 Sylvie Hermouet1,2*
Sylvie Hermouet1,2*- 1Nantes Université, INSERM, Immunology and New Concepts in ImmunoTherapy, INCIT, UMR 1302, Nantes, France
- 2Laboratoire d’Hématologie, CHU Nantes, Nantes, France
Knowledge on the myeloproliferative neoplasms (MPNs) – polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (PMF) – has accumulated since the discovery of the JAK/STAT-activating mutations associated with MPNs: JAK2V617F, observed in PV, ET and PMF; and the MPL and CALR mutations, found in ET and PMF. The intriguing lack of disease specificity of these mutations, and of the chronic inflammation associated with MPNs, triggered a quest for finding what precisely determines that MPN patients develop a PV, ET or PMF phenoptype. The mechanisms of action of MPN-driving mutations, and concomitant mutations (ASXL1, DNMT3A, TET2, others), have been extensively studied, as well as the role played by these mutations in inflammation, and several pathogenic models have been proposed. In parallel, different types of drugs have been tested in MPNs (JAK inhibitors, interferons, hydroxyurea, anagrelide, azacytidine, combinations of those), some acting on both JAK2 and inflammation. Yet MPNs remain incurable diseases. This review aims to present current, detailed knowledge on the pathogenic mechanisms specifically associated with PV, ET or PMF that may pave the way for the development of novel, curative therapies.
Introduction
Normal myelopoiesis depends on the activation of the JAK2/STAT5 pathway by hematopoietic cytokines and their receptors. The JAK2/STAT5 pathway and myelopoiesis are physiologically hyper-stimulated in case of bleeding, hypoxia, or inflammation (1). Other causes of hyperstimulation of the JAK2/STAT5 pathway and myelopoiesis include the chronic Philadelphia-negative myeloproliferative neoplasms (MPNs). MPNs are characterized by an excessive production of mature cells of the three myeloid lineages. They arise from the acquisition in a multipotent hematopoietic progenitor of a JAK2/STAT5-activating mutation in one of three genes – JAK2, MPL, CALR – and thus can be seen as clonal versions of myelopoiesis (2–8). Three subtypes of MPNs are distinguished: essential thrombocythemia (ET), where overproduction of megakaryocytes and platelets is predominant; polycythemia vera (PV), which concerns predominantly the erythroid lineage; and primary myelofibrosis (PMF), characterized by severe fibrosis of the bone marrow and splenomegaly (7–9). Among MPN-driving mutations, the V617F mutation of JAK2 exon 14 (JAK2V617F) was discovered first, rapidly followed by the MPL exon 10 (W515L, W515K) and CALR exon 9 mutations (2–6). JAK2V617F is detected in >95% PV cases and in 50-60% of ET and PMF cases, while CALR mutations characterize 25-30% ET and PMF cases; MPL mutations are found in 5-10% ET and PMF cases. In addition, MPN patients typically present with chronic inflammation (10–13). Logically, numerous inflammatory cytokines are overexpressed by MPN patients; some activate the JAK2/STAT5 pathway (G-CSF, GM-CSF, interleukin 6 (IL-6)) and further increase myelopoiesis, while others activate the JAK1/STAT1-STAT3 pathways (IL-6, interferons (IFN)) and thus enhance cytokine production and facilitate cell survival (13–19). The severity of MPN clinical symptoms – fatigue, fever, night sweats, weight loss, itching – and complications – thrombosis (arterial, venous), splenomegaly, bone marrow fibrosis – typically increase with the level of inflammation, mild in ET, moderate in PV, and severe in PMF (20).
The lack of disease specificity of JAK2/STAT5-activating mutations triggered a quest for finding what precisely determines that a patient develops a PV, ET or PMF phenotype. Over the last decade, the mechanisms of action of MPN-driving mutations, as well as co-occurring mutations (ASXL1, EZH2, DNMT3A, TET2), in MPN disease initiation and progression have been extensively studied, in vitro and in murine models (21–30). The roles played in inflammation by driving and non-driving mutations, and their chronology, have also been investigated (13–19, 31, 32). In parallel, clinical trials have tested different drugs in MPNs (hydroxyurea, anagrelide, interferons (IFN), azacytidine, JAK inhibitors, some blocking only JAK2, or JAK1 and inflammation, or both), sometimes with unexpected results (33–41). Logically, the JAK inhibitors that significantly inhibited inflammation reduced clinical symptoms and spleen size (34, 36, 42–45). However, JAK inhibitors suppress the MPN clone and mutation load only partially, whereas IFN-α2 therapy leads to durable clinical and hematological remission for >75% MPN patients, as well as molecular remission for ~10% JAK2V617F-mutated PV, ET and PMF patients (33, 37, 46–48). Interestingly, IFN-α2 and JAK inhibitors reportedly act in synergy in MPNs (49, 50).
Despite major advances in knowledge and in therapy, MPNs remain incurable. Indeed, to be curative, treatments must counter the initial and other main events responsible for a particular disease. This review summarizes the present knowledge on the pathogenic events associated with the PV, ET or PMF phenotypes, with the aim to identify new therapeutic targets that could lead to curative treatments in the different MPN subtypes.
JAK2/STAT5-activating mutations and MPN phenotype
Certain MPN phenotypes are associated with specific driving mutations or/and mutant allele burden, but none can be explained solely by the patient’s JAK2V617F load nor by the presence of CALR or MPL mutation(s). MPN phenotypes clearly do not depend on JAK2V617F, since this mutation is found in all MPN subtypes (PV, ET, PMF), as well as in refractory anemia with ring sideroblasts and thrombocytosis (RARS-T) and in splanchnic vein thrombosis (SVT) (Figure 1A). MPN clones can be heterozygous or homozygous for the JAK2V617F mutation, after recombination or gain of mutated chromosome 9, and the allele count and JAK2V617F load can also increase due to the amplification of the whole chromosome 9 (trisomy 9). Consequently, the size of JAK2V617F-mutated clones and the percentage (%) of JAK2V617F-mutated alleles varies widely, from 1% to 100%. Of note, 25-50% JAK2V617F-mutated alleles are observed in all MPN subtypes. Moreover, if homozygous JAK2V617F-mutated clones (JAK2V617F load ≥50%) are typical of PV, they are also found in PMF. Heterozygous JAK2V617F-mutated clones (JAK2V617F loads <50%) are typical of ET and RARS-T, but also observed in PMF, in SVT, and more rarely, in PV (Figure 1A). Yet the JAK2V617F mutant load affects clinical presentation: high JAK2V617F-mutated allele burdens are associated with increased hematocrit and leukocyte numbers, and more venous thrombotic events (51). In contrast, MPN patients with mutations in JAK2 exon 12 develop erythrocytosis only.
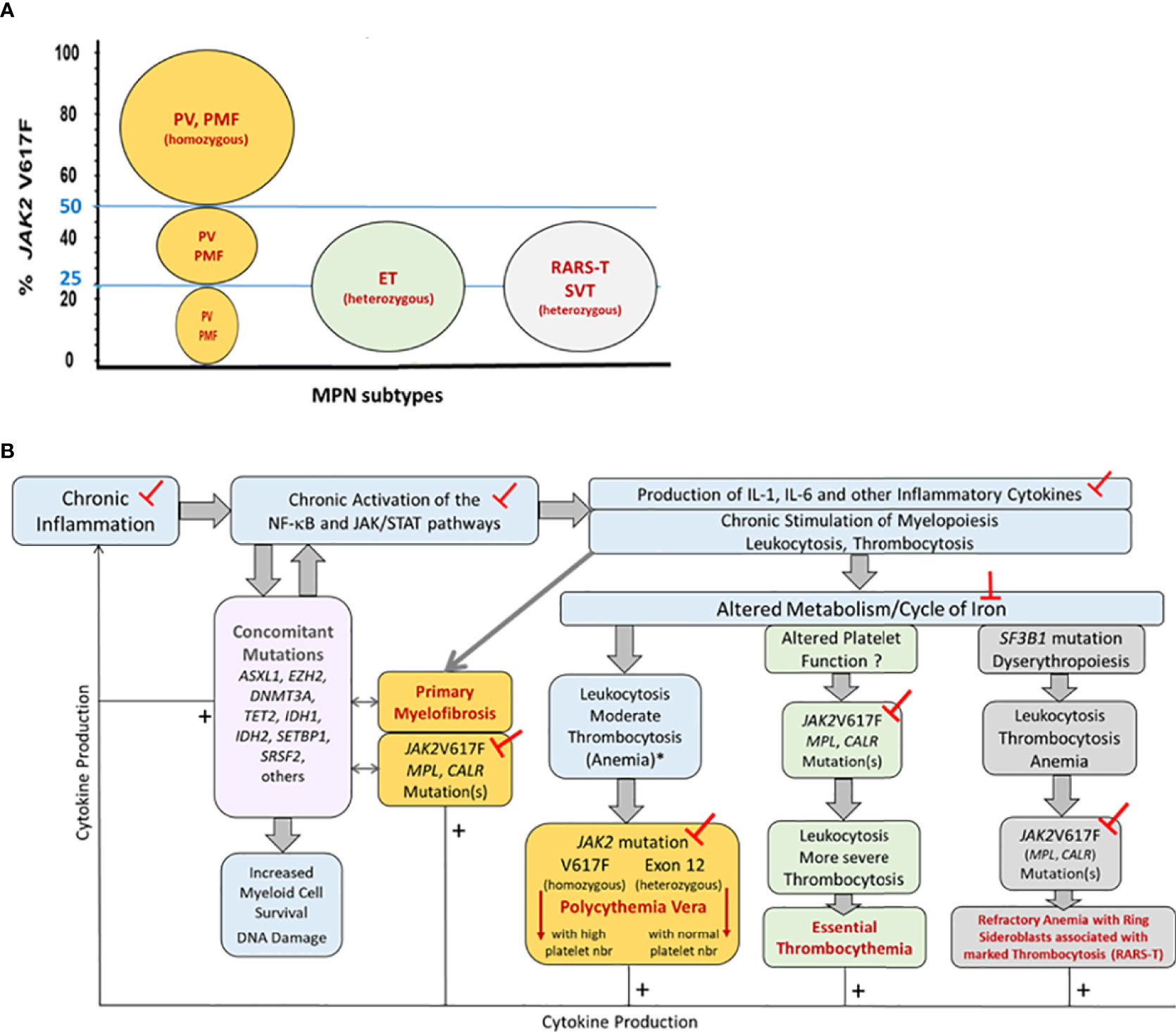
Figure 1 Representation of the pathogenesis of MPNs, associated mutations and inflammation, and impact on MPN phenotype. Part (A) JAK2V617F-mutant allele burdens in the different MPN phenotypes. Patients diagnosed with classic MPNs (PV, ET, PMF) or with refractory anemia with ring sideroblasts and thrombocytosis (RARS-T) or splanchnic vein thrombosis (SVT) can present with 25-50% JAK2V617F-mutated alleles in blood cells. The size of JAK2V617F-mutated clones varies: MPN clones are typically heterozygous for the JAK2V617F mutation in ET and RARS-T, and homozygous in PV and PMF. Note that in PV and in PMF with <50% JAK2V617F, clones may be heterozygous or homozygous for JAK2V617F. Part (B) Pathogenesis of MPNs and impact on MPN phenotype. Chronic inflammation of various causes (smoking, inflammatory diseases, auto-immunity, high-fat diet, metabolism disorders, genetic pre-disposition to inflammation) leads to chronic activation of the NF-κB and JAK/STAT pathways, hereby further increasing the production of inflammatory cytokines and myeloid cells, and facilitating the acquisition of non-driving, concomitant mutations (in the DNMT3A, TET2, SRSF2, SF3B1 genes) as well as driving JAK/STAT-activating mutations (in the JAK2, CALR or MPL genes) in myeloid progenitors, resulting in the development of a MPN. The MPN phenotype (ET, PV, PMF, RARS-T) depends in part from the driving JAK2, CALR or MPL mutation(s) and the presence of concomitant mutation(s), and in part from the level and type of inflammation, iron stocks, and metabolism. The mutation-dependent production of cytokines is indicated by thin arrows and + signs. Concomitant mutations can be found in all MPN phenotypes but are more frequent in PMF, which is indicated by arrows. The ┬ symbols indicate the different targets potentially useful in MPN prevention and therapy. (*) In case of inflammation and iron deficit.
MPN clones are typically heterozygous for the other driving mutations – MPL exon 10 (W515L, W515K) and CALR exon 9 mutations – with mutated allele loads close to 50%. Again, MPL and CALR mutations are found in both ET and PMF, in 5-10% MPN cases for MPL mutations, and 25-30% MPN cases for CALR mutations (Figure 1A). Compared to JAK2V617F-mutated ET or PMF, the presence of CALR mutations in ET or PMF is linked to a younger age and high platelet counts (51). The JAK2/CALR/MPL mutational status does not affect median survival in ET (19-20 years) (51). However, in PMF, the median survival is longest for patients with CALR mutations (15.9 years) compared to patients with MPL or JAK2V617F mutation (9.9 and 5.9 years, respectively), and worse for patients with no mutation in the JAK2/CALR/MPL genes (2.3 years) (52).
Other genetic alterations and MPN phenotype
Genetic predisposition to MPNs
Genetic predisposition to MPN is now established: relatives of MPN patients have about 6-8 fold higher risk of developing a MPN (53–55). Genetic predisposition to MPNs include the 46/1 or GGCC haplotype of JAK2, germline ATG2B and GSKIP duplication or mutations in RBBP6 or EPOR (EPOR-p.P488S), and single nucleotide polymorphisms (SNPs) in the TERT, MECOM, and CHEK2 genes (56–63). Genetic loci associated with a high risk of MPN typically affect the self-renewal of hematopoietic stem cells (ZNF521, GATA2, MECOM, HMGA1, ATM, FOXO1) (64). As in sporadic MPNs, mutations in JAK2, CALR or MPL are observed in individuals predisposed to MPNs, JAK2V617F being the most frequent driving mutation. The different germline variants or mutations that increase the risk of MPN are not associated with a specific MPN phenotype.
Concomitant mutations
Concomitant mutations found in MPN clones concern mostly the DNMT3A, TET2, ASXL1, EZH2, SRSF2 and SF3B1 genes (65). These mutations are not specific for MPNs, and their frequency is low in MPNs compared to other blood malignancies and solid cancers. Mutations in one or more of the DNMT3A, TET2, ASXL1, EZH2, SRSF2 and SF3B1 genes concern up to 20% PV, 20% ET, and 40% PMF. They typically occur after acquisition of a MPN-driving mutation, but also occur as early events that facilitate clonal emergence, followed by the acquisition of mutation(s) in JAK2, MPL or CALR. Concomitant mutations do not directly influence the MPN phenotype but are associated with clonal expansion and disease progression, notably secondary myelofibrosis and leukemic transformation (52, 65–67). DNMT3A, TET2, ASXL1 and EZH2 mutations alter epigenetic regulation; they are more frequent in PMF than in PV and ET. DNMT3A and TET2 mutations appear to lead to the activation of inflammatory pathways, notably NF-κB signalling (68, 69). In PMF, ASXL1 mutations are associated with increased white blood cell counts, and reduced survival (70). Mutations in the SRSF2 gene, which encodes a splicing factor, cause aberrant splicing that enhances differentiation towards the monocyte and megakaryocyte lineages (71, 72). SRSF2 mutations do not alter MPN phenotype but they are associated with inferior survival in PV, ET and PMF (70–73). Mutations in the SF3B1 gene, which encodes a splicing factor subunit, alter RNA splicing and are associated with the presence of ringed sideroblasts. SF3B1 mutations are frequent in patients with refractory anemia with ringed sideroblasts (RARS), with myelodysplastic/myeloproliferative neoplasms with ringed sideroblasts and thrombocytosis (RARS-T), and also in up to 14% PMF patients (73–76). Like DNMT3A, TET2, ASXL1 and EZH2 mutations, SRSF2 and SF3B1 mutations are more frequent in PMF than in PV and ET.
Chronic inflammation in MPNs
Chronic inflammation is a long-established hallmark of all MPN subtypes, and PMF is associated with the most severe level of inflammation. Pro-inflammatory cytokines IL-1 and IL-6 stimulate the production of leukocytes and megakaryocytes, which in turn secrete a number of pro-inflammatory molecules (including IL-6), thus reinforcing chronic inflammation and the production of myeloid cells, and increasing the risk of mutation of myeloid progenitors (77, 78).
Mutation-dependent inflammation
The discovery of driving and non-driving mutations in MPNs prompted researchers to investigate whether these mutations could explain the inflammation associated with MPNs. Then JAK inhibitors tested in PMF patients showed efficacy on clinical symptoms, spleen size and inflammation cytokine levels. Of note, most JAK inhibitors block JAK1 as well as JAK2, and JAK1 activation is required for the production of major inflammation cytokines, particularly IL-1 and IL-6 (79). Different pathogenic models were proposed, where MPN-associated inflammation could be either the consequence of the JAK2V617F mutation in the MPN clone (i.e. “clonal inflammation”), or an early event predisposing patients to the acquisition of JAK/STAT-activating mutations in myeloid progenitors and the development of MPN (10–13, 31, 32).
In recent years it has been demonstrated that most inflammation-linked cytokines or receptors produced in excess in MPNs were not directly linked to JAK2V617F, nor to CALR mutations; in vitro only IL-1β, IL-1Rα and IP-10 were induced by JAK2V617F (17). In turn, increased levels of IL-1β in blood or bone marrow presumably enhance the production of inflammatory cytokines, notably by monocytes and macrophages. This model has been validated in JAK2V617F-expressing mice, where the knockout of IL-1β resulted in reduced inflammatory cytokine levels, and decreased megakaryopoeisis and myelofibrosis (80, 81). In contrast, there is no evidence that CALR or MPL mutations can induce cytokine production: the main cytokines found in excess in CALR-mutated ET (IL-4, IL-9, IL-26) are typically produced by non-mutated T-cells (17). Thus mutation-independent inflammation is likely more important in CALR/MPL-mutated MPNs than in JAK2V617F-mutated MPNs, and possibly more frequently an early event in CALR/MPL-mutated MPNs.
The role played by non-driving mutations in the inflammation associated with MPNs has also be investigated. Several groups reported that mutations in the DNMT3A, TET2, SRSF2, SF3B1 genes could all result, indirectly, in the activation of the NF-κB signaling pathway (13, 68, 69). NF-κB is a major inducer of inflammatory cytokines (IL-1β, TNFα, TGF-β), and the crosstalk of NF-kB with other signaling pathways and the inflammasome is important (82). In addition, NF-κB regulates essential functions of monocytes and macrophages (M1/M2 polarization, activation, apoptosis). Thus DNMT3A, TET2, SRSF2 and SF3B1 mutations may contribute to increase inflammation in the subsets of MPN patients who carry these mutations. Of note, inflammation linked to concomitant mutations may precede the acquisition of mutations in the JAK2/CALR/MPL genes.
These findings do not explain MPN phenotype, but they have important consequences for therapy: they imply that in addition to JAK inhibitors, blocking major inflammatory cytokines in MPNs should be considered (80–84). The efficacy of this approach has been proven in JAK2V617F-expressing mice, where inhibition of IL-1β with anti-IL-1β antibody alone or in combination with ruxolitinib had beneficial effects on myelofibrosis and osteosclerosis (81). In fact, important mechanisms of action of IFN-α therapy include the repression of IL-1β and IL-1β-induced cytokines, as well as the NF-κB and c-MET/HGF pathways, which explains that long-term complete remissions can be obtained with IFN-α in both JAK2V617F- and CALR-mutated MPNs (13, 33, 46–48, 85–88). Consistently, IFN-α2 and JAK inhibitors were reported to act in synergy in MPNs (49, 50, 88). However, TET2, DNMT3A, ASLX1, EZH2 mutations are associated with inferior responses to IFN-α therapy (88).
Mutation-independent inflammation anterior to MPN
The link between inflammation and cancer is proven, especially for inflammation due to chronic infections (89, 90). Indeed, chronic inflammation may be a consequence of infection, lipid oxidation, metabolism disorders, auto-immunity. In older individuals, clonal inflammation may exist, linked to certain early genetic events (for instance, mutations in JAK2, TET2, DNMT3A, SRSF2, SF3B1…). Causes of inflammation other than genetic alterations have been investigated in myeloid malignancies. These include smoking, chronic inflammatory diseases, auto-immunity, metabolism disorders (13, 31, 32, 91–95). This field of research is important since specific causes of mutation-independent inflammation could become useful new targets in MPN therapy for subsets of patients.
Chronic inflammatory conditions or diseases
As a major risk of cell transformation, chronic inflammation likely facilitates the development of subsets of MPNs. During chronic inflammation, high levels of IL-6 stimulate the production of leukocytes and platelets, and increase the levels of hepcidin, a molecule that binds to ferroportin and inhibits iron absorption, thus decreasing the iron level in blood. The iron cycle is significantly disturbed during inflammation, notably via the repression of ferroportin expression, and altered synthesis of ferritin (increased) and transferrin (decreased). Thus, chronic inflammation is characterized by mild elevations of leukocyte and platelet counts, and impaired erythropoiesis despite important iron stocks, eventually resulting in anemia (Figure 1B). Acquisition of the JAK2V617F mutation in the context of chronic inflammation may counter or correct anemia, and increase leukocytosis and thrombocytosis. In contrast, the effect of CALR or MPL mutations would be restricted to a strong increase in thrombo-cytosis. Of note, iron deficiency is typically observed at the time of diagnosis in PV patients, but not in ET patients, and iron depletion is achieved in low-risk PV with phlebotomies (96).
It is now demonstrated that certain chronic inflammatory conditions can precede the development of a MPN: those inflammatory conditions include smoking, obesity, chronic inflammatory diseases such as Crohn disease, inflammatory bowel disease (IBD), polymyalgia rheumatica, giant cell arteritis (31, 32, 91–95, 97). Moreover, the 46/1 haplotype of JAK2, possibly a marker of inappropriate myeloid cell response to cytokine stimulation, has been shown to pre-dispose carriers to IBD and myeloid malignancies, notably MPNs (with or without mutation of JAK2) and acute myeloid leukemia (98, 99). Interestingly, the JAK2 46/1 haplotype contains two other genes, INSL6 and INSL4, in addition to JAK2. INSL6 and INSL4 encode insulin-like peptides, expressed in brain, gonads, placenta, not in healthy hematopoietic stem cells. In non-hematopoietic cancer cells, INSL4 expression can result in an autocrine loop, and INSL4 has been proposed as a cancer prognostic marker (100).
Inflammation may also be due to chronic infection, and infections have been shown to be associated with myeloid malignancies, including MPNs (cellulitis) (101). In addition, chronic infection may lead to myeloid malignancy by facilitating the acquisition of DNMT3A mutations, hereby causing clonal myelopoiesis and further inflammation (68, 69, 102).
Auto-immunity
Non-genetic pathogenic mechanisms such as chronic antigen stimulation and antigen-driven selection are implicated in the pathogenesis of blood malignancies. Prior history of any autoimmune disease confers a significant risk of developing a myeloid malignancy, notably a MPN; the autoimmune diseases concerned include immune thrombocytopenic purpura and aplastic anemia (91–93). In MPNs, chronic immune stimulation may facilitate clonal evolution and/or progression toward myelofibrosis.
Recently, autoantibodies reactive against pro-inflammatory glucosylsphingoside (GlcSph), also called lysoglucosylceramide (LGL1), were detected in 20% MPN (especially ET and PMF) patients, and 40% myeloma patients, which implied that an auto-immune process accompanied the development of MPN or myeloma disease in these patients (17, 103, 104). Accumulation of GlcSph is a hallmark of Gaucher disease (GD), where it is a consequence of germline mutations in the glucocerebrosidase (GBA) gene; subsets of GD patients develop GlcSph-reactive autoantibodies. Interestingly, GD patients present with chronic inflammation (with high levels of IL-1β, HGF, IL-8, MIP-1β, TNF-α), various clinical manifestations, and an increased risk of blood malignancies (105, 106). Intriguingly, MPN patients have slightly elevated GlcSph levels compared to healthy controls (17). One hypothesis is that anti-GlcSph autoantibodies contribute to reduce the GlcSph level in blood.
Diet and metabolism
Inflammation may also be diet-induced. The influence of dietary factors on the risk of MPN has been investigated: the only finding was that a high intake of caffeine protects against PV (107). In contrast, obesity elevates the risk for clonal hematopoiesis and MPN, especially ET (108, 109). A high-fat diet predisposes to chronic inflammation, leukocytosis and thrombocytosis, whereas adherence to a Mediterranean diet has been shown to reduce symptoms in MPN patients (107–110). Moreover, as stated above, high levels of certain glucolipids in blood are associated with an increased risk of MPN (105, 106).
Discussion
According to present knowledge, MPNs result from the combination of acquired mutations (JAK2/STAT5 activating driving mutations and/or concomitant mutations), chronic inflammation (of various origins, mutation-dependent or independent) and for a minority of individuals, of germline genetic pre-disposition to MPNs. Hence, whenever possible, better addressing the causes of mutation-independent inflammation (smoking, high-fat diet, inflammatory and autoimmune diseases) and iron deficiency) should prevent or reduce the risk of MPN. Moreover, to be curative MPN treatments should target mutations, eliminate disease-initiating stem cells, and suppress the production of inflammatory cytokines and causes of mutation-independent inflammation. Among present treatments, IFN-α2 and JAK inhibitors counter both inflammation and JAK2/STAT5 driving mutations, with partial results for JAK1/2 inhibitors (which do not act on NF-κB-dependent inflammation) and complete remissions for IFN-α2 (which counters inflammation more broadly) (33, 37, 47, 48, 86). Importantly, JAK inhibitors and IFN-α2 can act in synergy (49, 50). Further studies are needed to demonstrate the interest of using a JAK inhibitor/IFN-α2 combination therapy to eliminate the MPN clone in the early stages of MPN disease.
Presented in Table 1, the different pathogenic events, cytokines and other molecules associated with increased erythropoiesis (increased hematocrit, possible PV phenotype), thrombocytosis (possible ET phenotype), myelofibrosis (primary or secondary), or overproduction of specific inflammatory cytokines, constitute new potential therapeutic targets for those MPN patients who present with such characteristics. For instance, a better knowledge of the iron stocks and iron metabolism of patients, and the correction of iron deficiency, could help prevent the development of ET. Inversely, depletion of iron stocks or reduction of iron availability have been part of the treatment of PV for decades via phlebotomies; hepcidin mimetics and ferroportin inhibitors offer new therapeutic options (96).
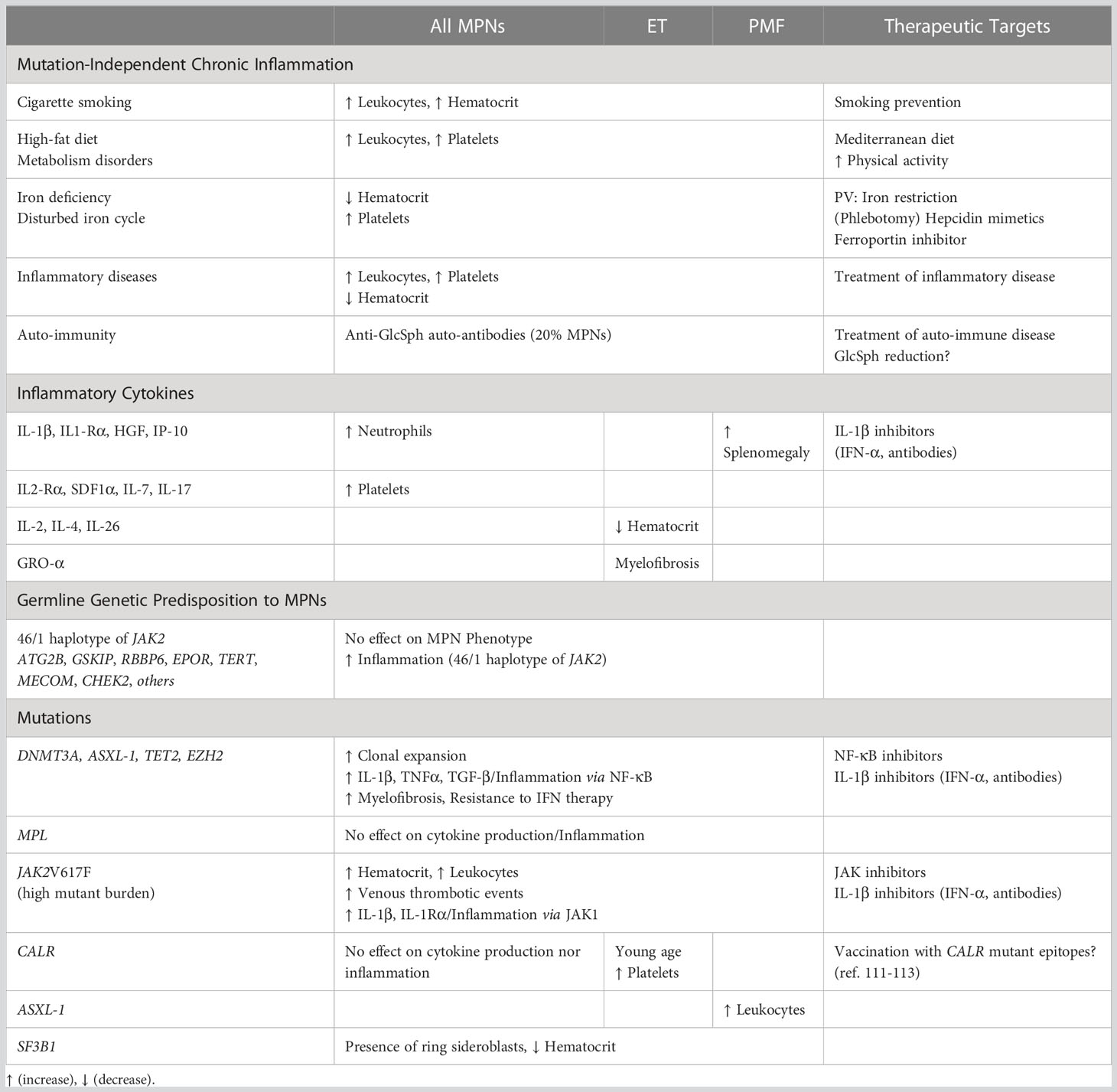
Table 1 Impact of mutations, cytokines and inflammatory conditions on MPN phenotype.
Importantly, searching for causes of inflammation in patients other than mutations may contribute to improve their response to treatment in case of established MPN, and help reduce the risk of developing a MPN in older individuals. For instance, prevention of smoking should reduce the risk for PV, and prevention of obesity, via increased physical activity and an improved diet, would be expected to reduce the risk for ET. A more systematic search for and treatment of undiagnosed chronic inflammatory or/and autoimmune diseases should help reduce inflammation and the associated risk of acquired MPN-driving mutations in healthy myeloid cells, or additional mutations in the MPN clone. In patients with proven autoimmunity against GlcSph, GlcSph could become a new target in MPN therapy, since GlcSph can be reduced with existing treatments (105, 106).
Other new potential therapeutic targets in MPNs include certain cytokines, particularly IL-1β, which can be inhibited efficiently with IFN-α, and also with anti-IL-1β antibodies or NF-κB inhibitors. Finally, because of the immunogenicity of CALR exon 9 mutants, patients with CALR-mutated ET or PMF may benefit from CALR mutant peptide vaccination (111–113).
Author contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Conflict of interest
The author declares the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Staerk J, Constantinescu SN. The JAK-STAT pathway and hematopoietic stem cells from the JAK2 V617F perspective. JAKSTAT (2012) 1:184–90. doi: 10.4161/jkst.22071
2. James C, Ugo V, Le Couédic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature (2005) 434:1144–8. doi: 10.1038/nature03546
3. Kralovics R, Passamonti F, Buser AS, Teo SS, Tiedt R, Passweg JP, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. New Engl J Med (2005) 352:1779–90. doi: 10.1056/NEJMoa051113
4. Pikman Y, Lee BH, Mercher T, McDowell E, Ebert BL, Gozo M, et al. MPLW515L is a novel somatic activating mutation in myelofibrosis with myeloid metaplasia. PloS Med (2006) 3(7):e270. doi: 10.1371/journal.pmed.0030270
5. Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. New Engl J Med (2013) 369:2379–90. doi: 10.1056/NEJMoa1311347
6. Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with non-mutated JAK2. New Engl J Med (2013) 369:2391–405. doi: 10.1056/NEJMoa1312542
7. Skoda RC, Duek A, Grisouard J. Pathogenesis of myeloproliferative neoplasms. Exp Hematol (2015) 43(8):599–608. doi: 10.1016/j.exphem.2015.06.007
8. Kleppe JM, Kwak M, Koppikar P, Riester M, Keller M, Bastian L, et al. JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response. Cancer Discovery (2015) 5:316–31. doi: 10.1158/2159-8290.CD-14-0736
9. Greenfield G, McMullin MF, Mills K. Molecular pathogenesis of the myeloproliferative neoplasms. J Hematol Oncol (2021) 14:103. doi: 10.1186/s13045-021-01116-z
10. Hasselbalch HC, Bjørn ME. MPNs as inflammatory diseases: the evidence, consequences, and perspectives. Mediators Inflammation (2015) 2015:102476. doi: 10.1155/2015/102476
11. Hasselbalch HC. Chronic inflammation as a promotor of mutagenesis in essential thrombo-cythemia, polycythemia vera and myelofibrosis. a human inflammation model for cancer development? Leukemia Res (2013) 37:214–20. doi: 10.1016/j.leukres.2012.10.020
12. Hermouet S. Pathogenesis of myeloproliferative neoplasms: more than mutations. Exp Hematol (2015) 43:993–4. doi: 10.1016/j.exphem.2015.08.014
13. Hermouet S, Bigot-Corbel E, Gardie B. Pathogenesis of myeloproliferative neoplasms: role and mechanisms of chronic inflammation. Mediators Inflammation (2015) 2015:145293. doi: 10.1155/2015/145293
14. Vaidya R, Gangat N, Jimma T, Finke CM, Lasho TL, Pardanani A, et al. Plasma cytokines in polycythemia vera: phenotypic correlates, prognostic relevance, and comparison with myelofibrosis. Am J Hematol (2012) 87:1003–5. doi: 10.1002/ajh.23295
15. Pardanani A, Finke C, Abdelrahman RA, Lasho TL, Hanson CA, Tefferi A. Increased circulating IL-2Rα (CD25) predicts poor outcome in both indolent and aggressive forms of mastocytosis: a comprehensive cytokine-phenotype study. Leukemia (2013) 27:1430–3. doi: 10.1038/leu.2013.11
16. Hasselbalch HC. The role of cytokines in the initiation and progression of myelofibrosis. Cytokine Growth Factor Rev (2013) 24:133–45. doi: 10.1016/j.cytogfr.2013.01.004
17. Allain-Maillet S, Bosseboeuf A, Mennesson N, Bostoën M, Dufeu L, Choi E-H, et al. Anti-glucosylsphingosine autoimmunity, JAK2V617F-dependent interleukin-1β and JAK2V617F-independent cytokines in myeloproliferative neoplasms. Cancers (Basel) (2020) 12:E2446. doi: 10.3390/cancers12092446
18. Hasselbalch HC. Cytokine profiling as a novel complementary tool to predict prognosis in MPNs? Hemasphere (2020) 4:e407. doi: 10.1097/HS9.0000000000000407
19. Øbro NF, Grinfeld J, Belmonte M, Irvine M, Shepherd MS, Rao TN, et al. Longitudinal cytokine profiling identifies GRO-α and EGF as potential biomarkers of disease progression in essential thrombocythemia. Hemasphere (2020) 4:e371. doi: 10.1097/HS9.0000000000000371
20. Geyer HL, Dueck AC, Scherber RM, Mesa RA. Impact of inflammation on myeloproliferative neoplasm symptom development. Mediators Inflammation (2015) 2015:284706. doi: 10.1155/2015/284706
21. Vainchenker W, Kralovics R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood (2017) 129:667–79. doi: 10.1182/blood-2016-10-695940
22. Challen GA, Sun D, Jeong M, Luo M, Jelinek J, Berg JS, et al. DNMT3A is essential for hematopoietic stem cell differentiation. Nat Genet (2011) 44:23–31. doi: 10.1038/ng.1009
23. Kameda T, Shide K, Yamaji T, Kamiunten A, Sekine M, Taniguchi Y, et al. Loss of TET2 has dual roles in murine myeloproliferative neoplasms: disease sustainer and disease accelerator. Blood (2015) 125:304–15. doi: 10.1182/blood-2014-04-555508
24. Chen E, Schneider RK, Breyfogle LJ, Rosen EA, Poveromo L, Elf S, et al. Distinct effects of concomitant Jak2V617F expression and Tet2 loss in mice promote disease progression in myeloproliferative neoplasms. Blood (2015) 125:327–35. doi: 10.1182/blood-2014-04-567024
25. Shimizu T, Kubovcakova L, Nienhold R, Zmajkovic J, Meyer SC, Hao-Shen H, et al. Loss of Ezh2 synergizes with JAK2-V617F in initiating myeloproliferative neoplasms and promoting myelofibrosis. J Exp Med (2016) 213:1479–96. doi: 10.1084/jem.20151136
26. Shi H, Yamamoto S, Sheng M, Bai J, Zhang P, Chen R, et al. ASXL1 plays an important role in erythropoiesis. Sci Rep (2016) 6:28789. doi: 10.1038/srep28789
27. Sano S, Oshima K, Wang Y, Katanasaka Y, Sano M, Walsh K. CRISPR-mediated gene editing to assess the roles of Tet2 and Dnmt3a in clonal hematopoiesis and cardiovascular disease. Circ Res (2018) 123:335–41. doi: 10.1161/CIRCRESAHA.118.313225
28. Asada S, Fujino T, Goyama S, Kitamura T. The role of ASXL1 in hematopoiesis and myeloid malignancies. Cell Mol Life Sci (2019) 76:2511–23. doi: 10.1007/s00018-019-03084-7
29. Wang B, Xia M, Chen T, Li M, Shi D, Wang X, et al. Loss of Tet2 affects platelet function but not coagulation in mice. Blood Sci (2020) 2:129–36. doi: 10.1097/BS9.0000000000000055
30. Ferrone CK, Blydt-Hansen M, Rauh MJ. Age-associated TET2 mutations: common drivers of myeloid dysfunction, cancer and cardiovascular disease. Int J Mol Sci (2020) 21:626. doi: 10.3390/ijms21020626
31. Andersen M, Sajid Z, Pedersen RK, Gudmand-Hoeyer J, Ellervik C, Skov V, et al. Mathematical modelling as a proof of concept for MPNs as a human inflammation model for cancer development. PloS One (2017) 12:e0183620. doi: 10.1371/journal.pone.0183620
32. Perner F, Perner C, Ernst T, Heidel FH. Roles of JAK2 in aging, inflammation, hematopoiesis and malignant transformation. Cells (2019) 8:854. doi: 10.3390/cells8080854
33. Kiladjian JJ, Cassinat B, Chevret S, Turlure P, Cambier N, Roussel M, et al. Pegylated interferon-alfa-2a induces complete hematologic and molecular responses with low toxicity in polycythemia vera. high molecular response rate of polycythemia vera patients treated with pegylated interferon alpha-2a. Blood (2008) 112:3065–72. doi: 10.1182/blood-2008-03-143537
34. Verstovsek S, Mesa RA, Gotlib J, Levy RS, Gupta V, DiPersio JF, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. New Engl J Med (2012) 366:799–807. doi: 10.1056/NEJMoa1110557
35. Pemmaraju N, Kantarjian H, Kadia T, Cortes J, Borthakur G, Newberry G, et al. Phase I/II study of the janus kinase (JAK)1 and 2 inhibitor ruxolitinib in patients with relapsed or refractory acute myeloid leukemia. Clin Lymphoma Myeloma Leukemia (2015) 15:171–6. doi: 10.1016/j.clml.2014.08.003
36. Greenfield G, McPherson S, Mills K, McMullin MF. The ruxolitinib effect: understanding how molecular pathogenesis and epigenetic dysregulation impact therapeutic efficacy in myeloproliferative neoplasms. J Transl Med (2018) 16:360. doi: 10.1186/s12967-018-1729-7
37. Pedersen RK, Andersen M, Knudsen TA, Sajid Z, Gudmand-Hoeyer J, Dam MJB, et al. Data-driven analysis of JAK2V617F kinetics during interferon-alpha2 treatment of patients with polycythemia vera and related neoplasms. Cancer Med (2020) 9:2039–51. doi: 10.1002/cam4.2741
38. Waksal JA, Douglas A Tremblay DA. Contemporary and future strategies in polycythemia vera. Review. Best Pract Res Clin Haematol (2022) 35(2):101370. doi: 10.1016/j.beha.2022.101370
39. Tefferi A, Tiziano Barbui T. Polycythemia vera and essential thrombocythemia: 2021 update on diagnosis, risk-stratification and management. Review. Am J Hematol (2020) 95(12):1599–613. doi: 10.1002/ajh.26008
40. Tefferi A. Primary myelofibrosis: 2023 update on diagnosis, risk-stratification, and management. Am J Hematol (2023) 98(5):801–21. doi: 10.1002/ajh.26857
41. Zhou S, Tremblay D, Hoffman R, Kremyanskaya M, Najfeld V, Li L, et al. Clinical benefit derived from decitabine therapy for advanced phases of myeloproliferative neoplasms. Acta Haematol (2021) 144(1):48–57. doi: 10.1159/000506146
42. Tefferi A, Gangat N, Pardanani A, Crispino JD. Myelofibrosis: genetic characteristics and the emerging therapeutic landscape. Cancer Res (2022) 82:749–63. doi: 10.1158/0008-5472.CAN-21-2930
43. Hasselbalch HC. Perspectives on the impact of JAK-inhibitor therapy upon inflammation-mediated comorbidities in myelofibrosis and related neoplasms. Exp Rev Hematol (2014) 7:203–16. doi: 10.1586/17474086.2013.876356
44. Li B, Rampal RK, Xiao Z. Targeted therapies for myeloproliferative neoplasms. biomark Res (2019) 7:15. doi: 10.1186/s40364-019-0166-y
45. Hobbs GS, Rozelle S, Mullally A. The development and use of janus kinase 2 inhibitors for the treatment of myeloproliferative neoplasms. Hematol Oncol Clin North Am (2017) 31:613–26. doi: 10.1016/j.hoc.2017.04.002
46. Kiladjian JJ, Chomienne C, Fenaux P. Interferon-alpha therapy in bcr-abl-negative myelo-proliferative neoplasms. Leukemia (2008) 22:1990–8. doi: 10.1038/leu.2008.280
47. Larsen TS, Katrine F Iversen KF, Esben Hansen E, Mathiasen AB, Marcher C, Frederiksen M, et al. Long term molecular responses in a cohort of Danish patients with essential thrombocythemia, polycythemia vera and myelofibrosis treated with recombinant interferon alpha. Leuk Res (2013) 37(9):1041–5. doi: 10.1016/j.leukres.2013.06.012
48. Ianotto JC, Chauveau A, Boyer-Perrard F, Gyan E, Laribi K, Cony-Makhoul P, et al. Benefits and pitfalls of pegylated interferon-α2a therapy in patients with myeloproliferative neoplasm-associated myelofibrosis: a French intergroup of myeloproliferative neoplasms (FIM) study. Haematologica (2018) 103(3):438–46. doi: 10.3324/haematol.2017.181297
49. Bjørn ME, Hasselbalch HC. Minimal residual disease or cure in MPNs? rationales and perspectives on combination therapy with interferon-alpha2 and ruxolitinib. Expert Rev Hematol (2017) 10:393–404. doi: 10.1080/17474086.2017.1284583
50. Mikkelsen SU, Kjaer L, Bjørn ME, Knudsen TA, Sørensen AL, Andersen CBL, et al. Safety and efficacy of combination therapy of interferon-α2 and ruxolitinib in polycythemia vera and myelofibrosis. Cancer Med (2018) 7:3571–81. doi: 10.1002/cam4.1619
51. Rolles B, Mullally A. Molecular pathogenesis of myeloproliferative neoplasms. Curr Hematologic Malignancy Rep (2022) 17(6):319–29. doi: 10.1007/s11899-022-00685-1
52. Tefferi A, Guglielmelli P, Larson DR, Finke C, Wassie EA, Pieri L, et al. Long-term survival and blast transformation in molecularly annotated essential thrombocythemia, polycythemia vera, and myelofibrosis. Blood (2014) 124(16):2507–13. doi: 10.1182/blood-2014-05-579136
53. Landgren O, Goldin LR, Kristinsson SY, Helgadottir EA, Samuelsson J, Bjorkholm M. Increased risks of polycythemia vera, essential thrombocythemia, and myelofibrosis among 24,577 first-degree relatives of 11,039 patients with myeloproliferative neoplasms in Sweden. Blood (2008) 112:2199–204. doi: 10.1182/blood-2008-03-143602
54. Kralovics R, Stockton DW, Prchal JT. Clonal hematopoiesis in familial polycythemia vera suggests the involvement of multiple mutational events in the early pathogenesis of the disease. Blood (2003) 102:3793–7. doi: 10.1182/blood-2003-03-0885
55. Bellanne-Chantelot C, Chaumarel I, Labopin M, Bellanger F, Barbu V, De Toma C, et al. Genetic and clinical implications of the Val617Phe JAK2 mutation in 72 families with myeloproliferative disorders. Blood (2006) 108:346–52. doi: 10.1182/blood-2005-12-4852
56. Olcaydu D, Harutyunyan A, Jager R, Berg T, Gisslinger B, Pabinger I, et al. A common JAK2 haplotype confers susceptibility to myeloproliferative neoplasms. Nat Genet (2009) 41(4):450–4. doi: 10.1038/ng.341
57. Jones AV, Chase A, Silver RT, Oscier D, Zoi K, Wang YL, et al. JAK2 haplotype is a major risk factor for the development of myeloproliferative neoplasms. Nat Genet (2009) 41(4):446–9. doi: 10.1038/ng.334
58. Kilpivaara O, Mukherjee S, Schram AM, Wadleigh M, Mullally A, Ebert BL, et al. A germline JAK2 SNP is associated with predisposition to the development of JAK2(V617F)-positive myeloproliferative neoplasms. Nat Genet (2009) 41(4):455–9. doi: 10.1038/ng.342
59. Saliba J, Saint-Martin C, Di Stefano A, Lenglet G, Marty C, Keren B, et al. Germline duplication of ATG2B and GSKIP predisposes to familial myeloid malignancies. Nat Genet (2015) 47(10):1131–40. doi: 10.1038/ng.3380
60. Harutyunyan AS, Giambruno R, Krendl C, Stukalov A, Klampfl T, Berg T, et al. Germline RBBP6 mutations in familial myeloproliferative neoplasms. Blood (2016) 127(3):362–5. doi: 10.1182/blood-2015-09-668673
61. Rabadan Moraes G, Pasquier F, Marzac C, Deconinck E, Damantii CC, Leroy G, et al. An inherited gain-of-function risk allele in EPOR predisposes to familial JAK2(V617F) myeloproliferative neoplasms. Br J Haematol (2022) 198(1):131–6. doi: 10.1111/bjh.18165
62. Oddsson A, Kristinsson SY, Helgason H, Gudbjartsson DF, Masson G, Sigurdsson A, et al. The germline sequence variant rs2736100_C in TERT associates with myeloproliferative neoplasms. Leukemia (2014) 28(6):1371–4. doi: 10.1038/leu.2014.48
63. Tapper W, Jones AV, Kralovics R, Harutyunyan AS, Zoi K, Leung W, et al. Genetic variation at MECOM, TERT, JAK2 and HBS1L-MYB predisposes to myeloproliferative neoplasms. Nat Commun (2015) 6:6691. doi: 10.1038/ncomms7691
64. Bao EL, Nandakumar SK, Liao X, Bick AG, Karjalainen J, Tabaka M, et al. Inherited myeloproliferative neoplasm risk affects haematopoietic stem cells. Nature (2020) 586:769–75. doi: 10.1038/s41586-020-2786-7
65. Lundberg P, Karow A, Nienhold R, Looser R, Hao-Shen H, Nissen I, et al. Clonal evolution and clinical correlates of somatic mutations in myeloproliferative neoplasms. Blood (2014) 123:2220–8. doi: 10.1182/blood-2013-11-537167
66. Song J, Hussaini M, Zhang H, Shao H, Qin D, Zhang X, et al. Comparison of the mutational profiles of primary myelofibrosis, polycythemia vera, and essential thrombocytosis. Am J Clin Pathol (2017) 147:444–52. doi: 10.1093/ajcp/aqw222
67. Dunbar AJ, Rampal RK, Levine R. Leukemia secondary to myeloproliferative neoplasms. Blood (2020) 136:61–70. doi: 10.1182/blood.2019000943
68. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med (2014) 371(26):2488–98. doi: 10.1056/NEJMoa1408617
69. Grabek J, Straube J, Bywater M, Lane SW. MPN: the molecular drivers of disease initiation, progression and transformation and their effect on treatment. Cells (2020) 9(8):1901. doi: 10.3390/cells9081901
70. Vannucchi AM, Lasho TL, Guglielmelli P, Biamonte F, Pardanani A, Pereira A, et al. Mutations and prognosis in primary myelofibrosis. Leukemia (2013) 27:1861–9. doi: 10.1038/leu.2013.119
71. Liang Y, Tebaldi T, Rejeski K, Joshi P, Stefani G, Taylor A, et al. SRSF2 mutations drive oncogenesis by activating a global program of aberrant alternative splicing in hematopoietic cells. Leukemia (2018) 32:2659–71. doi: 10.1038/s41375-018-0152-7
72. Smeets MF, Tan SY, Xu JJ, Anande G, Unnikrishnan A, Chalk AM, et al. SRSF2P95H initiates myeloid bias and myelodysplastic/myeloproliferative syndrome from hemopoietic stem cells. Blood (2018) 132:608–21. doi: 10.1182/blood-2018-04-845602
73. Guglielmelli P, Gangat N, Coltro G, Lasho TL, Loscocco GG, Finke CM, et al. Mutations and thrombosis in essential thrombocythemia. Blood Cancer J (2021) 11(4):77. doi: 10.1038/s41408-021-00470-y
74. Obeng EA, Chappell RJ, Seiler M, Chen MC, Campagna DR, Schmidt PJ, et al. Physiologic expression of Sf3b1K700E causes impaired erythropoiesis, aberrant splicing, and sensitivity to therapeutic spliceosome modulation. Cancer Cell (2016) 30:404–17. doi: 10.1016/j.ccell.2016.08.006
75. Tefferi A, Lasho TL, Guglielmelli P, Finke CM, Rotunno G, Elala Y, et al. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv (2016) 1:21–30. doi: 10.1182/bloodadvances.2016000216
76. Schischlik F, Jäger R, Rosebrock F, Hug E, Schuster M, Holly R, et al. Mutational landscape of the transcriptome offers putative targets for immunotherapy of myeloproliferative neoplasms. Blood (2019) 134(2):199–210. doi: 10.1182/blood.2019000519
77. Coudwell G, Machlus KR. Modulation of megakaryopoiesis and platelet production during inflammation. Review. Thromb Res (2019) 179:114–20. doi: 10.1016/j.thromres.2019.05.008
78. Varricchio L, Hoffman R. Megakaryocytes are regulators of the tumor microenvironment and malignant hematopoietic progenitor cells in myelofibrosis. Front Oncol (2022) 12:906698. doi: 10.3389/fonc.2022.906698
79. Čokić VP, Mitrović-Ajtić O, Beleslin-Čokić BB, Marković D, Buač M, Diklić M, et al. Proinflammatory cytokine IL-6 and JAK-STAT signaling pathway in myeloproliferative neoplasms. Mediators Inflammation (2015) 2015:453020. doi: 10.1155/2015/453020
80. Rai S, Hansen N, Hao-Shen H, Dirnhofer S, Tata NR, Skoda RC. IL-1β secreted from mutant cells carrying JAK2-V617F favors early clonal expansion and promotes MPN disease initiation and progression. Blood (2019) 134(Suppl. 1):307. doi: 10.1182/blood-2019-129800
81. Rai S, Grockowiak E, Hansen N, Luque Paz D, Stoll CB, Hao-Shen H, et al. Inhibition of interleukin-1β reduces myelofibrosis and osteosclerosis in mice with JAK2-V617F driven myeloproliferative neoplasm. Nat Commun (2022) 13(1):5346. doi: 10.1038/s41467-022-32927-4
82. Mussbacher M, Derler M, Basílio J, Schmid JA. NF-κB in monocytes and macrophages – an inflammatory master regulator in multitalented immune cells. Front Immunol (2023) 14:1134661. doi: 10.3389/fimmu.2023.1134661
83. Dinarello CA, van der Meer JW. Treating inflammation by blocking interleukin-1 in humans. Semin Immunol (2013) 25:469–84. doi: 10.1016/j.smim.2013.10.008
84. Arranz L, Arriero MDM, Villatoro A. Interleukin-1β as emerging therapeutic target in hematological malignancies and potentially in their complications. Blood Rev (2017) 31:306–17. doi: 10.1016/j.blre.2017.05.001
85. Boissinot M, Cleyrat C, Vilaine M, Y Jacques Y, I Corre I, S Hermouet S. Anti-inflammatory cytokines hepatocyte growth factor and interleukin-11 are over-expressed in polycythemia vera and contribute to the growth of clonal erythroblasts independently of JAK2V617F. Oncogene (2011) 30(8):990–1001. doi: 10.1038/onc.2010.479
86. Radaeva S, Jaruga B, Hong F, Kim WH, Fan S, Cai H, et al. Interferon-alpha activates multiple STAT signals and down-regulates c-met in primary human hepatocytes. Gastroenterol (2002) 122:1020–34. doi: 10.1053/gast.2002.32388
87. Verger E, Cassinat B, Chauveau A, Dosquet C, Giraudier S, Schlageter MH, et al. Clinical and molecular response to interferon-α therapy in essential thrombocythemia patients with CALR mutations. Blood (2015) 126(24):2585–91. doi: 10.1182/blood-2015-07-659060
88. Hasselbalch HC, Silver RT. New perspectives of interferon-alpha2 and inflammation in treating Philadelphia-negative chronic myeloproliferative neoplasms. HemaSphere (2021) 5:12:e645. doi: 10.1097/HS9.0000000000000645
89. Hussain SP, Harris CC. Inflammation and cancer: an ancient link with novel potentials. Int J Cancer (2007) 121(11):2373–80. doi: 10.1002/ijc.23173
90. Mantovani A, Garlanda C, Allavena P. Molecular pathways and targets in cancer-related inflammation. Ann Med (2010) 42(3):161–70. doi: 10.3109/07853890903405753
91. Anderson LA, Pfeiffer RM, Landgren O, Gadalla S, Berndt SI, Engels EA. Risks of myeloid malignancies in patients with autoimmune conditions. Br J Cancer (2009) 100(5):822–8. doi: 10.1038/sj.bjc.6604935
92. Kristinsson SY, Landgren O, Samuelsson J, Björkholm M, Goldin LR. Autoimmunity and the risk of myeloproliferative neoplasms. Haematologica (2010) 95, 7:1216–20. doi: 10.3324/haematol.2009.020412
93. Kristinsson SY, Björkholm M, Hultcrantz M, Derolf ÅR, Landgren O, Goldin LR. Chronic immune stimulation might act as a trigger for the development of acute myeloid leukemia or myelodysplastic syndromes. J Clin Oncol (2011) 29(21):2897–903. doi: 10.1200/JCO.2011.34.8540
94. Hasselbalch HC. Perspectives on chronic inflammation in essential thrombocythemia, polycythemia vera, and myelofibrosis: is chronic inflammation a trigger and driver of clonal evolution and development of accelerated atherosclerosis and second cancer? Blood (2012) 119(14):3219–25. doi: 10.1182/blood-2011-11-394775
95. Pedersen KM, Bak M, Sørensen AL, Zwisler AD, Ellervik C, Larsen MK, et al. Smoking is associated with increased risk of myeloproliferative neoplasms: a general population-based cohort study. Cancer Med (2018) 7:5796–802. doi: 10.1002/cam4.1815
96. Stetka J, Usart M, Kubovcakova L, Rai S, Nageswara Rao T, Sutter J, et al. Iron is a modifier of the phenotypes of JAK2-mutant myeloproliferative neoplasms. Blood (2023) 141(17):2127–40. doi: 10.1182/blood.2022017976
97. Marnell CS, Bick A, Natarajan P. Clonal hematopoiesis of indeterminate potential (CHIP): linking somatic mutations, hematopoiesis, chronic inflammation and cardiovascular disease. J Mol Cell Cardiol (2021) 161:98–105. doi: 10.1016/j.yjmcc.2021.07.004
98. Hermouet S, Vilaine M. The JAK2 46/1 haplotype: a marker of inappropriate myelomonocytic response to cytokine stimulation, leading to increased risk of inflammation, myeloid neoplasm, and impaired defense against infection? Haematologica (2011) 96(11):1575–9. doi: 10.3324/haematol.2011.055392
99. Ferguson LR, Han DY, Fraser AG, Huebner C, Lam WJ, Morgan AR, et al. Genetic factors in chronic inflammation: single nucleotide polymorphisms in the STAT-JAK pathway, susceptibility to DNA damage and crohn’s disease in a new Zealand population. Mutat Res (2010) 690(1-2):108–15. doi: 10.1016/j.mrfmmm.2010.01.017
100. Scopetti D, Piobbico D, Brunacci C, Pieroni S, Bellezza G, Castelli M, et al. INSL4 as prognostic marker for proliferation and invasiveness in non-Small-Cell lung cancer. J Cancer (2021) 12(13):3781–95. doi: 10.7150/jca.51332
101. Titmarsh GJ, McMullin MF, McShane CM, Clarke M, Engels EA, Anderson LA. Community-acquired infections and their association with myeloid malignancies. Cancer Epidemiol (2014) 38(1):56–61. doi: 10.1016/j.canep.2013.10.009
102. Hormaechea-Agulla D, Matatall KA, Le DT, Kain B, Long X, Kus P, et al. Chronic infection drives DNMT3A-loss-of-function clonal hematopoiesis via IFNγ signaling. Cell Stem Cell (2021) 28:1428–1442.e6. doi: 10.1016/j.stem.2021.03.002
103. Nair S, Branagan A, Liu J, Boddupalli CS, Mistry PK, Dhodapkar MV. Clonal immunoglobulin against lysolipids in the origin of myeloma. N Engl J Med (2016) 374:555–61. doi: 10.1056/NEJMoa1508808
104. Bosseboeuf A, Mennesson N, Allain-Maillet S, Tallet A, Piver E, Moreau C, et al. Characteristics of MGUS and multiple myeloma according to the target of monoclonal immunoglobulins, glucosyl-sphingosine, or Epstein-Barr virus EBNA-1. Cancers (Basel) (2020) 12(5):E1254. doi: 10.3390/cancers12051254
105. Mistry P, Taddei TH, Dahl SV, Rosenbloom BE. Gaucher disease and malignancy: a model for cancer pathogenesis in an inborn error of metabolism. Crit Rev Oncol (2013) 18:235–46. doi: 10.1615/critrevoncog.2013006145
106. Linari S, Castaman G. Hematological manifestations and complications of gaucher disease. Expert Rev Hematol (2015) 9:51–8. doi: 10.1586/17474086.2016.1112732
107. Ramanathan G, Hoover BM, Fleischman AG. Impact of host, lifestyle and environmental factors in the pathogenesis of MPN. Cancers (Basel) (2020) 12(8):2038. doi: 10.3390/cancers12082038
108. Duncombe AS, Anderson LA, James G, de Vocht F, Fritschi L, Mesa R, et al. Modifiable lifestyle and medical risk factors associated with myeloproliferative neoplasms. HemaSphere (2020) 4:e327. doi: 10.1097/HS9.0000000000000327
109. Pasupuleti SK, Ramdas B, Burns SS, Palam LR, Kanumuri R, Kumar R, et al. Obesity induced inflammation exacerbates clonal hematopoiesis. J Clin Invest (2023) e163968. doi: 10.1172/JCI163968
110. Valet C, Batut A, Vauclard A, Dortignac A, Bellio M, Payrastre B, et al. Adipocyte fatty acid transfer supports megakaryocyte maturation. Cell Rep (2020) 32:107875. doi: 10.1016/j.celrep.2020.107875
111. Holmström MO, Martinenaite E, Ahmad SM, Met Ö, Friese C, Kjaer L, et al. The calreticulin (CALR) exon 9 mutations are promising targets for cancer immune therapy. Leukemia (2018) 32:429–37. doi: 10.1038/leu.2017.214
112. Gigoux M, Holmström MO, Zappasodi R, Park JJ, Pourpe S, Bozkus CC, et al. Calreticulin mutant myeloproliferative neoplasms induce MHC-I skewing, which can be overcome by an optimized peptide cancer vaccine. Sci Transl Med (2022) 14(649):eaba4380. doi: 10.1126/scitranslmed.aba4380
Keywords: myeloproliferative neoplasms (MPN), polycythemia vera, essential thrombocythemia inflammation, therapeutic targets, primary fibrosis (PMF), mutations
Citation: Hermouet S (2023) Mutations, inflammation and phenotype of myeloproliferative neoplasms. Front. Oncol. 13:1196817. doi: 10.3389/fonc.2023.1196817
Received: 30 March 2023; Accepted: 09 May 2023;
Published: 22 May 2023.
Edited by:
Elisabeth Oppliger Leibundgut, University Hospital Bern, SwitzerlandReviewed by:
Anna Marneth, Radboud University Medical Centre, NetherlandsCopyright © 2023 Hermouet. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sylvie Hermouet, c3lsdmllLmhlcm1vdWV0QHVuaXYtbmFudGVzLmZy