 Caleb L. Lines
Caleb L. Lines Morgan J. McGrath
Morgan J. McGrath Tanis Dorwart
Tanis Dorwart Crystal S. Conn
Crystal S. Conn
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol., 03 August 2023
Sec. Cancer Molecular Targets and Therapeutics
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1206561
This article is part of the Research TopicCancer Plasticity and The Microenvironment: Implications for Immunity and Therapy Response: Volume IIView all 13 articles
During their quest for growth, adaptation, and survival, cancer cells create a favorable environment through the manipulation of normal cellular mechanisms. They increase anabolic processes, including protein synthesis, to facilitate uncontrolled proliferation and deplete the tumor microenvironment of resources. As a dynamic adaptation to the self-imposed oncogenic stress, cancer cells promptly hijack translational control to alter gene expression. Rewiring the cellular proteome shifts the phenotypic balance between growth and adaptation to promote therapeutic resistance and cancer cell survival. The integrated stress response (ISR) is a key translational program activated by oncogenic stress that is utilized to fine-tune protein synthesis and adjust to environmental barriers. Here, we focus on the role of ISR signaling for driving cancer progression. We highlight mechanisms of regulation for distinct mRNA translation downstream of the ISR, expand on oncogenic signaling utilizing the ISR in response to environmental stresses, and pinpoint the impact this has for cancer cell plasticity during resistance to therapy. There is an ongoing need for innovative drug targets in cancer treatment, and modulating ISR activity may provide a unique avenue for clinical benefit.
Cancer therapeutics and diagnostics have expanded considerably over the past few decades, yet cellular adaptations persist, enabling resistance to chemotherapeutics, targeted therapies, and immunotherapies alike. This is partly due to the heterogeneity that arises within tumor populations, but limitations in widely utilized experimental approaches also hinder the identification of novel targets for the treatment of resistant tumors. The majority of clinical efforts focus on identifying therapies and biomarkers based on chromosomal alterations and RNA sequencing data which highlights global transcriptional changes. However, mRNA transcript abundance does not faithfully portray the phenotypical representation of gene expression as functional protein within a cell (1–3). This discrepancy partially emerges from the regulation of mRNA translation– the process by which mRNAs are selectively bound and deciphered to produce protein (4). Once thought of as a housekeeping process, protein synthesis is now known to be highly coordinated and subject to modulation to introduce additional layers of gene regulation that can also increase proteomic diversity (5, 6). Nearly 60% of our protein variations can be attributed to post-transcriptional processes, including RNA splicing, RNA epigenetics, and distinct utilization of 5’ and 3’ untranslated regions (UTR) along mRNA (1). Through these mechanisms and others, modulators of protein synthesis are now recognized as major contributing factors of altered gene expression and increased phenotypic diversity— including therapeutic resistance-enabling heterogeneity which arises between cancer cells of a single tumor.
The regulation of mRNA translation allows for rapid responses to pathological challenges that enable cells with dynamic survival advantages to swiftly alter their phenotype. This translational rewiring allows for cellular plasticity— the ability of cells to assume a range of diverse phenotypes in response to their environment (7). Cell state transitions are essential during development and for tissue regeneration, but cancer cells can abuse this plasticity to thrive in adverse conditions (whether in response to intrinsic oncogenic stresses or extrinsic clinical therapies) through non-transcriptional mechanisms (8). Understanding the ability of cancer cells to evade therapy through enhanced plastic tendencies for dormancy, stemness, or other advantageous states is quickly becoming a major focus for battling cancer progression. The key role in which translational regulation facilitates cancer plasticity is gaining interest as it may create a unique therapeutic opportunity (9).
One distinct translational program frequently activated in response to oncogenic events is the integrated stress response (ISR). In eukaryotes, the ISR has been implicated in a wide range of physiological events beyond cancer including metabolic reprogramming, memory formation, neurodegenerative diseases, and aspects of aging— emphasizing the pathway’s roles in various contexts (10–17). The ISR is an adaptive signaling response activated by a wide array of physiological or pathological stimuli in order to cope with stress and attempt to restore homeostasis (18). Cell intrinsic stressors can include depleted nutrients, low ATP, increased reactive oxygen species (ROS), and unfolded protein aggregates in the lumen of the endoplasmic reticulum (ER). Extrinsic triggers include hypoxia, further nutrient deprivation, DNA damaging agents, altered pH, and/or viral infection. Clinical therapies that create similar internal cell stress can also act as external triggers to induce the ISR. As such, the ISR is a highly conserved signaling cascade that is central for sensing these stressors. At the core of the ISR lies the phosphorylation of the eukaryotic initiation factor 2α (P-eIF2α), which is catalyzed independently by four unique serine/threonine kinases. In short, this single phosphorylation event downregulates global cap-dependent protein synthesis while selectively upregulating the translation of specific mRNA transcripts which function to restore cellular homeostasis. Severe stress and prolonged activation of the ISR can bypass homeostasis measures and activate separate factors to promote cell death. In this review, we discuss the mechanisms by which cancer cells hijack the ISR to rewire translation initiation in response to oncogenic stress and consider the potential for ISR-targeting therapeutics to combat cancer plasticity, progression, and treatment resistance.
When a cell is faced with antagonizing stress stimuli, an initial mitigation strategy is to conserve energy and resources by decreasing the rate of global mRNA translation. The ISR is a major regulatory network that initiates this drop in global translation through P-eIF2α while allowing for enhanced expression of select genes to promote cell adaptation (18). There are four kinases which phosphorylate eIF2α on serine 51, each activated by a specific set of cellular stresses through distinct regulatory domains (Figure 1). These domains activate the conserved kinase to promote trans-autophosphorylation. The ISR kinases include heme-regulated inhibitor (HRI), double-stranded RNA-dependent protein kinase (PKR), PKR-like ER kinase (PERK, also known as pancreatic ER kinase), and general control non-derepressible 2 (GCN2). HRI, a kinase predominantly expressed in erythroid cells, functions by adjusting the synthesis of globin to correspond with heme availability in response to oxidative stress (19, 20). It is similarly responsive to mitochondrial stress and contributes to clearing cytotoxic protein aggregates (21, 22). During viral infections, dsRNA activates PKR, downregulating the translation of virus-derived transcripts (23, 24). PKR activation has also been observed in response to ER stress, DNA damage, and uncharged mitochondria tRNAs (25). The kinase activity of PERK, an ER resident transmembrane protein, is induced through the unfolded protein response (UPR) and can be activated by oxidative stress from DNA damage, mitochondrial stress during times of nutrient deprivation, and hypoxia (26, 27). Under deprivation of charged tRNAs, GCN2 kinase activity is activated by two amino acid-sensing His-tRNA-like domains in its carboxy terminus functioning as cytoplasmic sensors of amino acid levels (28, 29). As such, a variety of nutrient deprivation conditions will activate GCN2 (30). GCN2 also binds the ribosomal complex P-stalk of the large ribosomal subunit and is activated during ribosome stalling, suggesting additional roles in monitoring ribosome stress (31, 32). In subsequent sections, we expand upon the specificity of these kinases with an emphasis on their identified roles in oncogenic signaling and how their adaptive responses can be targeted in support of cancer remission.
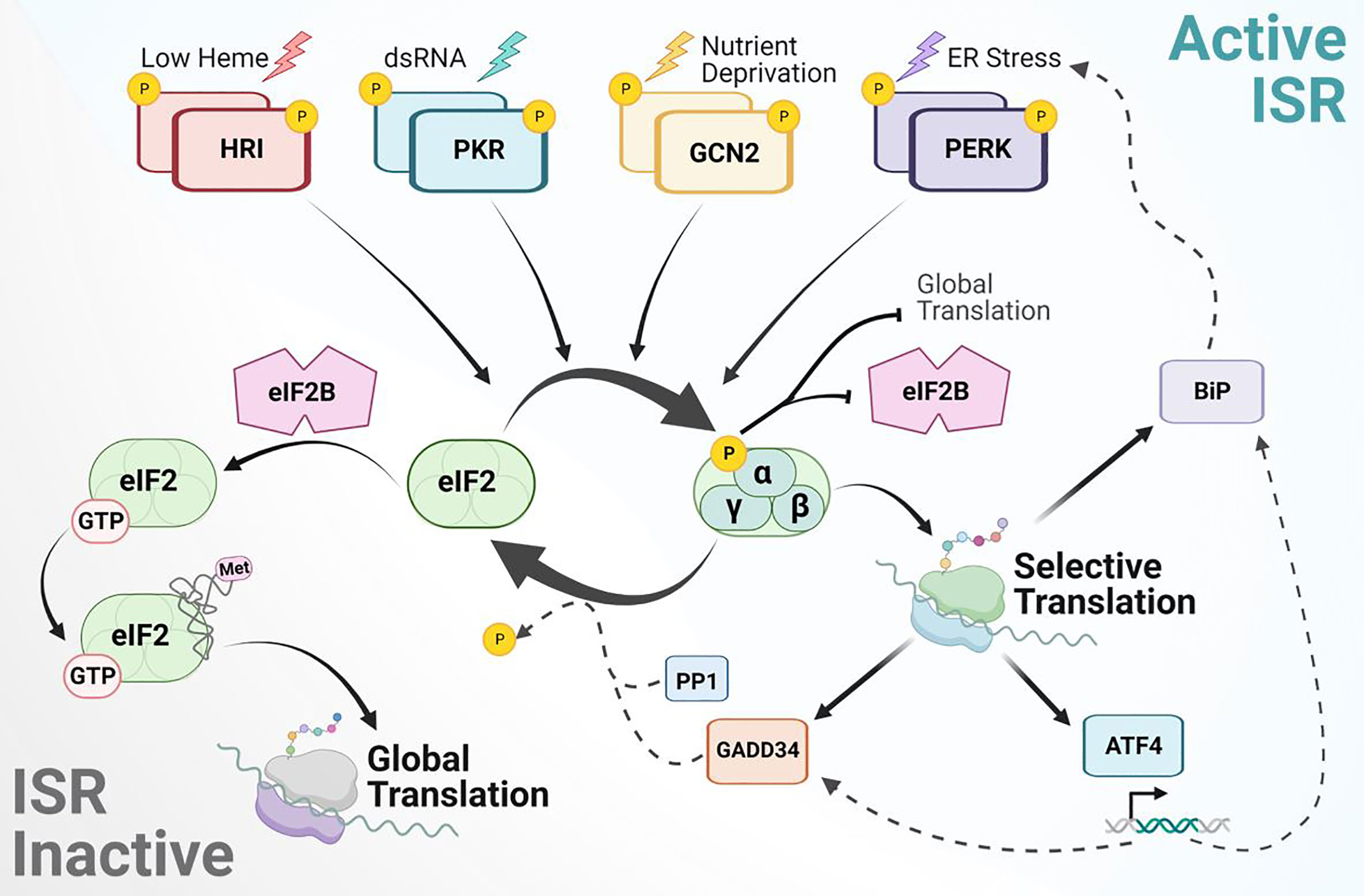
Figure 1 Mechanism of integrated stress response signaling to rewire protein synthesis. The ISR is activated by various stress stimuli recognized by four independent kinases to phosphorylate eIF2α. This blocks eIF2B GEF activity, leading to global attenuation of cap-dependent protein synthesis and activation of selective mRNA translation. Transcripts translationally upregulated during the ISR include BiP, ATF4, GADD34 and others not depicted. ATF4 is a main target that balances transcriptional regulation for adaptive response. ISR signaling is inactivated by stress-inducible phosphatases and their co-activators, such as PP1 with GADD34. Upon eIF2α dephosphorylation, eIF2B can bind eIF2 to catalyze GDP-GTP exchange and promote ternary complex formation for global protein synthesis.
Upon phosphorylation of eIF2α at serine 51, the binding affinity to eukaryotic initiation factor 2B (eIF2B) increases (33, 34). eIF2B is a heterodecamer guanine nucleotide exchange factor (GEF) which binds to the γ-subunit of eIF2 to catalyze GDP-GTP exchange on the un-phosphorylated pool of eIF2-GDP (35, 36). When eIF2B is sequestered by P-eIF2α, P-eIF2α acts as a competitive inhibitor and prevents the activation of eIF2 required for global translation initiation. Canonical translation in eukaryotes typically initiates when a charged initiator methionine-transfer RNA (Met-tRNAi) recognizes an AUG start codon following delivery to the 40S ribosomal subunit by eIF2-GTP (37). The term “ternary complex” refers to this association of Met-tRNAi, eIF2, and GTP. The ternary complex assembles with the 40S ribosomal subunit and other eIF components to form the 43S preinitiation complex (PIC) (38). Recruitment of the 43S PIC onto mRNA by the cap binding complex (eIF4F: consisting of eIF4E, eIF4G, and eIF4A) initiates global protein synthesis and scanning of the mRNA 5’untranslated region (UTR). When formation of the PIC is uninterrupted and the complementary start codon sequence is recognized with Met-tRNAi, the 60S ribosomal subunit joins and releases select eIFs to promote translation elongation in addition to recruitment of other elongation factors. However, upon activation of the ISR, sequestering of eIF2B by P-eIF2α inhibits ternary complex formation (39). It is through this mechanism that the ISR causes a reduction in global protein synthesis (Figure 1).
The interactions between major ISR factors are highly complex and context specific. For example, the kinases and phosphatases involved in the phosphorylation and dephosphorylation of eIF2α, respectively, rely on specific higher order contacts for enzymatic activity to take place. This is due to eIF2α substrate residues establishing analogous contacts with both the phosphatases and kinases (40). Additionally, as noted above, eIF2B serves as a GEF for eIF2 enabling activation of the initiation factor. When eIF2α becomes phosphorylated, an S-loop in its protein structure becomes altered, subsequently transforming the factor into a high-affinity inhibitor of eIF2B by sequestering the catalytic domain (41). A small molecule inhibitor of the ISR, ISRIB, can reverse the effect that P-eIF2α has on global translation not by altering P-eIF2α itself, but by binding to and enabling higher-order assembly of the decameric eIF2B holoenzyme. This enhances eIF2B’s stability and enzymatic activity to overcome the ISR and restore global translation (42–44). Targeting eIF2 interactions has promoted a better comprehension of the ISR function and role in altering mRNA translation between states of growth and adaptation with promise for potential therapeutics.
While the ISR is associated with downregulation of global-canonical translation, numerous transcripts are preferentially translated for cell survival in adverse conditions. A variety of cis-regulatory domains have been found to contribute to this translational selectivity, including upstream open reading frames (uORFs), internal ribosome entry sites (IRES), RNA tertiary structures, regulatory protein binding sites, and epitranscriptomic modifications (45, 46). The most notable mechanism utilized during the ISR is regulation through uORFs. An uORF is a translatable sequence with its initiation codon upstream of the main ORF (mORF). Under normal conditions, the presence of uORFs within the 5’ UTR of an mRNA transcript antagonize translation of its mORF (47, 48). This reduction occurs due to the 5’-3’ scanning involved in cap-dependent translation. Once the PIC reaches an initiation codon, the eIF2-bound GTP is hydrolyzed, and the 60S ribosomal subunit joins. This displaces the initiation factors and forms the complete ribosome, prompting translation to begin. However, induced phosphorylation of eIF2α— and the resulting reduction in the ternary complex— can overcome these inhibitory effects and allow reinitiation at the mORF. The most well-known translational target downstream of ISR signaling is the activating transcription factor 4 (ATF4), which contains two uORFs; the second uORF overlaps with the start of the protein coding sequence in the mORF (49). The first uORF promotes ribosome scanning and re-initiation of the ribosome downstream at the second uORF whose translation prevents synthesis at the mORF. Upon stress, ISR activation slows the turnover of the ternary complex allowing the 40S subunit to scan through the second uORF to reinitiate at the start codon of ATF4’s mORF (50). This translationally controlled increase in ATF4 expression via the ISR leads to downstream transcription of pro-survival genes to respond to cellular stress (30, 51, 52). Interestingly, ATF4 promotes transcription of the Growth Arrest and DNA-Damage-Inducible 34 protein (GADD34). GADD34 is the regulatory factor of eIF2α’s protein phosphatase (PP1) and is also regulated through selective translation of uORFs (53, 54). Together, GADD34:PP1 deactivate the ISR by dephosphorylating eIF2α and promote a return of global protein synthesis (Figure 1).
Notably, one of the striking ways in which the ISR rewires initiation is through the utilization of non-canonical translation start sites (55, 56). Translational tracing in T-cells showed that during ISR activation the translation of BiP, a molecular chaperone of the HSP70 family, was sustained using uORFs in the 5’UTR of BiP mRNA. Translation at the uORFs was not initiated by the canonical AUG start codon, but rather upstream at UUG or CUG initiation sites, requiring the assistance of eIF2A (45). eIF2A binds to non-canonical initiator tRNAs and delivers them to the small ribosome subunit without requiring the same GTPase activity necessary for eIF2 function. This ultimately aids in initiation during stress conditions when eIF2 is inhibited. eIF2A functions synergistically with eIF5B— the latter of which provides ribosome-binding and GTPase functions (57). The usage of uORFs for alternate translation initiation during ISR activation is also subject to regulation via mRNA modifications, most notably N6-methyladenosine (m6A) methylation. m6A is asymmetrically distributed in mammalian mRNA transcripts. Under heat shock, however, m6A is preferentially deposited within the 5’ UTR of nascent transcripts (58). This increased methylation can modulate start codon selection and promote cap-independent translation initiation of HSP70 (59). In response to GCN2 activation by nutrient deprivation, the demethylase ALKBH5 is recruited to ATF4 mRNA to erase m6A. This reduction of uORF methylation reduces translation of the uORF to enable selective translation of the ATF4 mORF (60). The methods by which the ISR can rewire translation initiation are diverse, and cancer cells ultimately use ISR signaling during tumor progression to overcome oncogenic and therapeutic stresses.
The multistep acquisition of tumorigenic traits as cells undergo neoplastic transition is highly heterogeneous due to the continuous alteration of gene expression to balance the demands of growth and adaptation for survival. This varies significantly between cancer types/subtypes and is further dependent on tissue specificity and site of transition (61). The use of single-cell RNA sequencing analysis has clearly demonstrated vast tumor heterogeneity (62). Multiple sources contribute to these heterogenetic populations. Advantageous mutations, chromosomal alterations, and epigenetic modifications may rise to prevalence within a tumor cell subset, producing a population evolved to combat the specific intrinsic and extrinsic stresses of the microenvironment. Other contributors to tumor heterogeneity, operating at post-transcriptional and translational levels, are often overlooked. Together, these reversible adaptive mechanisms constitute cell plasticity. While adaptive mechanisms may give rise to evolutionary alterations, both can confer therapeutic resistance to tumors. Here we focus on the capacity of oncogenic stress to activate adaptive pathways. In order to prevent and counter treatment resistance, one must ask: what are the mechanisms by which individual tumor cells and broader tumor subpopulations acquire resistant phenotypes?
One fundamental characteristic during cancer initiation is the deregulation of cell division, which eventually leads to uncontrolled cellular proliferation usually coupled to aberrant global protein synthesis (Figure 2) (63). Rapid oncogenic growth that occurs during tumor formation leads to a higher demand on the translational machinery (64, 65), resulting in a greater metabolic burden overall. These demands, in turn, place enhanced pressure on the proteostasis network (including ISR signaling) to balance nutrient demand for increased protein synthesis. This includes maintaining protein quality and proper peptide folding to prevent ER stress and activation of the UPR (66). Genomic instability is an enabling characteristic of cancer progression, and it synergizes with an increased metabolic rate and decreased oxygen availability. This leads to an increase in ROS due to increased oxidative and mitochondrial stress (67). These intrinsic tumorigenic pressures often result in a reconfiguration of the tumor microenvironment (TME). The rapid oncogenic growth during tumor formation also drains the local environment of nutrients and oxygen while limiting blood vessel perfusion to the core of the tumor, resulting in arid conditions (68). This challenges cancer cells to ration their resources, often perturbing metabolic usage towards aerobic glycolysis leading to decreased pH in the TME (69). Combined, these continuous stresses should disrupt the ability of the cell to regain homeostasis; however, activating adaptive pathways to promote dynamic cell state changes can promote survival. Active ISR results in lower global translation, decreased nutrient waste, enhanced pro-homeostasis factors, and improved survival. ISR signaling for adaptive mRNA translation can enable cancer cells to not only survive, but to enhance proliferation and progression within a stress-filled TME. The stress signals may vary by tumor type and location, but these adaptations are crucial for oncogenic survival.
The ISR is often exploited during oncogenesis, from cancer initiation to advanced metastasis and evasion of immunosurveillance. One seminal paper in the field identified that translational rewiring through ISR signaling, originating at pre-neoplastic stages, is a central player in skin cancer (70). Utilizing ribosome sequencing alongside an RNA interference-based screen, they observed that global mRNA translation was surprisingly lower than in normal cells due to ISR signaling for selective translation through uORFs requiring eIF2A for tumor initiation and progression. The ISR is also activated in pre-adenocarcinomas downstream of two major oncogenic lesions that promote metastatic-lethal prostate cancer (PTEN loss with MYC amplification) (71). In this context, eIF2α is phosphorylated in early neoplasia and signaling through PERK is required for tumor development. Inhibiting the ISR with ISRIB directly restores global protein synthesis, causing tumor regression and cell death in aggressive prostate cancer within genetic murine models and patient derived xenografts. The direct mechanism of activation is still not fully understood in these contexts, but PERK signaling is often observed for initiation and tumor progression due to oxidative stress, ER stress, and DNA damage (72–74). In lung adenocarcinoma, increased PERK activation likewise correlates with regions of higher proliferation, invasiveness, and tumor growth in patients. Specifically, P-eIF2α results in translational repression of the phosphatase DUSP6, promoting KRAS tumorigenesis and in turn a worse prognosis. ISRIB inhibits this translational repression and promotes tumor regression (75).
GCN2 has also been identified to promote prostate cancer by maintaining nutrient homeostasis (76). Intracellular amino acid levels are disrupted by GCN2 inhibition in metastatic prostate cancer cell lines, as GCN2 is necessary for transporter gene expression downstream of ATF4 (77). In the case of pancreatic cancer, cells relying on glycolytic pathways for energy have perpetually active ISR through GCN2-mediated ATF4 expression (78). Subsequently, ATF4 expression increases asparagine synthetase activity and the resulting asparagine is released into the TME. This asparagine can be taken up by cancer cells relying on cellular respiration and enable their proliferation even when respiration is blocked. In non-small cell lung cancer, KRAS also promotes asparagine biosynthesis in response to nutrient stress by regulating ATF4 transcription. However, ISR activity is still necessary to enhance ATF4 expression via translational regulation (79). The deletion of ATF4 alone, a single downstream ISR target, has been successful in significantly slowing MYC-driven tumor progression that relies on both GCN2 and PERK signaling in lymphomas (80).
The influence of the ISR is not solely confined to individual cancer cells. Rather, the ISR is used throughout the heterogeneous TME to enhance broader tumor resistance and persistence. In the context of melanoma and pancreatic tumors, a conditional knockout of ATF4 results in delayed tumor growth due to deficient vascularization (81). ATF4 was identified for regulating major amino acids of collagen, thus driving cancer-associated fibroblasts’ function for shaping the extracellular matrix to support tumor progression and metastasis. Independently, the TME cues ATF4 transcription of phosphoglycerate de-hydrogenase (PHGDH) in endothelial cells triggering altered metabolism towards glycolysis and aberrant over-sprouting vascularization in glioblastoma (82). This hostile vascularization presents a physical barrier to immune cells, hindering immunotherapies. ISR signaling also directly promotes non-canonical translation of programmed death ligand 1 (PD-L1) to escape immunosurveillance (83). Like ATF4, PD-L1 has an uORF inhibiting translation that is bypassed due to ISR. Utilizing an aggressive model of liver cancer, the authors showed that the non-canonical translation of PD-L1 is directly required for metastasis to the lung. The same immunosurveillance was observed in lung cancer, where a striking impairment of heme production resulted in the activation of HRI and enabled inhibitory uORFs of PD-L1 to be bypassed by the cancer cells’ translational machinery (84). Enhanced translation of PD-L1 decreases the ability of the local immune system to recognize and destroy the rapidly growing cancer. The increased presence of PD-L1 results in an overall suppression of T cell activity in the TME and prohibits T cell proliferation. The non-canonical translation necessary for the targeted translation of PD-L1 during ISR activation requires the activity of the alternative initiation factor, eIF5B. Understanding the broader role of eIF5B for ISR activity and immune regulation may present therapeutic opportunities to increase the susceptibility of immunologically ‘cold’ tumors (85).
It is understood that cells are pre-programmed to differentiate to a particular cell type (“a cell fate”). However, cells which were once differentiated possess the ability to phenotypically change their characteristics as a means of survival (86). This plasticity allows cells to evade apoptosis and obtain favorable traits to aid in their progression. During tumor development, individual oncogenic lesions or adaptations can push a population of cells into cell cycle arrest, promoting quiescence and often drug resistance (Figure 2). Activation of ISR signaling is recognized for selectively up-regulating translational expression of a p21 transcript variant that contributes to cell cycle arrest and promotes cell survival (87). Recent work discovered that distinct nutrient deprivation in liver cancer induces cell-cycle arrest and quiescence through GCN2-mediated translation of p21, leading to therapy resistance (88). The amino acid transporter SLC7A1 was specifically required for arginine import and decreased with GCN2 inhibition, indicating that SLC7A1 may be a downstream component of the ISR. Inhibiting GCN2 in this arginine-deprived environment drove a senescent phenotype, restoring the hepatocellular carcinoma cells’ vulnerability to further treatments. Likewise, PERK signaling aids in the maintenance of quiescence in stem cells and is essential for the survival of progenitor cells (89, 90). The ability of cancer cells to transition phenotypes to promote tumor plasticity and survival often relies on the GCN2 and PERK pathways.
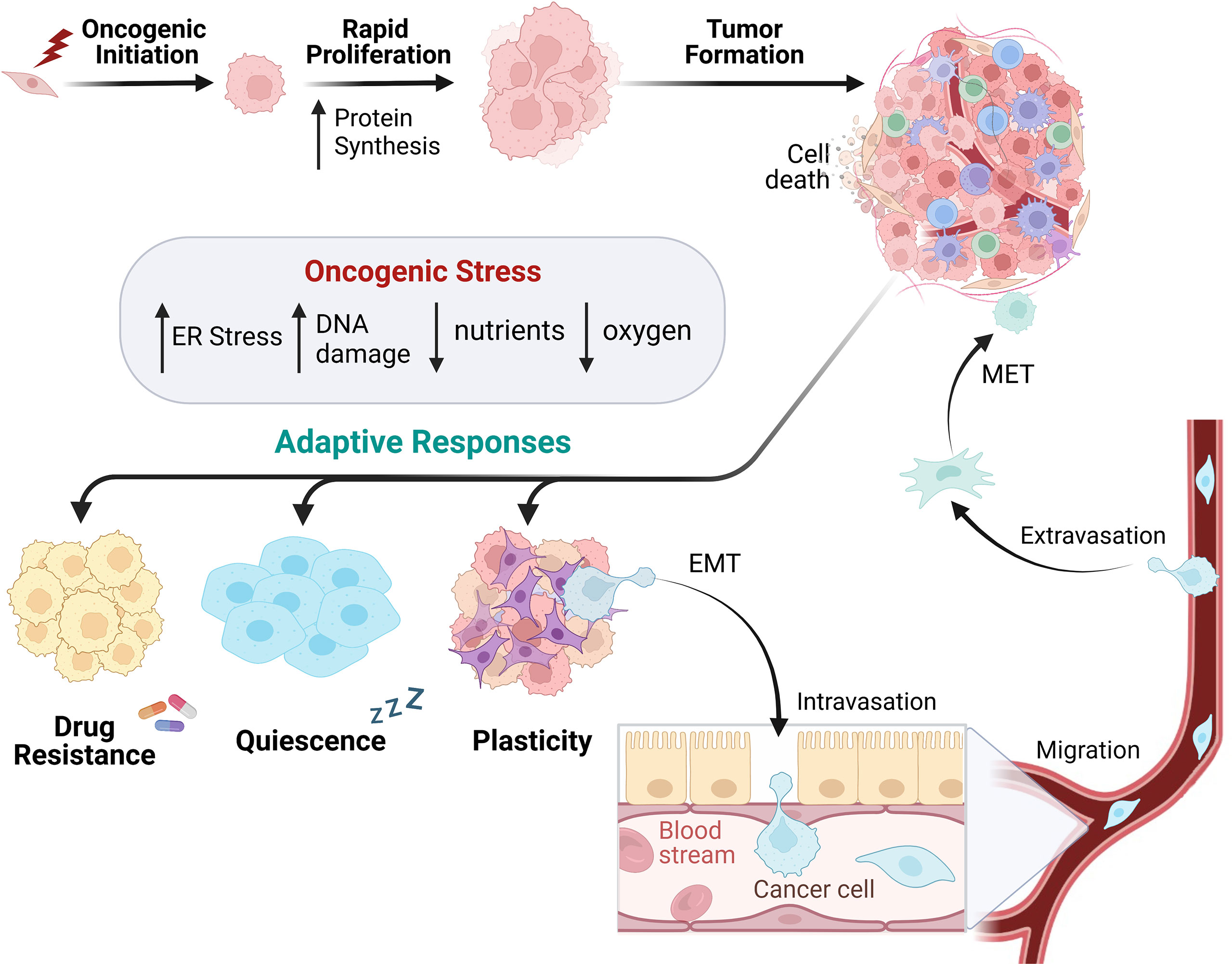
Figure 2 Cancer cell development utilizes heterogenetic adaption for tumor progression. In response to oncogenic lesions, protein synthesis is remodeled to enable rapid growth supporting tumor formation. Intracellular and extracellular oncogenic stressors subsequently alter the cancer cell phenotype, leading to the utilization of adaptive responses to promote survival. This can occur via drug resistance, cellular quiescence, and/or plasticity. Cells can promote a plastic phenotype by utilizing epithelial to mesenchymal transition (EMT) to migrate through the blood or lymphatic system and form metastases in other parts of the body.
Cancer cells are also capable of transiting from an epithelial phenotype to a mesenchymal one. This phenomenon, termed epithelial-to-mesenchymal transition (EMT), is utilized in normal development and exploited by developing cancers to invade surrounding tissues and metastasize (Figure 2). Many carcinomas will separate from neighboring cells via loss of cadherin junctions and obtain mesenchymal characteristics to metastasize to secondary locations within the body. During this transition, cells may have a combination of both epithelial and mesenchymal characteristics, existing in a “partial-EMT” state where their cellular structure is plastic. This plasticity facilitates migration and survival (91). Several EMT transcription factors are capable of silencing epithelial gene expression and promoting mesenchymal gene transcription. Examples include SNAI1, ZEB1, and TWIST1, all of which have been shown to drive this transition (92). Characterizing this process has proven difficult due to the cells’ plasticity and capability to revert to an epithelial state once they have reached their new metastatic site via MET (mesenchymal to epithelial transition). This reverse process of EMT allows cells to regain the ability to anchor within their new environment and restart their growth phase (Figure 2) (93). The EMT-MET spectrum allows cells to gain favorable traits under an array of environments, stressors, and functional demands. Upon arrival at a secondary site, cells need to regress back to an epithelial state, attach to the basement membrane, and begin the process of colonization.
The ISR can enable cancer cells to dynamically alter their cellular state depending on recognition of specific cues, whether at the cell-intrinsic or broader TME level. It has previously been stated that the UPR, another important cellular stress response that overlaps with the ISR through PERK phosphorylation of eIF2α, plays an important role in recognizing the intensity and duration of ER stress caused by an overburden of unfolded proteins. This pathway’s ability to identify stress stimuli is critical in determining cell fate, driving the cells towards either homeostatic activation of survival pathways or maladaptive activation of apoptosis (94). Similar to the UPR, the ISR has been increasingly implicated in modulating cell fate and cell state, leading to increased survival in the context of disease and providing mechanisms for resistance to cancer therapy (95). A hallmark of UPR and ISR activity, PERK signaling, is activated during EMT progression (96). PERK activation was identified for selective translation of genes which enables the cell to combat stress, transition toward a mesenchymal phenotype, and resist chemotherapy. Additionally, unique mRNA isoforms of EMT transcription factors (SNAIL, NANOG, and NODAL) were identified in breast cancer and are selectively translated under ISR activation through their 5’UTR (97). These transcriptional repressors are crucial drivers of EMT, implying that ISR signaling may be required for survival through cell plasticity.
ISR activation not only enhances the ability of tumors to successfully enter a mesenchymal state but also confers resistance to therapeutic interventions. Poorly differentiated tumors (portraying a mesenchymal phenotype) are better suited to tolerate chemotherapy, while well-differentiated tumors are more sensitive to treatment (86). Furthermore, the ISR has been directly linked to preventing chemotherapeutics from having their intended effect during cancer treatment. In pancreatic cancer, the ISR plays a critical role in providing resistance to gemcitabine— a common chemotherapy for pancreatic cancer. Gemcitabine treatment resulted in P-eIF2α, which leads to the downstream induction of ATF4. Successive inhibition of ATF4 expression in gemcitabine-treated cells enhances apoptosis (98). In BRAF-mutated melanoma, the presence of chronic ER stress leads to ISR activation by enhancing P-eIF2α expression and contributes to chemoresistance through a dysregulation of autophagy. When ER stress is inhibited using the induction of chemical chaperones, autophagic activity is reduced and apoptosis increases in the BRAF-mutated melanoma (99). Furthermore, it has been demonstrated that induction of the ISR in human gastric cancer cells provides extra chemotherapeutic protection from the apoptotic capabilities of cisplatin and requires the presence of the cystine/glutamate antiporter, xCT, for the development of cisplatin resistance (100). In tumor cells, utilization of ISR activity is able to induce plastic tendencies, ultimately preventing the effects of therapeutic treatments intended to inhibit oncogenic growth and induce cancer death. Paradoxically, the same ISR mechanisms that contribute to highly resistant tumor phenotypes can be either exploited or inhibited to provide therapeutic relief.
Ongoing therapeutic efforts aim to manipulate cancer cells’ intrinsic adaptive mechanisms to combat tumor growth (101–103). While the ISR often functions to restore homeostasis by promoting adaptations during cancer progression for survival, robust and prolonged activation of ISR can drive cell death. It has been suggested that the eIF2α kinases which initiate ISR may also have a role in determining whether the response encourages adaptive survival or cell death, with PKR primarily noted as pro-apoptotic (104, 105). Because of its capacity to both maintain and shift the balance between cell survival and cell death mechanisms, the ISR has garnered significant attention as a potential target for modulation and exploitation of cellular adaptation (17, 95). Manipulating ISR activity in combination with other therapeutics may be useful in the treatment of cancer and is becoming an attractive target for pharmacologic intervention. Several drugs that activate or inhibit the ISR are being used actively in both primary research and clinical trials for cancer treatment (Figure 3; Table 1).
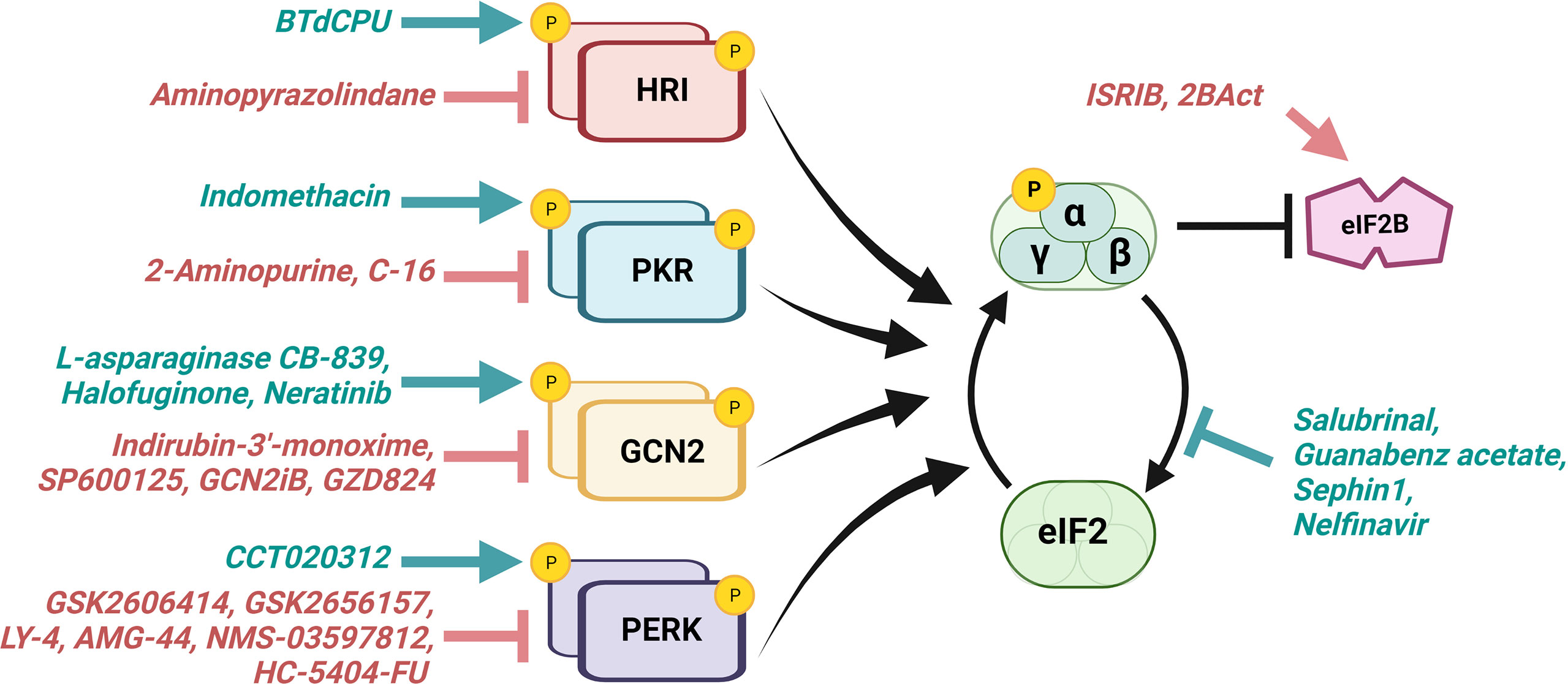
Figure 3 ISR-directed drugs target eIF2α kinases, eIF2α phosphatases, and eIF2B. Various therapeutics aim to activate (teal) or inhibit (red) the ISR for the treatment of cancer and other diseases. Those which promote the ISR aim to excessively activate a cell’s innate response mechanisms, driving that cell towards death, whereas those which inhibit the ISR aim to hinder the cell’s ability to adapt for survival under stress. eIF2α kinases serve as targets for both activators and inhibitors. Other current ISR-targeting drugs include eIF2α phosphatase inhibitors and eIF2B activators. Several of these therapeutics have shown promise in preclinical and/or clinical research described in Table 1.
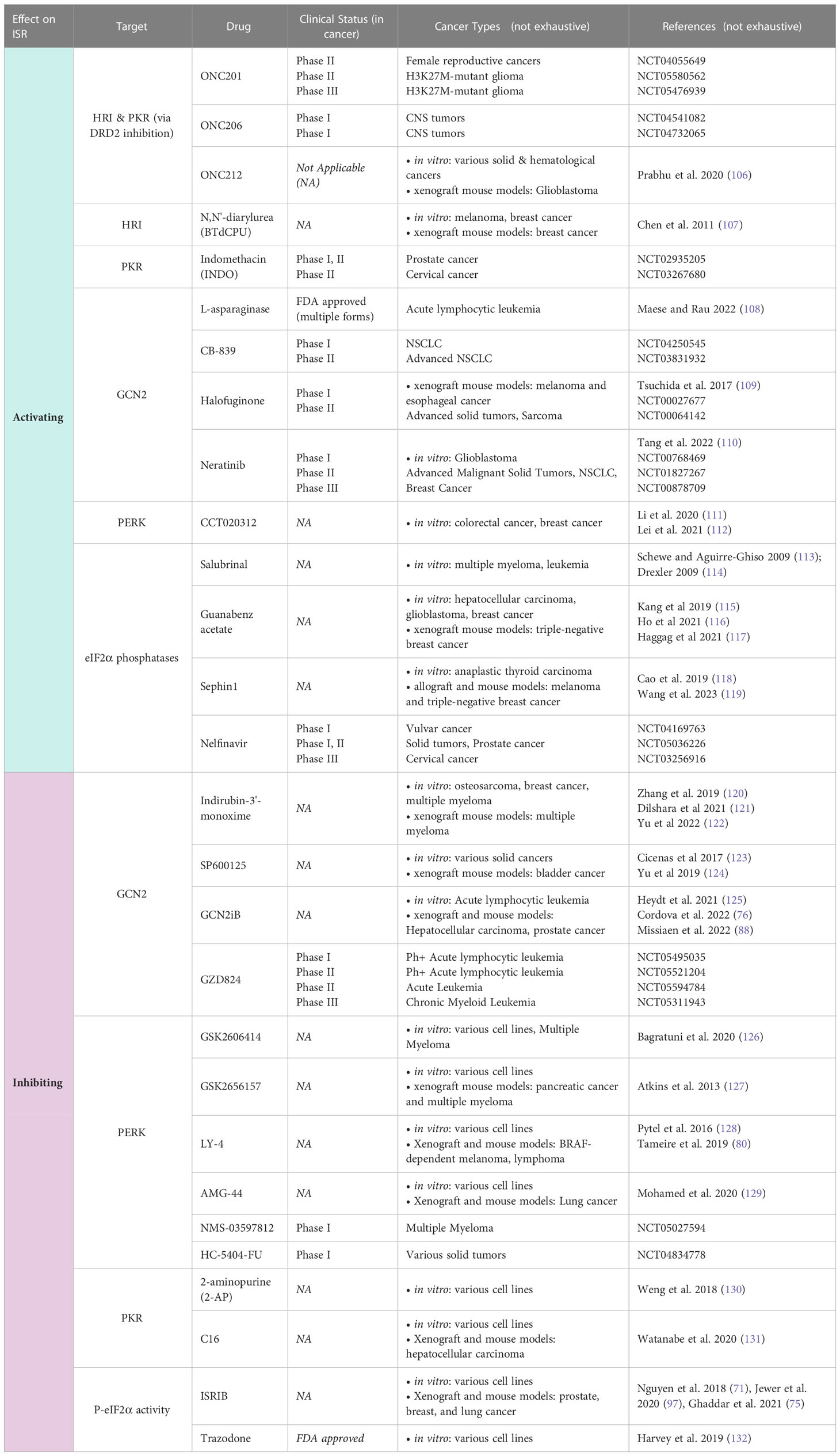
Table 1 ISR compounds in cancer treatment.
Cancer therapies which activate the ISR to provoke cell death have been in development for several decades. In 1953, lymphomas transplanted into mice regressed upon intraperitoneal injection of guinea pig serum (133). In 1963, these effects were attributed to the L-asparaginase found at high concentrations within the serum (134). The first clinical trial involving L-asparaginase was completed three years later (135). Since then, several asparaginase preparations have been FDA (Food and Drug Administration) approved for treatment of pediatric and adult hematological malignancies, particularly acute lymphoblastic leukemia (136). These early therapeutic discoveries unknowingly enhanced the ISR activity: L-asparaginase converts asparagine to aspartic acid and ammonia, which triggers activation of GCN2 to drive cancer cells towards ISR-mediated apoptosis (30). The efficacy of L-asparaginase as a highly selective therapeutic agent is rooted in leukemic cells’ deficiency in asparagine synthetase. Therefore, these cancer cells are dependent on the availability of extracellular L-asparagine, which is depleted by L-asparaginase (137, 138). Importantly, normal cells synthesize enough L-asparagine to survive treatments creating an asparaginase deficiency in the TME. Additional therapies that induce nutrient stress are being created for solid tumors, including CB-839, which acts as a glutaminase inhibitor and is currently in phase II clinical trials. Depletion of intracellular glutamine by CB-839 also induces the GCN2 arm of ISR, killing glutamine-addicted tumor cells (139). While responses to CB-839 monotherapy are limited, combinatorial treatment with immunotherapeutics has demonstrated potential. Another nutrient stress activator derived from a natural quinazolinone alkaloid, halofuginone, has been shown to promote the amino acid starvation response through GCN2 activation (140, 141). This ultimately chemosensitizes esophageal and lung carcinoma cells that have high expression of NRF2 in vitro (109). The same study found that halofuginone had similar anti-cancer effects in vivo by enhancing cisplatin-mediated tumor death in an esophageal cancer xenograft model. Similarly, drugs that have shown resistance with poor efficacy in the clinic (such as Neratinib in Glioblastoma) are now being recognized for off target effects turning on adaptations through activating the ISR via GCN2 (110). These studies highlight the presence of redundant signaling pathways that can promote cancer cell survival and provide foundations for combinational therapy strategies targeting the ISR.
A new family of selective cancer therapies known as imipridones are showing promise in the clinic. These drugs target distinct G protein-coupled receptors (GPCRs) and the mitochondrial caseinolytic protease P (ClpP) responsible for degrading misfolded proteins. When an impridone molecule binds to ClpP, it hyperactivates its function. This induces proteolysis leading to a loss of mitochondrial function, increased oxidative phosphorylation, and the triggering of the ISR and cancer cell death (142). ONC201 (also known as TIC10) is an imipridone molecule which induces HRI- and PKR-dependent phosphorylation of eIF2α and inactivation of Akt and ERK signaling (143). These effects lead to cell death by activation of Foxo3a and TRAIL for apoptotic pathways (144). Multiple phase II clinical trials have been completed and are ongoing for the use of ONC201 in treating solid and hematological cancers (145, 146), and phase III clinical trials are currently recruiting patients with H3K27M-mutant glioma and diffuse intrinsic pontine glioma (DIPG) (NCT05580562, NCT05476939). In these gliomas, the GPCR directly antagonized by ONC201, dopamine receptor D2 (DRD2), acts to promote tumor growth (147, 148). Additionally, several in vitro and in vivo studies have demonstrated the potential of DRD2 antagonists in other cancer types characterized by DRD2 upregulation including breast, prostatic, pancreatic, blood, oral, lung, gastric, and renal malignancies (149). ONC206, an antagonist for D2-like dopamine receptors, is actively in clinical trials for the treatment of primary nervous system neoplasms (such as glioblastoma) and has shown anti-tumor success in melanoma, colorectal cancer, and endometrial cancer inducing cell death by activating the ISR (150). A therapeutic relative of ONC206, ONC212, has been shown to be effective at inducing apoptosis in acute myeloid leukemia (AML) through the induction of the ISR. The resulting ISR induction also makes B-cell leukemia 2 protein (BCL-2) inhibition therapy in AML more effective by decreasing the expression of a known resistance factor for BCL-2 inhibition, myeloid cell leukemia-1 (151). A separate study found that venetoclax, a BCL-2 inhibitor, synergizes with tedizolid, an inhibitor of mitochondrial protein synthesis, to activate the ISR causing cell death through an inhibition of glycolytic activity in venetoclax-resistant AML (152).
Domperidone, another drug that works as an antagonist of dopamine receptors, and multiple tricyclic antidepressants (TCAs) have been shown to be effective at causing colorectal cancer cell death when paired with niclosamide ethanolamine (NEN), a mitochondrial uncoupler (153). The cytotoxic effects of these drug combinations are reversed with the treatment of ISRIB, indicating that they rely on ISR signaling. In the same study, the combination therapy was extremely effective at sensitizing pancreatic ductal adenocarcinoma to standard-of-care paclitaxel treatment. PKR has been shown to be activated by the non-steroidal anti-inflammatory drug, indomethacin (INDO). Upon PKR activation by INDO in colorectal cancer, the cells become susceptible to cisplatin chemotherapy treatment. Despite these findings, more work is required to identify the direct pathway of INDO mediated PKR activation (154). Furthermore, N,N’-diarylureas (BtdCPU) have demonstrated the potential to activate the ISR through the HRI kinase (155). This has been demonstrated to inhibit breast cancer growth in mice carrying human breast cancer xenografts (107). Overactivation of PERK has also been a subject of interest for creating novel therapies (156). Enhanced PERK activity by the drug CCT020312 has been shown to lead to enhanced apoptosis and taxol chemosensitivity in colorectal cancer (112, 157). In addition, CCT020312 has antitumor effects in triple-negative breast cancer through cell cycle arrest, ultimately leading to apoptosis (111).
Much like activation of eIF2α kinases, inhibition of eIF2α phosphatases can extend ISR activity and promote cancer cell death. Salubrinal is a potent cell-permeable inhibitor of eIF2α phosphatases originally identified for its ability to protect against ER stress and modulate ER stress-related cell death by targeting the conserved PP1-interacting domain of phosphatase regulatory binding partners, including GADD34 and CReP (158–160). In recent years, Salubrinal and its derivatives have risen in popularity in preclinical research, both as monotherapy and in conjunction with other treatments. An in vitro study of hepatocellular carcinoma demonstrated a potentiation of cell death when cytotoxic agent Pterostilbene was combined with Salubrinal (124). Another study demonstrated the efficacy of Salubrinal as a treatment for inflammatory breast cancer, showing that Salubrinal increased production of ROS and reduced cell proliferation (161). In ovarian cancer, it was discovered that inhibition of the therapeutic target valosin-containing protein (VCP) enhances P-eIF2α and ATF4 expression. When these VCP inhibitors are combined with Salubrinal treatment, ATF4 expression is enhanced leading to a greater increase in cancer apoptosis than with VCP inhibition alone (162). In inflammatory breast cancer (IBC), there is a markedly high expression of genes involved with the ISR such as CCAAT enhancer-binding protein homologous protein (CHOP), PERK, and ATF4. When these IBC cells are treated with Salubrinal there is a notable increase in apoptosis and expression of ATF4 and CHOP, effects that are not observed in Salubrinal-treated control cells (161). Furthermore, Salubrinal treatment was shown to be effective at causing doxorubicin-resistant MCF-7 breast cancer cells to become more susceptible to doxorubicin-induced apoptosis through the inhibition of GADD34 (163). In complex with copper (as CoSAL), Salubrinal has been shown to promote cell death and the accumulation of DNA damage in ovarian cancer in vitro (164). To combat the off-target cytotoxicity of Salubrinal, the analog SAL003 was developed to show similar efficacy to its predecessor under lower concentrations (158, 165). Trastuzumab, an important chemotherapy drug in the treatment of HER2+ cancers, has been shown to have enhanced potency when paired with the analog SAL003 in resistant HER2+ gastric and breast cancer (166).
Additional eIF2α phosphatase inhibitors have been investigated in preclinical and clinical studies. Guanabenz acetate, originally marketed as an antihypertensive, has been repurposed as an anticancer drug for its inhibition of GADD34 (117, 167). Guanabenz has been shown to induce cell death in primary hepatocellular carcinoma cells and sensitize glioblastoma cells to Sunitinib treatment (115, 116). A 2015 clinical trial exploring the anti-metastatic activity of guanabenz acetate in bone cancer patients was terminated prematurely due to poor accrual (NCT024432013), and no cancer-related clinical trials for the drug are ongoing at this time. One study that showed guanabenz acetate’s ability to inhibit the growth of anaplastic thyroid carcinoma (ATC) also employed the use of Sephin1, a small molecule inhibitor of GADD34 more commonly used in neurological studies of ISR (118). Sephin1 was also able to inhibit the growth of ATC, showcasing its potential as an anti-cancer drug. However, a more recent study demonstrated that Sephin1-mediated inhibition of GADD34 may actually have protumorigenic effect by decreasing levels of antitumor immune cells such as tumor-specific T cells and a TCR+ macrophage subtype (119). The ISR is also activated via inhibition of CReP by Nelfinavir, first identified as a human immunodeficiency virus protease inhibitor (168, 169). Recently, Nelfinavir has been shown to inhibit proliferation in patient-derived small-cell lung cancer xenograft mouse models, both as a monotherapy and in combination with autophagy inhibitor Chloroquine (170, 171). Currently, over 20 Nelfinavir clinical trials have been completed or are ongoing, with most investigating the drug as a treatment for advanced solid tumors in combination with chemoradiotherapy.
The ISR enables cells to take dynamic countermeasures when faced with extrinsic and intrinsic stressors. This plasticity is a major promoter of cell survival under adverse conditions, including cancer cell survival under genomic instability, anticancer therapy, and the stresses of the TME. Because of this role in maintaining homeostasis and promoting cell survival, the ISR may also have significant potential as a target for inhibition in the context of cancer. Specifically inhibiting the ISR may block oncogenic cellular adaptation, leading to the same outcome of cell death as seen when pushing ISR activation in the context of tumor growth.
A common method to halt the ISR is to directly inhibit the eIF2α kinases, often through an ATP-competitor. Indirubin-3’-monoxime, SP600125, and a SyK (spleen tyrosine kinase) inhibitor were all identified in a single screen as inhibitors of GCN2 kinase activity in UV-treated mouse embryonic fibroblasts (172). Since then, indirubin-3’-monoxime has been demonstrated to inhibit osteosarcoma cell proliferation and migration (120), induce paraptosis in breast cancer cells (121), and sensitize multiple myeloma cells to bortezomib-induced cell death (122). SP600125, an inhibitor of c-Jun N-terminal kinase (JNK), has shown promise in combatting oral squamous carcinoma, lung adenocarcinoma, cholangiocarcinoma, colon carcinoma, pancreatic cancer, glioblastoma, and doxorubicin-resistant stomach cancer (123). SyK is primarily expressed in hematopoietic cells and is a key component of the B-cell receptor signaling pathway crucial for B cell survival and for antigen-mediated activation, proliferation, and differentiation. Several SyK inhibitors have been evaluated in clinical trials (173). Notably, none of these GCN2 inhibitors act exclusively on GCN2.
The ATP competitive GCN2 inhibitor, GCN2iB, reduces the ISR and prevents GCN2 activation upon nutrient limitations (174). This novel inhibitor demonstrates that inhibition of GCN2 sensitizes cancer cells with low basal-level expression of asparagine synthetase to other agents such as the antileukemic agent L-asparaginase in acute lymphocytic leukemia. Preclinical models of hepatocellular carcinoma confirm that with combined dietary arginine deprivation and senotherapy, GCN2 inhibition promotes tumor regression (88). Growth inhibition was also evident using GCN2iB in cell line-derived and patient-derived xenograft models of prostate cancer (76). GZD824, a multikinase inhibitor also known as Olverembatinib, has been shown to halt the GCN2 pathway in human fibrosarcoma and non-small cell lung cancer in vitro (175). While its ability to inhibit an array of kinases may allow GZD824 considerable potential to induce off-target effects, several clinical trials are currently active or recruiting patients with hematological cancers, including one phase III clinical trial for patients with tumors that are resistant to at least two second-generation tyrosine kinase inhibitors (NCT05311943). Initially, GZD824 was identified as an inhibitor of BCR-ABL, the fusion protein with constitutively active tyrosine kinase activity, commonly associated with chronic myeloid leukemia (176). Interestingly, BCR-ABL inhibitors have been shown to prevent the activation of both GCN2 and PERK, thereby inhibiting downstream ATF4 induction in chronic myeloid leukemia (177).
Direct PERK inhibitors include GSK2606414, GSK2656157, LY-4, and AMG-44 (127, 178, 179). Unfortunately, previous research has shown off-target toxicity of PERK inhibition, particularly in pancreatic cancer. This toxicity is mediated by type 1 interferon signaling, and neutralization of this signaling has been shown to protect the healthy pancreatic tissue against PERK-inhibitors (180). Currently, a Phase I clinical trial for PERK inhibitor NMS-03597812 is recruiting patients with relapsed or refractory multiple myeloma (NCT05027594). Also recruiting, is a Phase Ia clinical trial for HC-5404-FU, another PERK inhibitor, seeking patients with renal cell carcinoma, gastric cancer, metastatic breast cancer, small cell lung cancer, and other solid tumors (except rapidly progressing neoplasms, such as pancreatic cancer) (NCT04834778).
A widely used inhibitor for PKR is 2-aminopurine (2-AP) an analog of guanosine and adenosine typically used at millimolar concentrations (181). This has been seen to affect EMT in lung cancer cells by suppressing TGF-β signaling though the influence of PKR here remains unknown and being used at such high concentrations increases likely hood of targeting several other kinases (130). An alternative PKR inhibitor is the small molecule C16, an oxindole/imidazole derivative (182). Although primarily studied in neurological contexts, C16 has shown to suppress proliferation in hepatocellular carcinoma cell lines and xenograft mouse models partially by decreasing angiogenesis (131). This raises the idea that targeting angiogenesis via PKR might be useful in other cancer subtypes, though future studies should consider its potential to inhibit immunotherapies. Similar to PKR, HRI has few known inhibitors. Aminopyrazolindane was identified in an HRI-kinase assay to act as selective inhibitor (183, 184). Sadly, in vivo the drug was cleared quickly and showed limited bioavailability for future studies. While few known inhibitors are currently in development for HRI compared to other eIF2α kinases, HRI inhibitors like aminopyrazolindane could have specific potential to combat the responses triggered by oxidative stress and dysregulation of iron homeostasis that may otherwise contribute to anemia. Anemia is remarkably common in cancer patients and while the numbers vary widely by cancer type and disease stage (as well as variation in designation of what constitutes “low” hemoglobin), studies have generally found 30-90% of cancer patients to be anemic (185). Moreover, chemotherapy is known to induce anemia. This highlights the potential for combinatorial therapies that utilize HRI inhibitors to combat oxidative stress.
As highlighted throughout, ISRIB is one of the ISR inhibitors that works downstream of all four eIF2α kinases by binding to eIF2B. This binding triggers an inhibitory allosteric change at the P-eIF2α binding site of eIF2B, thereby allowing free eIF2α to bind and promote the ternary complex to relieve ISR inhibition (186, 187). Interestingly, ISRIB functions as an effective modulator of P-eIF2α-mediated responses, but it does so without the pancreatic toxicity of other ISR inhibitors (such as PERK inhibitor GSK2606414) (188). Combinatorial treatment with ISRIB and imatinib both attenuated resistance-driving signaling pathways and more effectively eradicated chronic myeloid leukemia cells— in vitro and in vivo— than either drug alone (189). In combination with bortezomib, ISRIB has been shown to both protect bortezomib-sensitive multiple myeloma cells against apoptosis and induce paraptosis in bortezomib-insensitive breast cancer cells (190). ISRIB will likely not be available for clinical use due to poor solubility. Nevertheless, new analogs are in development for improved potency and solubility (e.g. 2BAct) (191). Direct prevention of P-eIF2α mediated reductions in ternary complex formation may also be a novel way to inhibit the ISR. Trazodone, an FDA-approved antidepressant, was shown to have similar effects as ISRIB working in this manner, yet the exact mechanism of regulation remains to be defined (132, 192). Altogether, the exponential success in basic research supports the idea that ISR-targeting therapeutics may be particularly useful in combatting an array of diseases, including highly resistant tumors (193).
The ISR is evolutionarily conserved and essential for normal mammalian development (194–196). Mutations in ISR factors have been associated with developmental malformation, as well as, cognitive, metabolic, and immune dysfunction (193, 197). The consensus is that the ISR plays a pivotal role as a molecular rheostat to fine-tune cellular adaptation mediated by translational reprogramming. As a central regulator, the ISR can be a favorable target to counter various pathologies depending on the disease context.
A wealth of research has presented the ISR as an oncogenic stress-induced translational program which enables swift cell state transitions to facilitate tumor growth, metastasis, and resistance. There are numerous mechanisms by which the ISR dynamically modulates translation— ranging from the recruitment of alternative initiation factors, selective translation of distinct transcripts, to utilization of noncanonical start codons. While the ISR operates as a means by which normal cells can adapt rapidly to non-oncogenic stressors (misfolded proteins, oxidative stress, heme-imbalance, etc.), it also exemplifies the capacity of cancer cells to hijack the pathways, networks, and fail-safes of their healthy precursors. This exploitation of typical cell functionality serves to promote neoplastic transformation. From the earliest stages of a precancerous lesion to the latest stages of metastatic disease, the ISR plays a crucial role in enabling tumors to handle the various oncogenic insults produced by their own development. Outside these intrinsic stressors, clinically available therapeutics provide extrinsic stimuli leading to the activation of the ISR. As a result, ISR-induced cell plasticity can become a driving force for therapeutic resistance with a need for pharmacological remedy.
The development of highly plastic, drug-resistant tumors by activation of the ISR has proven to be a formidable barrier to cancer therapy. On the other hand, a tumor’s overreliance on the ISR presents the translational program as a therapeutic liability. Both agonists and antagonists of the ISR have shown promise in promoting cancer cell death when combined with existing therapies. As such, there is an incentive for the development of novel ISR-modulating therapies capable of preventing ISR-mediated adaptations. Since therapeutic resistance is a major functional output of the ISR in cancer, there is more work to be done in determining the optimal partners for ISR modulators in combinatorial treatment approaches.
As outlined in this review, there are several eIF2α kinase inhibitors actively enrolled in clinical trials for a variety of cancer types. It is imperative to consider and investigate the potential unintended effects of these (often nonspecific) inhibitors to ensure that any possible off-target effects are rigorously explored to prevent toxicity, similar to what has been observed with PERK inhibition in pancreatic cancer (198). In service of the same goal, developing analogs of current therapeutics with more specific activity may increase drug efficacy while decreasing effective concentrations; this may also reduce off-target cytotoxicity. It will be useful to limit the breadth of ISR-modulating therapies by targeting specific pathways further downstream of eIF2α phosphorylation to avoid the broader impact of upstream kinase inhibition and redundancy between kinases. The potential of small molecules like ISRIB have shown promise in mouse models without off target toxicity, though the bioavailability still needs to be improved (199). Further identification and characterization of downstream interactors will aid in this endeavor. Repurposing FDA approved drugs, such as Trazodone for inhibiting ISR activity, may also be fruitful in future clinical trials.
Another area of focus will be to elucidate under what circumstances therapeutics should aim to activate or inhibit the ISR in cancer. In certain instances, extended activation of the ISR overwhelms basic cellular functions, resulting in a push towards cell death. However, activation of the ISR can enhance cancer cells’ plasticity and prevent cell death under therapeutic stress. Because of this dual nature of the ISR, a prominent dilemma is determining the specific circumstances under which upregulation of the ISR is more beneficial than downregulation (and vice versa). This will be key for future therapies to control pro-death or pro-survival mechanisms through ISR signaling. There is value in further investigating the variable role of the ISR in different cancer types and subtypes, tumor stages, and beyond cancer– in exploring ISR modulating treatments for metabolic and age-related disorders. Therapeutically fine-tuning ISR signaling is a formidable approach to overcome environmental barriers and therapy resistance to provide improved cancer targeting strategies to the clinic.
The outline was organized by CSC. All authors contributed to the article and approved the submitted version.
CLL was partially funded by the NIH grant R25-CA140116-12. MJM is a NSF Graduate Research fellow (GRFP DGE-2236662). This work was supported by funds associated with NCI P30CA016520 and Department of Defense grants HT9425-23-1-0122 and HT9425-23-1-0082 (to CSC).
We express regret to all the authors that contributed to this field of research that we failed to cite within the context of our manuscript. We thank additional members of the Conn Lab, especially Nora Kiledjian, for carefully reviewing our manuscript. We are grateful for John Davis for providing clinical perspective during editing. Figures were adapted after creation using our licensed subscription at BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Schwanhausser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, et al. Global quantification of mammalian gene expression control. Nature (2011) 473(7347):337–42. doi: 10.1038/nature10098
2. Liu Y, Beyer A, Aebersold R. On the dependency of cellular protein levels on mRNA abundance. Cell (2016) 165(3):535–50. doi: 10.1016/j.cell.2016.03.014
3. Buccitelli C, Selbach M. mRNAs, proteins and the emerging principles of gene expression control. Nat Rev Genet (2020) 21(10):630–44. doi: 10.1038/s41576-020-0258-4
4. Hershey JWB, Sonenberg N, Mathews MB. Principles of translational control. Cold Spring Harb Perspect Biol (2019) 11(9). doi: 10.1101/cshperspect.a032607
5. Chen J, Brunner AD, Cogan JZ, Nunez JK, Fields AP, Adamson B, et al. Pervasive functional translation of noncanonical human open reading frames. Science (2020) 367(6482):1140–6. doi: 10.1126/science.aay0262
6. Tharakan R, Sawa A. Minireview: Novel micropeptide discovery by proteomics and deep sequencing methods. Front Genet (2021) 12:651485. doi: 10.3389/fgene.2021.651485
7. Torborg SR, Li Z, Chan JE, Tammela T. Cellular and molecular mechanisms of plasticity in cancer. Trends Cancer (2022) 8(9):735–46. doi: 10.1016/j.trecan.2022.04.007
8. Fabbri L, Chakraborty A, Robert C, Vagner S. The plasticity of mRNA translation during cancer progression and therapy resistance. Nat Rev Cancer (2021) 21(9):558–77. doi: 10.1038/s41568-021-00380-y
9. Kovalski JR, Kuzuoglu-Ozturk D, Ruggero D. Protein synthesis control in cancer: selectivity and therapeutic targeting. EMBO J (2022) 41(8):e109823. doi: 10.15252/embj.2021109823
10. Delepine M, Nicolino M, Barrett T, Golamaully M, Lathrop GM, Julier C. EIF2AK3, encoding translation initiation factor 2-alpha kinase 3, is mutated in patients with wolcott-rallison syndrome. Nat Genet (2000) 25(4):406–9. doi: 10.1038/78085
11. Zhang W, Feng D, Li Y, Iida K, McGrath B, Cavener DR. PERK EIF2AK3 control of pancreatic beta cell differentiation and proliferation is required for postnatal glucose homeostasis. Cell Metab (2006) 4(6):491–7. doi: 10.1016/j.cmet.2006.11.002
12. Ryan DG, Yang M, Prag HA, Blanco GR, Nikitopoulou E, Segarra-Mondejar M, et al. Disruption of the TCA cycle reveals an ATF4-dependent integration of redox and amino acid metabolism. Elife (2021) 10:e72593. doi: 10.7554/eLife.72593
13. Costa-Mattioli M, Gobert D, Stern E, Gamache K, Colina R, Cuello C, et al. eIF2alpha phosphorylation bidirectionally regulates the switch from short- to long-term synaptic plasticity and memory. Cell (2007) 129(1):195–206. doi: 10.1016/j.cell.2007.01.050
14. Sharma V, Ounallah-Saad H, Chakraborty D, Hleihil M, Sood R, Barrera I, et al. Local inhibition of PERK enhances memory and reverses age-related deterioration of cognitive and neuronal properties. J Neurosci (2018) 38(3):648–58. doi: 10.1523/JNEUROSCI.0628-17.2017
15. Zhu PJ, Khatiwada S, Cui Y, Reineke LC, Dooling SW, Kim JJ, et al. Activation of the ISR mediates the behavioral and neurophysiological abnormalities in down syndrome. Science (2019) 366(6467):843–9. doi: 10.1126/science.aaw5185
16. Moon SL, Sonenberg N, Parker R. Neuronal regulation of eIF2alpha function in health and neurological disorders. Trends Mol Med (2018) 24(6):575–89. doi: 10.1016/j.molmed.2018.04.001
17. Derisbourg MJ, Hartman MD, Denzel MS. Perspective: Modulating the integrated stress response to slow aging and ameliorate age-related pathology. Nat Aging (2021) 1(9):760–8. doi: 10.1038/s43587-021-00112-9
18. Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep (2016) 17(10):1374–95. doi: 10.15252/embr.201642195
19. Chen JJ, Pal JK, Petryshyn R, Kuo I, Yang JM, Throop MS, et al. Amino acid microsequencing of internal tryptic peptides of heme-regulated eukaryotic initiation factor 2 alpha subunit kinase: homology to protein kinases. Proc Natl Acad Sci USA (1991) 88(2):315–9. doi: 10.1073/pnas.88.2.315
20. Ricketts MD, Emptage RP, Blobel GA, Marmorstein R. The heme-regulated inhibitor kinase requires dimerization for heme-sensing activity. J Biol Chem (2022) 298(10):102451. doi: 10.1016/j.jbc.2022.102451
21. Guo X, Aviles G, Liu Y, Tian R, Unger BA, Lin YT, et al. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature (2020) 579(7799):427–32. doi: 10.1038/s41586-020-2078-2
22. Mukherjee T, Ramaglia V, Abdel-Nour M, Bianchi AA, Tsalikis J, Chau HN, et al. The eIF2alpha kinase HRI triggers the autophagic clearance of cytosolic protein aggregates. J Biol Chem (2021) 296:100050. doi: 10.1074/jbc.RA120.014415
23. Kostura M, Mathews MB. Purification and activation of the double-stranded RNA-dependent eIF-2 kinase DAI. Mol Cell Biol (1989) 9(4):1576–86. doi: 10.1128/mcb.9.4.1576-1586.1989
24. Balachandran S, Roberts PC, Brown LE, Truong H, Pattnaik AK, Archer DR, et al. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity (2000) 13(1):129–41. doi: 10.1016/S1074-7613(00)00014-5
25. Garcia MA, Gil J, Ventoso I, Guerra S, Domingo E, Rivas C, et al. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev (2006) 70(4):1032–60. doi: 10.1128/MMBR.00027-06
26. Shi Y, Vattem KM, Sood R, An J, Liang J, Stramm L, et al. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol (1998) 18(12):7499–509. doi: 10.1128/MCB.18.12.7499
27. Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature (1999) 397(6716):271–4. doi: 10.1038/16729
28. Dever TE, Feng L, Wek RC, Cigan AM, Donahue TF, Hinnebusch AG. Phosphorylation of initiation factor 2 alpha by protein kinase GCN2 mediates gene-specific translational control of GCN4 in yeast. Cell (1992) 68(3):585–96. doi: 10.1016/0092-8674(92)90193-G
29. Wek SA, Zhu S, Wek RC. The histidyl-tRNA synthetase-related sequence in the eIF-2 alpha protein kinase GCN2 interacts with tRNA and is required for activation in response to starvation for different amino acids. Mol Cell Biol (1995) 15(8):4497–506. doi: 10.1128/MCB.15.8.4497
30. Ye J, Kumanova M, Hart LS, Sloane K, Zhang H, De Panis DN, et al. The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J (2010) 29(12):2082–96. doi: 10.1038/emboj.2010.81
31. Ishimura R, Nagy G, Dotu I, Chuang JH, Ackerman SL. Activation of GCN2 kinase by ribosome stalling links translation elongation with translation initiation. Elife (2016) 5:e14295. doi: 10.7554/eLife.14295
32. Harding HP, Ordonez A, Allen F, Parts L, Inglis AJ, Williams RL, et al. The ribosomal p-stalk couples amino acid starvation to GCN2 activation in mammalian cells. Elife (2019) 8:e50149. doi: 10.7554/eLife.50149
33. Goss DJ, Parkhurst LJ, Mehta HB, Woodley CL, Wahba AJ. Studies on the role of eukaryotic nucleotide exchange factor in polypeptide chain initiation. J Biol Chem (1984) 259(12):7374–7. doi: 10.1016/S0021-9258(17)42798-0
34. Kimball SR, Heinzinger NK, Horetsky RL, Jefferson LS. Identification of interprotein interactions between the subunits of eukaryotic initiation factors eIF2 and eIF2B. J Biol Chem (1998) 273(5):3039–44. doi: 10.1074/jbc.273.5.3039
35. Yang W, Hinnebusch AG. Identification of a regulatory subcomplex in the guanine nucleotide exchange factor eIF2B that mediates inhibition by phosphorylated eIF2. Mol Cell Biol (1996) 16(11):6603–16. doi: 10.1128/MCB.16.11.6603
36. Pavitt GD. Regulation of translation initiation factor eIF2B at the hub of the integrated stress response. Wiley Interdiscip Rev RNA (2018) 9(6):e1491. doi: 10.1002/wrna.1491
37. Hinnebusch AG, Ivanov IP, Sonenberg N. Translational control by 5'-untranslated regions of eukaryotic mRNAs. Science (2016) 352(6292):1413–6. doi: 10.1126/science.aad9868
38. Shin BS, Kim JR, Walker SE, Dong J, Lorsch JR, Dever TE. Initiation factor eIF2gamma promotes eIF2-GTP-Met-tRNAi(Met) ternary complex binding to the 40S ribosome. Nat Struct Mol Biol (2011) 18(11):1227–34. doi: 10.1038/nsmb.2133
39. Krishnamoorthy T, Pavitt GD, Zhang F, Dever TE, Hinnebusch AG. Tight binding of the phosphorylated alpha subunit of initiation factor 2 (eIF2alpha) to the regulatory subunits of guanine nucleotide exchange factor eIF2B is required for inhibition of translation initiation. Mol Cell Biol (2001) 21(15):5018–30. doi: 10.1128/MCB.21.15.5018-5030.2001
40. Yan Y, Harding HP, Ron D. Higher-order phosphatase-substrate contacts terminate the integrated stress response. Nat Struct Mol Biol (2021) 28(10):835–46. doi: 10.1038/s41594-021-00666-7
41. Kenner LR, Anand AA, Nguyen HC, Myasnikov AG, Klose CJ, McGeever LA, et al. eIF2B-catalyzed nucleotide exchange and phosphoregulation by the integrated stress response. Science (2019) 364(6439):491–5. doi: 10.1126/science.aaw2922
42. Sidrauski C, Acosta-Alvear D, Khoutorsky A, Vedantham P, Hearn BR, Li H, et al. Pharmacological brake-release of mRNA translation enhances cognitive memory. Elife (2013) 2:e00498. doi: 10.7554/eLife.00498
43. Tsai JC, Miller-Vedam LE, Anand AA, Jaishankar P, Nguyen HC, Renslo AR, et al. Structure of the nucleotide exchange factor eIF2B reveals mechanism of memory-enhancing molecule. Science (2018) 359(6383):eaaq0939. doi: 10.1126/science.aaq0939
44. Zyryanova AF, Weis F, Faille A, Alard AA, Crespillo-Casado A, Sekine Y, et al. Binding of ISRIB reveals a regulatory site in the nucleotide exchange factor eIF2B. Science (2018) 359(6383):1533–6. doi: 10.1126/science.aar5129
45. Starck SR, Tsai JC, Chen K, Shodiya M, Wang L, Yahiro K, et al. Translation from the 5' untranslated region shapes the integrated stress response. Science (2016) 351(6272):aad3867. doi: 10.1126/science.aad3867
46. Silva J, Fernandes R, Romao L. Translational regulation by upstream open reading frames and human diseases. Adv Exp Med Biol (2019) 1157:99–116. doi: 10.1007/978-3-030-19966-1_5
47. Calvo SE, Pagliarini DJ, Mootha VK. Upstream open reading frames cause widespread reduction of protein expression and are polymorphic among humans. Proc Natl Acad Sci USA (2009) 106(18):7507–12. doi: 10.1073/pnas.0810916106
48. Johnstone TG, Bazzini AA, Giraldez AJ. Upstream ORFs are prevalent translational repressors in vertebrates. EMBO J (2016) 35(7):706–23. doi: 10.15252/embj.201592759
49. Vattem KM, Wek RC. Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc Natl Acad Sci USA (2004) 101(31):11269–74. doi: 10.1073/pnas.0400541101
50. Dey S, Baird TD, Zhou D, Palam LR, Spandau DF, Wek RC. Both transcriptional regulation and translational control of ATF4 are central to the integrated stress response. J Biol Chem (2010) 285(43):33165–74. doi: 10.1074/jbc.M110.167213
51. Neill G, Masson GR. A stay of execution: ATF4 regulation and potential outcomes for the integrated stress response. Front Mol Neurosci (2023) 16:1112253. doi: 10.3389/fnmol.2023.1112253
52. Wortel IMN, van der Meer LT, Kilberg MS, van Leeuwen FN. Surviving stress: Modulation of ATF4-mediated stress responses in normal and malignant cells. Trends Endocrinol Metab (2017) 28(11):794–806. doi: 10.1016/j.tem.2017.07.003
53. Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol (2001) 153(5):1011–22. doi: 10.1083/jcb.153.5.1011
54. Lee YY, Cevallos RC, Jan E. An upstream open reading frame regulates translation of GADD34 during cellular stresses that induce eIF2alpha phosphorylation. J Biol Chem (2009) 284(11):6661–73. doi: 10.1074/jbc.M806735200
55. Kearse MG, Wilusz JE. Non-AUG translation: a new start for protein synthesis in eukaryotes. Genes Dev (2017) 31(17):1717–31. doi: 10.1101/gad.305250.117
56. Wright BW, Yi Z, Weissman JS, Chen J. The dark proteome: translation from noncanonical open reading frames. Trends Cell Biol (2022) 32(3):243–58. doi: 10.1016/j.tcb.2021.10.010
57. Kim E, Kim JH, Seo K, Hong KY, An SWA, Kwon J, et al. eIF2A, an initiator tRNA carrier refractory to eIF2alpha kinases, functions synergistically with eIF5B. Cell Mol Life Sci (2018) 75(23):4287–300. doi: 10.1007/s00018-018-2870-4
58. Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature (2012) 485(7397):201–6. doi: 10.1038/nature11112
59. Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB. Dynamic m(6)A mRNA methylation directs translational control of heat shock response. Nature (2015) 526(7574):591–4. doi: 10.1038/nature15377
60. Zhou J, Wan J, Shu XE, Mao Y, Liu XM, Yuan X, et al. N(6)-methyladenosine guides mRNA alternative translation during integrated stress response. Mol Cell (2018) 69(4):636–47 e7. doi: 10.1016/j.molcel.2018.01.019
61. Hanahan D. Hallmarks of cancer: New dimensions. Cancer Discovery (2022) 12(1):31–46. doi: 10.1158/2159-8290.CD-21-1059
62. Suva ML, Tirosh I. Single-cell RNA sequencing in cancer: Lessons learned and emerging challenges. Mol Cell (2019) 75(1):7–12. doi: 10.1016/j.molcel.2019.05.003
63. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell (2011) 144(5):646–74. doi: 10.1016/j.cell.2011.02.013
64. Truitt ML, Ruggero D. New frontiers in translational control of the cancer genome. Nat Rev Cancer (2016) 16(5):288–304. doi: 10.1038/nrc.2016.27
65. Pavon-Eternod M, Gomes S, Rosner MR, Pan T. Overexpression of initiator methionine tRNA leads to global reprogramming of tRNA expression and increased proliferation in human epithelial cells. RNA (2013) 19(4):461–6. doi: 10.1261/rna.037507.112
66. Chen X, Cubillos-Ruiz JR. Endoplasmic reticulum stress signals in the tumour and its microenvironment. Nat Rev Cancer (2021) 21(2):71–88. doi: 10.1038/s41568-020-00312-2
67. Perillo B, Di Donato M, Pezone A, Di Zazzo E, Giovannelli P, Galasso G, et al. ROS in cancer therapy: the bright side of the moon. Exp Mol Med (2020) 52(2):192–203. doi: 10.1038/s12276-020-0384-2
68. Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell (2009) 136(5):823–37. doi: 10.1016/j.cell.2009.02.024
69. Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: a perfect storm for cancer progression. Nat Rev Cancer (2011) 11(9):671–7. doi: 10.1038/nrc3110
70. Sendoel A, Dunn JG, Rodriguez EH, Naik S, Gomez NC, Hurwitz B, et al. Translation from unconventional 5' start sites drives tumour initiation. Nature (2017) 541(7638):494–9. doi: 10.1038/nature21036
71. Nguyen HG, Conn CS, Kye Y, Xue L, Forester CM, Cowan JE, et al. Development of a stress response therapy targeting aggressive prostate cancer. Sci Transl Med (2018) 10(439):eaar2036. doi: 10.1126/scitranslmed.aar2036
72. Blais JD, Addison CL, Edge R, Falls T, Zhao H, Wary K, et al. Perk-dependent translational regulation promotes tumor cell adaptation and angiogenesis in response to hypoxic stress. Mol Cell Biol (2006) 26(24):9517–32. doi: 10.1128/MCB.01145-06
73. Hart LS, Cunningham JT, Datta T, Dey S, Tameire F, Lehman SL, et al. ER stress-mediated autophagy promotes myc-dependent transformation and tumor growth. J Clin Invest (2012) 122(12):4621–34. doi: 10.1172/JCI62973
74. Bobrovnikova-Marjon E, Grigoriadou C, Pytel D, Zhang F, Ye J, Koumenis C, et al. PERK promotes cancer cell proliferation and tumor growth by limiting oxidative DNA damage. Oncogene (2010) 29(27):3881–95. doi: 10.1038/onc.2010.153
75. Ghaddar N, Wang S, Woodvine B, Krishnamoorthy J, van Hoef V, Darini C, et al. The integrated stress response is tumorigenic and constitutes a therapeutic liability in KRAS-driven lung cancer. Nat Commun (2021) 12(1):4651. doi: 10.1038/s41467-021-24661-0
76. Cordova RA, Misra J, Amin PH, Klunk AJ, Damayanti NP, Carlson KR, et al. GCN2 eIF2 kinase promotes prostate cancer by maintaining amino acid homeostasis. Elife (2022) 11:e81083. doi: 10.7554/eLife.81083
77. Gold LT, Masson GR. GCN2: roles in tumour development and progression. Biochem Soc Trans (2022) 50(2):737–45. doi: 10.1042/BST20211252
78. Halbrook CJ, Thurston G, Boyer S, Anaraki C, Jimenez JA, McCarthy A, et al. Differential integrated stress response and asparagine production drive symbiosis and therapy resistance of pancreatic adenocarcinoma cells. Nat Cancer (2022) 3(11):1386–403. doi: 10.1038/s43018-022-00463-1
79. Gwinn DM, Lee AG, Briones-Martin-Del-Campo M, Conn CS, Simpson DR, Scott AI, et al. Oncogenic KRAS regulates amino acid homeostasis and asparagine biosynthesis via ATF4 and alters sensitivity to l-asparaginase. Cancer Cell (2018) 33(1):91–107 e6. doi: 10.1016/j.ccell.2017.12.003
80. Tameire F, Verginadis II, Leli NM, Polte C, Conn CS, Ojha R, et al. ATF4 couples MYC-dependent translational activity to bioenergetic demands during tumour progression. Nat Cell Biol (2019) 21(7):889–99. doi: 10.1038/s41556-019-0347-9
81. Verginadis II, Avgousti H, Monslow J, Skoufos G, Chinga F, Kim K, et al. A stromal integrated stress response activates perivascular cancer-associated fibroblasts to drive angiogenesis and tumour progression. Nat Cell Biol (2022) 24(6):940–53. doi: 10.1038/s41556-022-00918-8
82. Zhang D, Li AM, Hu G, Huang M, Yang F, Zhang L, et al. PHGDH-mediated endothelial metabolism drives glioblastoma resistance to chimeric antigen receptor t cell immunotherapy. Cell Metab (2023) 35(3):517–34 e8. doi: 10.1016/j.cmet.2023.01.010
83. Xu Y, Poggio M, Jin HY, Shi Z, Forester CM, Wang Y, et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat Med (2019) 25(2):301–11. doi: 10.1038/s41591-018-0321-2
84. Suresh S, Chen B, Zhu J, Golden RJ, Lu C, Evers BM, et al. eIF5B drives integrated stress response-dependent translation of PD-L1 in lung cancer. Nat Cancer (2020) 1(5):533–45. doi: 10.1038/s43018-020-0056-0
85. Vonderheide RH. The immune revolution: A case for priming, not checkpoint. Cancer Cell (2018) 33(4):563–9. doi: 10.1016/j.ccell.2018.03.008
86. Yuan S, Norgard RJ, Stanger BZ. Cellular plasticity in cancer. Cancer Discov (2019) 9(7):837–51. doi: 10.1158/2159-8290.CD-19-0015
87. Lehman SL, Cerniglia GJ, Johannes GJ, Ye J, Ryeom S, Koumenis C. Translational upregulation of an individual p21Cip1 transcript variant by GCN2 regulates cell proliferation and survival under nutrient stress. PloS Genet (2015) 11(6):e1005212. doi: 10.1371/journal.pgen.1005212
88. Missiaen R, Anderson NM, Kim LC, Nance B, Burrows M, Skuli N, et al. GCN2 inhibition sensitizes arginine-deprived hepatocellular carcinoma cells to senolytic treatment. Cell Metab (2022) 34(8):1151–67 e7. doi: 10.1016/j.cmet.2022.06.010
89. Zismanov V, Chichkov V, Colangelo V, Jamet S, Wang S, Syme A, et al. Phosphorylation of eIF2alpha is a translational control mechanism regulating muscle stem cell quiescence and self-renewal. Cell Stem Cell (2016) 18(1):79–90. doi: 10.1016/j.stem.2015.09.020
90. Xiong G, Hindi SM, Mann AK, Gallot YS, Bohnert KR, Cavener DR, et al. The PERK arm of the unfolded protein response regulates satellite cell-mediated skeletal muscle regeneration. Elife (2017) 6:e22871. doi: 10.7554/eLife.22871.015
91. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell (2009) 139(5):871–90. doi: 10.1016/j.cell.2009.11.007
92. Fazilaty H, Rago L, Kass Youssef K, Ocana OH, Garcia-Asencio F, Arcas A, et al. A gene regulatory network to control EMT programs in development and disease. Nat Commun (2019) 10(1):5115. doi: 10.1038/s41467-019-13091-8
93. van Zijl F, Krupitza G, Mikulits W. Initial steps of metastasis: cell invasion and endothelial transmigration. Mutat Res (2011) 728(1-2):23–34. doi: 10.1016/j.mrrev.2011.05.002
94. Hetz C, Papa FR. The unfolded protein response and cell fate control. Mol Cell (2018) 69(2):169–81. doi: 10.1016/j.molcel.2017.06.017
95. Tian X, Zhang S, Zhou L, Seyhan AA, Hernandez Borrero L, Zhang Y, et al. Targeting the integrated stress response in cancer therapy. Front Pharmacol (2021) 12:747837. doi: 10.3389/fphar.2021.747837
96. Feng YX, Sokol ES, Del Vecchio CA, Sanduja S, Claessen JH, Proia TA, et al. Epithelial-to-mesenchymal transition activates PERK-eIF2alpha and sensitizes cells to endoplasmic reticulum stress. Cancer Discovery (2014) 4(6):702–15. doi: 10.1158/2159-8290.CD-13-0945
97. Jewer M, Lee L, Leibovitch M, Zhang G, Liu J, Findlay SD, et al. Translational control of breast cancer plasticity. Nat Commun (2020) 11(1):2498. doi: 10.1038/s41467-020-16352-z
98. Palam LR, Gore J, Craven KE, Wilson JL, Korc M. Integrated stress response is critical for gemcitabine resistance in pancreatic ductal adenocarcinoma. Cell Death Dis (2015) 6(10):e1913. doi: 10.1038/cddis.2015.264
99. Corazzari M, Rapino F, Ciccosanti F, Giglio P, Antonioli M, Conti B, et al. Oncogenic BRAF induces chronic ER stress condition resulting in increased basal autophagy and apoptotic resistance of cutaneous melanoma. Cell Death Differ (2015) 22(6):946–58. doi: 10.1038/cdd.2014.183
100. Wang SF, Wung CH, Chen MS, Chen CF, Yin PH, Yeh TS, et al. Activated integrated stress response induced by salubrinal promotes cisplatin resistance in human gastric cancer cells via enhanced xCT expression and glutathione biosynthesis. Int J Mol Sci (2018) 19(11):3389. doi: 10.3390/ijms19113389
101. Fulda S. Targeting apoptosis signaling pathways for anticancer therapy. Front Oncol (2011) 1:23. doi: 10.3389/fonc.2011.00023
102. Pentimalli F, Grelli S, Di Daniele N, Melino G, Amelio I. Cell death pathologies: targeting death pathways and the immune system for cancer therapy. Genes Immun (2019) 20(7):539–54. doi: 10.1038/s41435-018-0052-x
103. Carneiro BA, El-Deiry WS. Targeting apoptosis in cancer therapy. Nat Rev Clin Oncol (2020) 17(7):395–417. doi: 10.1038/s41571-020-0341-y
104. Koromilas AE. Roles of the translation initiation factor eIF2alpha serine 51 phosphorylation in cancer formation and treatment. Biochim Biophys Acta (2015) 1849(7):871–80. doi: 10.1016/j.bbagrm.2014.12.007
105. Balachandran S, Kim CN, Yeh WC, Mak TW, Bhalla K, Barber GN. Activation of the dsRNA-dependent protein kinase, PKR, induces apoptosis through FADD-mediated death signaling. EMBO J (1998) 17(23):6888–902. doi: 10.1093/emboj/17.23.6888
106. Prabhu VV, Morrow S, Rahman Kawakibi A, Zhou L, Ralff M, Ray J, et al. ONC201 and imipridones: anti-cancer compounds with clinical efficacy. Neoplasia (2020) 22(12):725–44. doi: 10.1016/j.neo.2020.09.005
107. Chen T, Ozel D, Qiao Y, Harbinski F, Chen L, Denoyelle S, et al. Chemical genetics identify eIF2alpha kinase heme-regulated inhibitor as an anticancer target. Nat Chem Biol (2011) 7(9):610–6. doi: 10.1038/nchembio.613
108. Maese L, Rau RE. Current use of asparaginase in acute lymphoblastic 871 leukemia/lymphoblastic lymphoma. Front Pediatr (2022) 10:902117. doi: 10.3389/fped.2022.902117
109. Tsuchida K, Tsujita T, Hayashi M, Ojima A, Keleku-Lukwete N, Katsuoka F, et al. Halofuginone enhances the chemo-sensitivity of cancer cells by suppressing NRF2 accumulation. Free Radic Biol Med (2017) 103:236–47. doi: 10.1016/j.freeradbiomed.2016.12.041
110. Tang CP, Clark O, Ferrarone JR, Campos C, Lalani AS, Chodera JD, et al. GCN2 kinase activation by ATP-competitive kinase inhibitors. Nat Chem Biol (2022) 18(2):207–15. doi: 10.1038/s41589-021-00947-8
111. Li X, Yu X, Zhou D, Chen B, Li W, Zheng X, et al. CCT020312 inhibits triple-negative breast cancer through PERK pathway-mediated G1 phase cell cycle arrest and apoptosis. Front Pharmacol (2020) 11:737. doi: 10.3389/fphar.2020.00737
112. Lei Y, He L, Yan C, Wang Y, Lv G. PERK activation by CCT020312 chemosensitizes colorectal cancer through inducing apoptosis regulated by ER stress. Biochem Biophys Res Commun (2021) 557:316–22. doi: 10.1016/j.bbrc.2021.03.041
113. Schewe DM, Aguirre-Ghiso JA. Inhibition of eIF2alpha dephosphorylation maximizes bortezomib efficiency and eliminates quiescent multiple myeloma cells surviving proteasome inhibitor therapy. Cancer Res (2009) 69(4):1545–52. doi: 10.1158/0008-5472.CAN-08-3858
114. Drexler HC. Synergistic apoptosis induction in leukemic cells by the phosphatase inhibitor salubrinal and proteasome inhibitors. PloS One (2009) 4(1):e4161. doi: 10.1371/journal.pone.0004161
115. Kang HJ, Seol HS, Lee SE, Suh YA, Kim J, Jang SJ, et al. Guanabenz acetate induces endoplasmic reticulum stress-related cell death in hepatocellular carcinoma cells. J Pathol Transl Med (2019) 53(2):94–103. doi: 10.4132/jptm.2019.01.14
116. Ho KH, Lee YT, Chen PH, Shih CM, Cheng CH, Chen KC. Guanabenz sensitizes glioblastoma cells to sunitinib by inhibiting GADD34-mediated autophagic signaling. Neurotherapeutics (2021) 18(2):1371–92. doi: 10.1007/s13311-020-00961-z
117. Haggag YA, Yasser M, Tambuwala MM, El Tokhy SS, Isreb M, Donia AA. Repurposing of guanabenz acetate by encapsulation into long-circulating nanopolymersomes for treatment of triple-negative breast cancer. Int J Pharm (2021) 600:120532. doi: 10.1016/j.ijpharm.2021.120532
118. Cao X, Dang L, Zheng X, Lu Y, Lu Y, Ji R, et al. Targeting super-Enhancer-Driven oncogenic transcription by CDK7 inhibition in anaplastic thyroid carcinoma. Thyroid (2019) 29(6):809–23. doi: 10.1089/thy.2018.0550
119. Wang R, Zhang Y, Guo S, Pei S, Guo W, Wu Z, et al. Single-cell RNA sequencing reveals the suppressive effect of PPP1R15A inhibitor Sephin1 in antitumor immunity. iScience (2023) 26(2):105954. doi: 10.1016/j.isci.2023.105954
120. Zhang Y, Song L, Li J, Zhang Y, Lu X, Zhang B. Inhibitory effects of indirubin-3'-monoxime against human osteosarcoma. IUBMB Life (2019) 71(10):1465–74. doi: 10.1002/iub.2058
121. Dilshara MG, Neelaka Molagoda IM, Prasad Tharanga Jayasooriya RG, Choi YH, Park C, Kim GY. Prasad tharanga jayasooriya RG Indirubin-3'-monoxime induces paraptosis in MDA-MB-231 breast cancer cells by transmitting Ca2+ from endoplasmic reticulum to mitochondria. Arch Biochem Biophys (2021) 698:108723. doi: 10.1016/j.abb.2020.108723
122. Yu Z, Wei X, Liu L, Sun H, Fang T, Wang L, et al. Indirubin-3'-monoxime acts as proteasome inhibitor: Therapeutic application in multiple myeloma. EBioMedicine (2022) 78:103950. doi: 10.1016/j.ebiom.2022.103950
123. Cicenas J, Zalyte E, Rimkus A, Dapkus D, Noreika R, Urbonavicius SJNK. p38, ERK, and SGK1 inhibitors in cancer. Cancers (Basel) (2017) 10(1):1. doi: 10.3390/cancers10010001
124. Yu H, Wu CL, Wang X, Ban Q, Quan C, Liu M, et al. SP600125 enhances C-2-induced cell death by the switch from autophagy to apoptosis in bladder cancer cells. J Exp Clin Cancer Res. (2019) 38(1):448. doi: 10.1186/s13046-019-1467-6
125. Heydt Q, Xintaropoulou C, Clear A, Austin M, Pislariu I, Miraki-Moud F, et al. Adipocytes disrupt the translational programme of acute lymphoblastic leukaemia to favour tumour survival and persistence. Nat Commun (2021) 12(1):5507. doi: 10.1038/s41467-021-25540-4
126. Bagratuni T, Patseas D, Mavrianou-Koutsoukou N, Liacos CI, Sklirou AD, Rousakis P, et al. Characterization of a PERK kinase inhibitor with anti-myeloma activity. Cancers (Basel) (2020) 12(10):2864. doi: 10.3390/cancers12102864
127. Atkins C, Liu Q, Minthorn E, Zhang SY, Figueroa DJ, Moss K, et al. Characterization of a novel PERK kinase inhibitor with antitumor and antiangiogenic activity. Cancer Res (2013) 73(6):1993–2002. doi: 10.1158/0008-5472.CAN-12-3109
128. Pytel D, Gao Y, Mackiewicz K, Katlinskaya YV, Staschke KA, Paredes MC, et al. PERK is a haploinsufficient tumor suppressor: gene dose determines tumor-suppressive versus tumor promoting properties of PERK in melanoma. PloS Genet (2016) 12(12):e1006518. doi: 10.1371/journal.pgen.1006518
129. Mohamed E, Sierra RA, Trillo-Tinoco J, Cao Y, Innamarato P, Payne KK, et al. The unfolded protein response mediator PERK governs myeloid cell-driven immunosuppression in tumors through inhibition of STING signaling. Immunity (2020) 52(4):668–82.e7. doi: 10.1016/j.immuni.2020.03.004
130. Weng D, Chen JX, Li HH, Liu F, Zhou LD, Liu HP, et al. 2-aminopurine suppresses the TGF-beta1-induced epithelial-mesenchymal transition and attenuates bleomycin-induced pulmonary fibrosis. Cell Death Discov (2018) 4:17. doi: 10.1038/s41420-017-0016-3
131. Watanabe T, Ninomiya H, Saitou T, Takanezawa S, Yamamoto S, Imai Y, et al. Therapeutic effects of the PKR inhibitor C16 suppressing tumor proliferation and angiogenesis in hepatocellular carcinoma in vitro and in vivo. Sci Rep (2020) 10(1):5133. doi: 10.1038/s41598-020-61579-x
132. Harvey RF, Poyry TAA, Stoneley M, Willis AE. Signaling from mTOR to eIF2alpha mediates cell migration in response to the chemotherapeutic doxorubicin. Sci Signal (2019) 12(612):eaaw6763. doi: 10.1126/scisignal.aaw6763
133. Kidd JG. Regression of transplanted lymphomas induced in vivo by means of normal guinea pig serum. II. studies on the nature of the active serum constituent: histological mechanism of the regression: tests for effects of guinea pig serum on lymphoma cells in vitro: discussion. J Exp Med (1953) 98(6):583–606. doi: 10.1084/jem.98.6.583
134. Broome JD. Evidence that the l-asparaginase of guinea pig serum is responsible for its antilymphoma effects. II. lymphoma 6C3HED cells cultured in a medium devoid of l-asparagine lose their susceptibility to the effects of guinea pig serum in vivo. J Exp Med (1963) 118(1):121–48. doi: 10.1084/jem.118.1.99
135. Dolowy WC, Henson D, Cornet J, Sellin H. Toxic and antineoplastic effects of l-asparaginase. study of mice with lymphoma and normal monkeys and report on a child with leukemia. Cancer (1966) 19(12):1813–9. doi: 10.1002/1097-0142(196612)19:12<1813::aid-cncr2820191208>3.0.co;2-e
136. Muller HJ, Boos J. Use of l-asparaginase in childhood ALL. Crit Rev Oncol Hematol (1998) 28(2):97–113. doi: 10.1016/S1040-8428(98)00015-8
137. Prager MD, Bachynsky N. Asparagine synthetase in normal and malignant tissues: correlation with tumor sensitivity to asparaginase. Arch Biochem Biophys (1968) 127(1):645–54. doi: 10.1016/0003-9861(68)90273-7
138. Lomelino CL, Andring JT, McKenna R, Kilberg MS. Asparagine synthetase: Function, structure, and role in disease. J Biol Chem (2017) 292(49):19952–8. doi: 10.1074/jbc.R117.819060
139. Yerbes R, Mora-Molina R, Fernandez-Farran FJ, Hiraldo L, Lopez-Rivas A, Palacios C. Limiting glutamine utilization activates a GCN2/TRAIL-R2/Caspase-8 apoptotic pathway in glutamine-addicted tumor cells. Cell Death Dis (2022) 13(10):906. doi: 10.1038/s41419-022-05346-y
140. Juarez P, Mohammad KS, Yin JJ, Fournier PG, McKenna RC, Davis HW, et al. Halofuginone inhibits the establishment and progression of melanoma bone metastases. Cancer Res (2012) 72(23):6247–56. doi: 10.1158/0008-5472.CAN-12-1444
141. Keller TL, Zocco D, Sundrud MS, Hendrick M, Edenius M, Yum J, et al. Halofuginone and other febrifugine derivatives inhibit prolyl-tRNA synthetase. Nat Chem Biol (2012) 8(3):311–7. doi: 10.1038/nchembio.790
142. Ishizawa J, Zarabi SF, Davis RE, Halgas O, Nii T, Jitkova Y, et al. Mitochondrial ClpP-mediated proteolysis induces selective cancer cell lethality. Cancer Cell (2019) 35(5):721–37 e9. doi: 10.1016/j.ccell.2019.03.014
143. Kline CL, Van den Heuvel AP, Allen JE, Prabhu VV, Dicker DT, El-Deiry WS. ONC201 kills solid tumor cells by triggering an integrated stress response dependent on ATF4 activation by specific eIF2alpha kinases. Sci Signal (2016) 9(415):ra18. doi: 10.1126/scisignal.aac4374
144. Allen JE, Krigsfeld G, Patel L, Mayes PA, Dicker DT, Wu GS, et al. Identification of TRAIL-inducing compounds highlights small molecule ONC201/TIC10 as a unique anti-cancer agent that activates the TRAIL pathway. Mol Cancer (2015) 14:99. doi: 10.1186/s12943-015-0346-9
145. Stein MN, Bertino JR, Kaufman HL, Mayer T, Moss R, Silk A, et al. First-in-Human clinical trial of oral ONC201 in patients with refractory solid tumors. Clin Cancer Res (2017) 23(15):4163–9. doi: 10.1158/1078-0432.CCR-16-2658
146. Anderson PM, Trucco MM, Tarapore RS, Zahler S, Thomas S, Gortz J, et al. Phase II study of ONC201 in neuroendocrine tumors including pheochromocytoma-paraganglioma and desmoplastic small round cell tumor. Clin Cancer Res (2022) 28(9):1773–82. doi: 10.1158/1078-0432.CCR-21-4030
147. Caragher SP, Hall RR, Ahsan R, Ahmed AU. Monoamines in glioblastoma: complex biology with therapeutic potential. Neuro Oncol (2018) 20(8):1014–25. doi: 10.1093/neuonc/nox210
148. Free RB, Cuoco CA, Xie B, Namkung Y, Prabhu VV, Willette BKA, et al. Pharmacological characterization of the imipridone anticancer drug ONC201 reveals a negative allosteric mechanism of action at the D(2) dopamine receptor. Mol Pharmacol (2021) 100(4):372–87. doi: 10.1124/molpharm.121.000336
149. Weissenrieder JS, Neighbors JD, Mailman RB, Hohl RJ. Cancer and the dopamine D(2) receptor: A pharmacological perspective. J Pharmacol Exp Ther (2019) 370(1):111–26. doi: 10.1124/jpet.119.256818
150. Zhang Y, Huang Y, Yin Y, Fan Y, Sun W, Zhao X, et al. ONC206, an imipridone derivative, induces cell death through activation of the integrated stress response in serous endometrial cancer in vitro. Front Oncol (2020) 10:577141. doi: 10.3389/fonc.2020.577141
151. Nii T, Prabhu VV, Ruvolo V, Madhukar N, Zhao R, Mu H, et al. Imipridone ONC212 activates orphan g protein-coupled receptor GPR132 and integrated stress response in acute myeloid leukemia. Leukemia (2019) 33(12):2805–16. doi: 10.1038/s41375-019-0491-z
152. Sharon D, Cathelin S, Mirali S, Di Trani JM, Yanofsky DJ, Keon KA, et al. Inhibition of mitochondrial translation overcomes venetoclax resistance in AML through activation of the integrated stress response. Sci Transl Med (2019) 11(516):eaax2863. doi: 10.1126/scitranslmed.aax2863
153. Hartleben G, Schorpp K, Kwon Y, Betz B, Tsokanos FF, Dantes Z, et al. Combination therapies induce cancer cell death through the integrated stress response and disturbed pyrimidine metabolism. EMBO Mol Med (2021) 13(4):e12461. doi: 10.15252/emmm.202012461
154. Brunelli C, Amici C, Angelini M, Fracassi C, Belardo G, Santoro MG. The non-steroidal anti-inflammatory drug indomethacin activates the eIF2alpha kinase PKR, causing a translational block in human colorectal cancer cells. Biochem J (2012) 443(2):379–86. doi: 10.1042/BJ20111236
155. Denoyelle S, Chen T, Chen L, Wang Y, Klosi E, Halperin JA, et al. In vitro inhibition of translation initiation by N,N'-diarylureas–potential anti-cancer agents. Bioorg Med Chem Lett (2012) 22(1):402–9. doi: 10.1016/j.bmcl.2011.10.126
156. Grandjean JMD, Wiseman RL. Small molecule strategies to harness the unfolded protein response: where do we go from here? J Biol Chem (2020) 295(46):15692–711. doi: 10.1074/jbc.REV120.010218
157. Stockwell SR, Platt G, Barrie SE, Zoumpoulidou G, Te Poele RH, Aherne GW, et al. Mechanism-based screen for G1/S checkpoint activators identifies a selective activator of EIF2AK3/PERK signalling. PloS One (2012) 7(1):e28568. doi: 10.1371/journal.pone.0028568
158. Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science (2005) 307(5711):935–9. doi: 10.1126/science.1101902
159. Bryant KF, Macari ER, Malik N, Boyce M, Yuan J, Coen DM. ICP34.5-dependent and -independent activities of salubrinal in herpes simplex virus-1 infected cells. Virology (2008) 379(2):197–204. doi: 10.1016/j.virol.2008.06.028
160. Rojas M, Vasconcelos G, Dever TE. An eIF2alpha-binding motif in protein phosphatase 1 subunit GADD34 and its viral orthologs is required to promote dephosphorylation of eIF2alpha. Proc Natl Acad Sci USA (2015) 112(27):E3466–75. doi: 10.1073/pnas.1501557112
161. Alsterda A, Asha K, Powrozek O, Repak M, Goswami S, Dunn AM, et al. Salubrinal exposes anticancer properties in inflammatory breast cancer cells by manipulating the endoplasmic reticulum stress pathway. Front Oncol (2021) 11:654940. doi: 10.3389/fonc.2021.654940
162. Bastola P, Neums L, Schoenen FJ, Chien J. VCP inhibitors induce endoplasmic reticulum stress, cause cell cycle arrest, trigger caspase-mediated cell death and synergistically kill ovarian cancer cells in combination with salubrinal. Mol Oncol (2016) 10(10):1559–74. doi: 10.1016/j.molonc.2016.09.005
163. Jeon YJ, Kim JH, Shin JI, Jeong M, Cho J, Lee K. Salubrinal-mediated upregulation of eIF2alpha phosphorylation increases doxorubicin sensitivity in MCF-7/ADR cells. Mol Cells (2016) 39(2):129–35. doi: 10.14348/molcells.2016.2243
164. Masuri S, Cadoni E, Cabiddu MG, Isaia F, Demuru MG, Moran L, et al. The first copper(ii) complex with 1,10-phenanthroline and salubrinal with interesting biochemical properties. Metallomics (2020) 12(6):891–901. doi: 10.1039/d0mt00006j
165. Robert F, Kapp LD, Khan SN, Acker MG, Kolitz S, Kazemi S, et al. Initiation of protein synthesis by hepatitis c virus is refractory to reduced eIF2.GTP.Met-tRNA(i)(Met) ternary complex availability. Mol Biol Cell (2006) 17(11):4632–44. doi: 10.1091/mbc.e06-06-0478
166. Darini C, Ghaddar N, Chabot C, Assaker G, Sabri S, Wang S, et al. An integrated stress response via PKR suppresses HER2+ cancers and improves trastuzumab therapy. Nat Commun (2019) 10(1):2139. doi: 10.1038/s41467-019-10138-8
167. Tsaytler P, Harding HP, Ron D, Bertolotti A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science (2011) 332(6025):91–4. doi: 10.1126/science.1201396
168. Pai VB, Nahata MC. Nelfinavir mesylate: a protease inhibitor. Ann Pharmacother (1999) 33(3):325–39. doi: 10.1345/aph.18089
169. De Gassart A, Bujisic B, Zaffalon L, Decosterd LA, Di Micco A, Frera G, et al. An inhibitor of HIV-1 protease modulates constitutive eIF2alpha dephosphorylation to trigger a specific integrated stress response. Proc Natl Acad Sci USA (2016) 113(2):E117–26. doi: 10.1073/pnas.1514076113
170. Kawabata S, Connis N, Gills JJ, Hann CL, Dennis PA. Nelfinavir inhibits the growth of small-cell lung cancer cells and patient-derived xenograft tumors. Anticancer Res (2021) 41(1):91–9. doi: 10.21873/anticanres.14754
171. Lopiccolo J, Kawabata S, Gills JJ, Dennis PA. Combining nelfinavir with chloroquine inhibits In vivo growth of human lung cancer xenograft tumors. In Vivo (2021) 35(1):141–5. doi: 10.21873/invivo.12241
172. Robert F, Williams C, Yan Y, Donohue E, Cencic R, Burley SK, et al. Blocking UV-induced eIF2alpha phosphorylation with small molecule inhibitors of GCN2. Chem Biol Drug Des (2009) 74(1):57–67. doi: 10.1111/j.1747-0285.2009.00827.x
173. Liu D, Mamorska-Dyga A. Syk inhibitors in clinical development for hematological malignancies. J Hematol Oncol (2017) 10(1):145. doi: 10.1186/s13045-016-0386-7
174. Nakamura A, Nambu T, Ebara S, Hasegawa Y, Toyoshima K, Tsuchiya Y, et al. Inhibition of GCN2 sensitizes ASNS-low cancer cells to asparaginase by disrupting the amino acid response. Proc Natl Acad Sci USA (2018) 115(33):E7776–E85. doi: 10.1073/pnas.1805523115
175. Kato Y, Kunimasa K, Takahashi M, Harada A, Nagasawa I, Osawa M, et al. GZD824 inhibits GCN2 and sensitizes cancer cells to amino acid starvation stress. Mol Pharmacol (2020) 98(6):669–76. doi: 10.1124/molpharm.120.000070
176. Ren X, Pan X, Zhang Z, Wang D, Lu X, Li Y, et al. Identification of GZD824 as an orally bioavailable inhibitor that targets phosphorylated and nonphosphorylated breakpoint cluster region-abelson (Bcr-abl) kinase and overcomes clinically acquired mutation-induced resistance against imatinib. J Med Chem (2013) 56(3):879–94. doi: 10.1021/jm301581y
177. Kato Y, Kunimasa K, Sugimoto Y, Tomida A. BCR-ABL tyrosine kinase inhibition induces metabolic vulnerability by preventing the integrated stress response in K562 cells. Biochem Biophys Res Commun (2018) 504(4):721–6. doi: 10.1016/j.bbrc.2018.09.032
178. Axten JM, Medina JR, Feng Y, Shu A, Romeril SP, Grant SW, et al. Discovery of 7-methyl-5-(1-[3-(trifluoromethyl)phenyl]acetyl-2,3-dihydro-1H-indol-5-yl)-7H-pyrrolo[2,3-d]pyrimidin-4-amine (GSK2606414), a potent and selective first-in-class inhibitor of protein kinase r (PKR)-like endoplasmic reticulum kinase (PERK). J Med Chem (2012) 55(16):7193–207. doi: 10.1021/jm300713s
179. Smith AL, Andrews KL, Beckmann H, Bellon SF, Beltran PJ, Booker S, et al. Discovery of 1H-pyrazol-3(2H)-ones as potent and selective inhibitors of protein kinase r-like endoplasmic reticulum kinase (PERK). J Med Chem (2015) 58(3):1426–41. doi: 10.1021/jm5017494
180. Yu Q, Zhao B, Gui J, Katlinski KV, Brice A, Gao Y, et al. Type i interferons mediate pancreatic toxicities of PERK inhibition. Proc Natl Acad Sci USA (2015) 112(50):15420–5. doi: 10.1073/pnas.1516362112
181. Hu Y, Conway TW. 2-aminopurine inhibits the double-stranded RNA-dependent protein kinase both in vitro and in vivo. J Interferon Res (1993) 13(5):323–8. doi: 10.1089/jir.1993.13.323
182. Jammi NV, Whitby LR, Beal PA. Small molecule inhibitors of the RNA-dependent protein kinase. Biochem Biophys Res Commun (2003) 308(1):50–7. doi: 10.1016/S0006-291X(03)01318-4
183. Kanelakis KC, Pyati J, Wagaman PC, Chuang JC, Yang Y, Shankley NP. Functional characterization of the canine heme-regulated eIF2alpha kinase: Regulation of protein synthesis. Adv Hematol (2009) 2009:251915. doi: 10.1155/2009/251915
184. Rosen MD, Woods CR, Goldberg SD, Hack MD, Bounds AD, Yang Y, et al. Discovery of the first known small-molecule inhibitors of heme-regulated eukaryotic initiation factor 2alpha (HRI) kinase. Bioorg Med Chem Lett (2009) 19(23):6548–51. doi: 10.1016/j.bmcl.2009.10.033
185. Knight K, Wade S, Balducci L. Prevalence and outcomes of anemia in cancer: a systematic review of the literature. Am J Med (2004) 116(Suppl 7A):11S–26S. doi: 10.1016/j.amjmed.2003.12.008
186. Sidrauski C, McGeachy AM, Ingolia NT, Walter P. The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife (2015) 4:e05033. doi: 10.7554/eLife.05033
187. Zyryanova AF, Kashiwagi K, Rato C, Harding HP, Crespillo-Casado A, Perera LA, et al. ISRIB blunts the integrated stress response by allosterically antagonising the inhibitory effect of phosphorylated eIF2 on eIF2B. Mol Cell (2021) 81(1):88–103 e6. doi: 10.1016/j.molcel.2020.10.031
188. Halliday M, Radford H, Sekine Y, Moreno J, Verity N, le Quesne J, et al. Partial restoration of protein synthesis rates by the small molecule ISRIB prevents neurodegeneration without pancreatic toxicity. Cell Death Dis (2015) 6(3):e1672. doi: 10.1038/cddis.2015.49
189. Dudka W, Hoser G, Mondal SS, Turos-Korgul L, Swatler J, Kusio-Kobialka M, et al. Targeting integrated stress response with ISRIB combined with imatinib treatment attenuates RAS/RAF/MAPK and STAT5 signaling and eradicates chronic myeloid leukemia cells. BMC Cancer (2022) 22(1):1254. doi: 10.1186/s12885-022-10289-w
190. Lee DM, Seo MJ, Lee HJ, Jin HJ, Choi KS. ISRIB plus bortezomib triggers paraptosis in breast cancer cells via enhanced translation and subsequent proteotoxic stress. Biochem Biophys Res Commun (2022) 596:56–62. doi: 10.1016/j.bbrc.2022.01.082
191. Wong YL, LeBon L, Basso AM, Kohlhaas KL, Nikkel AL, Robb HM, et al. eIF2B activator prevents neurological defects caused by a chronic integrated stress response. Elife (2019) 8:e42940. doi: 10.7554/eLife.42940
192. Halliday M, Radford H, Zents KAM, Molloy C, Moreno JA, Verity NC, et al. Repurposed drugs targeting eIF2α-p-mediated translational repression prevent neurodegeneration in mice. Brain (2017) 140(6):1768–83. doi: 10.1093/brain/awx074
193. Costa-Mattioli M, Walter P. The integrated stress response: From mechanism to disease. Science (2020) 368(6489):eaat5314. doi: 10.1126/science.aat5314
194. Zhang P, McGrath B, Li S, Frank A, Zambito F, Reinert J, et al. The PERK eukaryotic initiation factor 2 alpha kinase is required for the development of the skeletal system, postnatal growth, and the function and viability of the pancreas. Mol Cell Biol (2002) 22(11):3864–74. doi: 10.1128/MCB.22.11.3864-3874.2002
195. Scheuner D, Song B, McEwen E, Liu C, Laybutt R, Gillespie P, et al. Translational control is required for the unfolded protein response and in vivo glucose homeostasis. Mol Cell (2001) 7(6):1165–76. doi: 10.1016/S1097-2765(01)00265-9
196. Harding HP, Zhang Y, Scheuner D, Chen JJ, Kaufman RJ, Ron D. Ppp1r15 gene knockout reveals an essential role for translation initiation factor 2 alpha (eIF2alpha) dephosphorylation in mammalian development. Proc Natl Acad Sci USA (2009) 106(6):1832–7. doi: 10.1073/pnas.0809632106
197. English AM, Green KM, Moon SLA. (dis)integrated stress response: Genetic diseases of eIF2alpha regulators. Wiley Interdiscip Rev RNA (2022) 13(3):e1689. doi: 10.1002/wrna.1689
198. Shin S, Solorzano J, Liauzun M, Pyronnet S, Bousquet C, Martineau Y. Translational alterations in pancreatic cancer: a central role for the integrated stress response. NAR Cancer (2022) 4(4):zcac031. doi: 10.1093/narcan/zcac031
Keywords: cancer, stress response, mRNA translation, adaptation, drug resistance
Citation: Lines CL, McGrath MJ, Dorwart T and Conn CS (2023) The integrated stress response in cancer progression: a force for plasticity and resistance. Front. Oncol. 13:1206561. doi: 10.3389/fonc.2023.1206561
Received: 17 April 2023; Accepted: 07 June 2023;
Published: 03 August 2023.
Edited by:
Hasem Habelhah, The University of Iowa, United StatesReviewed by:
Luigi Alfano, G. Pascale National Cancer Institute Foundation (IRCCS), ItalyCopyright © 2023 Lines, McGrath, Dorwart and Conn. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Crystal S. Conn, Q3J5c3RhbC5Db25uQHBlbm5tZWRpY2luZS51cGVubi5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.