 Qiao-Yan Zhu1,2†
Qiao-Yan Zhu1,2† Wen-Ming Cao
Wen-Ming Cao Bei Li
Bei Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Oncol. , 12 May 2023
Sec. Breast Cancer
Volume 13 - 2023 | https://doi.org/10.3389/fonc.2023.1188371
TSC2 is a tumor suppressor gene as well as a disease-causing gene for autosomal dominant disorder tuberous sclerosis complex (TSC). Research has found that some tumor tissues have lower TSC2 expression levels than normal tissues. Furthermore, low expression of TSC2 is associated with poor prognosis in breast cancer. TSC2 acts as a convergence point of a complex network of signaling pathways and receives signals from the PI3K, AMPK, MAPK, and WNT pathways. It also regulates cellular metabolism and autophagy through inhibition of a mechanistic target of rapamycin complex, which are processes relevant to the progression, treatment, and prognosis of breast cancer. In-depth study of TSC2 functions provides significant guidance for clinical applications in breast cancer, including improving the treatment efficacy, overcoming drug resistance, and predicting prognosis. In this review, protein structure and biological functions of TSC2 were described and recent advances in TSC2 research in different molecular subtypes of breast cancer were summarized.
Loss-of-function mutations in TSC1 or TSC2 can give rise to an autosomal dominant disorder of tuberous sclerosis complex (TSC) that invades multiple organs, such as brain, skin, heart, lungs, and kidneys (1, 2). Moreover, TSC1/2 are significant anti-oncogenes that are mutated in breast (3), liver (4–6), lung (7, 8), kidney (9, 10) and other (11) cancers. Allison et al. (3) have reported on the prevalence of TSC1 germline mutations in 5,041 patients with breast cancer (pathogenic variants (PVs) vs. variants of uncertain significance (VUS): 0.00% vs. 0.30%) and the prevalence of TSC2 germline mutations in 5,040 patients with breast cancer (PVs vs. VUS: 0.00% vs. 1.20%). Mehta et al. (12) have shown that TSC1 rs7874234 was associated with delayed age at diagnosis of estrogen receptor (ER)-positive breast cancer (P = 0.0049). Daniel et al. have reported on 13 TSC2 mutations and six TSC1 mutations in 111 hepatocellular carcinomas cases (6). Moreover, 22% of 86 human lung cancer specimens were identified as those with TSC1 and/or TSC2 loss of heterozygosity (7). Furthermore, Yong et al. have shown that TSC2 rs30259G>A was associated with worse overall survival and disease-free survival (DFS) in patients with non-small cell lung cancer after curative surgery (P < 0.05) (8). These findings suggested that TSC1/2 may be candidates for a prognostic marker for several tumors.
Evidence of low TSC1/2 expression in prostate cancer (13) is lacking. However, studies have reported that the level of TSC1/2 expression is lower in breast cancer (12, 14) and oral squamous cell carcinoma (15) than in normal tissues. Data from a 10-year follow-up study have indicated that tumor tissues from breast cancer patients who relapsed or died had significantly low levels of TSC2 compared to patients who did not experience recurrence (P = 0.03 and 0.05, respectively). This suggested that low TSC2 expression was associated with aggressiveness and unfavorable prognosis in patients with breast cancer (14). TSC2-null breast cancer cells are sustained mechanistic targets of rapamycin complex 1 (mTORC1) activity that regulates essential cellular activities. Hyperactivated mTORC1 promotes the growth and metastasis of breast cancer (16–18). There is also growing evidence that TSC2 plays an essential role in breast cancer. Hence, the present review summarizes the functions of TSC2 and recent advances in TSC2 research in different subtypes of breast cancer in order to provide new directions for breast cancer clinical applications.
TSC2 in chromosome 16p13.3 encodes a 200-kDa protein tuberin containing 1,807 amino acids (19). The GTPase-activating protein (GAP) catalytic domain located on the TSC2 C-terminal is a highly conserved domain that can combine with two Ras homologs enriched in the brain (Rheb) to convert Rheb from GTP-bound to GDP-bound (20, 21). This domain is homologous to Ras-proximate1 (Rap1) GAPs, suggesting that the mechanisms of Rheb-GTP hydrolysis are identical to those of the Rap-Rap1GAP system, which may rely on the asparagine thumb (N1643) to stabilize the γ-phosphate of GTP and promote GTP hydrolysis of Rheb (22, 23). The presence of a structural domain bound to TSC1 at the N-terminal of TSC2 and TSC1 plays a role in stabilizing TSC2 and preventing its degradation (22, 24, 25). TSC1, TSC2, and Tre2-Bub2-Cdc16 (TBC) 1 domain member 7 (TBC1D7) assemble to form the TSC multiprotein complex (TSCC) with a 2:2:1 stoichiometry (23, 26). One TBC1D7 concurrently interacts with two parallel TSC1 helices, indicating that TBC1D7 may react by binding with the C-terminal ends of TSC1 to stabilize the TSCC (27). TBC1D7 knockdown affects the link between TSC1 and TSC2, causing a decrease in GAP activity, but has no effect on the localization of TSC2 in the lysosome (26). Thus, the function of TSC2 is closely related to the structural integrity of TSCC.
The mTORC1 acts as a protein kinase complex and is almost ubiquitous in eukaryotic cells. It regulates the nutritional status, cellular metabolism, and cell growth, including the synthesis of proteins, lipids, and nucleic acids (28, 29). The mTORC1 promotes mRNA transcription via phosphorylation of ribosomal protein S6 kinase and eukaryotic initiation factor 4E-binding protein 1 (30). It also prevents autophagy by regulating the lysosomal degradative capacity of cells or direct inhibition of the early autophagy process stages (31). TSC2 acts as an inhibitor of mTORC1 because its activation is dependent on Rheb-GTP (32). When TSC2 is absent or inactivated, the process of Rheb-GTP conversion to Rheb-GDP is blocked, leading to an increase in the level of Rheb-GTP to activate mTORC1 (33). Lysosomes are generally recognized as the main signaling platform for TSCC to inhibit mTORC1. In the absence of stress granules (SGs), the ras-GTPase-activating protein SH3-domain-binding protein (G3BP1/2) resides on the surface of the lysosomes and acts as a tether connecting TSCC (34). The N-terminal of the NTF2L domain of G3BP1 bonds with the lysosome-associated membrane proteins 1/2, which bridge the TSCC to the lysosomal surface (35). Thus, when conditions are beneficial for cell growth, TSC2 is diffusely localized in the cytoplasm. The amount of TSCC in the lysosome then decreases, while the abundance of Rheb-GTP increases, giving rise to the activated mTORC1 (33, 36). A single inhibitory stress factor, such as hypoxia and low amino acid levels, can cause lysosomal TSC2 repositioning to inhibit mTORC1 activation toward suppression of cell growth and promotion of autophagy (37). Compared to mTORC1, there are fewer studies related to the mechanism and function of TSC2 regulation of mTORC2. Some studies have found that TSC2 deletion can induce mTORC2-specific upregulation of Ras homolog family member A (38). Some studies have also found that TSC2 deletion impaired the enzymatic activity of mTORC2 (26). However, how TSC2 is involved in the regulation of mTORC2 is unclear and requires in-depth study.
TSC2 is the convergence point of a complex network of signaling pathways, where almost all upstream stimuli (including amino acids) that regulate mTORC1 activity converge on TSC2 (Figure 1) (37). The most well-known is the phosphoinositide-3-kinase (PI3K)/protein kinase B (AKT)/TSC2/mTOR pathway that insulin and other growth factors are dependent on (39, 40). The serine/threonine kinase AKT is located upstream of TSC2 and can directly phosphorylate the Ser939 and Thr1462 of TSC2. TSC2 is bound by the 14-3-3 protein in response to AKT phosphorylation and is then sequestered away from TSC1 (39). The 14-3-3 protein mediates the subcellular TSC2 translocation and protein hydrolytic degradation to increase the abundance of Rheb-GTP in the lysosome and thus promote mTORC1 activation (41). Hypoxia induces the regulated in development and DNA damage response (REDD1) to bind the 14-3-3 protein, releasing TSC2 from 14-3-3 (42). In addition, TSC2 is also involved in some classical signaling pathways, such as RAS and WNT signaling. The extracellular signal-regulated kinase 1/2 (ERK1/2) downstream of RAS signaling can phosphorylate the S664 of TSC2, leading to TSC1-TSC2 dissociation (43). In addition, S664 is a marker for ERK-mediated (rather than AKT-mediated) mTOR activation, which can be used to screen patients who may benefit from mTOR inhibitor (44). The p90 ribosomal S6 kinase acts as a downstream effector of ERK1/2 that inactivates TSC2 by phosphorylating it at a regulatory site Ser1798 (45). In hypoxic conditions and when energy deficiency was present, decreasing levels of intracellular ATP led to a rapid activation of AMP-activated protein kinase (AMPK) and TSC2 phosphorylation and enhancement of its activity (46, 47). This is the opposite of the effects of PI3K-AKT signaling in causing phosphorylation and inactivation of TSC2. Glycogen synthase kinase 3 (GSK3) downstream of WNT acts synergistically with AMPK. It can also phosphorylate TSC2 to increase its activity to inhibit mTOR (48). Based on this evidence, TSC2 plays an important role in cellular activities.
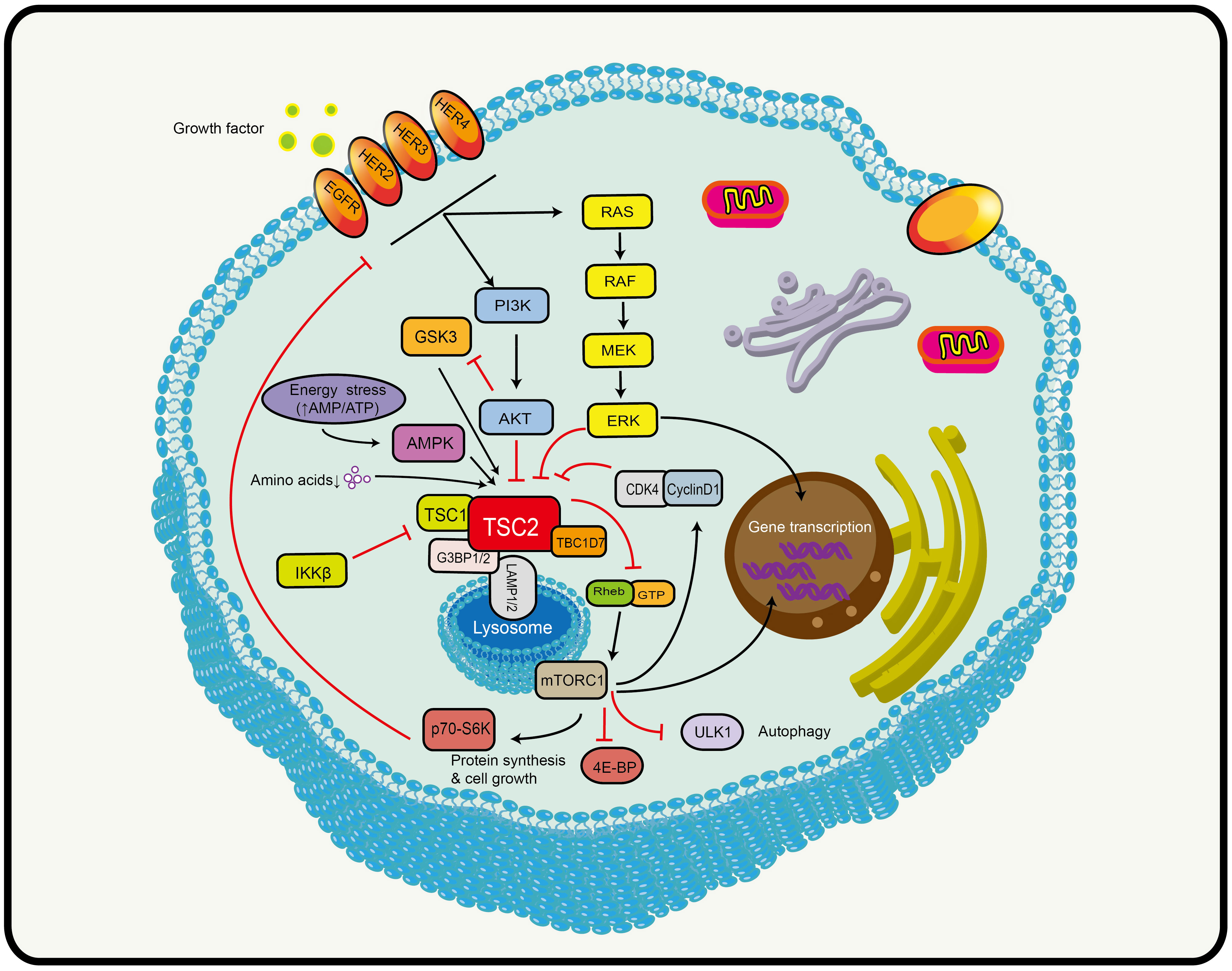
Figure 1 Schematic model depicting the TSC2-mTOR axis. TSC2 is the convergence point of a complex network of signaling pathways that receives signals from multiple signaling pathways, including PI3K, AMPK (AMP activated protein kinase), MAPK (mitogen-activated protein kinase, and WNT (see text for details). Black arrows indicate stimulation and red blocked arrows indicate inhibition. Abbreviations: HER2/3/4, human epidermal growth factor receptor 2/3/4; PI3K, phosphatidylinositol 3-kinase; AKT, protein kinase B; TSC1/2, tuberous sclerosis protein complex 1 and 2; TBC1D7, Tre2-Bub2-Cdc16 (TBC) 1 domain member 7; G3BP1/2, ras-GTPase-activating protein SH3-domain-binding protein; LAMP1/2, lysosomal-associated membrane proteins 1/2; GTP, guanosine triphosphate; Rheb, Ras homolog enriched in brain; mTORC1, mechanistic target of rapamycin complex 1; ULK1, unc-51-like kinase 1; 4E-BP1, 4E binding protein 1; p70s6k, ribosomal protein kinase S6; AMPK, AMP-activated protein kinase; GSK3, glycogen synthase kinase 3; RAS, rat sarcoma; MEK, mitogen-activated protein kinase; ERK, extracellular signal-regulated kinase; CDK4, cyclin D-dependent kinase 4.
There are many forms of post-translational protein modification. Based on the above evidence, it is clear that TSC2 activity is regulated through phosphorylation, but is not completely dependent on it. TSC2 can also be acetylated by arrest defective 1 (ARD1), also known as N-α-acetyltransferase 10. ARD1 has a role in inhibiting cell proliferation, inducing autophagy, and suppressing tumor growth, which is associated with ARD1 acetylation, stabilization, and degradation inhibition of TSC2 (49). Seishu et al. (50) have found that protein arginine methyltransferase 1 methylates TSC2 at R1457 and R1459. The methylation of two residues inhibits phosphorylation of TSC2 at T1462, which is critical for TSC2 stability. Therefore, studies on the regulation of TSC2 activity might play a role in the development of treatment strategies.
TSC2 is a multifunctional protein that also regulates pathways beyond the mTOR pathway (51, 52). Loss-of-function TSC2 is able to activate Rac1 and thereby increase ROS production (53). TSC2 also plays a part in cell cycle regulation. It has been shown that TSC2 positively correlates with the expression of cyclin-dependent kinase inhibitor p27 in the epithelium (54). Down-regulation of TSC2 expression induced quiescent fibroblasts in G0 phase to re-enter the cell cycle (55). This may be related to the fact that TSC2 affects the stability and intracellular spatial distribution of p27 (56, 57). In addition, TSC2 can play the role of a transcription factor, inhibiting the expression of epidermal growth factor family member epidermal regulator epiregulin by binding to its promoter (58). TSC2 is extremely significant in modulating cell growth and proliferation, while its nuclear function remains to be further explored.
Breast cancer is the most common malignant tumor in females worldwide, with its incidence surpassing that of lung cancer in 2020 (59). Based on the expression of ER, progesterone receptor (PR), and human epidermal growth factor receptor 2 (HER2), breast cancer can be classified into three major subtypes that include hormone receptor (HR)-positive breast cancer, HER2-positive breast cancer, and triple-negative breast cancer (TNBC). There are significant differences in biological behavior, clinical manifestations, and treatment strategies among these subtypes, providing important information for clinical antitumor therapy (60). The TSC/mTOR pathway is frequently deregulated in breast cancers and is associated with tumorigenesis (61). The hypermethylation of TSC1 promoters can be observed in breast cancer (14). Lee et al. (62) have found that IKKbeta phosphorylates TSC1 at Ser487 and Ser511 to activate mTOR. The expression of activated IKKbeta correlates with TSC1 Ser511 phosphorylation and VEGF production in breast cancer patients and is associated with poor prognosis in breast cancer. It follows that TSC1 plays an important role in breast cancer. TSC2 acts as an integrator of various signaling cues and regulates cellular metabolism and autophagy through inhibition of mTORC1, which are processes relevant to the progression, treatment (Figure 2), and prognosis of breast cancer (37). Therefore, it is important to explore the function of TSC2 in breast cancer.
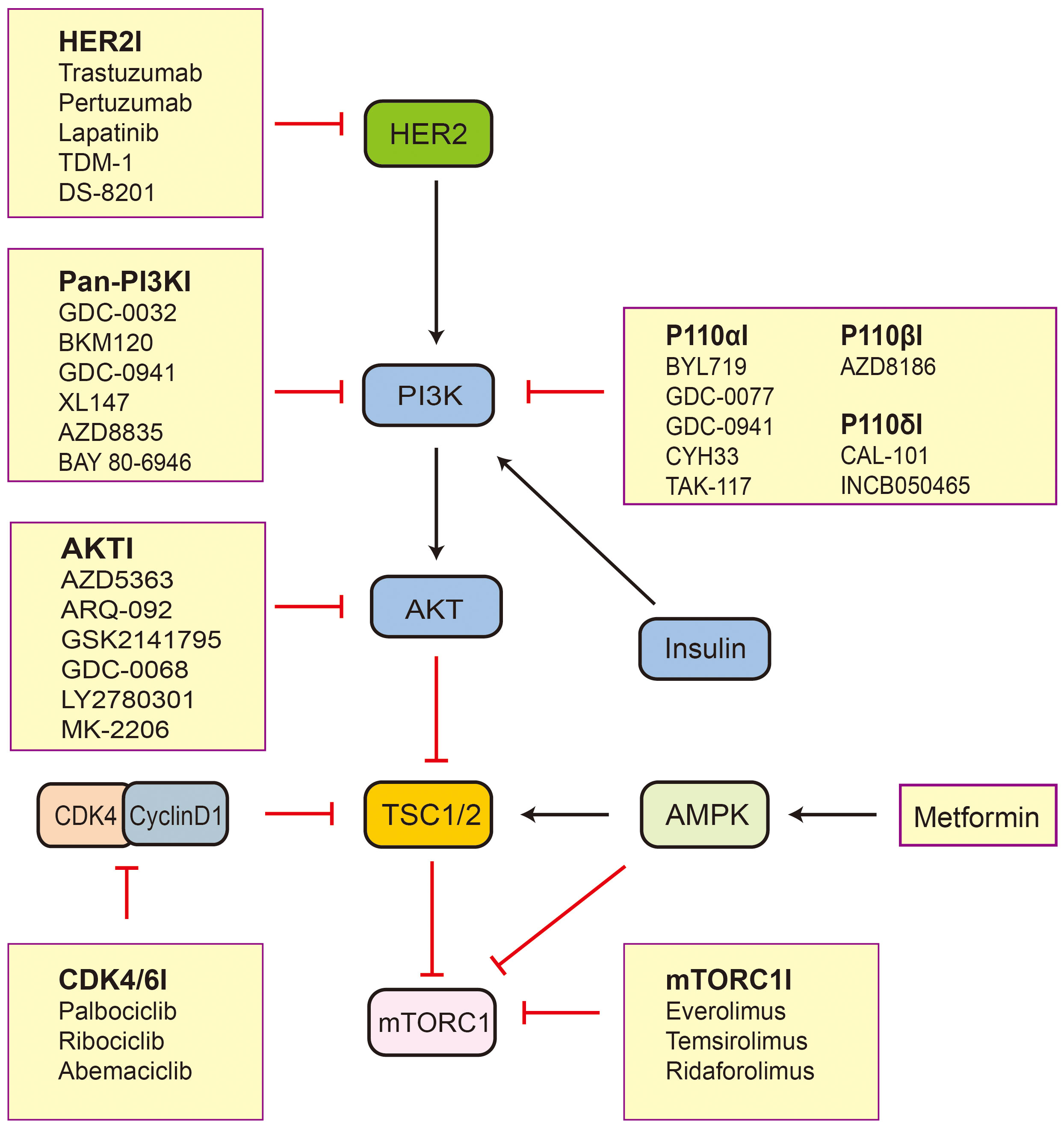
Figure 2 The inhibitors related to TSC2-mTOR axis active in preclinical and clinical trials. Black arrows indicate stimulation and red blocked arrows indicate inhibition. Abbreviations: HER2, human epidermal growth factor receptor 2; PI3K, phosphatidylinositol 3-kinase; AKT, protein kinase B; TSC1/2, tuberous sclerosis protein complex 1 and 2; mTORC1, mechanistic target of rapamycin complex 1; AMPK, AMP-activated protein kinase; CDK4, cyclin D-dependent kinase 4.
The ERBB family includes epidermal growth factor receptor (EGFR) (ERBB1), HER2 (ERBB2), HER3 (ERBB3), and HER4 (ERBB4), which are stimulated by ligands to form homodimers or heterodimers to activate intracellular pathways that are part of the RAS/RAF/MEK/ERK and PI3K/AKT/TSC2/mTOR pathways, promoting proliferation and metastasis of tumor cells (63). HER2 is overexpressed in approximately 15–20% of invasive breast cancers (64). HER2 does not directly bind with any ERBB ligands, but its conformation favors dimerization. Therefore, when HER2 is overexpressed, it can activate the downstream pathways through its own dimerization or heterodimerization of the other members of the ERBB family that bind to the ligands (65–67). EGFR can signal to PI3K through GAB1 in a HER3-independent manner, phosphorylating TSC2 and activating mTOR (68). There is a variety of clinical anti-HER2-targeting drugs. Multiple mechanisms of anti-HER2 therapy resistance have been demonstrated, with overactivation of the downstream PI3K/AKT/TSC2/mTOR pathway being the most common (69). Therefore, it is extremely significant to inhibit this pathway in anti-HER2 therapy. Inhibitors of cyclin D-dependent kinase 4/6 (CDK4/6) have been shown to reverse resistance to anti-HER2 therapy. CDK4 promotes cell proliferation both directly through retinoblastoma phosphorylation and indirectly through mTORC1 activation by binding and phosphorylating TSC2 on Ser1217 and Ser1452 to promote cell growth (70). CDK4/6 inhibition partially attenuates mTORC1 activity by reducing TSC2 phosphorylation, thereby relieving feedback inhibition of upstream ERBB family kinases and making tumors re-sensitized to EGFR/HER2 blocking. Therefore, dual inhibition of EGFR/HER2 and CDK4/6 more effectively inhibits TSC2 phosphorylation and blocks mTORC1 activity (71). Clinical research on CDK4/6 inhibitors combined with trastuzumab is underway (72). Metformin is an antidiabetic agent that also holds great promise in breast cancer treatment (73). It can not only indirectly lower the internal insulin levels but can also activate the AMPK pathway in the liver by depleting energy. Insulin relies on AKT-mediated phosphorylation of TSC2 to activate mTORC1, while AMPK enhances TSC2 activity and phosphorylates raptor on Ser722 and Ser792 in inhibiting mTORC1 activity (74–76). Wang et al. have found that a combination of mTOR inhibitor everolimus and metformin had a synergistic effect on inhibiting breast cancer cell growth (77). In a phase III randomized controlled study of early-stage breast cancer (78), metformin combined with a standard treatment showed significant overall survival and invasive DFS benefits over the placebo with a standard treatment in HER2-positive breast cancer (P = 0.038 and 0.0026, respectively). This indicates that TSC2 has significant potential in the treatment of HER2-positive breast cancer.
HR+/HER2- breast cancer accounts for approximately 60−75% of all breast cancers and is the most common breast cancer subtype. The ER pathway drives its development and progression and is directly and indirectly associated with TSC2 (79). Estrogen rapidly (within 5 min) stimulates TSC2 phosphorylation at T1462, one of the sites at which Akt phosphorylates and inactivates TSC2, suggesting that estrogen-induced cell proliferation is related to TSC2 inactivation (80). Some studies have also shown that TSC2 bound specifically to ERα and inhibited estrogen-induced proliferation by attenuating activation of platelet-derived growth factor receptor β and ERK1/2 (81). Therefore, it is clear that TSC2 plays an inhibitory role in the proliferation of cells in HR+ breast cancer.
In contrast to primary breast cancer (PBC), metastatic breast cancer (MBC) often has mutations in specific genes. Cha et al. have analyzed the data for 4,268 MBC and 5,217 PBC patients from eight different cohorts and found 11 genes containing TSC2 that were more frequently changed in MBC samples from pan-metastatic sites (82). Savas et al. have also found that mutations in TSC2 were significantly enriched in MBC compared to PBC (83). Lefebvre et al. have performed whole-exome sequencing of 216 tumor-blood pairs from MBC patients who provided a biopsy sample. There were 143 HR+/HER2- breast cancer samples, and TSC2 was mutated in four (2.8%) of them. In contrast, according to the Cancer Genome Atlas data, the mutation rate of TSC1/TSC2 was only 0.7% (P = 0.0004) in patients with HR+/HER2- early-stage breast cancer (84). This suggests that HR+/HER2- advanced breast cancer with a higher rate of TSC2 mutations compared to HR+/HER2- early breast cancer may be associated with tumor progression or resistance to therapy.
PIK3CA mutations occur in nearly 40% of patients with HR+/HER2- breast cancer (85, 86). Inhibitors of PI3K p110α (PI3Kα) are highly effective in patients with PIK3CA-mutated tumors, especially in combination with endocrine therapy (85, 87). However, there is a subset of patients who remain resistant to PI3K inhibitors. It has been suggested that when PI3K is inhibited, 3-phosphoinositide-dependent protein kinase-1 can activate serum- and glucocorticoid-inducible kinase (SGK1), which is highly homologous to AKT. SGK1 overcomes PI3K inhibition via direct phosphorylation and inhibition of TSC2 to maintain mTORC1 activity (88). Cai et al. have analyzed 1,918 tumors from 1,756 breast cancer patients (89), of which 443 samples of ER+/HER2- breast cancer had PIK3CA mutations. They have found that about 1.35% of the ER+ tumors with PIK3CA mutations carried genomic alterations of TSC2, which may have led to resistance to PI3K inhibitors (90). In addition, Yuan et al. have analyzed 28,847 single nucleotide polymorphisms (SNPs) in 61 genes that participate in the mTOR pathway from 3,663 (1,983 ER+ and 1,098 ER-) breast cancer patients and 4,687 healthy women. The results have demonstrated that the TSC2 gene was associated with the risk of overall and ER+ breast cancers (P = 0.009 and 0.012, respectively). However, TSC2 SNP rs181088346 was significantly associated with a reduced overall risk of breast cancer (odds ratio = 0.77, 95% confidence interval = 0.65–0.88, P adj = 0.035) (91). Thus, the TSC2 gene may be connected with genetic susceptibility of HR+/HER2- breast cancer.
TNBC accounts for 10–20% of all breast cancers. It can be classified into the following subtypes based on multi-omics data: luminal androgen receptor (LAR) and its most noteworthy features associated with androgen receptor signaling; immunomodulatory (IM) with high expression of cytokine gene signaling and activation of immune cell signaling; basal-like and immune-suppressed with cell cycle upregulation, DNA repair activation, and immune response gene downregulation; and mesenchymal-like abundant in mammary stem cell pathways (92). The altered TSC2 gene was observed in both metaplastic breast cancer (a rare special histological type of TNBC) and LAR cohorts (2/17, 2/8, respectively) (93, 94). TP53 is one of the most frequent mutated genes in TNBC somatic cells (95). Analyzing several data sets for breast cancer patients suggested that high expression of death-associated protein kinase 1 (DAPK1) is associated with a worse prognosis in patients with TP53-mutant cancers. Research has shown that DAPK1 overexpression increases the phosphorylation of TSC2 on Ser939, leading to the activation of the mTOR pathway (96). In addition, Huang et al. have found that TSC1/TSC2 inactivation may up-regulate programmed cell death receptor ligand 1 (PD-L1) at the transcriptional level and readjust the immune microenvironment by increasing the recruitment of cluster of differentiation (CD) 8+ T cells (97). In a case report, immunohistochemistry results for TNBC from a patient with TSC2 mutation showed PD-L1 and CD8 T cells diffuse positivity (98). This suggested that loss-of-function TSC2 may alter the TNBC tumor microenvironment and that immunotherapy may be effective for patients with TNBC. In addition, the IM subtype expresses higher levels of immune-related genes, including PD1, PD-L1, and T lymphocyte-associated antigen 4. Immune-related markers with high levels of expression indicate that individuals with this type of tumor may benefit from using immune checkpoint inhibitors (92, 99). The benefits of immunotherapy have been demonstrated in several clinical studies, including Keynote-355 (100, 101).
TSC2 is the main negative regulatory protein of mTORC1. Mutation, down-regulation, and protein inactivation of TSC2 in breast cancer cause over-activation of mTORC1, which leads to treatment resistance and metastasis. Therefore, it is extremely important to inhibit mTORC1. There are no drugs that directly target TSC2. However, an mTOR inhibitor (everolimus) has been approved by the U.S. Food and Drug Administration for treatment of breast cancer (102).
HR+/HER2- advanced breast cancer has a higher rate of TSC2 mutations compared to HR+/HER2- early breast cancer (83, 84). The BOLERO-2 study (103) has shown that everolimus combined with exemestane improved progression-free survival (PFS) compared to placebo and exemestane in postmenopausal patients with HR+ advanced breast cancer and resistance to aromatase inhibitor (AI). Everolimus also enhances the treatment efficacy of fulvestrant in AI-resistant, ER-positive metastatic breast cancer (104). In patients with AI-resistant HR+/HER2- breast cancer, the combination of everolimus with endocrine therapy has demonstrated a clinically significant benefit.
Miller et al. (105) have reported that HER2 inhibitor lapatinib reduces the cell growth and inhibits the activation of downstream effector mTORC1 in TSC2-expressing cells, but not in TSC2-knockdown cells. A combination of mTOR inhibition greatly improved the antitumor effects of trastuzumab (105). The BOLERO-3 study (106) has shown that the addition of everolimus to trastuzumab-based therapy in patients with HER2-positive breast cancer leads to a statistically significant prolonged PFS. Furthermore, everolimus in combination with antibody–drug conjugate T-DM1 had a synergistic antitumor effect in HER2-positive breast cancer (107). These outcomes suggested that the mTOR inhibitor plays a significant role in anti-HER2 therapy.
Everolimus is also used for patients with advanced TNBC. A Phase 1 trial for tamirolimus or everolimus in combination with doxorubicin and bevacizumab for patients with metastatic TNBC has shown that mTOR inhibitors extended the objective response rate to 21% and clinical benefit rate to 40%. Tissue samples were available for testing the genes in the PI3K pathway in 43 participants, but TSC2 mutations were not detected (108). In addition, the mTOR inhibitor rapamycin was shown to decrease the PD-L1 expression in PTEN-mutant TNBC cell lines (109). Although conclusive clinical evidence is lacking, mTOR inhibition combined with immunotherapy may be effective in patients with TNBC (110).
TSC2 is the hub of multiple signaling pathway networks. It can regulate cell cycle, autophagy, and other physiological functions and is closely related to the development, treatment, and prognosis of breast cancer. In recent years, immunotherapy and targeted drug use, including CDK4/6 and PI3K pathway inhibitors, have been on the rise in breast cancer treatment. They are being continuously explored in combination with chemotherapy and endocrine therapy. TSC2 involves mechanisms of action of many antitumor drugs. In addition, low expression of TSC2 is associated with poor prognosis. Therefore, in-depth study of TSC2 functions provides important clinical guidance for improving treatment efficacy, overcoming drug resistance, and predicting prognosis. It can also suggest new directions for breast cancer drug development in the future.
Q-YZ and Z-MH contributed equally to this work. Q-YZ and Z-MH wrote and edited the manuscript. W-MC and BL edited the manuscript and designed and supervised the work. All the authors read and approved the final version of the manuscript.
This research was funded by Natural Science Foundation of Zhejiang Province, China (grant number: LY17H160038, LY21H160005) and the Construction Fund of Key Medical Disciplines of Hangzhou (No.OO20200055).
We thank all those who participated in this study.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Henske EP, Jozwiak S, Kingswood JC, Sampson JR, Thiele EA. Tuberous sclerosis complex. Nat Rev Dis Primers (2016) 2:16035. doi: 10.1038/nrdp.2016.35
2. Jin F, Wienecke R, Xiao GH, Maize JC Jr., DeClue JE, Yeung RS. Suppression of tumorigenicity by the wild-type tuberous sclerosis 2 (Tsc2) gene and its c-terminal region. Proc Natl Acad Sci U S A (1996) 93:9154–9. doi: 10.1073/pnas.93.17.9154
3. Kurian AW, Ward KC, Abrahamse P, Bondarenko I, Hamilton AS, Deapen D, et al. Time trends in receipt of germline genetic testing and results for women diagnosed with breast cancer or ovarian cancer, 2012-2019. J Clin Oncol (2021) 39:1631–40. doi: 10.1200/jco.20.02785
4. Ye Q, Ling S, Jiang G, Shan Q, Xu S, Zhan Q, et al. Sirolimus-based immunosuppression improves the prognosis of liver transplantation recipients with low TSC1/2 expression in hepatocellular carcinoma beyond the Milan criteria. Eur J Surg Oncol (2021) 47:2533–42. doi: 10.1016/j.ejso.2021.04.001
5. Song K, He F, Xin Y, Guan G, Huo J, Zhu Q, et al. TSC2 mutations were associated with the early recurrence of patients with HCC underwent hepatectomy. Pharmgenom Pers Med (2021) 14:269–78. doi: 10.2147/pgpm.S294307
6. Ho DWH, Chan LK, Chiu YT, Xu IMJ, Poon RTP, Cheung TT, et al. TSC1/2 mutations define a molecular subset of HCC with aggressive behaviour and treatment implication. Gut (2017) 66:1496–506. doi: 10.1136/gutjnl-2016-312734
7. Liang MC, Ma J, Chen L, Kozlowski P, Qin W, Li D, et al. TSC1 loss synergizes with KRAS activation in lung cancer development in the mouse and confers rapamycin sensitivity. Oncogene (2010) 29:1588–97. doi: 10.1038/onc.2009.452
8. Lee YH, Do SK, Lee SY, Kang HG, Choi JE, Hong MJ, et al. TSC2 genetic variant and prognosis in non-small cell lung cancer after curative surgery. Thorac Cancer (2019) 10:335–40. doi: 10.1111/1759-7714.12951
9. Kwiatkowski DJ, Choueiri TK, Fay AP, Rini BI, Thorner AR, de Velasco G, et al. Mutations in TSC1, TSC2, and MTOR are associated with response to rapalogs in patients with metastatic renal cell carcinoma. Clin Cancer Res (2016) 22:2445–52. doi: 10.1158/1078-0432.Ccr-15-2631
10. Roldan-Romero JM, Beuselinck B, Santos M, Rodriguez-Moreno JF, Lanillos J, Calsina B, et al. PTEN expression and mutations in TSC1, TSC2 and MTOR are associated with response to rapalogs in patients with renal cell carcinoma. Int J Cancer (2020) 146:1435–44. doi: 10.1002/ijc.32579
11. Mallela K, Kumar A. Role of TSC1 in physiology and diseases. Mol Cell Biochem (2021) 476:2269–82. doi: 10.1007/s11010-021-04088-3
12. Mehta MS, Vazquez A, Kulkarni DA, Kerrigan JE, Atwal G, Metsugi S, et al. Polymorphic variants in TSC1 and TSC2 and their association with breast cancer phenotypes. Breast Cancer Res Treat (2011) 125:861–8. doi: 10.1007/s10549-010-1062-1
13. Wang S, Zhang C, Xu Z, Chen MH, Yu H, Wang L, et al. Differential impact of PI3K/AKT/mTOR signaling on tumor initiation and progression in animal models of prostate cancer. Prostate (2023) 83:97–108. doi: 10.1002/pros.24441
14. Jiang WG, Sampson J, Martin TA, Lee-Jones L, Watkins G, Douglas-Jones A, et al. Tuberin and hamartin are aberrantly expressed and linked to clinical outcome in human breast cancer: the role of promoter methylation of TSC genes. Eur J Cancer (2005) 41:1628–36. doi: 10.1016/j.ejca.2005.03.023
15. Chakraborty S, Mohiyuddin SM, Gopinath KS, Kumar A. Involvement of TSC genes and differential expression of other members of the mTOR signaling pathway in oral squamous cell carcinoma. BMC Cancer (2008) 8:163. doi: 10.1186/1471-2407-8-163
16. Sridharan S, Basu A. Distinct roles of mTOR targets S6K1 and S6K2 in breast cancer. Int J Mol Sci (2020) 21:1199. doi: 10.3390/ijms21041199
17. Maskey RS, Wang F, Lehman E, Wang Y, Emmanuel N, Zhong W, et al. Sustained mTORC1 activity during palbociclib-induced growth arrest triggers senescence in ER+ breast cancer cells. Cell Cycle (2021) 20:65–80. doi: 10.1080/15384101.2020.1859195
18. Miricescu D, Totan A, Stanescu S II, Badoiu SC, Stefani C, Greabu M. PI3K/AKT/mTOR signaling pathway in breast cancer: from molecular landscape to clinical aspects. Int J Mol Sci (2020) 22:173. doi: 10.3390/ijms22010173
19. Rosner M, Hanneder M, Siegel N, Valli A, Hengstschlager M. The tuberous sclerosis gene products hamartin and tuberin are multifunctional proteins with a wide spectrum of interacting partners. Mutat Res (2008) 658:234–46. doi: 10.1016/j.mrrev.2008.01.001
20. Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev (2003) 17:1829–34. doi: 10.1101/gad.1110003
21. Yan L, Findlay GM, Jones R, Procter J, Cao Y, Lamb RF. Hyperactivation of mammalian target of rapamycin (mTOR) signaling by a gain-of-function mutant of the rheb GTPase. J Biol Chem (2006) 281:19793–7. doi: 10.1074/jbc.C600028200
22. European Chromosome 16 Tuberous Sclerosis C. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell (1993) 75:1305–15. doi: 10.1016/0092-8674(93)90618-z
23. Yang H, Yu Z, Chen X, Li J, Li N, Cheng J, et al. Structural insights into TSC complex assembly and GAP activity on rheb. Nat Commun (2021) 12:339. doi: 10.1038/s41467-020-20522-4
24. Chong-Kopera H, Inoki K, Li Y, Zhu T, Garcia-Gonzalo FR, Rosa JL, et al. TSC1 stabilizes TSC2 by inhibiting the interaction between TSC2 and the HERC1 ubiquitin ligase. J Biol Chem (2006) 281:8313–6. doi: 10.1074/jbc.C500451200
25. Huang J, Manning BD. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem J (2008) 412:179–90. doi: 10.1042/BJ20080281
26. Dibble CC, Elis W, Menon S, Qin W, Klekota J, Asara JM, et al. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol Cell (2012) 47:535–46. doi: 10.1016/j.molcel.2012.06.009
27. Qin J, Wang Z, Hoogeveen-Westerveld M, Shen G, Gong W, Nellist M, et al. Structural basis of the interaction between tuberous sclerosis complex 1 (TSC1) and Tre2-Bub2-Cdc16 domain family member 7 (TBC1D7). J Biol Chem (2016) 291:8591–601. doi: 10.1074/jbc.M115.701870
28. Dibble CC, Manning BD. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat Cell Biol (2013) 15:555–64. doi: 10.1038/ncb2763
29. Howell JJ, Ricoult SJ, Ben-Sahra I, Manning BD. A growing role for mTOR in promoting anabolic metabolism. Biochem Soc Trans (2013) 41:906–12. doi: 10.1042/BST20130041
30. Valvezan AJ, Manning BD. Molecular logic of mTORC1 signalling as a metabolic rheostat. Nat Metab (2019) 1:321–33. doi: 10.1038/s42255-019-0038-7
31. Deleyto-Seldas N, Efeyan A. The mTOR-autophagy axis and the control of metabolism. Front Cell Dev Biol (2021) 9:655731. doi: 10.3389/fcell.2021.655731
32. Long X, Ortiz-Vega S, Lin Y, Avruch J. Rheb binding to mammalian target of rapamycin (mTOR) is regulated by amino acid sufficiency. J Biol Chem (2005) 280:23433–6. doi: 10.1074/jbc.C500169200
33. Kwiatkowski DJ. Rhebbing up mTOR: new insights on TSC1 and TSC2, and the pathogenesis of tuberous sclerosis. Cancer Biol Ther (2003) 2:471–6. doi: 10.4161/cbt.2.5.446
34. Prentzell MT, Rehbein U, Cadena Sandoval M, De Meulemeester AS, Baumeister R, Brohee L, et al. G3BPs tether the TSC complex to lysosomes and suppress mTORC1 signaling. Cell (2021) 184:655–74 e27. doi: 10.1016/j.cell.2020.12.024
35. Rehbein U, Prentzell MT, Cadena Sandoval M, Heberle AM, Henske EP, Opitz CA, et al. The TSC complex-mTORC1 axis: from lysosomes to stress granules and back. Front Cell Dev Biol (2021) 9:751892. doi: 10.3389/fcell.2021.751892
36. Demetriades C, Doumpas N, Teleman AA. Regulation of TORC1 in response to amino acid starvation via lysosomal recruitment of TSC2. Cell (2014) 156:786–99. doi: 10.1016/j.cell.2014.01.024
37. Demetriades C, Plescher M, Teleman AA. Lysosomal recruitment of TSC2 is a universal response to cellular stress. Nat Commun (2016) 7:10662. doi: 10.1038/ncomms10662
38. Goncharova EA, Goncharov DA, Li H, Pimtong W, Lu S, Khavin I, et al. mTORC2 is required for proliferation and survival of TSC2-null cells. Mol Cell Biol (2011) 31:2484–98. doi: 10.1128/mcb.01061-10
39. Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell (2002) 10:151–62. doi: 10.1016/s1097-2765(02)00568-3
40. Menon S, Dibble CC, Talbott G, Hoxhaj G, Valvezan AJ, Takahashi H, et al. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell (2014) 156:771–85. doi: 10.1016/j.cell.2013.11.049
41. Cai SL, Tee AR, Short JD, Bergeron JM, Kim J, Shen J, et al. Activity of TSC2 is inhibited by AKT-mediated phosphorylation and membrane partitioning. J Cell Biol (2006) 173:279–89. doi: 10.1083/jcb.200507119
42. DeYoung MP, Horak P, Sofer A, Sgroi D, Ellisen LW. Hypoxia regulates TSC1/2-mTOR signaling and tumor suppression through REDD1-mediated 14-3-3 shuttling. Genes Dev (2008) 22:239–51. doi: 10.1101/gad.1617608
43. Ma L, Chen Z, Erdjument-Bromage H, Tempst P, Pandolfi PP. Phosphorylation and functional inactivation of TSC2 by erk implications for tuberous sclerosis and cancer pathogenesis. Cell (2005) 121:179–93. doi: 10.1016/j.cell.2005.02.031
44. Ma L, Teruya-Feldstein J, Bonner P, Bernardi R, Franz DN, Witte D, et al. Identification of S664 TSC2 phosphorylation as a marker for extracellular signal-regulated kinase mediated mTOR activation in tuberous sclerosis and human cancer. Cancer Res (2007) 67:7106–12. doi: 10.1158/0008-5472.CAN-06-4798
45. Roux PP, Ballif BA, Anjum R, Gygi SP, Blenis J. Tumor-promoting phorbol esters and activated ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A (2004) 101:13489–94. doi: 10.1073/pnas.0405659101
46. Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell (2003) 115:577–90. doi: 10.1016/s0092-8674(03)00929-2
47. Van Nostrand JL, Hellberg K, Luo EC, Van Nostrand EL, Dayn A, Yu J, et al. AMPK regulation of raptor and TSC2 mediate metformin effects on transcriptional control of anabolism and inflammation. Genes Dev (2020) 34:1330–44. doi: 10.1101/gad.339895.120
48. Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, et al. TSC2 integrates wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell (2006) 126:955–68. doi: 10.1016/j.cell.2006.06.055
49. Kuo HP, Lee DF, Chen CT, Liu M, Chou CK, Lee HJ, et al. ARD1 stabilization of TSC2 suppresses tumorigenesis through the mTOR signaling pathway. Sci Signal (2010) 3:ra9. doi: 10.1126/scisignal.2000590
50. Gen S, Matsumoto Y, Kobayashi KI, Suzuki T, Inoue J, Yamamoto Y. Stability of tuberous sclerosis complex 2 is controlled by methylation at R1457 and R1459. Sci Rep (2020) 10:21160. doi: 10.1038/s41598-020-78274-6
51. Xiao GH, Shoarinejad F, Jin F, Golemis EA, Yeung RS. The tuberous sclerosis 2 gene product, tuberin, functions as a Rab5 GTPase activating protein (GAP) in modulating endocytosis. J Biol Chem (1997) 272:6097–100. doi: 10.1074/jbc.272.10.6097
52. Astrinidis A, Cash TP, Hunter DS, Walker CL, Chernoff J, Henske EP. Tuberin, the tuberous sclerosis complex 2 tumor suppressor gene product, regulates rho activation, cell adhesion and migration. Oncogene (2002) 21:8470–6. doi: 10.1038/sj.onc.1205962
53. Suzuki T, Das SK, Inoue H, Kazami M, Hino O, Kobayashi T, et al. Tuberous sclerosis complex 2 loss-of-function mutation regulates reactive oxygen species production through Rac1 activation. Biochem Biophys Res Commun (2008) 368:132–7. doi: 10.1016/j.bbrc.2008.01.077
54. Dressler AC, Hudelist G, Fink-Retter A, Gschwantler-Kaulich D, Pfeiler G, Rosner M, et al. Tuberin and p27 expression in breast cancer patients with or without BRCA germline mutations. J Cancer Res Clin Oncol (2013) 139:1349–55. doi: 10.1007/s00432-013-1443-z
55. Soucek T, Pusch O, Wienecke R, DeClue JE, Hengstschläger M. Role of the tuberous sclerosis gene-2 product in cell cycle control. loss of the tuberous sclerosis gene-2 induces quiescent cells to enter s phase. J Biol Chem (1997) 272:29301–8. doi: 10.1074/jbc.272.46.29301
56. Soucek T, Yeung RS, Hengstschläger M. Inactivation of the cyclin-dependent kinase inhibitor p27 upon loss of the tuberous sclerosis complex gene-2. Proc Natl Acad Sci U S A (1998) 95:15653–8. doi: 10.1073/pnas.95.26.15653
57. Rosner M, Freilinger A, Hengstschlager M. The tuberous sclerosis genes and regulation of the cyclin-dependent kinase inhibitor p27. Mutat Res (2006) 613:10–6. doi: 10.1016/j.mrrev.2006.03.001
58. Pradhan SA, Rather MI, Tiwari A, Bhat VK, Kumar A. Evidence that TSC2 acts as a transcription factor and binds to and represses the promoter of epiregulin. Nucleic Acids Res (2014) 42:6243–55. doi: 10.1093/nar/gku278
59. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin (2021) 71:209–49. doi: 10.3322/caac.21660
60. Prat A, Perou CM. Deconstructing the molecular portraits of breast cancer. Mol Oncol (2011) 5:5–23. doi: 10.1016/j.molonc.2010.11.003
61. Dey N, De P, Leyland-Jones B. PI3K-AKT-mTOR inhibitors in breast cancers: from tumor cell signaling to clinical trials. Pharmacol Ther (2017) 175:91–106. doi: 10.1016/j.pharmthera.2017.02.037
62. Lee DF, Kuo HP, Chen CT, Hsu JM, Chou CK, Wei Y, et al. IKK beta suppression of TSC1 links inflammation and tumor angiogenesis via the mTOR pathway. Cell (2007) 130:440–55. doi: 10.1016/j.cell.2007.05.058
63. Yarden Y, Pines G. The ERBB network: at last, cancer therapy meets systems biology. Nat Rev Cancer (2012) 12:553–63. doi: 10.1038/nrc3309
64. Slamon DJ, Clark GM, Wong SG, Levin WJ, Ullrich A, McGuire WL. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science (1987) 235:177–82. doi: 10.1126/science.3798106
65. Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell (2014) 25:282–303. doi: 10.1016/j.ccr.2014.02.025
66. Goutsouliak K, Veeraraghavan J, Sethunath V, De Angelis C, Osborne CK, Rimawi MF, et al. Towards personalized treatment for early stage HER2-positive breast cancer. Nat Rev Clin Oncol (2020) 17:233–50. doi: 10.1038/s41571-019-0299-9
67. Gutierrez C, Schiff R. HER2: biology, detection, and clinical implications. Arch Pathol Lab Med (2011) 135:55–62. doi: 10.5858/2010-0454-rar.1
68. Mattoon DR, Lamothe B, Lax I, Schlessinger J. The docking protein Gab1 is the primary mediator of EGF-stimulated activation of the PI-3K/Akt cell survival pathway. BMC Biol (2004) 2:24. doi: 10.1186/1741-7007-2-24
69. Rexer BN, Arteaga CL. Optimal targeting of HER2-PI3K signaling in breast cancer: mechanistic insights and clinical implications. Cancer Res (2013) 73:3817–20. doi: 10.1158/0008-5472.Can-13-0687
70. Romero-Pozuelo J, Figlia G, Kaya O, Martin-Villalba A, Teleman AA. Cdk4 and Cdk6 couple the cell-cycle machinery to cell growth via mTORC1. Cell Rep (2020) 31:107504. doi: 10.1016/j.celrep.2020.03.068
71. Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, et al. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 inhibitors. Cancer Cell (2016) 29:255–69. doi: 10.1016/j.ccell.2016.02.006
72. Corona SP, Ravelli A, Cretella D, Cappelletti MR, Zanotti L, Dester M, et al. CDK4/6 inhibitors in HER2-positive breast cancer. Crit Rev Oncol Hematol (2017) 112:208–14. doi: 10.1016/j.critrevonc.2017.02.022
73. De A, Kuppusamy G. Metformin in breast cancer: preclinical and clinical evidence. Curr Probl Cancer (2020) 44:100488. doi: 10.1016/j.currproblcancer.2019.06.003
74. Zhang HH, Huang J, Düvel K, Boback B, Wu S, Squillace RM, et al. Insulin stimulates adipogenesis through the akt-TSC2-mTORC1 pathway. PloS One (2009) 4:e6189. doi: 10.1371/journal.pone.0006189
75. Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, et al. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell (2008) 30:214–26. doi: 10.1016/j.molcel.2008.03.003
76. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest (2001) 108:1167–74. doi: 10.1172/jci13505
77. Wang Y, Wei J, Li L, Fan C, Sun Y. Combined use of metformin and everolimus is synergistic in the treatment of breast cancer cells. Oncol Res (2014) 22:193–201. doi: 10.3727/096504015x14348950540999
78. Goodwin PJ, Dowling RJO, Ennis M, Chen BE, Parulekar WR, Shepherd LE, et al. Effect of metformin versus placebo on metabolic factors in the MA.32 randomized breast cancer trial. NPJ Breast Cancer (2021) 7:74. doi: 10.1038/s41523-021-00275-z
79. Osborne CK, Schiff R. Mechanisms of endocrine resistance in breast cancer. Annu Rev Med (2011) 62:233–47. doi: 10.1146/annurev-med-070909-182917
80. Yu J, Henske EP. Estrogen-induced activation of mammalian target of rapamycin is mediated via tuberin and the small GTPase ras homologue enriched in brain. Cancer Res (2006) 66:9461–6. doi: 10.1158/0008-5472.CAN-06-1895
81. Finlay GA, York B, Karas RH, Fanburg BL, Zhang H, Kwiatkowski DJ, et al. Estrogen-induced smooth muscle cell growth is regulated by tuberin and associated with altered activation of platelet-derived growth factor receptor-beta and ERK-1/2. J Biol Chem (2004) 279:23114–22. doi: 10.1074/jbc.M401912200
82. Cha S, Lee E, Won HH. Comprehensive characterization of distinct genetic alterations in metastatic breast cancer across various metastatic sites. NPJ Breast Cancer (2021) 7:93. doi: 10.1038/s41523-021-00303-y
83. van Geelen CT, Savas P, Teo ZL, Luen SJ, Weng CF, Ko YA, et al. Clinical implications of prospective genomic profiling of metastatic breast cancer patients. Breast Cancer Res (2020) 22:91. doi: 10.1186/s13058-020-01328-0
84. Lefebvre C, Bachelot T, Filleron T, Pedrero M, Campone M, Soria JC, et al. Mutational profile of metastatic breast cancers: a retrospective analysis. PloS Med (2016) 13:e1002201. doi: 10.1371/journal.pmed.1002201
85. André F, Ciruelos E, Rubovszky G, Campone M, Loibl S, Rugo HS, et al. Alpelisib for PIK3CA-mutated, hormone receptor-positive advanced breast cancer. N Engl J Med (2019) 380:1929–40. doi: 10.1056/NEJMoa1813904
86. Corné J, Le Du F, Quillien V, Godey F, Robert L, Bourien H, et al. Development of multiplex digital PCR assays for the detection of PIK3CA mutations in the plasma of metastatic breast cancer patients. Sci Rep (2021) 11:17316. doi: 10.1038/s41598-021-96644-6
87. Juric D, Janku F, Rodón J, Burris HA, Mayer IA, Schuler M, et al. Alpelisib plus fulvestrant in PIK3CA-altered and PIK3CA-Wild-Type estrogen receptor-positive advanced breast cancer: a phase 1b clinical trial. JAMA Oncol (2019) 5:e184475. doi: 10.1001/jamaoncol.2018.4475
88. Castel P, Ellis H, Bago R, Toska E, Razavi P, Carmona FJ, et al. PDK1-SGK1 signaling sustains AKT-independent mTORC1 activation and confers resistance to PI3Kalpha inhibition. Cancer Cell (2016) 30:229–42. doi: 10.1016/j.ccell.2016.06.004
89. Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell (2018) 34:427–38.e6. doi: 10.1016/j.ccell.2018.08.008
90. Cai Y, Xu G, Wu F, Michelini F, Chan C, Qu X, et al. Genomic alterations in PIK3CA-mutated breast cancer result in mTORC1 activation and limit the sensitivity to PI3Kα inhibitors. Cancer Res (2021) 81:2470–80. doi: 10.1158/0008-5472.Can-20-3232
91. Cheng TD, Ambrosone CB, Hong CC, Lunetta KL, Liu S, Hu Q, et al. Genetic variants in the mTOR pathway and breast cancer risk in African American women. Carcinogenesis (2016) 37:49–55. doi: 10.1093/carcin/bgv160
92. Jiang YZ, Ma D, Suo C, Shi J, Xue M, Hu X, et al. Genomic and transcriptomic landscape of triple-negative breast cancers: subtypes and treatment strategies. Cancer Cell (2019) 35:428–40.e5. doi: 10.1016/j.ccell.2019.02.001
93. Stella S, Vitale SR, Massimino M, Motta G, Longhitano C, Lanzafame K, et al. Molecular analysis of luminal androgen receptor reveals activated pathways and potential therapeutic targets in breast cancer. Cancer Genomics Proteomics (2022) 19:464–76. doi: 10.21873/cgp.20333
94. Piscuoglio S, Ng CKY, Geyer FC, Burke KA, Cowell CF, Martelotto LG, et al. Genomic and transcriptomic heterogeneity in metaplastic carcinomas of the breast. NPJ Breast Cancer (2017) 3:48. doi: 10.1038/s41523-017-0048-0
95. Lehmann BD, Pietenpol JA. Clinical implications of molecular heterogeneity in triple negative breast cancer. Breast (2015) 24 Suppl 2:S36–40. doi: 10.1016/j.breast.2015.07.009
96. Zhao J, Zhao D, Poage GM, Mazumdar A, Zhang Y, Hill JL, et al. Death-associated protein kinase 1 promotes growth of p53-mutant cancers. J Clin Invest (2015) 125:2707–20. doi: 10.1172/jci70805
97. Huang Q, Li F, Hu H, Fang Z, Gao Z, Xia G, et al. Loss of TSC1/TSC2 sensitizes immune checkpoint blockade in non-small cell lung cancer. Sci Adv (2022) 8:eabi9533. doi: 10.1126/sciadv.abi9533
98. Gonda K, Akama T, Nakamura T, Hashimoto E, Kyoya N, Rokkaku Y, et al. Cluster of differentiation 8 and programmed cell death ligand 1 expression in triple-negative breast cancer combined with autosomal dominant polycystic kidney disease and tuberous sclerosis complex: a case report. J Med Case Rep (2019) 13:381. doi: 10.1186/s13256-019-2274-6
99. Jiang YZ, Liu Y, Xiao Y, Hu X, Jiang L, Zuo WJ, et al. Molecular subtyping and genomic profiling expand precision medicine in refractory metastatic triple-negative breast cancer: the FUTURE trial. Cell Res (2021) 31:178–86. doi: 10.1038/s41422-020-0375-9
100. Planes-Laine G, Rochigneux P, Bertucci F, Chrétien AS, Viens P, Sabatier R, et al. PD-1/PD-L1 targeting in breast cancer: the first clinical evidences are emerging. a literature review. Cancers (Basel) (2019) 11:1033. doi: 10.3390/cancers11071033
101. Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med (2018) 379:2108–21. doi: 10.1056/NEJMoa1809615
102. Zhu K, Wu Y, He P, Fan Y, Zhong X, Zheng H, et al. PI3K/AKT/mTOR-targeted therapy for breast cancer. Cells (2022) 11:2508. doi: 10.3390/cells11162508
103. Baselga J, Campone M, Piccart M, Burris HA 3rd, Rugo HS, Sahmoud T, et al. Everolimus in postmenopausal hormone-receptor-positive advanced breast cancer. N Engl J Med (2012) 366:520–9. doi: 10.1056/NEJMoa1109653
104. Kornblum N, Zhao F, Manola J, Klein P, Ramaswamy B, Brufsky A, et al. Randomized phase II trial of fulvestrant plus everolimus or placebo in postmenopausal women with hormone receptor-positive, human epidermal growth factor receptor 2-negative metastatic breast cancer resistant to aromatase inhibitor therapy: results of PrE0102. J Clin Oncol (2018) 36:1556–63. doi: 10.1200/jco.2017.76.9331
105. Miller TW, Forbes JT, Shah C, Wyatt SK, Manning HC, Olivares MG, et al. Inhibition of mammalian target of rapamycin is required for optimal antitumor effect of HER2 inhibitors against HER2-overexpressing cancer cells. Clin Cancer Res (2009) 15:7266–76. doi: 10.1158/1078-0432.Ccr-09-1665
106. André F, O’Regan R, Ozguroglu M, Toi M, Xu B, Jerusalem G, et al. Everolimus for women with trastuzumab-resistant, HER2-positive, advanced breast cancer (BOLERO-3): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet Oncol (2014) 15:580–91. doi: 10.1016/s1470-2045(14)70138-x
107. Casadevall D, Hernández-Prat A, García-Alonso S, Arpí-Llucià O, Menéndez S, Qin M, et al. mTOR inhibition and T-DM1 in HER2-positive breast cancer. Mol Cancer Res (2022) 20:1108–21. doi: 10.1158/1541-7786.Mcr-21-0545
108. Basho RK, Gilcrease M, Murthy RK, Helgason T, Karp DD, Meric-Bernstam F, et al. Targeting the PI3K/AKT/mTOR pathway for the treatment of mesenchymal triple-negative breast cancer: evidence from a phase 1 trial of mTOR inhibition in combination with liposomal doxorubicin and bevacizumab. JAMA Oncol (2017) 3:509–15. doi: 10.1001/jamaoncol.2016.5281
109. Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S, et al. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res (2014) 2:361–70. doi: 10.1158/2326-6066.Cir-13-0127
Keywords: TSC2, breast cancer, signaling pathways, mTOR, PI3K
Citation: Zhu Q-Y, He Z-M, Cao W-M and Li B (2023) The role of TSC2 in breast cancer: a literature review. Front. Oncol. 13:1188371. doi: 10.3389/fonc.2023.1188371
Received: 22 March 2023; Accepted: 03 May 2023;
Published: 12 May 2023.
Edited by:
Nitin Telang, Palindrome Liaisons Consultants, Montvale, NJ, United StatesReviewed by:
Sang-Oh Yoon, University of Illinois Chicago, United StatesCopyright © 2023 Zhu, He, Cao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wen-Ming Cao, Y2Fvd21AempjYy5vcmcuY24=; Bei Li, bGliZWliZWk5NzAyQDEyNi5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.