 Hyun Jin Shin
Hyun Jin Shin Junjie T. Hua
Junjie T. Hua Haolong Li
Haolong Li- 1Helen Diller Family Comprehensive Cancer Center, University of California, San Francisco, San Francisco, CA, United States
- 2Department of Radiation Oncology, University of California, San Francisco, San Francisco, CA, United States
Epigenetic modifications, such as DNA methylation, is widely studied in cancer. DNA methylation patterns have been shown to distinguish between benign and malignant tumors in various cancers, including prostate cancer. It may also contribute to oncogenesis, as it is frequently associated with downregulation of tumor suppressor genes. Aberrant patterns of DNA methylation, in particular the CpG island hypermethylator phenotype (CIMP), have shown associative evidence with distinct clinical features and outcomes, such as aggressive subtypes, higher Gleason score, prostate-specific antigen (PSA), and overall tumor stage, overall worse prognosis, as well as reduced survival. In prostate cancer, hypermethylation of specific genes is significantly different between tumor and normal tissues. Methylation patterns could distinguish between aggressive subtypes of prostate cancer, including neuroendocrine prostate cancer (NEPC) and castration resistant prostate adenocarcinoma. Further, DNA methylation is detectable in cell-free DNA (cfDNA) and is reflective of clinical outcome, making it a potential biomarker for prostate cancer. This review summarizes recent advances in understanding DNA methylation alterations in cancers with the focus on prostate cancer. We discuss the advanced methodology used for evaluating DNA methylation changes and the molecular regulators behind these changes. We also explore the clinical potential of DNA methylation as prostate cancer biomarkers and its potential for developing targeted treatment of CIMP subtype of prostate cancer.
1 Introduction
Prostate cancer is the most common cancer in men, making up 29% of new cases, and one of the top cancer-related causes of death in men in the United States (1). In prostate cancer, androgen receptor (AR) is a key oncogenic driver, is often found amplified in the gene body and enhancer upstream of AR and is associated with aggressive progression of disease. While androgen-deprivation therapy (ADT) is the first line treatment for patients with locally advanced or metastatic prostate cancer, the disease often progresses to the castration resistant prostate cancer (CRPC) stage. Newly developed AR targeted therapies, such as enzalutamide and abiraterone, have shown promise as effective treatments for advanced prostate cancer. Yet, the resistant tumor invariably occurs, leading the disease to its terminal stage. To understand disease progression and treatment resistance, majority of studies have focused on genetic alterations of the key drivers such as AR gene, AR co-factors (e.g. NCOA2, EP300, and FOXA1), ETS gene fusions (e.g. TMPRSS2-ERG fusion), SPOP mutations, mutations affecting gene expression, and chromatin regulation (e.g. KDM6A/UTX, MLL2, MLL3, CHD1, and EZH2) (2, 3). Some of these genetic changes can be used as prognostic markers, such as mutations in BRCA1, BRCA2, HOXB13, ATM, and CHEK2, dysregulation of PTEN, TMPRSS2-ERG fusion, and high overall number of somatic copy number aberrations, which are associated with poor prognosis (4). In addition to AR signaling driven tumors, subtypes such as neuroendocrine prostate cancer (NEPC) are AR-independent, thus making ADT ineffective (5). NEPC is an aggressive histologic subtype of prostate cancer associated with poor prognosis. NEPC tumors share some common genetic aberrations as prostate adenocarcinoma, such as TMPRSS2-ERG fusion and loss of RB1 and TP53, but often do not express AR and downstream AR-regulated targets such as PSA and prostate-specific membrane antigen (PSMA) (6). Beyond genomic and transcriptomic subtypes of prostate cancer, epigenetic alterations are also found to play a critical role in prostate cancer progression and treatment resistance.
DNA methylation is an epigenetic process that involves the addition of methyl groups to DNA. The most predominant type of DNA methylation, termed 5-methylcytosine (5mC), typically happens on cytosines of CpG dinucleotide sequences and are usually associated with DNA inactivation. DNA methylation is modulated by DNA methyltransferase (DNMT) and ten eleven translocation (TET) enzymes, which are writers and erasers of 5mC, respectively. More specifically, DNMT1 maintains DNA methylation and prefers hemimethylated DNA, and DNMT3A and DNMT3B are responsible for de novo DNA methylation. Meanwhile, the TET family enzymes, TET1, TET2, and TET3, convert 5mC back to unmethylated cytosine in a series of steps with functionally important intermediates. 5mC is first converted to 5-hydroxymethylcytosine (5hmC), then 5-formylcytosine, followed by 5-carboxylcytosine, and finally back to cytosine. A portion of total methylation modifications consists of 5hmC. Unlike 5mC, 5hmC is enriched at transcriptionally active regions and associated with expression of many genes. Further, IDH1 and IDH2, which are not directly involved in methylation, modulates methylation by affecting TET2 function. IDH1 and IDH2 produces α-ketoglutarate, an obligatory substrate for TET. Most CpG dinucleotides in the genome are highly methylated, with the exception of CpG islands, CpG shores ( ± 2 kbp around islands), and CpG shelves ( ± 2kbp around shores), which show variable methylation level (7). CpG islands are regions of DNA with high concentration of CpG dinucleotides and are typically associated with cis-regulatory regions such as promoters. DNA methylation at promoters leads to gene repression by blocking transcription factors from binding and/or by recruiting methyl-CpG binding domain proteins that subsequently recruit and synergize with chromatin remodelers and histone deacetylases to establish a silenced chromatin state for long-term transcriptional repression (8–10). Similarly, DNA methylation at enhancers is reported to repress enhancer activity (11). While DNA methylation is essential for mammalian development and aging, aberrant methylation patterns are significant contributors to oncogenesis (12). Specifically, global DNA hypomethylation and hypermethylation of specific CpG islands is frequently observed in cancer, leading to expression of normally silenced repetitive elements and repression of tumor suppressors and DNA repair genes, respectively (13–15). Interestingly, these CpG hypermethylation profiles are found to be highly tumor-type-specific and may serve as potential biomarkers (15). By clustering cancer samples based on methylation levels at specific loci, a subtype of tumors characterized by hypermethylation of CpG island methylation has been identified and termed the CpG island methylator phenotype (CIMP).
The presence of a CIMP subtype is widely accepted in several cancer types, such as colorectal and breast cancer. However, whether CIMP is a pan-cancer phenomenon is still unclear, as are the exact molecular mechanisms driving CIMP (16). Most early findings on CIMP have been solely based on selective loci that lack consistencies between studies and cancer types, which hindered pan-cancer interpretation. Recent improvement of sequencing technologies and development of novel sequencing approaches, particularly whole-genome bisulfite sequencing, plays a significant role in providing a pan-cancer CIMP definition (17). Furthermore, integrative analyses across different sequencing approaches have accelerated our understanding of potential molecular mechanisms behind CIMP. Clinically, CIMP subtype is often associated with differential tumor prognosis, aggressiveness, and survival across different cancer types, which highlights the potential for the development of methylation-based prognostic biomarkers. Furthermore, demethylating agents are showing promise as novel cancer treatments (16, 18, 19). However, whether CIMP-associated hypermethylation is causal in tumorigenesis and cancer progression has remained largely elusive until recently (20). Further, other epigenetic changes are believed to be signs of disease progression in prostate cancer. For instance, changes like chromatin accessibility, SWI/SNF, histone marks, and DNA methylation, are distinguishing features of NEPC (5). This review will highlight recent biological and clinical findings of CIMP and other changes in methylation patterns in prostate cancer and discuss its clinical potentials.
2 DNA methylation and cancer
Aberrant DNA methylation patterns, specifically CIMP, is well established in multiple cancers, including colorectal cancer, gastric cancer, glioma, breast cancer, and leukemia. CIMP has first been identified in colorectal cancer through the detection of colorectal cancer-specific methylation in selective CpG island regions, including p16 and THBS1 (18). This concept has subsequently been validated by various studies which examined additional regions (21–25). CIMP is now a well-established molecular subtype of colorectal cancer that is associated with specific genetic and clinicopathological features and tumorigenic pathways. Notably, CIMP colorectal cancer is associated with BRAF V600E and KRAS mutations, CDX2 loss, as well as low chromosomal aberrations and high microsatellite instability (MSI), which is reported to be driven by methylation of hMLH1 gene (18, 21, 23–26). Colorectal cancer can be further categorized into three molecularly distinct subclasses based on CIMP status: CIMP-high (CIMP-H) tumors associated with MSI and BRAF mutations, CIMP-low tumors associated with KRAS mutations, and CIMP-negative tumors associated with high p53 mutations (20, 23). However, beyond these associations, the causal relationship between CIMP and these key driver mutations in colorectal cancer are largely unclear. Recent studies have found that aberrant DNA methylation occurs at early stages of colorectal cancer development and may sensitize colorectal cells to BRAF V600E-driven tumorigenic transformations into colorectal cancer (25, 26).
CIMP has been identified in other types of cancer but lacks a clear definition. In gastric cancer, CIMP is identified based on hypermethylation of specific genes, most commonly MINT1, MINT2, MINT12, MINT25, MINT31, hMLH1, and p16 (27–34). While many studies’ CIMP markers include these genes, others do not; and more recent studies use whole-genome sequencing to identify CIMP. Meanwhile, CIMP is identified in breast cancer by hypermethylation of a few genes, including tumor suppressor genes BRCA1, p16, APC (35, 36). Moreover, in blood cancer, CIMP has been initially identified in acute myeloid leukemia and acute lymphoblastic leukemia based on hypermethylation of specific genes such as CDH1, CDH13, and sFRP1 (37, 38). However, these sites, as well as few additional CpG sites, have only been validated in some but not all studies, suggesting extensive heterogeneity between studies (39–41). Overall, there has also been a lack of consensus on the definition of CIMP in leukemia in terms of which specific CpG sites were used as biomarkers to identify CIMP. More recently, several studies have applied genome-wide DNA methylation arrays along with a panel of 1,293 CpG sites to classify CIMP in leukemia to standardize the field (42, 43). Using this approach, CIMP has been defined by comparing methylation levels across a wider range of CpG sites rather than a methylation of specific genes, providing a clearer definition of CIMP. Overall, there is a lack of consensus on the definition of CIMP in various cancers in terms of specific sites used as CIMP markers, but new methods of detecting methylation allow identification of CIMP based on methylation of a greater number of CpG islands.
Despite the lack of a consistent definition of CIMP across cancer types in early studies, there is a consensus between multiple studies within each cancer type that CIMP is associated with distinct molecular features (Table 1). In gastric cancer, CIMP is associated with MSI, Epstein-Barr virus (EBV)-associated gastric cancer, and H. pylori infection (27, 28, 32–34, 44–47). Unlike colorectal cancer CIMP, gastric CIMP is not associated with p53 and KRAS mutations (28, 32, 48, 49). Methylation patterns in breast cancer show association with molecular subtypes of breast cancer. Most notably, CIMP is associated with the presence of estrogen receptor (ER) and progesterone receptor (PR) status (36, 50, 51). CIMP is also associated with invasive lobular breast cancer, which displays higher frequency of hypermethylation than invasive ductal carcinoma (52, 53). Also, CIMP shows association with copy number alterations, which could help further classify breast cancer subtypes (53, 54). CIMP in leukemia is associated with distinct molecular features, including TET2, IDH1 and IDH2 mutations in acute myeloid leukemia (43, 55–58). Furthermore, in acute myeloid leukemia, CIMP can be further divided into two categories, I-CIMP associated with IDH1/2 mutations and A-CIMP enriched in CEBPA and WT1 mutations (59). These molecular features suggest an association between hypermethylation and leukemogenesis, but a causal relationship has yet to be established. Moreover, in glioma, hypermethylation of promoter associated CpG islands of genes such as TMS1/ASC is commonly reported (60–62). CIMP has then been identified in grade IV gliomas, or glioblastomas, and in lower grade gliomas and found to be associated with specific molecular and clinical features (63–65). Significantly, CIMP is associated with methylation of O6-methylguanine DNA methyltransferase (MGMT) gene and IDH1 mutations (63, 65–67). Also, a study has found IDH1 can induce DNA hypermethylation that mimics CIMP subtypes in lower grade gliomas, suggesting a causal relationship (65). In addition, CIMP status is also associated with gene copy variations (68, 69).
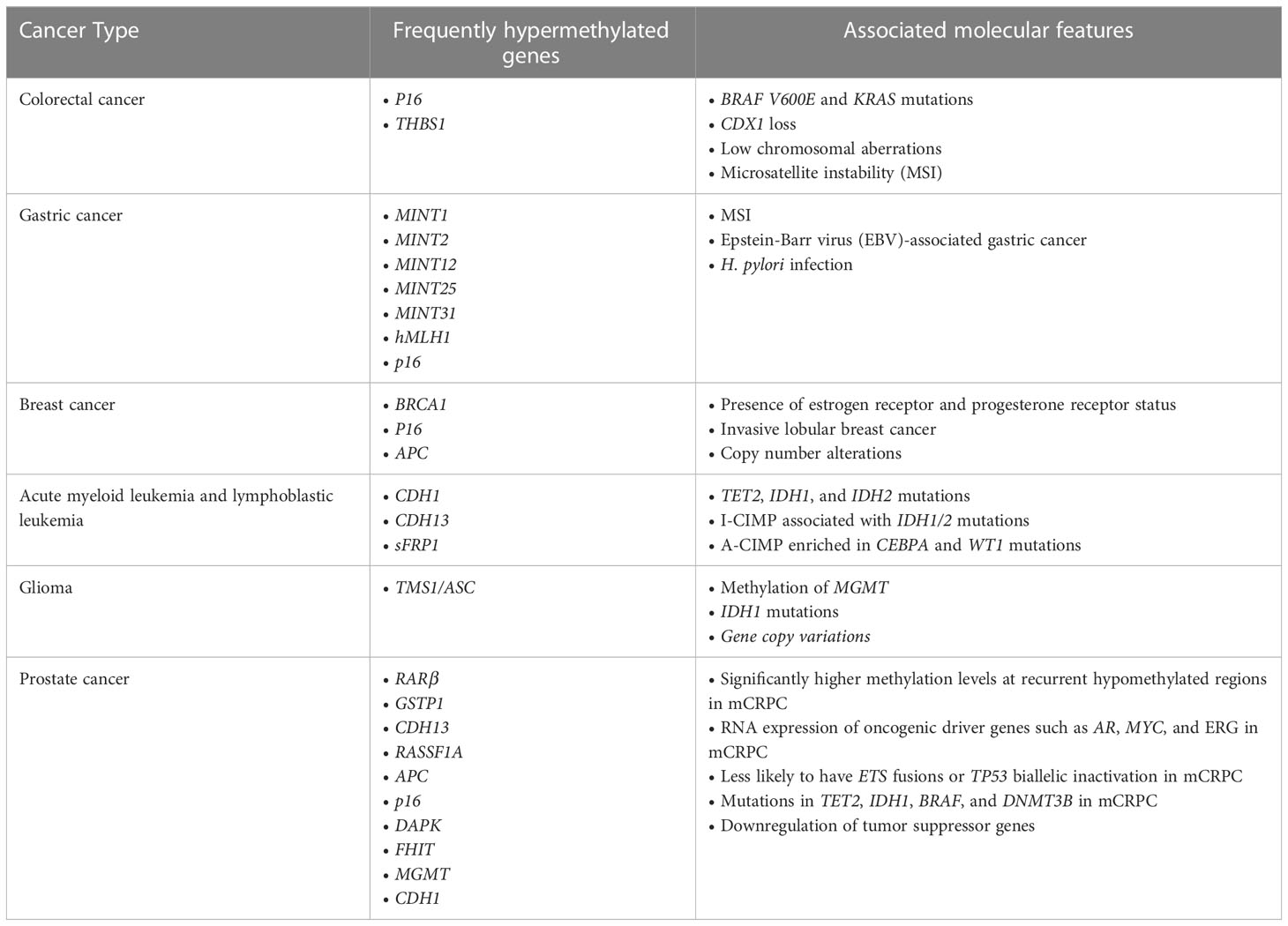
Table 1 Hypermethylated genes and associated molecular features of CIMP in various cancer types.
3 DNA methylation in prostate cancer
3.1 Methods for DNA methylation detection
Methylation-specific PCR (MS PCR) was most commonly used in early studies to analyze DNA methylation patterns in CpG island and determine CIMP status. For MS PCR, DNA is first purified and treated with sodium bisulfite, which converts cytosine cytosine to uracil but not 5-methylcytosine. Then, PCR is performed with two primer pairs for detectable methylated and unmethylated DNA. A different method to analyze DNA methylation patterns is using methyl-CpG binding domain (MBD). MBD preferentially binds methylated DNA and can be used to enrich methylated genomic DNA fragments and create libraries (70). This library can be analyzed using real-time PCR, tiling microarrays, and next-generation sequencing. DNA methylation alterations in prostate cancer samples can also be analyzed by sequencing sodium-bisulfite-converted genomic DNA (e.g. Illumina HumanMethylation27, MethylPlex-next-generation sequencing, MethylationEPIC Bead-Chip), which allows more quantitative accuracy and detection sensitivity, high efficiency, and a wide spectrum for analysis (71). Similar to MS PCR, for this method, DNA is treated with sodium bisulfite, then subsequent PCR and specific methylation primers are used to sequence and identify the methylated genomic regions (71). Another method to analyze DNA methylation patterns is using methylated DNA immunoprecipitation sequencing (MeDIP). In this method, genomic DNA is sonicated and immunoprecipitated using antibodies specific to 5mC, and resulting fragments are amplified, prepped, and sequenced. Similarly, hydroxymethylated DNA immunoprecipitation sequencing (hMeDIP-seq) can be used to analyze patterns of hydroxymethylation, using the same methods as MeDIP-seq, but using antibodies specific to 5hmC instead.
DNA methylation analysis methods each have their strengths and limitations. For example, array-based methods are useful for profiling DNA methylation changes across large regions of the genome, but they have limited coverage of CpG sites (72). Reduced representation bisulfite sequencing (RRBS) can provide high coverage of CpG sites, but its ability to read the entire genome is limited. Whole-genome bisulfite sequencing (WGBS) offers the highest resolution and can be performed on single nuclei, but the method would not distinguish 5mc and 5hmc (73). A combined 5mc and 5hmc detection method such as WGBS and oxidative WGBS (oxWGBS) could provide a more comprehensive view of DNA methylation changes, but this has not yet been extensively studied in the context of prostate cancer.
3.2 Patterns of CIMP in prostate cancer
Patterns in DNA methylation in prostate cancer are distinct from other cancer types mentioned above but share some common features. CIMP in prostate cancer is often determined by checking methylation status of several loci using MS PCR, then later confirmed using bilsulfite DNA sequencing (74). With newer methods of detecting DNA methylation at a more global scale, CIMP can be determined by cancer-specific differentially methylated regions rather than through checking DNA methylation of specific genes. As in leukemia, CIMP in prostate cancer can be identified as groups with higher methylation levels when comparing these differentially methylated regions. Genes commonly hypermethylated in prostate cancer include tumor suppressor genes involved in DNA damage repair, cell adhesion, apoptosis, cell cycle control, signal transduction, and hormonal responses, such as RARβ, GSTP1, CDH13, RASSF1A, APC, p16, DAPK, FHIT, MGMT, and CDH1 (70, 74–78). Some hypermethylated genes in prostate cancer are also hypermethylated in other types of cancer, such as p16 in colorectal cancer (18), gastric cancer (34), and leukemia; and CDH13, APC, and CDH1 in leukemia (35–38). When compared to samples from benign prostatic hyperplasia (BPH) and nonmalignant tissues, samples from prostate cancer showed higher levels of methylation (74, 76). Further, methylation of tumor suppressor genes GSTP1, APC, and MGMT is strongly associated with their downregulation, suggesting an important role for DNA methylation in driving carcinogenesis and disease progression (70, 78). Because increase in methylation may be an age-related event, Kang et al. have examined methylation status of APC, COX2, DAPK, CDH1, GSTP1, MGMT, p14, p16, RASSF1A, RUNX3, and THBS1 from non-neoplastic prostate samples of mostly older men. They have found that there is very low or no promoter methylation in these samples, suggesting hypermethylation of specific loci in prostate cancer is likely not an age-related event, but rather a tumor-related one (78).
While initial studies on methylation in prostate cancer have been limited due to the focus on evaluating methylation levels through MS PCR of select loci in a small number of prostate cancer samples, newer methods of analyzing methylation levels have emerged and allowed to analyze methylation in prostate cancer in a broader spectrum of an increasing number of tumor samples. Using the MBD approach, Aryee et al. have generated a library of methylated genomic DNA fragments and hybridized the library to Affymetrix SNP 6.0 high-density oligonucleotide microarrays and found DNA methylation alterations are maintained across all metastases within the same individual, and that regions with high consistency of hypermethylation across metastases within individuals show enrichment for cancer-related genes (70). Variability in genome-wide methylation patterns in benign, low-grade, and high-grade prostate cancer have also been analyzed using MDB-isolated genome sequencing (MiGS). This has revealed variations in methylation patterns that can distinguish between benign, low-grade, and high-grade prostate cancer samples. Further, by integrating DNA methylation data with RNA-seq and survival data, they have shown hypermethylation regions are in gene promoters and at intergenic regions that are enriched for DNA-protein binding sites (79). In addition, they have shown that downregulation of genes where DNA methylation and expression are well correlated is associated with poor outcome (79).
Using the sodium-bisulfite sequencing method, it has been shown that methylation pattern alterations are more frequent in prostate cancer and in benign prostate tissues adjacent to tumor, compared to age-matched organ-donor prostates (80). In addition, overall promoter CpG island methylation is significantly increased in localized and metastatic cancer tissues, and differentially methylated regions are cancer-specific (81). Also, by profiling DNA methylation in plasma samples of patients with metastatic prostate cancer over 9 months, Silva et al. show that methylation patterns within an individual are consistent with clinical progression, including disease progression and therapeutic response (82). By integrating methylome analysis with whole genome sequencing (WGS) and transcriptome sequencing (mRNA-seq), Gerhauser et al. describe four molecular subgroups of prostate cancer of different aggressiveness. Subgroup 1 represents normal basal and luminal prostate epithelium. Subgroup 2 is associated with high immune cell content but low T-luminal cell content, high GS, and shorter time to biochemical recurrence. Subgroup 3 represents an intermediate-risk group, and Subgroup 4 is associated with a high fraction of normal-like luminal cells and a known gene signature associated with less-aggressive prostate cancer (83). Hypermethylator phenotype has also been identified in metastatic castration resistant prostate cancer (mCRPC) using whole-genome bisulfite sequencing paired with deep whole-genome and transcriptome sequencing (84). This subtype has significantly higher methylation levels at recurrent hypomethylated regions (HMRs) and overall fewer HMRs at CpG island, shores, shelves, and in CpG open seas (regions outside islands, shelves, and shores) (84). There is also increased hypermethylation at differentiation and cancer genes (70). More specifically, in mCRPC, methylation is associated with RNA expression of oncogenic driver genes such as AR, MYC, and ERG (84). Moreover, key AR-associated genes, such as KLK3, NKX3-1, and FOLH1 are correlated with DNA methylation independent of DNA changes (84). They also have found that this subtype of tumors is less likely to have ETS fusions or TP53 biallelic inactivation, is not significantly associated with anatomic site of biopsy, and contains mutually exclusive mutations in TET2, IDH1, BRAF, and DNMT3B (84). In another study using enhanced reduced-representation bisulfite sequencing (eRRBS) on patient tumor samples, Beltran et al. show there is a strong epigenetic segregation between castration resistant neuroendocrine prostate cancer and castration resistant prostate adenocarcinoma. Notably, they have found hypermethylation and reduced expression of SPDEF, a tumor suppressor gene, in castration resistant neuroendocrine prostate cancer. This has been validated in the neuroendocrine prostate cancer cell line NCI-H660, as compared to prostate adenocarcinoma cell line LNCaP (85).
Furthermore, MeDIP sequencing of 51 tumor and 53 benign prostate samples has revealed there are more than 147,000 cancer-associated epigenetic alterations, there are significant global methylation pattern differences associated with TMPRSS2-ERG rearrangement status, and hypermethylation of miR-26a can be involved in ERG rearrangement-independent EZH2 activation (86). Further, another study using the same technique on samples from plasma DNA of patients with localized and metastatic prostate cancer has found that there is global hypermethylation in metastatic samples and hypomethylation in the pericentromeric regions (87). It also has shown that there is hypermethylation of the promoter of NR3C1, a glucocorticoid receptor gene, that is associated with decreased immune signature (87).
Beyond the hypermethylator phenotype, there are other methylation changes that may hold significance in prostate cancer. For example, somatic mutations and putative regulatory regions are frequently located in regions that are differentially hypomethylated (84). Not only is methylation silencing of tumor suppressors a significant event in progression of cancer, cancer-associated hypomethylation in oncogenic genes leading to their overexpression in mCRPC is also important (84). Multiple expression associated HMRs (eHMR) have been identified near AR, including AR promoter, AR enhancer, and additional loci upstream and downstream of AR (84). Although AR promoter is hypomethylated in all tissues, other eHMR are only identified in mCRPC samples but not in benign or primary PCa samples (84). The number of these hypomethylated eHMR loci is positively associated with AR expression. Additionally, eHMR loci found in AR gene body is positively associated with AR expression, representing novel intergenic regulatory regions of AR that can potentially contribute to ADT-resistance (84). 5hmC levels have also been shown to be associated with various clinical features of prostate cancer using hMeDIP-seq. 5hmC marks activation of cancer drivers and downstream targets such as AR, EZH2, CDK1, TBX3, HOXA13, FOXA1, and HOXB13 (88). There is also a progressive increase in 5hmC levels in genes in proliferative and oncogenic pathways during tumor progression, and 5hmC patterns can accurately track dedifferentiation and lineage plasticity to neuroendocrine and gastrointestinal lineages (88). Further, 5hmC patterns in cell-free DNA are able to be detected and used to accurately estimate ct-fraction and find specific gene activation of driver genes TOP2A and EZH2 that are not altered at the DNA level (88). Overall, in addition to patterns in 5mC methylation, hypomethylation and 5hmC patterns show potential to be used as a prognostic biomarker that can differentiate various subtypes of prostate cancer that genetic changes alone cannot.
4 Molecular drivers of CIMP
The development of CIMP in cancer has been attributed to various genomic and environmental factors, which differs depending on the cancer type. Most notably, protein-coding mutations in BRAF and IDH1 have been shown to establish CIMP (20, 65). In the case of CIMP-high colorectal cancer, spontaneous aging-like promoter hypermethylation makes organoids more sensitive to transformation by BRAF V600E mutation, which leads to CIMP (20). BRAF V600E mutation may lead to CIMP in a pathway that involves MAFG, which binds promoters of MLH1 and other CIMP-related genes and recruits corepressor complex, leading to hypermethylation and gene silencing (89). In CIMP-low colorectal cancer, KRAS upregulates zinc-finger DNA-binding protein, ZNF304, which binds promoters and recruits a corepressor complex with DNMT1, leading to DNA hypermethylation (90). Contrastingly, in glioma and leukemia, IDH1 mutations that result in 2-hydroxyglutarate production disrupts TET2 function and establishes CIMP and global DNA hypermethylation (58, 65). TET2 loss of function mutation itself is also associated with similar epigenetic defects as IDH1 mutants, and TET2 knockouts are also frequently associated with hypermethylation (58, 91–93). In addition, mutations in DNMT3A and DNMT3B and knock outs are also frequently associated with hypomethylation, while overexpression of DNMT3B is associated with hypermethylation in gastric and breast cancer cell lines (91, 94–100). As mentioned previously, mCRPC tumors of the hypermethylator subtype contain mutually exclusive mutations in TET2, IDH1, BRAF, and DNMT3B, suggesting mutations in these proteins may contribute to hypermethylation (84). Further, Kobayashi et al. have shown there is increased expression of DNMT3A2, DNMT3B, and EZH2 in tumors, and transient DNMT3B1 and DNMT3B2 overexpression in primary prostate cells results in increased methylation of some CpG sites that show increased methylation in tumors (101). Furthermore, in AML, TET2, IDH1, and DNMT3B do not seem to affect each other in terms of methylation pattern and regulation of downstream genes, but IDH1 and DNMT3A do (58, 102). More specifically, co-occurrence of DNMT3A and IDH1 mutations show epigenetic patterns different from those of either IDH1 or DNMT3A mutation, upregulation of RAS signaling and unique sensitivity to MEK inhibition and appear to be associated with either worse clinical outcome or show no difference in EFS or OS (103–105). In addition, DNMT3A and TET2 also seem to affect one another, showing different methylation patterns and phenotypes (106, 107). However, it is not clear if this is the case in prostate cancer, especially considering Zhao et al. have found mutations in TET2, IDH1, and BRAF were mutually exclusive in mCRPC samples. While mutations in TET2, IDH1, DNMT3A, and DNMT3B may play a role in establishing distinct methylation patterns found in prostate cancer, further study is required to establish a causal relationship and see how the various proteins involved in methylation interact with and affect one another.
Aside from gene mutations, several other factors such as EBV infection, aging and hypoxia also contribute to methylation changes. In multiple studies, EBV infection of epithelial cells in vitro directly induces global hypermethylation of the host genome, around the transcription start site, and results in gene silencing (108). It is partly driven by EBV latency protein, latent membrane protein 2A (LMP2A), which upregulates expression of DNMT1 and downregulates TET1 and TET2, as well as LMP1, which upregulates DNMT1, DNMT3A, and DNMT3B (108). In addition, it has been previously demonstrated that aging is associated with increased CpG island hypermethylation in colon mucosa (18, 109). Methylation of CpG island on the ER gene becomes progressively more pronounced with age, even in the early stages of tumor formation (109). This methylation of CpG island is associated with transcription repression, and in some cases, less ER expression (109). Moreover, re-expression of ER gene showed growth inhibition of colon carcinoma cells (109). Together, these findings suggest reduced ER expression, which is associated with age-related hypermethylation of CpG island on the ER gene, may be an early event that predisposes to sporadic colorectal tumorigenesis. Furthermore, hypoxia has been shown to increase hypermethylation at gene promoters in murine breast tumors (110). Mechanistically, hypoxia inhibits oxygen-dependent catalytic activities of the TET family methylation erasers, leading to the accumulation of methylation (110, 111). Oxidative stress from inflammation can also induce CpG island hypermethylation in tumors (112, 113). Specifically, oxidative stress generates 7,8-dihydro-8-guanine, which recruits DNMT1 that interacts with MSH2-MSH6 protein and methylates DNA promoters (112–114). JAK2, which is also associated with CIMP, localizes to the nucleus, interacts with MSH2-MSH6 upon oxidative stress induction and helps drive oxidative stress-induced interaction of MSH2-MSH6 with DNMT1 and consequently, global methylation (115).
5 Clinical implications
5.1 Biomarker
CIMP has been found to independently associate with patient survival in several cancer types, including kidney renal clear cell carcinoma, hepatocellular carcinoma, leukemia, gastric cancer, breast cancer, and adrenocortical carcinoma (30, 32, 39, 42, 43, 53, 116–119). CIMP is associated with worse prognosis in colorectal cancer, breast cancer, renal cell carcinoma, and adrenocortical carcinoma (26, 30, 41–43, 118, 120–124). CIMP status is also associated with other cancer type-specific clinical characteristics. In colorectal cancer, CIMP-H status is associated with female gender, proximal tumor location, higher tumor grade, older age, poor differentiation, and MSI (23, 26, 117). Moreover, colorectal cancer diagnosed within 5 years after colonoscopy is more likely to have CIMP and MSI than cancer diagnosed after 5 years (125). In breast cancer, CIMP is associated with high grade and increased metastatic risk (53, 118). Further, methylation in serum is also associated with breast cancer and recurrence risk of rural sporadic breast cancer, showing potential of CIMP to be detectable in serum of breast cancer patients and allow distinction between tumor and normal samples with at least 90% specificity and sensitivity (35, 36, 126). Classification of glioma based on combination of CIMP status and copy number alteration status is associated with survival (63, 68, 69). In addition, CIMP is associated with better overall survival, as well as low-grade glioma and improved outcome (63–65, 68, 69, 127, 128). Studies have also found that upon recurrence, there is a shift from CIMP high to CIMP low (26, 41).
However, clinical features of CIMP in gastric cancer and leukemia remain ambiguous. Some studies have shown that CIMP is associated with better overall survival and progression-free survival while others have concluded that CIMP is associated with higher stage, lymph node metastasis, and worse survival (27–34, 49, 120, 121, 129). This discrepancy is likely due to heterogeneity between studies in both CIMP markers and patient samples. Majority of studies showing better prognosis identifies CIMP by a set of genes that included MINT1, MINT2, MINT12, MINT25, and MINT31. In acute myeloid leukemia, some studies have found A-CIMP patients are associated with longer overall survival than CIMP-negative patients while I-CIMP patients are not (59, 130). There is also evidence of increase in methylation at relapse (38). Contrarily, recent studies using genome-wide approaches have found CIMP patients are associated with better overall and disease-free survival in both T and B cell acute lymphoblastic leukemia, with shorter response to treatments in T cell acute lymphoblastic leukemia (42, 43, 131, 132). Clinical features of CIMP acute lymphoblastic leukemia are ambiguous, possibly also due to the lack of consensus on the definition of CIMP. Early studies with CIMP defined by selective CpG sites found CIMP patients are associated with worse disease-free and overall survival (37, 39). Similarly, inconclusive results from gastric cancer perhaps are due to differences in what genes are used to identify CIMP, which further warrants meta-analysis of global methylation data in the future to identify CIMP. Regardless, specific differentially methylated regions can also be used to distinguish different subtypes (50, 53, 133). In gastric cancer, CIMP, as defined within each study, is associated with EBV, methylation increases with tumor progression, and CIMP status in combination with TP53 hotspot mutation status forms subgroups with distinct overall and progression free survival (28, 29, 31, 34, 47, 134).
In prostate cancer, the hypermethylator phenotype is associated with clinical features of poor prognosis. Multiple genes, such as GSTP1, APC, MDR1, MGMT, and RASSF1A, show higher methylation frequency in prostate cancer samples compared to BPH and non-neoplastic prostate samples (74, 78). Additionally, high methylation of RARβ, RASSF1A, GSTP1, CDH13, APC, RUNX3, MDR1, and cyclin D2 is associated with high Gleason score and high PSA (74–76, 78). Methylation score, determined by statistical analysis comparing methylation status of various genes of benign prostatic hyperplasia and prostate cancer, is also found to be associated with high pT and other advanced pathological features, and can also distinguish organ-confined cancers from locally advanced cancer (74). Through MS PCR, Yegnasubramanian et al. also show that hypermethylation patterns of GSTP1, APC, RASSF1A, PTGS2, and MDR1 is able to be used to distinguish primary prostate cancer from benign prostate tissues, hypermethylation of CpG island at EDNRB is correlated with tumor grade and stage of primary prostate cancers, and hypermethylation of CpG island of PTGS2 is associated with increased risk of recurrence (135). Beyond these specific loci, analysis using Illumina HumanMethylation27 platform has identified 87 CpG sites with increased DNA methylation in 83/87 tumor samples, making them the most predictive diagnostic methylation biomarkers that can predict either tumor state or benign adjacent state of prostate cancer (101). Also, by integrating clinical follow-up data, it has been shown that there are prognostic DNA methylation alterations that correlate with biochemical recurrence of tumor (101). Furthermore, hypermethylation changes are highly maintained across anatomically distinct metastases within an individual, highlighting the potential of methylation status to be used as a longitudinal biomarker for clinically advanced prostate cancer (70). Methylation patterns have also been shown to be able to distinguish between castration resistant adenocarcinoma and neuroendocrine prostate cancer, and Berchuck et al. has been able to build a model that predicts the presence of NEPC using MeDIP-seq with 100% sensitivity and 90% specificity (5, 136). Furthermore, methylation changes have also been detectable in cell-free DNA (cfDNA), showing potential of methylation patterns to be used to develop liquid biomarkers. Liquid biomarkers, including circulating tumor cells, tumor cell fragments, nucleic acids, and proteins, are more readily accessible through any bodily fluids such as urine and blood, making them more easily obtainable than biopsies of prostate cancer metastases. As such, ability to detect methylation patterns in cfDNA and use it to distinguish specific clinical features holds significant clinical implications. To do so, cfDNA are isolated from plasma samples of patients with localized and metastatic prostate cancer, isolated, then profiled using bisulfite sequencing, MeDIPseq, or 5hmC sequencing (5hmC-seq). cfDNA global methylation patterns within each individual are temporally stable throughout the disease course, can distinguish metastatic from localized samples with 0.989 prediction accuracy, and can be used to build a model that can predict presence of NEPC and discriminate NEPC from castration resistant prostate adenocarcinoma (82, 87, 136). Moreover, using methylation sensitive restriction enzyme-qPCR analyses in liquid biopsies from mCRPC patients responsive and non-responsive to different therapies, Dillinger et al. has found higher methylation of specific loci in non-responsive patients before and after abiraterone treatment and identified 23 individual marker genes for which methylation was a negative prognostic factor for disease recurrence (137). In addition, Wu et al. have shown by sequencing plasma DNA from mCRPC patients receiving abiraterone or enzalutamide pre and post chemotherapy, there is hypomethylation of segments of AR binding sequences that are associated with AR copy number gain and more aggressive clinical course (138). And as previously mentioned, 5hmC patterns with prognostic value can also be detected in cfDNA of mCRPC patients (88). Overall, methylation alterations show potential, even as a liquid biopsy, to serve to work in conjunction with genetic alterations for clinical biomarker development.
Using bisulfite sequencing, MeDIP-seq, or hMeDIP-seq are useful tools for scientific research. It allows exploration of mechanisms and its potential as a clinical prognostic biomarker. However, to be used in a clinical setting, the cost for running these sequencing methods should be considered. Currently, there is effort to develop targeted panels of DNA methylation to help reduce cost. In addition, there are commercial efforts to use different methods beyond pulling down methylated or hydroxymethylated DNA using antibodies to analyze DNA methylation patterns (139).
5.2 Treatment options
As CIMP tumors are hypermethylated, several DNA methyltransferase inhibitors, such as decitabine and azacitidine, are being evaluated in pre-clinical and clinical settings (34, 128, 140–142). DNA methyltransferase inhibitors azacitidine and decitabine are FDA-approved and show potential as a new therapeutic anticancer treatment. Treatment with decitabine has shown to slow tumor growth, decrease cell proliferation, and induce tumor suppressors in breast cancer cell lines in in vitro and in vivo studies using mice induced with human breast cancer cell lines (141, 142). Similarly, decitabine administered with talazoparib decreases tumor growth and increases overall survival in ovarian and breast cancer models (140). Further, in gastric cancer, as EBV-induced hypermethylation targets and silences key tumor suppressor genes including APC, RASSF1, BRCA1, THBS1, and CDKN2A, DNA methyltransferase inhibitors may also serve as a new therapeutic treatment for patients with EBV-positive gastric cancer (34, 129). There are also some concerns with using demethylation through the use of DNA methyltransferase inhibitors as a new treatment method, as demethylation induces pro-metastatic genes and increases invasiveness of non-invasive breast cancer (141, 142). However, an in vitro study by Chik et al. shows that depletion of DNMT1 suppresses cell growth but does not induce invasiveness while depletion of DNMT3a does not change cell transformation and increases cell invasiveness, demonstrating that specific DNMT1 inhibitors, azacitidine and decitabine, may avoid adverse effects (142). Current clinical trials on DNA methyltransferase inhibitors include studies on side effects and best dose of decitabine with nivolumab in treating colorectal cancer, efficacy of treatment of azacitidine in recurrent IDH1-mutant gliomas and finding maximum tolerated dose of azacitidine with capecitabine and oxaliplatin in treating metastatic colorectal cancer. They measure maximum tolerated dose, overall response rate, adverse events, and progression free and overall survival after treatment with decitabine or azacitidine for a month to 1 year.
Azacitidine has been explored as a new therapeutic drug for prostate cancer treatment in combination with chemotherapy or anti-androgen therapy. Azacitidine shows antiproliferative effects in 22Rv1 and PC3 cell lines, and in vivo, 0.8 mg/kg intraperitoneal injection of azacitidine reduced tumor proliferation and induced apoptosis in PC3 and 22Rv1 xenografts (143). Additionally, azacitidine shows synergistic effects with docetaxel and cisplatin, sensitizing both PC3 and 22Rv1 xenografts to docetaxel and cisplatin treatments and causing tumor growth delay without complete regression (143). This combination treatment was superior to either treatment alone and tolerable in mice (143). Azacitidine has also been tried in a clinical setting, to determine if it can reverse docetaxel resistance in mCRPC patients with disease progression during or within 6 months after cessation of minimum 6 weeks of docetaxel-based therapy (144). In this phase I/II study, azaciditine and docetaxel were alternately escalated with administration of prednisone. They found there was >50% decline in PSA in 10 out of 19 patients, favorable progression free survival and overall survival, and the common treatment-related adverse event was neutropenia (144). Furthermore, there is a Phase II trial to study effect of azacitidine in modulating PSA in patients continuing treatment with luteinizing hormone-releasing hormone and antiandrogen. This study plans to detect biological activity of azacitidine as well, by measuring fetal hemoglobin and plasma DNA methylation (145).
6 Discussion
Methylation changes in prostate cancer, including hypermethylation of CpG islands, hypomethylation patterns, and 5hmC patterns are reported in cancer transformation and progression. The CIMP subtype in prostate cancer shows decreased expression of tumor suppressor genes and has been associated with distinct clinical features, including higher Gleason score, higher PSA, higher tumor grade, and overall poor outcome. Tumors of this hypermethylation subtype can potentially benefit from FDA-approved demethylating agents, azacitidine and decitabine. There are also distinct patterns of methylation that can help distinguish benign prostate tissue from malignant prostate tumors, as well as the NEPC subtype from castration resistant adenocarcinoma. In addition, it can potentially be used to distinguish mCRPC. Further, the 5hmC landscape of prostate cancer also shows potential to serve as a marker of epigenetic activation throughout disease progression that can also identify distinct oncogenic signaling pathways that define subgroups of advanced prostate cancer and disease states. Prognostic DNA methylation patterns can also be detected in cell-free DNA isolated from plasma of patients with prostate cancer. Since biopsies of prostate cancer metastases can be difficult to obtain in comparison to more readily accessible plasma, analysis of methylation patterns in cfDNA can add to current analyses of cfDNA in advanced cancers to serve as a better liquid biomarker. Studies into methylation patterns in prostate cancer have also been improved with novel methods, such as whole genome bisulfite sequencing and MeDIP sequencing. Inclusion of DNA methylation data into future multi-omic studies of prostate cancer patient samples of different stages and clinical subtypes will allow better understanding of the molecular heterogeneity of prostate cancer. Delineating the relationship between the driving mutations (e.g. DNMT, IDH, TET, and BRAF genes) and aberrant methylation patterns in PCa can underlie the complex mechanism and help predict specific methylation subtypes. Future studies integrating methylation sequencing data with sequencing investigating chromatin structure such as chromatin immunoprecipitation sequencing (ChIP-seq) and chromatin interaction analysis by paired-end tag sequencing (ChIA-PET), could reveal complex 3D epigenetic regulation. Overall, DNA methylation analysis not only could elucidate mechanisms that drive cancer progression but also demonstrate potential for clinical biomarker and novel treatment plan development for prostate cancer.
Author contributions
HS, JH, and HL drafted the article or revised it critically for important intellectual content. All authors contributed to the article and approved the submitted version.
Funding
JH was funded by a Prostate Cancer Foundation Young Investigator Award. HL was supported by the Prostate Cancer Foundation Young Investigator Award.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Siegel RL, Miller KD, Wagle NS, Jemal A. Cancer statistics, 2023. CA Cancer J Clin (2023) 73:17–48. doi: 10.3322/caac.21763
2. Robinson D, Van Allen EM, Wu Y-M, Schultz N, Lonigro RJ, Mosquera J-M, et al. Integrative clinical genomics of advanced prostate cancer. Cell (2015) 161:1215–28. doi: 10.1016/j.cell.2015.05.001
3. Quigley DA, Dang HX, Zhao SG, Lloyd P, Aggarwal R, Alumkal JJ, et al. Genomic hallmarks and structural variation in metastatic prostate cancer. Cell (2018) 174:758–769.e9. doi: 10.1016/j.cell.2018.06.039
4. Wang G, Zhao D, Spring DJ, DePinho RA. Genetics and biology of prostate cancer. Genes Dev (2018) 32:1105–40. doi: 10.1101/gad.315739.118
5. Beltran H, Demichelis F. Therapy considerations in neuroendocrine prostate cancer: what next? endocr. Relat Cancer (2021) 28:T67–78. doi: 10.1530/ERC-21-0140
6. Yamada Y, Beltran H. Clinical and biological features of neuroendocrine prostate cancer. Curr Oncol Rep (2021) 23:15. doi: 10.1007/s11912-020-01003-9
7. Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature (2009) 462:315–22. doi: 10.1038/nature08514
8. Domcke S, Bardet AF, Adrian Ginno P, Hartl D, Burger L, Schübeler D. Competition between DNA methylation and transcription factors determines binding of NRF1. Nature (2015) 528:575–9. doi: 10.1038/nature16462
9. Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet (2003) 33:245–54. doi: 10.1038/ng1089
11. Song Y, van den Berg PR, Markoulaki S, Soldner F, Dall’Agnese A, Henninger JE, et al. Dynamic enhancer DNA methylation as basis for transcriptional and cellular heterogeneity of ESCs. Mol Cell (2019) 75:905–920.e6. doi: 10.1016/j.molcel.2019.06.045
12. Okano M, Bell DW, Haber DA, Li E. DNA Methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell (1999) 99:247–57. doi: 10.1016/S0092-8674(00)81656-6
13. Rasmussen KD, Helin K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev (2016) 30:733–50. doi: 10.1101/gad.276568.115
14. Ehrlich M. DNA Hypomethylation in cancer cells. Epigenomics (2009) 1:239–59. doi: 10.2217/epi.09.33
15. Esteller M. Epigenetic gene silencing in cancer: the DNA hypermethylome. Hum Mol Genet (2007) 16:50–9. doi: 10.1093/hmg/ddm018
16. Miller BF, Sánchez-Vega F, Elnitski L. The emergence of pan-cancer CIMP and its elusive interpretation. Biomolecules (2016) 6:1–14. doi: 10.3390/biom6040045
17. Moarii M, Reyal F, Vert J-P. Integrative DNA methylation and gene expression analysis to assess the universality of the CpG island methylator phenotype. Hum Genomics (2015) 9:26. doi: 10.1186/s40246-015-0048-9
18. Toyota M, Ahuja N, Ohe-Toyota M, Herman JG, Baylin SB, Issa J-PJ. CpG island methylator phenotype in colorectal cancer. Proc Natl Acad Sci USA (1999) 96:8681–6. doi: 10.1073/pnas.96.15.8681
19. Linnekamp JF, Butter R, Spijker R, Medema JP, van Laarhoven HWM. Clinical and biological effects of demethylating agents on solid tumours – a systematic review. Cancer Treat Rev (2017) 54:10–23. doi: 10.1016/j.ctrv.2017.01.004
20. Tao Y, Kang B, Petkovich DA, Bhandari YR, In J, Stein-O’Brien G, et al. Aging-like spontaneous epigenetic silencing facilitates wnt activation, stemness, and BrafV600E-induced tumorigenesis. Cancer Cell (2019) 35:315–328.e6. doi: 10.1016/j.ccell.2019.01.005
21. Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet (2006) 38:787–93. doi: 10.1038/ng1834
22. Shen L, Toyota M, Kondo Y, Lin E, Zhang L, Guo Y, et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci USA (2007) 104:18654–9. doi: 10.1073/pnas.0704652104
23. Samowitz WS, Albertsen H, Herrick J, Levin TR, Sweeney C, Murtaugh MA, et al. Evaluation of a large, population-based sample supports a CpG island methylator phenotype in colon cancer. Gastroenterology (2005) 129:837–45. doi: 10.1053/j.gastro.2005.06.020
24. De Sousa E Melo F, Wang X, Jansen M, Fessler E, Trinh A, De Rooij LPMH, et al. Poor-prognosis colon cancer is defined by a molecularly distinct subtype and develops from serrated precursor lesions. Nat Med (2013) 19:614–8. doi: 10.1038/nm.3174
25. Cheng YW, Pincas H, Bacolod MD, Schemmann G, Giardina SF, Huang J, et al. CpG island methylator phenotype associates with low-degree chromosomal abnormalities in colorectal cancer. Clin Cancer Res (2008) 14:6005–13. doi: 10.1158/1078-0432.CCR-08-0216
26. Baba Y, Nosho K, Shima K, Freed E, Irahara N, Philips J, et al. Relationship of CDX2 loss with molecular features and prognosis in colorectal cancer. Clin Cancer Res (2009) 15:4665–73. doi: 10.1158/1078-0432.CCR-09-0401
27. An C, Choi IS, Yao JC, Worah S, Xie K, Mansfield PF, et al. Prognostic significance of CpG island methylator phenotype and microsatellite instability in gastric carcinoma. Clin Cancer Res (2005) 11:656–63. doi: 10.1158/1078-0432.656.11.2
28. Kusano M, Toyota M, Suzuki H, Akino K, Aoki F, Fujita M, et al. Genetic, epigenetic, and clinicopathologic features of gastric carcinomas with the CpG island methylator phenotype and an association with Epstein-Barr virus. Cancer (2006) 106:1467–79. doi: 10.1002/cncr.21789
29. Enomoto S, Maekita T, Tsukamoto T, Nakajima T, Nakazawa K, Tatematsu M, et al. Lack of association between CpG island methylator phenotype in human gastric cancers and methylation in their background non-cancerous gastric mucosae. Cancer Sci (2007) 98:1853–61. doi: 10.1111/j.1349-7006.2007.00625.x
30. Shigeyasu K, Nagasaka T, Mori Y, Yokomichi N, Kawai T, Fuji T, et al. Clinical significance of MLH1 methylation and CpG island methylator phenotype as prognostic markers in patients with gastric cancer. PloS One (2015) 10:e0130409. doi: 10.1371/journal.pone.0130409
31. Oue N, Mitani Y, Motoshita J, Matsumura S, Yoshida K, Kuniyasu H, et al. Accumulation of DNA methylation is associated with tumor stage in gastric cancer. Cancer (2006) 106:1250–9. doi: 10.1002/cncr.21754
32. Tahara T, Tahara S, Horiguchi N, Okubo M, Terada T, Yamada H, et al. Molecular subtyping of gastric cancer combining genetic and epigenetic anomalies provides distinct clinicopathological features and prognostic impacts. Hum Mutat (2019) 40:347–54. doi: 10.1002/humu.23700
33. Chang MS, Uozaki H, Chong JM, Ushiku T, Sakuma K, Ishikawa S, et al. CpG island methylation status in gastric carcinoma with and without infection of Epstein-Barr virus. Clin Cancer Res (2006) 12:2995–3002. doi: 10.1158/1078-0432.CCR-05-1601
34. He D, Zhang Y, Zhang N, Zhou L, Chen J, Jiang Y, et al. Aberrant gene promoter methylation of p16, FHIT, CRBP1, WWOX, and DLC-1 in Epstein–Barr virus-associated gastric carcinomas. Med Oncol (2015) 32:92. doi: 10.1007/s12032-015-0525-y
35. Jing F, Yuping W, Yong C, Jie L, Jun L, Xuanbing T, et al. CpG island methylator phenotype of multigene in serum of sporadic breast carcinoma. Tumor Biol (2010) 31:321–31. doi: 10.1007/s13277-010-0040-x
36. Fang F, Turcan S, Rimner A, Kaufman A, Giri D, Luc GT, et al. Breast cancer methylomes establish an epigenomic foundation for metastasis. Sci Transl Med (2011) 3:75ra25. doi: 10.1126/scitranslmed.3001875
37. Roman-Gomez J, Jimenez-Velasco A, Agirre X, Castillejo JA, Navarro G, Calasanz MJ, et al. CpG island methylator phenotype redefines the prognostic effect of t(12;21) in childhood acute lymphoblastic leukemia. Clin Cancer Res (2006) 12:4845–50. doi: 10.1158/1078-0432.CCR-05-2592
38. Kroeger H, Jelinek J, Estecio MRH, He R, Kondo K, Chung W, et al. Aberrant CpG island methylation in acute myeloid leukemia is accentuated at relapse. Blood (2008) 112:1366–73. doi: 10.1182/blood-2007-11-126227
39. Sato H, Oka T, Shinnou Y, Kondo T, Washio K, Takano M, et al. Multi-step aberrant CpG island hyper-methylation is associated with the progression of adult T–cell Leukemia/Lymphoma. Am J Pathol (2010) 176:402–15. doi: 10.2353/ajpath.2010.090236
40. Kraszewska MD, Dawidowska M, Larmonie NSD, Kosmalska M, Sędek Ł, Szczepaniak M, et al. DNA Methylation pattern is altered in childhood T-cell acute lymphoblastic leukemia patients as compared with normal thymic subsets: insights into CpG island methylator phenotype in T-ALL. Leukemia (2012) 26:367–71. doi: 10.1038/leu.2011.208
41. Fu HY, Wu D, Sen, Zhou HR, Shen JZ. CpG island methylator phenotype and its relationship with prognosis in adult acute leukemia patients. Hematology (2014) 19:329–37. doi: 10.1179/1607845413Y.0000000137
42. Borssén M, Palmqvist L, Karrman K, Abrahamsson J, Behrendtz M, Heldrup J, et al. Promoter DNA methylation pattern identifies prognostic subgroups in childhood T-cell acute lymphoblastic leukemia. PloS One (2013) 8:e65373. doi: 10.1371/journal.pone.0065373
43. Haider Z, Larsson P, Landfors M, Köhn L, Schmiegelow K, Flaegstad T, et al. An integrated transcriptome analysis in T-cell acute lymphoblastic leukemia links DNA methylation subgroups to dysregulated TAL1 and ANTP homeobox gene expression. Cancer Med (2019) 8:311–24. doi: 10.1002/cam4.1917
44. Toyota M, Ahuja N, Suzuki H, Itoh F, Ohe-Toyota M, Imai K, et al. Aberrant methylation in gastric cancer associated with the CpG island methylator phenotype. Cancer Res (1999) 59:5438–42.
45. Kim H, Kim YH, Kim SE, Kim N-G, Noh SH, Kim H. Concerted promoter hypermethylation of hMLH1,p16INK4A, andE-cadherin in gastric carcinomas with microsatellite instability. J Pathol (2003) 200:23–31. doi: 10.1002/path.1325
46. Kondo T, Oka T, Sato H, Shinnou Y, Washio K, Takano M, et al. Accumulation of aberrant CpG hypermethylation by helicobacter pylori infection promotes development and progression of gastric MALT lymphoma. Int J Oncol (2009) 35:547–57. doi: 10.3892/ijo_00000366
47. Zong L, Seto Y. CpG island methylator phenotype, helicobacter pylori, Epstein-Barr virus, and microsatellite instability and prognosis in gastric cancer: a systematic review and meta-analysis. PloS One (2014) 9:e86097. doi: 10.1371/journal.pone.0086097
48. Oshimo Y, Oue N, Mitani Y, Nakayama H, Kitadai Y, Yoshida K, et al. Frequent epigenetic inactivation of RIZ1 by promoter hypermethylation in human gastric carcinoma. Int J Cancer (2004) 110:212–8. doi: 10.1002/ijc.20090
49. Kim JG, Takeshima H, Niwa T, Rehnberg E, Shigematsu Y, Yoda Y, et al. Comprehensive DNA methylation and extensive mutation analyses reveal an association between the CpG island methylator phenotype and oncogenic mutations in gastric cancers. Cancer Lett (2013) 330:33–40. doi: 10.1016/j.canlet.2012.11.022
50. Hill VK, Ricketts C, Bieche I, Vacher S, Gentle D, Lewis C, et al. Genome-wide DNA methylation profiling of CpG islands in breast cancer identifies novel genes associated with tumorigenicity. Cancer Res (2011) 71:2988–99. doi: 10.1158/0008-5472.CAN-10-4026
51. Li L, Lee K-M, Han W, Choi J-Y, Lee J-Y, Kang GH, et al. Estrogen and progesterone receptor status affect genome-wide DNA methylation profile in breast cancer. Hum Mol Genet (2010) 19:4273–7. doi: 10.1093/hmg/ddq351
52. Bae YK, Brown A, Garrett E, Bornman D, Fackler MJ, Sukumar S, et al. Hypermethylation in histologically distinct classes of breast cancer. Clin Cancer Res (2004) 10:5998–6005. doi: 10.1158/1078-0432.CCR-04-0667
53. Roessler J, Ammerpohl O, Gutwein J, Steinemann D, Schlegelberger B, Weyer V, et al. The CpG island methylator phenotype in breast cancer is associated with the lobular subtype. Epigenomics (2015) 7:187–99. doi: 10.2217/epi.14.74
54. Wang H, Yan W, Zhang S, Gu Y, Wang Y, Wei Y, et al. Survival differences of CIMP subtypes integrated with CNA information in human breast cancer. Oncotarget (2017) 8:48807–19. doi: 10.18632/oncotarget.16178
55. Damm F, Markus B, Thol F, Morgan M, Göhring G, Schlegelberger B, et al. TET2 mutations in cytogenetically normal acute myeloid leukemia: clinical implications and evolutionary patterns. Genes Chromosom Cancer (2014) 53:824–32. doi: 10.1002/gcc.22191
56. Tian X, Xu Y, Yin J, Tian H, Chen S, Wu D, et al. TET2 gene mutation is unfavorable prognostic factor in cytogenetically normal acute myeloid leukemia patients with NPM1+ and FLT3-ITD– mutations. Int J Hematol (2014) 100:96–104. doi: 10.1007/s12185-014-1595-x
57. Kroeze LI, Aslanyan MG, van Rooij A, Koorenhof-Scheele TN, Massop M, Carell T, et al. Characterization of acute myeloid leukemia based on levels of global hydroxymethylation. Blood (2014) 124:1110–8. doi: 10.1182/blood-2013-08-518514
58. Figueroa ME, Abdel-Wahab O, Lu C, Ward PS, Patel J, Shih A, et al. Leukemic IDH1 and IDH2 mutations result in a hypermethylation phenotype, disrupt TET2 function, and impair hematopoietic differentiation. Cancer Cell (2010) 18:553–67. doi: 10.1016/j.ccr.2010.11.015
59. Kelly AD, Kroeger H, Yamazaki J, Taby R, Neumann F, Yu S, et al. A CpG island methylator phenotype in acute myeloid leukemia independent of IDH mutations and associated with a favorable outcome. Leukemia (2017) 31:2011–9. doi: 10.1038/leu.2017.12
60. Martinez R, Schackert G, Esteller M. Hypermethylation of the proapoptotic gene TMS1/ASC: prognostic importance in glioblastoma multiforme. J Neurooncol (2007) 82:133–9. doi: 10.1007/s11060-006-9264-4
61. Stone AR, Bobo W, Brat DJ, Devi NS, Van Meir EG, Vertino PM. Aberrant methylation and down-regulation of TMS1/ASC in human glioblastoma. Am J Pathol (2004) 165:1151–61. doi: 10.1016/S0002-9440(10)63376-7
62. Kim T-Y, Zhong S, Fields CR, Kim JH, Robertson KD. Epigenomic profiling reveals novel and frequent targets of aberrant DNA methylation-mediated silencing in malignant glioma. Cancer Res (2006) 66:7490–501. doi: 10.1158/0008-5472.CAN-05-4552
63. Noushmehr H, Weisenberger DJ, Diefes K, Phillips HS, Pujara K, Berman BP, et al. Identification of a CpG island methylator phenotype that defines a distinct subgroup of glioma. Cancer Cell (2010) 17:510–22. doi: 10.1016/j.ccr.2010.03.017
64. Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, et al. Molecular profiling reveals biologically discrete subsets and pathways of progression in diffuse glioma. Cell (2016) 164:550–63. doi: 10.1016/j.cell.2015.12.028
65. Turcan S, Rohle D, Goenka A, Walsh LA, Fang F, Yilmaz E, et al. IDH1 mutation is sufficient to establish the glioma hypermethylator phenotype. Nature (2012) 483:479–83. doi: 10.1038/nature10866
66. van den Bent MJ, Gravendeel LA, Gorlia T, Kros JM, Lapre L, Wesseling P, et al. A hypermethylated phenotype is a better predictor of survival than MGMT methylation in anaplastic oligodendroglial brain tumors: a report from EORTC study 26951. Clin Cancer Res (2011) 17:7148–55. doi: 10.1158/1078-0432.CCR-11-1274
67. Wiestler B, Capper D, Hovestadt V, Sill M, Jones DTW, Hartmann C, et al. Assessing CpG island methylator phenotype, 1p/19q codeletion, and MGMT promoter methylation from epigenome-wide data in the biomarker cohort of the NOA-04 trial. Neuro Oncol (2014) 16:1630–8. doi: 10.1093/neuonc/nou138
68. Wiestler B, Capper D, Sill M, Jones DTW, Hovestadt V, Sturm D, et al. Integrated DNA methylation and copy-number profiling identify three clinically and biologically relevant groups of anaplastic glioma. Acta Neuropathol (2014) 128:561–71. doi: 10.1007/s00401-014-1315-x
69. Li KK-W, Shi Z-F, Malta TM, Chan AK-Y, Cheng S, Kwan JSH, et al. Identification of subsets of IDH-mutant glioblastomas with distinct epigenetic and copy number alterations and stratified clinical risks. Neuro-Oncol Adv (2019) 1:1–11. doi: 10.1093/noajnl/vdz015
70. Aryee MJ, Liu W, Engelmann JC, Nuhn P, Gurel M, Haffner MC, et al. DNA Methylation alterations exhibit intraindividual stability and interindividual heterogeneity in prostate cancer metastases. Sci Transl Med (2013) 5:1–14. doi: 10.1126/scitranslmed.3005211
71. Li Y, Tollefsbol TO. DNA Methylation detection: bisulfite genomic sequencing analysis. Methods Mol Biol (2011) 791:11–21. doi: 10.1007/978-1-61779-316-5_2
72. Li S, Tollefsbol TO. DNA Methylation methods: global DNA methylation and methylomic analyses. Methods (2021) 187:28–43. doi: 10.1016/j.ymeth.2020.10.002
73. Li L, Gao Y, Wu Q, Cheng ASL, Yip KY. New guidelines for DNA methylome studies regarding 5-hydroxymethylcytosine for understanding transcriptional regulation. Genome Res (2019) 29:543–53. doi: 10.1101/gr.240036.118
74. Enokida H, Shiina H, Urakami S, Igawa M, Ogishima T, Li LC, et al. Multigene methylation analysis for detection and staging of prostate cancer. Clin Cancer Res (2005) 11:6582–8. doi: 10.1158/1078-0432.CCR-05-0658
75. Florl AR, Steinhoff C, Müller M, Seifert HH, Hader C, Engers R, et al. Coordinate hypermethylation at specific genes in prostate carcinoma precedes LINE-1 hypomethylation. Br J Cancer (2004) 91:985–94. doi: 10.1038/sj.bjc.6602030
76. Maruyama R, Toyooka S, Toyooka KO, Virmani AK, Zöchbauer-Müller S, Farinas AJ, et al. Aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features. Clin Cancer Res (2002) 8:514–9.
77. Padar A, Sathyanarayana UG, Suzuki M, Maruyama R, Hsieh JT, Frenkel EP, et al. Inactivation of cyclin D2 gene in prostate cancers by aberrant promoter methylation. Clin Cancer Res (2003) 9:4730–4.
78. Kang GH, Lee S, Lee HJ, Hwang KS. Aberrant CpG island hypermethylation of multiple genes in prostate cancer and prostatic intraepithelial neoplasia. J Pathol (2004) 202:233–40. doi: 10.1002/path.1503
79. Bhasin JMM, Lee BHH, Matkin L, Taylor MGG, Hu B, Xu Y, et al. Methylome-wide sequencing detects DNA hypermethylation distinguishing indolent from aggressive prostate cancer. Cell Rep (2015) 13:2135–46. doi: 10.1016/j.celrep.2015.10.078
80. Yu YP, Ding Y, Chen R, Liao SG, Ren BG, Michalopoulos A, et al. Whole-genome methylation sequencing reveals distinct impact of differential methylations on gene transcription in prostate cancer. Am J Pathol (2013) 183:1960–70. doi: 10.1016/j.ajpath.2013.08.018
81. Kim JH, Dhanasekaran SM, Prensner JR, Cao X, Robinson D, Kalyana-Sundaram S, et al. Deep sequencing reveals distinct patterns of DNA methylation in prostate cancer. Genome Res (2011) 21:1028–41. doi: 10.1101/gr.119347.110
82. Silva R, Moran B, Baird AM, O’Rourke CJ, Finn SP, McDermott R, et al. Longitudinal analysis of individual cfDNA methylome patterns in metastatic prostate cancer. Clin Epigenet (2021) 13:1–10. doi: 10.1186/s13148-021-01155-w
83. Gerhauser C, Favero F, Risch T, Simon R, Feuerbach L, Assenov Y, et al. Molecular evolution of early-onset prostate cancer identifies molecular risk markers and clinical trajectories. Cancer Cell (2018) 34:996–1011.e8. doi: 10.1016/j.ccell.2018.10.016
84. Zhao SG, Chen WS, Li H, Foye A, Zhang M, Sjöström M, et al. DNA Methylation landscapes in advanced prostate cancer. Nat Genet (2020) 52:778–89. doi: 10.1038/s41588-020-0648-8
85. Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, et al. Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat Med (2016) 22:298–305. doi: 10.1038/nm.4045
86. Börno ST, Fischer A, Kerick M, Fälth M, Laible M, Brase JC, et al. Genome-wide DNA methylation events in TMPRSS2-ERG fusion-negative prostate cancers implicate an EZH2-dependent mechanism with miR-26a hypermethylation. Cancer Discov (2012) 2:1025–35. doi: 10.1158/2159-8290.CD-12-0041
87. Chen S, Petricca J, Ye W, Guan J, Zeng Y, Cheng N, et al. The cell-free DNA methylome captures distinctions between localized and metastatic prostate tumors. Nat Commun (2022) 13:6467. doi: 10.1038/s41467-022-34012-2
88. Sjöström M, Zhao SG, Levy S, Zhang M, Ning Y, Shrestha R, et al. The 5-hydroxymethylcytosine landscape of prostate cancer. Cancer Res (2022) 82:3888–902. doi: 10.1158/0008-5472.CAN-22-1123
89. Fang M, Ou J, Hutchinson L, Green MR. The BRAF oncoprotein functions through the transcriptional repressor MAFG to mediate the CpG island methylator phenotype. Mol Cell (2014) 55:904–15. doi: 10.1016/j.molcel.2014.08.010
90. Serra RW, Fang M, Park SM, Hutchinson L, Green MR. A KRAS-directed transcriptional silencing pathway that mediates the CpG island methylator phenotype. Elife (2014) 3:1–22. doi: 10.7554/eLife.02313
91. Ponciano-Gómez A, Martínez-Tovar A, Vela-Ojeda J, Olarte-Carrillo I, Centeno-Cruz F, Garrido E. Mutations in TET2 and DNMT3A genes are associated with changes in global and gene-specific methylation in acute myeloid leukemia. Tumor Biol (2017) 39:101042831773218. doi: 10.1177/1010428317732181
92. Rasmussen KD, Jia G, Johansen JV, Pedersen MT, Rapin N, Bagger FO, et al. Loss of TET2 in hematopoietic cells leads to DNA hypermethylation of active enhancers and induction of leukemogenesis. Genes Dev (2015) 29:910–22. doi: 10.1101/gad.260174.115
93. Kamdar S, Isserlin R, Van der Kwast T, Zlotta AR, Bader GD, Fleshner NE, et al. Exploring targets of TET2-mediated methylation reprogramming as potential discriminators of prostate cancer progression. Clin Epigenet (2019) 11:54. doi: 10.1186/s13148-019-0651-z
94. Hájková H, Marková J, Haškovec C, Šárová I, Fuchs O, Kostečka A, et al. Decreased DNA methylation in acute myeloid leukemia patients with DNMT3A mutations and prognostic implications of DNA methylation. Leuk Res (2012) 36:1128–33. doi: 10.1016/j.leukres.2012.05.012
95. Holz-Schietinger C, Matje DM, Reich NO. Mutations in DNA methyltransferase (DNMT3A) observed in acute myeloid leukemia patients disrupt processive methylation. J Biol Chem (2012) 287:30941–51. doi: 10.1074/jbc.M112.366625
96. Jin B, Tao Q, Peng J, Soo HM, Wu W, Ying J, et al. DNA Methyltransferase 3B (DNMT3B) mutations in ICF syndrome lead to altered epigenetic modifications and aberrant expression of genes regulating development, neurogenesis and immune function. Hum Mol Genet (2008) 17:690–709. doi: 10.1093/hmg/ddm341
97. Duymich CE, Charlet J, Yang X, Jones PA, Liang G. DNMT3B isoforms without catalytic activity stimulate gene body methylation as accessory proteins in somatic cells. Nat Commun (2016) 7:11453. doi: 10.1038/ncomms11453
98. Liao J, Karnik R, Gu H, Ziller MJ, Clement K, Tsankov AM, et al. Targeted disruption of DNMT1, DNMT3A and DNMT3B in human embryonic stem cells. Nat Genet (2015) 47:469–78. doi: 10.1038/ng.3258
99. Roll JD, Rivenbark AG, Jones WD, Coleman WB. DNMT3b overexpression contributes to a hypermethylator phenotype in human breast cancer cell lines. Mol Cancer (2008) 7:15. doi: 10.1186/1476-4598-7-15
100. Kanai Y, Ushijima S, Kondo Y, Nakanishi Y, Hirohashi S. DNA Methyltransferase expression and DNA methylation of CPG islands and peri-centromeric satellite regions in human colorectal and stomach cancers. Int J Cancer (2001) 91:205–12. doi: 10.1002/1097-0215(200002)9999:9999<::AID-IJC1040>3.0.CO;2-2
101. Kobayashi Y, Absher DM, Gulzar ZG, Young SR, McKenney JK, Peehl DM, et al. DNA Methylation profiling reveals novel biomarkers and important roles for DNA methyltransferases in prostate cancer. Genome Res (2011) 21:1017–27. doi: 10.1101/gr.119487.110
102. Lamim Lovatel V, de Souza Fernandez C, Ferreira Rodrigues E, de Cassia Tavares R, Sobral da Costa E, Abdelhay E, et al. Expression profiles of DNA methylation and demethylation machinery components in pediatric myelodysplastic syndrome: clinical implications. Cancer Manage Res (2020) 12:543–56. doi: 10.2147/CMAR.S219026
103. Glass JL, Hassane D, Wouters BJ, Kunimoto H, Avellino R, Garrett-Bakelman FE, et al. Epigenetic identity in AML depends on disruption of nonpromoter regulatory elements and is affected by antagonistic effects of mutations in epigenetic modifiers. Cancer Discov (2017) 7:868–83. doi: 10.1158/2159-8290.CD-16-1032
104. Ahr K, Famulare C, Koche R, Spitzer B, Levine RL, Tallman MS, et al. AML with mutations in IDH1 and DNMT3A exhibits a distinct epigenetic signature with poorer overall survival. Blood (2018) 132:1471–1. doi: 10.1182/blood-2018-99-118407
105. Zhang X, Shi J, Zhang J, Yang X, Zhang G, Yang S, et al. Clinical and biological implications of IDH1/2 in acute myeloid leukemia with DNMT3Amut. Cancer Manage Res (2018) 10:2457–66. doi: 10.2147/CMAR.S157632
106. Rinaldi L, Datta D, Serrat J, Morey L, Solanas G, Avgustinova A, et al. Dnmt3a and Dnmt3b associate with enhancers to regulate human epidermal stem cell homeostasis. Cell Stem Cell (2016) 19:491–501. doi: 10.1016/j.stem.2016.06.020
107. Zhang X, Su J, Jeong M, Ko M, Huang Y-HH, Park HJ, et al. DNMT3A and TET2 compete and cooperate to repress lineage-specific transcription factors in hematopoietic stem cells. Nat Genet (2016) 48:1014–23. doi: 10.1038/ng.3610
108. Stanland LJ, Luftig MA. The role of EBV-induced hypermethylation in gastric cancer tumorigenesis. Viruses (2020) 12:1222. doi: 10.3390/v12111222
109. Issa JPJ, Ottaviano YL, Celano P, Hamilton SR, Davidson NE, Baylin SB. Methylation of the oestrogen receptor CpG island links ageing and neoplasia in human colon. Nat Genet (1994) 7:536–40. doi: 10.1038/ng0894-536
110. Thienpont B, Steinbacher J, Zhao H, D’Anna F, Kuchnio A, Ploumakis A, et al. Tumour hypoxia causes DNA hypermethylation by reducing TET activity. Nature (2016) 537:63–8. doi: 10.1038/nature19081
111. Burr S, Caldwell A, Chong M, Beretta M, Metcalf S, Hancock M, et al. Oxygen gradients can determine epigenetic asymmetry and cellular differentiation via differential regulation of tet activity in embryonic stem cells. Nucleic Acids Res (2018) 46:1210–26. doi: 10.1093/nar/gkx1197
112. O’Hagan HM, Wang W, Sen S, DeStefano Shields C, Lee SS, Zhang YW, et al. Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG islands. Cancer Cell (2011) 20:606–19. doi: 10.1016/j.ccr.2011.09.012
113. Maiuri AR, Peng M, Podicheti R, Sriramkumar S, Kamplain CM, Rusch DB, et al. Mismatch repair proteins initiate epigenetic alterations during inflammation-driven tumorigenesis. Cancer Res (2017) 77:3467–78. doi: 10.1158/0008-5472.CAN-17-0056
114. Ding N, Bonham EM, Hannon BE, Amick TR, Baylin SB, O’Hagan HM. Mismatch repair proteins recruit DNA methyltransferase 1 to sites of oxidative DNA damage. J Mol Cell Biol (2016) 8:244–54. doi: 10.1093/jmcb/mjv050
115. Ding N, Miller SA, Savant SS, O’Hagan HM. JAK2 regulates mismatch repair protein-mediated epigenetic alterations in response to oxidative damage. Environ Mol Mutagen (2019) 60:308–19. doi: 10.1002/em.22269
116. Ricketts CJ, Hill VK, Linehan WM. Tumor-specific hypermethylation of epigenetic biomarkers, including SFRP1, predicts for poorer survival in patients from the TCGA kidney renal clear cell carcinoma (KIRC) project. PloS One (2014) 9:e85621. doi: 10.1371/journal.pone.0085621
117. Li G, Xu W, Zhang L, Liu T, Jin G, Song J, et al. Development and validation of a CIMP-associated prognostic model for hepatocellular carcinoma. EBioMedicine (2019) 47:128–41. doi: 10.1016/j.ebiom.2019.08.064
118. Van der Auwera I, Yu W, Suo L, Van Neste L, van Dam P, Van Marck EA, et al. Array-based DNA methylation profiling for breast cancer subtype discrimination. PloS One (2010) 5:e12616. doi: 10.1371/journal.pone.0012616
119. Barreau O, Assié G, Wilmot-Roussel H, Ragazzon B, Baudry C, Perlemoine K, et al. Identification of a CpG island methylator phenotype in adrenocortical carcinomas. J Clin Endocrinol Metab (2013) 98:174–84. doi: 10.1210/jc.2012-2993
120. Park S-Y, Kook MC, Kim YW, Cho N-Y, Jung N, Kwon H-J, et al. CpG island hypermethylator phenotype in gastric carcinoma and its clinicopathological features. Virchows Arch (2010) 457:415–22. doi: 10.1007/s00428-010-0962-0
121. Chen HY, Zhu BH, Zhang CH, Yang DJ, Peng JJ, Chen JH, et al. High CpG island methylator phenotype is associated with lymph node metastasis and prognosis in gastric cancer. Cancer Sci (2012) 103:73–9. doi: 10.1111/j.1349-7006.2011.02129.x
122. Arai E, Chiku S, Mori T, Gotoh M, Nakagawa T, Fujimoto H, et al. Single-CpG-resolution methylome analysis identifies clinicopathologically aggressive CpG island methylator phenotype clear cell renal cell carcinomas. Carcinogenesis (2012) 33:1487–93. doi: 10.1093/carcin/bgs177
123. Cancer Genome Atlas Research Network, Linehan WM, Spellman PT, Rickeets CJ, Creighton CJ, Fei SS, et al. Comprehensive molecular characterization of papillary renal cell carcinoma. N Engl J Med (2016) 374:135–45. doi: 10.1056/NEJMoa1505917
124. Assié G, Letouzé E, Fassnacht M, Jouinot A, Luscap W, Barreau O, et al. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet (2014) 46:607–12. doi: 10.1038/ng.2953
125. Nishihara R, Wu K, Lochhead P, Morikawa T, Liao X, Qian ZR, et al. Long-term colorectal-cancer incidence and mortality after lower endoscopy. N Engl J Med (2013) 369:1095–105. doi: 10.1056/NEJMoa1301969
126. Radpour R, Barekati Z, Kohler C, Lv Q, Bürki N, Diesch C, et al. Hypermethylation of tumor suppressor genes involved in critical regulatory pathways for developing a blood-based test in breast cancer. PloS One (2011) 6:e16080. doi: 10.1371/journal.pone.0016080
127. Mur P, Rodríguez de Lope Á, Díaz-Crespo FJ, Hernández-Iglesias T, Ribalta T, Fiaño C, et al. Impact on prognosis of the regional distribution of MGMT methylation with respect to the CpG island methylator phenotype and age in glioma patients. J Neurooncol (2015) 122:441–50. doi: 10.1007/s11060-015-1738-9
128. de Souza CF, Sabedot TS, Malta TM, Stetson L, Morozova O, Sokolov A, et al. A distinct DNA methylation shift in a subset of glioma CpG island methylator phenotypes during tumor recurrence. Cell Rep (2018) 23:637–51. doi: 10.1016/j.celrep.2018.03.107
129. Zouridis H, Deng N, Ivanova T, Zhu Y, Wong B, Huang D, et al. Methylation subtypes and Large-scale epigenetic alterations in gastric cancer. Sci Transl Med (2012) 4:156ra140–156ra140. doi: 10.1126/scitranslmed.3004504
130. Bolouri H, Farrar JE, Triche T, Ries RE, Lim EL, Alonzo TA, et al. The molecular landscape of pediatric acute myeloid leukemia reveals recurrent structural alterations and age-specific mutational interactions. Nat Med (2018) 24:103–12. doi: 10.1038/nm.4439
131. Borssén M, Haider Z, Landfors M, Norén-Nyström U, Schmiegelow K, Åsberg AE, et al. DNA Methylation adds prognostic value to minimal residual disease status in pediatric T-cell acute lymphoblastic leukemia. Pediatr Blood Cancer (2016) 63:1185–92. doi: 10.1002/pbc.25958
132. Borssén M, Nordlund J, Haider Z, Landfors M, Larsson P, Kanerva J, et al. DNA Methylation holds prognostic information in relapsed precursor b-cell acute lymphoblastic leukemia. Clin Epigenet (2018) 10:1–7. doi: 10.1186/s13148-018-0466-3
133. Bediaga NG, Acha-Sagredo A, Guerra I, Viguri A, Albaina C, Ruiz Diaz I, et al. DNA Methylation epigenotypes in breast cancer molecular subtypes. Breast Cancer Res (2010) 12:R77. doi: 10.1186/bcr2721
134. Michigami Y, Watari J, Ito C, Nakai K, Yamasaki T, Kondo T, et al. Long-term effects of h. pylori eradication on epigenetic alterations related to gastric carcinogenesis. Sci Rep (2018) 8:14369. doi: 10.1038/s41598-018-32717-3
135. Yegnasubramanian S, Kowalski J, Gonzalgo ML, Zahurak M, Piantadosi S, Walsh PC, et al. Hypermethylation of CpG islands in primary and metastatic human prostate cancer. Cancer Res (2004) 64:1975–86. doi: 10.1158/0008-5472.CAN-03-3972
136. Berchuck JE, Baca SC, McClure HM, Korthauer K, Tsai HK, Nuzzo PV, et al. Detecting neuroendocrine prostate cancer through tissue-informed cell-free DNA methylation analysis. Clin Cancer Res (2022) 28:928–38. doi: 10.1158/1078-0432.CCR-21-3762
137. Dillinger T, Sheibani-Tezerji R, Pulverer W, Stelzer I, Hassler MR, Scheibelreiter J, et al. Identification of tumor tissue-derived DNA methylation biomarkers for the detection and therapy response evaluation of metastatic castration resistant prostate cancer in liquid biopsies. Mol Cancer (2022) 21:1–8. doi: 10.1186/s12943-021-01445-0
138. Wu A, Cremaschi P, Wetterskog D, Conteduca V, Franceschini GM, Kleftogiannis D, et al. Genome-wide plasma DNA methylation features of metastatic prostate cancer. J Clin Invest (2020) 130:1991–2000. doi: 10.1172/JCI130887
139. Guler GD, Ning Y, Ku C-J, Phillips T, McCarthy E, Ellison CK, et al. Detection of early stage pancreatic cancer using 5-hydroxymethylcytosine signatures in circulating cell free DNA. Nat Commun (2020) 11:5270. doi: 10.1038/s41467-020-18965-w
140. Pulliam N, Fang F, Ozes AR, Tang J, Adewuyi A, Keer H, et al. An effective epigenetic-PARP inhibitor combination therapy for breast and ovarian cancers independent of BRCA mutations. Clin Cancer Res (2018) 24:3163–75. doi: 10.1158/1078-0432.CCR-18-0204
141. Ateeq B, Unterberger A, Szyf M, Rabbani SA. Pharmacological inhibition of DNA methylation induces proinvasive and prometastatic genes. In Vitro In Vivo Neoplasia (2008) 10:266–78. doi: 10.1593/neo.07947
142. Chik F, Szyf M. Effects of specific DNMT gene depletion on cancer cell transformation and breast cancer cell invasion; toward selective DNMT inhibitors. Carcinogenesis (2011) 32:224–32. doi: 10.1093/carcin/bgq221
143. Festuccia C, Gravina GL, D’Alessandro AM, Muzi P, Milimaggi D, Dolo V, et al. Azacitidine improves antitumor effects of docetaxel and cisplatin in aggressive prostate cancer models. Endocr Relat Cancer (2009) 16:401–13. doi: 10.1677/ERC-08-0130
144. Singal R, Ramachandran K, Gordian E, Quintero C, Zhao W, Reis IM. Phase I/II study of azacitidine, docetaxel, and prednisone in patients with metastatic castration-resistant prostate cancer previously treated with docetaxel-based therapy. Clin Genitourin Cancer (2015) 13:22–31. doi: 10.1016/j.clgc.2014.07.008
Keywords: epigenetic regulation, DNA methylation, 5mC, CIMP, mCRPC
Citation: Shin HJ, Hua JT and Li H (2023) Recent advances in understanding DNA methylation of prostate cancer. Front. Oncol. 13:1182727. doi: 10.3389/fonc.2023.1182727
Received: 09 March 2023; Accepted: 24 April 2023;
Published: 10 May 2023.
Edited by:
Sifeng Qu, Shandong University, ChinaReviewed by:
Hongtao Xiao, University of Electronic Science and Technology of China, ChinaAnqi Li, The Ohio State University, United States
Copyright © 2023 Shin, Hua and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haolong Li, SGFvbG9uZy5MaUB1Y3NmLmVkdQ==