
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Oncol., 19 July 2023
Sec. Breast Cancer
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1203483
This article is part of the Research TopicEpigenetic and Metabolic Regulators of Breast CarcinogenesisView all 13 articles
 Yosra Bejaoui1
Yosra Bejaoui1 Sara Alresheq1Sophie Durand2Marie Vilaire-Meunier2Louise Maillebouis2
Sara Alresheq1Sophie Durand2Marie Vilaire-Meunier2Louise Maillebouis2 Ayman Al Haj Zen1
Ayman Al Haj Zen1 André Mégarbané2,3
André Mégarbané2,3 Nady El Hajj1,4*
Nady El Hajj1,4*Background: Down Syndrome (DS) is the most common chromosome anomaly in humans and occurs due to an extra copy of chromosome 21. The malignancy profile in DS is unique, since DS patients have a low risk of developing solid tumors such as breast cancer however they are at higher risk of developing acute myeloid leukemia and acute lymphoblastic leukemia.
Methods: In this study, we investigated DNA methylation signatures and epigenetic aging in DS individuals with and without breast cancer. We analyzed DNA methylation patterns in Trisomy 21 (T21) individuals without breast cancer (T21-BCF) and DS individuals with breast cancer (T21-BC), using the Infinium Methylation EPIC BeadChip array.
Results: Our results revealed several differentially methylated sites and regions in the T21-BC patients that were associated with changes in gene expression. The differentially methylated CpG sites were enriched for processes related to serine-type peptidase activity, epithelial cell development, GTPase activity, bicellular tight junction, Ras protein signal transduction, etc. On the other hand, the epigenetic age acceleration analysis showed no difference between T21-BC and T21-BCF patients.
Conclusions: This is the first study to investigate DNA methylation changes in Down syndrome women with and without breast cancer and it could help shed light on factors that protect against breast cancer in DS.
Down syndrome (DS) is a genetic disorder caused by an additional copy of all or part of chromosome 21 resulting in 47 chromosomes instead of the typical 46 chromosomes. The etiology of DS was identified following the discovery of karyotyping techniques when the French geneticist Jérôme Lejeune reported that an extra chromosome 21 results in the phenotypic features and intellectual disability associated with DS (1). DS is considered the most common chromosomal condition in humans occurring in 1 out of every 700 newborn babies (2). DS has three different forms including Trisomy 21 (nondisjunction), mosaicism, and translocation. Nondisjunction of chromosome 21, also called standard trisomy 21, is the most common DS type and accounts for ~ 95% of all cases. The cause of this chromosomal non-disjunction occurs mainly during maternal meiotic division (~88% of the cases). Whereas, ~ 5-10% of the cases are caused by non-disjunction during spermatogenesis and a small percentage of cases are due to mitotic error or occur during the first mitotic divisions of the embryo (3–5).
Trisomy 21 is associated with more than 100 features including intellectual disability, distinctive facial features, early aging, neurodegeneration, and muscle hypotonia during childhood (6). Intellectual disability is the most common feature in DS patients, where it usually ranges from mild to moderate. Besides, DS patients have a high incidence of congenital heart disease, early onset Alzheimer’s disease, gastrointestinal and skeletal malformations, and a diversity of neurobehavioral abnormalities (7–9). Even though DS patients are predisposed to developing acute lymphoblastic and myeloblastic leukemia during childhood, solid tumors seem to be extremely rare in both children and adults (10–16). Several epidemiological studies suggested that the risk of developing solid tumors in DS patients is at least 12 times lower than that of the general population (16, 17). For example, breast cancer (BC) is almost non-existent in DS females, despite genetic instability, deficiencies in DNA repair, increased oxidative stress, sedentary lifestyle, higher obesity rates, and increased DNA damage. Environmental factors including decreased exposure to estrogens and low alcohol consumption are not sufficient by themselves to explain the low rate of BC in DS females (17–19). Therefore, it would be important to study possible molecular mechanisms that protect against the development of breast cancer in Down syndrome.
Epigenetic dysregulation in response to an additional copy of chromosome 21 has been reported to affect the entire genome and not only genes located on chromosome 21 (20–24). Those changes arise during development and systemically affect multiple tissues (21, 25). Epigenetic clocks based on DNA methylation measurements have been used to estimate a person’s biological age and epigenetic aging acceleration. Epigenetic age acceleration has been reported to be associated with cancer risk, prognosis, and survival (26). Furthermore, patients with Down syndrome were reported to have drastic epigenetic age acceleration that was even higher than in certain progeroid syndromes (27, 28). Taking into account the occurrence of epigenetic alterations in most cancers and that they act as drivers to cancer progression, it would be important to study whether DNA methylation alterations affecting certain genes/pathways confers protection against breast cancer in DS. Therefore, we performed a genome-wide DNA methylation analysis in DS females with and without BC to determine epigenetically dysregulated regions linked to the lower BC frequency in DS. In addition, we compared epigenetic age acceleration in DS females with and without BC.
A total of 5532 files were screened at the Jérôme Lejeune Institute (CRB BioJeL, Paris, France) to identify two DS females with homogeneous Trisomy 21 (T21) diagnosed with breast cancer (no mosaicism or translocation cases were included). Sequencing analysis revealed no pathogenic or likely pathogenic variants in genes associated with an increased risk of breast cancer in the selected samples. A total of 10 age matched DS females with homogeneous T21 and without breast cancer (or any mammary lesion) were selected as controls (Supplementary Table 1). All the recruited DS women were > 34 years old, without any chronic medications or social problems. No breast cancer was recorded in the families of the DS women in this study. Whole blood samples were collected from all the patients and human peripheral blood mononuclear cells (PBMCs) were isolated. DNA was extracted from both whole blood and PBMCs. Written informed consent was obtained from the parents or guardians for all participants included in the research study.
DNA methylation profiling was performed for two T21 females with breast cancer (referred to as T21-BC) and for 10 T21 females without breast cancer (T21-BCF) (n=10) using the Illumina Infinium Epic array. DNA samples were processed on Illumina Infinium Epic array according to the manufacturer’s protocol. Briefly, 500 ng DNA for each sample was bisulfite converted using the EZ DNA Methylation Kit (Zymo Research, Irvine, CA, USA). Afterwards, bisulfite converted DNA was whole-genome amplified, enzymatically fragmented, and hybridized to Infinium Methylation EPIC BeadChips. Array scanning was performed via the Illumina iScan. To avoid batch effects, all samples were processed simultaneously and measured samples were randomly hybridized on the arrays. Idat files were exported and analyzed with the R software package (version 3.2.2) and the BioConductor platform (version 3.2).
The RnBeads package was used for differential methylation analysis (29). First, the data quality was assessed and probes mapping to multiple regions in the genome (Cross-reactive) or overlapping SNPs were removed. Furthermore, probes with unreliable measurements were removed via greedycut prior to further analysis. Next, additional filtering of polymorphic probes in the European, admixed American, South and East Asian, and African was applied using “filtering.blacklist” option (30). Data was normalized using Dasen and probes located on the X chromosome were retained because only females were analyzed. A total of 534862 (whole blood) and 534049 (PBMC) probes were finally retained for differential DNA methylation analysis. Inference for blood cell composition was performed using the Houseman method (31). Next, a limma based approach was used to correct for cell type composition, age, and surrogate variables. Differential methylation analysis was performed at the single CpG site level and at the level of promoters, CpG islands, and tiling windows (5Kb). Combined p-values were calculated and adjusted for multiple testing using false discovery rate (FDR) correction. Gene Ontology (GO) enrichment analysis was performed via the methylglm function from the methylGSA package (32).
Epigenetic age acceleration was measured using several epigenetic clocks that utilize different CpG sites to estimate DNA methylation (DNAm) age using the DNAm age calculator (https://dnamage.genetics.ucla.edu/) with the normalization option selected.
DNA methylation profiles of women with normal karyotype diagnosed with breast cancer (n=43) were downloaded from NCBI’s Gene Expression Omnibus (GEO Series accession: GSE104942). The Raw (IDAT) files were processed as previously described in the “Differential DNA methylation analysis” section. In total, the studied dataset was comprised of blood DNA methylation data measured via the Illumina EPIC arrays on 43 Breast cancer patients and 12 controls.
To identify epigenetically altered regions associated with BC in DS, we measured DNA methylation levels in T21 breast cancer patients (T21-BC, n=2) vs T21 breast cancer-free patients (T21-BCF, n=10) using the Illumina EPIC arrays. DNA methylation was profiled in DNA isolated from both whole blood and from PBMCs. The number of T21-BC samples was limited because only two T21 females with breast cancer were identified after screening 5532 files at Jérôme Lejeune Institute. For this reason, we decided to measure DNA methylation in duplicates across both whole blood and PBMC samples. First, we compared the deconvoluted blood cell proportions in T21-BC vs. T21-BCF as estimated by the Houseman method, which revealed no change in immune blood cell proportions (Figure 1A).
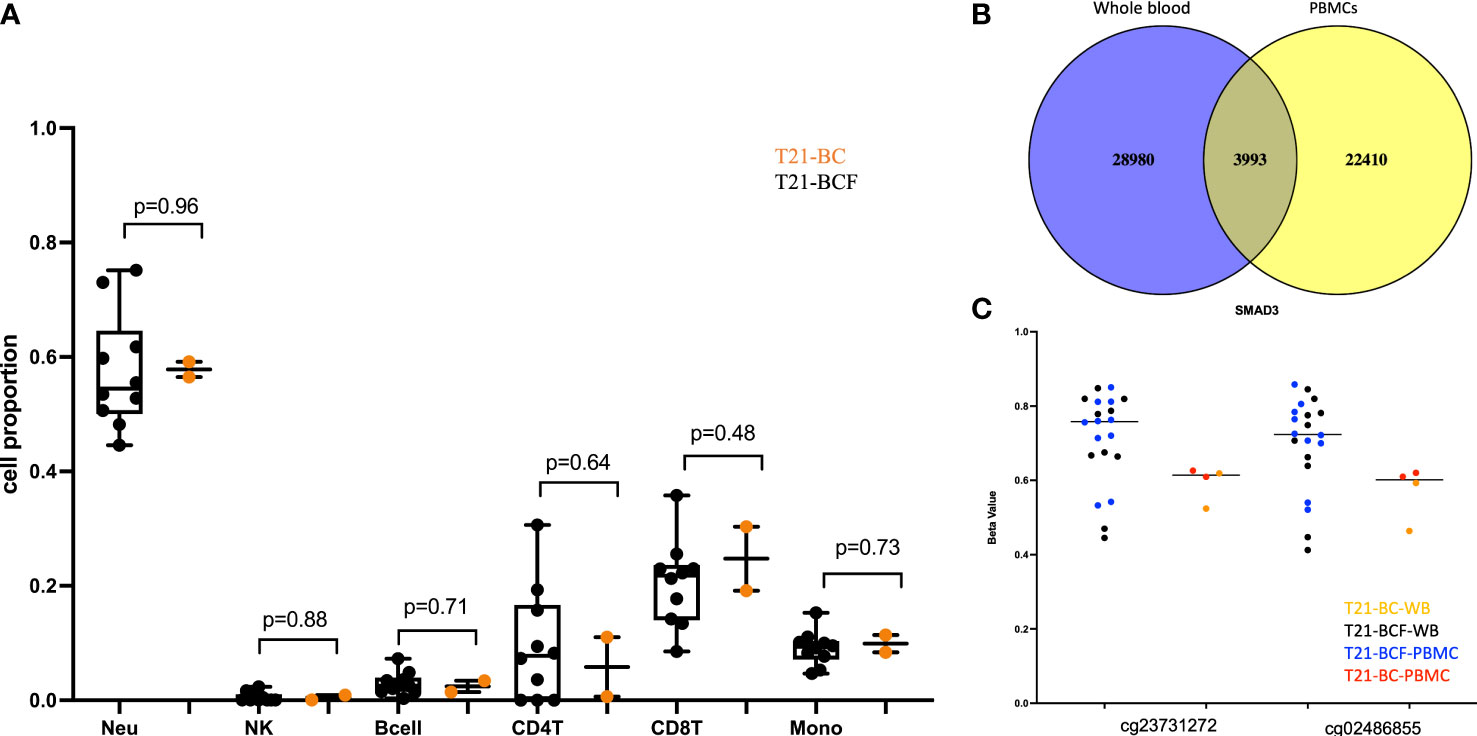
Figure 1 (A) Comparison of deconvoluted cell proportions measured via the Houseman method in whole blood of Trisomy 21 (T21) individuals with breast cancer (T21-BC) vs T21 without breast cancer (T21-BCF); (B) Ven-diagram of significant sites with unadjusted p-value <0.05 when comparing whole blood and PBMCs of T21-BC vs T21BCF; (C) differentially methylated probes s in SMAD3 with more than 9% methylation differences in whole blood and PBMC and present in the list of differentially expressed genes in T21-BC performed on the same samples. T21-BC-WB: Trisomy 21 (T21) individuals with breast cancer whole blood analysis. T21-BCF-WB: Trisomy 21 (T21) individuals without breast cancer whole blood DNA methylation analysis. T21-BCF-PBMC and T21-BC-PBMC indicate the DNA methylation analysis in peripheral blood mononuclear cells.
Next, differential DNA methylation analysis was performed to compare T21-BC vs. T21-BCF. The differential methylation was assessed primarily at the CpG sites level in addition to the region level including promoter, CpG Island, and tiling regions using a 5Kb sliding window. We did not observe any significance at the CpG site or the region level after FDR adjustment when adjusting for age, gender, cell type composition, and surrogate variables. This could be related to the low sample number in the T21-BC group. For this reason, we looked at common significant sites/regions with unadjusted p-value <0.05 between T21-BC and T21-BCF. In total, 32973 and 26403 CpG sites were significant before FDR adjustment in WB and PBMCs, respectively. Out of which, 3993 CpG sites were common between the whole blood and PBMC samples (Figure 1B). Out of those, 3087 had a similar direction of DNA methylation change when comparing T21-BC and T21-BCF in both WB and PBMC samples. When we filtered for ≥ 3% methylation in both tissues, a total of 1601 CpG sites were retained. Next, we applied the methylGSA package to test for gene ontology (GO) and KEGG pathway enrichment in those CpG sites, after adjusting for probe bias distribution across genes in the EPIC arrays. The GO enrichment analysis revealed several significant terms including serine-type peptidase activity, exopeptidase activity, serine hydrolase activity, epithelial cell development, etc (Supplementary Table 2). On the other hand, the KEGG analysis did not reveal any pathway enrichment for the 1601 CpG sites. We additionally investigated epigenetic age acceleration in T21-BC vs T21-BCF using the Horvath clock, GrimAge and PhenoAge, which revealed no DNA methylation age acceleration difference T21-BC in whole blood (Figure 2) and PBMC samples.
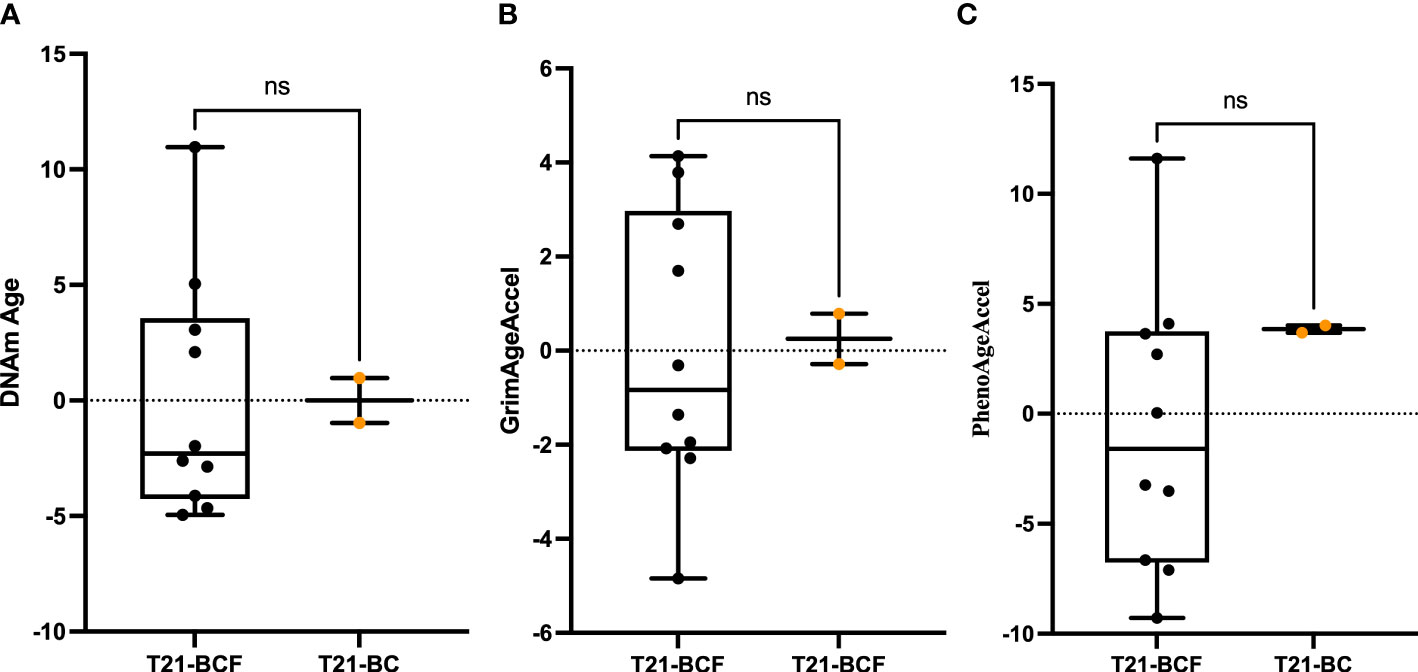
Figure 2 Epigenetic age acceleration in whole blood DNA of T21-BC vs T21-BCF using the (A) Horvath, (B) PhenoAge and, (C) GrimAge clocks. DNA methylation (DNAm) age acceleration, which represents the residual of regressing epigenetic age on chronological age is shown on the y-axes. ns, not significant.
Next, we looked at the promoter region where we could identify 832 significant promoters (unadjusted p-value <0.05) in whole blood and 744 significant promoters in PBMCs. A total of 78 promoters were significant in both analyses when comparing whole blood and PBMCs from T21-BC vs T21-BCF with the same direction of methylation change. Out of which, 22 promoters had > 2 CpG sites and ≥ 3% methylation in both whole blood and PBMC samples (Supplementary Table 3). For the CpG Island analysis, we could observe 131 common significant CGIs with the same direction of methylation change, including 43 with > 2 CpG sites and ≥ 3% methylation (Supplementary Table 4). For the tiling analysis, we could identify 677 regions (5Kb) differentially methylated in a similar direction in both datasets, however only 79 remained after filtering using the previously defined criteria (≥ 2 CpG sites, ≥ 3% methylation). Next, we tested whether the identified DMPs/DMRs are similarly epigenetically dysregulated in blood DNA of breast cancer patients. The studied dataset (GSE104942) contained blood DNA methylation data of 43 Breast cancer patients and 12 healthy controls. This analysis revealed no common significant DMRs between the T21-BC list and the differentially methylated genes in breast cancer patients. Two DMPs (cg05997779 and cg26845300) were similarly epigenetically altered in both datasets, however, they exhibited different direction of DNA methylation change.
Finally, we compared the differentially methylated sites/regions to the list of 183 differentially expressed genes in T21-BC identified following RNA-seq on the same samples (33). Here, we could observe 37 differentially methylated probes (DMPs) associated with differentially expressed genes and same direction of methylation change in both DNA methylation datasets. When we filtered for ≥ 3% methylation difference, we could only detect 12 CpG sites that fit this criteria including two close DMPs in SMAD3 with more than 9% methylation differences in all comparisons (Figure 1C). In addition, there was a single CpG site located on chromosome 21 in the BACH1 gene. Next, we checked the promoter and tiling differentially methylated regions (DMR), which revealed one DMR in the promoter analysis and two in the tiling region analysis. The common promoter was located in the gene TNFAIP3 Interacting Protein 1 (TNIP1) (Figure 3A), whereas the 5Kb tiling regions were located in the KRAB box domain containing 4 (KRBOX4) and Target Of Myb1 Membrane Trafficking Protein (TOM1) gene (Figures 3B, C). None of the CpG Island associated genes were differentially expressed in T21-BC.
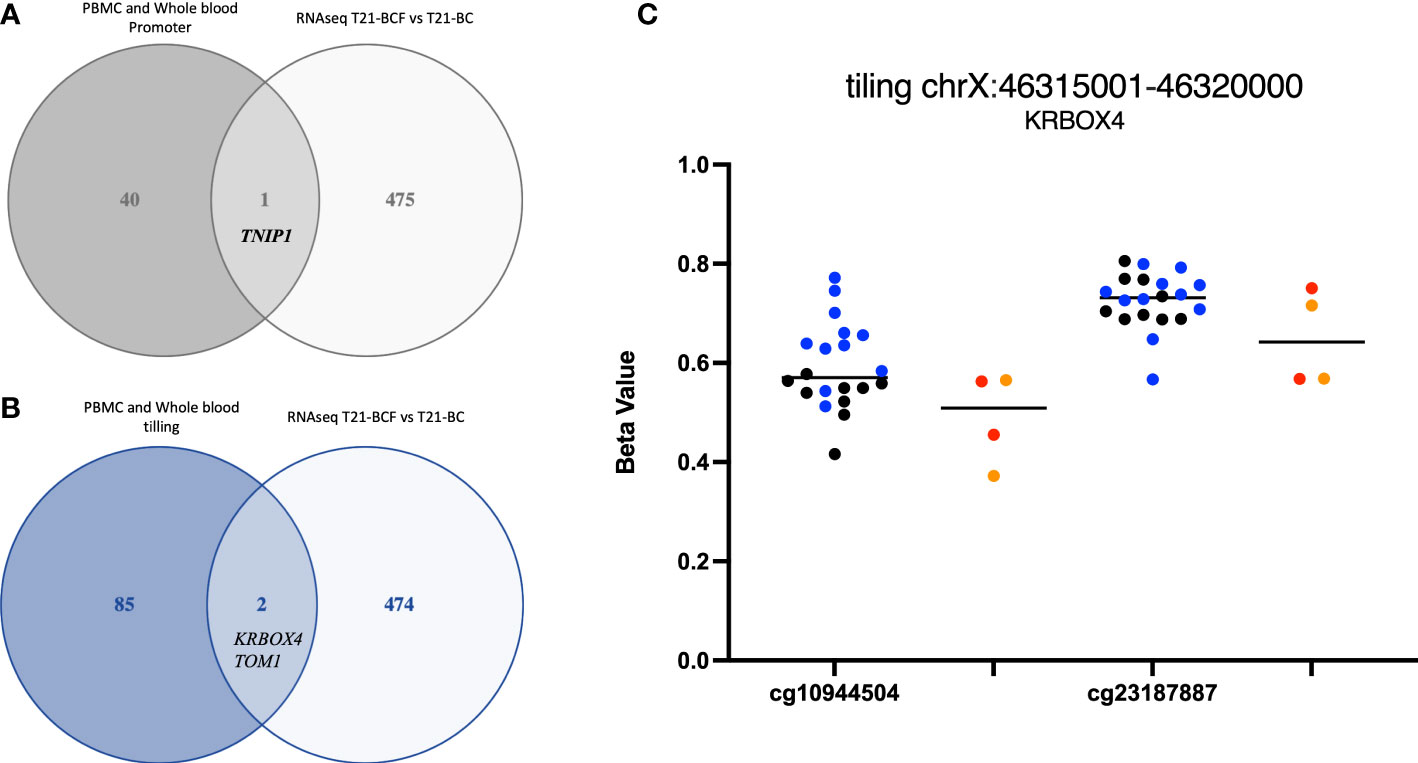
Figure 3 Ven-diagram displaying (A) differentially methylated promoters and (B) differentially methylated regions following a tiling analysis of differentially expressed genes in Trisomy 21 (T21) individuals with breast cancer (T21-BC) vs T21 without breast cancer (T21-BCF). (C) KRBOX4 DNA methylation distribution in T21 BC vs T21 BCF. Genomic coordinates based on genome assembly GRCh37 (hg19).
To understand whether epigenetic dysregulation might explain the lower frequency of breast cancer in DS, we profiled DNA methylation in 12 women with T21 including two with breast cancer and 10 without breast cancer. DNA methylation was measured in both whole blood DNA and PBMCs as replicates due to the small number of breast cancer patients with T21.
The differential methylation analysis at the single CpG site level revealed 1601 DMPs with the same direction in methylation change. The gene ontology analysis revealed enrichment for serine-type peptidase activity, exopeptidase activity, serine hydrolase activity, epithelial cell development, endothelium development, transcription coactivator activity, GTPase activity, GTP binding, bicellular tight junction, and Ras protein signal transduction. Cell surface anchored serine proteases are deregulated in cancer cells and contribute to tumour invasion and metastasis (34). Evidently, Ras protein signal transduction is extremely important in cancer where mutations in the RAS genes were the first mutations reported in human cancers (35–37). The expression and activity small GTPases subfamily of “Ras-homology” (Rho) GTPase are known to be linked with breast tumour progression, angiogenesis, and metastasis (38). Similarly, bicellular tight junctions play a role in the epithelial-mesenchymal transition, which is essential in cancer progression (39). Excessive angiogenesis is a crucial component of tumour growth, invasiveness, and metastasis (40). The individuals with DS showed an elevated expression of DSCR1 on the extra copy of chromosome 21, which is known to inhibit the growth of new blood vessels “angiogenesis” by suppressing vascular endothelial growth factor (VEGF)-mediated angiogenic signalling (41). In the current study, GO was enriched for endothelium development in which many genes overlap with angiogenesis. In addition, the crosstalk between several Rho GTPases and VEGF signalling is essential to control the process of angiogenesis (42–45). The epigenetic dysregulation in DS due to dosage imbalance of an additional chromosome 21 has been reported to occur extensively throughout the genome and is not restricted to genes located on chromosome 21 (21). This might lead to DNA methylation alterations in genes associated with the previously mentioned GO terms. This epigenetic dysregulation might confer protection to DS patients from breast cancer, which might help explain its reduced risk.
Furthermore, the comparison of the differentially methylated sites to differentially expressed genes following RNA-seq on the same set of samples identified 12 DMPs including two CpG sites in SMAD3 with > 9% methylation difference. Smad3 is a major transcription factor mediating transforming growth factor-β (TGF-β) signaling (46). The TGF‐β-Smad3 signaling has important roles in differentiation, apoptosis, and epithelial‐mesenchymal transition (EMT) (46, 47). SMAD-dependent signaling mediated by TGF-β has two opposing roles in cancer, where it first acts as a tumor suppressor in the initial phase, however in more advanced stages it is involved in inducing invasion and metastasis (48, 49). The regulation of estrogen receptor signaling pathways via TGF-beta was shown to be mediated by SMAD3, which indicates a role of SMAD3 in breast cancer progression (50). Furthermore, we identified a DMP located on chromosome 21 in the BTB and CNC homology1 (BACH1) gene. BACH1 encodes a transcription factor that is upregulated in tumours from triple-negative breast cancer (TNBCs) patients (51). BACH1 has been previously reported as a regulator of metastasis in TNBCs and its gene signature was shown to predict poor outcomes in breast cancer (52). The promoter of TNIP1 was differentially methylated and transcriptionally dysregulated in the T21-BC group. The tumor necrosis factor α–induced protein 3–interacting protein 1 (TNIP1) is part of the NF-κB and RAR signaling pathways (53, 54). TNIP1 was one of the stromal genes exhibiting expression changes when comparing adenomas vs cancer-associated stroma (55). Two DMRs were identified in KRBOX4 and TOM1 in the tiling analysis located in promoter flanking regions. KRBOX4 is located on the X chromosome and no studies so far have provided any link to breast cancer. TOM1 is required for autophagosome maturation and endosomal trafficking (56). TOM1 additionally represses Toll-like receptor signalling and plays a role in immune receptor recycling (57, 58). Mutations in TOM1 have been recently shown to be associated with early-onset autoimmunity and combined immunodeficiency (59). In addition, it is important to mention that we did not observe any DNA methylation changes at the region level in GTPases of the immunity-associated proteins (GIMAPs) despite their recently identified tumour suppressive role against breast cancer in DS (33). Therefore, it seems that upregulation of GIMAPs in T21 women is not associated with changes in DNA methylation.
DS patients are known to exhibit strong epigenetic age acceleration and for this reason we tested T21-BC patients age acceleration in comparison to the T21-BCF group. Epigenetic age acceleration have been previously shown to occur in several diseases including cancer, and can be used as a potential biomarker for early disease detection (26). Furthermore, a longitudinal study reported epigenetic age acceleration measured on blood DNA to be associated with a higher risk of developing breast cancer (60). The PhenoAge clock was also shown to measure increased epigenetic age acceleration in breast tissue of from breast cancer patients. However, our analysis revealed no difference in epigenetic age acceleration between T21-BC and T21-BCF using various clocks. This might be related to the drastic increase in epigenetic age acceleration in DS, which masks the effects of breast cancer on DNA methylation age.
The limitations of our study are the small sample size, however this is related to the uniqueness of the condition since breast cancer is almost non-existent in T21 patients. Furthermore, we have only looked at blood DNA in this study, which might not reflect similar epigenetic changes to target tissues involved in disease pathogenesis. Therefore, future studies should include additional tissues to determine whether the observed epigenetic changes related to breast cancer in DS are systemic or only restricted to one tissue.
In conclusion, this is the first study to investigate the DNA methylation profile in Down syndrome women suffering from breast cancer. The identified differentially methylated genes/regions could help us better understand factors that protect against breast cancer, which can provide new avenues for potential therapeutic targets or preventive approaches.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Institutional Review Board Statement. This research was conducted according to the guidelines of the Declaration of Helsinki. It was approved by the Institute Jérôme Lejeune Committee on Clinical Investigation and the Institutional Review Board (IRB) of the Qatar Biomedical Research Institute (reference number: QBRI-IRB 2021-07-098). Written informed consent was obtained from the parents or guardians prior to the participants being included in the research study. The patients/participants provided their written informed consent to participate in this study.
AM and NH designed the study; YB and SA analyzed the data, performed experiments, and prepared figures; SD, MV, LM, and AM collected the samples and clinical data; NH and AA wrote the manuscript, NH, AM, and AA reviewed and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported with funding by Qatar Foundation to the College of Health and Life Sciences, Hamad Bin Khalifa (NH).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fonc.2023.1203483/full#supplementary-material
1. Lejeune J, Gauthier M, Turpin R. [Human chromosomes in tissue cultures]. C R Hebd Seances Acad Sci (1959) 248:602–3.
2. Mai CT, Isenburg JL, Canfield MA, Meyer RE, Correa A, Alverson CJ, et al. National population-based estimates for major birth defect-2014. Birth Defects Res (2019) 111:1420–35. doi: 10.1002/bdr2.1589
3. Antonarakis SE. Parental origin of the extra chromosome in trisomy 21 as indicated by analysis of DNA polymorphisms. down syndrome collaborative group. N Engl J Med (1991) 324:872–6. doi: 10.1056/NEJM199103283241302
4. Sherman SL, Takaesu N, Freeman SB, Grantham M, Phillips C, Blackston RD, et al. Trisomy 21: association between reduced recombination and nondisjunction. Am J Hum Genet (1991) 49:608–20.
5. Mantikou E, Wong KM, Repping S, Mastenbroek S. Molecular origin of mitotic aneuploidies in preimplantation embryos. Biochim Biophys Acta (2012) 1822:1921–30. doi: 10.1016/j.bbadis.2012.06.013
6. Megarbane A, Ravel A, Mircher C, Sturtz F, Grattau Y, Rethore MO, et al. The 50th anniversary of the discovery of trisomy 21: the past, present, and future of research and treatment of down syndrome. Genet Med (2009) 11:611–6. doi: 10.1097/GIM.0b013e3181b2e34c
7. Epstein CJ. The consequences of chromosome imbalance. Am J Med Genet Suppl (1990) 7:31–7. doi: 10.1002/ajmg.1320370706
8. Nelson L, Johnson JK, Freedman M, Lott I, Groot J, Chang M, et al. Learning and memory as a function of age in down syndrome: a study using animal-based tasks. Prog Neuropsychopharmacol Biol Psychiatry (2005) 29:443–53. doi: 10.1016/j.pnpbp.2004.12.009
9. Dierssen M, Herault Y, Estivill X. Aneuploidy: from a physiological mechanism of variance to down syndrome. Physiol Rev (2009) 89:887–920. doi: 10.1152/physrev.00032.2007
10. Satge D, Sommelet D, Geneix A, Nishi M, Malet P, Vekemans M. A tumor profile in down syndrome. Am J Med Genet (1998) 78:207–16. doi: 10.1002/(SICI)1096-8628(19980707)78:3<207::AID-AJMG1>3.0.CO;2-M
11. Hasle H, Clemmensen IH, Mikkelsen M. Risks of leukaemia and solid tumours in individuals with down's syndrome. Lancet (2000) 355:165–9. doi: 10.1016/S0140-6736(99)05264-2
12. Patja K, Pukkala E, Sund R, Iivanainen M, Kaski M. Cancer incidence of persons with down syndrome in Finland: a population-based study. Int J Cancer (2006) 118:1769–72. doi: 10.1002/ijc.21518
13. Sullivan SG, Hussain R, Glasson EJ, Bittles AH. The profile and incidence of cancer in down syndrome. J Intellect Disabil Res (2007) 51:228–31. doi: 10.1111/j.1365-2788.2006.00862.x
14. Nizetic D, Groet J. Tumorigenesis in down's syndrome: big lessons from a small chromosome. Nat Rev Cancer (2012) 12:721–32. doi: 10.1038/nrc3355
15. Hasle H, Friedman JM, Olsen JH, Rasmussen SA. Low risk of solid tumors in persons with down syndrome. Genet Med (2016) 18:1151–7. doi: 10.1038/gim.2016.23
16. Rethore MO, Rouesse J, Satge D. Cancer screening in adults with down syndrome, a proposal. Eur J Med Genet (2020) 63:103783. doi: 10.1016/j.ejmg.2019.103783
17. Satge D, Sasco AJ, Pujol H, Rethore MO. Breast cancer in women with trisomy 21. Bull Acad Natl Med (2001) 185:1239–52.
18. Ayed W, Gouas L, Penault-Llorca F, Amouri A, Tchirkov A, Vago P. [Trisomy 21 and cancers]. Morphologie (2012) 96:57–66. doi: 10.1016/j.morpho.2012.10.001
19. Osuna-Marco MP, Lopez-Barahona M, Lopez-Ibor B, Tejera AM. Ten reasons why people with down syndrome are protected from the development of most solid tumors -a review. Front Genet (2021) 12:749480. doi: 10.3389/fgene.2021.749480
20. Mendioroz M, Do C, Jiang X, Liu C, Darbary HK, Lang CF, et al. Trans effects of chromosome aneuploidies on DNA methylation patterns in human down syndrome and mouse models. Genome Biol (2015) 16:263. doi: 10.1186/s13059-015-0827-6
21. Do C, Xing Z, Yu YE, Tycko B. Trans-acting epigenetic effects of chromosomal aneuploidies: lessons from down syndrome and mouse models. Epigenomics (2017) 9:189–207. doi: 10.2217/epi-2016-0138
22. El Hajj N, Dittrich M, Haaf T. Epigenetic dysregulation of protocadherins in human disease. Semin Cell Dev Biol (2017) 69:172–82. doi: 10.1016/j.semcdb.2017.07.007
23. Almenar-Queralt A, Merkurjev D, Kim HS, Navarro M, Ma Q, Chaves RS, et al. Chromatin establishes an immature version of neuronal protocadherin selection during the naive-to-primed conversion of pluripotent stem cells. Nat Genet (2019) 51:1691–701. doi: 10.1038/s41588-019-0526-4
24. Haertle L, Muller T, Lardenoije R, Maierhofer A, Dittrich M, Riemens RJM, et al. Methylomic profiling in trisomy 21 identifies cognition- and alzheimer's disease-related dysregulation. Clin Epigenet (2019) 11:195. doi: 10.1186/s13148-019-0787-x
25. El Hajj N, Dittrich M, Bock J, Kraus TF, Nanda I, Muller T, et al. Epigenetic dysregulation in the developing down syndrome cortex. Epigenetics (2016) 11:563–78. doi: 10.1080/15592294.2016.1192736
26. Salameh Y, Bejaoui Y, El Hajj N. DNA Methylation biomarkers in aging and age-related diseases. Front Genet (2020) 11:171. doi: 10.3389/fgene.2020.00171
27. Horvath S, Garagnani P, Bacalini MG, Pirazzini C, Salvioli S, Gentilini D, et al. Accelerated epigenetic aging in down syndrome. Aging Cell (2015) 14:491–5. doi: 10.1111/acel.12325
28. Bejaoui Y, Razzaq A, Yousri NA, Oshima J, Megarbane A, Qannan A, et al. DNA Methylation signatures in blood DNA of Hutchinson-gilford progeria syndrome. Aging Cell (2022) 21:e13555. doi: 10.1111/acel.13555
29. Muller F, Scherer M, Assenov Y, Lutsik P, Walter J, Lengauer T, et al. RnBeads 2.0: comprehensive analysis of DNA methylation data. Genome Biol (2019) 20:55. doi: 10.1186/s13059-019-1664-9
30. Mccartney DL, Walker RM, Morris SW, Mcintosh AM, Porteous DJ, Evans KL. Identification of polymorphic and off-target probe binding sites on the illumina infinium MethylationEPIC BeadChip. Genom Data (2016) 9:22–4. doi: 10.1016/j.gdata.2016.05.012
31. Houseman EA, Accomando WP, Koestler DC, Christensen BC, Marsit CJ, Nelson HH, et al. DNA Methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinf (2012) 13:86. doi: 10.1186/1471-2105-13-86
32. Ren X, Kuan PF. methylGSA: a bioconductor package and shiny app for DNA methylation data length bias adjustment in gene set testing. Bioinformatics (2019) 35:1958–9. doi: 10.1093/bioinformatics/bty892
33. Megarbane A, Piquemal D, Rebillat AS, Stora S, Pierrat F, Bruno R, et al. Transcriptomic study in women with trisomy 21 identifies a possible role of the GTPases of the immunity-associated proteins (GIMAP) in the protection of breast cancer. Sci Rep (2020) 10:9447. doi: 10.1038/s41598-020-66469-w
34. Martin CE, List K. Cell surface-anchored serine proteases in cancer progression and metastasis. Cancer Metastasis Rev (2019) 38:357–87. doi: 10.1007/s10555-019-09811-7
35. Der CJ, Krontiris TG, Cooper GM. Transforming genes of human bladder and lung carcinoma cell lines are homologous to the ras genes of Harvey and kirsten sarcoma viruses. Proc Natl Acad Sci USA (1982) 79:3637–40. doi: 10.1073/pnas.79.11.3637
36. Parada LF, Tabin CJ, Shih C, Weinberg RA. Human EJ bladder carcinoma oncogene is homologue of Harvey sarcoma virus ras gene. Nature (1982) 297:474–8. doi: 10.1038/297474a0
37. Stites EC, Ravichandran KS. A systems perspective of ras signaling in cancer. Clin Cancer Res (2009) 15:1510–3. doi: 10.1158/1078-0432.CCR-08-2753
38. Van Golen KL. Inflammatory breast cancer: relationship between growth factor signaling and motility in aggressive cancers. Breast Cancer Res (2003) 5:174–9. doi: 10.1186/bcr598
39. Kyuno D, Takasawa A, Kikuchi S, Takemasa I, Osanai M, Kojima T. Role of tight junctions in the epithelial-to-mesenchymal transition of cancer cells. Biochim Biophys Acta Biomembr (2021) 1863:183503. doi: 10.1016/j.bbamem.2020.183503
40. Folkman J. Role of angiogenesis in tumor growth and metastasis. Semin Oncol (2002) 29:15–8. doi: 10.1053/sonc.2002.37263
41. Baek KH, Zaslavsky A, Lynch RC, Britt C, Okada Y, Siarey RJ, et al. Down's syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature (2009) 459:1126–30. doi: 10.1038/nature08062
42. Adini I, Rabinovitz I, Sun JF, Prendergast GC, Benjamin LE. RhoB controls akt trafficking and stage-specific survival of endothelial cells during vascular development. Genes Dev (2003) 17:2721–32. doi: 10.1101/gad.1134603
43. Kim C, Yang H, Fukushima Y, Saw PE, Lee J, Park JS, et al. Vascular RhoJ is an effective and selective target for tumor angiogenesis and vascular disruption. Cancer Cell (2014) 25:102–17. doi: 10.1016/j.ccr.2013.12.010
44. Fantin A, Lampropoulou A, Gestri G, Raimondi C, Senatore V, Zachary I, et al. NRP1 regulates CDC42 activation to promote filopodia formation in endothelial tip cells. Cell Rep (2015) 11:1577–90. doi: 10.1016/j.celrep.2015.05.018
45. Hoeppner LH, Sinha S, Wang Y, Bhattacharya R, Dutta S, Gong X, et al. RhoC maintains vascular homeostasis by regulating VEGF-induced signaling in endothelial cells. J Cell Sci (2015) 128:3556–68. doi: 10.1242/jcs.167601
46. Millet C, Zhang YE. Roles of Smad3 in TGF-beta signaling during carcinogenesis. Crit Rev Eukaryot Gene Expr (2007) 17:281–93. doi: 10.1615/CritRevEukarGeneExpr.v17.i4.30
47. Hao Y, Baker D, Ten Dijke P. TGF-beta-Mediated epithelial-mesenchymal transition and cancer metastasis. Int J Mol Sci (2019) 20. doi: 10.3390/ijms20112767
48. Nawshad A, Lagamba D, Polad A, Hay ED. Transforming growth factor-beta signaling during epithelial-mesenchymal transformation: implications for embryogenesis and tumor metastasis. Cells Tissues Organs (2005) 179:11–23. doi: 10.1159/000084505
49. Moustakas A, Heldin CH. Signaling networks guiding epithelial-mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci (2007) 98:1512–20. doi: 10.1111/j.1349-7006.2007.00550.x
50. Matsuda T, Yamamoto T, Muraguchi A, Saatcioglu F. Cross-talk between transforming growth factor-beta and estrogen receptor signaling through Smad3. J Biol Chem (2001) 276:42908–14. doi: 10.1074/jbc.M105316200
51. Lee J, Yesilkanal AE, Wynne JP, Frankenberger C, Liu J, Yan J, et al. Effective breast cancer combination therapy targeting BACH1 and mitochondrial metabolism. Nature (2019) 568:254–8. doi: 10.1038/s41586-019-1005-x
52. Elbasateeny SS, Yassin MA, Mokhtar MM, Ismail AM, Ebian HF, Hussein S, et al. Prognostic implications of MALAT1 and BACH1 expression and their correlation with CTCs and Mo-MDSCs in triple negative breast cancer and surgical management options. Int J Breast Cancer (2022) 2022:8096764. doi: 10.1155/2022/8096764
53. Heyninck K, De Valck D, Vanden Berghe W, Van Criekinge W, Contreras R, Fiers W, et al. The zinc finger protein A20 inhibits TNF-induced NF-kappaB-dependent gene expression by interfering with an RIP- or TRAF2-mediated transactivation signal and directly binds to a novel NF-kappaB-inhibiting protein ABIN. J Cell Biol (1999) 145:1471–82. doi: 10.1083/jcb.145.7.1471
54. Gurevich I, Aneskievich BJ. Liganded RARalpha and RARgamma interact with but are repressed by TNIP1. Biochem Biophys Res Commun (2009) 389:409–14. doi: 10.1016/j.bbrc.2009.08.159
55. Amini P, Nassiri S, Malbon A, Markkanen E. Differential stromal reprogramming in benign and malignant naturally occurring canine mammary tumours identifies disease-modulating stromal components. Sci Rep (2020) 10:5506. doi: 10.1038/s41598-020-62354-8
56. Tumbarello DA, Waxse BJ, Arden SD, Bright NA, Kendrick-Jones J, Buss F. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol (2012) 14:1024–35. doi: 10.1038/ncb2589
57. Katoh Y, Shiba Y, Mitsuhashi H, Yanagida Y, Takatsu H, Nakayama K. Tollip and Tom1 form a complex and recruit ubiquitin-conjugated proteins onto early endosomes. J Biol Chem (2004) 279:24435–43. doi: 10.1074/jbc.M400059200
58. Xiao S, Brannon MK, Zhao X, Fread KI, Ellena JF, Bushweller JH, et al. Tom1 modulates binding of tollip to phosphatidylinositol 3-phosphate via a coupled folding and binding mechanism. Structure (2015) 23:1910–20. doi: 10.1016/j.str.2015.07.017
59. Keskitalo S, Haapaniemi EM, Glumoff V, Liu X, Lehtinen V, Fogarty C, et al. Dominant TOM1 mutation associated with combined immunodeficiency and autoimmune disease. NPJ Genom Med (2019) 4:14. doi: 10.1038/s41525-019-0088-5
Keywords: Down syndrome, breast cancer, DNA methylation, epigenetics, Trisomy 21
Citation: Bejaoui Y, Alresheq S, Durand S, Vilaire-Meunier M, Maillebouis L, Zen AAH, Mégarbané A and Hajj NE (2023) DNA methylation profiling in Trisomy 21 females with and without breast cancer. Front. Oncol. 13:1203483. doi: 10.3389/fonc.2023.1203483
Received: 10 April 2023; Accepted: 03 July 2023;
Published: 19 July 2023.
Edited by:
Iman Mamdouh Talaat, University of Sharjah, United Arab EmiratesReviewed by:
Sung Hoon Sim, National Cancer Center, Republic of KoreaCopyright © 2023 Bejaoui, Alresheq, Durand, Vilaire-Meunier, Maillebouis, Zen, Mégarbané and Hajj. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nady El Hajj, bmVsaGFqakBoYmt1LmVkdS5xYQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.