 Sara Tirendi1,2†
Sara Tirendi1,2† Barbara Marengo1,2†
Barbara Marengo1,2† Cinzia Domenicotti1,2†
Cinzia Domenicotti1,2† Anna M. Bassi1,2
Anna M. Bassi1,2 Vanessa Almonti1
Vanessa Almonti1 Stefania Vernazza1,2*†
Stefania Vernazza1,2*†- 1Department of Experimental Medicine, University of Genoa, Genoa, Italy
- 2Inter-University Center for the Promotion of the 3Rs Principles in Teaching & Research (Centro 3R), Genoa, Italy
Introduction: The latest GLOBOCAN 2021 reports that colorectal cancer (CRC) is the second leading cause of cancer-related death worldwide. Most CRC cases are sporadic and associated with several risk factors, including lifestyle habits, gut dysbiosis, chronic inflammation, and oxidative stress.
Aim: To summarize the biology of CRC and discuss current therapeutic interventions designed to counteract CRC development and to overcome chemoresistance.
Methods: Literature searches were conducted using PubMed and focusing the attention on the keywords such as “Current treatment of CRC” or “chemoresistance and CRC” or “oxidative stress and CRC” or “novel drug delivery approaches in cancer” or “immunotherapy in CRC” or “gut microbiota in CRC” or “systematic review and meta-analysis of randomized controlled trials” or “CSCs and CRC”. The citations included in the search ranged from September 1988 to December 2022. An additional search was carried out using the clinical trial database.
Results: Rounds of adjuvant therapies, including radiotherapy, chemotherapy, and immunotherapy are commonly planned to reduce cancer recurrence after surgery (stage II and stage III CRC patients) and to improve overall survival (stage IV). 5-fluorouracil-based chemotherapy in combination with other cytotoxic drugs, is the mainstay to treat CRC. However, the onset of the inherent or acquired resistance and the presence of chemoresistant cancer stem cells drastically reduce the efficacy. On the other hand, the genetic-molecular heterogeneity of CRC often precludes also the efficacy of new therapeutic approaches such as immunotherapies. Therefore, the CRC complexity made of natural or acquired multidrug resistance has made it necessary the search for new druggable targets and new delivery systems.
Conclusion: Further knowledge of the underlying CRC mechanisms and a comprehensive overview of current therapeutic opportunities can provide the basis for identifying pharmacological and biological barriers that render therapies ineffective and for identifying new potential biomarkers and therapeutic targets for advanced and aggressive CRC.
1 Introduction
Colorectal cancer (CRC) is the third most diagnosed cancer and the second leading cause of death worldwide regardless of gender (1). Approximately 90% of CRCs are adenocarcinoma originating from epithelial cells of the colorectal mucosa, whilst the remaining 10% are represented by rare CRC types (i.e., squamous cell carcinoma, adenosquamous carcinoma, spindle cell carcinoma, and undifferentiated carcinoma) (2).
Most CRC cases are sporadically and associated with non-hereditary spontaneous mutations and epigenetic aberrations arising from several risk factors, including dysregulation of the gut microbiome, obesity, sedentary lifestyle, excess intake of meats, fats, starches, and sugars, folate deficiency, alcohol, cigarette smoking, and so on (3). However, a lower percentage of cases (about 30%) is represented by familial cases, of which approximately 5% present specific genetic signatures, penetrance, and transmission due to germline variants in CRC predisposing genes, e.g., adenomatous polyposis coli (APC), mismatch repair (MMR) genes, epithelial cell adhesion molecule (EPCAM), SMAD4/BMPR1A, and MUTYH (4–7).
Data report that the highest CRC incidence rates are recorded in developed countries and the incidence of early-onset CRC in individuals younger than 50 continues to rise (2, 4). Therefore, to facilitate diagnosis of CRC cancer in earlier stages, the recommended screening age was recently lowered to 45.
Early-stage colon cancer may be asymptomatic and often become symptomatic late in the disease. Indeed, about >25% of patients are diagnosed with advanced disease, i.e., extensive or metastatic colorectal cancer (mCRC), at the time of diagnosis, while more than 50% of patients with the initially localized disease develop metastases during or after therapies (8, 9). As known, metastasis poses a huge clinical challenge because only 20% of mCRC patients survive (10).
When neoadjuvant therapy is not included in the treatment plan, surgical resection is performed as the first curative intent in patients with localized and locoregional CRC (stages I, II, and III), as well as for those with resectable distant metastases (5, 11–13). However, the National Comprehensive Cancer Network (NCCN) guidelines recommend neoadjuvant oxaliplatin-based chemotherapy for patients with “bulky nodal disease or clinical T4b” colon cancer to decrease the size of the tumor before surgery (14). To reduce the risk of cancer recurrence and improve patient outcomes, an adjuvant postoperative chemotherapy regimen is routinely employed in stage III patients (i.e., localized tumor with lymph node invasion) and, in some cases, in stage II patients (i.e., localized tumor w/o lymph node invasion) (15, 16). Moreover, chemotherapy is the first-line therapy also for mCRC treatment.
The genetic variability of CRC makes necessary to identify the tumor subtypes (e.g., mismatch repair or microsatellite instability status, mutations in KRAS, NRAS, BRAF) to set the most suitable adjuvant therapy (i.e., systemic chemotherapy alone or with other FDA-approved drugs).
The CRC prognosis depends essentially on comorbid conditions, the frailty of patients, and drug resistance promoted by cancer stem cells and/or genetic mutations in key driver genes (e.g., KRAS, p53, BRAF) (17, 18). Therefore, this present review aims to summarize the mechanisms that characterize the stepwise nature of CRC, its genetic landscape, and the current and future approaches for CRC management.
2 Methods
The data in this present systematic review were collected using two different searches: PubMed (https://pubmed.ncbi.nlm.nih.gov/) and an online bioinformatic database (http://clinicaltrials.gov). The search of the references using PubMed identified a total of 144 hits from 1988 to 2022 relatively to keywords such as “Current treatment of CRC” or “immunotherapy in CRC” or “gut microbiota in CRC” or “chemoresistance and CRC” or “oxidative stress and CRC” or “novel drug delivery approaches in cancer” or “CSCs and CRC” or “systematic review and meta-analysis of randomized controlled trials”. Thus, the combined information obtained from the two data sources has represented the basis for writing the review.
3 Results
3.1 Mechanisms of CRC initiation and progression
The stepwise nature of sporadic CRC is still poorly understood, even though several mechanisms have been described to be involved in its initiation and progression. Epidemiological studies have found a relationship between CRC and chronic exposure to environmental risk factors (see above section) with strong pro-inflammatory potential. Moreover, increasing evidence suggests that intestinal microbiota and its products (e.g., butyrate and bacterial toxins) play a pivotal role in all CRC steps (initiation, progression, and metastasis) (19–21) (Figure 1). CRC patients display a reduced bacterial diversity and richness compared to healthy individuals, reflecting a distinctive intestinal microbial dysbiosis (22). Dysbiosis causes alteration in gut mucosa integrity and permeability, due to alteration of intercellular tight junctions. This condition, enhancing the colocyte susceptibility to mutagenic/carcinogenic factors and pathogenic bacteria, also promotes the activation of Mucosal Associated Lymphatic Tissue. Moreover, Th2-derived cytokines induced by pathogens or autoantigens may result in myeloid cell recruitment (neutrophils and macrophages) and, consequently in Reactive Oxygen Species (ROS) production.
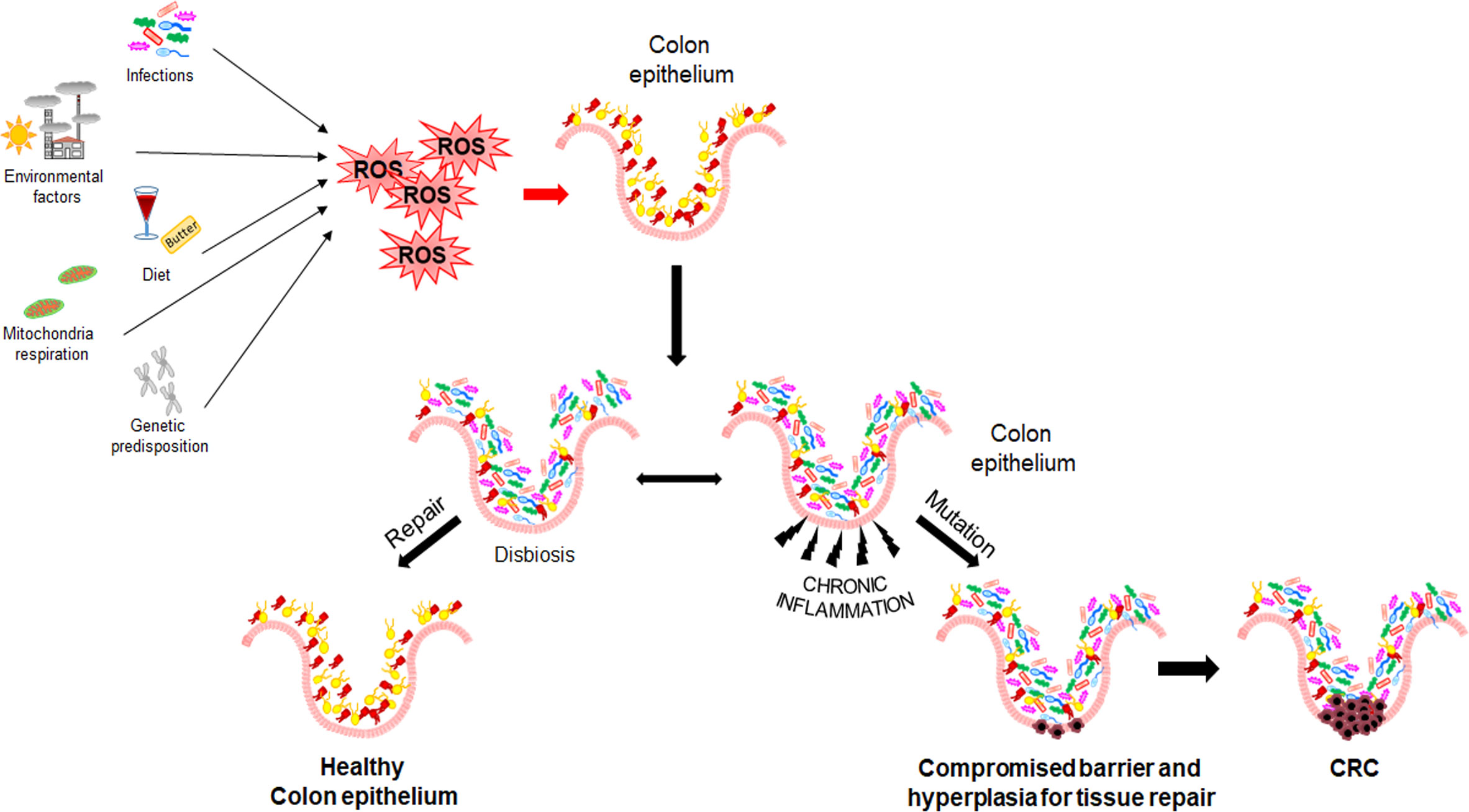
Figure 1 Mechanisms of CRC initiation and progression. Environmental and genetic risk factors promoting Reactive Oxygen Species production, dysbiosis, and inflammation.
As known, inflammation and oxidative stress are tightly coupled; in fact, a chronic activation of inflammatory cells and production of pro-inflammatory mediators (e.g., cyclooxygenase 2, prostaglandin E2, tumor necrosis factor α, and transforming growth factor β) enhance ROS generation and dysregulate the activity of signal transduction pathways, including Transducer and Activator of Transcription 3, Nuclear Factor-kappa B (NF-kB), hypoxia-inducible factor-1α (HIF1α), NF-E2 related factor-2 (NRF2) (23–25). In addition, it has been reported that ROS overproduction may result in genetic/epigenetic changes, such as single-strand cleavage, point mutations, miscoding, abnormal amplification, oncogene activation, and immune suppression, leading to possible precancerous lesions (i.e., adenomatous polyps) (26, 27).
Mutations providing a selective growth advantage to the cells within their microenvironment can potentially drive cancer development. Generally, two mutated gene drivers can lead to a net cell gain and detectable benign lesions, but over three gene mutations promote the invasion through the basement membrane, thereby leading to malignancy. In CRC, the first mutations usually concern the APC gene resulting in a proliferative advantage of epithelial cells promoting benign lesions (small adenomas) (Figure 2). APC mutated small adenomas have a slow growth rate, but further mutations of the KRAS gene can increase their proliferation. However, the mutational status may be worsen by sequential mutations of genes such as PIK3CA, SMAD4, and TP53 that promote the onset of malignant tumors capable of infiltrating surrounding tissue and metastasizing distant organs (Figure 2) (28). In addition, both colorectal adenomas and CRC are linked to epigenetic alterations such as aberrant DNA methylation in key tumor-suppressor and oncogenes and dysregulation of miRNA expression (29, 30).
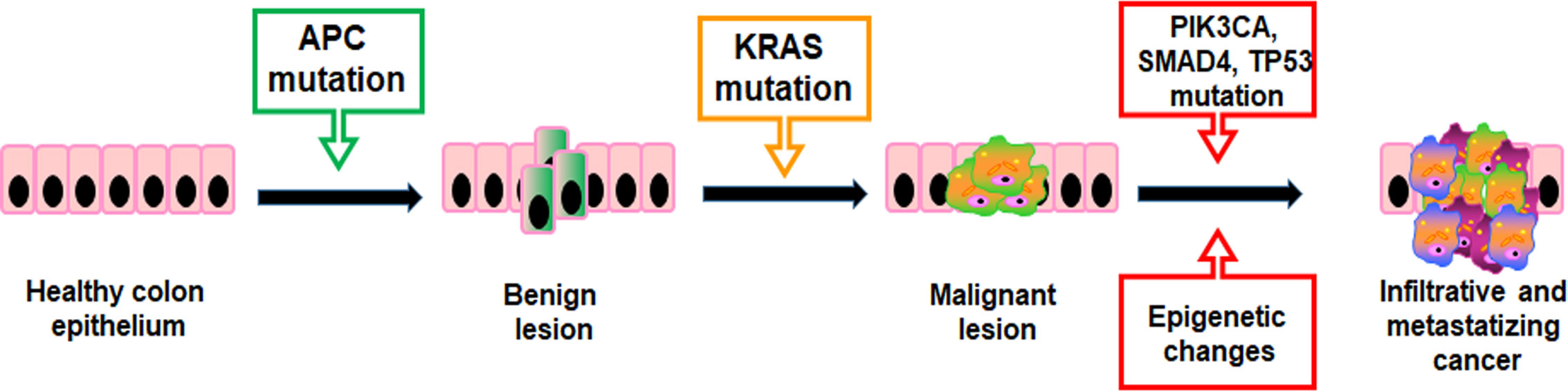
Figure 2 Stepwise molecular/genetic events that underlie the initiation and progression of CRC.
Well-known examples of the carcinogenic role of ROS in CRC are missense mutations at p53 suppressor gene, activation of canonical Wnt signaling pathway (Wnt/β-catenin), which is involved in cancer stem cell renewal process, and PI3K/Akt signaling pathway, which regulates cell proliferation (31, 32). Moreover, the most frequent ROS-dependent pre-mutagenic DNA lesion is represented by 8-oxoguanine (8-oxoG) (33).
Notably, oxidative stress, due to either ROS overproduction or a reduced activity of the enzymatic and non-enzymatic antioxidant systems, is often involved in development and progression of several cancers via activation of redox-responsive signaling pathways leading to uncontrolled cell growth and oxidation of lipids, carbohydrates, and proteins (i.e., cancer initiation, promotion, and progression stages). Accordingly, oxidative stress is one of the main cancer research topics given its involvement in both genetic and metabolic cell damage (34–36). Increasing evidence showed that oxidative stress control biogenesis of cancer-associated microRNA (miRNA) via targeting various transcription and epigenetic factors. Recently, CRC-associated miRNAs (e.g., miR-106b-5p, miR-335-5p, miR-193a-5p, miR-378a-3p and miR-423-5p) are becoming attractive biomarkers as they are expressed from the early stage of tumor development (37).
Moreover, either up-regulation or down-regulation of miRNAs also known as “onco-miRNAs” are involved in CRC progression and metastasis contributing to dysregulate several signaling pathways, including mitogen-activated protein kinases (e.g., miR-422a, miR-195), Wnt (e.g., miR-135a, miR-135b, miR-155, miR-17–5p, miR-224), transforming growth factor-β (e.g., miR-224, miR-20a-5p) and epithelial-to-mesenchymal transition (EMT) (e.g., miRNA-155, miR-34) (38–40).
Oncogene, oncosuppressor and metabolic gene mutations contribute to the profound metabolic alterations found in cancer cells, i.e., impaired respiration, increased fermentation and anabolism (41). In most cases, the metabolism of cancer cells favors aerobic glycolysis (the Warburg effect) rather than oxidative metabolism to fulfill their biosynthetic and bioenergetic demands of rapid and sustained proliferation. Mitochondrial Oxidative Phosphorylation System (OXPHOS) is not necessarily defective in tumorigenic cells, and it can take place proportionally to the oxygen supply. Indeed, it has been shown that cancer stem cells are able to revert glycolysis to TCA cycle to better satisfy their metabolic needs and overproduce ROS (42). The Warburg phenotype has been demonstrated to be driven by overexpression of oncogenes such as c-Myc and HIF-1α (43). The inhibition of pyruvate dehydrogenase activity and the increase of lactate dehydrogenase activity lead to the conversion of pyruvate to lactate following the mass action law (44). Moreover, lactate is also generated from catabolism of glutamine and it is considered a metabolite eliciting a broad spectrum of effects useful to sustain cancer progression and metastasis (45). Cancer cells are capable of adapting to metabolic-derived acidosis via monocarboxylate transporters (MCTs), which export lactate and favor intracellular alkalinization. Thus, the lactate exported by tumor cells can be imported by cells of tumor microenvironmental where it acts as important intracellular signaling for angiogenesis (46, 47).
Notably, cancer cells well adapt to ROS by triggering a powerful antioxidant response mainly driven by glutathione (GSH) and antioxidant enzymes, such as superoxide dismutase, catalase, peroxiredoxins, GSH peroxidases, and thioredoxins (48). Thus, the maintenance of the oxidative balance enables cancer cells to perform their biological functions such as proliferation, differentiation, and migration (49–51).
3.1.1 Genetic- molecular heterogeneity of CRC
CRC from a genetic-molecular standpoint is extremely diversified. In fact, there are four main mechanisms of gene alteration: (i) microsatellite instability (MSI), (ii) chromosomal instability (CIN), (iii) CpG island methylator phenotype (CIMP), (iv) and BRAF or KRAS mutations (52). Another aspect concerning the molecular and phenotypic differences is the tumor localization (i.e., the right or left side), which leads to different gene expression and mutation profiles. Right-sided CRC occurs mainly in patients with genetic predisposition and is characterized by hypermethylation, higher frequency of BRAF mutation, and, in some cases, MSI (53). Instead, left-sided CRC is characterized by CIN and the activation of the EGFR pathway (54). Moreover, differences in tumor microenvironment components (e.g., tumor epithelial cells, immune cells, and cancer-associated fibroblasts) play a critical role in defining CRC with a positive or poor prognosis and in maintaining immune surveillance (through the increase in tumor T-lymphocyte subset density) or in promoting immune escape (55). For example, a high density of specific tumor-infiltrating lymphocytes (i.e. cytotoxic and memory T-cells) in MSI-high CRC can be considered a favorable prognostic marker, because it counteracts the establishment of the “immunoediting” process and reduces the tumor spread (56–58). Furthermore, Canna et al. (59), found a relationship between systemic inflammatory response and local inflammatory response in patients undergoing resection for CRC, demonstrating that a high concentration of C-reactive protein and low tumor-infiltrating CD4+ are predictive of poor cancer-specific survival.
The analysis of genetic profiles cannot be used for clinical purposes due to a discrepancy in results (e.g., sample preparation methods, use of different data processing and algorithms among different patient cohorts, gene expression platforms, and so on). However, the consensus molecular subtypes (CMS), which represent a transcriptome-based classification of CRC, include some superficial similarities useful for predictable CRC prognosis (60, 61). The first called CMS1 (MSI immune) is characterized by hypermutation, frequent BRAF mutation, MSI, and strong immune activation, the CMS2 (canonical) by CIN and marked WNT and MYC signaling, and the CM3 (metabolic) by evident metabolic dysregulation and KRAS-mutated tumors. Lastly, the CM4 (mesenchymal), includes tumors characterized by prominent TGFβ activation, epithelial-mesenchymal transition gene up-regulation, angiogenesis, and matrix remodeling.
3.2 Adjuvant treatments of CRC
Chemotherapy agents are usually used after a surgical excision as the treatment of choice to eradicate Minimal Residual Disease (MRD) in high-risk stage II and stage III patients and to increase the overall survival rate in stage IV patients (62–65). However, the only use of chemotherapy as standard-of-care (Table 1) can represent a limit due to the high systemic toxicity, unsatisfactory response rate, the onset of drug resistance, and the low tumor-specific selectivity. Therefore, massive investments have been earmarked to develop new approaches to improve patient outcomes. The identification of point mutations in specific oncogenes (KRAS, NRAS, and BRAF), amplification of human epidermal growth factor receptor 2 (HER2), the MSI status, the DNA mismatch repair status (deficiency or proficiency), has provided a framework for finding additional approaches, as well as new prognostic perspective (66). Up to today it is possible to hit the cancer more effectively, by administering the most suitable biological agents with the standard chemotherapy taking into account the genetic setting of patients (67, 68). In this regard, targeted therapies (i.e., antibodies and small molecules) and immunotherapies, which actively or passively target the patient’s immune system, are widely used in combination with FOLFOX or FOLFIRI as a first-/second-line setting or alone as a third-line setting to improve the overall survival (OS) and progression-free survival (PFS) of advanced/metastatic cancer patients (69, 70). Moreover, antitumor immunity exerted by vaccines, specialized dendritic cells or new generation of cytotoxic T cells are currently under investigation in clinical trials (71, 72) (Table 2).
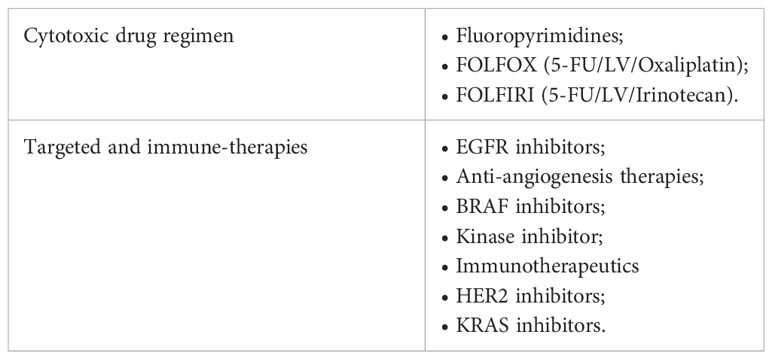
Table 1 The main therapeutic approaches in the CRC treatment.
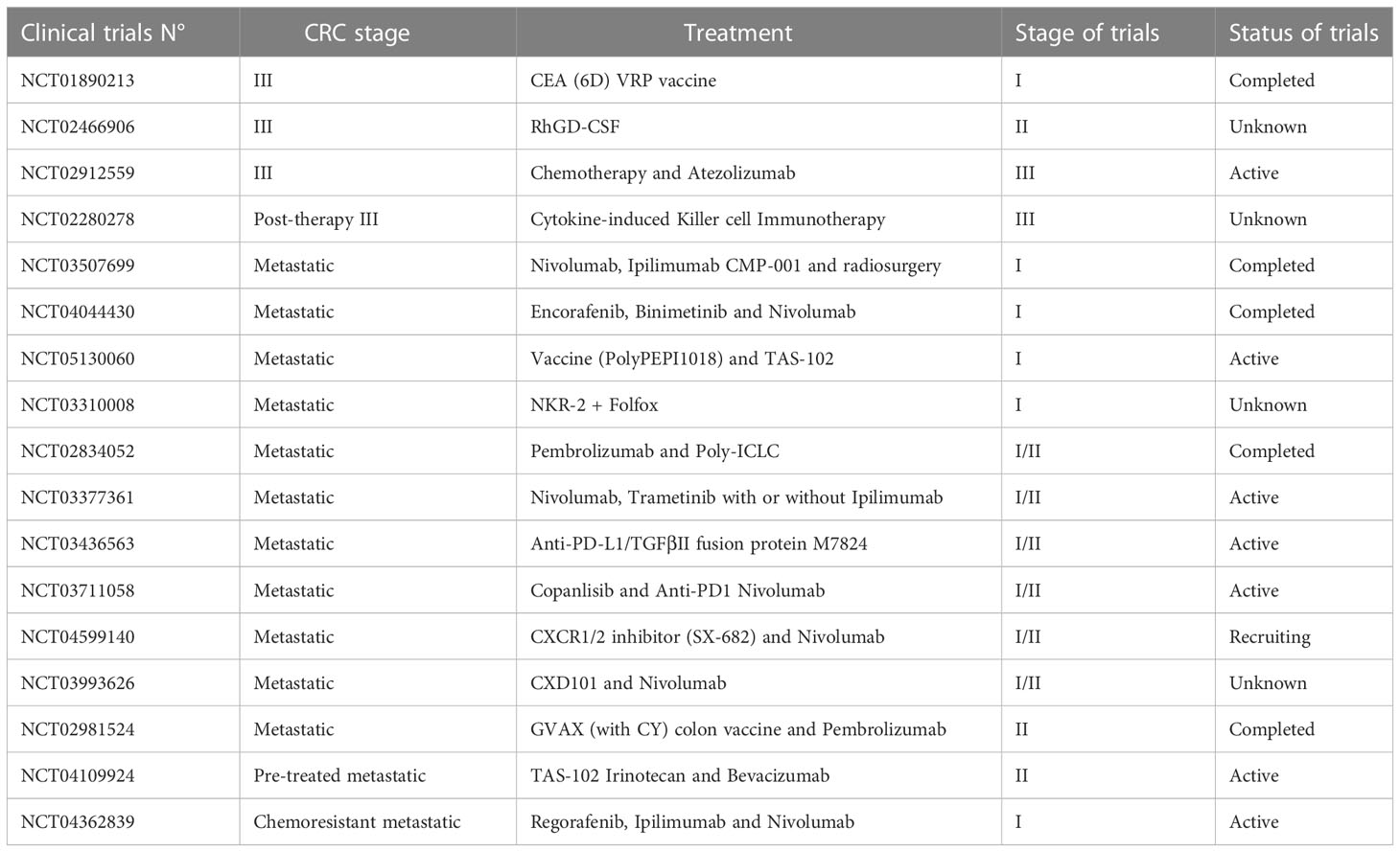
Table 2 Current clinical trials based on immunotherapy.
In patients with left-sided KRAS wild-type tumors, for instance, the administration of anti-EGFR (i.e., cetuximab or panitumumab) in combination with standard-of-care chemotherapy as a first-line setting shows improvement in both OS and progression-free survival (PFS) (73–75). Additionally, anti-EGFR can be used alone in chemo-refractory patients with advanced CRC (76).
Recently, in patients with BRAF-mutated mCRC, the use of anti-EGFR in combination with a selective inhibitor of BRAF kinase (encorafenib) and a reversible inhibitor of the kinase activity of mitogen-activated extracellular signal-regulated kinase 1 (MEK1) and MEK2 (binimetinib) has been proposed as the third line treatment to improve the prognosis (77, 78).
Among the first- and second line interventions for mCRC, also the VEGF inhibitors (i.e., Bevacizumab, Aflibercept) in combination with standard-of-care chemotherapy contribute to improve OS and PFS in patients (79, 80).
Another targeted therapy to treat advanced/metastatic CRC refractory to all standard treatments is represented by the diphenylurea-based multikinase inhibitor (i.e., Regorafenib). This anti-tumoral drug targeting multiple protein kinases regulating angiogenesis, proliferation, immunity, and metastases, increases OS of heavily pre-treated patients (79, 81–83).
KRAS-targeted drugs, such as sotorasib and adagrasib, are emerging for their anti-cancer activity in heavily pre-treated patients harboring the KRASG12C mutation. These drugs are small molecules that keep KRAS in its inactive state, allowing apoptosis. However, their use in CRC treatment is still under investigation (84).
3.2.1 5- fluorouracil
5-FU is a fluorinated analogue of uracil that belongs to fluoropyrimidines. It was developed in 1957 and still today, it represents the mainstay of systemic combination chemotherapy for the treatment of CRC (85, 86). Although several 5-FU administration schedules were used (e.g., bolus intravenous, bolus plus intermittent intravenous infusion), today the standard of care is represented by continuous or intermittent intravenous infusion (87). Moreover, for around twenty years, also oral 5-FU prodrugs (e.g., Capecitabine, Tegafur, 5′-deoxy-5-fluorouridine) are commonly used as part of combination regimens or as monotherapy (88, 89).
5-FU is easily incorporated into DNA and RNA where it acts as an antimetabolite because shares a common structure with pyrimidines (90). After administration, 5-FU is converted via anabolic pathways into fluorodeoxyuridine monophosphate (FdUMP), fluorodeoxyuridine triphosphate (FdUTP), and fluorouridine triphosphate (FUTP) (91). Stable complex between FdUMP and thymidylate synthase (TS) inhibits deoxythymidine mono-phosphate production, which consequently results in severe disruption of DNA synthesis and repair. Leucovorin (LV, Folinic acid), and Methotrexate (MTX, Folate analogue) are preferably used in combination with 5-FU to improve its antitumor activity (92, 93). Moreover, metabolites of 5-FU produce also alterations in the cellular membrane (89).
Approximately 80% of the total 5-FU dose is metabolized primarily in the liver by dihydropyrimidine dehydrogenase (DPD) (94), an enzyme that catalyzes the rate-limiting step in its metabolism.
Severe 5-FU-associated toxicity (e.g., leukopenia, neutropenia, thrombocytopenia, anemia, neuropathy, skin rash, hand-foot syndrome, and so on) is mainly due to a partial or complete DPD deficiency (95–99). In particular, different rare variants in the gene encoding DPD (DPYD) have been identified as validated risk variants for drug toxicity (86). Therefore, FDA-approved drug label prevents the use of 5-FU in individuals with absent DPD activity (88), while the Clinical Pharmacogenetics Implementation Consortium and the Dutch Pharmacogenetics Working Groups report dosing recommendations for 5-FU-based chemotherapy, based on DPYD genotype (88, 100, 101).
Although 5-FU-based chemotherapy combining with oxaliplatin or irinotecan has improved the response rate in patients with advanced CRC, primary or acquired chemoresistance is the leading cause of unsatisfactory outcomes in over 90% of patients with metastatic disease (93, 102). Indeed, intratumoral heterogeneity due to genetic mutations, tumor microenvironment (TME), and the presence of cancer stem cells, and the molecular complexity of CRC as well, making it necessary to develop other novel therapeutic strategies to overcome drug resistance and improve drug response rates.
3.3 The presence of cancer stem cells limits therapy efficacy against CRC
The main limiting factor for cancer patients is the onset of multi-drug resistance (MDR), which makes cancer cells tolerant to anti-cancer drugs. In fact, the combined chemotherapy and the development of different administration schedules are not always sufficient to avoid these issues due to the biological complexity of the tumor (103).
Tumor MDR is a highly-complex phenomenon that encompasses a plethora of molecular mechanisms involving not only cancer cells, but also infiltrating cells (e.g., endothelial, hematopoietic, and stromal cells) and the resulting tumor microenvironment (104). The constant interactions between tumor cells and their surrounding stroma result in alterations of many different cellular processes. Moreover, the presence of a sub clonal variation among cancer cells allows greater adaptability of the tumor to therapy, promoting its evolution.
Multiple molecular mechanisms have been identified as contributing factors to MDR development. Among these, the interplay between pre-existing and drug-induced mechanisms, including defects in the apoptotic machinery, mitochondrial dysfunction, altered autophagy activity, aberrant cell signaling, reduction in drug concentration and genetic and epigenetic changes, plays a significant role (105–108).
Moreover, the major cause of primary therapy resistance is represented by unresponsive subpopulations, such as cancer stem cells (CSCs) that can increase by up to 30% following long-term drug treatment (109).
Stochastically CSCs are distributed within tumors, but preferably they reside in specific niches, characterized by hypoxia, low pH, and fewer nutrients, which in addition to conferring them stemness features, allow the generation of differentiated progenies (110, 111).
CSCs are frequently quiescent and poorly differentiated cell populations with a lower level of intracellular ROS that share with normal stem cells both properties (i.e., self-renewal, self-sufficiency, and differentiation), and stemness signaling pathways (e.g., Notch, Sonic hedgehog, WNT/β-Catenin, JAK/STAT, and NF-kB). Their origin is still debated but it has been suggested that, at the moment of tumor initiation, the acquisition of CSC phenotype from either transformed differentiated cells (stochastic model) or transformed tissue-resident stem cells (hierarchical model) is promoted by the overexpression of oncogenes and the inhibition of tumor suppressor genes (e.g., APC, TP53, TGFBR2, SMAD4, PTEN, and RAS). Instead, following chemotherapy or radiotherapy regimen, new CSCs derive from either non-CSC subpopulations or therapy-induced senescent tumor cells (112).
Standard chemotherapy is not a valid therapeutic option for CSCs because they can effectively counteract the chemotherapy-induced oxidative stress through their free radical scavenging systems, such as GSH, and overexpression of the anti-apoptotic protein B-cell lymphoma 2 (Bcl2) (113–115). Moreover, the enhancement of ATP-binding cassette (ABC) transporters and aldehyde dehydrogenase (ALDH) expression, the increased resistance to apoptosis, and the activation of DNA damage sensor and repair machinery contribute to give to CSCs a survival advantage against anti-cancer therapy (116). Additionally, CSCs can transiently and reversibly switch between epithelial and mesenchymal states and vice versa (i.e., epithelial-mesenchymal plasticity) via Wnt/b-catenin signaling (117, 118). Such versatility, consequently, results in metabolic reprogramming in cellular bioenergetics, where energy supply can alternatively depend on aerobic glycolysis or mitochondrial OXPHOS.
It has been shown that metformin potentially offers therapeutic advantages by inhibiting the mitochondrial respiration, forces CSCs to a metabolic shift from OXPHOS to glycolysis (119, 120). The temporary OXPHOS suppression renders CSCs more prone to apoptosis. However, tumor relapse under metformin treatment cannot be excluded since CSCs can acquire resistance mainly due to MYC overexpression, promoting a Warburg-like glycolytic phenotype (120). Also, progressive increase in glycolysis-derived lactate may promote the activation of proteases, leading to ECM degradation, and resistance to chemotherapy (121).
The release of IL-4 from colorectal CSCs promotes their survival and hampers the CD8+T cell-mediated antitumor immune response, while the presence of inflammatory cytokines, including IL-1, IL-4, IL-6, IL-8, IL-10, and TGF-β, fuels an inflammatory loop, via Stat3/NF-κB pathways, for stimulating the self-renewal of CSCs (118, 122). Moreover, the tumorigenic and self-renewal capacity of CSCs also depend on the hyperactivation of β-catenin, Notch, and Hedgehog signaling pathways (123, 124).
Although several CSC biomarkers have been identified for CRC, their preclinical application is still unavailable due to the intrinsic features of CSCs, i.e., phenotypic heterogeneity, and the influence of the TME or CSC behavior. In this regard, previous studies have focused the attention on both the CSC-related signature and immune cell infiltration as important prognostic factors. The correlation between infiltrating portion of immune cells, i.e., tumor immune microenvironment (TIME), and hallmark gene sets may represent a possible starting point for developing CSC-targeted therapeutic strategies (122, 125).
3.4 New drug delivery approaches and latest strategies implemented in the treatment of CRC
Increasing evidence suggests that the use of nanoscale nanoparticles (NPs) as drug delivery systems (DDS), including liposomes, nanoemulsions, hydrogels, multifunctional inorganic materials (e.g., carbon nanotubes, gold nanoparticles, quantum dots), and peptides, could provide a novel therapeutic approach useful in overcoming MDR and improving the pharmacokinetics and biodistribution of anticancer compounds, resulting in reduced side effects (126). These NPs refers to nanometer scale systems (10-1000 nm) capable of protecting encapsuled molecules from degradation and passively or actively delivering drugs, small molecules, proteins, peptides, DNA and RNA into specific targets. However, their bio-distribution and clearance in the body depend not only by the NP chemical, physical and biological properties (e.g., size, stability, surface charge, solubility, and so on) but also by factors, such as the administration route (e.g., intravenous, oral, pulmonary and dermal administration) and host environment (e.g., pre-existing inflammation) (127).
Recently, it has been reported that NPs can accumulate in tumor tissues by passive or active delivery. Passive delivery of drug-loaded nanoparticles (i.e., the Enhanced Permeability and Retention, EPR, effect) is mainly due to fenestrated and immature new tumor vessels (128–131) while, the active delivery is due to a ligand-binding mechanism (e.g., nanoparticles targeting EpCAM, the folate receptor, EGFR and CD44) (132–134) (Figure 3). However, regardless of NP delivery, nanoparticle-protein complex, namely protein corona (PC), can permanently change the NP fate. The protein profile of the corona complex does not have a standard composition, because it varies not only among NPs of different chemical designs but even across the NPs of the same type. This latter is explained by the so-called Vroman effect, in which protein turnover depends not only by the high-affinity binding of proteins, but also on their exchange kinetics (135). In general, on the basis of the binding affinity between plasma proteins and NP surfaces a “hard corona” and a “soft corona” are distinguished, respectively (136). Moreover, proteins participating in the complex influence the cell recognition pathway by the reticuloendothelial system (RES) and promote biological processes against NPs, including aggregation, opsonization, and phagocytosis. Therefore, either second-generation NPs or PEGylation technique enhance the effect of cancer therapy by ensuring drug delivery within the tumor and evading phagocytosis (137). In this regard, emerging self-assembly pH-sensitive pegylated nano drug delivery systems, namely HA-mPEG-Cis NPs, are able to target CD44-CRC-positive cells and dissolve the hydrated PEG in the acidic tumor environment. These drug delivery systems improve drug circulation time and tumor targeting while reducing the side effects of the loaded drug (138) (Figure 3).
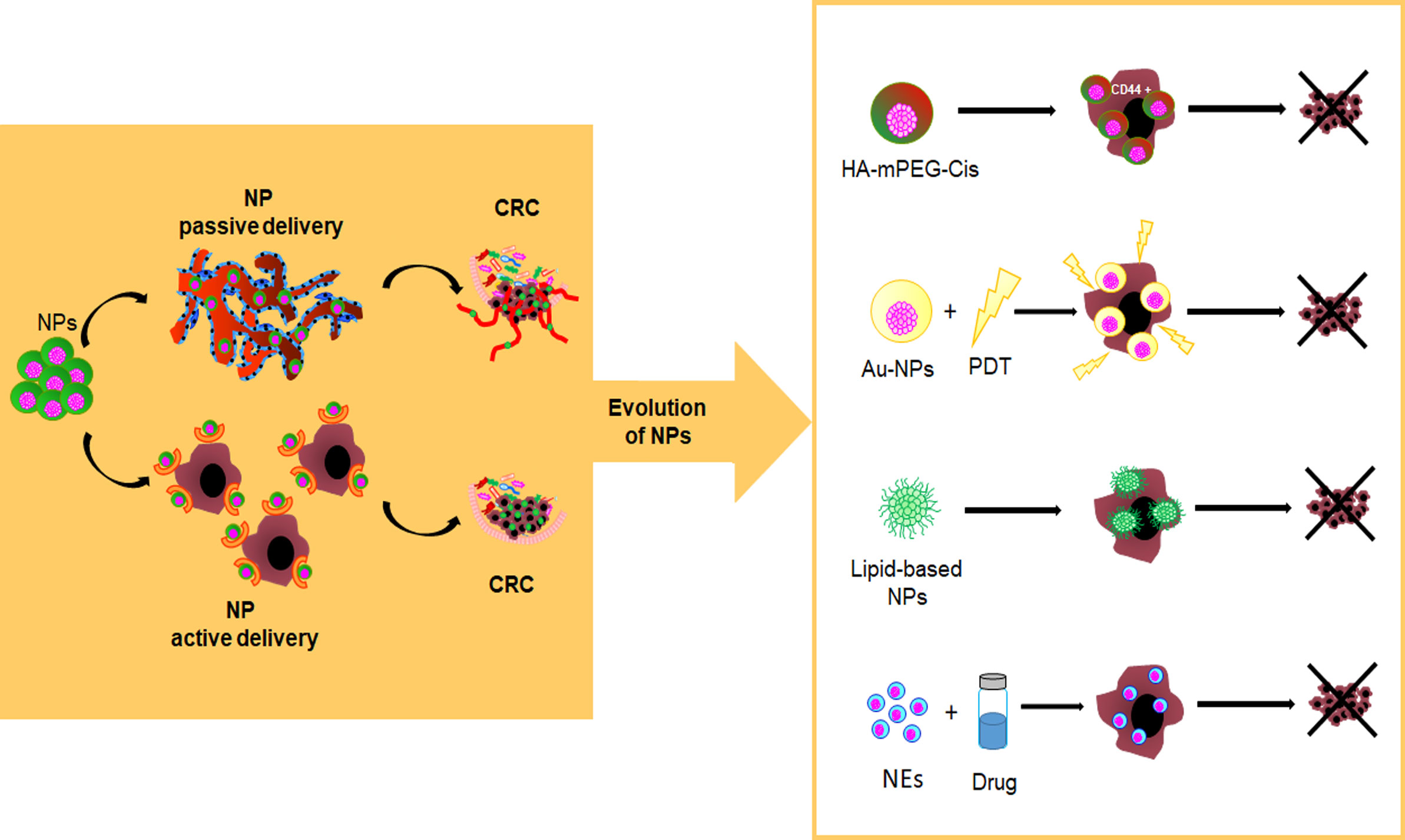
Figure 3 Evolution of nanoparticles as innovative drug delivery systems. pH-sensitive pegylated nano drug delivery systems (HA-mPEG-Cis NPs) are able to target CD44+ cells; Gold nanoparticles (AuNPs) find application in photodynamic therapy (PDT); Lipid-based NPs; Nanoemulsions (NEs) are a system to deliver hydrophobic drugs and hydrophilic or hydrophobic compounds.
Inorganic nanocarrier, of controlled size and shape, such as gold NPs (AuNPs), show a certain versatility of use, including chemical sensing, imaging, and drug delivery due to their favourable optical and physical properties coupled with a reasonable biocompatibility with regard to biological environment (139). Interestingly, AuNPs, including gold nanorods, nanocages, nanostars, nanocubes, and nanospheres, find application in photodynamic therapy (PDT) for their specific physical features (i.e., optical and Surface Plasmon Resonance properties, proton-capture cross-section) (140) (Figure 3).
Lipid-based NPs are already FDA-approved for various therapeutic purposes, including cancer treatment (e.g., Doxil®, DaunoXome®, Myocet®, DepoCyt®, Marqibo® and Onivyde®), severe infections or immunocompromised conditions (e.g. AmBisome®) and RNAi therapeutic (Onpattro®) (127, 141) (Figure 3).
Liposomes are small-size vesicles consisting of an outer lipid bilayer, synthetic or natural, and an aqueous core, widely used to encapsulate/entrap drugs or nucleic acids (i.e. gene therapy) (142). Currently, the manipulation of liposome lipid membrane components (e.g., neutral and/or negatively charged lipids plus cholesterol, sphingomyelin plus cholesterol, hydrogenated soy phosphatidylcholine plus cholesterol) as well as specific key parameters (e.g., size and shape) has improved their biological performance, in term of enhanced delivery efficiency, maximizing so-called nano-bio interactions (143).
Nanoemulsions (NEs) are another system to deliver hydrophobic drugs and hydrophilic or hydrophobic compounds through different routes of administration (e.g., aerosols, ingestion, and injections). NEs are made as single (i.e., oil-in-water [o/w], water-in-oil [w/o]) or dual (w/o/w, o/w/o) emulsions with biocompatible and FDA-approved biodegradable oils (143). Previous in vitro studies have shown that natural active compounds encapsulated within NEs, acting synergistically with chemotherapy, can improve the therapeutic value of treatment despite the use of a lower dosage of drug (144, 145). Also, the entrapment of active or cytotoxic drugs within nanoemulsions can be useful to sensitize CSCs to apoptosis (146) (Figure 3).
Over the past decade, nanotechnology has been widely explored to develop cytotoxic drug carriers. Although further improvements are needed, different types of NPs are already considered reliable systems for drug delivery due to their ability in targeting the tumor before releasing the drug.
4 Conclusion
Considering the critical nature of this review, and the variety of the included studies, it highlights that the sporadic CRC is a multi-stage and multi-step process in which the early mutational events seem to be driven by dysbiosis, chronic inflammation, and ROS. Moreover, treatments with standard cytotoxic agents, such as FOLFOX and FOLFIRI regimens, also contribute to the variation in the molecular profile of CRC in the advanced stage. Furthermore, this review also highlights that the limitation in treatment approaches for advanced CRC patients is mainly represented by both extrinsic (chemotherapy) and intrinsic mutation burden in cancer subpopulations (CSCs) developing MDR phenotype. In this regard, many strategies have been studied to overcome this issue, including the inhibition of crucial signaling involved in the self-renewal and metabolism of CSCs, as well as the redox-targeting approach. Moreover, using anti-vasculature therapies (e.g., bevacizumab and cetuximab) to modulate the tumor microenvironment represents a valid approach for enhancing cytotoxic drug uptake. Lastly, the development of novel DDS and promoter drugs can improve the delivery and the effectiveness of anti-cancer agents, opening up to personalized treatment protocols for CRC.
Author contributions
Conceptualization: SV. Investigation: SV, ST. Writing – original draft: SV, ST, VA. Supervision: SV, AMB, CD, BM. Writing – review & editing: SV, BM, CD. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the review was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: A Cancer J Clin (2018) 68(6):394–424. doi: 10.3322/caac.21492
2. Keum N, Giovannucci E. Global burden of colorectal cancer: emerging trends, risk factors and prevention strategies. Nat Rev Gastroenterol Hepatol (2019) 16(12):713–32. doi: 10.1038/s41575-019-0189-8
3. Giovannucci E. Modifiable risk factors for colon cancer. Gastroenterol Clinics North A (2002) 31(4):925–43. doi: 10.1016/S0889-8553(02)00057-2
4. Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal cancer statistics, 2020. CA A Cancer J Clin (2020) 70(3):145–64. doi: 10.3322/caac.21601
5. Vogel JD, Felder SI, Bhama AR, Hawkins AT, Langenfeld SJ, Shaffer VO, et al. The American society of colon and rectal surgeons clinical practice guidelines for the management of colon cancer. Dis Colon Rectum (2022) 65(2):148–77. doi: 10.1097/DCR.0000000000002323
6. Giglia M, Chu D. Familial colorectal cancer: understanding the alphabet soup. Clinics Colon Rectal Surg (2016) 29(03):185–95. doi: 10.1055/s-0036-1584290
7. Miao B, Skopelitou D, Srivastava A, Giangiobbe S, Dymerska D, Paramasivam N, et al. Whole-exome sequencing identifies a novel germline variant in PTK7 gene in familial colorectal cancer. IJMS (2022) 23(3):1295. doi: 10.3390/ijms23031295
8. Ciombor KK, Wu C, Goldberg RM. Recent therapeutic advances in the treatment of colorectal cancer. Annu Rev Med (2015) 66(1):83–95. doi: 10.1146/annurev-med-051513-102539
9. Fakih MG. Metastatic colorectal cancer: current state and future directions. JCO (2015) 33(16):1809–24. doi: 10.1200/JCO.2014.59.7633
10. Fan A, Wang B, Wang X, Nie Y, Fan D, Zhao X, et al. Immunotherapy in colorectal cancer: current achievements and future perspective. Int J Biol Sci (2021) 17(14):3837–49. doi: 10.7150/ijbs.64077
11. O’Connor ES, Greenblatt DY, LoConte NK, Gangnon RE, Liou JI, Heise CP, et al. Adjuvant chemotherapy for stage II colon cancer with poor prognostic features. JCO (2011) 29(25):3381–8. doi: 10.1200/JCO.2010.34.3426
12. Cheong CK, Nistala KRY, Ng CH, Syn N, Chang HSY, Sundar R, et al. Neoadjuvant therapy in locally advanced colon cancer: a meta-analysis and systematic review. J Gastrointest Oncol (2020) 11(5):847–57. doi: 10.21037/jgo-20-220
13. Sun W, Li J, Zhou L, Han J, Liu R, Zhang H, et al. The c-Myc/miR-27b-3p/ATG10 regulatory axis regulates chemoresistance in colorectal cancer. Theranostics (2020) 10(5):1981–96. doi: 10.7150/thno.37621
14. Benson AB, Venook AP, Al-Hawary MM, Arain MA, Chen YJ, Ciombor KK, et al. Colon cancer, version 2.2021, NCCN clinical practice guidelines in oncology. J Natl Compr Cancer Network (2021) 19(3):329–59. doi: 10.6004/jnccn.2021.0012
15. Shinji S, Yamada T, Matsuda A, Sonoda H, Ohta R, Iwai T, et al. Recent advances in the treatment of colorectal cancer: a review. J Nippon Med Sch (2022) JNMS.2022:89–310. doi: 10.1272/jnms.JNMS.2022_89-310
16. Assed Bastos D, Coelho Ribeiro S, de Freitas D, Hoff PM. Review: combination therapy in high-risk stage II or stage III colon cancer: current practice and future prospects. Ther Adv Med Oncol (2010) 2(4):261–72. doi: 10.1177/1758834010367905
17. Huang D, Sun W, Zhou Y, Li P, Chen F, Chen H, et al. Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev (2018) 37(1):173–87. doi: 10.1007/s10555-017-9726-5
18. Wong CC, Xu J, Bian X, Wu JL, Kang W, Qian Y, et al. In colorectal cancer cells with mutant KRAS, SLC25A22-mediated glutaminolysis reduces DNA demethylation to increase WNT signaling, stemness, and drug resistance. Gastroenterology (2020) 159(6):2163–80.e6. doi: 10.1053/j.gastro.2020.08.016
19. Fang Y, Yan C, Zhao Q, Xu J, Liu Z, Gao J, et al. The roles of microbial products in the development of colorectal cancer: a review. Bioengineered (2021) 12(1):720–35. doi: 10.1080/21655979.2021.1889109
20. Sears CL, Pardoll DM. Perspective: alpha-bugs, their microbial partners, and the link to colon cancer. J Infect Diseases (2011) 203(3):306–11. doi: 10.1093/jinfdis/jiq061
21. Tjalsma H, Boleij A, Marchesi JR, Dutilh BE. A bacterial driver–passenger model for colorectal cancer: beyond the usual suspects. Nat Rev Microbiol (2012) 10(8):575–82. doi: 10.1038/nrmicro2819
22. Cheng Y, Ling Z, Li L. The intestinal microbiota and colorectal cancer. Front Immunol (2020) 11:615056. doi: 10.3389/fimmu.2020.615056
23. Tomasello G. Intestinal microbiota mutualism and gastrointestinal diseases. EuroMediterranean Biomed J (2015) 10):65–75. doi: 10.3269/1970-5492.2015.10.1
24. El-Kenawi A, Ruffell B. Inflammation ROS. And mutagenesis. Cancer Cell (2017) 32(6):727–9. doi: 10.1016/j.ccell.2017.11.015
25. Carini F, Mazzola M, Rappa F, Jurjus A, Geagea AG, Al Kattar S, et al. Colorectal carcinogenesis: role of oxidative stress and antioxidants. Anticancer Res (2017) 37(9):4759–66. doi: 10.21873/anticanres.11882
26. Limoli CL, Giedzinski E. Induction of chromosomal instability by chronic oxidative stress. Neoplasia (2003) 5(4):339–46. doi: 10.1016/S1476-5586(03)80027-1
27. Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, et al. Genetic alterations during colorectal-tumor development. N Engl J Med (1988) 319(9):525–32. doi: 10.1056/NEJM198809013190901
28. Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Kinzler KW. Cancer genome landscapes. Science (2013) 339(6127):1546–58. doi: 10.1126/science.1235122
29. Okugawa Y, Grady WM, Goel A. Epigenetic alterations in colorectal cancer: emerging biomarkers. Gastroenterology (2015) 149(5):1204–25.e12. doi: 10.1053/j.gastro.2015.07.011
30. Beggs AD, Jones A, El-Bahwary M, Abulafi M, Hodgson SV, Tomlinson IP. Whole-genome methylation analysis of benign and malignant colorectal tumours. J Pathol (2013) 229(5):697–704. doi: 10.1002/path.4132
31. Dong S, Liang S, Cheng Z, Zhang X, Luo L, Li L, et al. ROS/PI3K/Akt and wnt/β-catenin signalings activate HIF-1α-induced metabolic reprogramming to impart 5-fluorouracil resistance in colorectal cancer. J Exp Clin Cancer Res (2022) 41(1):15. doi: 10.1186/s13046-021-02229-6
32. Jelic MD, Mandic AD, Maricic SM, Srdjenovic BU. Oxidative stress and its role in cancer. J Cancer Res Ther (2021) 17(1):22–8. doi: 10.4103/jcrt.JCRT_862_16
33. Li C, Xue Y, Ba X, Wang R. The role of 8-oxoG repair systems in tumorigenesis and cancer therapy. Cells (2022) 11(23):3798. doi: 10.3390/cells11233798
34. Mena S, Ortega A, Estrela JM. Oxidative stress in environmental-induced carcinogenesis. Mutat Research/Genetic Toxicol Environ Mutagenesis (2009) 674(1–2):36–44. doi: 10.1016/j.mrgentox.2008.09.017
35. Berghella AM, Aureli A, Canossi A, Beato TD, Colanardi A, Pellegrini P. Redox, immune and genetic biomarker system for personalized treatments in colorectal cancer. WJGO (2019) 11(2):117–38. doi: 10.4251/wjgo.v11.i2.117
36. Akbari A, Majd HM, Rahnama R, Heshmati J, Morvaridzadeh M, Agah S, et al. Cross-talk between oxidative stress signaling and microRNA regulatory systems in carcinogenesis: focused on gastrointestinal cancers. Biomedicine Pharmacother (2020) 131:110729. doi: 10.1016/j.biopha.2020.110729
37. Zanutto S, Ciniselli CM, Belfiore A, Lecchi M, Masci E, Delconte G, et al. Plasma miRNA-based signatures in CRC screening programs. Int J Cancer (2020) 146(4):1164–73. doi: 10.1002/ijc.32573
38. Huang X, Zhu X, Yu Y, Zhu W, Jin L, Zhang X, et al. Dissecting miRNA signature in colorectal cancer progression and metastasis. Cancer Letters (2021) 501:66–82. doi: 10.1016/j.canlet.2020.12.025
39. Cheng D, Zhao S, Tang H, Zhang D, Sun H, Yu F, et al. MicroRNA-20a-5p promotes colorectal cancer invasion and metastasis by downregulating Smad4. Oncotarget (2016) 7(29):45199–213. doi: 10.18632/oncotarget.9900
40. Rokavec M, Öner MG, Li H, Jackstadt R, Jiang L, Lodygin D, et al. IL-6R/STAT3/miR-34a feedback loop promotes EMT-mediated colorectal cancer invasion and metastasis. J Clin Invest (2014) 124(4):1853–67. doi: 10.1172/JCI73531
41. Erez A, DeBerardinis RJ. Metabolic dysregulation in monogenic disorders and cancer [[/amp]]mdash; finding method in madness. Nat Rev Cancer (2015) 15(7):440–8. doi: 10.1038/nrc3949
42. Pecqueur C, Oliver L, Oizel K, Lalier L, Vallette FM. Targeting metabolism to induce cell death in cancer cells and cancer stem cells. Int J Cell Biol (2013) 2013:1–13. doi: 10.1155/2013/805975
43. Gwangwa MV, Joubert AM, Visagie MH. Crosstalk between the warburg effect, redox regulation and autophagy induction in tumourigenesis. Cell Mol Biol Lett (2018) 23(1):20. doi: 10.1186/s11658-018-0088-y
44. Cairns RA, Papandreou I, Sutphin PD, Denko NC. Metabolic targeting of hypoxia and HIF1 in solid tumors can enhance cytotoxic chemotherapy. Proc Natl Acad Sci USA (2007) 104(22):9445–50. doi: 10.1073/pnas.0611662104
45. Brown TP, Ganapathy V. Lactate/GPR81 signaling and proton motive force in cancer: role in angiogenesis, immune escape, nutrition, and warburg phenomenon. Pharmacol Ther (2020) 206:107451. doi: 10.1016/j.pharmthera.2019.107451
46. Sun S, Li H, Chen J, Qian Q. Lactic acid: no longer an inert and end-product of glycolysis. Physiology (2017) 32(6):453–63. doi: 10.1152/physiol.00016.2017
47. Végran F, Boidot R, Michiels C, Sonveaux P, Feron O. Lactate influx through the endothelial cell monocarboxylate transporter MCT1 supports an NF-κB/IL-8 pathway that drives tumor angiogenesis. Cancer Res (2011) 71(7):2550–60. doi: 10.1158/0008-5472.CAN-10-2828
48. Traverso N, Ricciarelli R, Nitti M, Marengo B, Furfaro AL, Pronzato MA, et al. Role of glutathione in cancer progression and chemoresistance. Oxid Med Cell Longevity (2013) 2013:1–10. doi: 10.1155/2013/972913
49. Tsang CK, Liu Y, Thomas J, Zhang Y, Zheng XFS. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat Commun (2014) 5(1):3446. doi: 10.1038/ncomms4446
50. Liu Y, Guo JZ, Liu Y, Wang K, Ding W, Wang H, et al. Nuclear lactate dehydrogenase a senses ROS to produce α-hydroxybutyrate for HPV-induced cervical tumor growth. Nat Commun (2018) 9(1):4429. doi: 10.1038/s41467-018-06841-7
51. Gorrini C, Harris IS, Mak TW. Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discovery (2013) 12(12):931–47. doi: 10.1038/nrd4002
52. Zhu X, Tian X, Ji L, Zhang X, Cao Y, Shen C, et al. A tumor microenvironment-specific gene expression signature predicts chemotherapy resistance in colorectal cancer patients. NPJ Precis Onc (2021) 5(1):7. doi: 10.1038/s41698-021-00142-x
53. The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature (2012) 487(7407):330–7. doi: 10.1038/nature11252
54. Molinari C, Marisi G, Passardi A, Matteucci L, De Maio G, Ulivi P. Heterogeneity in colorectal cancer: a challenge for personalized medicine? IJMS (2018) 19(12):3733. doi: 10.3390/ijms19123733
55. Whiteside TL. The tumor microenvironment and its role in promoting tumor growth. Oncogene (2008) 27(45):5904–12. doi: 10.1038/onc.2008.271
56. Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med (2005) 353(25):2654–66. doi: 10.1056/NEJMoa051424
57. Prall F. Prognostic role of CD8+ tumor-infiltrating lymphocytes in stage III colorectal cancer with and without microsatellite instability. Hum Pathol (2004) 35(7):808–16. doi: 10.1016/j.humpath.2004.01.022
58. Pagès F, Kirilovsky A, Mlecnik B, Asslaber M, Tosolini M, Bindea G, et al. In situ cytotoxic and memory T cells predict outcome in patients with early-stage colorectal cancer. JCO (2009) 27(35):5944–51. doi: 10.1200/JCO.2008.19.6147
59. Canna K, McArdle PA, McMillan DC, McNicol AM, Smith GW, McKee RF, et al. The relationship between tumour T-lymphocyte infiltration, the systemic inflammatory response and survival in patients undergoing curative resection for colorectal cancer. Br J Cancer (2005) 92(4):651–4. doi: 10.1038/sj.bjc.6602419
60. Lenz HJ, Ou FS, Venook AP, Hochster HS, Niedzwiecki D, Goldberg RM, et al. Impact of consensus molecular subtype on survival in patients with metastatic colorectal cancer: results from CALGB/SWOG 80405 (Alliance). JCO (2019) 37(22):1876–85. doi: 10.1200/JCO.18.02258
61. Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, et al. The consensus molecular subtypes of colorectal cancer. Nat Med (2015) 21(11):1350–6. doi: 10.1038/nm.3967
62. Benson AB, Schrag D, Somerfield MR, Cohen AM, Figueredo AT, Flynn PJ, et al. American Society of clinical oncology recommendations on adjuvant chemotherapy for stage II colon cancer. JCO (2004) 22(16):3408–19. doi: 10.1200/JCO.2004.05.063
63. Luo XJ, Zhao Q, Liu J, Zheng JB, Qiu MZ, Ju HQ, et al. Novel genetic and epigenetic biomarkers of prognostic and predictive significance in stage II/III colorectal cancer. Mol Ther (2021) 29(2):587–96. doi: 10.1016/j.ymthe.2020.12.017
64. Akgül Ö. Role of surgery in colorectal cancer liver metastases. WJG (2014) 20(20):6113. doi: 10.3748/wjg.v20.i20.6113
65. Onate-Ocana LF, Montesdeoca R, Lopez-Graniel CM, Aiello-Crocifoglio V, Mondragon-Sanchez R, Cortina-Borja M, et al. Identification of patients with high-risk lymph node-negative colorectal cancer and potential benefit from adjuvant chemotherapy. Japanese J Clin Oncol (2004) 34(6):323–8. doi: 10.1093/jjco/hyh054
66. Afrăsânie VA, Marinca MV, Alexa-Stratulat T, Gafton B, Păduraru M, Adavidoaiei AM, et al. KRAS, NRAS, BRAF, HER2 and microsatellite instability in metastatic colorectal cancer – practical implications for the clinician. Radiol Oncol (2019) 53(3):265–74. doi: 10.2478/raon-2019-0033
67. Ducreux M, Bouche O, Pignon JP, Mousseau M, Raoul JL, Cassan P, et al. Randomised trial comparing three different schedules of infusional 5FU and raltitrexed alone as first-line therapy in metastatic colorectal cancer. Oncology (2006) 70(3):222–30. doi: 10.1159/000094357
68. Maughan T, James R, Kerr D, Ledermann J, McArdle C, Seymour M, et al. Comparison of survival, palliation, and quality of life with three chemotherapy regimens in metastatic colorectal cancer: a multicentre randomised trial. Lancet (2002) 359(9317):1555–63. doi: 10.1016/S0140-6736(02)08514-8
69. Johdi NA, Sukor NF. Colorectal cancer immunotherapy: options and strategies. Front Immunol (2020) 11:1624. doi: 10.3389/fimmu.2020.01624
70. Lesterhuis WJ, Haanen JBAG, Punt CJA. Cancer immunotherapy – revisited. Nat Rev Drug Discovery (2011) 10(8):591–600. doi: 10.1038/nrd3500
71. Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol (2020) 20(1):7–24. doi: 10.1038/s41577-019-0210-z
72. Raskov H, Orhan A, Christensen JP, Gögenur I. Cytotoxic CD8+ T cells in cancer and cancer immunotherapy. Br J Cancer (2021) 124(2):359–67. doi: 10.1038/s41416-020-01048-4
73. Fornasier G, Francescon S, Baldo P. An update of efficacy and safety of cetuximab in metastatic colorectal cancer: a narrative review. Adv Ther (2018) 35(10):1497–509. doi: 10.1007/s12325-018-0791-0
74. Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. JCO (2010) 28(31):4697–705. doi: 10.1200/JCO.2009.27.4860
75. Kim K, Kim YW, Shim H, Kim BR, Kwon HY. Differences in clinical features and oncologic outcomes between metastatic right and left colon cancer. J BUON (2018) 23(7):11–8.
76. Jonker DJ, O’Callaghan CJ, Karapetis CS, Zalcberg JR, Tu D, Au HJ, et al. Cetuximab for the treatment of colorectal cancer. N Engl J Med (2007) 357(20):2040–8. doi: 10.1056/NEJMoa071834
77. Tabernero J, Grothey A, Van Cutsem E, Yaeger R, Wasan H, Yoshino T, et al. Encorafenib plus cetuximab as a new standard of care for previously treated BRAF V600E–mutant metastatic colorectal cancer: updated survival results and subgroup analyses from the BEACON study. JCO (2021) 39(4):273–84. doi: 10.1200/JCO.20.02088
78. Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E–mutated colorectal cancer. N Engl J Med (2019) 381(17):1632–43. doi: 10.1056/NEJMoa1908075
79. Grothey A. VEGF inhibition beyond tumour progression. Lancet Oncol (2013) 14(1):2–3. doi: 10.1016/S1470-2045(12)70516-8
80. Van Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausová J, Macarulla T, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. JCO (2012) 30(28):3499–506. doi: 10.1200/JCO.2012.42.8201
81. Ettrich TJ, Seufferlein T. Regorafenib. In: Martens UM, editor. Small molecules in oncology. Cham: Springer International Publishing (2018). p. 45–56 211. Available at: http://link.springer.com/10.1007/978-3-319-91442-8_3.
82. Arai H, Battaglin F, Wang J, Lo JH, Soni S, Zhang W, et al. Molecular insight of regorafenib treatment for colorectal cancer. Cancer Treat Rev (2019) 81:101912. doi: 10.1016/j.ctrv.2019.101912
83. Mross K, Frost A, Steinbild S, Hedbom S, Büchert M, Fasol U, et al. A phase I dose–escalation study of regorafenib (BAY 73–4506), an inhibitor of oncogenic, angiogenic, and stromal kinases, in patients with advanced solid tumors. Clin Cancer Res (2012) 18(9):2658–67. doi: 10.1158/1078-0432.CCR-11-1900
84. Akhave NS, Biter AB, Hong DS. Mechanisms of resistance to KRASG12C-targeted therapy. Cancer Discov (2021) 11(6):1345–52. doi: 10.1158/2159-8290.CD-20-1616
85. Gustavsson B, Carlsson G, Machover D, Petrelli N, Roth A, Schmoll HJ, et al. A review of the evolution of systemic chemotherapy in the management of colorectal cancer. Clin Colorectal Cancer (2015) 14(1):1–10. doi: 10.1016/j.clcc.2014.11.002
86. Diasio RB, Innocenti F, Offer SM. Pharmacogenomic-guided therapy in colorectal cancer. Clin Pharma Ther (2021) 110(3):616–25. doi: 10.1002/cpt.2334
87. Lee JJ, Beumer JH, Chu E. Therapeutic drug monitoring of 5-fluorouracil. Cancer Chemother Pharmacol (2016) 78(3):447–64. doi: 10.1007/s00280-016-3054-2
88. García-Alfonso P, Muñoz Martín AJ, Ortega Morán L, Soto Alsar J, Torres Pérez-Solero G, Blanco Codesido M, et al. Oral drugs in the treatment of metastatic colorectal cancer. Ther Adv Med Oncol (2021) 13:175883592110090. doi: 10.1177/17588359211009001
89. McQuade RM, Stojanovska V, Bornstein JC, Nurgali K. Colorectal cancer chemotherapy: the evolution of treatment and new approaches. CMC (2017) 24(15):1537–57. doi: 10.2174/0929867324666170111152436
90. Blondy S, David V, Verdier M, Mathonnet M, Perraud A, Christou N. 5-fluorouracil resistance mechanisms in colorectal cancer: from classical pathways to promising processes. Cancer Sci (2020) 111(9):3142–54. doi: 10.1111/cas.14532
91. van Kuilenburg ABP. Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5-fluorouracil. Eur J Cancer (2004) 40(7):939–50. doi: 10.1016/j.ejca.2003.12.004
92. Zhang N, Yin Y, Xu SJ, Chen WS. 5-fluorouracil: mechanisms of resistance and reversal strategies. Molecules (2008) 13(8):1551–69. doi: 10.3390/molecules13081551
93. André T, de Gramont A, Vernerey D, Chibaudel B, Bonnetain F, Tijeras-Raballand A, et al. Adjuvant fluorouracil, leucovorin, and oxaliplatin in stage II to III colon cancer: updated 10-year survival and outcomes according to BRAF mutation and mismatch repair status of the MOSAIC study. JCO (2015) 33(35):4176–87. doi: 10.1200/JCO.2015.63.4238
94. Sharma V, Gupta SK, Verma M. Dihydropyrimidine dehydrogenase in the metabolism of the anticancer drugs. Cancer Chemother Pharmacol (2019) 84(6):1157–66. doi: 10.1007/s00280-019-03936-w
95. Liu JH, Cheng YY, Hsieh CH, Tsai TH. The herb–drug pharmacokinetic interaction of 5-fluorouracil and its metabolite 5-Fluoro-5,6-Dihydrouracil with a traditional Chinese medicine in rats. IJMS (2017) 19(1):25. doi: 10.3390/ijms19010025
96. Forouzesh DC, Moran GR. Mammalian dihydropyrimidine dehydrogenase. Arch Biochem Biophysics (2021) 714:109066. doi: 10.1016/j.abb.2021.109066
97. Ibrahim B, Mady OY, Tambuwala MM, Haggag YA. pH-sensitive nanoparticles containing 5-fluorouracil and leucovorin as an improved anti-cancer option for colon cancer. Nanomedicine (2022) 17(6):367–81. doi: 10.2217/nnm-2021-0423
98. Vodenkova S, Buchler T, Cervena K, Veskrnova V, Vodicka P, Vymetalkova V. 5-fluorouracil and other fluoropyrimidines in colorectal cancer: past, present and future. Pharmacol Ther (2020) 206:107447. doi: 10.1016/j.pharmthera.2019.107447
99. Vainchtein LD, Rosing H, Schellens JHM, Beijnen JH. A new, validated HPLC-MS/MS method for the simultaneous determination of the anti-cancer agent capecitabine and its metabolites: 5′-deoxy-5-fluorocytidine, 5′-deoxy-5-fluorouridine, 5-fluorouracil and 5-fluorodihydrouracil, in human plasma. BioMed Chromatogr (2009) 24:374–86. doi: 10.1002/bmc.1302
100. Dean L, Kane M. Fluorouracil therapy and DPYD genotype. In: Pratt VM, Scott SA, Pirmohamed M, Esquivel B, Kattman BL, Malheiro AJ, editors. Medical genetics summaries. Bethesda (MD: National Center for Biotechnology Information (US (2012). Available at: http://www.ncbi.nlm.nih.gov/books/NBK395610/.
101. Kindler HL, Schilsky RL. Eniluracil: an irreversible inhibitor of dihydropyrimidine dehydrogenase. Expert Opin Investigational Drugs (2000) 9(7):1635–49. doi: 10.1517/13543784.9.7.1635
102. Longley DB, Harkin DP, Johnston PG. 5-fluorouracil: mechanisms of action and clinical strategies. Nat Rev Cancer (2003) 3(5):330–8. doi: 10.1038/nrc1074
103. Vasan N, Baselga J, Hyman DM. A view on drug resistance in cancer. Nature (2019) 575(7782):299–309. doi: 10.1038/s41586-019-1730-1
104. Kreso A, Dick JE. Evolution of the cancer stem cell model. Cell Stem Cell (2014) 14(3):275–91. doi: 10.1016/j.stem.2014.02.006
105. Peetla C, Vijayaraghavalu S, Labhasetwar V. Biophysics of cell membrane lipids in cancer drug resistance: implications for drug transport and drug delivery with nanoparticles. Advanced Drug Delivery Rev (2013) 65(13–14):1686–98. doi: 10.1016/j.addr.2013.09.004
106. Samuel P, Fabbri M, Carter DRF. Mechanisms of drug resistance in cancer: the role of extracellular vesicles. Proteomics (2017) 17(23–24):1600375. doi: 10.1002/pmic.201600375
107. Brasseur K, Gévry N, Asselin E. Chemoresistance and targeted therapies in ovarian and endometrial cancers. Oncotarget (2017) 8(3):4008–42. doi: 10.18632/oncotarget.14021
108. Lu C, Shervington A. Chemoresistance in gliomas. Mol Cell Biochem (2008) 312(1–2):71–80. doi: 10.1007/s11010-008-9722-8
109. Najafi M, Mortezaee K, Majidpoor J. Cancer stem cell (CSC) resistance drivers. Life Sci (2019) 234:116781. doi: 10.1016/j.lfs.2019.116781
110. Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell (2008) 132(4):598–611. doi: 10.1016/j.cell.2008.01.038
111. Barbato L, Bocchetti M, Di Biase A, Regad T. Cancer stem cells and targeting strategies. Cells (2019) 8(8):926. doi: 10.3390/cells8080926
112. Walcher L, Kistenmacher AK, Suo H, Kitte R, Dluczek S, Strauß A, et al. Cancer stem cells–origins and biomarkers: perspectives for targeted personalized therapies. Front Immunol (2020) 11:1280. doi: 10.3389/fimmu.2020.01280
113. Baumann M, Krause M, Hill R. Exploring the role of cancer stem cells in radioresistance. Nat Rev Cancer (2008) 8(7):545–54. doi: 10.1038/nrc2419
114. Eyler CE, Rich JN. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. JCO (2008) 26(17):2839–45. doi: 10.1200/JCO.2007.15.1829
115. Ding S, Li C, Cheng N, Cui X, Xu X, Zhou G. Redox regulation in cancer stem cells. Oxid Med Cell Longevity (2015) 2015:1–11. doi: 10.1155/2015/750798
116. Zhao J. Cancer stem cells and chemoresistance: the smartest survives the raid. Pharmacol Ther (2016) 160:145–58. doi: 10.1016/j.pharmthera.2016.02.008
117. Eun K, Ham SW, Kim H. Cancer stem cell heterogeneity: origin and new perspectives on CSC targeting. BMB Rep (2017) 50(3):117–25. doi: 10.5483/BMBRep.2017.50.3.222
118. Sulaiman A, McGarry S, Li L, Jia D, Ooi S, Addison C, et al. Dual inhibition of wnt and yes-associated protein signaling retards the growth of triple-negative breast cancer in both mesenchymal and epithelial states. Mol Oncol (2018) 12(4):423–40. doi: 10.1002/1878-0261.12167
119. Banerjee A, Birts CN, Darley M, Parker R, Mirnezami AH, West J, et al. Stem cell-like breast cancer cells with acquired resistance to metformin are sensitive to inhibitors of NADH-dependent CtBP dimerization. Carcinogenesis (2019) 40(7):871–82. doi: 10.1093/carcin/bgy174
120. Sancho P, Burgos-Ramos E, Tavera A, Bou Kheir T, Jagust P, Schoenhals M, et al. MYC/PGC-1α balance determines the metabolic phenotype and plasticity of pancreatic cancer stem cells. Cell Metab (2015) 22(4):590–605. doi: 10.1016/j.cmet.2015.08.015
121. Chen C, Bai L, Cao F, Wang S, He H, Song M, et al. Targeting LIN28B reprograms tumor glucose metabolism and acidic microenvironment to suppress cancer stemness and metastasis. Oncogene (2019) 38(23):4527–39. doi: 10.1038/s41388-019-0735-4
122. Zheng H, Liu H, Li H, Dou W, Wang J, Zhang J, et al. Characterization of stem cell landscape and identification of stemness-relevant prognostic gene signature to aid immunotherapy in colorectal cancer. Stem Cell Res Ther (2022) 13(1):244. doi: 10.1186/s13287-022-02913-0
123. Vermeulen L, Snippert HJ. Stem cell dynamics in homeostasis and cancer of the intestine. Nat Rev Cancer (2014) 14(7):468–80. doi: 10.1038/nrc3744
124. Zeuner A, Todaro M, Stassi G, De Maria R. Colorectal cancer stem cells: from the crypt to the clinic. Cell Stem Cell (2014) 15(6):692–705. doi: 10.1016/j.stem.2014.11.012
125. Wang W, Xu C, Ren Y, Wang S, Liao C, Fu X, et al. A novel cancer stemness-related signature for predicting prognosis in patients with colon adenocarcinoma. Stem Cells Int (2021) 2021:1–23. doi: 10.1155/2021/7625134
126. Sun Y, Ma X, Hu H. Application of nano-drug delivery system based on cascade technology in cancer treatment. IJMS (2021) 22(11):5698. doi: 10.3390/ijms22115698
127. Garbayo E, Pascual-Gil S, Rodríguez-Nogales C, Saludas L, Estella-Hermoso de Mendoza A, Blanco-Prieto MJ. Nanomedicine and drug delivery systems in cancer and regenerative medicine. WIREs Nanomed Nanobiotechnol (2020) 12(5):e1637. doi: 10.1002/wnan.1637
128. Wang K, Shen R, Meng T, Hu F, Yuan H. Nano-drug delivery systems based on different targeting mechanisms in the targeted therapy of colorectal cancer. Molecules (2022) 27(9):2981. doi: 10.3390/molecules27092981
129. Maeda H, Sawa T, Konno T. Mechanism of tumor-targeted delivery of macromolecular drugs, including the EPR effect in solid tumor and clinical overview of the prototype polymeric drug SMANCS. J Controlled Release (2001) 74(1–3):47–61. doi: 10.1016/S0168-3659(01)00309-1
130. Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharmaceutics Biopharmaceutics (2009) 71(3):409–19. doi: 10.1016/j.ejpb.2008.11.010
131. Wang Y, Ma J, Qiu T, Tang M, Zhang X, Dong W. In vitro and in vivo combinatorial anticancer effects of oxaliplatin- and resveratrol-loaded N,O-carboxymethyl chitosan nanoparticles against colorectal cancer. Eur J Pharm Sci (2021) 163:105864. doi: 10.1016/j.ejps.2021.105864
132. Ge P, Niu B, Wu Y, Xu W, Li M, Sun H, et al. Enhanced cancer therapy of celastrol in vitro and in vivo by smart dendrimers delivery with specificity and biosafety. Chem Eng J (2020) 383:123228. doi: 10.1016/j.cej.2019.123228
133. Soe ZC, Poudel BK, Nguyen HT, Thapa RK, Ou W, Gautam M, et al. Folate-targeted nanostructured chitosan/chondroitin sulfate complex carriers for enhanced delivery of bortezomib to colorectal cancer cells. Asian J Pharm Sci (2019) 14(1):40–51. doi: 10.1016/j.ajps.2018.09.004
134. Bhattacharya S. Anti-EGFR-mAb and 5-fluorouracil conjugated polymeric nanoparticles for colorectal cancer. PRA (2021) 16(1):84–100. doi: 10.2174/1574892815666201221121859
135. Huang J, Yue Y, Zheng C. [Vroman effect of plasma protein adsorption to biomaterials surfaces]. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi. (1999) 16(3):371–6.
136. Gunawan C, Lim M, Marquis CP, Amal R. Nanoparticle–protein corona complexes govern the biological fates and functions of nanoparticles. J Mater Chem B (2014) 2(15):2060. doi: 10.1039/c3tb21526a
137. Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Advanced Drug Delivery Rev (2016) 99:28–51. doi: 10.1016/j.addr.2015.09.012
138. Li W, Sun Y, Chen J, Jiang Z, Yang J. PEGylated cisplatin nanoparticles for treating colorectal cancer in a pH-responsive manner. Wang F curatore J Immunol Res (2022) 2022:1–11. doi: 10.1155/2022/8023915
139. Alkilany AM, Lohse SE, Murphy CJ. The gold standard: gold nanoparticle libraries to understand the nano–bio interface. Acc Chem Res (2013) 46(3):650–61. doi: 10.1021/ar300015b
140. Singh P, Pandit S, Mokkapati VRSS, Garg A, Ravikumar V, Mijakovic I. Gold nanoparticles in diagnostics and therapeutics for human cancer. IJMS (2018) 19(7):1979. doi: 10.3390/ijms19071979
141. Shah S, Dhawan V, Holm R, Nagarsenker MS, Perrie Y. Liposomes: advancements and innovation in the manufacturing process. Advanced Drug Delivery Rev (2020) 154–155:102–22. doi: 10.1016/j.addr.2020.07.002
142. Amreddy N, Babu A, Muralidharan R, Panneerselvam J, Srivastava A, Ahmed R, et al. Recent advances in nanoparticle-based cancer drug and gene delivery. In: Advances in cancer research. Elsevier (2018). p. 115–70. Available at: https://linkinghub.elsevier.com/retrieve/pii/S0065230X17300519.
143. Li M, Du C, Guo N, Teng Y, Meng X, Sun H, et al. Composition design and medical application of liposomes. Eur J Medicinal Chem (2019) 164:640–53. doi: 10.1016/j.ejmech.2019.01.007
144. Raviadaran R, Ng MH, Chandran D, Ooi KK, Manickam S. Stable W/O/W multiple nanoemulsion encapsulating natural tocotrienols and caffeic acid with cisplatin synergistically treated cancer cell lines (A549 and HEP G2) and reduced toxicity on normal cell line (HEK 293). Materials Sci Engineering: C (2021) 121:111808. doi: 10.1016/j.msec.2020.111808
145. Vernazza S, Dellacasa E, Tirendi S, Pastorino L, Bassi AM. Lipoperoxide nanoemulsion as adjuvant in cisplatin cancer therapy: In vitro study on human colon adenocarcinoma DLD-1 cells. Nanomaterials (2021) 11(6):1365. doi: 10.3390/nano11061365
Keywords: CRC, adjuvant treatments, chemoresistance, CSCs, drug delivery system
Citation: Tirendi S, Marengo B, Domenicotti C, Bassi AM, Almonti V and Vernazza S (2023) Colorectal cancer and therapy response: a focus on the main mechanisms involved. Front. Oncol. 13:1208140. doi: 10.3389/fonc.2023.1208140
Received: 18 April 2023; Accepted: 19 June 2023;
Published: 19 July 2023.
Edited by:
Che-Pei Kung, Washington University in St. Louis, United StatesCopyright © 2023 Tirendi, Marengo, Domenicotti, Bassi, Almonti and Vernazza. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Stefania Vernazza, c3RlZmFuaWEudmVybmF6emFAdW5pZ2UuaXQ=
†These authors contributed equally to this work