 Eric S. Christenson1,2*
Eric S. Christenson1,2* Hua-Ling Tsai1,3Dung T. Le1,2Elizabeth M. Jaffee1,2Jonathan Dudley1,4Rena R. Xian1,4Christopher D. Gocke1,4James R. Eshleman1,4
Hua-Ling Tsai1,3Dung T. Le1,2Elizabeth M. Jaffee1,2Jonathan Dudley1,4Rena R. Xian1,4Christopher D. Gocke1,4James R. Eshleman1,4 Ming-Tseh Lin4
Ming-Tseh Lin4- 1Department of Oncology, Sidney Kimmel Comprehensive Cancer Center at Johns Hopkins, Baltimore, MD, United States
- 2The Cancer Convergence Institute at Johns Hopkins, Johns Hopkins University School of Medicine, Baltimore, MD, United States
- 3Division of Quantitative Sciences, Johns Hopkins University, Baltimore, MD, United States
- 4Department of Pathology, Johns Hopkins University, Baltimore, MD, United States
Introduction: The highest incidence of colorectal cancer (CRC) is in patients diagnosed at 80 years or older highlighting a need for understanding the clinical and molecular features of these tumors. Methods. In this retrospective cohort study, 544 CRCs underwent next generation sequencing and mismatch repair (MMR) evaluation. Molecular and clinical features were compared between 251 patients with traditional-onset CRC (50-69 years at diagnosis) and 60 with late-onset CRC (>80 years at diagnosis).
Results: Late-onset CRC showed a significantly higher rate of right-sided tumors (82% vs 35%), MMR deficiency (35% vs. 8%) and BRAF p.V600E mutations (35% vs. 8%) and a significantly lower rate of stage IV disease (15% vs 28%) and APC mutations (52% vs. 78%). Association of these features with advanced age was supported by stratifying patients into 6 age groups (<40, 40-49, 50-59, 60-69, 70-79 and >80 years). However, the age-related rise in MMR deficient (dMMR) CRC was only seen in the female patients with an incidence of 48% (vs. 10% in the male patient) in the >80y group. In addition, BRAF p.V600E was significantly enriched in MMR deficient CRC of advanced age (67% in late-onset CRC). Categorizing CRC by mutational profiling, late-onset CRC revealed a significantly higher rate of dMMR/BRAF+APC- (18% vs. 2.0%), dMMR/BRAF-APC- (8.3% vs. 1.2%) and MMR proficient (pMMR)/BRAF+APC- (12% vs. 4.0%) as compared to traditional-onset CRC.
Discussion: In summary, there was a higher rate of dMMR and BRAF p.V600E in late-onset CRC, independently or in combination. The higher incidence of dMMR in late-onset CRC in females is most likely predominantly driven by BRAF p.V600E induced hypermethylation. Prospective studies with treatment plans designed specifically for these older patients are warranted to improve their outcomes.
Introduction
While important progress has been made in the treatment of colorectal cancer (CRC) over the last decade, CRC still represents the second leading cause of cancer related death in the United States (1, 2). Recently, a disturbing rise in CRC incidence amongst young-adults has increased emphasis on understanding age-related differences in clinical presentation and molecular features of CRC (3). Investigations in this area have primarily focused on exploring the differences between these early-onset CRC and those that occur in individuals diagnosed at age 50 and above. However, the highest incidence of CRC is seen in patients at age 80 and older highlighting the importance of also understanding how tumors that occur in this age group might differ from the general population (2).
Several prior groups have sought to address this question with some cohorts reporting BRAF and KRAS mutations to be more common in CRC seen in patients with later age of diagnosis which is particularly relevant given the implications of these mutations for targeted therapies (4–8). Several of these cohorts additionally suggested that this higher rate of BRAF mutations translated into an elevated rate of mismatch repair deficient (dMMR) disease while another did not show any increase in dMMR disease (9–11). Given the higher risk and toxicity with surgery and chemotherapy in patients of advanced age, understanding the prevalence of targetable and/or prognostic genomic markers, such as KRAS, NRAS and BRAF p.V600E mutations and MMR deficiency is critical to guide management (12, 13).
Materials and methods
To investigate how age impacts the clinical and molecular features of CRC in the Johns Hopkins University (JHU) catchment area, all CRC tumors which underwent molecular testing following the launch of our solid tumor next-generation sequencing (NGS) panel in September 2017 through January 2020 were systematically collected along with the patient’s clinical data. The Johns Hopkins Medicine institutional review board granted approval to this study.
Clinical features
Patient’s age at diagnosis and gender as well as other clinical features of the tumor were recorded. These included primary site of disease, histologic grade and TNM staging as determined by the seventh or eighth edition of the American Joint Committee on Cancer staging system as recommended at the time of pathologic evaluation.
Mismatch repair deficiency
MMR status was determined by using immunohistochemistry (IHC) stains and/or microsatellite instability assay. IHC results for MMR proteins (MLH1, MSH2, MSH and PMS2) were obtained from surgical pathology reports. Microsatellite stability assay was determined by comparing 5 mononucleotide microsatellite loci between neoplastic and nonneoplastic tissues using the MSI Analysis System Version 1.2 (Promega, Madison, WI) as described previously (14).
Molecular testing by next generation sequencing
All patients underwent testing on an internal solid tumor NGS panel as described previously (15–17). Briefly, DNA was extracted from formalin fixed, paraffin embedded tissues using the Tissue Preparation System (Siemens, Berlin, German), and measured by Qubit 2.0 Fluorometer (Life Technologies, Carlsbad, California). Libraries were prepared using the SureSelect-XT Target Enrichment System (Agilent Technologies Santa Clara, CA) and an Agilent custom panel covering full coding regions of over 400 cancer-associated genes (https://pathology.jhu.edu/jhml-services/assets/test-directory/SolidTumorPanel-II_GeneList_v5.0). NGS was performed to an average 500-1000x read depth by the HiSeq 2500 or NovaSeq 6000 system platform using Illumina paired end technology (Illumina, San Diego, CA, USA). All reads were aligned to human reference sequence genome assembly hg19 (NCBI build GRCh37) using the Burrows-Wheeler alignment (BWA) algorithm. BMA files were generated using Picard Tools v1.119 and variant calling was performed using an in-house variant caller algorithm as well as a third-party variant caller (Haplotyper Genome Analysis TK-3.3). Variants were reviewed using the Integrated Genomics Viewer (Broad Institute, Cambridge, MA) and annotated utilizing the COSMIC, dbSNP and Annovar databases (18). The limit of detection was 5% mutant allele. A panel of hotspot mutations was reviewed using the Integrated Genomics Viewer at a threshold of 2% mutant allele. This included codons 12, 13, 59, 61, 117 and 146 of the KRAS and NRAS gene and codon 600 of the BRAF gene. The assay was validated for clinical reporting at a Clinical Laboratory Improvement Amendments (CLIA) certified clinical laboratory.
To minimize multiple comparisons, molecular markers analyzed in the current study included only those recommended as standard-of-care (BRAF, KRAS, NRAS mutations and MMR deficiency) according to the guidelines from the American Society for Clinical Pathology, College of American Pathologists, Association for Molecular Pathology and the American Society of Clinical Oncology and those with a higher incidence (>20%) of mutations in CRC (APC, PIK3CA and TP53) (12, 19).
Statistical analysis
To determine whether patients with advanced age (80 years or older) at the time of diagnosis of CRC had molecularly and clinically distinct tumors, we compared this cohort of patients which we referred to as late-onset (LO) disease, to patients diagnosed with CRC from 50 to 69 years of age which we referred to as traditional-onset (TO) disease via Fisher exact testing or Chi-square testing.
In addition, the age-related changes on the molecular features across the entire cohort were assessed by stratification of age at the time of diagnosis as 6 groups (<40, 40-49, 50-59, 60-69, 70-79, and 80+), or by age at the time of diagnosis as a continuous variable. Patient characteristic and mutation proportion differences across age cohorts were evaluated via Chi-square testing.
Mutation profile at a patient level was presented with the correlations between mutations estimated via Cramér’s phi coefficient and tested via Pearson’s Chi-square test. Model based regression analyses were conducted via logistic regression with two‐way interaction effects between patient characteristics and age in relation to mutations examined. Non-linear relationships between age and mutations were also evaluated. In the case of significant interactions between age and mutations, the regression models were stratified by the corresponding variable (gender in our study). In the case of significant non-linear associations between age and mutations, age was categorized into three groups (≤ 49, 50-69, ≥ 70). Otherwise, the association between age and mutations was assessed by increasing decade of age at diagnosis. Multivariable analysis (MVA) was conducted with adjustments selected based on testing results between age cohorts and patient characteristics. P-values of 0.05 or lower were considered statistically significant without multiplicity adjustment. Analyses were compiled in Microsoft excel (Microsoft, Redmond, USA) and R version 4.2.2 (R Foundation for Statistical Computing, Vienna, Austria).
Results
Association of aging with clinical and molecular features
A total of 566 CRC specimens were analyzed using the solid tumor NGS panel. Twenty-two patients did not have corresponding MMR results and were excluded from statistical evaluation. Of these 22 patients, 9 were from the late-onset CRC (≥80y) representing 13% (9/69) of this population compared to 0%-2.5% of the other age groups (P = 0.001). Among the late-onset CRC with no known MMR status, wild-type APC was seen in 5 of 9 patients, including 3 patients with BRAF p.V600E.
CRC patients (n = 544) with NGS and MMR results were compared between late-onset (LO) CRC arising in older individuals (80 years and older) and traditional-onset (TO) CRC occurring between 50 to 69 years of age. In late-onset CRC, there was a significantly higher rate of female gender (LO: 67% vs. TO: 49%), right-sided primary CRCs (LO: 82% vs. TO: 35%) and a lower rate of stage IV disease at diagnosis (LO: 15%, TO: 28%) and a trend of a higher rate of high-grade histomorphology (LO:29% vs. TO:17%) (Figure 1A).
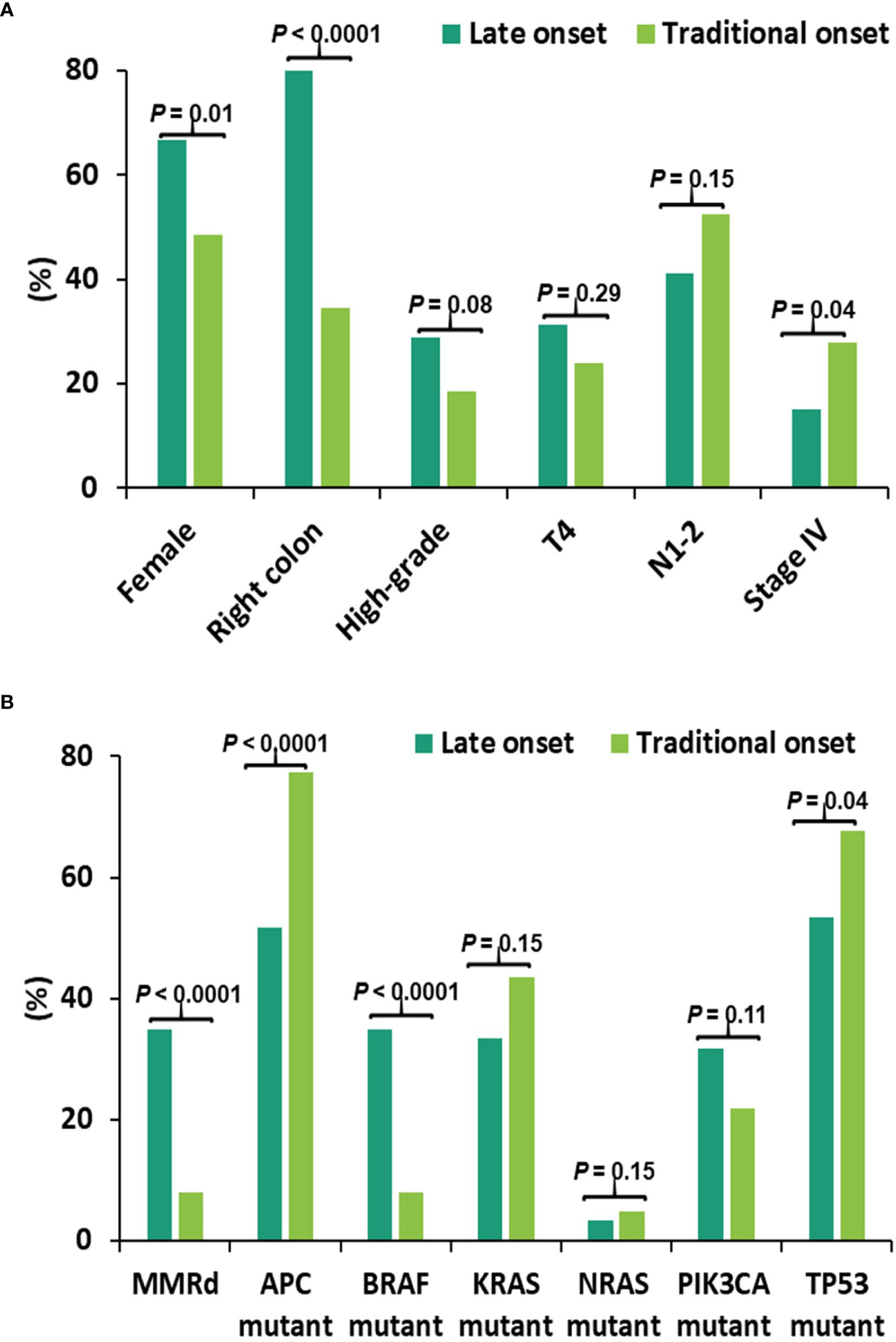
Figure 1 Clinical and molecular features in late-onset CRC (≥80y, n = 60) and traditional-onset CRC (50-69y, n = 251). (A) Clinical Features. High-grade: histomorphology of the primary tumor. T4, N1-2 and stage IV: TNM staging at diagnosis. Late-onset CRC showed a significantly higher rate of female gender and primary CRCs within right colon and a significantly lower incidence of stage IV disease at presentation. (B) Molecular Features. Late-onset CRC showed a significantly higher rate of mismatch repair deficiency (dMMR) and BRAF p.V600E mutation (BRAF mutant) and a significantly lower rate of APC and TP53 mutations.
Molecular features between these two age cohorts were also compared, identifying a higher rate of MMR deficiency (LO: 35% vs. TO: 8.0%) and BRAF p.V600E mutation (LO: 35% vs. TO: 8.0%), and a lower rate of APC (LO: 52% vs. TO: 78%) and TP53 mutations (LO: 53% vs. TO: 68%) in late-onset CRC (Figure 1B).
To determine whether these associations were discrete changes once patients reached a particular age of diagnosis or represented a continuum of clinical characteristics and molecular changes, we stratified patients according to the decade of life in which CRC had been diagnosed and patient sex. Association with clinical and molecular features was tested across each age cohort (Figure 2). This investigation revealed that there was an age-related rise in dMMR CRC only in female patients while an increase in BRAF p.V600E in older patients was present in both male and female patients. In contrast, APC mutations decreased with age in both male and female patients. In the 80y group, dMMR was detected in 19 (48%) of 40 female patients and in 2 (10%) of 20 male patients (P = 0.004), while BRAF p.V600E was seen in 16 (40%) female patients and 5 (25%) male patients (P = 0.39). Aging was significantly associated with primary CRCs within right colon, absence of regional nodal metastasis (N0) and absence of systemic metastasis at diagnosis (stage I-III) (Table 1) as well as MMR deficiency, BRAF p.V600E mutation and wild-type APC (Table 2).
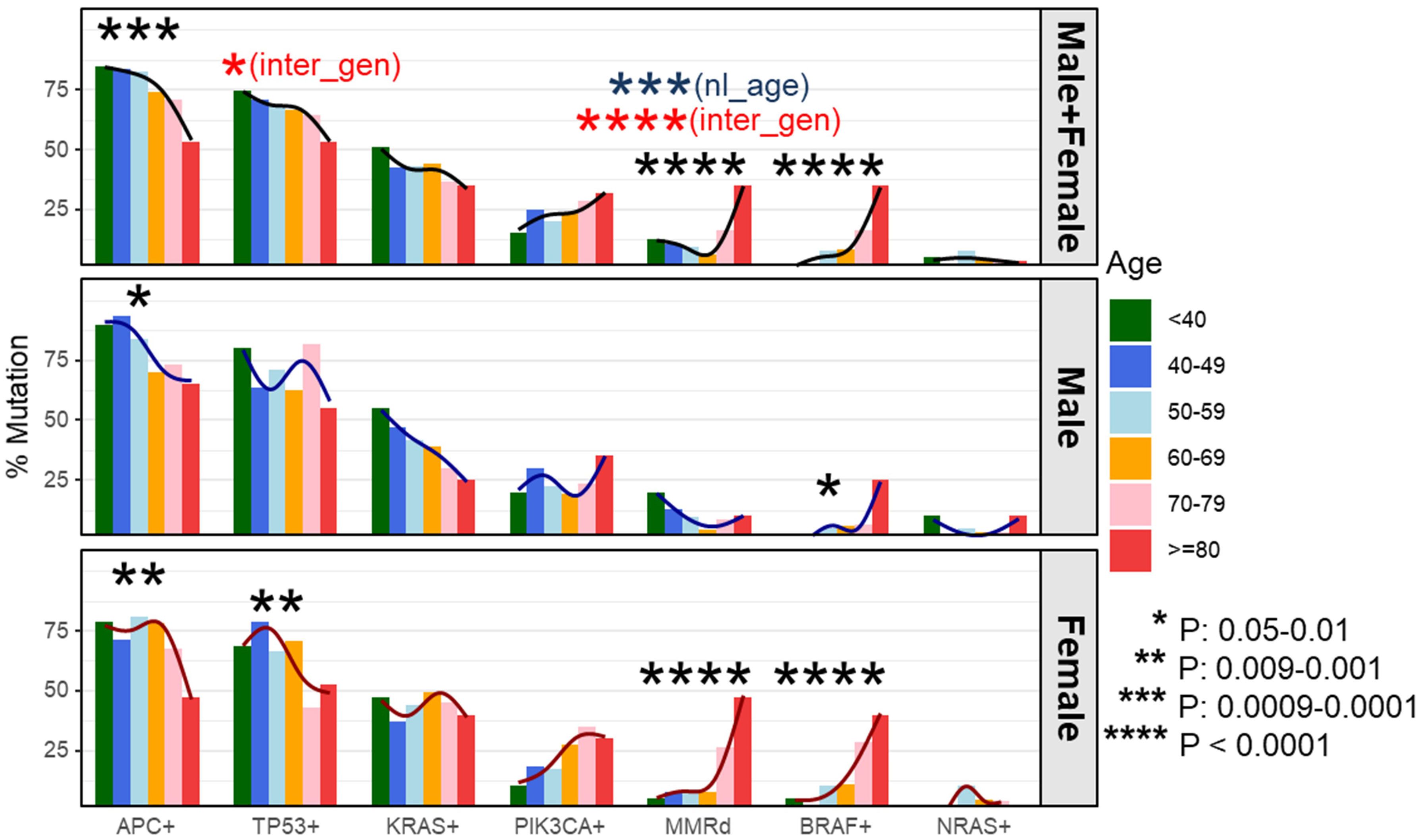
Figure 2 Percentage of dMMR/mutation by age cohorts and stratified by gender. We compared the incidence of each molecular alteration stratified by patient sex and decade of colorectal (CRC) diagnosis. This identified a negative correlation between incidence of APC mutations and age of CRC diagnosis in both male and female patients whereas a decreased incidence of TP53 mutations was only seen in female patients. In contrast, the incidence of dMMR disease and BRAF p.V600E increased with patient age at CRC diagnosis. Stratifying by patient sex, both male and female patients had a rise in BRAF p.V600E incidence with increasing age whereas the increase in dMMR disease was confined to female patients. Black curve represents a smoothing trend by age cohorts at the corresponding mutation. dMMR - mismatch repair deficiency; nl_age - testing result in the entire cohort for nonlinearity effect of age at the corresponding mutation; inter_gen - testing result in the entire cohort for interaction effect between age and gender at the corresponding mutation.
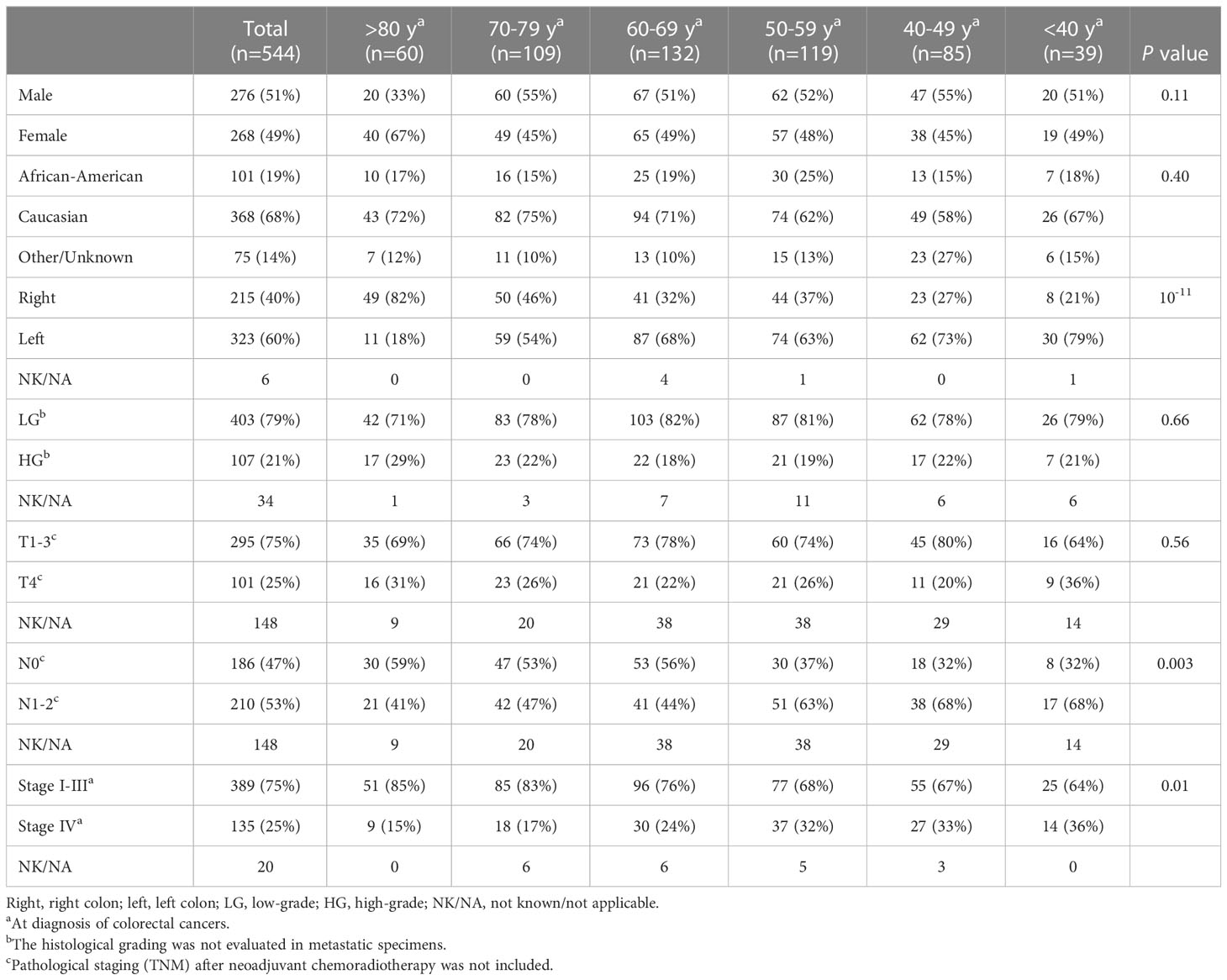
Table 1 Association of advanced age with clinical features.

Table 2 Association of advanced age with mutational profiling.
Association with dMMR, BRAF p.V600E and wild-type APC
As shown in previous cohort studies, dMMR CRCs demonstrated a significantly higher incidence of BRAF p.V600E (48% vs. 5.7%, P < 0.0001) and wild-type APC (56% vs. 22%, P < 0.0001) as compared to pMMR CRCs in the current cohort (20). To assess whether this elevated rate of BRAF p.V600E mutations might be responsible for the increase in dMMR disease amongst the late-onset CRC population, the co-occurrence between BRAF p.V600E mutations and MMR deficiency was assessed. The proportion of dMMR tumors with BRAF p.V600E was enriched with advanced age (67% in 80y group and 70-79y group, 63% in 60-69y group, 25% in 50-59y group, 11% in 40-49y group and 0% in <40y group, P = 0.003). In the overall cohort, BRAF p.V600E mutated CRC showed a significantly higher incidence of MMR deficiency compared to CRC without BRAF p.V600E (56% vs. 7.9%, P < 0.0001). BRAF p.V600E tumors also exhibited a lower rate of APC mutations (16% vs. 80%, P < 0.0001) demonstrating a strong co-segregation among these three variables (Figure 3).
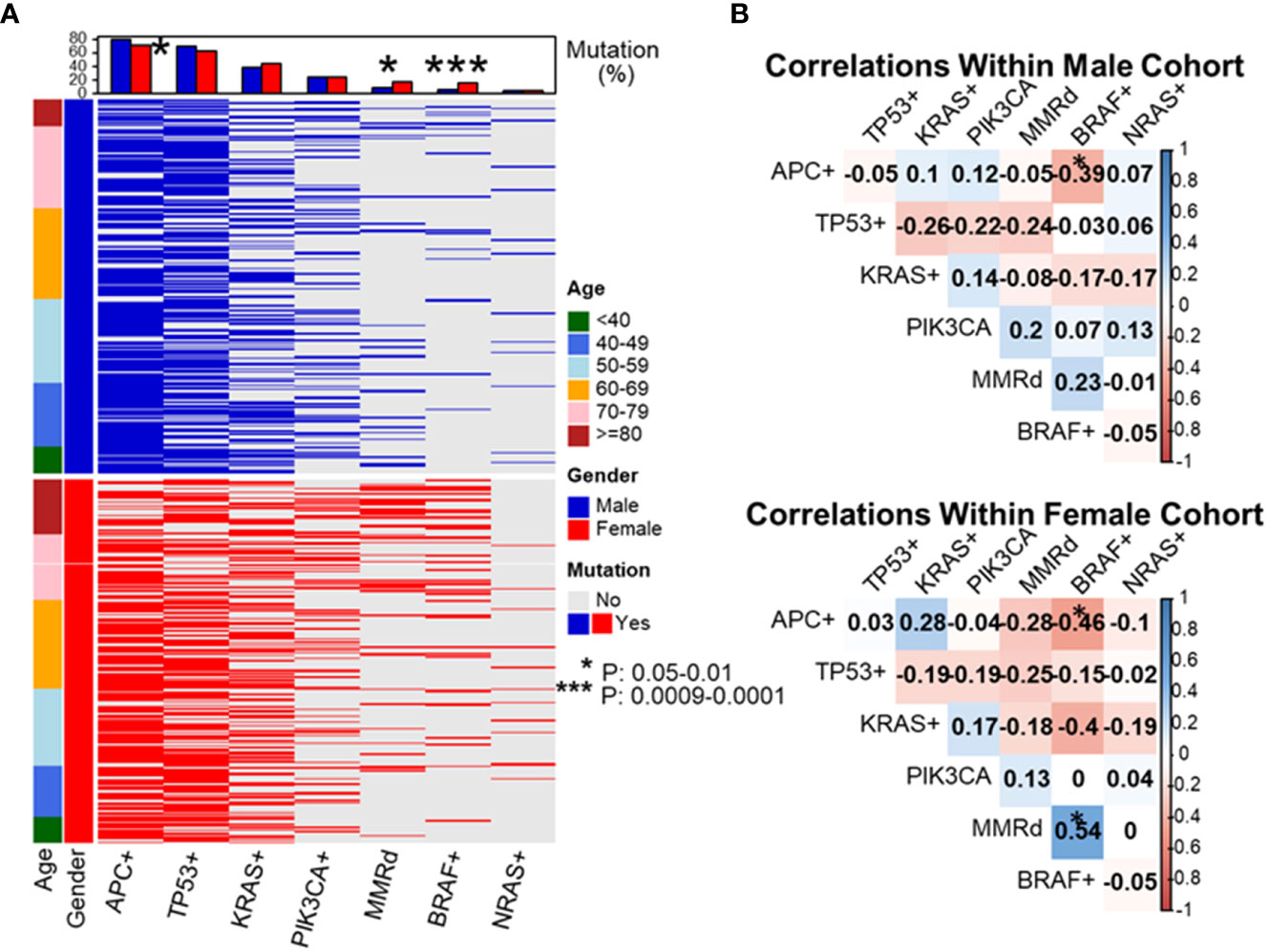
Figure 3 Evaluation of molecular features of patient tumors by decade of diagnosis. To dissect which genetic alterations tend to co-segregate or be mutual exclusive as a function of age and gender, we created a mutational profile of our patient cohort separated by gender and listing patients in ascending order of age at diagnosis (A). We then performed pairwise correlation coefficients by gender looking at the association between each individual molecular alteration and the others listed (B). These data revealed APC+ was associated with a lower rate of BRAF+ disease in both male and female patients. BRAF+ disease was associated with a statistically higher rate of mismatch repair deficiency (dMMR) in female patients and a trend towards higher rate of BRAF+ disease in dMMR colorectal cancer in male patients. *P-value: 0.05-0.01, ***P-value: 0.0009-0.0001.
As MMR deficiency, BRAF p.V600E, and wild-type APC CRC were all noted to increase in incidence as patients got older at their age of diagnosis, the relationship between these 3 variables was simultaneously assessed. APC mutational status was found to be strongly affected by the presence of BRAF p.V600E. APC and BRAF p.V600E mutations were nearly mutually exclusive in pMMR CRC (3/471, 0.6%), though there was a significant higher rate of co-existence of APC and BRAF p.V600E mutations in dMMR CRC (8 in 73, 11%, P < 0.0001).
In contrast BRAF p.V600E was seen at a significant higher rate in dMMR CRCs with either mutant APC (25% vs. 0.8%, Figure 4) or wild-type APC (65% vs. 23%, Figure 4), indicating the association of BRAF p.V600E with dMMR is independent of APC mutational status. Wild-type APC was seen in a significantly higher incidence in BRAF p.V600E mutated CRCs with either deficient (77% vs. 37%, Figure 4) or proficient MMR (89% vs. 18%, Figure 4), indicating the association of BRAF p.V600E with wild-type APC is independent from MMR status.
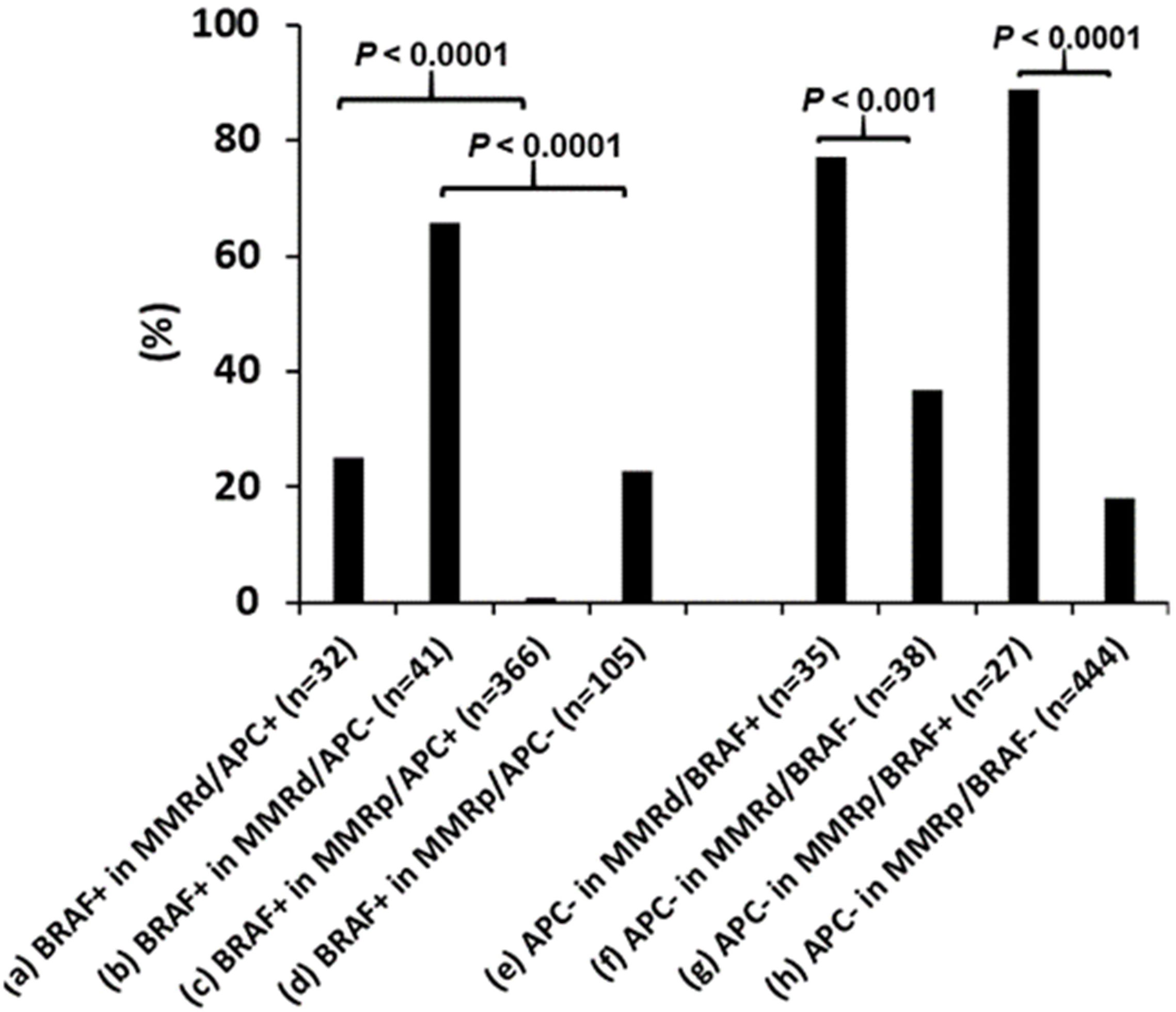
Figure 4 Co-existence of mismatch repair deficiency (dMMR), BRAF p.V600E and APC mutations. BRAF p.V600E (BRAF+) association with dMMR is independent of APC mutations and the BRAF p.V600E association with wild-type APC is independent of MMR status. pMMR: mismatch repair proficiency.
Categorization of CRC according to mutational profiling and its association with aging
CRCs were segregated into 8 subgroups according to MMR deficiency, BRAF p.V600E and APC mutations. The dominant population was pMMR CRC with wild-type BRAF and mutant APC (pMMR/BRAF-APC+) (67%), followed by pMMR/BRAF-APC- CRC (15%). Advanced age was significantly associated with a higher rate of BRAF+APC- in both dMMR CRC and pMMR CRC, a lower rate of pMMR/BRAF-APC+ CRC, and a trend of a higher rate of dMMR/BRAF-APC- CRC (Table 3). When the late-onset CRC (≥ 80y) was compared to the traditional-onset CRC (50-69y), there was also a significantly higher rate of dMMR/BRAF+APC- CRC (18% vs. 2.0%), dMMR/BRAF-APC- CRC (8.3% vs. 1.2%) and pMMR/BRAF+APC- CRC (12% vs. 4.0%) and a significantly lower incidence of pMMR/BRAF-APC+ CRC (43% vs. 72%) (Figure 5).
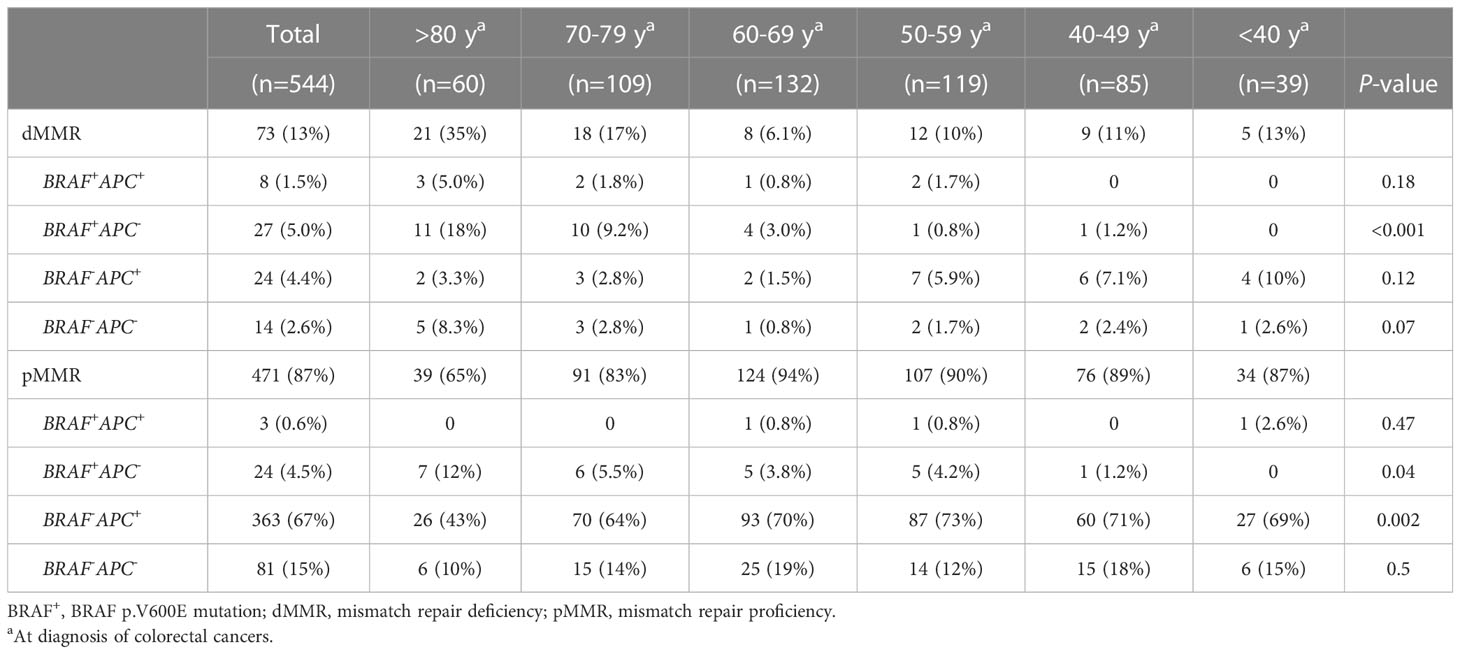
Table 3 Association of advanced age with CRCs categorized by mismatch repair deficiency, BRAF p.V600E mutation and APC mutations.
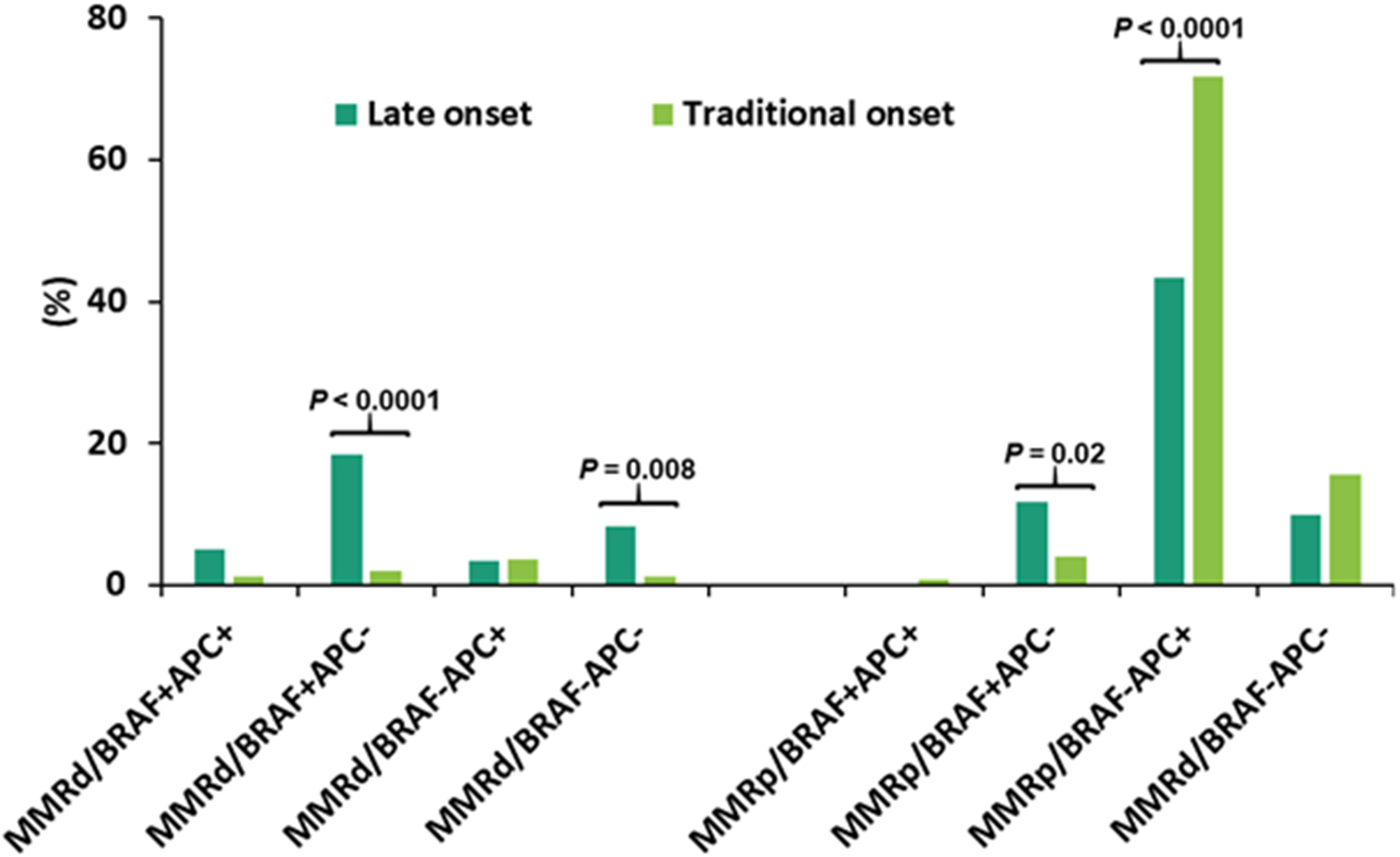
Figure 5 Categorization of CRC according to mismatch repair (MMR) status, BRAF p.V600E and APC mutations in late-onset CRC (≥80y, n = 60) and traditional-onset CRC (50-69y, n = 251). Late-onset CRC showed a significantly higher incidence of dMMR/BRAF+APC-, dMMR/BRAF-APC- and pMMR/BRAF+APC- and a significantly lower incidence of pMMR/BRAF-APC+. dMMR: mismatch repair deficiency; pMMR: mismatch repair proficiency, BRAF+: BRAF p.V600E mutant, BRAF-: BRAF p.V600E wild type, APC+: APC mutant, APC-: APC wild type.
Multivariate analyses by clinically significant features
Since disease stage at diagnosis and primary location of tumors were significantly associated with advanced age (Table 1), we next performed multivariate analysis as an exploratory investigation to understand how molecular features were influenced by age across the entire cohort when controlling for these clinical features (Table 4). When compared to younger patients with similar disease stage and primary location, older patients were less likely to have APC or KRAS mutations but more likely to have BRAF p.V600E. (Table 4A; per decade of age increase, odds ratio [OR]= 0.70 [95% confidence interval [CI]: 0.60-0.82] for APC+; OR=0.86 [95% CI: 0.76-0.98] for KRAS+; OR = 1.70 [95% CI: 1.34-2.16] for BRAF+). In addition to BRAF p.V600E (OR=4.76; 95% CI: [2.35-9.67], P < 0.001), right-sided CRC also harbored a higher incidence of KRAS mutations (OR=1.41; 95% CI: [0.96-2.08], P = 0.08) and PIK3CA mutations (OR=2.18; 95% CI: [1.41-3.39], P < 0.001).
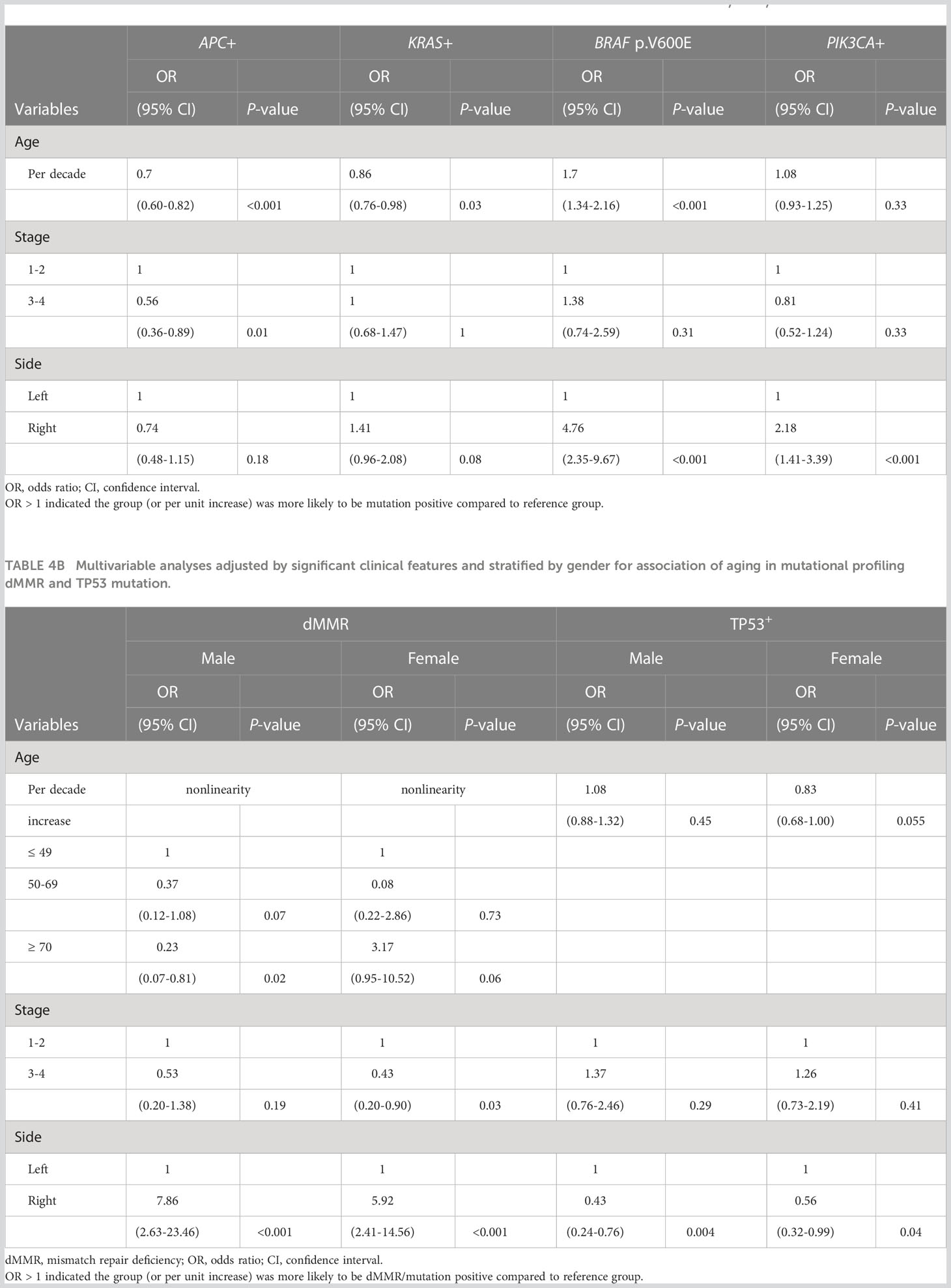
Table 4A Multivariable analyses adjusted by significant clinical features for association of aging in mutational profiling (APC, KRAS, BRAF and PIK3CA mutations).
Since age-related increased incidence of dMMR and decreased incidence of TP53 mutations were only seen in female patients (Figure 2), gender was also included for analyses (Table 4B). The relationship between dMMR incidence and patient age differed between male and female patients. In male patients, older patients with CRC had a lower rate of dMMR. Specifically, older male patients with age of diagnosis ≥70 showed a 77% (95% CI: 19-93%) lower rate of dMMR when compared to younger male patients with similar disease stage and side of colon. By contrast, older female patients ≥70 had a higher rate of dMMR disease when compared to younger female patients age <50 with similar disease stage and side of colon (Table 4B; OR=3.17; 95% CI: [0.95-10.52], P = 0.06). In female patients, there was a trend toward a lower rate of TP53 mutated disease in older patients compared to younger patients with similar disease stage and colon side (Table 4B; per decade age increase, OR=0.83, 95% CI [0.68-1.00], P = 0.055). This relationship was not observed in male patients. The incidence of TP53 mutations was significantly lower in both male (OR=0.43; 95% CI: [0.24-0.76], P = 0.004) and female (OR=0.56; 95% CI: [0.32-0.99], P = 0.004) right-sided CRC.
Discussion
This investigation adds to a growing body of literature suggesting age-dependent shifts in the clinical and molecular features of CRC (4, 6, 7). The current cohort demonstrated that CRC diagnosed in patients who were 80 years or older (late-onset CRC), as compared to the traditional-onset CRC diagnosed at 50-69 years, had a substantially higher rate of dMMR disease (35%) and BRAF p.V600E mutation (35%) and increased incidences of other clinicopathological features associated with MMR deficiency. These included female gender, right-sided primary, high-grade histomorphology and absence of systemic metastasis at diagnosis which confirms findings noted by other groups (7, 8, 21, 22). Given the low number (n=10) of African-American patients in our age 80 or older cohort we did not evaluate the influence of this variable on tumor-specific features. The association of aging with a higher rate of MMR deficiency (P = 4x10-6) and BRAF p.V600E mutation (P = 2x10-9) were also supported by stratifying patients into 6 groups based on the age. While the higher rate of BRAF p.V600E mutations was seen in both male and female elderly patients, the age-related higher rate of dMMR disease was seen exclusively in female patients. BRAF pV600E disease, as previously reported, was more common in cancer of the right colon, suggesting that right and left-sided colonic epithelium produce distinct CRC molecular phenotypes (7, 8). In addition to the findings of MMR deficiency and BRAF p.600E mutations, multivariate analyses also demonstrated a decrease of APC and KRAS mutations with aging and a higher incidence of KRAS and PIK3CA mutations and a low incidence of TP53 mutations in the right-sided CRC, consistent with the previous reports (23–25).
The current study confirmed a positive correlation between BRAF p.V600E mutation and dMMR CRC shown in the previous studies (7, 8, 20, 26). In addition, the proportion of dMMR tumors carrying BRAF p.V600E was significantly enriched with advanced age. In late-onset (≥80y) group and the 70-79y group, two-thirds of dMMR CRC harbored BRAF p.V600E. Sporadic MMR deficiency usually arises from epigenetic silencing of the MLH1 promoter, a global hypermethylation in CpG islands, and is associated with BRAF p.V600E mutation (26–29). The findings suggested that the higher incidence of MMR deficiency in late-onset CRC is most likely predominantly driven by hypermethylation of MLH1 promoter resulting from BRAF mutations (26–29).
Approximately 10-15% of CRC are MMR deficient and 3-5% of CRC are associated with Lynch syndrome (30–32). The recommended paradigm for screening of Lynch syndrome is to examine for BRAF p.V600E and/or MLH1 promoter methylation to select dMMR CRC for further germline testing (33, 34). In the NGS era, testing tumor tissues by a comprehensive NGS panel to include BRAF, KRAS, NRAS, MMR genes (MLH1, MSH2, MSH6, PMS2, EPCAM) and a panel of genes associated with other hereditary CRC not only identifies somatic mutations for targeted therapy and prognostication prediction, but also detect double somatic mutations of the MMR genes and potential germline mutations for further confirmation by testing non-neoplastic tissues (34, 35). In this cohort, potential germline mutations were seen in 50% of dMMR/BRAF- CRC, but none of MMRd/BRAF+ CRC. Among the dMMR/BRAF- CRC, patients with potential germline mutations were significantly younger (data not shown). However, obtaining germline testing results for confirmation was not feasible in this retrospective study.
The increased incidence of dMMR with advancing age was only statistically significant in female patients within this cohort while male patients showed a similar dMMR incidence in late-onset CRC as in traditional-onset CRC despite both sexes showing an age-related rise in the incidence of BRAF p.V600E mutated CRC. The mechanism for this sex-related difference in the incidence of dMMR CRC in older patients is not entirely clear but it is notable that prior investigations with hormone replacement therapy (HRT) observed a reduction in CRC risk in those that received estrogen replacement (36). The shift in the molecular profile of CRC that develops post-HRT differs between cohorts but a case-control study that stratified patients by age noted female patients age 71 or greater who had previously been on HRT saw the greatest reductions in CRC incidence specifically for microsatellite unstable and/or BRAF mutant disease (36, 37). Given the previously established role for estrogen in modifying the epigenetic landscape of cells through regulation of DNA methyltransferases it is possible that menopause-related hormone changes might facilitate BRAF pV600E associated MLH1 silencing in female patients (38). In this retrospective study, however, obtaining the hormone replacement therapy history in a high proportion of patients was not feasible. Further prospective studies are warranted to elucidate the effect of hormone replacement therapy on colorectal tumorigenesis.
The co-association of BRAF p.V600E mutation, dMMR disease, and wild-type APC may make it hard to decipher if one or several of these factors is responsible for the increased proportion of tumors with these features in late-onset CRC. To delineate these relationships, the proportion of patients with and without each variable was evaluated, and showed association of BRAF p.V600E with MMR deficiency was independent of APC mutational status and association of BRAF p.V600E with wild-type APC is independent of MMR deficiency. APC mutations and BRAF p.V600E were essentially mutually exclusive in pMMR CRC as reported previously, though 11% dMMR CRC harbored concurrent APC and BRAF p.V600E mutations (39–41). Further categorization of CRC according to these 3 genomic alterations revealed a significantly higher rate of dMMR/BRAF+APC- in late-onset CRC (18% vs. 2.0% in traditional-onset CRC), supporting MLH1 promoter hypermutation driven by BRAF p.V600E as the predominant mechanism of the increased rate of dMMR disease in late-onset CRC. However, the incidences of dMMR/BRAF-APC- (8.3% vs. 1.2%) and pMMR/BRAF+APC- (12% vs. 4.0%) were also significantly higher in the late-onset CRC as compared to traditional-onset CRC, suggesting additional mechanisms independently contribute to a higher rate of MMR deficiency and BRAF p.V600E mutation in late-onset CRC.
Testing for KRAS, NRAS and BRAF p.V600E mutations as well as MMR deficiency are recommended as standard-of-care for CRC (12, 19). BRAF p.V600E is tested not only for prognostic stratification and evaluation of risk of Lynch syndrome, but also to allow for treatment with recently approved targeted therapy (12, 19). BRAF p.V600E, as a prognostic marker, is associated with poor outcomes in CRC (42, 43). Lynch syndrome occurs in approximately only 1% of BRAF p.V600E mutated CRC. The NCCN recommended germline testing in BRAF p.V600E mutated CRC only for young patients and patients with a significant family history (44). In 2021, the Food and Drug Administration (FDA) in the United States approved combined BRAF inhibitor (encorafenib) plus anti-EGFR monoclonal antibody (cetuximab) for BRAF p.V600E mutated metastatic CRC after prior therapy (45, 46). KRAS and NRAS mutations are predictive for a lack of benefit to epidermal growth factor receptor (EGFR) inhibitor therapy (47, 48).
MMR deficiency is a molecular marker for screening for Lynch syndrome, prognostic stratification and prediction of immune checkpoint therapy efficacy (12, 21, 22). Prior to identification of its therapeutic implications, MMR testing was primarily focused on the identification of patients with Lynch Syndrome in younger patient (<50 years) according to the revised Bethesda guideline in 2004, which was extended to <70 years in 2014 guideline from the National Comprehensive Cancer Network (NCCN) (49, 50). This initial emphasis on screening younger patients for Lynch syndrome may be part of the reason for reduced MMR evaluation in patients diagnosed at 80 year or older as seen in the current cohort. Before the era of targeted therapy and immune checkpoint therapy, BRAF p.V600E was associated with inferior outcomes while MMR deficiency was associated with superior outcomes (20, 43, 51, 52). Stratification by BRAF p.V600E and MMR status showed the worst prognosis in pMMR/BRAF+ patients and the best prognosis in dMMR/BRAF- patients, though results from different studies were not completely consistent (20, 43, 51, 52).
In 2017, the FDA granted approval of immune checkpoint therapy (pembrolizumab) in pediatric and adult patients with dMMR solid tumors agnostic of tumor type (53–55). This included dMMR CRC that has progressed following treatment with fluoropyrimidine, oxaliplatin, and irinotecan. Recent clinical trials, including phase 3 studies, have shown improved survival rates with immune checkpoint therapy as the first-line treatment for dMMR metastatic CRC, which was independent of BRAF p.V600E mutation status (56–58). In 2020, pembrolizumab was approved by the FDA for the first-line treatment of patients with unresectable or metastatic dMMR CRC (57). The presence of a BRAF pV600E mutation does not appear to impact the responsiveness of these tumors to immune checkpoint therapy (56, 57). These recent advances of targeted therapy for BRAF p.V600E mutated CRC and immune checkpoint therapy for dMMR CRC highlight the importance of molecular testing, especially in late-onset CRC which harbors the highest incidence of BRAF p.V600E mutation and MMR deficiency.
The increase in dMMR disease is particularly interesting given the higher prevalence of co-morbidities in this older age group leading to an elevated risk of complications from surgical interventions. Recent investigations suggest localized dMMR CRC responds well to immunotherapy which obviated the need for surgery in a small cohort of locally advanced rectal cancer patients recently published by Cercek et al. (59, 60) Given the high rate of dMMR disease in patients ≥80 years old, clinical trials investigating the use of immunotherapy as an alternative to surgery in high-risk older patients may be warranted.
This retrospective cohort has several important limitations that must be considered when interpreting the outlined results. Foremost, this is a retrospective study of patients that underwent NGS and dMMR testing at JHH. While this represents a diverse group of patients across age groups and ethnicities, there may be important unrealized factors that skew the prevalence of different genomic alterations between this cohort and other patient populations such as those that did not undergo NGS and/or dMMR testing. In addition, most patients have their examination of MMR status by either IHC stains or MSI assay in this retrospective study. Although this could lead to a potential bias, IHC and MSI assays is expected to be highly concordant in CRC (21). Furthermore our number of patients over 80 was only 60 patients (20 males and 40 females) making our conclusions hypothesis generating rather than confirmatory. It is notable that a statistically higher rate of patients with late-onset CRC (≥80y) than younger age groups with NGS did not undergo dMMR testing. It is possible that a lower percentage of these patients would have been dMMR than the patients that were examined although it is notable that 5 of the 9 were wild type APC and 3 were BRAF p.V600E which both co-segregated with dMMR in our dataset.
In this retrospective study, we report on 544 cases of CRC with a particular focus on how the clinical and molecular features of these tumors shift with patient age at diagnosis. These investigations identified a higher rate of BRAF p.V600E mutated CRC in older patients along with a higher rate of dMMR disease specifically in female patients. These findings provide important insight into the biology of cancer in older patients in particular in those 80 years and older. Further studies with larger cohorts are needed to elucidate the genomic landscape of late-onset CRC. Prospective studies with treatment plans designed specifically for these older patients are also warranted to evaluate whether this improves clinical outcomes.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The studies involving human participants were reviewed and approved by Johns Hopkins University IRB. Written informed consent for participation was not required for this study in accordance with the national legislation and the institutional requirements.
Author contributions
EC, DL, EJ, and M-TL contributed to conception and design of the study. EC and M-TL organized the database. EC, H-LT, and M-TL performed the statistical analysis. EC, H-LT, DL, EJ, JD, RX, CG, JE, M-TL provided input of manuscript writing, manuscript revision, read, and approved the submitted version.
Funding
This work was generously supported by Swim Across America (EC), Bloomberg-Kimmel Institute for Cancer Immunotherapy (All authors), Cancer Convergence Institute (All authors).
Acknowledgments
Thank you to Daniel Laheru and Ben Park for your discussion and insights into this data.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin (2021) 71:7–33. doi: 10.3322/caac.21654
2. Siegel RL, Miller KD, Goding Sauer A, Fedewa SA, Butterly LF, Anderson JC, et al. Colorectal cancer statistics, 2020. CA A Cancer J Clin (2020) 70:145–64. doi: 10.3322/caac.21601
3. Akimoto N, Ugai T, Zhong R, Hamada T, Fujiyoshi K, Giannakis M, et al. Rising incidence of early-onset colorectal cancer–a call to action. Nat Rev Clin Oncol (2021) 18:230–43. doi: 10.1038/s41571-020-00445-1
4. Kotake K, Asano M, Ozawa H, Kobayashi H, Sugihara K. Tumour characteristics, treatment patterns and survival of patients aged 80 years or older with colorectal cancer. Colorectal Dis (2015) 17:205–15. doi: 10.1111/codi.12826
5. Berg M, Danielsen SA, Ahlquist T, Merok MA, Ågesen TH, Vatn MH, et al. DNA Sequence profiles of the colorectal cancer critical gene set KRAS-BRAF-PIK3CA-PTEN-TP53 related to age at disease onset. PloS One (2010) 5:e13978. doi: 10.1371/journal.pone.0013978
6. Iyer P, Wong K, Lieu CH, Henry J, Davis SL, Goodman KA, et al. Clinical and molecular characteristics of younger versus older patients with colorectal cancer. J Clin Orthod (2018) 36:630–0. doi: 10.1200/JCO.2018.36.4_suppl.630
7. Fanelli GN, Dal Pozzo CA, Depetris I, Schirripa M, Brignola S, Biason P, et al. The heterogeneous clinical and pathological landscapes of metastatic braf-mutated colorectal cancer. Cancer Cell Int (2020) 20:30. doi: 10.1186/s12935-020-1117-2
8. Nakayama I, Hirota T, Shinozaki E. BRAF mutation in colorectal cancers: from prognostic marker to targetable mutation. Cancers (2020) 12:3236. doi: 10.3390/cancers12113236
9. Koketsu S, Watanabe T, Tada T, Kanazawa T, Ueda E, Nagawa H. Sporadic colorectal cancer in elderly people. Hepatogastroenterology (2003) 50:1749–52.
10. Malkhosyan SR, Yamamoto H, Piao Z, Perucho M. Late onset and high incidence of colon cancer of the mutator phenotype with hypermethylated hMLH1 gene in women. Gastroenterology (2000) 119:598. doi: 10.1053/gast.2000.16154
11. Kakar S, Burgart LJ, Thibodeau SN, Rabe KG, Petersen GM, Goldberg RM, et al. Frequency of loss of hMLH1 expression in colorectal carcinoma increases with advancing age. Cancer (2003) 97:1421–7. doi: 10.1002/cncr.11206
12. Sepulveda AR, Hamilton SR, Allegra CJ, Grody W, Cushman-Vokoun AM, Funkhouser WK, et al. Molecular biomarkers for the evaluation of colorectal cancer: guideline from the American society for clinical pathology, college of American pathologists, association for molecular pathology, and the American society of clinical oncology. J Clin Oncol (2017) 35:1453–86. doi: 10.1200/JCO.2016.71.9807
13. Morris VK, Bekaii-Saab T. Improvements in clinical outcomes for BRAFV600E -mutant metastatic colorectal cancer. Clin Cancer Res (2020) 26:4435–41. doi: 10.1158/1078-0432.CCR-19-3809
14. Lin M-T, Zheng G, Tseng L-H, Zhang P, Ling H, Azad N, et al. Multiclonal colorectal cancers with divergent histomorphological features and RAS mutations: one cancer or separate cancers? Hum Pathol (2020) 98:120–8. doi: 10.1016/j.humpath.2020.03.002
15. Huang J, Tseng L-H, Parini V, Lokhandwala PM, Pallavajjala A, Rodriguez E, et al. IDH1 and IDH2 mutations in colorectal cancers. Am J Clin Pathol (2021) 156:777–86. doi: 10.1093/ajcp/aqab023
16. Fischer CG, Pallavajjala A, Jiang L, Anagnostou V, Tao J, Adams E, et al. Artificial intelligence-assisted serial analysis of clinical cancer genomics data identifies changing treatment recommendations and therapeutic targets. Clin Cancer Res (2022) 28:2361–72. doi: 10.1158/1078-0432.CCR-21-4061
17. Xian RR, Xie Y, Haley LM, Yonescu R, Pallavajjala A, Pittaluga S, et al. CREBBP and STAT6 co-mutation and 16p13 and 1p36 loss define the t(14;18)-negative diffuse variant of follicular lymphoma. Blood Cancer J (2020) 10:69. doi: 10.1038/s41408-020-0335-0
18. Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform (2013) 14:178–92. doi: 10.1093/bib/bbs017
19. Morris VK, Kennedy EB, Baxter NN, Benson AB 3rd, Cercek A, Cho M, et al. Treatment of metastatic colorectal cancer: ASCO guideline. J Clin Oncol (2022) 3:JCO2201690. doi: 10.1200
20. Lochhead P, Kuchiba A, Imamura Y, Liao X, Yamauchi M, Nishihara R, et al. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst (2013) 105:1151–6. doi: 10.1093/jnci/djt173
21. Funkhouser WK, Lubin IM, Monzon FA, Zehnbauer BA, Evans JP, Ogino S, et al. Relevance, pathogenesis, and testing algorithm for mismatch repair–defective colorectal carcinomas: a report of the association for molecular pathology. J Mol Diagn (2012) 14:91–103. doi: 10.1016/j.jmoldx.2011.11.001
22. Dudley JC, Lin M-T, Le DT, Eshleman JR. Microsatellite instability as a biomarker for PD-1 blockade. Clin Cancer Res (2016) 22:813–20. doi: 10.1158/1078-0432.CCR-15-1678
23. Lieu CH, Golemis EA, Serebriiskii IG, Newberg J, Hemmerich A, Connelly C, et al. Comprehensive genomic landscapes in early and later onset colorectal cancer. Clin Cancer Res (2019) 25:5852–8. doi: 10.1158/1078-0432.CCR-19-0899
24. Haley L, Tseng L-H, Zheng G, Dudley J, Anderson DA, Azad NS, et al. Performance characteristics of next-generation sequencing in clinical mutation detection of colorectal cancers. Mod Pathol (2015) 28:1390–9. doi: 10.1038/modpathol.2015.86
25. Loree JM, Pereira AAL, Lam M, Willauer AN, Raghav K, Dasari A, et al. Classifying colorectal cancer by tumor location rather than sidedness highlights a continuum in mutation profiles and consensus molecular subtypes. Clin Cancer Res (2018) 24:1062–72. doi: 10.1158/1078-0432.CCR-17-2484
26. Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE. Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature (2002) 418:934. doi: 10.1038/418934a
27. Fang M, Ou J, Hutchinson L, Green MR. The BRAF oncoprotein functions through the transcriptional repressor MAFG to mediate the CpG island methylator phenotype. Mol Cell (2014) 55:904–15. doi: 10.1016/j.molcel.2014.08.010
28. Deng G, Bell I, Crawley S, Gum J, Terdiman JP, Allen BA, et al. BRAF mutation is frequently present in sporadic colorectal cancer with methylated hMLH1, but not in hereditary nonpolyposis colorectal cancer. Clin Cancer Res (2004) 10:191–5. doi: 10.1158/1078-0432.CCR-1118-3
29. Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet (2006) 38:787–93. doi: 10.1038/ng1834
30. Dominguez-Valentin M, Sampson JR, Seppälä TT, Ten Broeke SW, Plazzer J-P, Nakken S, et al. Cancer risks by gene, age, and gender in 6350 carriers of pathogenic mismatch repair variants: findings from the prospective lynch syndrome database. Genet Med (2020) 22:15–25. doi: 10.1038/s41436-019-0596-9
31. Boland CR, Goel A. Microsatellite instability in colorectal cancer. Gastroenterology (2010) 138:2073–2087.e3. doi: 10.1053/j.gastro.2009.12.064
32. Rebuzzi F, Ulivi P, Tedaldi G. Genetic predisposition to colorectal cancer: how many and which genes to test? Int J Mol Sci (2023) 24:2137. doi: 10.3390/ijms24032137
33. Adar T, Rodgers LH, Shannon KM, Yoshida M, Ma T, Mattia A, et al. A tailored approach to BRAF and MLH1 methylation testing in a universal screening program for lynch syndrome. Mod Pathol (2017) 30:440–7. doi: 10.1038/modpathol.2016.211
34. Hampel H, Pearlman R, Beightol M, Zhao W, Jones D, Frankel WL, et al. Assessment of tumor sequencing as a replacement for lynch syndrome screening and current molecular tests for patients with colorectal cancer. JAMA Oncol (2018) 4:806–13. doi: 10.1001/jamaoncol.2018.0104
35. Daca Alvarez M, Quintana I, Terradas M, Mur P, Balaguer F, Valle L. The inherited and familial component of early-onset colorectal cancer. Cells (2021) 10:710. doi: 10.3390/cells10030710
36. Nelson HD, Humphrey LL, Nygren P, Teutsch SM, Allan JD. Postmenopausal hormone replacement therapy. JAMA (2002) 288:872. doi: 10.1001/jama.288.7.872
37. Amitay EL, Carr PR, Jansen L, Alwers E, Roth W, Herpel E, et al. Postmenopausal hormone replacement therapy and colorectal cancer risk by molecular subtypes and pathways. Int J Cancer (2020) 147:1018–26. doi: 10.1002/ijc.32868
38. Kovács T, Szabó-Meleg E, Ábrahám IM. Estradiol-induced epigenetically mediated mechanisms and regulation of gene expression. Int J Mol Sci (2020) 21:3177. doi: 10.3390/ijms21093177
39. TCGA. Findings spotlight drivers for colorectal cancer. Cancer Discovery (2012) 2:OF5–5. doi: 10.1158
40. Nguyen LH, Goel A, Chung DC. Pathways of colorectal carcinogenesis. Gastroenterology (2020) 158:291–302. doi: 10.1053/j.gastro.2019.08.059
41. Kambara T, Matsubara N, Nagao A, Uchida T, Nakagawa H, Tanaka N, et al. Mtations in BRAF, KRAS, and APC, and CpG island methylation: alternative pathways to colorectal cancer. Cancer Res (2006) 66:81–1.
42. Cremolini C, Loupakis F, Antoniotti C, Lupi C, Sensi E, Lonardi S, et al. FOLFOXIRI plus bevacizumab versus FOLFIRI plus bevacizumab as first-line treatment of patients with metastatic colorectal cancer: updated overall survival and molecular subgroup analyses of the open-label, phase 3 TRIBE study. Lancet Oncol (2015) 16:1306–15. doi: 10.1016/S1470-2045(15)00122-9
43. Venderbosch S, Nagtegaal ID, Maughan TS, Smith CG, Cheadle JP, Fisher D, et al. Mismatch repair status and BRAF mutation status in metastatic colorectal cancer patients: a pooled analysis of the CAIRO, CAIRO2, COIN, and FOCUS studies. Clin Cancer Res (2014) 20:5322–30. doi: 10.1158/1078-0432.CCR-14-0332
44. Gupta S, Provenzale D, Llor X, Halverson AL, Grady W, Chung DC, et al. NCCN guidelines insights: Genetic/Familial high-risk assessment: colorectal, version 2.2019. J Natl Compr Canc Netw (2019) 17:1032–41. doi: 10.6004/jnccn.2019.0044
45. Tabernero J, Grothey A, Van Cutsem E, Yaeger R, Wasan H, Yoshino T, et al. Encorafenib plus cetuximab as a new standard of care for previously treated BRAF V600E-mutant metastatic colorectal cancer: updated survival results and subgroup analyses from the BEACON study. J Clin Oncol (2021) 39:273–84. doi: 10.1200/JCO.20.02088
46. Kopetz S, Grothey A, Yaeger R, Van Cutsem E, Desai J, Yoshino T, et al. Encorafenib, binimetinib, and cetuximab in BRAF V600E-mutated colorectal cancer. N Engl J Med (2019) 381:1632–43. doi: 10.1056/NEJMoa1908075
47. Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ, et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol (2008) 26:1626–34. doi: 10.1200/JCO.2007.14.7116
48. De Roock W, Piessevaux H, De Schutter J, Janssens M, De Hertogh G, Personeni N, et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann Oncol (2008) 19:508–15. doi: 10.1093/annonc/mdm496
49. Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Rüschoff J, et al. Revised Bethesda guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst (2004) 96:261–8. doi: 10.1093/jnci/djh034
50. Hampel H. NCCN increases the emphasis on genetic/familial high-risk assessment in colorectal cancer. J Natl Compr Canc Netw (2014) 12:829–31. doi: 10.6004/jnccn.2014.0200
51. French AJ, Sargent DJ, Burgart LJ, Foster NR, Kabat BF, Goldberg R, et al. Prognostic significance of defective mismatch repair and BRAF V600E in patients with colon cancer. Clin Cancer Res (2008) 14:3408–15. doi: 10.1158/1078-0432.CCR-07-1489
52. Phipps AI, Limburg PJ, Baron JA, Burnett-Hartman AN, Weisenberger DJ, Laird PW, et al. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology (2015) 148:77–87.e2. doi: 10.1053/j.gastro.2014.09.038
53. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. N Engl J Med (2015) 372:2509–20. doi: 10.1056/NEJMoa1500596
54. Lemery S, Keegan P, Pazdur R. First FDA approval agnostic of cancer site - when a biomarker defines the indication. N Engl J Med (2017) 377:1409–12. doi: 10.1056/NEJMp1709968
55. Marcus L, Lemery SJ, Keegan P, Pazdur R. FDA Approval summary: pembrolizumab for the treatment of microsatellite instability-high solid tumors. Clin Cancer Res (2019) 25:3753–8. doi: 10.1158/1078-0432.CCR-18-4070
56. Lenz H-J, Van Cutsem E, Luisa Limon M, Wong KYM, Hendlisz A, Aglietta M, et al. First-line nivolumab plus low-dose ipilimumab for microsatellite instability-High/Mismatch repair-deficient metastatic colorectal cancer: the phase II CheckMate 142 study. J Clin Oncol (2022) 40:161–70. doi: 10.1200/JCO.21.01015
57. Casak SJ, Marcus L, Fashoyin-Aje L, Mushti SL, Cheng J, Shen Y-L, et al. FDA Approval summary: pembrolizumab for the first-line treatment of patients with MSI-H/dMMR advanced unresectable or metastatic colorectal carcinoma. Clin Cancer Res (2021) 27:4680–4. doi: 10.1158/1078-0432.CCR-21-0557
58. André T, Shiu K-K, Kim TW, Jensen BV, Jensen LH, Punt C, et al. Pembrolizumab in microsatellite-Instability-High advanced colorectal cancer. N Engl J Med (2020) 383:2207–18. doi: 10.1056/NEJMoa2017699
59. Cercek A, Lumish M, Sinopoli J, Weiss J, Shia J, Lamendola-Essel M, et al. PD-1 blockade in mismatch repair-deficient, locally advanced rectal cancer. N Engl J Med (2022) 386:2363–76. doi: 10.1056/NEJMoa2201445
Keywords: mismatch repair (MMR) deficiency, colorectal cancer, BRAF mutation V600E, late onset colorectal cancer, APC mutations
Citation: Christenson ES, Tsai H-L, Le DT, Jaffee EM, Dudley J, Xian RR, Gocke CD, Eshleman JR and Lin M-T (2023) Colorectal cancer in patients of advanced age is associated with increased incidence of BRAF p.V600E mutation and mismatch repair deficiency. Front. Oncol. 13:1193259. doi: 10.3389/fonc.2023.1193259
Received: 24 March 2023; Accepted: 19 May 2023;
Published: 07 June 2023.
Edited by:
Tiziana Venesio, Institute for Cancer Research and Treatment (IRCC), ItalyReviewed by:
Francesca Rebuzzi, Scientific Institute of Romagna for the Study and Treatment of Tumors (IRCCS), ItalyElisabetta Fenocchio, IRCCS Candiolo Cancer Institute, Italy
Cristina Oliani, Azienda Ulss 5 Polesana, Italy
Copyright © 2023 Christenson, Tsai, Le, Jaffee, Dudley, Xian, Gocke, Eshleman and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eric S. Christenson, ZWNocmlzMTRAamhtaS5lZHU=