nanoparticles promote macrophage reversal from M2 to M1 phenotypes in the tumor microenvironment. Biomaterials (2017) 112:153–63. doi: 10.1016/j.biomaterials.2016.09.034
122. Henry CJ, Ornelles DA, Mitchell LM, Brzoza-Lewis KL, Hiltbold EM. IL-12 produced by dendritic cells augments CD8+ T cell activation through the production of the chemokines CCL1 and CCL17. J Immunol (2008) 181:8576–84. doi: 10.4049/jimmunol.181.12.8576
123. Watkins SK, Egilmez NK, Suttles J, Stout RD. IL-12 rapidly alters the functional profile of tumor-associated and tumor-infiltrating macrophages in vitro and in vivo. J Immunol (2007) 178:1357–62. doi: 10.4049/jimmunol.178.3.1357
124. Chaudhuri AA, So AY-L, Sinha N, Gibson WSJ, Taganov KD, O’Connell RM, et al. MicroRNA-125b potentiates macrophage activation. J Immunol (2011) 187:5062–8. doi: 10.4049/jimmunol.1102001
125. Parayath NN, Parikh A, Amiji MM. Repolarization of tumor-associated macrophages in a genetically engineered nonsmall cell lung cancer model by intraperitoneal administration of hyaluronic acid-based nanoparticles encapsulating microRNA-125b. Nano Lett (2018) 18:3571–9. doi: 10.1021/acs.nanolett.8b00689
126. Zanganeh S, Hutter G, Spitler R, Lenkov O, Mahmoudi M, Shaw A, et al. Iron oxide nanoparticles inhibit tumour growth by inducing pro-inflammatory macrophage polarization in tumour tissues. Nat Nanotechnol (2016) 11:986–94. doi: 10.1038/nnano.2016.168
127. Parayath NN, Gandham SK, Leslie F, Amiji MM. Improved anti-tumor efficacy of paclitaxel in combination with MicroRNA-125b-based tumor-associated macrophage repolarization in epithelial ovarian cancer. Cancer Lett (2019) 461:1–9. doi: 10.1016/j.canlet.2019.07.002
128. Qian B-Z, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell (2010) 141:39–51. doi: 10.1016/j.cell.2010.03.014
129. Serafini P, Meckel K, Kelso M, Noonan K, Califano J, Koch W, et al. Phosphodiesterase-5 inhibition augments endogenous antitumor immunity by reducing myeloid-derived suppressor cell function. J Exp Med (2006) 203:2691–702. doi: 10.1084/jem.20061104
130. Wiers KM, Lathers DM, Wright MA, Young MR. Vitamin D3 treatment to diminish the levels of immune suppressive CD34+ cells increases the effectiveness of adoptive immunotherapy. J Immunother (2000) 23:115–24. doi: 10.1097/00002371-200001000-00014
131. Hengesbach LM, Hoag KA. Physiological concentrations of retinoic acid favor myeloid dendritic cell development over granulocyte development in cultures of bone marrow cells from mice. J Nutr (2004) 134:2653–9. doi: 10.1093/jn/134.10.2653
132. Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, et al. Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res (2009) 15:2148–57. doi: 10.1158/1078-0432.CCR-08-1332
133. Lu P, Yu B, Xu J. Cucurbitacin B regulates immature myeloid cell differentiation and enhances antitumor immunity in patients with lung cancer. Cancer Biother Radiopharm (2012) 27(8):495–503. doi: 10.1089/cbr.2012.1219
134. Suzuki E, Kapoor V, Jassar AS, Kaiser LR, Albelda SM. Gemcitabine selectively eliminates splenic Gr-1+/CD11b+ myeloid suppressor cells in tumor-bearing animals and enhances antitumor immune activity. Clin Cancer Res (2005) 11:6713–21. doi: 10.1158/1078-0432.CCR-05-0883
135. Le HK, Graham L, Cha E, Morales JK, Manjili MH, Bear HD. Gemcitabine directly inhibits myeloid derived suppressor cells in BALB/c mice bearing 4T1 mammary carcinoma and augments expansion of T cells from tumor-bearing mice. Int Immunopharmacol (2009) 9:900–9. doi: 10.1016/j.intimp.2009.03.015
136. Plebanek MP, Bhaumik D, Bryce PJ, Thaxton CS. Scavenger receptor type B1 and lipoprotein nanoparticle inhibit myeloid-derived suppressor cells. Mol Cancer Ther (2018) 17:686–97. doi: 10.1158/1535-7163.MCT-17-0981
137. Kong M, Tang J, Qiao Q, Wu T, Qi Y, Tan S, et al. Biodegradable hollow mesoporous silica nanoparticles for regulating tumor microenvironment and enhancing antitumor efficiency. Theranostics (2017) 7:3276–92. doi: 10.7150/thno.19987
138. Curti B, Daniels GA, McDermott DF, Clark JI, Kaufman HL, Logan TF, et al. Improved survival and tumor control with Interleukin-2 is associated with the development of immune-related adverse events: data from the PROCLAIMSM registry. J Immunother Cancer (2017) 5:102. doi: 10.1186/s40425-017-0307-5
139. Hamson EJ, Keane FM, Tholen S, Schilling O, Gorrell MD. Understanding fibroblast activation protein (FAP): substrates, activities, expression and targeting for cancer therapy. Proteomics Clin Appl (2014) 8:454–63. doi: 10.1002/prca.201300095
140. Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, et al. Stromal biology and therapy in pancreatic cancer. Gut (2011) 60:861–8. doi: 10.1136/gut.2010.226092
141. Ernsting MJ, Hoang B, Lohse I, Undzys E, Cao P, Do T, et al. Targeting of metastasis-promoting tumor-associated fibroblasts and modulation of pancreatic tumor-associated stroma with a carboxymethylcellulose-docetaxel nanoparticle. J Control Release (2015) 206:122–30. doi: 10.1016/j.jconrel.2015.03.023
142. Liu C, Lai H, Chen T. Boosting natural killer cell-based cancer immunotherapy with selenocystine/transforming growth factor-beta inhibitor-encapsulated nanoemulsion. ACS Nano (2020) 14:11067–82. doi: 10.1021/acsnano.9b10103
143. Lai H, Zeng D, Liu C, Zhang Q, Wang X, Chen T. Selenium-containing ruthenium complex synergizes with natural killer cells to enhance immunotherapy against prostate cancer via activating TRAIL/FasL signaling. Biomaterials (2019) 219:119377. doi: 10.1016/j.biomaterials.2019.119377
144. Hou L, Liu Q, Shen L, Liu Y, Zhang X, Chen F, et al. Nano-delivery of fraxinellone remodels tumor microenvironment and facilitates therapeutic vaccination in desmoplastic melanoma. Theranostics (2018) 8:3781–96. doi: 10.7150/thno.24821
145. Hu T, Gong H, Xu J, Huang Y, Wu F, He Z. Nanomedicines for overcoming cancer drug resistance. Pharmaceutics (2022) 14:1606. doi: 10.3390/pharmaceutics14081606
146. Dietl K, Renner K, Dettmer K, Timischl B, Eberhart K, Dorn C, et al. Lactic acid and acidification inhibit TNF secretion and glycolysis of human monocytes. J Immunol (2010) 184:1200–9. doi: 10.4049/jimmunol.0902584
147. Singer K, Kastenberger M, Gottfried E, Hammerschmied CG, Büttner M, Aigner M, et al. Warburg phenotype in renal cell carcinoma: high expression of glucose-transporter 1 (GLUT-1) correlates with low CD8(+) T-cell infiltration in the tumor. Int J Cancer (2011) 128:2085–95. doi: 10.1002/ijc.25543
148. Bonatelli M, Silva ECA, Cárcano FM, Zaia MG, Lopes LF, Scapulatempo-Neto C, et al. The warburg effect is associated with tumor aggressiveness in testicular germ cell tumors. Front Endocrinol (Lausanne) (2019) 10:417. doi: 10.3389/fendo.2019.00417
149. Chang C-H, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell (2015) 162:1229–41. doi: 10.1016/j.cell.2015.08.016
150. Platten M, Wick W, Van den Eynde BJ. Tryptophan catabolism in cancer: beyond IDO and tryptophan depletion. Cancer Res (2012) 72:5435–40. doi: 10.1158/0008-5472.CAN-12-0569
151. Ikonen E. Cellular cholesterol trafficking and compartmentalization. Nat Rev Mol Cell Biol (2008) 9:125–38. doi: 10.1038/nrm2336
152. Jakobsson T, Treuter E, Gustafsson J-Å, Steffensen KR. Liver X receptor biology and pharmacology: new pathways, challenges and opportunities. Trends Pharmacol Sci (2012) 33:394–404. doi: 10.1016/j.tips.2012.03.013
153. Li J, Gu D, Lee SS-Y, Song B, Bandyopadhyay S, Chen S, et al. Abrogating cholesterol esterification suppresses growth and metastasis of pancreatic cancer. Oncogene (2016) 35:6378–88. doi: 10.1038/onc.2016.168
154. Yang W, Bai Y, Xiong Y, Zhang J, Chen S, Zheng X, et al. Potentiating the antitumour response of CD8(+) T cells by modulating cholesterol metabolism. Nature (2016) 531:651–5. doi: 10.1038/nature17412
Keywords: cancer, microenvironment, immunotherapy, drug resistance, nanotechnology
Citation: Shao J, Jin Y and Jin C (2023) A new approach to overcoming resistance to immunotherapy: nanotechnology. Front. Oncol. 13:1210245. doi: 10.3389/fonc.2023.1210245
Received: 27 April 2023; Accepted: 27 July 2023;
Published: 10 August 2023.
Edited by:
Shu Wang, Tulane University, United StatesReviewed by:
Runzi Sun, University of Pittsburgh, United StatesCopyright © 2023 Shao, Jin and Jin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chunxiang Jin, amluY3g3MjJAamx1LmVkdS5jbg==; Ying Jin, amlueWluZzAyMTZAamx1LmVkdS5jbg==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.