 Lasse Kjær1*
Lasse Kjær1* Vibe Skov1
Vibe Skov1 Morten Kranker Larsen1
Morten Kranker Larsen1 Tobias Idor Boklund2Morten Andersen2Maria Kefala3Trine A. Knudsen1Christina Schjellerup Eickhardt-Dalbøge1
Tobias Idor Boklund2Morten Andersen2Maria Kefala3Trine A. Knudsen1Christina Schjellerup Eickhardt-Dalbøge1 Thomas Stiehl2,4Johanne Gudmand-Høyer2
Thomas Stiehl2,4Johanne Gudmand-Høyer2 Jordan Snyder2
Jordan Snyder2 Morten Holmström5
Morten Holmström5 Mads H. Andersen5
Mads H. Andersen5 Johnny T. Ottesen2Christina Ellervik6
Johnny T. Ottesen2Christina Ellervik6 Hans C. Hasselbalch1
Hans C. Hasselbalch1- 1Department of Hematology, Zealand University Hospital, Roskilde, Denmark
- 2Centre for Mathematical Modeling - Human Health and Disease, IMFUFA, Department of Science and Environment, Roskilde University, Roskilde, Denmark
- 3Department of Surgical Pathology, Zealand University Hospital, Roskilde, Denmark
- 4Institute for Computational Biomedicine – Disease Modeling, RWTH Aachen University, Aachen, Germany
- 5National Center for Cancer Immune Therapy, Copenhagen University Hospital Herlev, Herlev, Denmark
- 6Department of Data and Data Support, Sorø, Region Zealand, Denmark
Initial diagnosis of overt myeloproliferative neoplasms (MPNs) represents the juncture during clonal evolution when symptoms or complications prompt an afflicted individual to seek medical attention. In 30-40% of the MPN subgroups essential thrombocythemia (ET) and myelofibrosis (MF), somatic mutations in the calreticulin gene (CALR) are drivers of the disease resulting in constitutive activation of the thrombopoietin receptor (MPL). In the current study, we describe a healthy CALR mutated individual during a 12 year follow-up from initial identification of CALR clonal hematopoiesis of indeterminate potential (CHIP) to the diagnosis of pre-MF. The pre-diagnostic exponential development dynamics of the malignant clone demonstrated close correlation with the platelet counts, neutrophil-to-lymphocyte (NLR) ratio, and inversely correlated to hemoglobin and erythrocyte counts. Backward extrapolation of the growth rate indicated the potential for discovery of the malignant clone many years prior to presentation of overt disease, opening a window of opportunity for early treatment intervention. We did not find any additional mutations associated with MPNs and the current case report provides novel information regarding the development of a driver mutation and the association with blood cell counts prior to clinical manifestation of symptoms suggesting that pre-diagnostic dynamics may supplement future diagnostic criteria for early diagnosis and intervention in MPN patients.
Introduction
The Philadelphia negative myeloproliferative neoplasms (MPNs) are clonally derived entities sub-grouped into essential thrombocythemia (ET), polycythemia vera (PV), and myelofibrosis (MF) according to their clinical features, genetic aberrations, and preferably - bone marrow morphology (1). The most prevalent somatic Janus kinase (JAK2) mutation - JAK2V617F - is the ‘driver’ of more than 95% of cases with PV, in addition to 50-60% of cases with ET and MF (2–4). Mutations in the calreticulin gene (CALR) are present in 80-90% of the JAK2V617F-negative ET and MF patients (5, 6) introducing a novel C-terminus enabling the protein to activate the thrombopoietin receptor (MPL) by homo-multimerization (7–9). The World Health Organization (WHO) has added clonal hematopoiesis (CH) to its classification system recognizing a precursor state for myeloid disease based on the entity clonal hematopoiesis of indeterminate potential (CHIP) where somatic mutations with a frequency of 2% or higher without other indications of hematological neoplasm, is identified as a pre-leukemic clone (10, 11). Clonal hematopoiesis is widely observed in the elderly and is furthermore associated with increased risk for cardiovascular disease (CVD) (12, 13). Alarmingly, we recently identified the MPN driver mutation JAK2V617F in 3% of the background population of which 42% resembled an early MPN phenotype even though the mutant allele burden in the vast majority was below 2% (14). Interestingly, the CALR type 1 and type 2 mutations appeared to be around 19 fold less frequent than JAK2V617F in the background population compared to the expected 5.6 fold difference estimated for MPNs. The CALR mutated individuals had a higher mean allele burden and accordingly 60% displayed the early MPN phenotype suggesting shorter latency for development of disease compared to the JAK2V617F mutation (5, 6, 14). In MPNs, clonal CALR mutations are considered an early event (5) and, compared to JAK2V617F, the clones have been suspected of a rapid growth rate as the proportion of diseased CALR positive individuals is higher, patients rarely present with a low mutant allele burden at the time of diagnosis, and are diagnosed at a younger age (14–17). The notion was recently further supported by a study investigating disease initiation and dynamics of clonal expansion by mathematical modeling (18). In MPNs, the dynamics of the mutant allele burden and the blood cell counts appear to be associated (19) but, to our knowledge, the association of the CALR dynamics with blood cell counts in the pre-MPN state has not been described by longitudinal studies so far.
The discrepancy between the prevalence of driver mutations in the background population and MPN diagnoses might involve the expected long latency from acquisition of the mutation until diagnosis (20). Increased death rate from thrombotic events during the pre-diagnostic phase because of late detection of developing MPNs would result in the disease eluding discovery suggesting a possible massive under-diagnosis of the disease (21, 22).
In this case report, we present for the first time a healthy CALR mutated individual with 12-year longitudinal follow-up until development of pre-fibrotic MPN, where early treatment could be initiated, demonstrating consistent exponential growth of the mutant clone independent of other somatic mutations associated with hematological neoplasms. The expansion rate of the clone suggests a narrow time window from diagnosis to full dominance of the mutated clone. Close correlation of pre-diagnostic mutant allele burden-and platelet count dynamics combined with backward extrapolation of the growth rate suggested the potential for discovery of the malignant clone many years prior to presentation of overt disease, opening a window of opportunity for early treatment intervention.
Case presentation
In 2017, a screening study for JAK2V617F and CALR mutations was performed on samples collected in 2010 for the Danish General Suburban Population Study (GESUS) – a cross sectional cohort of the Danish background population (14). Here, a CALR type 1 mutation was identified in a 63-year-old male, with a mutant allele burden in peripheral blood of 0.071%.
In 1976, at the age of 29 years he had a pacemaker implantation because of intermittent bradyarrhythmias, was diagnosed with essential hypertension in 2010, and hiatus hernia in 2012. In these years, the blood cell counts were normal (Table 1). The patient quit smoking in 1970 at the age of 23 but information on smoking pack years was not available. From the time of identification of the CALR mutation, until diagnosis of pre-MF, the patient experienced no MPN related symptoms other than the elevated platelet counts, including absence of splenomegaly. Before the time of diagnosis, the patient was treated for elevated blood pressure form June 2021 with 5 mg Amlopidin daily, which was changed in January 2022 to 100 mg Losartan daily. The patient furthermore received 0.1% Mometason cream for psoriasis in February 2022 and 500 mg Metformin daily for diabetes in November 2022. At the end of November 2022, the patient was treated with 45 µg/week pegylated interferon-alfa-2a.
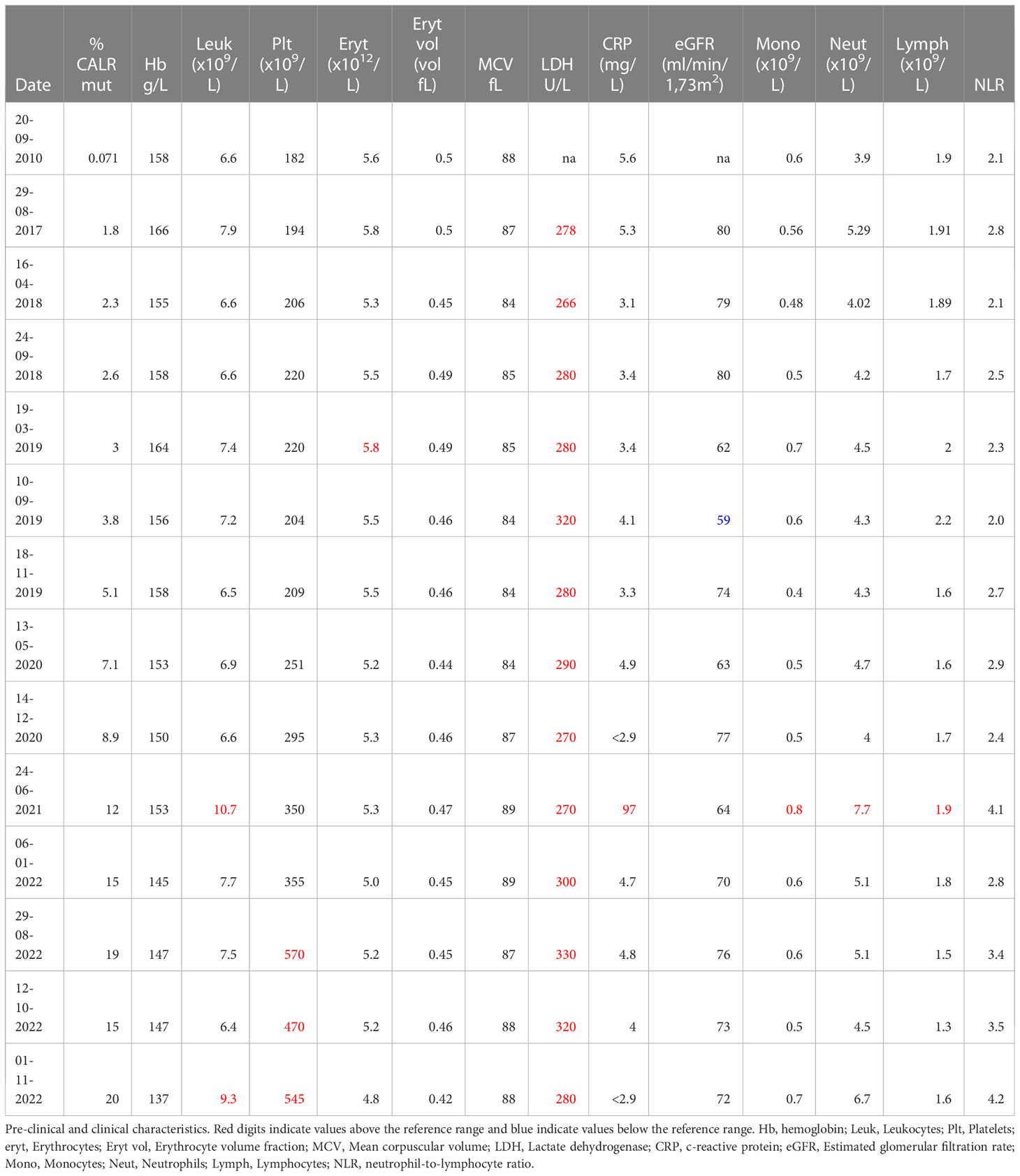
Table 1 Pre-clinical and clinical characteristics.
Subsequent to identification of the mutation in 2017 - seven years after the initial GESUS blood sample - follow-up samples and a bone marrow biopsy analysis were performed on the patient who was 70 years old at the time. The blood cell counts and bone marrow biopsy were normal although, when reviewed, early MPN features could be discerned (Figures 1A, C). Plasma lactate dehydrogenase (LDH) was slightly elevated, and qPCR determined that the CALR allele burden had increased to 1.8% (19). Thereafter, the patient was followed twice yearly with routine blood tests and a CALR mutation analysis. In 2018, the CALR mutant allele burden had increased to 2.3% and elevated plasma LDH remained the only abnormal blood parameter (Table 1), but at this point, the exponential growth of the mutated clone became evident (Figure 2A). The normal blood parameters remained within the reference range until October 2019. The Leukocyte count was briefly elevated in 2021 and at the end of 2022. In August 2022, the platelet count had increased to the diagnostic criteria for MPN and the mutant allele burden was 15-19%. A bone marrow biopsy was performed in November showing hyperplastic granulopoiesis and hyperplastic megakaryopoiesis with dense and loose small clusters of megakaryocytes demonstrating clear myeloproliferative features. This included forms with hyper-lobulated, as well as bulbous and hyperchromatic, nuclei (Figure 1B). In addition, silver staining revealed focally lightly accentuated reticulin fibrosis grade 0-1 reminiscent of an early pre-fibrotic myelofibrosis (Figure 1D). Although within the reference range, the platelet count clearly increased alongside the mutant allele burden demonstrating significant correlation (Pearson: R2 = 0.9259, P<0.0001) (Figures 2A, 3A). Interestingly, the platelet count dynamics resembled the mutant allele burden dynamics several years prior to diagnosis with an initial increase in platelet count around 2% mutated alleles that reached a plateau phase and subsequently decreased, suggesting an intrinsic regulation of the platelet number. However, this appeared to be overwhelmed when the mutant allele burden reached 5% and platelets increased, exceeding the reference range when the mutant allele burden was above 15% (Figure 2A). Fitting the exponential expression , where is the time measured in years after the first measurement, to the CALR allele burden (using the fit function in MATLAB’s curve fitting toolbox, version R2021b) results in (95% CI ) and, (95% CI , with , corresponding to a doubling time of (95% CI ) and a yearly growth rate of (95% CI ). Assuming the assay for determination of mutant allele burden is performed in duplicate with 150 ng DNA (2 x 150 ng DNA containing 100.000 copies of CALR), and that the assay has the ability to detect 3 mutated copies, the detection limit will correspond to approximately 0.003% mutated alleles. To determine the ‘window of opportunity’ for detection of the mutation, we assume that the exponential fit also describes the growth of the mutated clone prior to the first measurement, and we extrapolate the exponential fit backwards in time to the detection limit of the assay (0.003%) (14). Solving the equation , we find , i.e. that the mutation was detectable (95% CI based on the 95% CI of ) before the first measurement in 2010, thus enabling detection (95% CI prior to the MPN diagnosis in August 2022. In addition, assuming that the allele burden in the peripheral blood equals the allele burden in the stem cell compartment (or at least that the time delay between the mutation happening at stem cell level and subsequently appearing in the peripheral blood is negligible on a time scale of years), and neglecting stochasticity, stem cell-specific regulations, and anti-cancer immunity, backward idealized extrapolation can be used to estimate the time of the initial mutation. If we assume that the number of stem cells is and that the mutation is heterozygous as it normally is for CALR mutations, then the estimated time of mutation from extrapolation can be found by solving the equation , where the factor of 100% converts the fraction of cells to an allele burden measured as a percentage, and the factor of is due to the heterozygosity. Different sources have different estimates for the number of hematopoietic stem cells in a human, and therefore we have calculated the estimated time of mutation for a wide range of the assumed number of stem cells (Figure 2B). A heterozygous mutation in one out of N = stem cells (23) (corresponding to an allele burden of ) is estimated to have occurred (95% CI based on the 95% CI of ) before the first measurement, and a heterozygous mutation in one out of N = stem cells (24) (corresponding to an allele burden of ) is estimated to have occurred (95% CI based on the 95% CI of ) before the first measurement, i.e. somewhere between the age of 45 and 53 for the patient based on the assumed number of stem cells (Figure 2B).
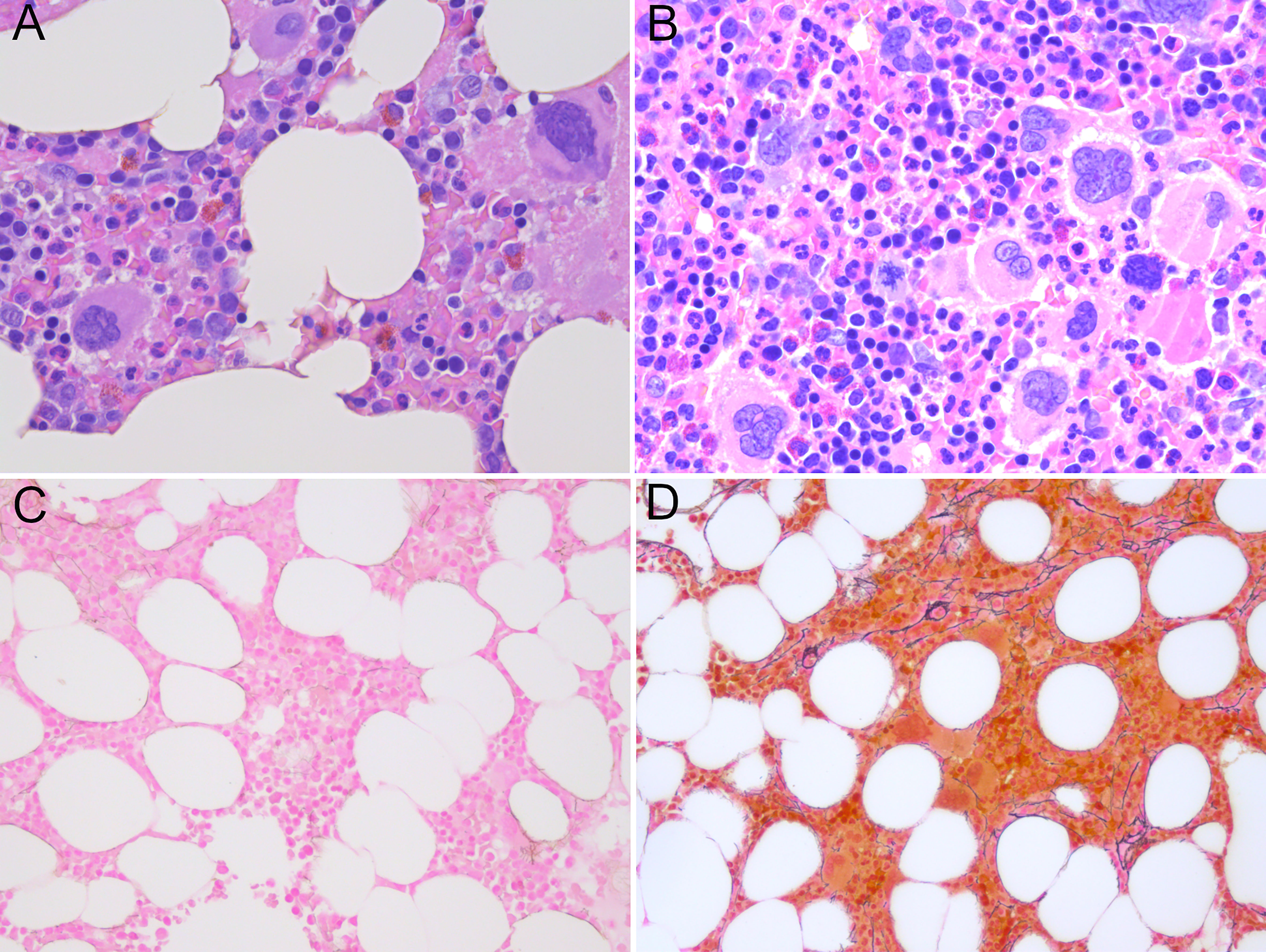
Figure 1 (A) Normocellular bone marrow not diagnostic for myeloproliferative neoplasia, only a few megakaryocytes with myeloproliferative features in sample from 2017. (B) Hypercellular bone marrow with clusters of atypical megakaryocytes with myeloproliferative features without fibrosis in sample from 2022. (C) Reticulin staining in sample from 2017 reflecting a normal bone marrow. (D) Loose networks of reticulin staining grade 0-1 in bone marrow sample from 2022. (A, B) Hematoxylin and eosin staining; (C, D) Reticulin staining; magnification x400).
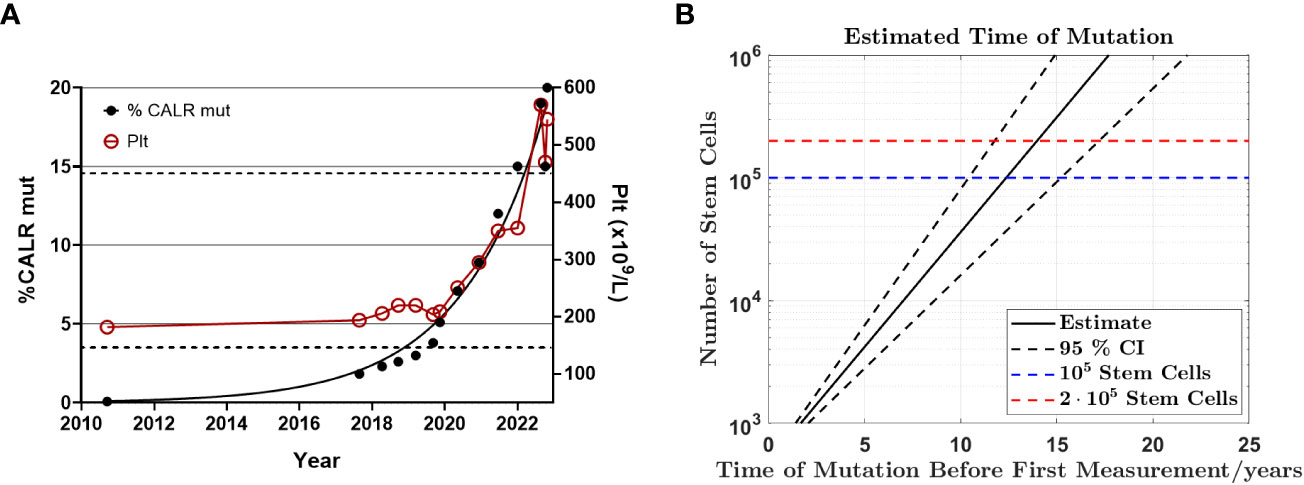
Figure 2 Exponential growth of CALR mutant allele burden from CH to MPN (A) Graph illustrating the mutant allele burden of CALR type 1 from September 2010 to November 2022 demonstrating exponential growth of the mutant allele burden and the platelet count. Note the close correlation between the CALR mutant allele burden and the platelet count once it exceeds 5%, including the transient drop even from 19% mutant alleles to 15% indicating a biological phenomenon rather than a technical discrepancy. The left Y-axis indicates the mutant allele burden, where black dots indicate CALR data points. The right Y-axis describes the number of platelets/µl and the red circles measurements of platelets. The dashed lines indicate the upper and lower boundary for the reference range of platelet count. (B) Backward extrapolation used to estimate when the mutation occurred. A heterozygous mutation in 1 out of 100.000 (dashed blue line) or 200.000 (dashed red line) stem cells.
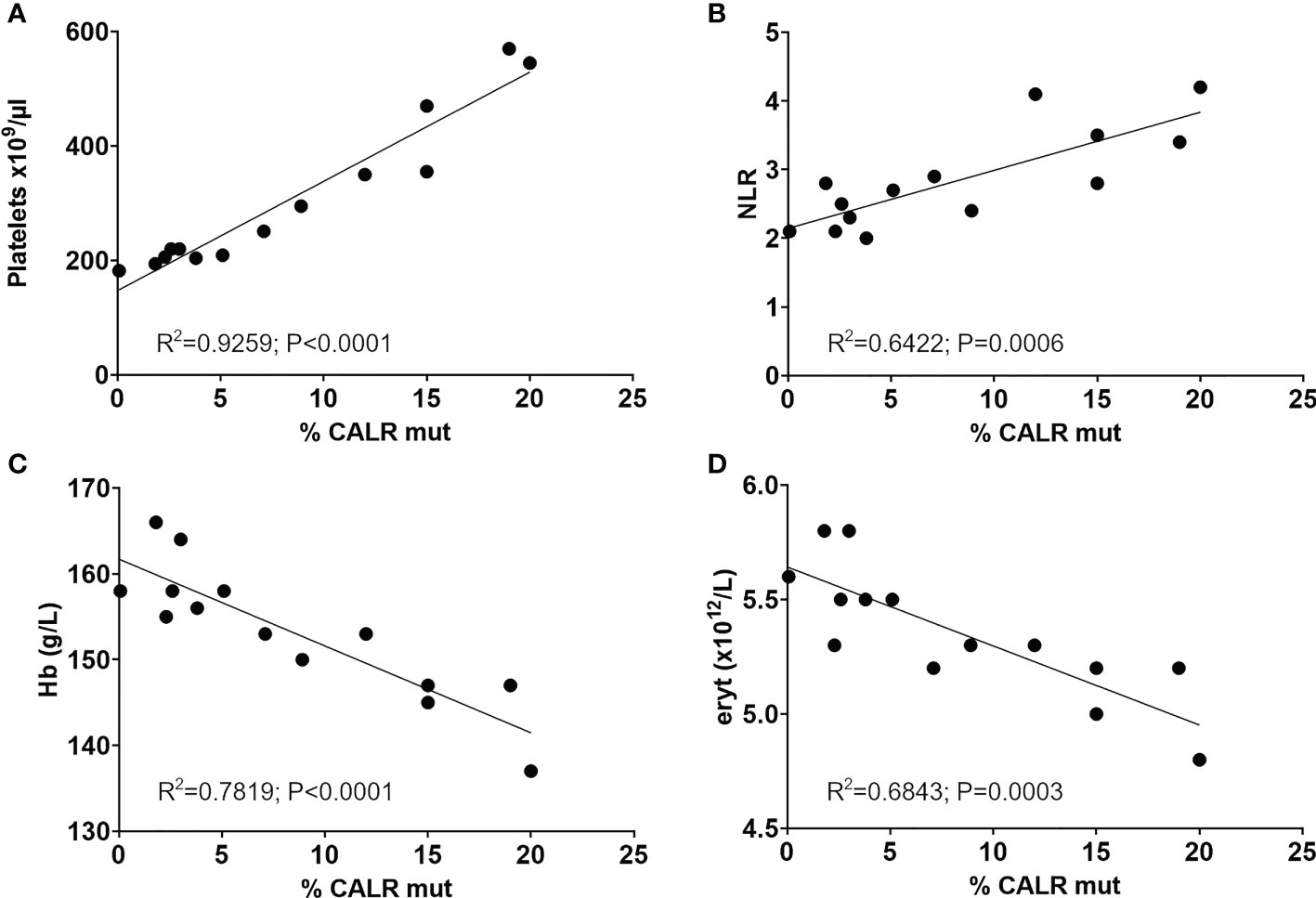
Figure 3 Significant correlation of longitudinal CALR mutant allele burden with (A) platelet counts, (B) the neutrophil-to-lymphocyte ratio, (C) hemoglobin and (D) erythrocyte count. R squared and p values from Person correlation are shown in graphs. NLR, Neutrophil-to-lymphocyte ratio; Hb, Hemoglobin; eryt, Erythrocyte count.
The mutant allele burden demonstrated a significant correlation with the neutrophil-to-lymphocyte ratio (NLR) (Pearson: R2 = 0.6422, P=0.0006) (Figure 3B). An inverse correlation was observed between the mutant allele burden and both hemoglobin (Hb) (Pearson: R2 = 0.7819, P<0.0001) and erythrocytes (Pearson: R2 = 0.6843, P=0.0003) (Figures 3C, D). There was no correlation between mutant allele burden and monocytes, leucocytes, plasma LDH, or renal function as assessed by eGFR (data not shown). Chromosome analysis revealed loss of the Y-chromosome in single metaphases as the only cytogenetic aberration. No additional mutations were detected in the last available peripheral blood sample by next generation sequencing (NGS) analysis of peripheral blood using the custom panel of 42 genes implicated in myeloid malignancies described by Grauslund et al. (25, 26).
Discussion
To our knowledge, this is the first observation of a CALR mutation detected at a very low mutant allele burden in an individual longitudinally monitored over a 12-year period prior to developing pre-fibrotic myelofibrosis. Absence of known co-mutations involved in myeloid malignancies suggested that CALR was the only genetic cause of the expansion.
Recently, a study investigated the longitudinal dynamics of clonal hematopoiesis suggesting that the large majority of clones expanded at a stable exponential rate with average annual growth rates ranging from 5% to 50% per year (24). The CALR growth rate of approximately 54%, in the current study, exceeds the rate of expansion for the fastest clones such as SRSF2P95H and mutant U2AF1 (24). Initial longitudinal observations of slow exponential growth for JAK2V617F mutated clones suggested complex growth dynamics (27) confirmed by a wide range of clonal expansion rates increasing with additional mutations (12). The growth dynamics for CALR remain to be studied in larger cohorts, but at least for the patient in the current study, the rapid growth rate appears to be independent of additional mutations in genes typically associated with hematological cancers and may involve other factors such as mutated calreticulin acting as a rogue cytokine (28). The JAK2V617F mutation is acquired early in life (29, 30) and even in utero (20, 31). Considering the estimated midlife CALR acquisition estimated both by mathematical modeling (18) and the current study, the reported in utero origin of CALR, recently confirmed in a set of monozygotic twins (32), suggests that the growth dynamics for CALR may be as irregular as seen for JAK2V617F (20). Assessing clonal growth rates requires a set of assumptions. For predicting the time of mutant acquisition of DNMT3A variants (20), the assumption that the clones expanded at a steady rate meant that the mutations initially appeared to have occurred several years before birth. However, the underlying age dependent deceleration in growth rate for DNMT3A clones was not observed for clones with variants in TET2 (20) highlighting the importance of longitudinal monitoring of clone expansion. In the current study, we make necessity assumptions when neglecting stochasticity, stem cell specific regulations, and anti-cancer immunity when determining the time of mutation, but we are reasonably confident about our assumptions as for the current patient the growth rate appears constant over the entire 12-year period and thus we simplify the potentially complex growth of the clone. However, we acknowledge that this may not necessarily reflect the CALR development in other patients. Of note, the bone marrow biopsy appeared normal 5 years prior to the MPN diagnosis when the mutant allele burden was around 2%, although with rare MPN features. Prominent MPN features were present when the allele burden had exceeded 15% corresponding to 30% of nucleated cells as CALR mutated individuals rarely present with dominant homozygous clones (15). The estimated doubling time of the CALR clone of 1.61 years suggests that the vast majority of nucleated cells in the blood will consist of the mutated clone 1-2 years after initial clinical presentation, although differences between cell lineages are expected (33).
We have previously observed a close association between the dynamics of the blood cell counts and the mutant allele burden in CALR mutated MPN patients (19). In the current patient, the dynamics of the platelet count bore a striking resemblance to the mutant allele burden and indicated disease development even when parameters were still within the reference interval. Consequently, such pre-diagnostic alterations might supplement future diagnostic criteria, enabling an even earlier MPN -diagnosis prior to potential thrombotic events.
The MPNs are driving – and driven by – inflammation, reflected in the positive correlation between the CALR mutant allele burden and the chronic inflammation proxy: NLR, in this case study. This supports findings of increased NLR in CALR positive individuals compared to mutation negative and further suggests persistent correlation over several years during development of the mutated clone (34). We did not observe the previously reported negative associations with eGFR suggesting either a mild impairment, not yet reflected by eGFR, or the presence of additional factors having impact on kidney function.
Although CH is recognized as a clinical entity associated with increased overall mortality, CVD and myeloid disease, treatment in the absence of a hematologic disorder according to current guidelines is a controversial subject. Clonal hematopoiesis is not a single entity but is associated with a wide variety of mutations some clearly representing a critical precancerous state – especially MPN driver mutations in JAK2, CALR, and MPL genes require extra scrutiny (7). It is becoming increasingly evident that the MPN diagnosis only reflects the juncture during the clonal evolution where symptoms or complications prompts an afflicted individual to seek medical attention (35). Here, we detected a CALR mutation many years prior to clinical manifestation and were able to initiate early intervention and treatment at the very instant the criteria for the MPN diagnosis were achieved, prior to potential thrombotic – and thromboembolic complications. Recently, eradication of a double mutant-driven pre-leukemic clone prevented leukemic onset in a mouse model (36) proving proof of principle for the elimination of the malignant clone prior to overt disease and extending the concept of early intervention in MPN patients (37). Transition from ET to post-ET MF is associated with increased mutant load of CALR (38) providing further incentive for reducing the mutant allele burden, keeping in mind that its reduction is much more difficult and time-consuming when the mutated fraction is high, exposing the patient to unnecessary complications. It is likely that there will be a wide variety of developmental trajectories for CALR mutated clones depending on intrinsic and extrinsic factors where some carriers may never develop MPN. In the current case, presence of the mutation in 30% of circulating nucleated cells at clinical manifestation, and the observed growth rate of the clone, suggested a very short time-period from MPN diagnosis until eclipse of the normal cells. Considering the aggressive expansion, the early intervention may have prevented thromboembolic events, but understanding risk stratification requires further studies on how the development of CALR clones associate with clinical events. We need to start the discussion on how to handle the driver mutation CHIP patients with either dormant clones only requiring regular monitoring or rapid clonal expansions prompting ‘preventive’ medicine interventions such as aspirin to reduce the risk of thromboembolic events or even attempting to deplete the malignant clone by interferon treatment (19, 39). Therefore, the current case report highlights the urgent matter of re-considering early intervention and disease prevention when driver mutation and the dynamics of the blood cell counts, even within the reference interval, suggest progression.
Data availability statement
The original contributions presented in the study are included in the article/supplementary material. Further inquiries can be directed to the corresponding author.
Ethics statement
The study was approved by the Regional Committee on Health Research Ethics and the Danish Data Protection agency. The patients/participants provided their written informed consent to participate in this study. Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
Author contributions
LK, VS, ML, TK, CS, MH, MA, and HH conceived and designed the study. HH and CE managed the patient. MK carried out the pathological analysis. VS carried out the NS analysis. TB, MA, TS, JG, JA and JO did the mathematical calculations. LK wrote the manuscript. All authors contributed to the article and approved the submitted version.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the world health organization classification of myeloid neoplasms and acute leukemia. Blood (2016) 127(20):2391–405. doi: 10.1182/blood-2016-03-643544
2. Baxter EJ, Scott LM, Campbell PJ, East C, Fourouclas N, Swanton S, et al. Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet (2005) 365(9464):1054–61. doi: 10.1016/S0140-6736(05)71142-9
3. Levine RL, Wadleigh M, Cools J, Ebert BL, Wernig G, Huntly BJ, et al. Activating mutation in the tyrosine kinase JAK2 in polycythemia vera, essential thrombocythemia, and myeloid metaplasia with myelofibrosis. Cancer Cell (2005) 7(4):387–97. doi: 10.1016/j.ccr.2005.03.023
4. James C, Ugo V, Le Couedic JP, Staerk J, Delhommeau F, Lacout C, et al. A unique clonal JAK2 mutation leading to constitutive signalling causes polycythaemia vera. Nature (2005) 434(7037):1144–8. doi: 10.1038/nature03546
5. Klampfl T, Gisslinger H, Harutyunyan AS, Nivarthi H, Rumi E, Milosevic JD, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med (2013) 369(25):2379–90. doi: 10.1056/NEJMoa1311347
6. Nangalia J, Massie CE, Baxter EJ, Nice FL, Gundem G, Wedge DC, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med (2013) 369(25):2391–405. doi: 10.1056/NEJMoa1312542
7. Kjaer L. Clonal hematopoiesis and mutations of myeloproliferative neoplasms. Cancers (Basel) (2020) 12(8):2100. doi: 10.3390/cancers12082100
8. Araki M, Yang Y, Masubuchi N, Hironaka Y, Takei H, Morishita S, et al. Activation of the thrombopoietin receptor by mutant calreticulin in CALR-mutant myeloproliferative neoplasms. Blood (2016) 127(10):1307–16. doi: 10.1182/blood-2015-09-671172
9. Chachoua I, Pecquet C, El-Khoury M, Nivarthi H, Albu RI, Marty C, et al. Thrombopoietin receptor activation by myeloproliferative neoplasm associated calreticulin mutants. Blood (2016) 127(10):1325–35. doi: 10.1182/blood-2015-11-681932
10. Khoury JD, Solary E, Abla O, Akkari Y, Alaggio R, Apperley JF, et al. The 5th edition of the world health organization classification of haematolymphoid tumours: myeloid and Histiocytic/Dendritic neoplasms. Leukemia (2022) 36(7):1703–19. doi: 10.1038/s41375-022-01613-1
11. Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood (2015) 126(1):9–16. doi: 10.1182/blood-2015-03-631747
12. Jaiswal S, Fontanillas P, Flannick J, Manning A, Grauman PV, Mar BG, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med (2014) 371(26):2488–98. doi: 10.1056/NEJMoa1408617
13. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, et al. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med (2017) 377(2):111–21. doi: 10.1056/NEJMoa1701719
14. Cordua S, Kjaer L, Skov V, Pallisgaard N, Hasselbalch HC, Ellervik C. Prevalence and phenotypes of JAK2 V617F and calreticulin mutations in a Danish general population. Blood (2019) 134(5):469–79. doi: 10.1182/blood.2019001113
15. Jeromin S, Kohlmann A, Meggendorfer M, Schindela S, Perglerova K, Nadarajah N, et al. Next-generation deep-sequencing detects multiple clones of CALR mutations in patients with BCR-ABL1 negative MPN. Leukemia (2016) 30:973–976. doi: 10.1038/leu.2015.207
16. Tefferi A, Thiele J, Vannucchi AM, Barbui T. An overview on CALR and CSF3R mutations and a proposal for revision of WHO diagnostic criteria for myeloproliferative neoplasms. Leukemia (2014) 28(7):1407–13. doi: 10.1038/leu.2014.35
17. Rumi E, Pietra D, Ferretti V, Klampfl T, Harutyunyan AS, Milosevic JD, et al. JAK2 or CALR mutation status defines subtypes of essential thrombocythemia with substantially different clinical course and outcomes. Blood (2014) 123(10):1544–51. doi: 10.1182/blood-2013-11-539098
18. Hermange G, Rakotonirainy A, Bentriou M, Tisserand A, El-Khoury M, Girodon F, et al. Inferring the initiation and development of myeloproliferative neoplasms. Proc Natl Acad Sci U S A (2022) 119(37):e2120374119. doi: 10.1073/pnas.2120374119
19. Kjaer L, Cordua S, Holmstrom MO, Thomassen M, Kruse TA, Pallisgaard N, et al. Differential dynamics of CALR mutant allele burden in myeloproliferative neoplasms during interferon Alfa treatment. PLoS One (2016) 11(10):e0165336. doi: 10.1371/journal.pone.0165336
20. Williams N, Lee J, Mitchell E, Moore L, Baxter EJ, Hewinson J, et al. Life histories of myeloproliferative neoplasms inferred from phylogenies. Nature (2022) 602(7895):162–8. doi: 10.1038/s41586-021-04312-6
21. Cordua S, Kjaer L, Skov V, Pallisgaard N, Kefala M, Gjerdrum LMR, et al. Early detection of myeloproliferative neoplasms in a Danish general population study. Leukemia (2021) 35:2706–2709. doi: 10.1038/s41375-021-01159-8
22. Hasselbalch HC, Elvers M, Schafer AI. The pathobiology of thrombosis, microvascular disease, and hemorrhage in the myeloproliferative neoplasms. Blood (2021) 137(16):2152–60. doi: 10.1182/blood.2020008109
23. Lee-Six H, Obro NF, Shepherd MS, Grossmann S, Dawson K, Belmonte M, et al. Population dynamics of normal human blood inferred from somatic mutations. Nature (2018) 561(7724):473–8. doi: 10.1038/s41586-018-0497-0
24. Fabre MA, de Almeida JG, Fiorillo E, Mitchell E, Damaskou A, Rak J, et al. The longitudinal dynamics and natural history of clonal haematopoiesis. Nature (2022) 606(7913):335–42. doi: 10.1038/s41586-022-04785-z
25. Handlos Grauslund J, Holmstrom MO, Jorgensen NG, Klausen U, Weis-Banke SE, El Fassi D, et al. Therapeutic cancer vaccination with a peptide derived from the calreticulin exon 9 mutations induces strong cellular immune responses in patients with CALR-mutant chronic myeloproliferative neoplasms. Front Oncol (2021) 11:637420. doi: 10.3389/fonc.2021.637420
26. Skov V. Next generation sequencing in MPNs. lessons from the past and prospects for use as predictors of prognosis and treatment responses. Cancers (Basel) (2020) 12(8):2194. doi: 10.3390/cancers12082194
27. Nielsen C, Bojesen SE, Nordestgaard BG, Kofoed KF, Birgens HS. JAK2V617F somatic mutation in the general population: myeloproliferative neoplasm development and progression rate. Haematologica (2014) 99(9):1448–55. doi: 10.3324/haematol.2014.107631
28. Pecquet C, Papadopoulos N, Balligand T, Chachoua I, Tisserand A, Vertenoeil G, et al. Secreted mutant calreticulins as rogue cytokines in myeloproliferative neoplasms. Blood (2023) 141(8):917–29. doi: 10.1182/blood.2022016846
29. Van Egeren D, Escabi J, Nguyen M, Liu S, Reilly CR, Patel S, et al. Reconstructing the lineage histories and differentiation trajectories of individual cancer cells in myeloproliferative neoplasms. Cell Stem Cell (2021) 28(3):514–23.e9. doi: 10.1016/j.stem.2021.02.001
30. McKerrell T, Park N, Chi J, Collord G, Moreno T, Ponstingl H, et al. JAK2 V617F hematopoietic clones are present several years prior to MPN diagnosis and follow different expansion kinetics. Blood Adv (2017) 1(14):968–71. doi: 10.1182/bloodadvances.2017007047
31. Hirsch P, Mamez AC, Belhocine R, Lapusan S, Tang R, Suner L, et al. Clonal history of a cord blood donor cell leukemia with prenatal somatic JAK2 V617F mutation. Leukemia (2016) 30(8):1756–9. doi: 10.1038/leu.2016.31
32. Sousos N, Ni Leathlobhair M, Simoglou Karali C, Louka E, Bienz N, Royston D, et al. In utero Origin of myelofibrosis presenting in adult monozygotic twins. Nat Med (2022) 28(6):1207–11. doi: 10.1038/s41591-022-01793-4
33. Kjaer L, Holmstrom MO, Cordua S, Andersen MH, Svane IM, Thomassen M, et al. Sorted peripheral blood cells identify CALR mutations in b- and T-lymphocytes. Leuk Lymphoma (2018) 59(4):973–7. doi: 10.1080/10428194.2017.1359743
34. Larsen MK, Skov V, Kjaer L, Moller-Palacino NA, Pedersen RK, Andersen M, et al. Clonal haematopoiesis of indeterminate potential and impaired kidney function-a Danish general population study with 11 years follow-up. Eur J Haematol (2022) 109(5):576–85. doi: 10.1111/ejh.13845
35. Frederiksen H, Farkas DK, Christiansen CF, Larsen TS, Hasselbalch HC, Stentoft J, et al. Survival of patients with chronic myeloproliferative neoplasms and new primary cancers: a population-based cohort study. Lancet Haematol (2015) 2(7):e289–96. doi: 10.1016/S2352-3026(15)00092-7
36. Uckelmann HJ, Kim SM, Wong EM, Hatton C, Giovinazzo H, Gadrey JY, et al. Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science (2020) 367(6477):586–90. doi: 10.1126/science.aax5863
37. Hasselbalch HC, Silver RT. New perspectives of interferon-alpha2 and inflammation in treating Philadelphia-negative chronic myeloproliferative neoplasms. Hemasphere (2021) 5(12):e645. doi: 10.1097/HS9.0000000000000645
38. Andrikovics H, Krahling T, Balassa K, Halm G, Bors A, Koszarska M, et al. Distinct clinical characteristics of myeloproliferative neoplasms with calreticulin mutations. Haematologica (2014) 99(7):1184–90. doi: 10.3324/haematol.2014.107482
Keywords: case report, MPN, CALR, clonal hematopoiesis, myelofibrosis
Citation: Kjær L, Skov V, Larsen MK, Boklund TI, Andersen M, Kefala M, Knudsen TA, Schjellerup Eickhardt-Dalbøge C, Stiehl T, Gudmand-Høyer J, Snyder J, Holmström M, Andersen MH, Ottesen JT, Ellervik C and Hasselbalch HC (2023) Case Report: First longitudinal study of a patient with CALR positive clonal hematopoiesis of indeterminate potential developing into pre-fibrotic myelofibrosis. Front. Oncol. 13:1176173. doi: 10.3389/fonc.2023.1176173
Received: 28 February 2023; Accepted: 11 April 2023;
Published: 08 May 2023.
Edited by:
Raajit Rampal, Memorial Sloan Kettering Cancer Center, United StatesReviewed by:
Ann Mullally, Harvard Medical School, United StatesGonzalo Carreño-Tarragona, University Hospital October 12, Spain
Copyright © 2023 Kjær, Skov, Larsen, Boklund, Andersen, Kefala, Knudsen, Schjellerup Eickhardt-Dalbøge, Stiehl, Gudmand-Høyer, Snyder, Holmström, Andersen, Ottesen, Ellervik and Hasselbalch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lasse Kjær, bGFza2pAcmVnaW9uc2phZWxsYW5kLmRr